 Ji Wang1†
Ji Wang1† Yiyuan Xia
Yiyuan Xia- 1Hubei Key Laboratory of Cognitive and Affective Disorders, Jianghan University, Wuhan, Hubei, China
- 2School of Fine Arts and Design, Hunan City University, Yiyang, Hunan, China
- 3Postdoctoral Mobile Station of Journalism and Communication, Hunan Normal University, Changsha, Hunan, China
- 4Hubei Provincial Demonstration Center for Experimental Medicine Education, School of Medicine, Jianghan University, Wuhan, Hubei, China
CCAAT/enhancer-binding protein beta (C/EBPβ), a key transcription factor, plays a central role in regulating inflammasome signaling in neurodegenerative diseases (NDs). This review synthesizes the mechanisms by which C/EBPβ modulates neuroinflammation and its potential as a therapeutic target. We conducted a comprehensive systematic review spanning January 1995 to June 2025, systematically querying Google Scholar and PubMed with the following keywords: neuroinflammation, inflammasome activation, C/EBPβ, therapeutic targeting, and neurodegenerative diseases. C/EBPβ exists in three isoforms-LAP1, LAP2, and LIP-each with distinct functions in inflammasome activation. In Alzheimer’s disease (AD), C/EBPβ drives tau cleavage and Aβ pathology through the AEP axis and exacerbates neuroinflammation by upregulating APOE4. In Parkinson’s disease (PD), C/EBPβ silencing reduces α-synuclein aggregation and dopaminergic neuron loss by suppressing the NLRP3 inflammasome. In Amyotrophic Lateral Sclerosis (ALS), C/EBPβ is hypothesized to contribute to TDP-43-associated inflammasome activation, though this requires further validation. In Multiple Sclerosis (MS), C/EBPβ may influence microglial activation and neuroinflammation, as shown in experimental autoimmune encephalomyelitis models. Modulators of the C/EBPβ-inflammasome axis include endogenous regulators like gut-derived metabolites and pharmacological interventions such as small-molecule inhibitors. Therapeutic strategies targeting C/EBPβ hold promise for mitigating neuroinflammation and neurodegeneration, though challenges remain in achieving isoform-specific targeting and blood-brain barrier penetration. Future directions include CRISPR-based editing and biomarker development for personalized therapies.
1 Introduction
1.1 Overview of neuroinflammation and inflammasomes in neurodegenerative diseases
Neurodegenerative diseases (NDs), including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS), are characterized by progressive neuronal loss and functional decline in the central nervous system (CNS). A hallmark shared across these disorders is chronic neuroinflammation, driven by dysregulated immune responses and sustained activation of innate immune pathways (1–3). Central to this process are inflammasomes, multiprotein complexes that orchestrate inflammatory signaling and contribute to neuronal damage (4).
Neuroinflammation initially serves as a protective mechanism aimed at eliminating pathogens and cellular debris. Resident CNS immune cells, such as microglia and astrocytes, detect danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and Nod-like receptors (NLRs) (5). Acute activation of these pathways promotes tissue repair and homeostasis. However, persistent stimuli-such as misfolded protein aggregates (e.g., amyloid-β [Aβ] in AD, α-synuclein in PD), oxidative stress, or mitochondrial dysfunction-trigger chronic neuroinflammation. This leads to the sustained release of pro-inflammatory cytokines (e.g., IL-1β, IL-18), chemokines, and reactive oxygen species (ROS) (6–8). This chronic state exacerbates neuronal death, synaptic dysfunction, and blood-brain barrier (BBB) disruption, creating a self-perpetuating cycle of neurodegeneration (9–13).
In AD, the presence of abnormally phosphorylated tau protein and extracellular deposits of Aβ peptide are key pathological features (14). These deposits activate microglia and astrocytes, leading to the release of pro-inflammatory cytokines and neurotoxicity (15–17). In PD, the misfolding and aggregation of α-synuclein due to oxidative stress result in the accumulation of toxic protein aggregates (18, 19). This triggers a cascade of pro-inflammatory events in microglia and astrocytes, amplifying neuronal loss and persistent neurodegeneration (20, 21).
The NLRP3 inflammasome is a critical player in neuroinflammation, activated by DAMPs and PAMPs in microglia, astrocytes, and neurons (22–24). Activation of the NLRP3 inflammasome leads to the maturation and release of pro-inflammatory cytokines such as IL-1β and IL-18, contributing to neuroinflammation and neuronal damage (25). Inhibiting the NLRP3 inflammasome has been proposed as a potential therapeutic strategy to counteract neurodegenerative diseases (1, 26). Most evidence and therapeutic concepts presented here derive from in vitro systems or animal models. While indispensable for mechanistic insight and proof-of-concept, these data must be validated in rigorously designed clinical trials before any therapeutic claims can be extended to human disease.
1.2 Introduction to C/EBPβ: structure, isoforms, and physiological roles
CCAAT/enhancer-binding protein beta (C/EBPβ) is a member of the C/EBP family of transcription factors that play crucial roles in various physiological processes (27). As a leucine-zipper (bZIP) transcription factor, C/EBPβ binds DNA as dimers and regulates the transcription of genes containing specific T(TG)NNGNAA(TG) motifs (28, 29). The uniqueness of the C/EBPβ gene lies in its lack of introns and its ability to be alternatively translated into three major isoforms: liver activator protein 1(LAP1), liver activator protein 2(LAP2), and liver inhibitor protein (LIP) (30). These isoforms arise through alternative translation initiation sites, leading to differences in their N-terminal regions (30, 31).
LAP1 and LAP2 are both transcriptional activators, with LAP2 being a stronger transactivator than LAP1. This difference is attributed to the regulation of C/EBPβ protein tertiary structure and unique N-terminal protein-protein interactions (28, 32). LAP1 and LAP2 differ in their first 21 N-terminal amino acids due to internal translation initiation from the downstream LAP2 start codon (28, 30). In contrast, the LIP isoform lacks the N-terminal transactivation domain (TAD) and most of the negative regulatory domain, functioning primarily as a dominant-negative regulator of transcription. However, in some cellular contexts, LIP can act as a transcriptional activator by interacting with other cofactors (30, 33).
C/EBPβ is expressed in various tissues, including the liver, brain, intestine, and skin, and is involved in multiple physiological processes (34). It is essential for the differentiation of mammary epithelial and granulosa cells, macrophage function, and brown adipose tissue formation (35–37). C/EBPβ also plays a role in cell cycle arrest and differentiation in hepatocytes, hematopoietic cells, and adipocytes (37–39). Additionally, it is involved in apoptosis and senescence in microglia and neurons. For instance, methamphetamine (METH) upregulates C/EBPβ expression, thereby activating Lipocalin2 (an apoptosis-inducing factor) and leading to apoptosis in microglial cells. Silencing C/EBPβ can reverse this process (40, 41). METH induces neuronal autophagy and apoptosis through the C/EBPβ-DDIT4/TSC2/mTOR signaling axis and the Trib3/Parkin/α-syn pathway. Inhibition of C/EBPβ can mitigate neurotoxicity (42, 43). C/EBPβ regulates the pro-inflammatory program in microglia and is involved in the expression of several inflammatory genes in astrocytes (44, 45). It also plays a role in the regulation of the complement component 3 gene in neural cells, contributing to its pro-inflammatory effects (46).
1.3 Rationale for focusing on C/EBPβ-inflammasome axis: bridging transcriptional regulation and chronic inflammation in NDs
The C/EBPβ-inflammasome axis has emerged as a critical pathway linking transcriptional regulation to chronic inflammation in NDs (Table 1). This axis is particularly relevant in conditions such as AD, PD, ALS, and MS, where chronic neuroinflammation plays a significant role in disease progression (54).

Table 1. The molecular pathways of neurodegenerative diseases are regulated by C/EBPβ.
C/EBPβ plays a pivotal role in regulating the expression of genes involved in inflammasome activation, thereby linking transcriptional regulation to chronic inflammation. Inflammasomes, such as the NLRP3 inflammasome, are activated by various stimuli, including misfolded protein aggregates, oxidative stress, and mitochondrial dysfunction (55–58). For instance, C/EBPβ has been shown to directly bind to the promoter region of the SerpinB2 gene, which is crucial for LPS-induced transcription in macrophages. This binding is essential for driving transcription from the SerpinB2 promoter in response to LPS stimulation (59). Additionally, C/EBPβ regulates the expression of caspase-1, a key component of the non-canonical inflammasome pathway (60, 61). This regulation is critical for understanding how chronic inflammation is sustained in NDs.
C/EBPβ is also implicated in the regulation of mitochondrial function and the expression of mitochondrial transcription factor A (TFAM). In a cellular model of Parkinson’s disease using SH-SY5Y dopaminergic cells treated with the neurotoxin 6-hydroxydopamine (6-OHDA), C/EBPβ levels increased over time, reaching a peak at 18 hours when cells began to die due to stress. In contrast, TFAM expression decreased after 4 hours of treatment, followed by a partial recovery. This recovery is likely due to C/EBPβ’s activation of the TFAM promoter (62). Mitochondrial dysfunction, a common feature of NDs, contributes to chronic inflammation through the release of damage-associated molecular patterns (DAMPs) (63, 64). By regulating mitochondrial function, C/EBPβ can influence the inflammatory response and contribute to the pathogenesis of NDs.
C/EBPβ has been shown to regulate autophagy, a process crucial for the degradation of damaged mitochondria. In C/EBPβ-silenced cells, there is an accumulation of autophagic markers under oxidative stress and inflammatory conditions, indicating that C/EBPβ is involved in the regulation of autophagy. This accumulation is not due to increased autophagy induction but rather to decreased autophagosome degradation (62, 65). This finding suggests that C/EBPβ may play a role in maintaining the balance between mitochondrial biogenesis and degradation, which is essential for cellular homeostasis.
2 C/EBPβ in inflammasome activation
2.1 Transcriptional control of inflammasome components
C/EBPβ is a key transcription factor that directly regulates the expression of critical inflammasome components by binding to their promoter regions. This direct regulation is essential for the expression and activation of inflammasomes, which play a crucial role in the inflammatory response in various diseases (66). For instance, C/EBPβ has been shown to regulate the expression of NLRP3, a key component of the inflammasome complex (60, 67). NLRP3 is activated by various stimuli, including misfolded proteins and oxidative stress, leading to the release of pro-inflammatory cytokines like IL-1β and IL-18 (68–70). Similarly, C/EBPβ also regulates the expression of AIM2, another inflammasome sensor that recognizes cytosolic DNA and forms a caspase-1-activating inflammasome (71, 72). Additionally, C/EBPβ directly controls the expression of caspase-1, a crucial enzyme in the inflammasome pathway that processes pro-IL-1β and pro-IL-18 into their active forms (60). The specific mechanism is illustrated in Figure 1A.
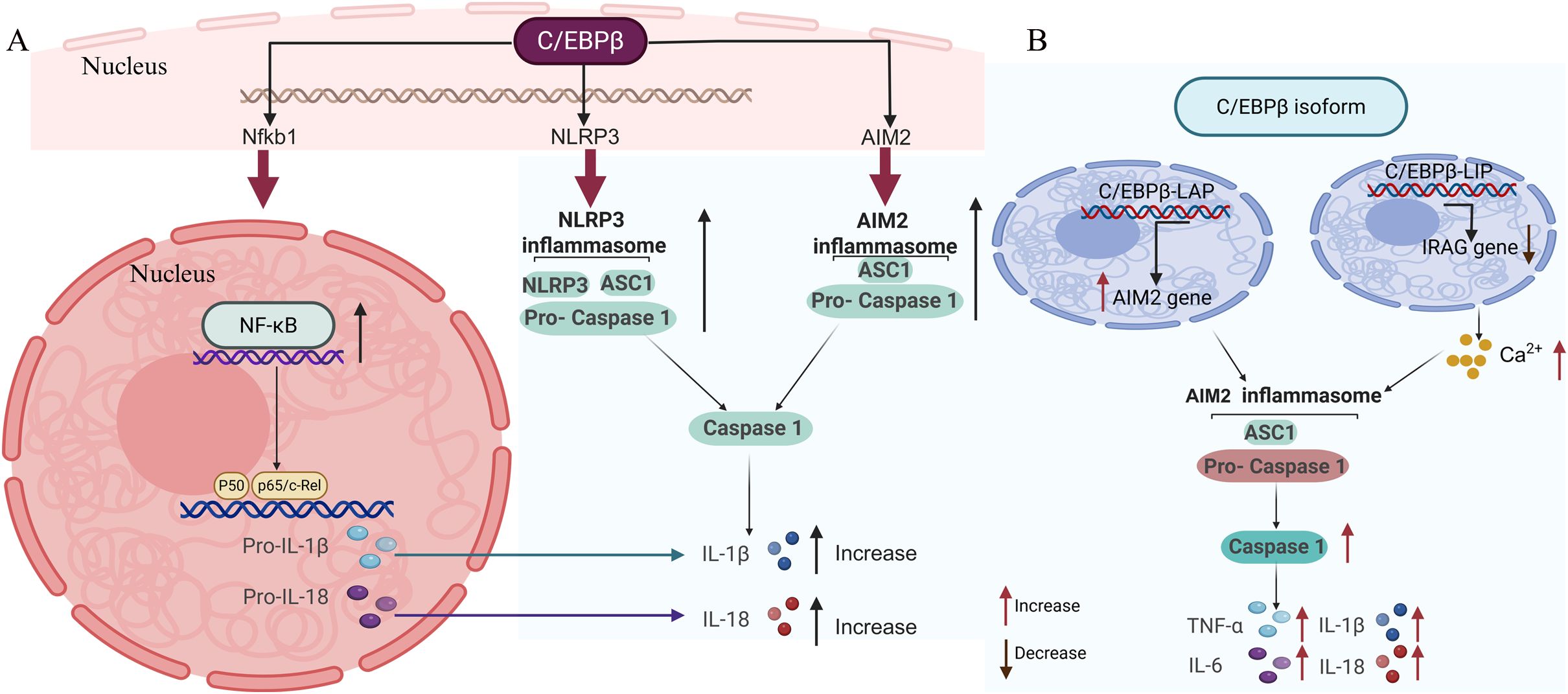
Figure 1. Transcriptional regulation of inflammasome activation by C/EBPb. C/EBPb governs the expression of NLRP3 and AIM2, which are pivotal inflammasome components responsible for triggering the cleavage of latent procaspase-1 into its active form, caspase-1. This activation facilitates the conversion of the cytokine precursors pro-IL-1b and pro-IL-18 into their mature, bioactive forms, IL-1b and IL-18, respectively. Beyond this, C/EBPb directly regulates caspase-1 expression and influences NF-kB expression, which in turn modulates the activation of pro-IL-1b and pro-IL-18. (B) The C/EBPb isoforms, C/EBPb-LAP and C/EBPb-LIP, drive the production of inflammatory factors by activating the AIM2 inflammasome. Created with BioRender.com.
C/EBPβ not only acts independently but also synergizes with other transcription factors, such as NF-κB, to amplify the production of pro-inflammatory cytokines (Figure 1A). This synergy is particularly evident in the regulation of IL-1β and IL-18, two key cytokines involved in the inflammatory response (73, 74). NF-κB is a well-known regulator of inflammatory genes, and its cooperation with C/EBPβ enhances the transcriptional activation of these genes. For example, studies have shown that C/EBPβ and NF-κB can bind to adjacent sites on the promoters of IL-1β and IL-18 genes, leading to a coordinated upregulation of these cytokines (60). This synergistic effect is crucial for the robust inflammatory response observed in conditions such as sepsis and neurodegenerative diseases (46, 62, 75, 76).
2.2 Isoform-specific mechanisms
C/EBPβ-LAP, the transcriptionally active isoform of C/EBPβ, is crucial for activating the AIM2 inflammasome by enhancing the expression of AIM2 and caspase-1 (Table 2). This mechanism has been observed in several contexts.

Table 2. The role of the C/EBPβ isoform in inflammation.
In Lupus Nephritis (LN), C/EBPβ-LAP activates the AIM2 inflammasome and induces podocyte pyroptosis. This activation is achieved by binding to the promoters of AIM2 and CASPASE1, thereby enhancing their expression (Figure 1B). Knockdown of AIM2 or caspase-1 reversed the effects of C/EBPβ-LAP overexpression, highlighting the critical role of these interactions in inflammasome activation (71). In the context of liver inflammation, C/EBPβ-LAP activates the transcription of various pro-inflammatory genes, including IL-6 and TNF. This activation is crucial for the acute phase response and the recruitment of immune cells to the site of inflammation (66, 77, 78).
In contrast to C/EBPβ-LAP, the C/EBPβ-LIP isoform functions primarily as a transcriptional inhibitor and has been shown to promote Ca²+-mediated inflammasome assembly by suppressing the expression of inositol-1,3,4-phosphat receptor associated G-kinase substrate (IRAG). Overexpression of C/EBPβ-LIP in LN transcriptionally inhibits IRAG, leading to increased Ca²+ release. This increase in Ca²+ levels facilitate the assembly and activation of the AIM2 inflammasome (71, 79, 80). This finding suggests that C/EBPβ-LIP not only regulates the expression of key inflammasome proteins but also affects their polymerization through the regulation of Ca²+ release (Figure 1B). In Myeloid-Derived Suppressor Cells (MDSCs), C/EBPβ-LIP suppresses the expression of immunosuppressive genes such as arginase-1 (Arg-1) and inducible nitric oxide synthase (iNOS). This suppression is achieved by blocking the activity of the transcriptionally active LAP-1 and LAP-2 isoforms (78). The balance between LIP and LAP isoforms is crucial for the regulation of MDSC function and the inflammatory response. In hepatocytes, C/EBPβ-LIP has been shown to downregulate the expression of cytochrome P450 enzymes (e.g., CYP3A4) by antagonizing the transactivation activity of LAP. This mechanism involves the binding of LIP to LAP (81, 82), preventing it from initiating transcription. This regulation is important for the metabolic response to inflammatory stimuli.
3 Role in specific neurodegenerative diseases
3.1 Alzheimer’s disease
C/EBPβ has been identified as a key player in the progression of AD. Over the past decade, numerous studies have elucidated its multifaceted role in the pathogenesis of AD. For instance, C/EBPβ has been shown to be a crucial transcription factor for APOE, preferentially mediating the expression of ApoE4, which is associated with an increased risk of AD (47). In addition to its role in Aβ production, C/EBPβ is implicated in the neurofibrillary pathology of AD. It has been demonstrated that C/EBPβ can upregulate the expression of certain proteins that mediate the cleavage of tau and APP, proteins implicated in the development of AD (48, 49). This suggests that C/EBPβ may contribute to both major pathological features of AD.
Moreover, C/EBPβ is involved in neuroinflammation, a critical component of AD pathology (83, 84). It is a key regulator of pro-inflammatory genes in microglia and is overexpressed in AD animal models and AD patients. There is a positive feedback loop between C/EBPβ and inflammatory components-inflammatory factors can activate C/EBPβ, which in turn further promotes the production of inflammatory factors (46, 66). This inflammatory response can further exacerbate neuronal damage and contribute to the progression of the disease. Chronic neuroinflammation activates the transcription factor C/EBPβ, which in turn up-regulates the cysteine protease asparagine endopeptidase (AEP) (85). Clinically, heightened AEP activity is documented in post-mortem AD brains, while mechanistic studies reveal that genetic ablation of C/EBPβ attenuates AD pathology via AEP suppression in animal models (86, 87). Importantly, AEP truncates tau at N368 and N255, yielding aggregation-prone fragments that precipitate neurofibrillary tangle formation—a defining feature of AD neurodegeneration (88).
In AD mouse models, knockdown of C/EBPβ significantly reduces the levels of inflammatory factors and the number of activated microglia. Conversely, overexpression of C/EBPβ exacerbates these pathological features (48). C/EBPβ isoforms can bind the promoter regions of inflammasome genes (e.g., caspase-1, NLRP3, and AIM2) via their DNA-binding domains and enhance transcription. While this suggests a potential regulatory role in inflammasome-mediated processes in AD, direct experimental confirmation is still lacking.
Recent studies have also highlighted the potential therapeutic implications of targeting C/EBPβ. For example, inhibiting the C/EBPβ/δ-secretase axis has been shown to reduce Aβ levels and improve cognitive function in animal models of AD (89). Decrease FOXO inhibition, reverse GABA neuron degeneration, maintaining the homeostasis of excitation inhibition balance (90). These findings suggest that interventions aimed at modulating C/EBPβ activity could represent a promising strategy for treating AD (Table 1).
3.2 Parkinson’s disease
Over the past decade, research has illuminated the multifaceted role of C/EBPβ in PD (51, 62). C/EBPβ is involved in the regulation of AEP, also known as legumain, a cysteine protease highly activated in the brains of PD patients (91). AEP cleaves α-synuclein (α-syn), promoting its aggregation and neurotoxicity, which contributes to the loss of dopaminergic neurons and motor deficits characteristic of PD (52, 92). Additionally, C/EBPβ acts as a transcription factor to upregulate α-syn and monoamine oxidase B (MAOB), both of which are implicated in PD pathogenesis in human wild-type α-Syn transgenic mice (51). This transcription factor can be activated by lipopolysaccharide (LPS) and inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α) (50). Therefore, gut microbiota dysbiosis and inflammation activation contribute to PD pathology through the C/EBPβ/AEP signaling pathway (52). In a study using a rotenone-induced PD mouse model, combined with antibiotic-induced microbiome depletion and fecal microbiota transplantation, it was found that gut microbiota dysbiosis, along with leaky gut-induced bacterial endotoxins, activates C/EBPβ/AEP signaling and α-syn pathology, ultimately leading to neurodegeneration in PD (52). This suggests that the gut microbiota may play a significant role in the activation of C/EBPβ/AEP signaling and the progression of PD.
Furthermore, silencing C/EBPβ has been shown to reduce α-synuclein aggregation and dopaminergic neuron loss. This effect is mediated through the suppression of the NLRP3 inflammasome (93, 94). By downregulating C/EBPβ, the expression of NLRP3 and other inflammasome components is reduced, thereby attenuating the inflammatory response and mitigating neuronal damage in an MPTP neurotoxic model of PD (55, 95). This finding suggests that targeting C/EBPβ could be a promising therapeutic strategy for reducing neuroinflammation and neurodegeneration in PD (Table 1).
3.3 Amyotrophic lateral sclerosis
TDP-43 (TAR DNA-binding protein 43) is a nuclear protein that regulates several RNA metabolic pathways. Dysregulation of TDP-43 induces its cytoplasmic accumulation and aggregation, which is a hallmark of ALS (96). The C/EBPβ expression in microglia has indeed been observed to increase in spinal cord of ALS animal models and human ALS patients (97).TDP-43 interacts with NF-κB, a key factor contributing to the inflammatory response, and activates NF-κB in microglia (98). Activated NF-κB induces the production of pro-inflammatory cytokines, contributing to neuroinflammation (97, 99). Considering that C/EBPβ is a key transcription factor for NF-κB,it may play a role in regulating these inflammatory pathways, thereby contributing to the neuroinflammatory response in ALS (100).
3.4 Multiple sclerosis
C/EBPβ has been shown to be involved in the inflammatory response in MS. Specifically, myelin basic protein-specific T cells, an autoantigen in MS, induce the expression of IL-1β, IL-1α, TNF-α, and IL-6 in microglial cells through a mechanism dependent on C/EBPβ activation (53). This suggests that C/EBPβ plays a crucial role in mediating the inflammatory response in MS (53). In the context of MS, C/EBPβ may contribute to the activation of the NLRP3 inflammasome, which is associated with the production of IL-1β and IL-18, key cytokines in neuroinflammation (101). This activation can lead to further recruitment of immune cells and exacerbation of the inflammatory process in MS. Additionally, studies have shown that C/EBPβ deficiency in myeloid cells can reshape microglial gene expression and is protective in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS (102). However, additional direct evidence is necessary to demonstrate that C/EBPβ regulates the inflammasome in MS pathogenesis. Furthermore, C/EBPβ has been implicated in the regulation of other inflammatory mediators such as HMGB1. HMGB1, a damage-associated molecular pattern (DAMP) protein, is known to interact with various receptors including RAGE, TLR2, TLR4, and TLR9, and can induce inflammatory responses (103–105), C/EBPβ can be activated by inflammatory stimuli such as lipopolysaccharide (LPS), and it has been shown to upregulate the expression of IL-1β, a cytokine that can be further enhanced by HMGB1 through its interaction with transcription factors like PU.1 (106–109). This suggests that C/EBPβ and HMGB1 can act in concert to amplify inflammatory signaling.
4 Modulators of the C/EBPβ-inflammasome axis
4.1 Endogenous regulators
Gut-derived metabolites, such as 12-HHTrE, have been identified as activators of the C/EBPβ-inflammasome axis. These metabolites can induce oxidative stress, which in turn activates C/EBPβ and promotes the expression of pro-inflammatory cytokines (110). Additionally, oxidative stress itself can act as an activator of this axis, contributing to the production of reactive oxygen species (ROS) and subsequent inflammatory responses (111–113). On the other hand, COP1-mediated ubiquitination and degradation of C/EBPβ in microglia have been reported as a mechanism to control the activity of this transcription factor (114). This process helps to regulate the inflammatory response by reducing the levels of active C/EBPβ, thereby limiting the activation of the inflammasome.
4.2 Pharmacological interventions
Small-molecule inhibitors have been developed to target components of the C/EBPβ-inflammasome axis (115, 116). For instance, an AEP inhibitor, Compound #11A, has been shown to reduce the activation of the inflammasome and mitigate neuroinflammation (117, 118). This pharmacological approach aims to block the downstream effects of C/EBPβ activation, thereby reducing the production of pro-inflammatory cytokines. Among pharmacologic NLRP3 inhibitors, the orally bioavailable agent ZYIL1 prevents ASC oligomerization; its Phase II trial in ALS has recently concluded. Likewise, VTX3232—an orally active, CNS-penetrant, and selective NLRP3 inhibitor—is currently undergoing Phase II evaluation in early-stage Parkinson’s disease (119).
Another strategy involves the use of lentiviral shRNA delivery systems to silence C/EBPβ expression. This method has been employed to specifically target and reduce the levels of C/EBPβ in cells, thereby attenuating the activation of the inflammasome and associated inflammatory responses (120, 121). This approach provides a potential therapeutic avenue for conditions where excessive activation of the C/EBPβ-inflammasome axis contributes to pathology.
5 Therapeutic implications and challenges
5.1 Preclinical success
Knockdown of C/EBPβ has shown promising results in preclinical models of AD and PD. Studies have demonstrated that reducing C/EBPβ levels can improve cognitive function and reduce pathological markers in these models. For instance, in PD models, C/EBPβ reduction has been shown to mitigate dopaminergic neuronal damage and glial activation, suggesting that C/EBPβ depletion could be a valuable therapeutic target for PD (52). Similarly, in AD models, the anti-inflammatory cytokine interferon-gamma acts as a potential therapeutic target of AD (122), and C/EBPβ knockdown has been associated with reduced amyloid-beta (Aβ) pathology and improved cognitive outcomes (47).
5.2 Translational barriers
Despite these encouraging preclinical findings, several translational barriers must be addressed to develop effective therapies targeting the C/EBPβ-inflammasome axis. One significant challenge is achieving isoform-specific targeting of C/EBPβ to avoid off-target effects. C/EBPβ has multiple isoforms, and targeting the wrong isoform could lead to unintended consequences (47, 123). Therefore, developing strategies to specifically target the pathogenic isoforms of C/EBPβ is crucial. High-throughput small-molecule screening can now be coupled with AI/ML-driven chemical-structure modelling, similarity-based target prediction, and cross-species transcriptomics to markedly reduce off-target liabilities.
Another major challenge is the blood-brain barrier (BBB) penetration for inhibitors. The BBB restricts the entry of many drugs into the central nervous system, making it difficult to deliver effective concentrations of therapeutic agents to the brain (124). Overcoming this barrier is essential for the successful translation of C/EBPβ inhibitors from preclinical to clinical settings. Nanobodies—single-domain antibodies of ~15 kDa—readily access cryptic epitopes inaccessible to conventional antibodies and can stabilize distinct protein conformations with exquisite specificity. Several nanobodies have already demonstrated brain penetrance (125) and the platform is rapidly gaining traction as a therapeutic modality for CNS disorders (126, 127).
5.3 Future directions
Looking forward, several innovative approaches hold promise for addressing these challenges. CRISPR-based isoform editing is a cutting-edge technology that could enable precise targeting of specific C/EBPβ isoforms, thereby reducing the risk of off-target effects (128–130). This approach could be particularly useful in selectively targeting the pathogenic isoforms of C/EBPβ in neurodegenerative diseases.
Additionally, the development of biomarkers for C/EBPβ activity could facilitate patient stratification and personalized medicine. Identifying reliable biomarkers that reflect C/EBPβ activity in vivo would allow for better selection of patients who are most likely to benefit from C/EBPβ-targeted therapies (131). This could enhance the success rate of clinical trials and improve patient outcomes.
6 Conclusion
In conclusion, the C/EBPβ-inflammasome axis provides a critical link between transcriptional regulation and chronic inflammation in NDs. Understanding this axis is essential for developing therapeutic strategies aimed at mitigating neuroinflammation and neurodegeneration. Future research should focus on elucidating the specific mechanisms by which C/EBPβ regulates inflammasome activation and identifying potential therapeutic targets within this axis.
C/EBPβ, a transcription factor, plays a vital role in regulating immune and inflammatory responses. It has been shown to directly target the promoter region of various genes, such as IL-1β, thereby contributing to the activation of the NLRP3 inflammasome (106, 132). This activation leads to the production of pro-inflammatory cytokines, which are key drivers of neuroinflammation in NDs. For instance, in models of Alzheimer’s disease and Parkinson’s disease, C/EBPβ knockdown has been shown to reduce pathological markers and improve cognitive function (133–136).
However, targeting C/EBPβ for therapeutic purposes presents several challenges. One major issue is achieving isoform-specific targeting to avoid off-target effects, as C/EBPβ has multiple isoforms with distinct functions. Additionally, delivering therapeutic agents across the blood-brain barrier remains a significant hurdle, as the BBB restricts the entry of many drugs into the central nervous system.
Future directions in research should include the development of isoform-specific inhibitors and strategies to enhance BBB penetration. CRISPR-based isoform editing could enable precise targeting of specific C/EBPβ isoforms, thereby reducing the risk of off-target effects. Moreover, the development of biomarkers for C/EBPβ activity could facilitate patient stratification and personalized medicine, allowing for better selection of patients who are most likely to benefit from C/EBPβ-targeted therapies.
In summary, the dual role of C/EBPβ as a transcriptional orchestrator and inflammasome amplifier underscores its potential as a therapeutic target for NDs. Emphasis on personalized therapeutic strategies targeting this axis could lead to more effective treatments for mitigating neuroinflammation and neurodegeneration. Future research should aim to overcome current translational barriers and explore innovative approaches to harness the therapeutic potential of targeting the C/EBPβ -inflammasome axis.
Author contributions
YX: Visualization, Conceptualization, Writing – original draft, Writing – review & editing, Funding acquisition. JW: Conceptualization, Investigation, Writing – original draft. YL: Writing – original draft, Visualization, Conceptualization.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. Hubei Provincial Natural Science Foundation of China for Distinguished Young Scholars (Grant No. 2022CFA104), Key Research and Development Program of Wuhan (2024020802030159) and the Scientific Research Project Funding of Jianghan University (2023ZDRC01) to YX.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Piancone F, La Rosa F, Marventano I, Saresella M, and Clerici M. The role of the inflammasome in neurodegenerative diseases. Molecules. (2021) 26:953. doi: 10.3390/molecules26040953
2. Chen Z, Shi J, and Li L. Application of single-cell sequencing technology and its clinical implications in Parkinson’s disease and Alzheimer’s disease: a narrative review. Advanced Technol Neurosci. (2025) 2:9–15. doi: 10.4103/ATN.ATN-D-24-00015
3. Elyaman W, Stern LJ, Jiang N, Dressman D, Bradley P, Klatzmann D, et al. Exploring the role of T cells in Alzheimer's and other neurodegenerative diseases: Emerging therapeutic insights from the T Cells in the Brain symposium. Alzheimers Dement. (2025) 21:e14548. doi: 10.1002/alz.14548
4. Wang Q, Yang S, Zhang X, Zhang S, Chen L, Wang W, et al. Inflammasomes in neurodegenerative diseases. Transl Neurodegener. (2024) 13:65. doi: 10.1186/s40035-024-00459-0
5. Suleiman Khoury Z, Sohail F, Wang J, Mendoza M, Raake M, Tahoor Silat M, et al. Neuroinflammation: A critical factor in neurodegenerative disorders. Cureus. (2024) 16:e62310. doi: 10.7759/cureus.62310
6. Jurcău MC, Andronie-Cioara FL, Jurcău A, Marcu F, Ţiţ DM, Paşcalău N, et al. The link between oxidative stress, mitochondrial dysfunction and neuroinflammation in the pathophysiology of alzheimer’s disease: therapeutic implications and future perspectives. Antioxidants. (2022) 11:2167. doi: 10.3390/antiox11112167
7. Picca A, Guerra F, Calvani R, Romano R, Coelho-Júnior HJ, Bucci C, et al. Mitochondrial dysfunction, protein misfolding and neuroinflammation in parkinson’s disease: roads to biomarker discovery. Biomolecules. (2021) 11:1508. doi: 10.3390/biom11101508
8. Pisanti S, Rimondi E, Pozza E, Melloni E, Zauli E, Bifulco M, et al. Prenylation defects and oxidative stress trigger the main consequences of neuroinflammation linked to mevalonate pathway deregulation. Int J Environ Res Public Health. (2022) 19:9061. doi: 10.3390/ijerph19159061
9. Frank-Cannon TC, Alto LT, McAlpine FE, and Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegeneration. (2009) 4:47. doi: 10.1186/1750-1326-4-47
10. Gao H-M and Hong J-S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. (2008) 29:357–65. doi: 10.1016/j.it.2008.05.002
11. Amelimojarad M, Amelimojarad M, and Cui X. The emerging role of brain neuroinflammatory responses in Alzheimer’s disease. Front Aging Neurosci. (2024) 16. doi: 10.3389/fnagi.2024.1391517
12. Yang J, Ran M, Li H, Lin Y, Ma K, Yang Y, et al. New insight into neurological degeneration: Inflammatory cytokines and blood–brain barrier. Front Mol Neurosci. (2022) 15. doi: 10.3389/fnmol.2022.1013933
13. Mou Y, Du Y, Zhou L, Yue J, Hu X, Liu Y, et al. Gut microbiota interact with the brain through systemic chronic inflammation: implications on neuroinflammation, neurodegeneration, and aging. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.796288
14. Liu E, Zhang Y, and Wang J-Z. Updates in Alzheimer's disease: from basic research to diagnosis and therapies. Trans Neurodegeneration. (2024) 13:45. doi: 10.1186/s40035-024-00432-x
15. Dhapola R, Hota SS, Sarma P, Bhattacharyya A, Medhi B, and Reddy DH. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology. (2021) 29:1669–81. doi: 10.1007/s10787-021-00889-6
16. Zhang H, Wei W, Zhao M, Ma L, Jiang X, Pei H, et al. Interaction between Aβ and tau in the pathogenesis of alzheimer's disease. Int J Biol Sci. (2021) 17:2181–92. doi: 10.7150/ijbs.57078
17. Wang X, Liu P, Alterovitz G, Zhou S, Grinstaff MW, Brody DL, et al. Tryptophan metabolism in alzheimer’s disease with the involvement of microglia and astrocyte crosstalk and gut-brain axis. Aging Dis. (2024) 15:2168–90. doi: 10.14336/AD.2024.0134
18. Ganguly G, Chakrabarti S, Chatterjee U, and Saso L. Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer's disease and Parkinson's disease. Drug Design Dev Ther. (2017) 11:797–810. doi: 10.2147/DDDT.S130514
19. Dong-Chen X, Yong C, Yang X, Chen-Yu S, and Li-Hua P. Signaling pathways in Parkinson’s disease: molecular mechanisms and therapeutic interventions. Signal Transduction Targeted Ther. (2023) 8:73. doi: 10.1038/s41392-023-01353-3
20. Rasheed M, Liang J, Wang C, Deng Y, and Chen Z. Epigenetic regulation of neuroinflammation in parkinson’s disease. Int J Mol Sci. (2021) 22:4956. doi: 10.3390/ijms22094956
21. Palumbo L, Carinci M, Guarino A, Asth L, Zucchini S, Missiroli S, et al. The NLRP3 inflammasome in neurodegenerative disorders: insights from epileptic models. Biomedicines. (2023) 11:2825. doi: 10.3390/biomedicines11102825
22. Yu J, Zhao Z, Li Y, Chen J, Huang N, and Luo Y. Role of NLRP3 in Parkinson's disease: Specific activation especially in dopaminergic neurons. Heliyon. (2024) 10:e28838. doi: 10.1016/j.heliyon.2024.e28838
23. Freeman L, Guo H, David CN, Brickey WJ, Jha S, and Ting JPY. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. (2017) 214:1351–70. doi: 10.1084/jem.20150237
24. Zhang M, Guo B, Zhang X, Han D, Lv L, Yan X, et al. IFP35, a novel DAMP, aggravates neuroinflammation following acute ischemic stroke via TLR4/NF-kappaB/NLRP3 signaling. J Neuroinflamm. (2025) 22:164. doi: 10.1186/s12974-025-03492-6
25. Han Q-Q and Le W. NLRP3 inflammasome-mediated neuroinflammation and related mitochondrial impairment in parkinson’s disease. Neurosci Bull. (2023) 39:832–44. doi: 10.1007/s12264-023-01023-y
26. Satoh T, Trudler D, Oh C-K, and Lipton SA. Potential therapeutic use of the rosemary diterpene carnosic acid for alzheimer’s disease, parkinson’s disease, and long-COVID through NRF2 activation to counteract the NLRP3 inflammasome. Antioxidants. (2022) 11:124. doi: 10.3390/antiox11010124
27. Pulido-Salgado M, Vidal-Taboada JM, and Saura J. C/EBPβ and C/EBPδ transcription factors: Basic biology and roles in the CNS. Prog Neurobiol. (2015) 132:1–33. doi: 10.1016/j.pneurobio.2015.06.003
28. Belluti S, Rigillo G, and Imbriano C. Transcription factors in cancer: when alternative splicing determines opposite cell fates. Cells. (2020) 9:760. doi: 10.3390/cells9030760
29. Lountos GT, Cherry S, Tropea JE, Wlodawer A, and Miller M. Structural basis for cell type specific DNA binding of C/EBPβ: The case of cell cycle inhibitor p15INK4b promoter. J Struct Biol. (2022) 214:107918. doi: 10.1016/j.jsb.2022.107918
30. Zahnow CA. CCAAT/enhancer-binding protein β: its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev Mol Med. (2009) 11:e12. doi: 10.1017/S1462399409001033
31. Tolomeo M and Grimaudo S. The “Janus” Role of C/EBPs family members in cancer progression. Int J Mol Sci. (2020) 21:4308. doi: 10.3390/ijms21124308
32. Sterken BA, Ackermann T, Müller C, Zuidhof HR, Kortman G, Hernandez-Segura A, et al. C/EBPβ isoform-specific regulation of migration and invasion in triple-negative breast cancer cells. NPJ Breast Cancer. (2022) 8:11. doi: 10.1038/s41523-021-00372-z
33. Park B-H, Kook S, Lee S, Jeong J-H, Brufsky A, and Lee B-C. An isoform of C/EBPβ, LIP, regulates expression of the chemokine receptor CXCR4 and modulates breast cancer cell migration. J Biol Chem. (2013) 288:28656–67. doi: 10.1074/jbc.M113.509505
34. Zahnow CA. CCAAT/enhancer binding proteins in normal mammary development and breast cancer. Breast Cancer Res. (2002) 4:113–21. doi: 10.1186/bcr428
35. Rojo MD, Bandyopadhyay I, Burke CM, Sturtz AD, Phillips ES, Matherne MG, et al. C/EBPβ deletion in macrophages impairs mammary gland alveolar budding during the estrous cycle. Life Sci Alliance. (2024) 7:e202302516. doi: 10.26508/lsa.202302516
36. Wessells J, Yakar S, and Johnson PF. Critical prosurvival roles for C/EBPβ and insulin-like growth factor I in macrophage tumor cells. Mol Cell Biol. (2023) 24:3238–50. doi: 10.1128/MCB.24.8.3238-3250.2004
37. Guo L, Li X, and Tang Q-Q. Transcriptional regulation of adipocyte differentiation: A central role for CCAAT/enhancer-binding protein (C/EBP) β. J Biol Chem. (2015) 290:755–61. doi: 10.1074/jbc.R114.619957
38. Liu S, Wang J, Chen S, Han Z, Wu H, Chen H, et al. C/EBPβ Coupled with E2F2 Promoted the Proliferation of hESC-Derived Hepatocytes through Direct Binding to the Promoter Regions of Cell-Cycle-Related Genes. Cells. (2023) 12:497. doi: 10.3390/cells12030497
39. Sato A, Kamio N, Yokota A, Hayashi Y, Tamura A, Miura Y, et al. C/EBPβ isoforms sequentially regulate regenerating mouse hematopoietic stem/progenitor cells. Blood Adv. (2020) 4:3343–56. doi: 10.1182/bloodadvances.2018022913
40. Chen X, Lu J, Zhao X, Chen C, Qiao D, Wang H, et al. Role of C/EBP-beta in methamphetamine-mediated microglial apoptosis. Front Cell Neurosci. (2019) 13:366. doi: 10.3389/fncel.2019.00366
41. Samuel Olajide T, Oyerinde TO, Omotosho OI, Okeowo OM, Olajide OJ, and Ijomone OM. Microglial senescence in neurodegeneration: Insights, implications, and therapeutic opportunities. Neuroprotection. (2024) 2:182–95. doi: 10.1002/nep3.v2.3
42. Jayanthi S, Daiwile AP, and Cadet JL. Neurotoxicity of methamphetamine: Main effects and mechanisms. Exp Neurol. (2021) 344:113795. doi: 10.1016/j.expneurol.2021.113795
43. Ma J, Wan J, Meng J, Banerjee S, Ramakrishnan S, and Roy S. Methamphetamine induces autophagy as a pro-survival response against apoptotic endothelial cell death through the Kappa opioid receptor. Cell Death Dis. (2014) 5:e1099–9. doi: 10.1038/cddis.2014.64
44. Straccia M, Gresa-Arribas N, Dentesano G, Ejarque-Ortiz A, Tusell JM, Serratosa J, et al. Pro-inflammatory gene expression and neurotoxic effects of activated microglia are attenuated by absence of CCAAT/enhancer binding protein β. J Neuroinflamm. (2011) 8:156. doi: 10.1186/1742-2094-8-156
45. Fields J and Ghorpade A. C/EBPβ regulates multiple IL-1β-induced human astrocyte inflammatory genes. J Neuroinflamm. (2012) 9:177. doi: 10.1186/1742-2094-9-177
46. Yao Q, Long C, Yi P, Zhang G, Wan W, Rao X, et al. C/EBPβ: A transcription factor associated with the irreversible progression of Alzheimer's disease. CNS Neurosci Ther. (2024) 30:e14721. doi: 10.1111/cns.14721
47. Xia Y, Wang ZH, Zhang J, Liu X, Yu SP, Ye KX, et al. C/EBPbeta is a key transcription factor for APOE and preferentially mediates ApoE4 expression in Alzheimer's disease. Mol Psychiatry. (2021) 26:6002–22. doi: 10.1038/s41380-020-00956-4
48. Wang ZH, Xia Y, Wu Z, Kang SS, Zhang JC, Liu P, et al. Neuronal ApoE4 stimulates C/EBPbeta activation, promoting Alzheimer's disease pathology in a mouse model. Prog Neurobiol. (2022) 209:102212. doi: 10.1016/j.pneurobio.2021.102212
49. Qian Z, Wang Z, Li B, Meng X, Kuang Z, Li Y, et al. Thy1-ApoE4/C/EBPbeta double transgenic mice act as a sporadic model with Alzheimer's disease. Mol Psychiatry. (2024) 29:3040–55. doi: 10.1038/s41380-024-02565-x
50. Liu P, Wang ZH, Kang SS, Liu X, Xia Y, Chan CB, et al. High-fat diet-induced diabetes couples to Alzheimer's disease through inflammation-activated C/EBPβ/AEP pathway. Mol Psychiatry. (2022) 27:3396–409. doi: 10.1038/s41380-022-01600-z
51. Wu Z, Xia Y, Wang Z, Su Kang S, Lei K, Liu X, et al. C/EBPβ/δ-secretase signaling mediates Parkinson's disease pathogenesis via regulating transcription and proteolytic cleavage of α-synuclein and MAOB. Mol Psychiatry. (2021) 26:568–85. doi: 10.1038/s41380-020-0687-7
52. Fang X, Liu S, Muhammad B, Zheng M, Ge X, Xu Y, et al. Gut microbiota dysbiosis contributes to α-synuclein-related pathology associated with C/EBPβ/AEP signaling activation in a mouse model of Parkinson's disease. Neural Regener Res. (2024) 19:2081–8. doi: 10.4103/1673-5374.391191
53. Dasgupta S, Jana M, Liu X, and Pahan K. Role of very-late antigen-4 (VLA-4) in myelin basic protein-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. J Biol Chem. (2003) 278:22424–31. doi: 10.1074/jbc.M301789200
54. Thiruvengadam M. Neuroinflammation: Unraveling its role in neurodegenerative diseases. Brain Spine. (2024) 4:102866. doi: 10.1016/j.bas.2024.102866
55. Wu D, Zhang Y, Zhao C, Li Q, Zhang J, Han J, et al. Disruption of C/EBPβ-Clec7a axis exacerbates neuroinflammatory injury via NLRP3 inflammasome-mediated pyroptosis in experimental neuropathic pain. J Trans Med. (2022) 20:583. doi: 10.1186/s12967-022-03779-9
56. Baragetti A, Catapano AL, and Magni P. Multifactorial activation of NLRP3 inflammasome: relevance for a precision approach to atherosclerotic cardiovascular risk and disease. Int J Mol Sci. (2020) 21:4459. doi: 10.3390/ijms21124459
57. Hamilton C and Anand PK. Right place, right time: localisation and assembly of the NLRP3 inflammasome. F1000Research. (2019) 8:F1000 Faculty Rev-676. doi: 10.12688/f1000research
58. Halliday M and Mallucci GR. Review: Modulating the unfolded protein response to prevent neurodegeneration and enhance memory. Neuropathology Appl Neurobiol. (2015) 41:414–27. doi: 10.1111/nan.2015.41.issue-4
59. Spilianakis CB, Udofa EA, Stringer BW, Gade P, Mahony D, Buzza MS, et al. The transcription factor C/EBP-β Mediates constitutive and LPS-inducible transcription of murine serpinB2. PloS One. (2013) 8:e57855. doi: 10.1371/journal.pone.0057855
60. Ma J, Liu C, Yang Y, Yu J, Yang J, Yu S, et al. C/EBPβ Acts upstream of NF-κB P65 subunit in ox-LDL-induced IL-1β Production by macrophages. Cell Physiol Biochem. (2018) 48:1605–15. doi: 10.1159/000492282
61. Endo M, Mori M, Akira S, and Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammatio. J Immunol. (2006) 176:6245–53. doi: 10.4049/jimmunol.176.10.6245
62. Sierra-Magro A, Bartolome F, Lozano-Muñoz D, Alarcón-Gil J, Gine E, Sanz-SanCristobal M, et al. C/EBPβ Regulates TFAM expression, mitochondrial function and autophagy in cellular models of parkinson’s disease. Int J Mol Sci. (2023) 24:1459. doi: 10.3390/ijms24021459
63. Bajwa E, Pointer CB, and Klegeris A. The role of mitochondrial damage-associated molecular patterns in chronic neuroinflammation. Mediators Inflammation. (2019) 2019:1–11. doi: 10.1155/2019/4050796
64. Lin M-M, Liu N, Qin Z-H, and Wang Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacologica Sin. (2022) 43:2439–47. doi: 10.1038/s41401-022-00879-6
65. Barakat DJ, Mendonca J, Barberi T, Zhang J, Kachhap SK, Paz-Priel I, et al. C/EBPβ regulates sensitivity to bortezomib in prostate cancer cells by inducing REDD1 and autophagosome–lysosome fusion. Cancer Lett. (2016) 375:152–61. doi: 10.1016/j.canlet.2016.03.005
66. Ma J, Yang X, and Chen X. C/EBPβ is a key transcription factor of ox-LDL inducing THP-1 cells to release multiple pro-inflammatory cytokines. Inflammation Res. (2021) 70:1191–9. doi: 10.1007/s00011-021-01509-3
67. Wang X, Cheng W, Chen X, Gong Y, Wang G, Zhang X, et al. Inhibition of CEBPB attenuates lupus nephritis via regulating pim-1 signaling. Mediators Inflammation. (2022) 2022:1–14. doi: 10.1155/2022/2298865
68. Jones GH, Vecera CM, Pinjari OF, and MaChado-Vieira R. Inflammatory signaling mechanisms in bipolar disorder. J Biomed Sci. (2021) 28:45. doi: 10.1186/s12929-021-00742-6
69. Wang K, Gao Y, Wang C, Liang M, Liao Y, and Hu K. Role of oxidative stress in varicocele. Front Genet. (2022) 13. doi: 10.3389/fgene.2022.850114
70. Jiang C, Xie S, Yang G, and Wang N. Spotlight on NLRP3 inflammasome: role in pathogenesis and therapies of atherosclerosis. J Inflammation Res Volume. (2021) 14:7143–72. doi: 10.2147/JIR.S344730
71. Zou H, Chen M, Wang X, Yu J, Li X, Xie Y, et al. C/EBPβ isoform-specific regulation of podocyte pyroptosis in lupus nephritis-induced renal injury. J Pathol. (2023) 261:269–85. doi: 10.1002/path.v261.3
72. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
73. Barnes PJ and Adcock IM. Transcription factors and asthma. Eur Respir J. (1998) 12:221–34. doi: 10.1183/09031936.98.12010221
74. Hirai H, Yokota A, Tamura A, Sato A, and Maekawa T. Non-steady-state hematopoiesis regulated by the C/EBPβ transcription factor. Cancer Sci. (2015) 106:797–802. doi: 10.1111/cas.2015.106.issue-7
75. McClure C, McPeak MB, Youssef D, Yao ZQ, McCall CE, and El Gazzar M. Stat3 and C/EBPβ synergize to induce miR-21 and miR-181b expression during sepsis. Immunol Cell Biol. (2016) 95:42–55. doi: 10.1038/icb.2016.63
76. McPeak MB, Youssef D, Williams DA, Pritchett CL, Yao ZQ, McCall CE, et al. Frontline Science: Myeloid cell-specific deletion of Cebpb decreases sepsis-induced immunosuppression in mice. J Leukocyte Biol. (2017) 102:191–200. doi: 10.1189/jlb.4HI1216-537R
77. Wang W, Xia X, Mao L, and Wang S. The CCAAT/enhancer-binding protein family: its roles in MDSC expansion and function. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.01804
78. Yan D, Wang J, Sun H, Zamani A, Zhang H, Chen W, et al. TIPE2 specifies the functional polarization of myeloid-derived suppressor cells during tumorigenesis. J Exp Med. (2020) 217:e20182005. doi: 10.1084/jem.20182005
79. Chen M, Rong R, and Xia X. Spotlight on pyroptosis: role in pathogenesis and therapeutic potential of ocular diseases. J Neuroinflamm. (2022) 19:183. doi: 10.1186/s12974-022-02547-2
80. Shao R, Wang X, Xu T, Xia Y, and Cui D. The balance between AIM2-associated inflammation and autophagy: the role of CHMP2A in brain injury after cardiac arrest. J Neuroinflamm. (2021) 18:257. doi: 10.1186/s12974-021-02307-8
81. de Jong LM, Jiskoot W, Swen JJ, and Manson ML. Distinct effects of inflammation on cytochrome P450 regulation and drug metabolism: lessons from experimental models and a potential role for pharmacogenetics. Genes. (2020) 11:1509. doi: 10.3390/genes11121509
82. Li Y, Bevilacqua E, Chiribau C-B, Majumder M, Wang C, Croniger CM, et al. Differential control of the CCAAT/enhancer-binding protein β (C/EBPβ) products liver-enriched transcriptional activating protein (LAP) and liver-enriched transcriptional inhibitory protein (LIP) and the regulation of gene expression during the response to endoplasmic reticulum stress. J Biol Chem. (2008) 283:22443–56. doi: 10.1074/jbc.M801046200
83. Chen C, Liao J, Xia Y, Liu X, Jones R, Haran J, et al. Gut microbiota regulate Alzheimer's disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut. (2022) 71:2233–52. doi: 10.1136/gutjnl-2021-326269
84. Ndoja A, Reja R, Lee SH, Webster JD, Ngu H, Rose CM, et al. Ubiquitin ligase COP1 suppresses neuroinflammation by degrading c/EBPβ in microglia. Cell. (2020) 182:1156–1169.e12. doi: 10.1016/j.cell.2020.07.011
85. Adhikary K, Mohanty S, Bandyopadhyay B, Maiti R, Bhattacharya K, and Karak P. β-Amyloid peptide modulates peripheral immune responses and neuroinflammation in rats. Biomolecular Concepts. (2024) 15:15. doi: 10.1515/bmc-2022-0042
86. Wang ZH, Gong K, Liu X, Zhang Z, Sun X, Wei ZZ, et al. C/EBPbeta regulates delta-secretase expression and mediates pathogenesis in mouse models of Alzheimer's disease. Nat Commun. (2018) 9:1784. doi: 10.1038/s41467-018-04120-z
87. Muller UC, Deller T, and Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. (2017) 18:281–98. doi: 10.1038/nrn.2017.29
88. Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat Med. (2014) 20:1254–62. doi: 10.1038/nm.3700
89. Xiong J, Zhang Z, and Ye K. C/EBPbeta/AEP signaling drives alzheimer's disease pathogenesis. Neurosci Bull. (2023) 39:1173–85. doi: 10.1007/s12264-023-01025-w
90. Xia Y, Qadota H, Wang ZH, Liu P, Liu X, Ye KX, et al. Neuronal C/EBPbeta/AEP pathway shortens life span via selective GABAnergic neuronal degeneration by FOXO repression. Sci Adv. (2022) 8:eabj8658. doi: 10.1126/sciadv.abj8658
91. Wang H, Chen G, Ahn EH, Xia Y, Kang SS, Liu X, et al. C/EBPβ/AEP is age-dependently activated in Parkinson's disease and mediates α-synuclein in the gut and brain. NPJ Parkinsons Dis. (2023) 9:1. doi: 10.1038/s41531-022-00430-8
92. Li L, Dawson VL, and Dawson TM. Gastrointestinal tract cleavage of α-synuclein by asparaginyl endopeptidase leads to Parkinson's disease. Neuron. (2024) 112:3516–8. doi: 10.1016/j.neuron.2024.10.015
93. Morales-Garcia JA, Gine E, Hernandez-Encinas E, Aguilar-Morante D, Sierra-Magro A, Sanz-SanCristobal M, et al. CCAAT/Enhancer binding protein β silencing mitigates glial activation and neurodegeneration in a rat model of Parkinson's disease. Sci Rep. (2017) 7:13526. doi: 10.1038/s41598-017-13269-4
94. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med. (2018) 10:eaah4066. doi: 10.1126/scitranslmed.aah4066
95. Amo-Aparicio J, Daly J, Højen JF, and Dinarello CA. Pharmacologic inhibition of NLRP3 reduces the levels of α-synuclein and protects dopaminergic neurons in a model of Parkinson's disease. J Neuroinflamm. (2023) 20:147. doi: 10.1186/s12974-023-02830-w
96. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. (2020) 183:636–649.e18. doi: 10.1016/j.cell.2020.09.020
97. Kumar S, Phaneuf D, and Julien JP. Withaferin-A treatment alleviates TAR DNA-binding protein-43 pathology and improves cognitive function in a mouse model of FTLD. Neurotherapeutics. (2021) 18:286–96. doi: 10.1007/s13311-020-00952-0
98. Zhao W, Beers DR, Bell S, Wang J, Wen S, Baloh RH, et al. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp Neurol. (2015) 273:24–35. doi: 10.1016/j.expneurol.2015.07.019
99. Thammisetty SS, Pedragosa J, Weng YC, Calon F, Planas A, and Kriz J. Age-related deregulation of TDP-43 after stroke enhances NF-κB-mediated inflammation and neuronal damage. J Neuroinflamm. (2018) 15:312. doi: 10.1186/s12974-018-1350-y
100. Valente T, Mancera P, Tusell JM, Serratosa J, and Saura J. C/EBPβ expression in activated microglia in amyotrophic lateral sclerosis. Neurobiol Aging. (2012) 33:2186–99. doi: 10.1016/j.neurobiolaging.2011.09.019
101. Cui Y, Yu H, Bu Z, Wen L, Yan L, and Feng J. Focus on the role of the NLRP3 inflammasome in multiple sclerosis: pathogenesis, diagnosis, and therapeutics. Front Mol Neurosci. (2022) 15:894298. doi: 10.3389/fnmol.2022.894298
102. Pulido-Salgado M, Vidal-Taboada JM, Garcia Diaz-Barriga G, Serratosa J, Valente T, Castillo P, et al. Myeloid C/EBPβ deficiency reshapes microglial gene expression and is protective in experimental autoimmune encephalomyelitis. J Neuroinflamm. (2017) 14:54. doi: 10.1186/s12974-017-0834-5
103. Andersson U, Tracey KJ, and Yang H. Post-translational modification of HMGB1 disulfide bonds in stimulating and inhibiting inflammation. Cells. (2021) 10:3323. doi: 10.3390/cells10123323
104. Pandolfi F, Altamura S, Frosali S, and Conti P. Key role of DAMP in inflammation, cancer, and tissue repair. Clin Ther. (2016) 38:1017–28. doi: 10.1016/j.clinthera.2016.02.028
105. De Luca G, Goette NP, Lev PR, Baroni Pietto MC, Marin Oyarzún CP, Castro Ríos MA, et al. Elevated levels of damage-associated molecular patterns HMGB1 and S100A8/A9 coupled with toll-like receptor-triggered monocyte activation are associated with inflammation in patients with myelofibrosis. Front Immunol. (2024) 15:1365015. doi: 10.3389/fimmu.2024.1365015
106. Luo Y, Ge P, Wen H, Zhang Y, Liu J, Dong X, et al. C/EBPβ Promotes LPS-induced IL-1β Transcription and secretion in alveolar macrophages via NOD2 signaling. J Inflammation Res. (2022) 15:5247–63. doi: 10.2147/JIR.S377499
107. Dai XG, Li Q, Li T, Huang WB, Zeng ZH, Yang Y, et al. The interaction between C/EBPβ and TFAM promotes acute kidney injury via regulating NLRP3 inflammasome-mediated pyroptosis. Mol Immunol. (2020) 127:136–45. doi: 10.1016/j.molimm.2020.08.023
108. Wei Z, Li C, Zhang Y, Lin C, Zhang Y, Shu L, et al. Macrophage-derived IL-1β Regulates emergency myelopoiesis via the NF-κB and C/ebpβ in zebrafish. J Immunol. (2020) 205:2694–706. doi: 10.4049/jimmunol.2000473
109. Zhang G, Zhou B, Li S, Yue J, Yang H, Wen Y, et al. Allele-specific induction of IL-1β expression by C/EBPβ and PU.1 contributes to increased tuberculosis susceptibility. PloS Pathog. (2014) 10:e1004426. doi: 10.1371/journal.ppat.1004426
110. Xia Y, Xiao Y, Wang ZH, Liu X, Alam AM, Haran JP, et al. Bacteroides Fragilis in the gut microbiomes of Alzheimer's disease activates microglia and triggers pathogenesis in neuronal C/EBPβ transgenic mice. Nat Commun. (2023) 14:5471. doi: 10.1038/s41467-023-41283-w
111. Kim KS, Lee D, Song CG, and Kang PM. Reactive oxygen species-activated nanomaterials as theranostic agents. Nanomedicine (Lond). (2015) 10:2709–23. doi: 10.2217/nnm.15.108
112. Sharma A, Singh S, Ahmad S, Gulzar F, Schertzer JD, and Tamrakar AK. NOD1 activation induces oxidative stress via NOX1/4 in adipocytes. Free Radic Biol Med. (2021) 162:118–28. doi: 10.1016/j.freeradbiomed.2020.11.036
113. Villarreal-García V, Estupiñan-Jiménez JR, Vivas-Mejía PE, Gonzalez-Villasana V, Vázquez-Guillén JM, and Reséndez-Pérez D. A vicious circle in breast cancer: The interplay between inflammation, reactive oxygen species, and microRNAs. Front Oncol. (2022) 12:980694. doi: 10.3389/fonc.2022.980694
114. Guo Y, Zhang Y, Guan Y, Chen N, Zhao M, Li Y, et al. IL-37d enhances COP1-mediated C/EBPβ degradation to suppress spontaneous neutrophil migration and tumor progression. Cell Rep. (2024) 43:113787. doi: 10.1016/j.celrep.2024.113787
115. Singh S, Sharma S, and Sharma H. Potential impact of bioactive compounds as NLRP3 inflammasome inhibitors: an update. Curr Pharm Biotechnol. (2024) 25:1719–46. doi: 10.2174/0113892010276859231125165251
116. Haque I, Thapa P, Burns DM, Zhou J, Sharma M, Sharma R, et al. NLRP3 inflammasome inhibitors for antiepileptogenic drug discovery and development. Int J Mol Sci. (2024) 25:6078. doi: 10.3390/ijms25116078
117. Cheng Q, Ma X, Liu J, Feng X, Liu Y, Wang Y, et al. Pharmacological inhibition of the asparaginyl endopeptidase (AEP) in an alzheimer's disease model improves the survival and efficacy of transplanted neural stem cells. Int J Mol Sci. (2023) 24:7739. doi: 10.3390/ijms24097739
118. He C, Wang T, Han Y, Zuo C, and Wang G. E3 ubiquitin ligase COP1 confers neuroprotection in cerebral ischemia/reperfusion injury via regulation of transcription factor C/EBPβ in microglia. Int J Biol Macromol. (2022) 222:1789–800. doi: 10.1016/j.ijbiomac.2022.09.264
119. Ramachandran R, Manan A, Kim J, and Choi S. NLRP3 inflammasome: a key player in the pathogenesis of life-style disorders. Exp Mol Med. (2024) 56:1488–500. doi: 10.1038/s12276-024-01261-8
120. Guo DK, Zhu Y, Sun HY, Xu XY, Zhang S, Hao ZB, et al. Pharmacological activation of REV-ERBα represses LPS-induced microglial activation through the NF-κB pathway. Acta Pharmacol Sin. (2019) 40:26–34. doi: 10.1038/s41401-018-0064-0
121. Li X, Sun M, Men S, Shi Y, Ma L, An Y, et al. The inflammatory transcription factor C/EBPβ Plays a critical role in cardiac fibroblast differentiation and a rat model of cardiac fibrosis induced by autoimmune myocarditis. Int Heart J. (2018) 59:1389–97. doi: 10.1536/ihj.17-446
122. Li W, Huang X, Pang X, Sun Y, Zeng Z, Zheng P, et al. The anti-inflammatory cytokine interferon-gamma as a potential therapeutic target of Alzheimer’s disease: a retrospective observational study. Aging Adv. (2024) 1:105–11. doi: 10.4103/AGINGADV.AGINGADV-D-24-00006
123. Spike AJ and Rosen JM. C/EBPß Isoform Specific Gene Regulation: It's a Lot more Complicated than you Think! J Mammary Gland Biol Neoplasia. (2020) 25:1–12. doi: 10.1007/s10911-020-09444-5
124. Chen Y and Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Delivery Rev. (2012) 64:640–65. doi: 10.1016/j.addr.2011.11.010
125. Caljon G, Caveliers V, Lahoutte T, Stijlemans B, Ghassabeh GH, Van Den Abbeele J, et al. Using microdialysis to analyse the passage of monovalent nanobodies through the blood-brain barrier. Br J Pharmacol. (2012) 165:2341–53. doi: 10.1111/j.1476-5381.2011.01723.x
126. Butler YR, Liu Y, Kumbhar R, Zhao P, Gadhave K, Wang N, et al. alpha-Synuclein fibril-specific nanobody reduces prion-like alpha-synuclein spreading in mice. Nat Commun. (2022) 13:4060. doi: 10.1038/s41467-022-31787-2
127. Oosterlaken M, Rogliardo A, Lipina T, Lafon PA, Tsitokana ME, Keck M, et al. Nanobody therapy rescues behavioural deficits of NMDA receptor hypofunction. Nature. (2025). doi: 10.1038/s41586-025-09265-8
128. Sinclair F, Begum AA, Dai CC, Toth I, and Moyle PM. Recent advances in the delivery and applications of nonviral CRISPR/Cas9 gene editing. Drug Delivery Transl Res. (2023) 13:1500–19. doi: 10.1007/s13346-023-01320-z
129. Tripathi S, Sharma Y, Rane R, and Kumar D. CRISPR/cas9 gene editing: A novel approach towards alzheimer's disease treatment. CNS Neurol Disord Drug Targets. (2024) 23:1405–24. doi: 10.2174/0118715273283786240408034408
130. Laurent M, Geoffroy M, Pavani G, and Guiraud S. CRISPR-based gene therapies: from preclinical to clinical treatments. Cells. (2024) 13:800. doi: 10.3390/cells13100800
131. Cecchin E and Stocco G. Pharmacogenomics and personalized medicine. Genes (Basel). (2020) 11:679. doi: 10.3390/genes11060679
132. Wang H, Larris B, Peiris TH, Zhang L, Le Lay J, Gao Y, et al. C/EBPbeta activates E2F-regulated genes in vivo via recruitment of the coactivator CREB-binding protein/P300. J Biol Chem. (2007) 282:24679–88. doi: 10.1074/jbc.M705066200
133. Rui W, Xiao H, Fan Y, Ma Z, Xiao M, Li S, et al. Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer's disease. J Neuroinflamm. (2021) 18:280. doi: 10.1186/s12974-021-02329-2
134. Wang ZH, Gong K, Liu X, Zhang Z, Sun X, Wei ZZ, et al. Author Correction: C/EBPβ regulates delta-secretase expression and mediates pathogenesis in mouse models of Alzheimer's disease. Nat Commun. (2019) 10:5452. doi: 10.1038/s41467-019-13553-z
135. Lin Z, Huang L, Cao Q, Luo H, Yao W, and Zhang JC. Inhibition of abnormal C/EBPβ/α-Syn signaling pathway through activation of Nrf2 ameliorates Parkinson's disease-like pathology. Aging Cell. (2023) 22:e13958. doi: 10.1111/acel.v22.10
Keywords: neuroinflammation, inflammasome activation, C/EBPβ isoforms, neurodegenerative diseases, therapeutic targeting
Citation: Wang J, Li Y and Xia Y (2025) C/EBPβ as a master regulator of inflammasome signaling in neurodegenerative diseases: mechanisms and therapeutic implications. Front. Immunol. 16:1656165. doi: 10.3389/fimmu.2025.1656165
Received: 29 June 2025; Accepted: 18 August 2025;
Published: 05 September 2025.
Edited by:
Jinbiao Liu, Hubei University of Technology, ChinaReviewed by:
Lina Zhou, University of Tennessee Health Science Center (UTHSC), United StatesYongkui Li, Jinan University, China
Sandip Godse, University of Tennessee Health Science Center (UTHSC), United States
Copyright © 2025 Wang, Li and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiyuan Xia, eHl5QGpodW4uZWR1LmNu
†These authors have contributed equally to this work