 Lili Li1†
Lili Li1† Lijuan Qin
Lijuan Qin- 1Department of Radiotherapy, Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, Taiyuan, ShanXi, China
- 2Department of Pathology, Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, Taiyuan, Shanxi, China
- 3Shanxi Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, No.3 Zhigong Xincun Street, Xinghualing District, Taiyuan, Shanxi, China
Cervical cancer remains a leading cause of cancer-related mortality in women worldwide, particularly in regions with limited access to screening and vaccination. While immunotherapy has shown promise in treating advanced cervical cancer, immune evasion mechanisms within the tumor microenvironment continue to limit therapeutic efficacy. Ferroptosis, a form of iron-dependent regulated cell death characterized by lipid peroxidation, has recently been recognized as a crucial regulator of tumor progression and immune modulation. Emerging evidence suggests that ferroptosis interacts with immune signaling pathways, contributing to immune suppression, antigen presentation defects, and the remodeling of the tumor immune microenvironment in cervical cancer. This review highlights the current understanding of ferroptosis-related mechanisms underlying immune evasion in cervical cancer, including alterations in ferroptosis regulators, redox imbalance, and ferroptosis-induced release of immunomodulatory molecules. We further explore how targeting ferroptosis may enhance anti-tumor immunity and overcome resistance to immunotherapy. Finally, we discuss recent advances in ferroptosis-based therapeutic strategies and identify future directions for integrating ferroptosis modulation into cervical cancer treatment.
1 Introduction
Cervical cancer remains one of the most prevalent malignancies affecting women worldwide, with over 600,000 new cases and 340,000 deaths reported in 2022 from the World Health Organization (WHO), predominantly in low- and middle-income countries where access to HPV vaccination and screening remains limited. Despite significant progress in prevention and treatment, especially through the implementation of immunotherapeutic strategies such as PD-1/PD-L1 checkpoint blockade, durable responses are seen only in a subset of patients (1, 2). A major contributing factor to this limited efficacy is the ability of cervical cancer cells, especially those driven by high-risk human papillomavirus (HPV) strains such as HPV16 and HPV18, to escape immune surveillance through multiple mechanisms, including impaired antigen presentation, suppression of cytotoxic T cell infiltration, and recruitment of immunosuppressive myeloid populations (3, 4). The complexity of immune evasion in the cervical tumor microenvironment (TME) underscores the urgent need to identify novel regulatory pathways that modulate immune activity and determine therapeutic responsiveness (5, 6).
Ferroptosis, first defined in 2012 as an iron-dependent, lipid peroxidation-driven form of regulated cell death, has since emerged as a critical process in cancer biology with unique implications for immunogenicity (7, 8). Unlike apoptosis or necroptosis, ferroptosis is characterized by the accumulation of lethal lipid reactive oxygen species (ROS) and iron overload, and is tightly controlled by metabolic regulators such as glutathione peroxidase 4 (GPX4), SLC7A11, and ferroptosis suppressor protein 1 (FSP1) (9). Intriguingly, recent studies reveal that ferroptotic tumor cells can actively shape the immune landscape through the release of damage-associated molecular patterns (DAMPs), modulation of cytokine signaling, and alteration of antigen presentation machinery. In breast and pancreatic cancers, ferroptosis has been shown to either promote anti-tumor immunity or exacerbate immune escape, depending on the context. However, the role of ferroptosis in HPV-driven cancers, including cervical cancer, remains underexplored—particularly in how it interfaces with immune escape pathways.
Recent transcriptomic and single-cell analyses of cervical cancer tissues have begun to uncover ferroptosis-related gene signatures correlated with immune exclusion and poor prognosis, suggesting a functional crosstalk between ferroptotic signaling and immune evasion. For instance, elevated expression of SLC7A11 in cervical tumors has been associated with reduced CD8+ T cell infiltration and resistance to immune checkpoint therapy (10). Moreover, HPV oncoproteins influence ferroptosis sensitivity via regulation of p53 and NRF2 pathways, offering a mechanistic link between viral oncogenesis and ferroptotic control (11, 12). These emerging insights highlight the dual role of ferroptosis in cervical cancer, not only as a cell death modality but also as a pivotal modulator of tumor immunity. This mini-review aims to synthesize recent advances in our understanding of ferroptosis–immune crosstalk in cervical cancer, focusing on mechanistic underpinnings and therapeutic implications, with an eye toward novel combination strategies to overcome immune resistance.
2 Molecular mechanisms of ferroptosis in cervical cancer
Ferroptosis is a regulated form of cell death driven by iron-dependent lipid peroxidation and impaired redox homeostasis (7–13). It is morphologically and biochemically distinct from apoptosis, necroptosis, and other cell death modes. Canonical ferroptosis involves excessive accumulation of ROS, particularly lipid ROS, resulting from disturbed iron metabolism, depletion of glutathione (GSH), and inactivation of glutathione peroxidase 4 (GPX4). Additional protective systems, including the FSP1–CoQ10 axis and the recently discovered mitochondrial DHODH–CoQ10 pathway, also counteract ferroptotic lipid peroxidation. Key regulators of ferroptosis include GPX4, SLC7A11, ACSL4, and iron metabolism proteins such as TFR1, ferritin, and ferroportin. When these defense systems are disrupted, cells become highly sensitive to ferroptotic cell death, especially under oxidative stress. In the following sections, we focus on how these mechanisms are uniquely regulated or altered in cervical cancer, particularly under the influence of HPV oncoproteins (Table 1).
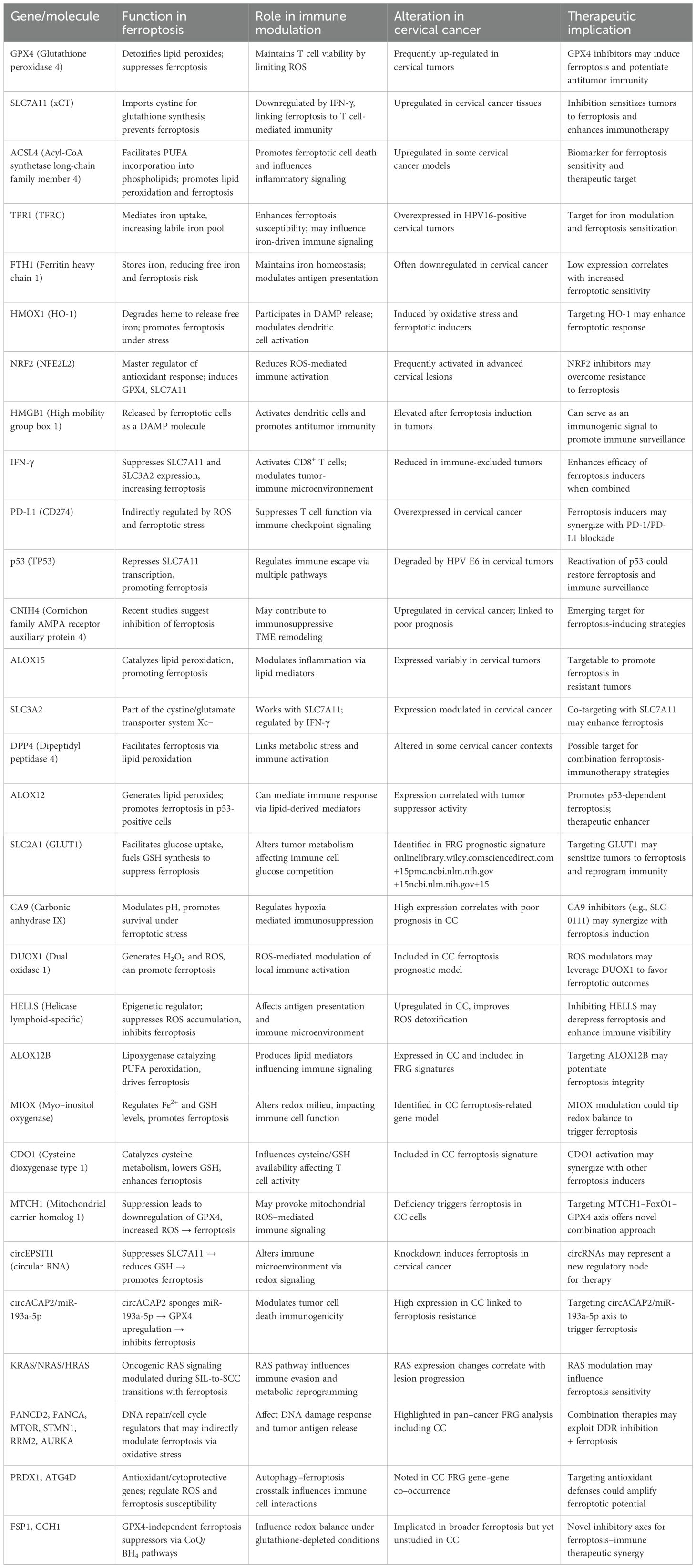
Table 1. Key ferroptosis-related genes and immune modulators in cervical cancer.
2.1 Iron metabolism drives ferroptosis through redox dysregulation
Ferroptosis in cervical cancer is tightly linked to altered iron metabolism, HPV-induced reprogramming of iron-regulatory genes, and enhanced ferritin degradation. Together, these factors converge to elevate intracellular iron and oxidative stress (Figure 1A).
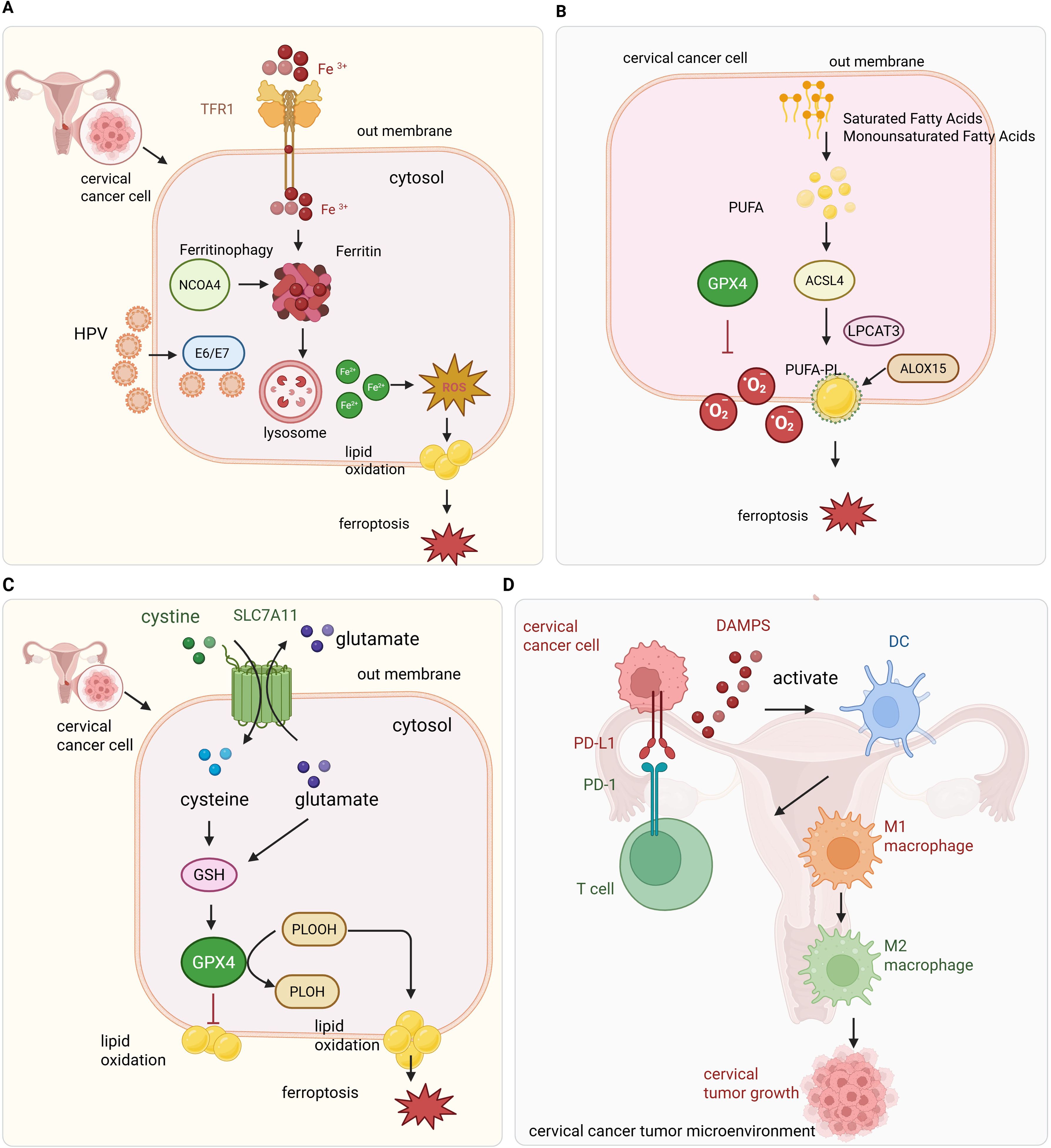
Figure 1. Overview of ferroptosis regulation and immune interactions in cervical cancer. (A) HPV oncoproteins dysregulate iron metabolism by increasing iron uptake through TFR1 and promoting ferritin degradation via NCOA4-mediated ferritinophagy, resulting in elevated Fe2+ and ROS production through the Fenton reaction. (B) Lipid metabolism enzymes such as ACSL4 facilitate incorporation of polyunsaturated fatty acids (PUFAs) into membranes, where ROS-induced lipid peroxidation triggers ferroptosis. Key antioxidant defenses including GPX4, SLC7A11, FSP1, and NRF2 mitigate lipid peroxidation to protect tumor cells. (C) Intracellular antioxidant defense against ferroptosis in cervical cancer cells. The cystine/glutamate antiporter SLC7A11 (system Xc−) imports extracellular cystine, which is reduced to cysteine inside the cell and used for glutathione (GSH) synthesis. GPX4 utilizes GSH to detoxify lipid peroxides (PLOOH) to non-toxic lipid alcohols (PLOH), preventing ferroptotic cell death. SLC7A11 exchanges intracellular glutamate for extracellular cystine, maintaining redox balance. This antioxidant system is critical for cervical cancer cell survival by suppressing lipid peroxidation and ferroptosis. (D) Ferroptotic tumor cells release immunogenic signals that activate immune cells; however, the oxidative microenvironment promotes immune suppression via checkpoint molecules like PD-L1, facilitating tumor immune evasion.
2.1.1 Cellular iron homeostasis and ferroptosis susceptibility
Iron metabolism is essential for cellular functions such as DNA synthesis, oxygen transport, and mitochondrial respiration (14). However, dysregulated iron homeostasis generates excessive ROS via the Fenton reaction, which forms highly reactive hydroxyl radicals (•OH) that initiate lipid peroxidation—central to ferroptosis (15). Ferroptosis is characterized by iron-dependent accumulation of lipid ROS leading to oxidative membrane damage and cell death (16).
The major contributors to intracellular iron regulation include transferrin (TF), transferrin receptor 1 (TFR1), divalent metal transporter 1 (DMT1), ferritin (FTH1 and FTL), and ferroportin (SLC40A1) (17). Under physiological conditions, transferrin binds extracellular Fe3+, which is internalized through TFR1-mediated endocytosis. Inside the endosome, Fe3+ is reduced to Fe2+ and released into the cytosol by DMT1, entering the labile iron pool (LIP). Ferritin stores excess iron, while ferroportin exports iron to maintain balance. Cancer cells demand high levels of iron for rapid growth, resulting in upregulated transferrin receptor 1 (TfR1) to increase iron uptake. This elevated TfR1 expression enables targeted therapeutic strategies, such as TransTACs, which exploit TfR1-mediated endocytosis to selectively degrade membrane proteins crucial for tumor survival and drug resistance, offering a novel approach for precise cancer treatment (18). Importantly, iron overload is not just a byproduct of tumor metabolism but a necessary driver of malignant transformation (19). High LIP levels enhance mitochondrial ROS production and lipid peroxidation. These redox-active conditions are particularly pronounced in rapidly proliferating cervical cancer cells, which require increased iron for nucleotide synthesis but become vulnerable to ferroptosis if antioxidant defenses are compromised.
2.1.2 HPV-driven alteration of iron-regulatory genes in cervical cancer
In cervical cancer, the iron input-export pathway is disrupted, HPV infection induces changes in iron regulatory proteins, leading to altered ferroptosis. While ferroptosis occurs during precancerous squamous intraepithelial lesions (SIL), cervical squamous cell carcinoma (CSCC) cells develop anti-ferroptotic mechanisms that enable survival and oncogenesis (20). In addition, HPV oncoproteins E6 and E7 not only interfere with p53 and retinoblastoma protein (pRb) but also impact metabolic gene expression, including iron-regulatory genes (21). E6 upregulates TFR1 and DMT1, while suppressing FTH1 expression via epigenetic silencing. This promotes iron accumulation in HPV-transformed cells, establishing a redox-unstable environment conducive to ferroptosis initiation (22). Furthermore, HPV-driven inflammation and oxidative stress activates the iron storage release mechanism through ferritin degradation (23). Thus, iron becomes more available for catalyzing lipid oxidation, increasing ferroptosis vulnerability. Yet, tumor cells may simultaneously activate compensatory antioxidant pathways to evade this outcome, forming the basis for ferroptosis resistance in advanced cervical tumors.
2.1.3 Ferritinophagy releases stored iron and amplifies ferroptotic potential
Ferritinophagy, the autophagic degradation of ferritin, is a selective process that liberates stored iron into the cytosol. This process is mediated by the cargo receptor NCOA4 (nuclear receptor coactivator 4), which binds ferritin and targets it to the lysosome (24, 25). Degradation of ferritin increases the intracellular Fe2+ pool, driving the Fenton reaction and ROS generation, thereby sensitizing cells to ferroptosis. In cervical cancer, NCOA4 promotes ferroptosis by mediating ferritinophagy, which increases the intracellular labile iron pool and reactive oxygen species (ROS) through enhanced Fenton reactions. This mechanism is activated by dihydroartemisinin (DHA), sensitizing cervical cancer cells to ferroptotic cell death and enhancing the efficacy of doxorubicin (26). Moreover, inhibition of autophagy with chloroquine abrogated this effect, confirming that ferritinophagy is necessary for this form of cell death (27). Interestingly, radiation and chemotherapeutic stress may activate NCOA4-mediated ferritinophagy in cervical tumors as a cellular attempt to recycle iron. When combined with ferroptosis inducers, this response becomes maladaptive, leading to extensive oxidative injury and cell death (28). These findings suggest that modulation of ferritinophagy could enhance the therapeutic index of ferroptosis-based interventions.
2.2 Lipid metabolism promotes ferroptosis through PUFA peroxidation
2.2.1 Polyunsaturated phospholipid biosynthesis and lipid ROS accumulation
The defining feature of ferroptosis is the peroxidation of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs) in cell membranes. These PUFA-PLs are highly susceptible to oxidative damage due to the presence of bis-allylic hydrogen atoms. The biosynthesis of PUFA-PLs involves the activation of free PUFAs by Acyl-CoA synthetase long-chain family member 4 (ACSL4) and their incorporation into membrane phospholipids via lysophosphatidylcholine acyltransferase 3 (LPCAT3) (29). ACSL4 catalyzes the esterification of PUFAs into their corresponding acyl-CoA derivatives. These activated fatty acids are then incorporated into phospholipids within cellular membranes, rendering them highly susceptible to oxidative attack during ferroptosis. ACSL4 determines the lipid composition of cell membranes by selectively enriching PUFA-phospholipids, particularly phosphatidylethanolamines, that serve as substrates for enzymatic peroxidation (Figure 1B). This biochemical specificity defines the ferroptosis sensitivity profile of cancer cells. Loss of ACSL4 function results in a phospholipid landscape dominated by monounsaturated species, which are comparatively inert to peroxidation and ferroptosis. ACSL4 also mediates the anticancer effects of oleanolic acid (OA) in cervical cancer by promoting ferroptosis (30). Cervical tumor cells may exploit this pathway for immune modulation or treatment resistance. For instance, cells with high PUFA content exhibit increased susceptibility to ferroptosis when Glutathione peroxidase 4 (GPX4) is inhibited (31). These observations underscore the importance of lipid metabolism in determining ferroptosis sensitivity in the tumor microenvironment.
2.2.2 Role of lipoxygenases in lipid peroxidation and ferroptosis execution
While ROS from mitochondrial metabolism or the Fenton reaction can initiate lipid oxidation, lipoxygenases (ALOXs) catalyze specific peroxidation of PUFA-PLs, playing a crucial role in ferroptosis execution (32). In particular, arachidonate 15-lipoxygenase (ALOX15) is highly expressed in cervical epithelial cells and has been shown to oxidize PE-AA to 15-HpETE-PE, a key lipid peroxide triggering ferroptosis. ALOX15 knockdown conferred resistance to RSL3-induced ferroptosis, while its overexpression enhanced lipid peroxidation and cell death. Moreover, upregulation of ALOX15 has been observed in cervical intraepithelial neoplasia, suggesting its involvement in the early stages of ferroptosis priming. ALOX15 promotes ferroptosis in cervical cancer by facilitating lipid peroxidation, and its expression is suppressed by tumor-associated macrophage-derived miRNA-660-5p, which inhibits ferroptosis and contributes to tumor progression. High ALOX15 levels correlate with better prognosis, making it a potential therapeutic and prognostic target (33).
2.3 Key regulators of ferroptosis and their mechanistic roles
2.3.1 GPX4 detoxifies lipid peroxides and suppresses ferroptosis
GPX4 serves as a central executor of ferroptosis resistance by directly catalyzing the reduction of lipid hydroperoxides (PLOOHs) to their corresponding alcohols (PLOHs), utilizing glutathione (GSH) as a cofactor (16). Unlike other GPX isoforms, GPX4 exhibits unique substrate specificity for complex membrane phospholipid peroxides, and its inactivation is both necessary and sufficient to initiate ferroptotic death. Structurally, GPX4 harbors a selenocysteine residue at its active site, which is essential for its peroxidase activity. The availability of reduced GSH and selenocysteine incorporation during translation tightly regulates its activity. Pharmacologic inhibitors such as RSL3 bind the active site and inhibit enzymatic function, leading to an unchecked accumulation of PLOOHs and subsequent ferroptotic collapse. Genetic depletion of GPX4 produces a similar phenotype, confirming its non-redundant role.
GPX4 acts as a key anti-ferroptosis enzyme in cervical cancer, and its downregulation—triggered by MTCH1 deficiency and impaired FoxO1 nuclear translocation—leads to elevated ROS and ferroptotic cell death (34). Targeting the MTCH1–FoxO1–GPX4 axis sensitizes cervical cancer cells to ferroptosis, offering a promising therapeutic strategy. GPX4 also protects cells from ferroptosis through the NRF2/GPX4/xCT antioxidant pathway. Triptolide induces ferroptosis in cervical cancer cells by downregulating NRF2, which leads to decreased GPX4 and xCT expression, resulting in increased lipid peroxidation and tumor growth inhibition (35). In cervical cancer, elevated GPX4 expression correlates with chemoradiotherapy resistance, particularly in HPV-positive tumors. Tumor cells exhibiting mesenchymal or hypoxic signatures demonstrate GPX4 dependency for survival. Inhibition of GPX4 in these contexts induces catastrophic lipid peroxidation and irreversible mitochondrial damage. These observations highlight the potential for GPX4-targeted strategies to overcome intrinsic resistance mechanisms in cervical malignancies. Moreover, metabolic stressors such as cystine deprivation, GSH depletion, or oxidative phosphorylation inhibitors synergistically impair GPX4 function, offering avenues for therapeutic combination approaches. The essentiality of GPX4 in protecting cancer cells from ferroptotic damage underscores its centrality as a druggable target.
2.3.2 SLC7A11 sustains GSH synthesis and inhibits ferroptosis
SLC7A11 encodes the light chain subunit of the cystine/glutamate antiporter system Xc−, which imports extracellular cystine in exchange for intracellular glutamate (Figure 1C). Cystine is rapidly reduced to cysteine intracellularly and serves as a rate-limiting substrate for GSH biosynthesis. Through this mechanism, SLC7A11 indirectly maintains GPX4 activity and redox equilibrium, thereby suppressing ferroptotic responses (36). Transcriptional regulation of SLC7A11 is mediated by stress-responsive transcription factors including NRF2 and ATF4. Under oxidative or metabolic stress, these factors promote SLC7A11 expression, increasing cystine uptake and buffering intracellular ROS. Conversely, tumor suppressor p53 represses SLC7A11 transcription under certain conditions, lowering cysteine levels and sensitizing cells to ferroptosis.
SLC7A11 plays a critical role in suppressing ferroptosis in cervical cancer by mediating cystine uptake for glutathione synthesis. The RACK1/miR-1275/FUT8 axis stabilizes SLC7A11 through core-fucosylation, preventing its degradation and thus inhibiting ferroptosis, which promotes cervical cancer cell survival and progression (37). In addition, in cervical cancer, fatty acid synthase (FASN) promotes cisplatin resistance by upregulating SLC7A11, which suppresses ferroptosis. Inhibition of FASN reduces SLC7A11 expression, thereby enhancing ferroptosis and restoring cisplatin sensitivity, suggesting that targeting the FASN/SLC7A11 axis may overcome chemotherapy resistance (38). Under hypoxia-like conditions, cervical cancer cells increase SLC7A11 expression through KDM4A SUMOylation at the K471 site, which reduces H3K9me3-mediated repression of SLC7A11, thereby enhancing GPX4 levels and promoting ferroptosis resistance (39). SLC7A11 promotes cervical cancer cell survival by mediating cystine uptake to maintain glutathione synthesis, thereby inhibiting ferroptosis (40). Targeting SLC7A11 with specific inhibitors disrupts redox balance, increases ROS, and induces ferroptotic cell death, representing a promising therapeutic strategy for cervical cancer. This mechanism contributes to the survival of cervical cancer cells by inhibiting ferroptotic cell death. Targeting SLC7A11 may potentiate ferroptosis-based therapeutic strategies and disrupt redox adaptation in treatment-resistant tumors.
2.3.3 FSP1 reduces CoQ10 and suppresses ferroptosis independently of GPX4
Ferroptosis suppressor protein 1 (FSP1), previously known as AIFM2, functions as an NAD(P)H-dependent oxidoreductase that reduces ubiquinone (CoQ10) to ubiquinol (CoQ10H2), a lipophilic radical-trapping antioxidant (41). This reaction occurs at the plasma membrane and is independent of the canonical GPX4-GSH axis, representing a parallel defense system against ferroptotic lipid damage. FSP1 is myristoylated at the N-terminus, allowing for membrane localization where lipid peroxidation is initiated. Upon CoQ10 reduction, ubiquinol neutralizes lipid radicals and halts the propagation of peroxidation chains, thus preventing membrane rupture and ferroptotic death. Cells with intact FSP1 can survive in the absence of GPX4 activity if sufficient CoQ10 and NAD(P)H are available.
In cervical cancer, treatments like propofol and paclitaxel can synergistically induce ferroptosis by modulating the ubiquinol/CoQ10/FSP1 pathway, promoting oxidative stress and ferroptotic mitochondrial damage, which enhances cancer cell death beyond apoptosis (42). In cervical cancer, high FSP1 expression has been observed in resistant tumor clones and is associated with recurrence after chemoradiotherapy. Genetic knockdown or pharmacologic inhibition of FSP1 enhances the efficacy of GPX4 inhibitors, suggesting that dual blockade of ferroptosis defense pathways can overcome therapeutic resistance. Furthermore, CoQ10 biosynthesis is tightly regulated by the mevalonate pathway, which is frequently upregulated in tumors (43). Statins or mevalonate pathway inhibitors may reduce CoQ10 availability and sensitize cervical cancer cells to ferroptosis through indirect impairment of the FSP1 pathway (44). These interactions highlight the potential of FSP1 as a therapeutic vulnerability in ferroptosis-based interventions.
2.3.4 The NRF2 signaling in cervical cancer and ferroptosis regulation
NRF2 (NFE2L2) is a master transcriptional regulator of antioxidant and detoxification pathways, safeguarding cells from oxidative stress (45). When Keap1 fails to sequester NRF2, NRF2 accumulates in the nucleus, reducing cancer cell sensitivity to ferroptosis [88]. Tossetta et al. demonstrated that the NRF2/Keap1 axis is critically involved in cervical and endometrial carcinogenesis by modulating gene expression profiles that contribute to chemotherapy resistance. Inhibiting inducible NRF2 may synergize with ferroptosis-inducing strategies in cancer therapy. Additionally, NRF2 activity is modulated by its interaction with prolyl isomerase PIN1 (46). In cervical cancer, HELLS promotes tumor cell proliferation by repressing NRF2 expression, suggesting that targeting HELLS could restore ferroptosis sensitivity (47). Another downstream effector, heme oxygenase–1 (HO–1), exhibits context-dependent roles in cervical cancer: while the NRF2/HO–1 pathway generally suppresses ferroptosis due to its antioxidative function, HO–1 can switch to a pro-ferroptotic role under certain stress conditions (48). For example, CENPF knockdown leads to decreased NRF2 activity and HO–1 expression, weakening the cellular antioxidant defense (49). Conversely, Erastin treatment increases ROS and upregulates both NRF2 and HO–1 in HeLa cells; knocking down HO–1 reduces Erastin’s inhibition of colony formation, migration, invasion, and ROS generation, implying HO–1’s facilitative role in ferroptosis-mediated anticancer effects (50). The dualistic nature of HO–1 highlights its importance in cervical cancer progression and therapy response. Moreover, the Wnt signaling pathway, which is frequently dysregulated in various cancers, can modulate NRF2 expression, identifying Wnt–NRF2 crosstalk as a promising target for ferroptosis-centric therapies in cervical cancer (51).
Besides all mentioned above, several additional regulators have been implicated in ferroptosis control. For example, mitochondrial enzyme DHODH was shown to suppress ferroptosis via an alternative CoQ10-dependent mechanism in mitochondria, offering protection when GPX4 or FSP1 is compromised (52). DHODH is upregulated in cervical cancer and acts as a ferroptosis defender (53); its inhibition promotes ferroptosis and suppresses tumor cell proliferation. Combined DHODH inhibition and cisplatin synergistically enhance ferroptosis by downregulating the mTOR pathway, offering a potential therapeutic strategy for cervical cancer (53). In addition, Cornichon family AMPA receptor auxiliary protein 4 (CNIH4) is upregulated in cervical cancer, CHIH4 promotes tumor progression by inhibiting ferroptosis by enhancing SLC7A11-mediated cystine import, boosting glutathione synthesis and GPX4 activity (54). Silencing CNIH4 or SLC7A11 restores ferroptotic sensitivity, identifying CNIH4 as a potential prognostic biomarker and therapeutic target in cervical cancer. Together, these emerging regulators illustrate the complexity of ferroptosis networks and highlight the potential for multi-targeted interventions in ferroptosis modulation.
3 Ferroptosis and immune regulation in cervical cancer
3.1 Ferroptosis disrupts antigen presentation and impairs immune recognition
Effective immune surveillance relies on tumor antigen presentation via major histocompatibility complex class I (MHC-I) molecules and subsequent recognition by cytotoxic T lymphocytes (55, 56). However, many cervical cancer cells, particularly those influenced by HPV oncogenes, downregulate MHC-I expression, impairing T cell-mediated cytotoxicity (57, 58). Recent studies have demonstrated that ferroptosis contributes to this immune escape process by modulating key components of antigen processing and presentation. Ferroptotic cancer cells impair dendritic cell maturation and antigen cross-presentation, thereby suppressing adaptive immune responses and weakening antitumor immunity (59).
Lipid peroxidation, the central biochemical hallmark of ferroptosis, has been shown to disrupt endoplasmic reticulum (ER) homeostasis and interfere with the biosynthesis and trafficking of MHC-I molecules (60, 61). Oxidized phospholipids impair peptide loading onto MHC-I, leading to suboptimal antigen presentation. In HPV-transformed cervical cancer cells, where oxidative stress and redox imbalance are already prominent, induction of ferroptosis further compromises antigen processing machinery, reinforcing immune escape. Furthermore, iron accumulation within ferroptosis-prone tumor cells generates ROS, which suppress proteasome activity and alter peptide repertoire generation. Reduced proteasomal degradation of viral and neoantigenic peptides weakens the formation of immunogenic MHC-I complexes (62). However, ferroptosis-related lipid peroxidation in post-synaptic dendritic cells (psDCs) enhances MHC-I expression and facilitates their licensing, thereby promoting effective CD8+ T cell activation and adaptive immunity (63).
3.2 Ferroptosis-derived DAMPs shape the immune microenvironment
Ferroptosis can release DAMPs that activate immune responses. However, in cervical cancer, this process tends to favor immunosuppressive remodeling rather than stimulate effective anti-tumor immunity (Figure 1D). This is largely due to the oxidative stress and HPV-induced immunomodulation that shift the TME toward immune tolerance. During ferroptosis, DAMPs such as high mobility group box 1 (HMGB1), ATP, and oxidized phospholipids are released into the extracellular space (64). These signals are typically recognized by pattern recognition receptors (PRRs) on dendritic cells and macrophages, initiating inflammatory signaling cascades. In cervical cancer, however, prolonged exposure to oxidized lipids promotes the differentiation of tolerogenic dendritic cells and the recruitment of regulatory T cells (Tregs), which suppress cytotoxic immune responses (65). In addition, ferroptosis-induced ROS stimulate the secretion of immunosuppressive cytokines, such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), further inhibiting effector T cell activity. Tumor-associated macrophages (TAMs), which are abundant in cervical cancer lesions, are skewed toward an M2-like phenotype under the influence of lipid-derived DAMPs. This polarization contributes to angiogenesis, extracellular matrix remodeling, and suppression of anti-tumor immunity (66). Thus, ferroptosis can paradoxically support immune evasion through inflammation-induced immune suppression.
3.3 Ferroptosis reshapes immune cell infiltration in the tumor microenvironment
The tumor immune microenvironment in cervical cancer is highly heterogeneous and shaped by multiple factors, including viral oncogenes, cytokine networks, and metabolic states (67, 68). Ferroptosis represents an additional layer of immune modulation, altering the composition and function of infiltrating immune cells. Ferroptosis inducers modify chemokine expression profiles in tumor cells, leading to increased recruitment of CD8+ T cells and natural killer (NK) cells. In models of ferroptosis-sensitive tumors, lipid peroxidation products enhance CXCL10 and CCL5 secretion, favoring cytotoxic immune infiltration. However, in cervical cancer, the effectiveness of this recruitment is often limited by concurrent immunosuppressive signals. In contrast, ferroptosis-induced inflammation may also recruit myeloid-derived suppressor cells (MDSCs) and Tregs, depending on the balance of DAMPs and cytokines released. For instance, 4-hydroxynonenal (4-HNE), a major lipid peroxidation product, induces prostaglandin E2 (PGE2) synthesis in TAMs, which supports MDSC expansion and inhibits dendritic cell maturation (69). In addition, immunotherapy-activated CD8+ T cells promote tumor ferroptosis by releasing IFN-γ, which suppresses SLC3A2 and SLC7A11 expression, reduces cystine uptake, and increases lipid peroxidation—thereby enhancing antitumor efficacy and suggesting ferroptosis as a synergistic mechanism with checkpoint blockade therapy (70).
3.4 Ferroptosis interacts with immune checkpoint pathways
Immune checkpoint inhibitors (ICIs), including PD–1/PD–L1 and CTLA–4 antagonists, have transformed cancer treatment by reinvigorating exhausted CD8+ T cells. However, many patients with cervical cancer still exhibit poor responses due to a “cold” TME and insufficient T–cell infiltration (71). Ferroptosis induction synergizes with ICIs by enhancing tumor immunogenicity and overcoming immune resistance. In multiple tumor models, combining GPX4 inhibitors with anti–PD-1 therapy improved tumor responses and increased activated CD8+ T cell and NK cell infiltration. Specifically, IFN–γ released from ICI–activated CD8+ T cells downregulates system xc– components (SLC3A2 and SLC7A11) in tumor cells, thereby promoting lipid peroxidation and ferroptosis (72). Critically, PD–1 pathway blockade also protects immune effector cells from ferroptosis. A recent study showed that dendritic cells (DCs) expressing PD–L1 are protected from ferroptotic cell death during chemotherapy; PD–L1 maintains SLC7A11 expression in DCs, preventing membrane lipid peroxidation and preserving T–cell priming capacity (73). This suggests dual roles for the PD–1/PD–L1 axis: promoting ferroptosis in tumor cells while safeguarding APCs.
HPV-driven modulation of ferroptosis pathways distinctly impacts ferroptosis-immune interactions compared to non-HPV cancers. HPV oncoproteins such as E6 and E7 suppress key ferroptosis regulators including ACSL4 and ALOX15, limiting lipid peroxidation and ferroptotic cell death, thereby facilitating immune escape and contributing to resistance to ICIs (33). Additionally, HPV-induced metabolic reprogramming and oxidative stress alter the tumor immune microenvironment by affecting immune cell susceptibility to ferroptosis, further skewing immune responses toward tolerance. These HPV-specific mechanisms underscore the challenges and opportunities for combining ferroptosis induction with immunotherapy in cervical cancer. High PD–L1 expression in cervical tumors correlates with poor prognosis and immune suppression. Preclinical data from other cancers hint that combining PD–1 blockade with ferroptosis inducers may reprogram TAMs and DCs toward pro-immunity, potentially overcoming HPV-mediated tolerance. Pembrolizumab, a PD–1 inhibitor, is FDA-approved for advanced cervical cancer, combining it with ferroptosis inducers may reprogram TAMs and DCs toward immune activation. Though clinical data in cervical cancer is still limited, preclinical studies support the rationale for combined immuno-ferroptotic therapy.
3.5 Ferroptosis of immune cells and its implications in cervical cancer
Ferroptosis is not limited to tumor cells—immune cells themselves can undergo ferroptotic death, which can either limit or facilitate anti-tumor immunity depending on context (74). CD8+ T cells and NK cells, particularly under oxidative stress or nutrient deprivation in the tumor microenvironment, are vulnerable to ferroptosis due to high metabolic demands and limited antioxidant capacity (8–75). Depletion of GPX4 or cystine transport leads to dysfunctional T cell responses, impaired cytokine secretion, and eventual cell death. In cervical cancer, HPV-induced metabolic remodeling and hypoxia may further predispose infiltrating lymphocytes to ferroptosis, weakening the immune response. Dendritic cells (DCs) also rely on SLC7A11 and GPX4 to maintain their function and survival during antigen presentation. Loss of ferroptosis protection in DCs impairs T cell priming, tipping the balance toward immune evasion (65). However, controlled ferroptosis in immunosuppressive cells may offer a therapeutic opportunity to remodel the TME toward immunogenicity. Thus, understanding the ferroptosis susceptibility of different immune cell subsets is essential for designing combination therapies that protect beneficial immune cells while sensitizing tumor and suppressive immune populations.
4 Therapeutic potential and future perspectives
4.1 Ferroptosis inducers offer a novel strategy for cervical cancer treatment
The pharmacologic induction of ferroptosis has emerged as a promising therapeutic strategy in malignancies characterized by resistance to conventional treatments. In cervical cancer, which often exhibits immune evasion and recurrence following chemoradiotherapy, ferroptosis induction represents a novel modality to overcome these therapeutic limitations.
Several ferroptosis inducers, including erastin, RSL3, and FIN56, target distinct molecular nodes such as system Xc-, GPX4, or CoQ10 pathways to induce lethal lipid peroxidation. Erastin, by inhibiting SLC7A11, depletes intracellular cystine and reduces glutathione synthesis, indirectly disabling GPX4 activity. RSL3 directly binds and inactivates GPX4, leading to accumulation of lipid peroxides. FIN56 promotes degradation of GPX4 and depletes CoQ10, disrupting antioxidant defense systems. In cervical cancer models, these agents have shown selective cytotoxicity in HPV-transformed cells, particularly those with aberrant redox metabolism and elevated iron pools. Such metabolic vulnerabilities sensitize cervical cancer cells to ferroptosis induction, highlighting a potential therapeutic window. Importantly, recent studies have demonstrated that ferroptosis inducers can sensitize cervical cancer cells to traditional treatments. For example, combining erastin with cisplatin enhances DNA damage and oxidative stress, leading to synergistic tumor cell death. Similarly, ionizing radiation upregulates ACSL4 and increases PUFA-phospholipid content, priming tumor cells for ferroptosis upon GPX4 inhibition (76). These synergistic effects support the rationale for integrating ferroptosis inducers into existing therapeutic regimens.
4.2 Challenges limiting the clinical translation of ferroptosis-based therapies
Despite the therapeutic promise, several challenges hinder the clinical translation of ferroptosis inducers in cervical cancer. Tumor heterogeneity, both genetic and metabolic, results in variable ferroptosis sensitivity (77, 78). Subpopulations of cervical cancer cells may harbor compensatory antioxidant pathways (e.g., FSP1, DHODH) that confer resistance to GPX4 inhibition. Therefore, patient stratification based on ferroptosis regulator expression profiles may be necessary. Another limitation involves the delivery and specificity of ferroptosis inducers, many existing compounds exhibit poor solubility, off-target toxicity, or lack tumor selectivity. Moreover, novel delivery systems, such as lipid nanoparticles or tumor-targeted prodrugs, are being explored to enhance drug bioavailability and reduce systemic toxicity. In cervical cancer, leveraging HPV-specific antigens or receptors may provide a basis for targeted delivery. Furthermore, ferroptosis inducers may activate systemic inflammation or trigger unintended ferroptosis in normal tissues, particularly in organs with high PUFA content such as the liver or brain.
4.3 Clinical perspectives and future directions
Although no ferroptosis-based therapy has yet received regulatory approval, multiple ferroptosis inducers are in early-phase clinical trials for solid tumors, including agents targeting GPX4, system Xc-, and iron metabolism. The development of companion diagnostics based on ACSL4, SLC7A11, or lipid peroxidation biomarkers may aid in identifying responsive patient subsets. In cervical cancer, future clinical studies should explore rational combinations of ferroptosis inducers with radiotherapy, chemotherapy, and immunotherapy. Personalized approaches integrating multi-omic profiling, including transcriptomics, lipidomics, and redox metabolomics, may optimize therapeutic strategies and predict ferroptosis susceptibility. Additionally, the temporal dynamics of ferroptosis induction—whether pulsed or sustained—may influence therapeutic outcomes and should be systematically evaluated.
5 Conclusion
Ferroptosis has emerged as a pivotal process influencing tumor biology and immune dynamics in cervical cancer. Here, we highlight how ferroptosis intersects with immune evasion mechanisms to shape tumor progression and treatment resistance. Specifically,
Dysregulated iron metabolism, aberrant lipid peroxidation, and impaired antioxidant defenses—shaped in part by HPV oncoproteins—modulate ferroptotic vulnerability and immune escape in cervical tumors. Key regulators such as GPX4, SLC7A11, ACSL4, and FSP1 define the ferroptosis threshold, while HPV-driven suppression of ACSL4 and ALOX15 contributes to resistance. The immune consequences of ferroptosis are context-dependent. Ferroptotic tumor cells can enhance antigenicity and support CD8+ T cell responses, yet also release DAMPs and ROS that promote immunosuppressive inflammation and recruitment of Tregs and MDSCs. Moreover, immune cells themselves, including T cells and dendritic cells, are vulnerable to ferroptosis, particularly in the oxidative and HPV-modulated tumor microenvironment.
Therapeutically, ferroptosis induction offers a novel strategy to combat cervical cancers that are refractory to conventional therapies. Combination approaches—such as ferroptosis inducers with immune checkpoint inhibitors—may restore anti-tumor immunity, reprogram suppressive myeloid cells, and overcome resistance to PD-1/PD-L1 blockade. Erastin, RSL3, and other small molecules have shown preclinical efficacy, particularly in HPV-transformed cells with elevated iron load and redox imbalance. Radiation and cisplatin may also sensitize tumors to ferroptosis through lipid remodeling.
In summary, ferroptosis represents both a vulnerability and a regulatory node in cervical cancer. Targeting ferroptosis—especially in combination with immunotherapy—may reshape the immune microenvironment and improve treatment outcomes. Future research should prioritize HPV-specific ferroptosis-immune interactions, immune cell protection strategies, and biomarker-driven patient selection to facilitate clinical translation.
Author contributions
LL: Writing – original draft, Formal Analysis, Project administration, Data curation, Methodology, Investigation. YB: Investigation, Writing – review & editing, Methodology, Data curation, Resources, Project administration. DX: Investigation, Writing – review & editing, Visualization, Supervision. LQ: Supervision, Visualization, Validation, Writing – review & editing, Conceptualization, Software.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank BioRender for generation of figures in this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ferrall L, Lin KY, Roden RBS, Hung CF, and Wu TC. Cervical cancer immunotherapy: facts and hopes. Clin Cancer Res. (2021) 27:4953–73. doi: 10.1158/1078-0432.CCR-20-2833
2. Monk BJ, Enomoto T, Kast WM, McCormack M, Tan DSP, Wu X, et al. Integration of immunotherapy into treatment of cervical cancer: Recent data and ongoing trials. Cancer Treat Rev. (2022) 106:102385. doi: 10.1016/j.ctrv.2022.102385
3. Ling J, Sun Q, Tian Q, Shi H, Yang H, and Ren J. Human papillomavirus 16 E6/E7 contributes to immune escape and progression of cervical cancer by regulating miR-142-5p/PD-L1 axis. Arch Biochem Biophys. (2022) 731:109449. doi: 10.1016/j.abb.2022.109449
4. Shamseddine AA, Burman B, Lee NY, Zamarin D, and Riaz N. Tumor immunity and immunotherapy for HPV-related cancers. Cancer Discov. (2021) 11:1896–912. doi: 10.1158/2159-8290.CD-20-1760
5. Qu X, Wang Y, Jiang Q, Ren T, Guo C, Hua K, et al. Interactions of Indoleamine 2, 3-dioxygenase-expressing LAMP3(+) dendritic cells with CD4(+) regulatory T cells and CD8(+) exhausted T cells: synergistically remodeling of the immunosuppressive microenvironment in cervical cancer and therapeutic implications. Cancer Commun (Lond). (2023) 43:1207–28. doi: 10.1002/cac2.12486
6. Li J, Cao Y, Liu Y, Yu L, Zhang Z, Wang X, et al. Multiomics profiling reveals the benefits of gamma-delta (gammadelta) T lymphocytes for improving the tumor microenvironment, immunotherapy efficacy and prognosis in cervical cancer. J Immunother Cancer. (2024) 12(1):e008355. doi: 10.1136/jitc-2023-008355
7. Jiang X, Stockwell BR, and Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
8. Liu H, Xue H, Guo Q, Xue X, Yang L, Zhao K, et al. Ferroptosis meets inflammation: A new frontier in cancer therapy. Cancer Lett. (2025) 620:217696. doi: 10.1016/j.canlet.2025.217696
9. Liu Y, Lu S, Wu LL, Yang L, Yang L, and Wang J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. (2023) 14:519. doi: 10.1038/s41419-023-06045-y
10. Guy MM, Bian T, Sun L, Hao Y, Jiao X, Zhang W, et al. SLC7A11 is a potential therapeutic target and prognostic biomarker correlated with immune cell infiltration in cervical cancer. Discov Oncol. (2025) 16:125. doi: 10.1007/s12672-025-01888-7
11. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, et al. Landscape of genomic alterations in cervical carcinomas. Nature. (2014) 506:371–5. doi: 10.1038/nature12881
12. Carrillo-Beltran D, Munoz JP, Guerrero-Vasquez N, Blanco R, Leon O, de Souza Lino V, et al. Human papillomavirus 16 E7 promotes EGFR/PI3K/AKT1/NRF2 signaling pathway contributing to PIR/NF-kappaB activation in oral cancer cells. Cancers (Basel). (2020) 12(7):1904. doi: 10.3390/cancers12071904
13. Cui K, Wang K, and Huang Z. Ferroptosis and the tumor microenvironment. J Exp Clin Cancer Res. (2024) 43:315. doi: 10.1186/s13046-024-03235-0
14. Bayir H, Dixon SJ, Tyurina YY, Kellum JA, and Kagan VE. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat Rev Nephrol. (2023) 19:315–36. doi: 10.1038/s41581-023-00689-x
15. Tang Z, Zhao P, Wang H, Liu Y, and Bu W. Biomedicine meets fenton chemistry. Chem Rev. (2021) 121:1981–2019. doi: 10.1021/acs.chemrev.0c00977
16. Xu L, Liu Y, Chen X, Zhong H, and Wang Y. Ferroptosis in life: To be or not to be. BioMed Pharmacother. (2023) 159:114241. doi: 10.1016/j.biopha.2023.114241
17. Shi JF, Liu Y, Wang Y, Gao R, Wang Y, and Liu J. Targeting ferroptosis, a novel programmed cell death, for the potential of alcohol-related liver disease therapy. Front Pharmacol. (2023) 14:1194343. doi: 10.3389/fphar.2023.1194343
18. Zhang D, Duque-Jimenez J, Facchinetti F, Brixi G, Rhee K, Feng WW, et al. Transferrin receptor targeting chimeras for membrane protein degradation. Nature. (2025) 638:787–95. doi: 10.1038/s41586-024-07947-3
19. Kim H, Villareal LB, Liu Z, Haneef M, Falcon DM, Martin DR, et al. Transferrin receptor-mediated iron uptake promotes colon tumorigenesis. Adv Sci (Weinh). (2023) 10:e2207693. doi: 10.1002/advs.202207693
20. Wang T, Gong M, Cao Y, Zhao C, Lu Y, Zhou Y, et al. Persistent ferroptosis promotes cervical squamous intraepithelial lesion development and oncogenesis by regulating KRAS expression in patients with high risk-HPV infection. Cell Death Discov. (2022) 8:201. doi: 10.1038/s41420-022-01013-5
21. Taghizadeh E, Jahangiri S, Rostami D, Taheri F, Renani PG, Taghizadeh H, et al. Roles of E6 and E7 human papillomavirus proteins in molecular pathogenesis of cervical cancer. Curr Protein Pept Sci. (2019) 20:926–34. doi: 10.2174/1389203720666190618101441
22. Chang X and Miao J. Ferroptosis: Mechanism and potential applications in cervical cancer. Front Mol Biosci. (2023) 10:1164398. doi: 10.3389/fmolb.2023.1164398
23. Porter VL and Marra MA. The drivers, mechanisms, and consequences of genome instability in HPV-driven cancers. Cancers (Basel). (2022) 14(19):4623. doi: 10.3390/cancers14194623
24. Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. (2021) 17:4266–85. doi: 10.1080/15548627.2021.1911016
25. Wang J, Wu N, Peng M, Oyang L, Jiang X, Peng Q, et al. Ferritinophagy: research advance and clinical significance in cancers. Cell Death Discov. (2023) 9:463. doi: 10.1038/s41420-023-01753-y
26. Shi H, Xiong L, Yan G, Du S, Liu J, and Shi Y. Susceptibility of cervical cancer to dihydroartemisinin-induced ferritinophagy-dependent ferroptosis. Front Mol Biosci. (2023) 10:1156062. doi: 10.3389/fmolb.2023.1156062
27. Xu J, Zheng B, Wang W, and Zhou S. Ferroptosis: a novel strategy to overcome chemoresistance in gynecological Malignancies. Front Cell Dev Biol. (2024) 12:1417750. doi: 10.3389/fcell.2024.1417750
28. Zhou H, Zhou YL, Mao JA, Tang LF, Xu J, Wang ZX, et al. NCOA4-mediated ferritinophagy is involved in ionizing radiation-induced ferroptosis of intestinal epithelial cells. Redox Biol. (2022) 55:102413. doi: 10.1016/j.redox.2022.102413
29. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
30. Xiaofei J, Mingqing S, Miao S, Yizhen Y, Shuang Z, Qinhua X, et al. Oleanolic acid inhibits cervical cancer Hela cell proliferation through modulation of the ACSL4 ferroptosis signaling pathway. Biochem Biophys Res Commun. (2021) 545:81–8. doi: 10.1016/j.bbrc.2021.01.028
31. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. (2019) 10:1617. doi: 10.1038/s41467-019-09277-9
32. Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, et al. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. (2017) 108:2187–94. doi: 10.1111/cas.13380
33. Luo Y, Chen Y, Jin H, Hou B, Li H, Li X, et al. The suppression of cervical cancer ferroptosis by macrophages: The attenuation of ALOX15 in cancer cells by macrophages-derived exosomes. Acta Pharm Sin B. (2023) 13:2645–62. doi: 10.1016/j.apsb.2023.03.025
34. Wang X, Ji Y, Qi J, Zhou S, Wan S, Fan C, et al. Mitochondrial carrier 1 (MTCH1) governs ferroptosis by triggering the FoxO1-GPX4 axis-mediated retrograde signaling in cervical cancer cells. Cell Death Dis. (2023) 14:508. doi: 10.1038/s41419-023-06033-2
35. Feng M, Wu H, Zhu L, Gao J, and Deng G. Triptolide promotes ferroptosis in cervical cancer cell via NRF2/xCT/GPX4. Phytother Res. (2025) 39:875–87. doi: 10.1002/ptr.8398
36. Wang X, Chen Y, Wang X, Tian H, Wang Y, Jin J, et al. Stem cell factor SOX2 confers ferroptosis resistance in lung cancer via upregulation of SLC7A11. Cancer Res. (2021) 81:5217–29. doi: 10.1158/0008-5472.CAN-21-0567
37. Yan A, Wu H, and Jiang W. RACK1 inhibits ferroptosis of cervical cancer by enhancing SLC7A11 core-fucosylation. Glycoconj J. (2024) 41:229–40. doi: 10.1007/s10719-024-10167-6
38. Wang X, Du Q, Mai Q, Zou Q, Wang S, Lin X, et al. Targeting FASN enhances cisplatin sensitivity via SLC7A11-mediated ferroptosis in cervical cancer. Transl Oncol. (2025) 56:102396. doi: 10.1016/j.tranon.2025.102396
39. Xiong J, Chen P, He L, Chai X, Zhang Y, and Sun S. Functional mechanism of hypoxia-like conditions mediating resistance to ferroptosis in cervical cancer cells by regulating KDM4A SUMOylation and the SLC7A11/GPX4 pathway. Environ Toxicol. (2024) 39:4207–20. doi: 10.1002/tox.24304
40. Yue J, Yin Y, Feng X, Xu J, Li Y, Li T, et al. Discovery of the inhibitor targeting the SLC7A11/xCT axis through in silico and in vitro experiments. Int J Mol Sci. (2024) 25(15):8284. doi: 10.3390/ijms25158284
41. Li W, Liang L, Liu S, Yi H, and Zhou Y. FSP1: a key regulator of ferroptosis. Trends Mol Med. (2023) 29:753–64. doi: 10.1016/j.molmed.2023.05.013
42. Zhao MY, Liu P, Sun C, Pei LJ, and Huang YG. Propofol augments paclitaxel-induced cervical cancer cell ferroptosis. In Vitro Front Pharmacol. (2022) 13:816432. doi: 10.3389/fphar.2022.816432
43. Kaymak I, Maier CR, Schmitz W, Campbell AD, Dankworth B, Ade CP, et al. Mevalonate pathway provides ubiquinone to maintain pyrimidine synthesis and survival in p53-deficient cancer cells exposed to metabolic stress. Cancer Res. (2020) 80:189–203. doi: 10.1158/0008-5472.CAN-19-0650
44. Alexandru I, Nistor D, Motofelea AC, Cadar Andone BA, Crintea A, Tatu C, et al. Vitamins, coenzyme Q10, and antioxidant strategies to improve oocyte quality in women with gynecological cancers: A comprehensive review. Antioxid (Basel). (2024) 13(12):1567. doi: 10.3390/antiox13121567
45. Bae T, Hallis SP, and Kwak MK. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp Mol Med. (2024) 56:501–14. doi: 10.1038/s12276-024-01180-8
46. Zhang Z, Hu Q, Ye S, and Xiang L. Inhibition of the PIN1-NRF2/GPX4 axis imparts sensitivity to cisplatin in cervical cancer cells. Acta Biochim Biophys Sin (Shanghai). (2022) 54:1325–35. doi: 10.3724/abbs.2022109
47. Tie W and Ge F. Lymphoid-specific helicase inhibits cervical cancer cells ferroptosis by promoting Nrf2 expression. PeerJ. (2023) 11:e16451. doi: 10.7717/peerj.16451
48. Chiang SK, Chen SE, and Chang LC. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci. (2018) 20(1):39. doi: 10.3390/ijms20010039
49. Tang XH, Zhao TN, Guo L, Liu XY, Zhang WN, and Zhang P. Cell-cycle-related protein centromere protein F deficiency inhibits cervical cancer cell growth by inducing ferroptosis via nrf2 inactivation. Cell Biochem Biophys. (2024) 82:997–1006. doi: 10.1007/s12013-024-01251-7
50. Wei X, Huang Q, Huang J, Yu L, and Chen J. Erastin induces ferroptosis in cervical cancer cells via Nrf2/HO-1 signaling pathway. Int J Immunopathol Pharmacol. (2023) 37:3946320231219348. doi: 10.1177/03946320231219348
51. Hushmandi K, Alimohammadi M, Heiat M, Hashemi M, Nabavi N, Tabari T, et al. Targeting Wnt signaling in cancer drug resistance: Insights from pre-clinical and clinical research. Pathol Res Pract. (2025) 267:155837. doi: 10.1016/j.prp.2025.155837
52. Mishima E, Nakamura T, Zheng J, Zhang W, Mourao ASD, Sennhenn P, et al. DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature. (2023) 619:E9–E18. doi: 10.1038/s41586-023-06269-0
53. Jiang M, Song Y, Liu H, Jin Y, Li R, and Zhu X. DHODH Inhibition Exerts Synergistic Therapeutic Effect with Cisplatin to Induce Ferroptosis in Cervical Cancer through Regulating mTOR Pathway. Cancers (Basel). (2023) 15(2):546. doi: 10.3390/cancers15020546
54. Yang JY, Ke D, Li Y, Shi J, Wan SM, Wang AJ, et al. CNIH4 governs cervical cancer progression through reducing ferroptosis. Chem Biol Interact. (2023) 384:110712. doi: 10.1016/j.cbi.2023.110712
55. Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang J, et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transd Target Ther. (2023) 8:104. doi: 10.1038/s41392-023-01365-z
56. Liu Y, Tan H, Dai J, Lin J, Zhao K, Hu H, et al. Targeting macrophages in cancer immunotherapy: Frontiers and challenges. J Adv Res. (2025) 2090-1232(24)00622-2. doi: 10.1016/j.jare.2024.12.043
57. Liu C, Li X, Huang Q, Zhang M, Lei T, Wang F, et al. Single-cell RNA-sequencing reveals radiochemotherapy-induced innate immune activation and MHC-II upregulation in cervical cancer. Signal Transd Target Ther. (2023) 8:44. doi: 10.1038/s41392-022-01264-9
58. Evans AM, Salnikov M, Tessier TM, and Mymryk JS. Reduced MHC class I and II expression in HPV-negative vs. HPV-positive cervical cancers. Cells. (2022) 11(23):3911. doi: 10.3390/cells11233911
59. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. (2022) 13:3676. doi: 10.1038/s41467-022-31218-2
60. Dixon SJ and Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. (2024) 25:424–42. doi: 10.1038/s41580-024-00703-5
61. Wang B, Wang Y, Zhang J, Hu C, Jiang J, Li Y, et al. ROS-induced lipid peroxidation modulates cell death outcome: mechanisms behind apoptosis, autophagy, and ferroptosis. Arch Toxicol. (2023) 97:1439–51. doi: 10.1007/s00204-023-03476-6
62. Wang C, Li P, Liu L, Pan H, Li H, Cai L, et al. Self-adjuvanted nanovaccine for cancer immunotherapy: Role of lysosomal rupture-induced ROS in MHC class I antigen presentation. Biomaterials. (2016) 79:88–100. doi: 10.1016/j.biomaterials.2015.11.040
63. Calzada-Fraile D, Iborra S, Ramirez-Huesca M, Jorge I, Dotta E, Hernandez-Garcia E, et al. Immune synapse formation promotes lipid peroxidation and MHC-I upregulation in licensed dendritic cells for efficient priming of CD8(+) T cells. Nat Commun. (2023) 14:6772. doi: 10.1038/s41467-023-42480-3
64. Wen Q, Liu J, Kang R, Zhou B, and Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
65. Dang Q, Sun Z, Wang Y, Wang L, Liu Z, and Han X. Ferroptosis: a double-edged sword mediating immune tolerance of cancer. Cell Death Dis. (2022) 13:925. doi: 10.1038/s41419-022-05384-6
66. Huang J, Pan H, Sun J, Wu J, Xuan Q, Wang J, et al. TMEM147 aggravates the progression of HCC by modulating cholesterol homeostasis, suppressing ferroptosis, and promoting the M2 polarization of tumor-associated macrophages. J Exp Clin Cancer Res. (2023) 42:286. doi: 10.1186/s13046-023-02865-0
67. Xu M, Cao C, Wu P, Huang X, and Ma D. Advances in cervical cancer: current insights and future directions. Cancer Commun (Lond). (2025) 45:77–109. doi: 10.1002/cac2.12629
68. Gu X, Liu Y, Dai X, Yang YG, and Zhang X. Deciphering the potential roles of ferroptosis in regulating tumor immunity and tumor immunotherapy. Front Immunol. (2023) 14:1137107. doi: 10.3389/fimmu.2023.1137107
69. Wang Y, Liu XY, Wang Y, Zhao WX, Li FD, Guo PR, et al. NOX2 inhibition stabilizes vulnerable plaques by enhancing macrophage efferocytosis via MertK/PI3K/AKT pathway. Redox Biol. (2023) 64:102763. doi: 10.1016/j.redox.2023.102763
70. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
71. Friedmann Angeli JP, Krysko DV, and Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. (2019) 19:405–14. doi: 10.1038/s41568-019-0149-1
72. Chattopadhyay S, Hazra R, Mallick A, Gayen S, and Roy S. A review on comprehending immunotherapeutic approaches inducing ferroptosis: Managing tumour immunity. Immunology. (2024) 172:547–65. doi: 10.1111/imm.13789
73. Xiao K, Zhang S, Peng Q, Du Y, Yao X, Ng II, et al. PD-L1 protects tumor-associated dendritic cells from ferroptosis during immunogenic chemotherapy. Cell Rep. (2024) 43:114868. doi: 10.1016/j.celrep.2024.114868
74. Gao J, Zhang X, Liu Y, and Gu X. Ferroptosis in immune cells: Implications for tumor immunity and cancer therapy. Cytokine Growth Factor Rev. (2025) S1359-6101(25)00081-4. doi: 10.1016/j.cytogfr.2025.06.007
75. Liu Y, Wu G, Feng L, Li J, Xia Y, Guo W, et al. Harnessing antioxidants in cancer therapy: opportunities, challenges, and future directions. Antioxid (Basel). (2025) 14(6):674. doi: 10.3390/antiox14060674
76. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. (2020) 30:146–62. doi: 10.1038/s41422-019-0263-3
77. Li C, Wu H, Guo L, Liu D, Yang S, Li S, et al. Single-cell transcriptomics reveals cellular heterogeneity and molecular stratification of cervical cancer. Commun Biol. (2022) 5:1208. doi: 10.1038/s42003-022-04142-w
Keywords: cervical cancer, ferroptosis, immune evasion, tumor microenvironment, immunotherapy
Citation: Li L, Bo Y, Xue D and Qin L (2025) Ferroptosis-immune crosstalk in cervical cancer: mechanisms and therapeutic implications. Front. Immunol. 16:1657905. doi: 10.3389/fimmu.2025.1657905
Received: 02 July 2025; Accepted: 25 July 2025;
Published: 15 August 2025.
Edited by:
Matheus Mattos, KU Leuven, BelgiumReviewed by:
Zhaohui Huang, Affiliated Hospital of Jiangnan University, ChinaTengfei Liu, Shanghai Jiao Tong University, China
Copyright © 2025 Li, Bo, Xue and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Xue, MTM1NDY3MjM5MzlAMTM5LmNvbQ==; Lijuan Qin, cWlubGptbUAxNjMuY29t
†These authors have contributed equally to this work