Abstract
Acellular pertussis (aP) vaccines have markedly reduced the global burden of severe pertussis. However, their limited ability to elicit mucosal and durable immunity has been linked to waning protection and sustained Bordetella pertussis circulation. Selective pressure exerted by widespread aP vaccination has contributed to the emergence and regional dissemination of pertactin-deficient (PRN−) strains, raising additional concerns regarding vaccine effectiveness. In this context, we investigated whether incorporating outer membrane vesicles (OMVs) derived from B. pertussis into the aP vaccine could enhance its immunological profile, specifically by promoting Th1/Th17 polarization, inducing tissue-resident memory (TRM) T cells, and broadening protective coverage to include PRN− isolates, while maintaining aP-induced immunity against lower respiratory tract colonization. Using a murine intranasal challenge model with a two-dose vaccination schedule, we assessed the safety, immunogenicity, and protective capacity of the OMV+aP vaccine prototype (combined) versus aP vaccine. The combined formulation was well tolerated and induced robust systemic and mucosal responses, characterized by higher IgG2a/IgG1 ratios, increased Th1/Th17 cytokine production (IFN-γ, IL - 17, and IL - 22), and elevated anti-B. pertussis IgA titers. Flow cytometric analyses revealed lung- and nasal-resident CD4+ TRM cells in the combined immunized mice, which were absent in those receiving aP alone. Functionally, OMV+aP formulation conferred superior protection in pulmonary and nasal compartments, significantly reducing lung bacterial loads (including against PRN− strains) and uniquely diminishing nasal colonization even under high-dose challenge conditions. Passive transfer experiments confirmed the role of cellular and humoral immunity in bacterial clearance. These results demonstrate that OMVs synergize with aP to enhance immune response magnitude and quality, addressing key gaps in current aP vaccines and offering a next-generation strategy to prevent both disease and transmission.
Introduction
Pertussis, a highly contagious respiratory disease caused by the Gram-negative bacterium Bordetella pertussis, remains a persistent and complex global health challenge, despite the availability of vaccines for over seven decades (1–4). While the disease can affect individuals of all ages, it is particularly severe in unvaccinated or under-vaccinated infants, often leading to hospitalization and, in many cases, death. Clinically characterized by paroxysmal coughing, cyanosis, and episodes of apnea, pertussis continues to impose a substantial burden on pediatric healthcare systems worldwide, including an increasing impact among older children and adolescents (5–10).
Global vaccination efforts using whole-cell (wP) and acellular (aP) pertussis vaccines have significantly reduced childhood morbidity and mortality. However, the resurgence of pertussis in recent decades, even in countries with consistently high aP vaccine coverage, such as the United States, the United Kingdom, and Australia, has raised major concerns about the effectiveness of current immunization strategies (1, 11, 12). Investigations into the causes of this resurgence suggest it is multifactorial, involving waning immunity (13, 14), pathogen evolution toward vaccine-resistant strains (15–20), particularly in the context of aP-induced immunity, and the limited capacity of existing vaccines to prevent nasopharyngeal colonization (21).
The COVID - 19 pandemic further exacerbated this epidemiologic situation by disrupting global immunization programs and eroding public trust in vaccines, contributing to dangerous declines in pertussis vaccine coverage (22, 23). According to WHO/UNICEF estimates, global coverage with the third dose of DTP (which includes pertussis) dropped from 86% in 2019 to 81% in 2021, with only partial recovery to 84% by 2023. Furthermore, the number of “zero-dose” children, those who received no vaccines at all, rose to 14.5 million in 2023, up from 12.9 million in 2019, with the highest impact observed in low- and middle-income countries. These trends represent a serious threat to global pertussis control and underscore the urgent need for more effective vaccines that provide durable immunity and enable simplified vaccination schedules (24–26).
Current pertussis vaccines include either whole-cell formulations, more reactogenic but immunologically robust, inducing mainly Th1/Th17 responses, or acellular formulations, which are safer but generate weaker and less durable immunity, skewed toward a Th2 profile (14, 24, 27, 28). Notably, aP vaccines are particularly ineffective at preventing nasal colonization, thereby allowing asymptomatic transmission (21). Moreover, the emergence and spread of pertactin-deficient (PRN−) B. pertussis strains prevalent in several region has further compromised aP vaccine effectiveness, as PRN remains a key antigen in most aP formulations (16, 29–31).
In response to these limitations, new strategies are being developed. One promising approach, which we explore in this study, involves the use of outer membrane vesicles (OMVs) derived from B. pertussis. OMVs naturally contain a wide array of immunogenic proteins and pathogen-associated molecular patterns (PAMPs) (32–35). Preclinical studies have shown that OMVs can elicit strong Th1/Th17 immune responses, significantly reduce bacterial loads in the lungs, and display lower reactogenicity compared to wP vaccines (31, 36–47). Intranasal administration has also been shown to reduce colonization in the upper respiratory tract (46, 48). In addition, OMVs possess intrinsic adjuvant properties, enhancing the immunogenicity of co-administered antigens and promoting a more protective immune profile (49, 50).
In this study, we evaluated the combination of OMVs with the commercial aP vaccine to assess whether this strategy could address the limitations of current aP vaccines and, importantly, facilitate future clinical evaluation through non-inferiority trials, given that the combined formulation includes components already licensed for human use. Specifically, we assessed whether the combined formulation could generate a more robust humoral immune response, modulate the aP-induced immune profile toward a protective Th1/Th17 bias, promote the induction of CD4+ TRM cell population, and enhance mucosal protection, including against nasal colonization. We also explored whether this approach could improve protection against strains more resistant to aP-induced immunity. To further characterize the quality of the induced immunity, we investigated immune cell memory recall using adoptive transfer models.
These efforts aim to provide a strong scientific foundation for the development of next-generation pertussis vaccines capable of inducing long-lasting immunity and restoring effective population-level protection.
Materials and methods
B. pertussis strains and growth conditions
B. pertussis gentamicin resistant strains used for challenge in the murine protection model included the reference Tohama phase I strain (CIP 8132, ptxP1-ptxA2-prn1), which expresses the adhesin pertactin (PRN), as well as clinical isolates (ptxP3-ptxA1-prn2) from the United States and Argentina that do not express the vaccine antigen pertactin (51–53). Bacteria were grown in Bordet–Gengou (BG) agar supplemented with 10% (v/v) defibrinated sheep blood (BGAS) and 50 μg/mL of gentamicin (BGAS-Gn)for 72 h at 36.5°C. Isolated colonies were replated in the same medium for 24 h and then resuspended in phosphate-buffered saline (PBS). The optical density at 650 nm was measured and serial 10-fold dilutions plated onto BG-blood agar to determine the number of bacteria in the challenge inoculum. To obtain OMVs, B. pertussis Tohama phase I strain CIP 8132, 24 h colonies grown in BGAS medium were used to seed liquid medium Stainer–Scholte (SS) (54).
Isolation and characterization of OMVs
OMVs used for vaccine formulation were isolated and characterized as previously described (37, 55). Briefly, culture samples from the decelerating growth phase in SS medium were centrifuged and the bacterial pellet obtained was resuspended in 20 mM Tris–HCl, 2 mM EDTA pH 8.5. The suspension was sonicated in cool water for 20 min. After two centrifugations at 10,000×g for 20 min at 4°C, the supernatant was pelleted at 100,000×g for 2 h at 4°C. This pellet was re-suspended in Tris buffer (20 mM pH 7.6). The samples obtained were negatively stained for electron microscope examination. Protein content was estimated by the Bradford method using bovine serum albumin as standard (56). The presence of the main immunogenic proteins in the OMVs was detected by immunoblot assays using specific antibodies as we previously described (not shown) (37, 40).
Combined OMV+aP vaccine composition
The vaccine formulation tested here combined 1/10 of the human dose of commercial ADACEL® vaccine (aP, Sanofi Pasteur Limited) and 3µg of formalin detoxified OMVs derived from B. pertussis (hereafter named as combined). The pertussis component of commercial aP vaccine per human dose consists of 2.5 µg detoxified pertussis toxin (PTx), 5 µg filamentous hemagglutinin (FHA), 3 µg pertactin (PRN), 5 µg fimbriae types 2 and 3 (FIM).
Mice immunization
Animal experiments were performed using BALB/c mice (4–6 weeks old), provided by the Faculty of Veterinary Sciences, La Plata, Argentina. Animals were kept in ventilated cages and housed under standardized conditions with regulated daylight, humidity, and temperature. They received food and water ad libitum. The animal protocols were authorized by the Ethical Committee for Animal Experiments of the Faculty of Science at La Plata National University (approval number 004 - 06-15, 003 - 06–15 extended its validity until August 10, 2027). A two-dose intramuscular immunization schedule, with doses administered 14 days apart, was used to immunize groups of 4–6 BALB/c mice with either the combined OMV+aP vaccine or 1/10 of the human dose of the commercial aP vaccine ADACEL® (Figure 1A). Non-immunized mice served as the control group.
Figure 1

(A) Schematic representation of experimental vaccination schedule. BALB/c mice (n=6/group) were intramuscularly immunized at days 0 and 14, with a 2-dose scheme using the combined vaccine formulation and aP commercial vaccine. Non-immunized mice were incorporated as controls. (B) Mouse weight gain test. Animals were monitored for 7 days, with body weights recorded at 24 hours, 3 days, and 7 days post-intervention. The graph displays the percentage of weight gained by each animal relative to its initial baseline weight. No statistically significant differences in weight gain were observed between immunized and non-immunized groups in each time point tested (p > 0.05). (C) Inflammatory marker upon systemic vaccination. IL - 6 levels were measured by ELISA in sera obtained from mice 4h after the 1st vaccine dose. Bars represent the means ± SEM of pg/mL. No statistically significant differences in IL - 6 levels were observed among all studied groups (p > 0.05).
Mouse weight gain test
The MWG-test was carried out using groups of 6–8 BALB/c mice (14–16 g) which were vaccinated with the here tested vaccine formulation. Animals were observed for 7 days and body weight was recorded after 16 h, 3 and 7 days. Vaccines were considered non-toxic when passing the WHO and EP requirements (57).
Expression of inflammatory marker upon systemic delivery of vaccines
In order to have an additional marker of pro-inflammatory capacity of the different formulations, serum was collected 4 h after each immunization by submandibular bleeding and serum separation. Serum IL - 6 was measured by ELISA using ELISA Flex: Mouse IL - 6 (HRP) Mabtech following manufacturer’s instructions.
Stimulation of human whole blood
The whole blood IL - 6 cytokine release assay was performed as described by Stoddard et al. (58). Briefly, a 245 μl of blood sample was dispensed into each well of a 96-well tissue culture plate. Serial dilutions of each vaccine were prepared in RPMI 1640 cell culture medium (GIBCO) in a range of 5–300 ng/ml, incubated with the whole blood and then briefly centrifuged. A sample of 55 μl of plasma from each well was removed and frozen pending quantification of the cytokines. IL - 6 levels were measured by ELISA using BD OptiEIA (BD Biosciences) following manufacturer instructions.
Intranasal challenge mouse model
For protection assays, mice were intranasally challenged with a sublethal dose of B. pertussis (as specified in the figure legends). Animals were euthanized one-week post-challenge, and bacterial loads in lungs and nasal tissue were quantified. Organs were aseptically excised, serially diluted, and plated for colony counting according to previously established protocols (31, 40, 48). Specifically, lungs and the nasal tissue was aseptically excised to minimize contamination from environmental bacteria.
Once extracted, the biological material was mechanically homogenized in a known volume of sterile phosphate-buffered saline (PBS). Subsequently, serial 10-fold dilutions of the homogenates were prepared and plated onto BGAS. The plates were incubated at 36.5°C for 72 hours. After incubation, colony-forming units (CFU) were counted to determine the bacterial load per lungs and nose. A minimum of three independent experiments were performed.
To study protection conferred by passive transfer, pooled serum (100 µl) or spleen cells (2.5 × 107) derived from non-immunized or immunized mice were transferred intraocularly into 4-week-old naïve recipient mice. Twenty-four hours later, the mice were infected with B. pertussis and protection assessed on day 7 as described above.
Enzyme-linked immunosorbent assay
Blood samples were collected two weeks after the final immunization and at sacrifice.
To detect antibody levels induced in mice by vaccination, Enzyme-linked immunosorbent assay (ELISA) was performed as we previously described (31, 40, 59). Plates (Nunc A/S, Roskilde, Denmark) were coated with sonicated B. pertussis Tohama phase I (whole cell lysates) or purified recombinant PTxA both at 3 µg/mL in 0.5 M carbonate buffer, pH 9.5 by overnight incubation at 4°C. Blocked plates with 3% milk in PBS (2 h 37°C) were incubated with serially diluted samples of mouse serum (1 h 37°C). Sera was obtained after leaving the blood samples to clot for 1 h at 37°C followed by centrifuging for 10 min at 6,000xg. Total IgG, IgG-isotypes and IgA from individual serum or pooled sera bound to the plates were detected after a 2-h incubation with goat anti–mouse-IgG–linked horseradish peroxidase (1:8,000 Invitrogen, USA) and goat anti–mouse-IgA–linked horseradish peroxidase (1:3,000) (Abcam). For measuring IgG isotypes, detection of bound antibody was determined using HRP labeled subclass-specific anti-mouse IgG1 (1:3,000), IgG2a (1:2,000) (Invitrogen USA). As substrate 1.0 mg/mL o-phenylendiamine (OPD, Bio Basic Canada Inc) in 0.1 M citrate-phosphate buffer, pH 5.0 containing 0.1% hydrogen peroxide was used. Optical densities (ODs) were measured with BioTek 800 TS microplate reader (BioTek, Agilent Technologies, US) at 490 nm. From the experimental protocol performed in triplicate, one representative experiment is presented in the Results.
Avidity assay
Avidity was measured by an ELISA elution assay as the overall strength of binding between antibody and antigen, using plates incubated for 10 min with increasing concentration of ammonium thiocyanate (NH4SCN) from 0 to 1 M. Antibody avidity was defined as the amount (percentage) of antibody retained for each increment of NH4SCN concentration relative to that measured in absence of NH4SCN.
Ag- specific IFN-γ, IL - 17, IL - 22 and IL - 5 production by spleen cells
After B. pertussis challenge, spleens from untreated and immunized mice were passed through a 40-mm cell strainer to obtain a single-cell suspension. Spleen cells were seeded in 48 well culture plates in a final volume of 500 µl/well RPMI 1640 with 10% fetal bovine serum, containing 100 IU/mL penicillin and 100 µg/mL streptomycin (44). All cell samples were stimulated with heat killed bacteria suspension 5x106 UFC/well (HKBp), PTx subunit A (PTxA1 μg/mL) or medium only. After 72 h of incubation (37 °C and 5% CO2), IFN-γ, IL - 17, IL - 22 and IL - 5 concentrations were quantified in supernatants by ELISA (Mabtech, USA) using conditions recommended by the manufacturer.
FACS analysis of tissue resident memory CD4+T cells in lungs and nose
To discriminate blood-borne circulating cells from tissue-localized cells we administered intravenously anti-mouse PE-CD45 Ab (eBioscience) 10 min before they were euthanized as previously described (52). After the enzymatic disruption of tissue for 1 h at 37 °C with Collagenase D (1 mg/mL; Sigma-Aldrich) and DNAse I (20 U/mL; Sigma-Aldrich) and the lysis of red blood cells, cells from lungs and noses were incubated with CD16/CD32 FcgRIII (1:100) to block IgG Fc receptors. Cells were incubated with LIVE/DEAD Aqua (Invitrogen), followed by specific surface TRM-CD4+ T cells staining with CD45.2-BV650 (BD), CD3-PeCy7 (BD), CD4-FITC (Invitrogen), CD44-PE (BD), CD62L-PE-CF594 (BD), CD103-APC-EF780 (Invitrogen), CD69-APC (Invitrogen) (Supplementary Figure S1).
Fluorescence minus one or non-specific isotype Abs were used as controls. Flow cytometric analysis was performed on an LSR Fortessa, and data were acquired using Diva software (BD Biosciences). The results were analyzed using FlowJo software (TreeStar) (60).
Statistical analysis
The data were evaluated statistically by t- Student test, two-way or one-way analysis of variance (ANOVA) followed by Bonferroni for multiple comparisons (via the GraphPad Prism® software). Differences were considered significant at a p <0.05.
Results
Preclinical evaluation of the safety of a combined B. pertussis vaccine based on OMVs and commercial acellular formulation
Although it may seem straightforward to assume that vaccine combinations offer clear benefits for both individuals and public health systems, formulations that include multiple antigens must overcome unforeseen challenges, even when combining vaccines with well-established efficacy. Immunological, physical, and/or chemical interactions among the combined components may alter the host immune response (61). Therefore, a key challenge in the development of combination vaccines is the potential reduction in efficacy or safety compared to the individual components. Accordingly, our first experiment aimed to evaluate the safety of a combination of the commercial acellular vaccine and outer membrane vesicles derived from B. pertussis (hereafter referred to as combined) using a murine intranasal challenge model. Since the aim of this study is to address the limitations of the commercial acellular vaccine (aP) through a combined formulation including both aP and OMVs, all experiments presented herein were conducted in direct comparison with the aP vaccine. As a negative control, we included a group of animals that received no immunogens.
For the combined formulation, we used doses previously shown to be safe and immunogenic when administered individually in the same animal model. As in subsequent experiments, groups of BALB/c mice were immunized intramuscularly (i.m.) following a two-dose schedule with a 14-day interval between the first and second doses (Figure 1A).
Vaccine safety in the murine model was assessed by both monitoring body weight gain and measuring serum levels of the pro-inflammatory cytokine IL - 6 at 4 hours post-administration of the first dose (Figures 1B, C). For the weight gain test, animals were weighed at 24 h, 3 days, and 7 days post-immunization. As expected, all PBS-treated mice steadily gained weight at all time points analyzed.
While the acellular vaccine did not cause any reduction in weight at any of the assessed time points, the combined vaccine induced a slight, non-significant decrease in body weight at 24 and 72 hours. By day 7, mice in both vaccinated groups had recovered and showed more than 60% of the weight gain observed in the non-immunized control group.
Taken together, these results indicate that both the combined and acellular vaccine formulations meet the safety criteria based on the weight gain curve and can be considered non-toxic in this model.
Furthermore, serum IL - 6 levels, used as a marker of pro-inflammatory response, reached 257.0 ± 47.5 pg/mL at 4 hours post-immunization with the combined vaccine. A similar level was observed with the commercial aP vaccine used at 1/10 of the human dose (405.7 ± 47.5 pg/mL). Importantly, both values were not significantly different from those measured in the untreated control group. Additionally, in in vitro human whole-blood assays, we observed that IL - 6 levels induced by OMV doses ranging from 0.25 to 3 µg (combined with 1/10 of the human aP dose) ranged from 2 ng/mL to 12 ng/mL for the lowest and highest OMV quantities tested. These levels were lower than those induced by an approved commercial whole-cell vaccine (~60 ng/mL). No cytokine stimulating activity was detected for the commercial aP vaccine. Moreover, no visible signs of local reactogenicity at the injection site, such as erythema, swelling, or induration, were observed in any of the vaccinated animals. This visual assessment, performed by trained personnel, further supports the safety profile of both the combined and aP formulations in this model.
Immunogenicity of combined vaccine
Given the known properties of OMVs to induce Th1, Th2, and Th17 immune profiles (31, 43, 44, 46, 62), as well as their adjuvant and immunomodulatory capabilities (49), we evaluated the humoral immune response specific to pertussis toxin (in particular PTxA) and a heat-killed B. pertussis lysate (HKBp). Specifically, we comparatively assessed the levels of IgG and the IgG1 and IgG2a isotypes specific to these antigens in response to the combined vaccine formulation and the commercial aP vaccine.
Using the previously described two-dose immunization schedule (Figure 1A), triplicate experiments demonstrated that the combined formulations elicited a more robust antibody response (Figure 2). Notably, mice immunized with the combined vaccine exhibited significantly higher serum IgG antibody titers against both PTxA (p < 0.05) and HKBp (p < 0.0001) compared to those receiving the aP treatment alone. As expected, IgG levels in unimmunized mice were markedly lower (data not shown). The NH4SCN avidity assay using the chaotropic agent showed that the antibodies induced by the combined vaccine (anti-PTxA IgG) were of slightly higher quality than those elicited by the aP vaccine, although the difference was not statistically significant (not shown).
Figure 2
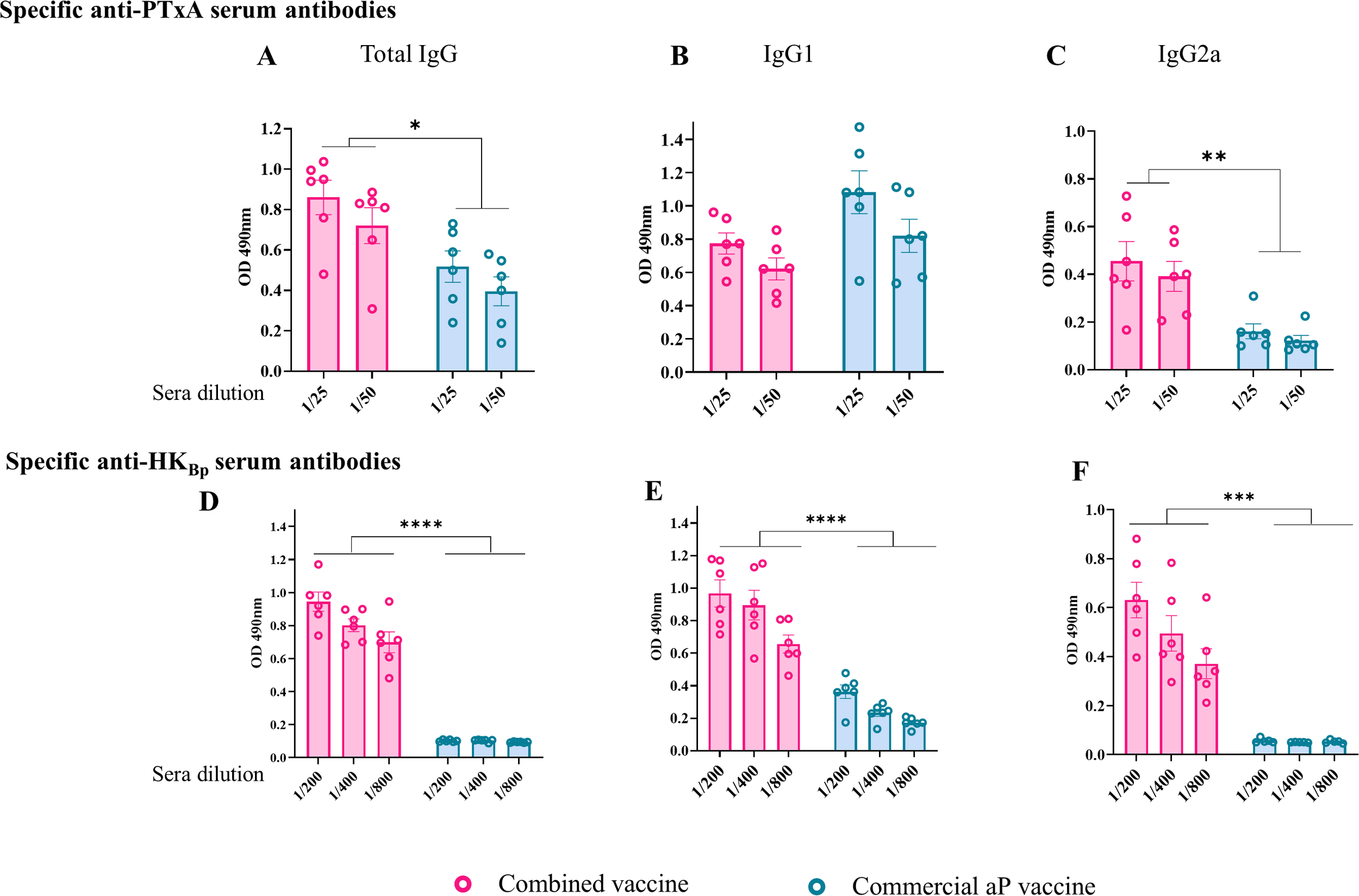
Characterization of the humoral immune response induced by the combined vaccine (OMVBp+aP). Antibody responses specific to pertussis toxin (PTxA; panels A–C) and heat-killed B. pertussis lysate (HKBp; panels D–F) were evaluated by ELISA in sera collected 14 days after the second vaccine dose. Absorbance values at 490 nm were measured for two or three serum dilutions. Sera from mice immunized with the combined vaccine were compared to those receiving the aP vaccine or non-immunized controls. Antigen-specific levels of total IgG (A, D), IgG1 (B, E), and IgG2a (C, F) are shown. Statistically significant differences are indicated (*p < 0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Regarding IgG isotype profiles, mice immunized with the aP vaccine exhibited marginally higher (though statistically insignificant) levels of PTxA-specific IgG1 compared to those receiving the combined formulation, while PTxA-specific IgG2a levels were significantly lower (p < 0.05). This pattern aligns with the established Th2-skewed response characteristic of aP vaccines, whereas the combined vaccine preferentially induced a Th1-biased response. For HKBp-specific antibodies, both IgG1 and IgG2a titers were significantly reduced in aP-vaccinated mice relative to the combined vaccine group (p< 0.001). Notably, the combined formulation elicited slightly higher IgG2a than IgG1 levels against HKBp, further supporting its Th1-polarizing capacity (Figures 2E, F).
These results are of particular interest as they demonstrate that the addition of OMVs to the aP vaccine enhances the humoral immune response and modulates the immune profile toward Th1 polarization.
To further investigate the ability of OMVs to modulate the immune response of combined vaccine formulations toward a Th1 profile, we performed spleen cell stimulation assays using splenocytes from mice immunized with different vaccine formulations, as well as from non-immunized controls (Figure 3). The stimuli used were the A subunit of pertussis toxin (PTxA) and the HKBp. After the incubation period, we evaluated IFN-γ levels as a marker of Th1-type response (Figure 3A). Additionally, we assessed IL - 17 [Th17 profile marker, Figure 3B)] and IL - 5 (Th2 profile marker, Figure 3C) levels to further characterize the immune response induced by the tested combined formulations.
Figure 3
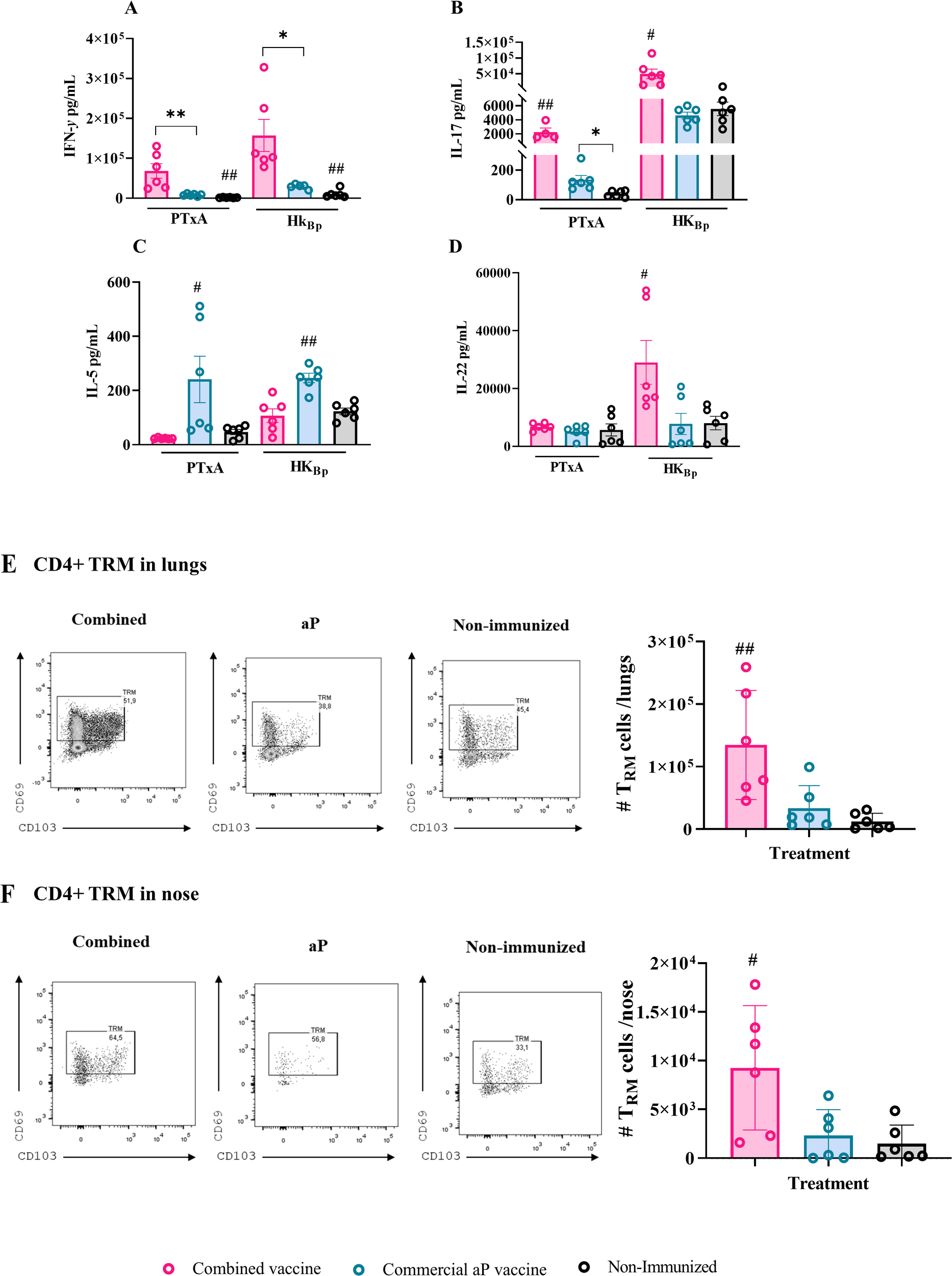
Cellular immune response induced by the combined vaccine formulation. Mice were immunized intramuscularly on Days 0 and 14 with the combined formulation (OMV+aP) as described in Figure 1. Seven days after challenge with a sublethal intranasal dose of B. pertussis (1×107–5×107 CFU in 40 µL), mice were euthanized, and spleens were harvested. Splenocytes were restimulated ex vivo with heat-killed B. pertussis (HKBp; 5×106 CFU/well), pertussis toxin subunit A (PTxA; 1 µg/mL), or medium only (non-stimulated, not shown for simplicity of the figures). Cytokine concentrations of IFN-γ (A), IL - 17 (B), IL - 5 (C), and IL - 22 (D) in culture supernatants were measured by ELISA. Bars represent mean ± SEM (pg/mL). Absolute counts of CD4+ TRM (CD45–, CD44+, CD62L−, CD69+, CD103+/–, CD4+) in lungs and nose are represented in panels (E, F) respectively. Bars represent mean ± SEM CD4+TRM cells count/lungs or nose. Statistical analysis was performed by two-way ANOVA followed by Bonferroni post hoc test. * and ** indicate significant pairwise differences (*p < 0.05, **p < 0.01). # and ## indicate significant differences between the treatment and the other groups (#p<0.05, ##p < 0.01).
The results were obtained from determinations performed after bacterial intranasal challenge, in alignment with the 3Rs principle, as the same challenged animals were used to evaluate protection against colonization.
Figure 3A shows that, for both stimuli, the presence of OMVs in the combined vaccines induced significantly higher IFN-γ levels compared to the aP formulation (68,415 ± 18,244 pg/mL vs. 8,217 ± 1,129 pg/mL, p<0.001 for PTxA stimulation; 157,291 ± 40,285 pg/mL vs. 29,610 ± 2,581 pg/mL for HKBp stimulation, p<0.05 between combined and aP, respectively). Similarly, IL - 17 levels (Figure 3B) were significantly higher in samples from mice immunized with the combined vaccine compared to those from mice vaccinated with aP (2,260 ± 568.8 pg/mL vs. 132.8 ± 30.67 pg/mL, p < 0.001 for PTxA stimulation; 49,772 ± 15,278 pg/mL vs. 4,625 ± 475.2 pg/mL for HKBp stimulation, p < 0.05).
In contrast, IL - 5 levels (Figure 3C) were significantly elevated in the aP group relative to the combined vaccine group (p < 0.001 for PTxA stimulation and p < 0.05 for HKBp stimulation).
We also measured IL - 22 levels (Figure 3D), given its recognized role as a key mediator of early mucosal defense and protection against epithelial lung damage (49, 50). Consistently, IL - 22 levels in samples from mice immunized with the combined vaccine were significantly higher than those from the aP group following HKBp stimulation (25,982 ± 7,104 pg/mL vs. 8,391 ± 3,153 pg/mL, p < 0.05). Notably, IL - 22 levels in aP-immunized mice were not significantly different from those in non-immunized controls. When the spleen cells were stimulated with PTxA, no differences were detected among the studied treatment.
Flow cytometry analysis revealed a significantly higher number of lung- (Figure 3E) and nasal-resident (Figure 3F) memory CD4+ T cells in OMV+aP-treated mice compared to those that received the aP vaccine alone. These findings indicate that the inclusion of OMVs in the aP formulation enhances the recruitment and establishment of memory T cells in the respiratory mucosa, a response not achieved with the aP vaccine alone.
Altogether, these results demonstrate that the addition of OMVs to combined vaccine formulation is advantageous over the standalone aP formulation, not only in terms of enhancing the humoral response but also by promoting a Th1/Th17/Th22 immune profile, increasing epitope diversity due to their complex composition and resident memory CD4+ T cells.
Protective capacity of the combined vaccine prototype against lung bacterial colonization (severe disease)
To evaluate the protective capacity of the different vaccine formulations tested in this study, groups of mice immunized with two-dose schedule were intranasally challenged with a sublethal dose of B. pertussis (1×107–5×107 CFU) 14 days after receiving the final dose (Figure 4A). Seven days post-challenge, all animals, including those in the non-immunized control group, were euthanized by cervical dislocation to assess bacterial colonization in the upper and lower respiratory tracts.
Figure 4
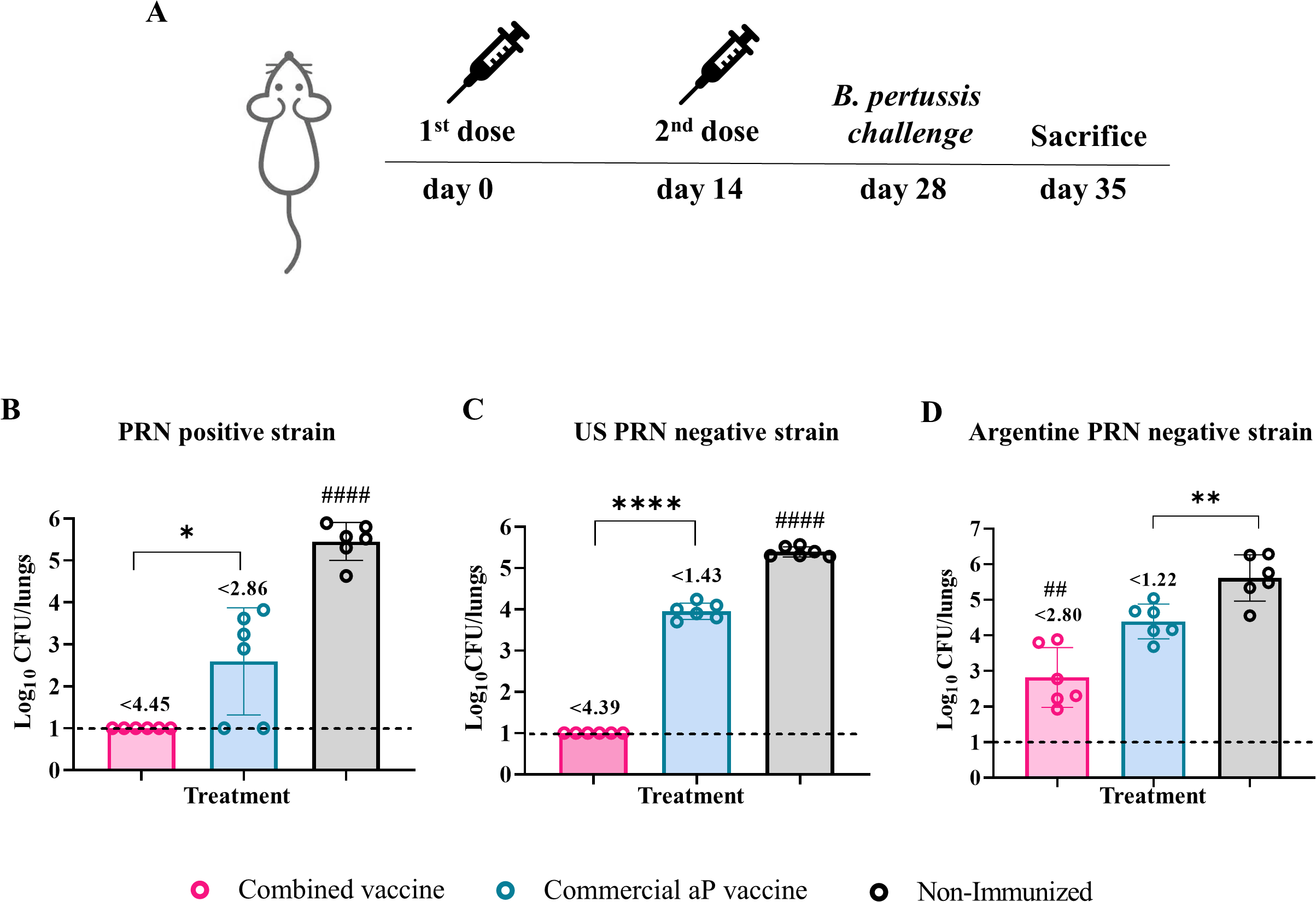
Protective capacity of the combined vaccine against PRN-positive and PRN-deficient B. pertussis strains. Mice were immunized on Days 0 and 14 with two doses of the indicated vaccine formulations and challenged intranasally on Day 28 with different B. pertussis strains (A): PRN-positive reference strain (B. pertussis Tohama pahase I) (B), PRN-deficient clinical isolate from the U.S. outbreak (C), and locally circulating PRN-deficient clinical isolate (D). Bacterial burden in the lungs was assessed 7 days post-challenge (Day 35). Results are expressed as log10 CFU per lungs. Numbers above the bars indicate the difference between the mean bacterial loads of each group and the non-immunized group, expressed in log10. Bars represent mean ± SEM. Statistical comparisons were performed using one-way ANOVA with Bonferroni correction. *, **, and **** indicate significant pairwise differences (*p<0.05, **p<0.01, ****p<0.0001). ## and #### indicate significant differences between the treatment and the other groups (##p<0.01, ####p<0.0001).
The challenge was performed using B. pertussis strains with distinct genotypic and phenotypic characteristics. These included the reference Tohama I phase I strain (ptxP1-ptxA2-prn1), which expresses the adhesin pertactin (PRN), and two ptxP3-ptxA1-prn2 strains that lack PRN expression. One of the PRN-deficient strains (PRN-) corresponds to a locally circulating isolate, while the other derives from the 2012 U.S. outbreak, kindly provided by Dr. Lucia Tondella (CDC).
As expected, the non-immunized control group exhibited the highest bacterial burden in the lungs (3.02×105 CFU/lungs, Figure 4B). Immunized groups showed significant reductions in lung colonization by the Tohama strain compared to controls (p < 0.0001). The highest reduction was observed in animals immunized with the combined OMV+aP formulation, which exhibited a 4.45-log decrease (complete bacterial clearance, Figure 4B). In contrast, mice immunized with the aP vaccine alone showed a 2.86-log10 reduction (Figure 4B).
When challenged with the U.S. PRN− strain, mice receiving the combined vaccine showed complete bacterial clearance from the lungs (4.39-log decrease, p< 0.0001 Figure 4C). In contrast, aP-immunized mice exhibited bacterial loads close to those of the non-immunized group (1.43-log decrease, Figure 4C). A similar trend was observed with the locally circulating PRN− strain (Figure 4D), though all groups showed higher residual bacterial loads compared to those seen with the U.S. PRN− strain, suggesting intrinsic differences in virulence or immune evasion capacity between the PRN-deficient isolates.
Passive transfer experiments
To dissect the respective contributions of humoral and cellular responses induced by the combined OMV+aP vaccine, passive transfer experiments were conducted using sera or splenocytes from immunized donor mice. Naïve recipient mice were subsequently challenged intranasally with B. pertussis Tohama phase I strain, and bacterial burden, antibody titers, and immune cell populations were assessed.
Transfer of sera (Figure 5A) from OMV+aP-immunized mice conferred significantly higher protection in the lungs compared to sera from aP-immunized or PBS-treated donors (Figure 5B, p<0.0001). Lung bacterial counts were undetectable in recipients of sera from OMV+aP-immunized mice, indicating complete protection. In contrast, mice receiving sera from aP-vaccinated donors showed residual bacterial loads in the lungs (4.1 102CFU/lungs), while PBS controls exhibited the highest bacterial burden (3.1 104CFU/lungs).
Figure 5
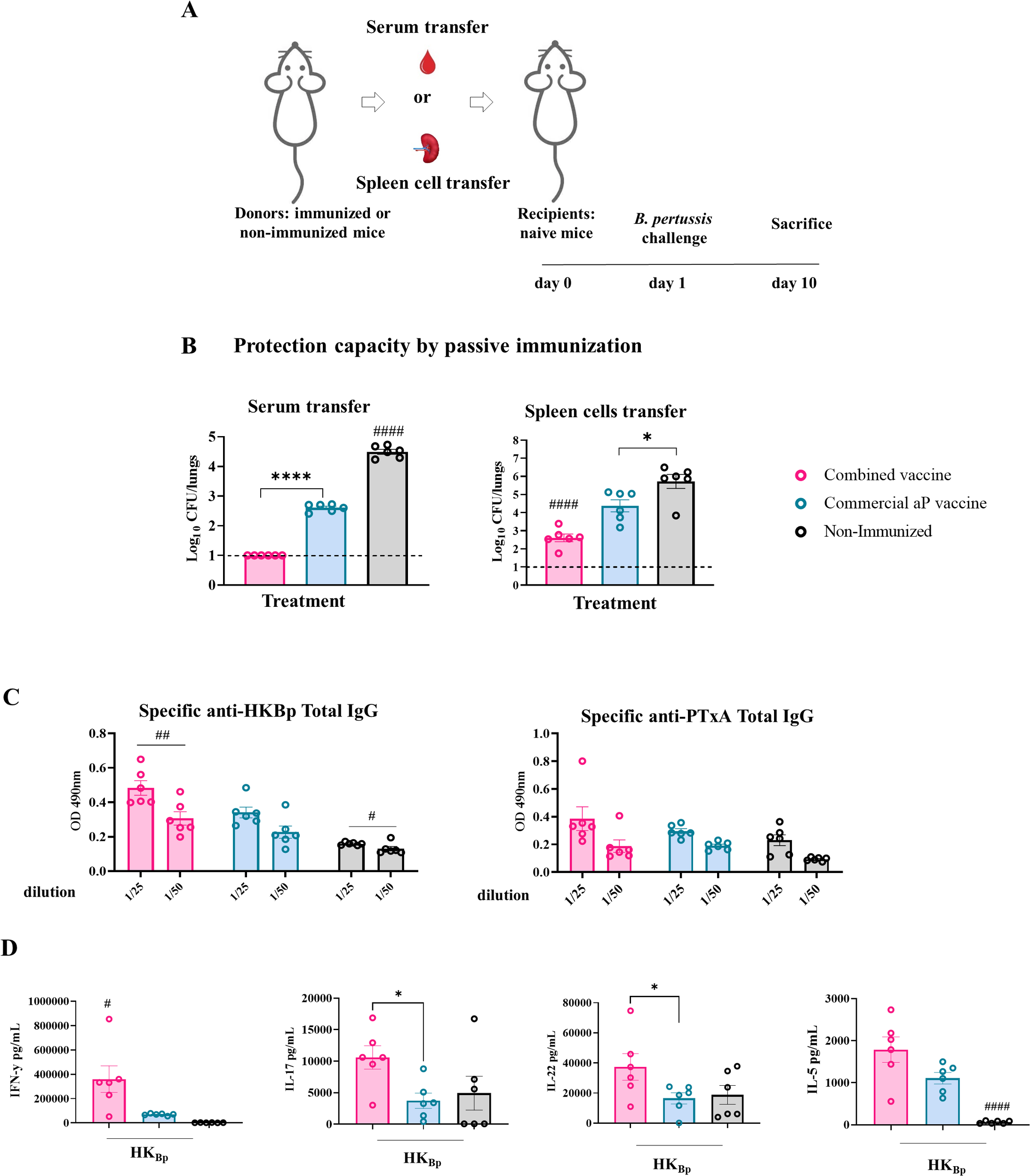
Protection capacity of passive transfer of serum or splenocytes from immunized mice (donors). (A) Experimental design: naïve recipient mice received either serum or splenocytes from donor mice immunized with 2-dose scheme with OMV+aP, aP, or PBS. One day later, recipients were challenged intranasally with a sublethal dose of (5x107 CFU) B. pertussis Tohama phase I strain, and bacterial burden in the lungs was quantified 10 days post-transfer. (B) Lungs bacterial load following either serum- or splenocyte- transfer. (C) IgG titers against HKBp and PTxA were measured in sera from recipient mice 7 days post-challenge. (D) Cellular immune response in receptor mice that received immune splenocytes cells. Seven days after challenge with a sublethal intranasal dose of B. pertussis (1×107–5×107 CFU in 40 µL), mice were euthanized, and spleens were harvested. Splenocytes were restimulated ex vivo with heat-killed B. pertussis (HKBp; 5×106 CFU/well) or medium only (non-stimulated, not shown for simplicity of the figures). Cytokine concentrations of IFN-γ, IL - 17, IL - 5, and IL - 22 in culture supernatants were measured by ELISA. Bars represent mean ± SEM (pg/mL). Statistical analysis was conducted using one-way ANOVA with post hoc testing. *p<0.05, ****p<0.0001, #p<0.05, ##p<0.01, and ####p<0.0001.
Transfer of splenocytes (Figure 5A) from OMV+aP-vaccinated donors resulted in enhanced protection in the lungs colonization (3.1 -log decrease in comparison with control group, p< 0.0001) compared to recipients of aP- (1.3-log decrease in comparison with control group, p< 0.01) or PBS- splenocytes (Figure 5B). Mice receiving aP splenocytes exhibited only partial protection.
Consistently, sera from mice that received OMV+aP-derived splenocytes showed significantly elevated anti-B. pertussis IgG titers 7 days post-challenge, as determined by ELISA (p < 0.01), indicating the ability of transferred lymphocytes to support antibody production upon infection (Figure 5C). We also observed that post-challenge, recipients of immune cells from donor animals vaccinated with the combined vaccine showed significantly higher IFN-γ levels compared to those receiving cells from the aP-vaccinated group (359,802 ± 133,612 pg/mL vs. 69,517± 7,314 pg/mL; p<0.05 between combined and aP groups, respectively) (Figure 5D). Similarly, IL - 17 and IL - 22 levels were significantly elevated in mice that received splenocytes from combined-vaccinated animals versus those receiving splenocytes from aP-vaccinated animals (p<0.05 Figure 5D). No significant differences were observed for IL - 5.
Collectively, these data highlight the immunological superiority of the OMV+aP vaccine, which elicits robust humoral and cellular responses and confers enhanced protection not only against PRN+ strains but also against PRN− isolates. Passive transfer studies support a mechanistic role for both antibodies and antigen-experienced lymphocytes in mediating protection, reinforcing the rationale for combined vaccine strategies.
The combined vaccine reduces bacterial colonization in the upper respiratory tract, a key factor in pertussis transmission
Reducing the transmission of B. pertussis has long been a central goal of vaccination strategies, particularly given the extremely high contagiousness of the pathogen. The basic reproduction number (R0) for B. pertussis has been estimated at approximately 17, making it one of the most transmissible bacterial infections.
Pioneering studies by Warfel et al. (21) using the non-human primate baboon model demonstrated that acellular pertussis vaccines, while effective at protecting against disease, fail to reduce bacterial colonization in the upper respiratory tract (URT), thereby allowing continued transmission despite immunization. This finding underscored the need to develop vaccines capable of inducing mucosal immunity and limiting nasopharyngeal colonization. Here we investigated whether the addition of OMVs to the aP vaccine formulation could enhance its ability to reduce B. pertussis colonization in the nasal cavity. Mice were immunized with a two-dose schedule, as previously described, and then challenged intranasally with the B. pertussis Tohama phase I strain using two different inoculum doses (Figure 6A):
-
A low dose (3 × 10³ CFU) delivered in a reduced volume to restrict bacterial colonization primarily to the nasal cavity.
-
A high dose (5 × 107 CFU) administered in a larger volume to ensure the bacteria also reached the lower respiratory tract.
Figure 6
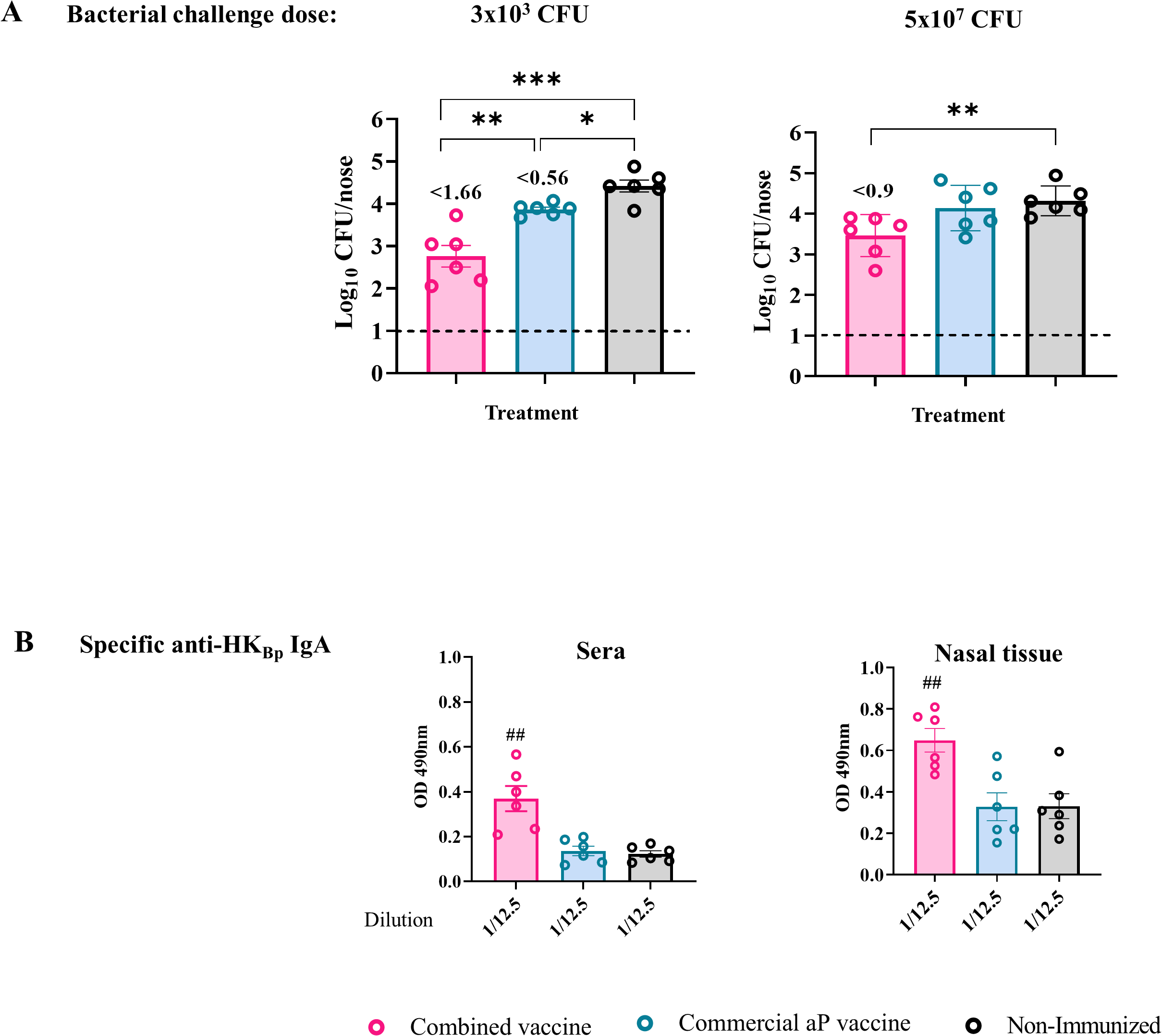
Protective capacity induced by the combined vaccine formulation against upper respiratory tract bacterial colonization. Mice were immunized on Days 0 and 14, then challenged intranasally with either a high dose (5×107 CFU in 40 µL) or a low dose (3×10³ CFU in 6 µL) of B. pertussis Tohama phase I in volumes targeting the lungs and nose or only nasal cavity, respectively. Nasal bacterial load was evaluated 7 days post-challenge (A). Numbers above the bars indicate the difference between the mean bacterial loads of each group and the non-immunized group, expressed in log10. Serum and nasal tissue IgA titers against HKBp were measured by ELISA at the same time point (B). Bars represent mean ± SEM. Statistical analysis was performed using one-way ANOVA. *p<0.05, **p<0.001, ***p<0.0001, ##p<0.01.
The high-dose challenge was included to assess the extent to which the treatments could reduce nasal colonization.
In non-immunized control animals, both doses resulted in comparable levels of colonization in the nasal cavity (~2.24 × 104 CFU/nose). This finding suggests a local niche limitation that constrains bacterial growth in the URT regardless of the inoculum size. Mice immunized with the aP vaccine alone showed practically no reduction in nasal colonization under either challenge condition. In contrast, the combined OMV+aP vaccine demonstrated significant protective efficacy in the URT, with bacterial reductions of 1.66 logs (p < 0.001) and 0.9 log (p < 0.001) following challenge with 3 × 10³ CFU and 5 × 107 CFU, respectively. These results indicate that the inclusion of OMVs may overcome the limitations of aP vaccines in controlling colonization and, by extension, transmission.
Given the established role of mucosal IgA in preventing colonization by B. pertussis, particularly in the nasal epithelium (63), we evaluated serum IgA levels post-vaccination. Elevated levels of antigen-specific IgA were observed in mice immunized with the combined formulation (Figure 6B, p< 0.001), both in sera and in nasal tissue homogenates, consistent with the improved control of bacterial colonization.
Together, these findings support the hypothesis that adding OMVs to aP vaccines enhances mucosal immunity, promoting IgA production and reducing colonization in the URT, a critical step toward curbing transmission of pertussis.
Discussion
Current acellular pertussis (aP) vaccines, while significantly safer than first-generation whole-cell (wP) formulations, exhibit critical immunological limitations (64, 65). They predominantly elicit Th2-skewed responses, induce short-lived immunity, and fail to prevent bacterial colonization of the upper respiratory tract (URT), thereby allowing asymptomatic transmission (21, 27, 28). The emergence and global spread of pertactin-deficient (PRN−) B. pertussis strains have further undermined the protective capacity of conventional aP vaccines (16, 51, 52). PRN is a key antigen in most licensed formulations, and the expansion of PRN− variants seem to reduce aP effectiveness (15, 66). However, post-pandemic surveillance has revealed heterogeneous trends: in some countries PRN− strains have markedly declined or even ceased to circulate, while in others, particularly certain regions of the Americas and Europe, they continue to predominate. This dynamic landscape underscores the need for next-generation vaccines that confer broader and more durable protection against both classical and immune-evasive B. pertussis lineages. Outer membrane vesicles (OMVs) derived from B. pertussis represent a compelling platform to address these shortcomings. OMVs inherently package multiple immunogenic proteins and pathogen-associated molecular patterns (PAMPs), providing intrinsic adjuvanticity that promotes robust Th1 and Th17 immunity (31, 46–49). Preclinical studies, including our own prior work, have consistently demonstrated that OMV-based vaccines can significantly reduce bacterial loads in the lungs, induce mucosal IgA, and exhibit markedly lower reactogenicity than wP (37, 40, 43, 46–48, 67). Importantly, intranasal OMV administration has been associated with URT colonization control, a key feature absent from standard aP vaccination (21, 65, 68, 69).
In this study, we leveraged these immunological attributes by combining B. pertussis-derived OMVs with a licensed aP vaccine to generate a combined formulation aimed at overcoming the two principal limitations of current aP vaccines: poor mucosal immunity and vulnerability to immune-evasive bacteria (28). The combined OMV+aP vaccine demonstrated an excellent safety profile across in vitro and in vivo models, with no signs of local or systemic toxicity even at the highest OMV dose tested (3 µg in combination with 1/10 of the human aP dose). Human whole-blood assays confirmed low cytokine induction relative to wP, supporting the translational potential of this approach (43).
Functionally, the combined vaccine reshaped the humoral immune response compared to the aP vaccine alone. Mice immunized with OMV+aP generated higher total IgG titers against PTxA and heat-killed B. pertussis lysate (HKBp) than those receiving aP alone. Isotype profiling revealed a shift toward a Th1-biased IgG2a response, both against purified antigens and whole-cell lysates, suggesting an enhanced potential for opsonophagocytosis and bacterial clearance.
Concomitantly, splenocytes from OMV+aP-vaccinated animals secreted markedly higher levels of IFN-γ and IL-17 following ex vivo antigenic stimulation, with the addition of a distinct IL-22 signal, a cytokine linked to mucosal barrier protection and epithelial antimicrobial responses (70, 71). Notably, IL-5 production remained unaltered, underscoring the transition from a Th2-dominated profile toward a functionally protective Th1/Th17 axis. These immune signatures translated into robust in vivo protection. The OMV + aP vaccine markedly reduced bacterial burden in both the lungs and the nasal cavity. In the lungs, colonization by the PRN+ strain (Tohama phase I) was decreased by 4.45 log10 CFU compared with non-immunized controls, surpassing the 2.86 log10 reduction achieved by the aP vaccine alone. In the nasal cavity, the combined formulation achieved a 1.66 log10 CFU reduction under low-dose challenge and a 0.9 log10 CFU reduction under the stringent high-dose model. Together, these results highlight the superior ability of the OMV + aP vaccine to limit both pulmonary infection and upper-airway colonization, addressing key limitations of current acellular vaccines. The ability to limit URT colonization is particularly noteworthy, as it directly addresses the Achilles’ heel of current aP vaccines: their inability to prevent transmission. Consistent with this mucosal effect, OMV+aP induced CD4+ tissue-resident memory (TRM) cells in both lungs and nasal mucosa, a cellular compartment that has emerged as a key correlate of near-sterilizing immunity against B. pertussis (24, 25, 72). The parallel increase in serum and nasal tissue IgA levels, which play a crucial role in URT protection, further reinforces the functional complementarity of humoral and cellular immunity elicited by this combined formulation (63, 73).
The breadth of protection was underscored by challenge experiments using PRN− clinical isolates. Whereas aP vaccination alone conferred minimal protection, OMV+aP immunization effectively eliminated or markedly reduced lung colonization by both the U.S. outbreak PRN− strain and a locally circulating Argentinean isolate. These findings underscore the combined vaccine’s ability to overcome antigenic drift and selective immune evasion, a critical feature for achieving long-term, population-level impact.
Passive transfer experiments provided mechanistic validation of the dual contribution of humoral and cellular immunity. Serum and splenocytes from OMV+aP-immunized donors conferred superior protection in naïve recipients, with sera achieving complete lung clearance of B. pertussis Tohama phase I strain and splenocyte transfer mediating profound reductions in bacterial burden (3.1-log decrease versus controls). Post-challenge analysis of cytokine production in recipient mice confirmed the transfer of Th1/Th17 functionality, including elevated IFN-γ, IL-17, and IL-22, linking these immune signatures to the observed protection.
Altogether, our data establish that OMV inclusion not only preserves the immunogenic properties previously described for this platform but also enhances the magnitude, quality, and mucosal reach of the immune response when combined with a licensed aP formulation. This strategy effectively addresses critical shortcomings of current aP vaccines, namely, weak mucosal immunity, limited durability, and susceptibility to PRN− variants. Importantly, the use of clinically approved aP components provides a regulatory bridge for non-inferiority trial designs, offering a practical and accelerated path toward clinical translation.
In conclusion, the OMV+aP vaccine represents a next-generation pertussis candidate that is safe, highly immunogenic, and functionally superior to existing aP formulations. By simultaneously eliciting potent systemic and mucosal immunity, including TRM induction and IL-22–mediated mucosal defense, this strategy offers a feasible and strategically sound approach to strengthen global pertussis control and reduce transmission.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal protocols were authorized by the Ethical Committee for Animal Experiments of the Faculty of Science at La Plata National University (approval number 004-06-15, 003-06-15 extended its validity until August 10, 2027). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
LL: Formal analysis, Methodology, Writing – review & editing. DB: Formal analysis, Methodology, Writing – review & editing, Investigation. FC: Methodology, Writing – review & editing. OL: Methodology, Writing – review & editing. BP: Methodology, Writing – review & editing. EZ: Methodology, Writing – review & editing, Supervision. PMA: Methodology, Writing – review & editing. MG: Methodology, Writing – review & editing. DH: Methodology, Writing – review & editing, Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Validation, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by ANCPyT (PICT 2020 3034) PIP 3022, BactiVac grants to DH. Since the beginning of 2024, the Argentinian government has decided to defund science, and as a result, the money from our grants has not been transferred to the funding units of our institutions so far. DH, DB, MG, and EZ are members of the Scientific Career of CONICET. BP, LL, OL, PMA and FC are felows from CONICET.
Acknowledgments
Alejandro Fernández, Luciana Cayuela and Magali Gabrielli provided excellent technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1655910/full#supplementary-material
Supplementary Figure 1Gating strategy for the flow cytometric analysis of tissue-resident memory (TRM) CD4+ T cells in lungs and nasal tissue. To discriminate tissue-localized lymphocytes from circulating cells, mice received an intravenous injection of PE-conjugated anti-mouse CD45 antibody (eBioscience) 10 min prior to euthanasia, as previously described (60). Lungs and nasal tissues were enzymatically digested for 1 h at 37 °C using Collagenase D (1 mg/mL; Sigma-Aldrich) and DNase I (20 U/mL; Sigma-Aldrich). Following red blood cell lysis, single-cell suspensions were incubated with CD16/CD32 FcγRIII (1:100) to block Fc receptors, stained with LIVE/DEAD Aqua (Invitrogen), and subsequently labeled with the following antibodies: CD45.2-BV650 (BD), CD3-PeCy7 (BD), CD4-FITC (Invitrogen), CD44-PE (BD), CD62L-PE-CF594 (BD), CD103-APC-EF780 (Invitrogen), and CD69-APC (Invitrogen). TRM CD4+ T cells were defined as CD45− CD44+ CD62L− CD69+ CD103+/− CD4+. Fluorescence minus one (FMO) and isotype control antibodies were used to validate gating. Flow cytometric acquisition was performed on an LSR Fortessa using Diva software (BD Biosciences), and data were analyzed with FlowJo (TreeStar).
References
1
Celentano LP Massari M Paramatti D Salmaso S Tozzi AE . Resurgence of pertussis in europe. Pediatr Infect Dis J. (2005) 24:761–5. doi: 10.1097/01.inf.0000177282.53500.77
2
Cherry JD . Epidemic pertussis in 2012 — The resurgence of a vaccine-preventable disease. New Engl J Med. (2012) 367:785–7. doi: 10.1056/NEJMp1209051
3
Zhang Y Bambrick H Mengersen K Tong S Feng L Zhang L et al . Resurgence of pertussis infections in Shandong, China: Space-time cluster and trend analysis. Am J Trop Med Hygiene. (2019) 100:1342–54. doi: 10.4269/ajtmh.19-0013
4
Grilc E Pirnat N . Pertussis outbreak in recently vaccinated children in a kindergarten in Ljubljana during a resurgence in pertussis incidence. Euro Surveill. (2005) 10:E050818.4. doi: 10.2807/esw.10.33.02779-en
5
Fedele G Stefanelli P . Pertussis in infants and the resurgence of a vaccine preventable disease: What to do? Ann Ist Super Sanita. (2017) 53:100–3. doi: 10.4415/ANN_17_02_04
6
Nordholm AC Emborg HD Norgaard SK Nygaard U Ronayne A Nielsen LB et al . Pertussis epidemic in Denmark, august 2023 to february 2024. Euro Surveill. (2024) 29:1–10. doi: 10.2807/1560-7917.ES.2024.29.14.2400160
7
Plotkin SA . The pertussis problem. Clin Infect Dis. (2014) 58:830–3. doi: 10.1093/CID/CIT934
8
Khalil A Samara A Campbell H Ladhani SN Amirthalingam G . Recent increase in infant pertussis cases in Europe and the critical importance of antenatal immunizations: We must do better … now. Int J Infect Dis. (2024) 146:148–153. doi: 10.1016/J.IJID.2024.107148
9
Meng Q Shi W Hu Y Yao K . Pertussis in infants: Alarm lights and amplifiers for persistent community transmission. J Infect. (2024) 89:62–64. doi: 10.1016/J.JINF.2024.106219
10
Kmietowicz Z . Whooping cough: Nine infants have died in England in latest outbreak. BMJ. (2024) 386:q1545. doi: 10.1136/BMJ.Q1545
11
Sabbe M Vandermeulen C . The resurgence of mumps and pertussis. Hum Vaccin Immunother. (2016) 12:955–9. doi: 10.1080/21645515.2015.1113357
12
Fullen AR Yount KS Dubey P Deora R . Whoop! There it is: The surprising resurgence of pertussis. PloS Pathog. (2020) 16:e1008625. doi: 10.1371/journal.ppat.1008625
13
Mooi FR van der Maas NAT De Melker HE . Pertussis resurgence: Waning immunity and pathogen adaptation - Two sides of the same coin. Epidemiol Infect. (2014) 142:685–94. doi: 10.1017/S0950268813000071
14
Szwejser-Zawislak E Wilk MM Piszczek P Krawczyk J Wilczyńska D Hozbor D . Evaluation of whole-cell and acellular pertussis vaccines in the context of long-term herd immunity. Vaccines (Basel). (2022) 11:1. doi: 10.3390/vaccines11010001
15
Martin SW Pawloski L Williams M Weening K Debolt C Qin X et al . Pertactin-negative bordetella pertussis strains: Evidence for a possible selective advantage. Clin Infect Dis. (2015) 60:223–7. doi: 10.1093/cid/ciu788
16
Lesne E Cavell BE Freire-Martin I Persaud R Alexander F Taylor S et al . Acellular pertussis vaccines induce anti-pertactin bactericidal antibodies which drives the emergence of pertactin-negative strains. Front Microbiol. (2020) 11:2108. doi: 10.3389/fmicb.2020.02108
17
Xu Z Octavia S Luu LDW Payne M Timms V Tay CY et al . Pertactin-negative and filamentous hemagglutinin-negative Bordetella pertussis, Australia, 2013 - 2017. Emerg Infect Dis. (2019) 25:1196–9. doi: 10.3201/eid2506.180240
18
Lam C Octavia S Ricafort L Sintchenko V Gilbert GL Wood N et al . Rapid increase in pertactin-deficient Bordetella pertussis isolates, Australia. Emerg Infect Dis. (2014) 20:626–33. doi: 10.3201/eid2004.131478
19
Ma L Caulfield A Dewan KK Harvill ET . Pertactin-deficient bordetella pertussis, vaccine-driven evolution, and reemergence of pertussis. Emerg Infect Dis. (2021) 27:1561–6. doi: 10.3201/EID2706.203850
20
Weigand MR Peng Y Cassiday PK Loparev VN Johnson T Juieng P et al . Complete genome sequences of bordetella pertussis isolates with novel pertactin-deficient deletions. Genome Announc. (2017) 5:1–2. doi: 10.1128/genomeA.00973-17
21
Warfel JM Zimmerman LI Merkel TJ . Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proc Natl Acad Sci U.S.A. (2014) 111:787–92. doi: 10.1073/pnas.1314688110
22
Causey K Fullman N Sorensen RJD Galles NC Zheng P Aravkin A et al . Estimating global and regional disruptions to routine childhood vaccine coverage during the COVID-19 pandemic in 2020: a modelling study. Lancet. (2021) 398(10299):522–534. doi: 10.1016/S0140-6736(21)01337-4
23
Aguinaga-Ontoso I Guillen-Aguinaga S Guillen-Aguinaga L Alas-Brun R Guillen-Aguinaga M Onambele L et al . The impact of COVID - 19 on DTP3 vaccination coverage in europe (2012–2023). Vaccines (Basel). (2025) 13:1–43. doi: 10.3390/VACCINES13010006
24
Hozbor D . New pertussis vaccines: A need and a challenge. Adv Exp Med Biol. (2019) 1183:115–26. doi: 10.1007/5584_2019_407
25
Locht C . Pertussis: Where did we go wrong and what can we do about it? J Infect. (2016) 72 Suppl:S34–40. doi: 10.1016/j.jinf.2016.04.020
26
Locht C . Live pertussis vaccines: will they protect against carriage and spread of pertussis? Clin Microbiol Infection. (2016) 22:S96–S102. doi: 10.1016/j.cmi.2016.05.029
27
Brady MT Mahon BP Mills KHG . Pertussis infection and vaccination induces Th1 cells [2. Immunol Today. (1998) 19:534. doi: 10.1016/S0167-5699(98)01359-0
28
Brummelman J Wilk MM Han WG van Els CA Mills KH . Roads to the development of improved pertussis vaccines paved by immunology. Pathog Dis. (2015) 73:ftv067. doi: 10.1093/FEMSPD/FTV067
29
Breakwell L Kelso P Finley C Schoenfeld S Goode B Misegades LK et al . Pertussis vaccine effectiveness in the setting of pertactin-deficient pertussis. Pediatrics. (2016) 137:e20153973. doi: 10.1542/PEDS.2015-3973
30
Safarchi A Octavia S Luu LDW Tay CY Sintchenko V Wood N et al . Pertactin negative Bordetella pertussis demonstrates higher fitness under vaccine selection pressure in a mixed infection model. Vaccine. (2015) 33:6277–81. doi: 10.1016/j.vaccine.2015.09.064
31
Zurita ME Wilk MM Carriquiriborde F Bartel E Moreno G Misiak A et al . A pertussis outer membrane vesicle-based vaccine induces lung-resident memory CD4 T cells and protection against bordetella pertussis, including pertactin deficient strains. Front Cell Infect Microbiol. (2019) 9:125. doi: 10.3389/fcimb.2019.00125
32
Peng Y Yin S Wang M . Extracellular vesicles of bacteria as potential targets for immune interventions. Hum Vaccin Immunother. (2021) 17:897–903. doi: 10.1080/21645515.2020.1799667
33
Curley SM Putnam D . Biological nanoparticles in vaccine development. Front Bioeng Biotechnol. (2022) 10:867119. doi: 10.3389/FBIOE.2022.867119
34
Demento SL Siefert AL Bandyopadhyay A Sharp FA Fahmy TM . Pathogen-associated molecular patterns on biomaterials: A paradigm for engineering new vaccines. Trends Biotechnol. (2011) 29:294–306. doi: 10.1016/J.TIBTECH.2011.02.004
35
Tiku V Tan MW . Host immunity and cellular responses to bacterial outer membrane vesicles. Trends Immunol. (2021) 42:1024–36. doi: 10.1016/j.it.2021.09.006
36
Rumbo M Hozbor D . Development of improved pertussis vaccine. Landes Bioscience. (2014) 10:2450–3. doi: 10.4161/hv.29253
37
Roberts R Moreno G Bottero D Gaillard ME Fingermann M Graieb A et al . Outer membrane vesicles as acellular vaccine against pertussis. Vaccine. (2008) 26:4639–4646. doi: 10.1016/j.vaccine.2008.07.004
38
Bottero D Gaillard MEE Errea A Moreno G Zurita E Pianciola L et al . Outer membrane vesicles derived from Bordetella parapertussis as an acellular vaccine against Bordetella parapertussis and Bordetella pertussis infection. Vaccine. (2013) 31:5262–8. doi: 10.1016/j.vaccine.2013.08.059
39
Elizagaray ML Gomes MTR Guimaraes ES Rumbo M Hozbor DF Oliveira SC et al . Canonical and non-canonical inflammasome activation by outer membrane vesicles derived from bordetella pertussis. Front Immunol. (2020) 11:1879. doi: 10.3389/fimmu.2020.01879
40
Asensio CJA Gaillard ME Moreno G Bottero D Zurita E Rumbo M et al . Outer membrane vesicles obtained from Bordetella pertussis Tohama expressing the lipid a deacylase PagL as a novel acellular vaccine candidate. Vaccine. (2011) 29:1649–1656. doi: 10.1016/j.vaccine.2010.12.068
41
Carriquiriborde F Martin Aispuro P Ambrosis N Zurita E Bottero D Gaillard ME et al . Pertussis vaccine candidate based on outer membrane vesicles derived from biofilm culture. Front Immunol. (2021) 12:730434. doi: 10.3389/fimmu.2021.730434
42
Ormazábal M Bartel E Gaillard ME Bottero D Errea A Zurita ME et al . Characterization of the key antigenic components of pertussis vaccine based on outer membrane vesicles. Vaccine. (2014) 32. doi: 10.1016/j.vaccine.2014.08.084
43
Bottero D Gaillard MEE Zurita E Moreno G Martinez DSS Bartel E et al . Characterization of the immune response induced by pertussis OMVs-based vaccine. Vaccine. (2016) 34:3303–9. doi: 10.1016/j.vaccine.2016.04.079
44
Gaillard ME Bottero D Errea A Ormazábal M Zurita ME Moreno G et al . Acellular pertussis vaccine based on outer membrane vesicles capable of conferring both long-lasting immunity and protection against different strain genotypes. Vaccine. (2014) 32:931–937. doi: 10.1016/j.vaccine.2013.12.048
45
Kanojia G Raeven RHM van der Maas L Bindels THE van Riet E Metz B et al . Development of a thermostable spray dried outer membrane vesicle pertussis vaccine for pulmonary immunization. J Controlled Release. (2018) 286:167–78. doi: 10.1016/j.jconrel.2018.07.035
46
Raeven RHM Rockx-Brouwer D Kanojia G van der Maas L Bindels THE ten Have R et al . Intranasal immunization with outer membrane vesicle pertussis vaccine confers broad protection through mucosal IgA and Th17 responses. Sci Rep. (2020) 10:7396. doi: 10.1038/S41598-020-63998-2
47
Soltani MS Noofeli M Banihashemi SR Shahcheraghi F Eftekhar F . Evaluation of outer membrane vesicles obtained from predominant local isolate of bordetella pertussis as a vaccine candidate. Iran BioMed J. (2021) 25:399. doi: 10.52547/IBJ.25.6.399
48
Rudi E Gaillard E Bottero D Ebensen T Guzman CA Hozbor D . Mucosal vaccination with outer membrane vesicles derived from Bordetella pertussis reduces nasal bacterial colonization after experimental infection. Front Immunol. (2024) 15:1506638/PDF. doi: 10.3389/FIMMU.2024.1506638/PDF
49
Pschunder B Locati L López O Martin Aispuro P Zurita E Stuible M et al . Outer membrane vesicles derived from Bordetella pertussis are potent adjuvant that drive Th1-biased response. Front Immunol. (2024) 15:1387534. doi: 10.3389/fimmu.2024.1387534
50
Galeas-Pena M Hirsch A Kuang E Hoffmann J Gellings P Brown JB et al . A novel outer membrane vesicle adjuvant improves vaccine protection against Bordetella pertussis. NPJ Vaccines. (2024) 9:190–198. doi: 10.1038/S41541-024-00990-1
51
Pawloski LC Queenan AM Cassiday PK Lynch AS Harrison MJ Shang W et al . Prevalence and molecular characterization of pertactin-deficient Bordetella pertussis in the United States. Clin Vaccine Immunol. (2014) 21:119–25. doi: 10.1128/CVI.00717-13
52
Bowden KE Williams MM Cassiday PK Milton A Pawloski L Harrison M et al . Molecular epidemiology of the pertussis epidemic in washington state in 2012. J Clin Microbiol. (2014) 52:3549–57. doi: 10.1128/JCM.01189-14
53
Carriquiriborde F Regidor V Aispuro PM Magali G Bartel E Bottero D et al . Rare detection of bordetella pertussis pertactin-deficient strains in Argentina. Emerg Infect Dis. (2019) 25:2043–2046. doi: 10.3201/eid2511.190329
54
Stainer DW Scholte MJ . A simple chemically defined medium for the production of phase I Bordetella pertussis. J Gen Microbiol. (1970) 63:211–20. doi: 10.1099/00221287-63-2-211
55
Hozbor D Rodriguez MEE Fernández J Lagares A Guiso N Yantorno O . Release of outer membrane vesicles from Bordetella pertussis. Curr Microbiol. (1999) 38:273–8. doi: 10.1007/PL00006801
56
Kielkopf CL Bauer W Urbatsch IL . Bradford assay for determining protein concentration. Cold Spring Harb Protoc. (2020) 2020:136–8. doi: 10.1101/pdb.prot102269
57
WHO Expert Committee on biological standardization . World Health Organ Tech Rep Ser (2007). Available online at (Accessed June 16, 2025).
58
Stoddard MB Pinto V Keiser PB Zollinger W . Evaluation of a whole-blood cytokine release assay for use in measuring endotoxin activity of group B neisseria meningitidis vaccines made from lipid A acylation mutants. Clin Vaccine Immunol. (2010) 17:98. doi: 10.1128/CVI.00342-09
59
Martin Aispuro P Ambrosis N Zurita ME Gaillard ME Bottero D Hozbor DF . Use of a neonatal-mouse model to characterize vaccines and strategies for overcoming the high susceptibility and severity of pertussis in early life. Front Microbiol. (2020) 11:723. doi: 10.3389/fmicb.2020.00723
60
Anderson KG Mayer-Barber K Sung H Beura L James BR Taylor JJ et al . Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. (2014) 9:209–22. doi: 10.1038/NPROT.2014.005
61
Bar-On ES Goldberg E Hellmann S Leibovici L . Combined DTP-HBV-HIB vaccine versus separately administered DTP-HBV and HIB vaccines for primary prevention of diphtheria, tetanus, pertussis, hepatitis B and Haemophilus influenzae B (HIB). Cochrane Database Systematic Rev. (2012) 2012:CD005530. doi: 10.1002/14651858.CD005530.PUB3
62
Hozbor DF . Outer membrane vesicles: an attractive candidate for pertussis vaccines. London: Taylor and Francis Ltd (2017). doi: 10.1080/14760584.2017.1276832
63
Solans L Debrie AS Borkner L Aguilo N Thiriard A Coutte L et al . IL - 17-dependent SIgA-mediated protection against nasal Bordetella pertussis infection by live attenuated BPZE1 vaccine. Mucosal Immunol. (2018) 11:1–10. doi: 10.1038/s41385-018-0073-9
64
Esposito S Principi N . Prevention of pertussis: An unresolved problem. Hum Vaccin Immunother. (2018) 14:2452–9. doi: 10.1080/21645515.2018.1480298
65
Ní Chasaide C Schmitt P Diallo BK Borkner L Leane CM Jazayeri SD et al . Acellular pertussis vaccines induce CD8+ and CD4+ Regulatory T cells that suppress protective tissue-resident memory CD4+ T cells, in part via IL - 10. Eur J Immunol. (2025) 55:e51630. doi: 10.1002/EJI.202451630
66
Prygiel M Mosiej E Wdowiak K Górska P Polak M Lis K et al . Effectiveness of experimental and commercial pertussis vaccines in the elimination of Bordetella pertussis isolates with different genetic profiles in murine model. Med Microbiol Immunol. (2021) 210:251–62. doi: 10.1007/S00430-021-00718-1
67
Raeven RH van der Maas L Tilstra W Uittenbogaard JP Bindels TH Kuipers B et al . Immunoproteomic profiling of bordetella pertussis outer membrane vesicle vaccine reveals broad and balanced humoral immunogenicity. J Proteome Res. (2015) 14:2929–42. doi: 10.1021/ACS.JPROTEOME.5B00258
68
Chasaide CN Mills KHG . Next-generation pertussis vaccines based on the induction of protective t cells in the respiratory tract. Vaccines (Basel). (2020) 8:1–28. doi: 10.3390/vaccines8040621
69
Wilk MM Borkner L Misiak A Curham L Allen AC Mills KHG . Immunization with whole cell but not acellular pertussis vaccines primes CD4 TRM cells that sustain protective immunity against nasal colonization with Bordetella pertussis. Emerg Microbes Infect. (2019) 8:169–85. doi: 10.1080/22221751.2018.1564630
70
Ngo VL Abo H Maxim E Harusato A Geem D Medina-Contreras O et al . A cytokine network involving IL - 36γ, IL - 23, and IL - 22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc Natl Acad Sci U.S.A. (2018) 115:E5076–85. doi: 10.1073/PNAS.1718902115
71
Corbière V Lambert EE Rodesch M van Gaans-van den Brink JAM Misiak A Simonetti E et al . A semi high-throughput whole blood-based flow cytometry assay to detect and monitor Bordetella pertussis-specific Th1, Th2 and Th17 responses. Front Immunol. (2023) 14:1101366/PDF. doi: 10.3389/FIMMU.2023.1101366/PDF
72
Wilk MM Mills KHG . CD4 TRM cells following infection and immunization: Implications for more effective vaccine design. Front Immunol. (2018) 9:1860. doi: 10.3389/fimmu.2018.01860
73
Solans L Locht C . The role of mucosal immunity in pertussis. Front Immunol. (2019) 10:3068. doi: 10.3389/fimmu.2018.03068
Summary
Keywords
Bordetella pertussis , outer-membrane vesicles, pertussis, combined vaccine, Th1, modulator, CD4+TRM cells
Citation
Locati L, Bottero D, Carriquiriborde F, López O, Pschunder B, Zurita E, Martin Aispuro P, Gaillard ME and Hozbor D (2025) Harnessing outer membrane vesicles derived from Bordetella pertussis to overcome key limitations of acellular pertussis vaccines. Front. Immunol. 16:1655910. doi: 10.3389/fimmu.2025.1655910
Received
28 June 2025
Accepted
19 August 2025
Published
02 September 2025
Volume
16 - 2025
Edited by
Leonard Peruski, Wadsworth Center, United States
Reviewed by
Peter Van Der Ley, Intravacc, Netherlands
Martine Chabaud-Riou, Sanofi Pasteur, France
Michelle Galeas-Pena, Tulane University School of Medicine, United States
Updates
Copyright
© 2025 Locati, Bottero, Carriquiriborde, López, Pschunder, Zurita, Martin Aispuro, Gaillard and Hozbor.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Hozbor, hozbor.daniela@gmail.com; hozbor@biol.unlp.edu.ar
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.