 Yujia Zhu1,2,3
Yujia Zhu1,2,3 Hongxiao Song
Hongxiao Song Guangyun Tan
Guangyun Tan- 1Department of Hepatology, Center of Infectious Diseases and Pathogen Biology, Institute of Translational Medicine, The First Hospital of Jilin University, Changchun, Jilin, China
- 2Jilin Provincial Key Laboratory of Metabolic Liver Diseases, Jilin University, Changchun, Jilin, China
- 3China-Singapore Belt and Road Joint Laboratory on Liver Disease Research, Changchun, Jilin, China
The hepatitis B core protein (HBc) is a multifunctional viral protein central to hepatitis B virus (HBV) replication, immune modulation, and capsid assembly. Structurally, HBc consists of an N-terminal domain (NTD) essential for capsid formation and a C-terminal domain (CTD) critical for RNA binding and genome packaging. Frequent HBc mutations, driven by HBV’s high mutation rate, enhance the virus’s ability to adapt to environmental pressures. HBc interacts with host factors to regulate viral transcription, stabilize capsids, and modulate immune responses, including the suppression of interferon signaling and promotion of immune exhaustion. Clinically, anti-HBc antibodies serve as key diagnostic markers, while HBc-targeting therapies, such as capsid assembly modulators (CAMs), represent promising strategies for achieving functional cure. This review uniquely integrates structural, functional, and clinical perspectives on HBc, providing a comprehensive understanding of its role in HBV biology and its potential as a therapeutic target. By highlighting recent advances in CAMs and the challenges of drug resistance, this work offers valuable insights for researchers and clinicians aiming to develop innovative HBV treatments.
1 Introduction
Chronic hepatitis B virus (HBV) infection remains a major global public health challenge. In 2019, an estimated 296 million individuals worldwide tested positive for hepatitis B surface antigen (HBsAg), with a global prevalence of chronic HBV infection of approximately 3.5%. High-endemic areas include China, Southeast Asia, and sub-Saharan Africa (1). HBV is a hepatotropic virus with a narrow host range, infecting only humans and a few non-human primates. HBV infection can lead to a spectrum of liver diseases, including acute and chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (HCC), and it is recognized as a leading cause of HCC worldwide (2–4).
There are three types of HBV particles present in the blood of infected individuals: 22 nm diameter spherical and filamentous subviral particles (SVPs) and the 44 nm diameter Dane particles. SVPs are non-infectious but highly immunogenic, whereas Dane particles are infectious (5). Dane particles consist of an envelope made of lipid bilayers embedded with HBsAg and a nucleocapsid. The nucleocapsid is a capsid composed of 180 or 240 hepatitis B core proteins (HBc), which encloses the viral DNA genome and the viral polymerase (Pol) (6). The HBV genome is a 3.2 kb partially double-stranded, relaxed circular DNA (rcDNA), containing a complete negative strand and an incomplete positive strand.
Upon infection, HBV initially binds with low affinity to heparan sulfate proteoglycans (HSPGs) on the host cell surface, facilitating viral concentration on the target cell and increasing the likelihood of receptor engagement (7, 8). High-affinity binding then occurs between the PreS1 domain of the HBV L envelope protein and the sodium taurocholate co-transporting polypeptide (NTCP), leading to NTCP oligomerization and promoting viral internalization and release of the nucleocapsid into the cytoplasm. Neuropilin-1 (NRP1) further enhances viral attachment and facilitates the interaction between PreS1 and NTCP, thus promoting HBV infection (9–11). Subsequently, the rcDNA is transported into the nucleus, where it is converted into covalently closed circular DNA (cccDNA) with the assistance of host DNA repair machinery (12, 13).
The HBV genome contains four overlapping open reading frames (ORFs)—S, C, P, and X—which, after infection and cccDNA formation, serve as transcriptional templates for six partially overlapping viral mRNAs: pregenomic RNA (pgRNA, 3.5 kb), preC RNA (3.5 kb), preS1 HBs RNA (2.4 kb), preS2/S HBs RNA (2.1 kb), and HBV X RNA (0.7 kb) (14). These transcripts encode various viral proteins: pgRNA encodes HBc and Pol; preC RNA encodes hepatitis B e antigen (HBeAg); preS1, preS2 and S HBs RNA encodes the L envelope protein; preS2 and S HBs RNA encodes the M envelope proteins, S HBs RNA encodes the S envelope proteins; and HBV X RNA encodes HBx protein.
HBc forms dimers that assemble into capsids, encapsidating Pol along with pgRNA. Inside the capsid, Pol catalyzes reverse transcription of pgRNA to synthesize rcDNA. Mature nucleocapsids can either be transported back into the nucleus to replenish the cccDNA pool or be enveloped by viral surface proteins within multivesicular bodies (MVBs) and subsequently released from hepatocytes as fully formed infectious virions (15–17).
2 Hepatitis B core protein
2.1 HBc protein structure
HBc protein consists of 183 or 185 amino acid residues (aa) and contains two structural domains: the N-terminal domain (NTD, aa 1-140) and the C-terminal domain (CTD, aa 150-183/185), which are connected by a linker peptide (aa 141-149) (18). HBc is primarily composed of α-helical structures, with five α-helices. The main feature is a long α-helix formed by residues 50–73 (α3) and 79–110 (α4), which forms a helical hairpin that dimerizes through α-helical interactions (19). HBc exhibits a flexible structure, and the conformation of free HBc dimers differs from the conformation of dimers that form the capsid. It is thus proposed that HBc exists in capsid assembly-active (HBcAct), capsid assembly-incompetent (HBcInc), or abnormal (HBcAbb) conformations, with only HBcAct capable of assembling into icosahedral capsids. In contrast, HBcInc/HBcAbb assemble into non-icosahedral forms, disrupting the HBV life cycle (20, 21). The homodimer of HBc contains two structural domains: the α3 and α4 helices of the monomer, which wrap around each other to form a four-helix bundle dimerization interface linked by disulfide bonds, and the α1, α2, and α5 helices, which surround the base of the four-helix bundle to form the hydrophobic core of the contact domain (21, 22) (Figure 1). HBc dimers serve as the building blocks of capsid assembly, which consists of a double-layer structure, with the NTD forming the outer layer and the CTD and its associated RNA forming the inner layer (23).
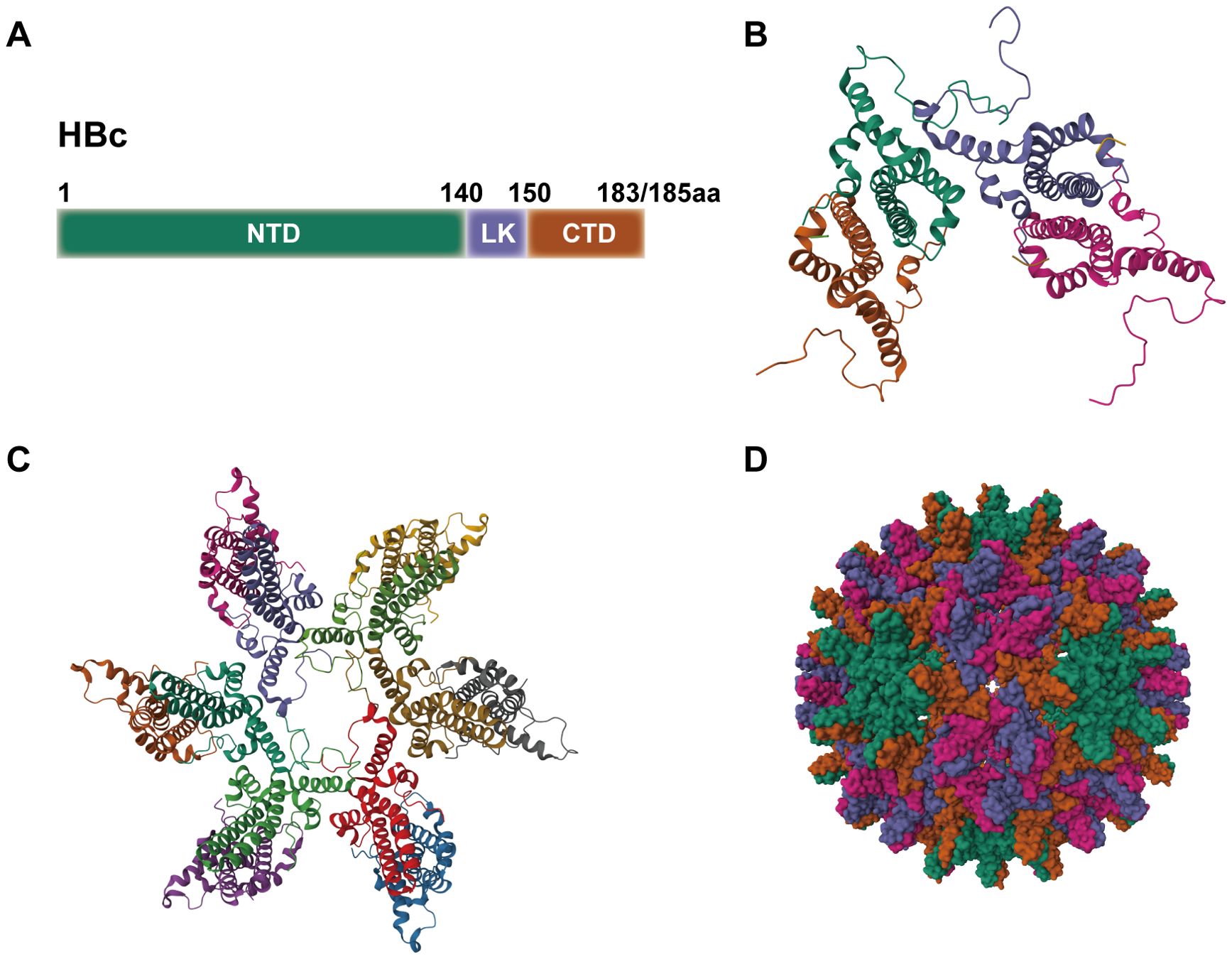
Figure 1. The structure of HBc. (A) Domains of the HBc protein. (B) Ribbon model of HBc protein. (C) Ribbon model of HBV capsid. (D) Spherical structure of HBV capsid.
2.2 HBc variants
HBV replicates through reverse transcription of its pgRNA. Due to the lack of proofreading activity in its reverse transcriptase, HBV has a significantly higher mutation rate than other DNA viruses (24). Studies have identified 54 missense mutation sites in the HBc protein in hepatocellular carcinoma tissues, mainly concentrated in three major (codons 83–87, 95–104, and 130–135) and three minor (codons 21–38, 59–63, and 151–155) mutation cluster regions (MCR), with the most frequent mutations being P130T, I97L, and S87G (25).
HBV can evolve through mutations, increasing its adaptability to environmental selection pressures. These mutations may influence various aspects of viral replication and pathogenesis. In the presence of HLA class I alleles, sequence analysis of HBc revealed nine mutated amino acid residues, seven of which are within the CD8+ T cell-targeted epitopes (26). For instance, the L60V mutation in HBc, where leucine is substituted with valine, is a typical HLA-A2-binding peptide, and its affinity for HLA-A*0201 is enhanced. The HLA molecule captures and presents this antigenic peptide to T cells, leading to T cell activation, proliferation, differentiation, and cytotoxic T lymphocyte (CTL) responses that contribute to liver damage. Furthermore, the HBc L60V mutation promotes capsid assembly and enhances HBV replication, but it results in reduced viral secretion. Additionally, a proline deletion at position 5 of HBc also causes low-level viral secretion (27, 28).
The most common natural mutation of HBc occurs at amino acid position 97, where isoleucine or phenylalanine is substituted with leucine. The surface spikes of the capsid contain a hydrophobic pocket formed largely by residues P5, L60, L95, K96, and I97/F97 (29, 30). Bioinformatics and cryo-EM studies revealed that the hydrophobic pocket surrounding the I97L mutation exhibits defects in maintaining tight coupling between genome maturation and viral secretion. The signal for genome maturation is transmitted from the interior of the capsid to its surface via this hydrophobic pocket, and thus the I97L mutant results in excessive secretion of virions containing immature genomes (31). Additionally, the F97L mutation enhances capsid assembly capability (32). Although the P130T mutation does not directly affect the pocket, it increases the maturation of the intracellular genome. Studies have shown that the immature secretion of the I97L mutant can be compensated by the P130T mutation, and the P130T mutation is often associated with the occurrence of I97L mutations (33) (Table 1).
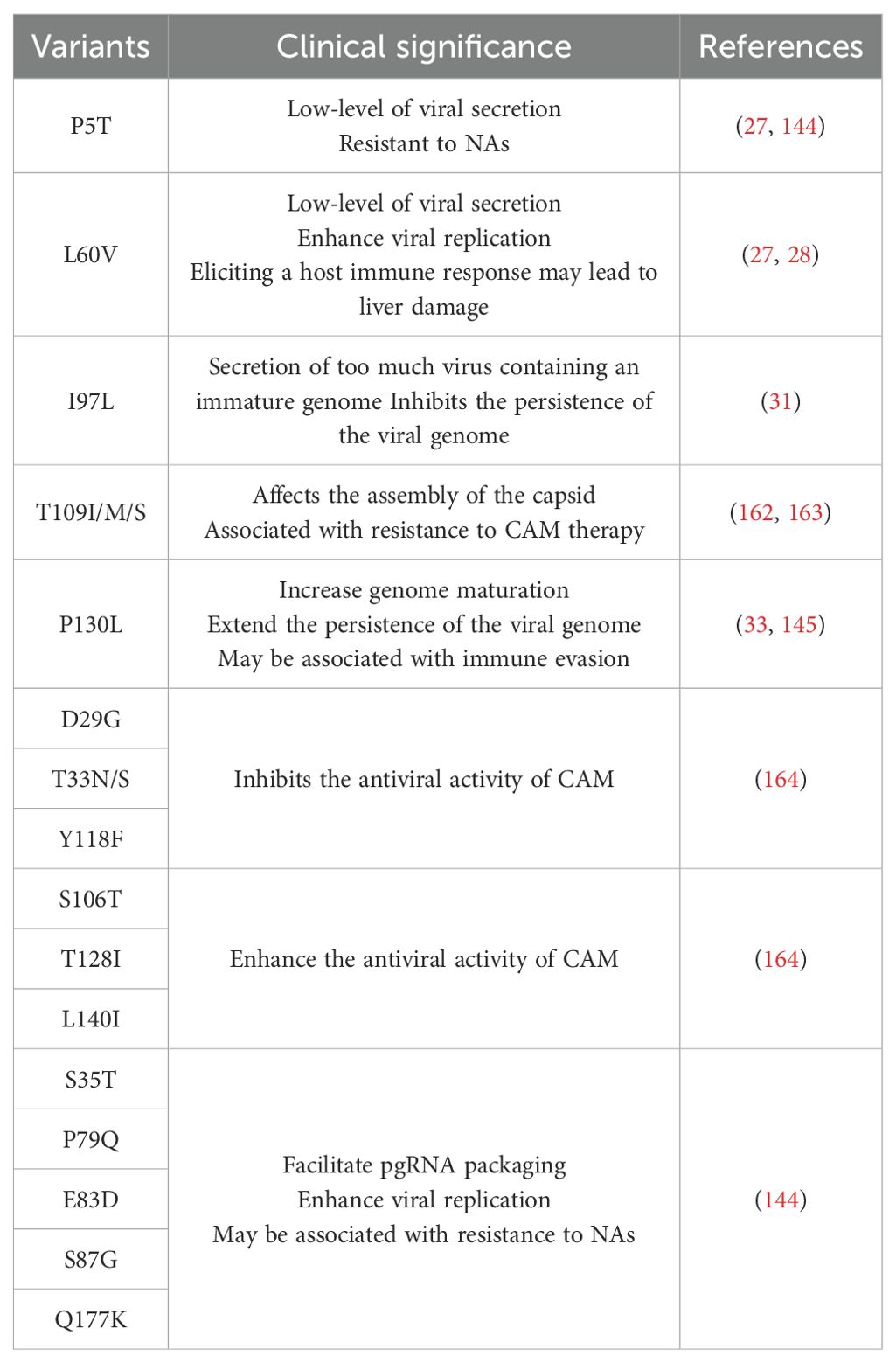
Table 1. HBc variants and clinical significance.
3 The role of core in viral particle formation
3.1 NTD-mediated capsid assembly
The NTD is essential for capsid formation, consisting of five α-helices. Among them, α1, α2, and α5 form a hydrophobic core that stabilizes the monomer structure, while the α3 and α4 helices of two monomers interact through hydrophobic forces to drive the formation of a four-helix bundle, resulting in core dimerization and the formation of a homodimer. This dimer then assembles into a capsid through hydrophobic interactions between dimer-dimer interfaces (34–36). The core dimer can form two types of icosahedral symmetry shells, namely T = 3 or T = 4. The T = 3 capsid, with a diameter of 30 nm, consists of 90 core dimers, while the T = 4 particle, with a diameter of 34 nm, consists of 120 core dimers (15). Studies have shown that the NTD alone is sufficient to assemble a capsid under high HBc and/or salt concentrations, but the stability of the capsid is reduced. Under low HBc concentrations, the NTD alone is insufficient to support capsid assembly, and the presence of the CTD is necessary (37, 38). In the absence of CTD, the linker peptide affects the assembly of the NTD and the type of capsid formed. Cp140 predominantly forms T = 3 capsids, whereas Cp149 primarily forms T = 4 capsids. When both NTD and CTD are present, capsid assembly can occur without the linker peptide (18, 39). Heat shock protein 90 (Hsp90) also participates in capsid assembly by binding to Cp149 dimers, which are then packaged into the capsid. The activation of Hsp90 promotes capsid assembly and enhances stability (40).
3.2 Functions of CTD in HBV life cycle
Although CTD is not essential for capsid assembly, it plays a crucial role in regulating several aspects of the HBV life cycle, including the packaging of pgRNA, genome reverse transcription, and intracellular transport (41). The CTD contains seven serine and one threonine residues that can be phosphorylated, namely Ser155, Ser162, Ser168, Ser170, Ser176, Ser178, Ser181, and Thr160 (42). CTD regulates HBV replication through phosphorylation and dephosphorylation. Serine/arginine protein kinase (SRPK), which binds to the unphosphorylated CTD, inhibits capsid assembly by preventing premature self-assembly. Upon phosphorylation, SRPK’s affinity for HBc decreases, releasing HBc to assemble the capsid, ensuring that HBc assembles at the appropriate time and location. SRPK is removed at the right time to allow capsid self-assembly (43, 44). CTD also interacts with RNA and is essential for the packaging of pgRNA. In vitro studies have shown that HBc lacking CTD cannot encapsidate pgRNA, but CTD does not have specificity for pgRNA (37, 41, 45). The Pol protein consists of four structural domains: the terminal protein (TP) domain, spacer domain (SD), reverse transcriptase (RT) domain, and RNase H domain. The tyrosine in the TP domain binds to the ϵ structure of pgRNA to form a P-ϵ ribonucleoprotein (RNP) complex, which recruits HBc protein to assemble the viral capsid. Phosphorylation of CTD facilitates the specific packaging of the RNP complex into the capsid, while preventing non-specific RNA packaging when the complex is absent or limited (16, 46). Studies have shown that CTD is phosphorylated in both dimer and pgRNA-containing capsid forms. Substitution of Ser155, Ser162, and Ser170 with alanine or aspartic acid to mimic dephosphorylated or phosphorylated states affects pgRNA packaging. Phosphorylation of Ser162 is necessary for pgRNA packaging, while phosphorylation of Ser170 optimizes the process (47). Substituting Thr160, Ser168, and Ser176 with alanine to mimic the dephosphorylated state severely impairs pgRNA packaging (42). Interestingly, studies have shown that HBc is phosphorylated in free dimers and empty capsids but is less phosphorylated in nucleocapsids containing pgRNA and DNA, suggesting that dephosphorylation may occur during pgRNA encapsidation (48). Pol recruits protein phosphatase 1 (PP1) (Figure 2), which dephosphorylates HBc-Ser170, and PP1 catalytic subunits α and β cooperate with pgRNA and Pol to package the nucleocapsid, promoting pgRNA packaging (49, 50).
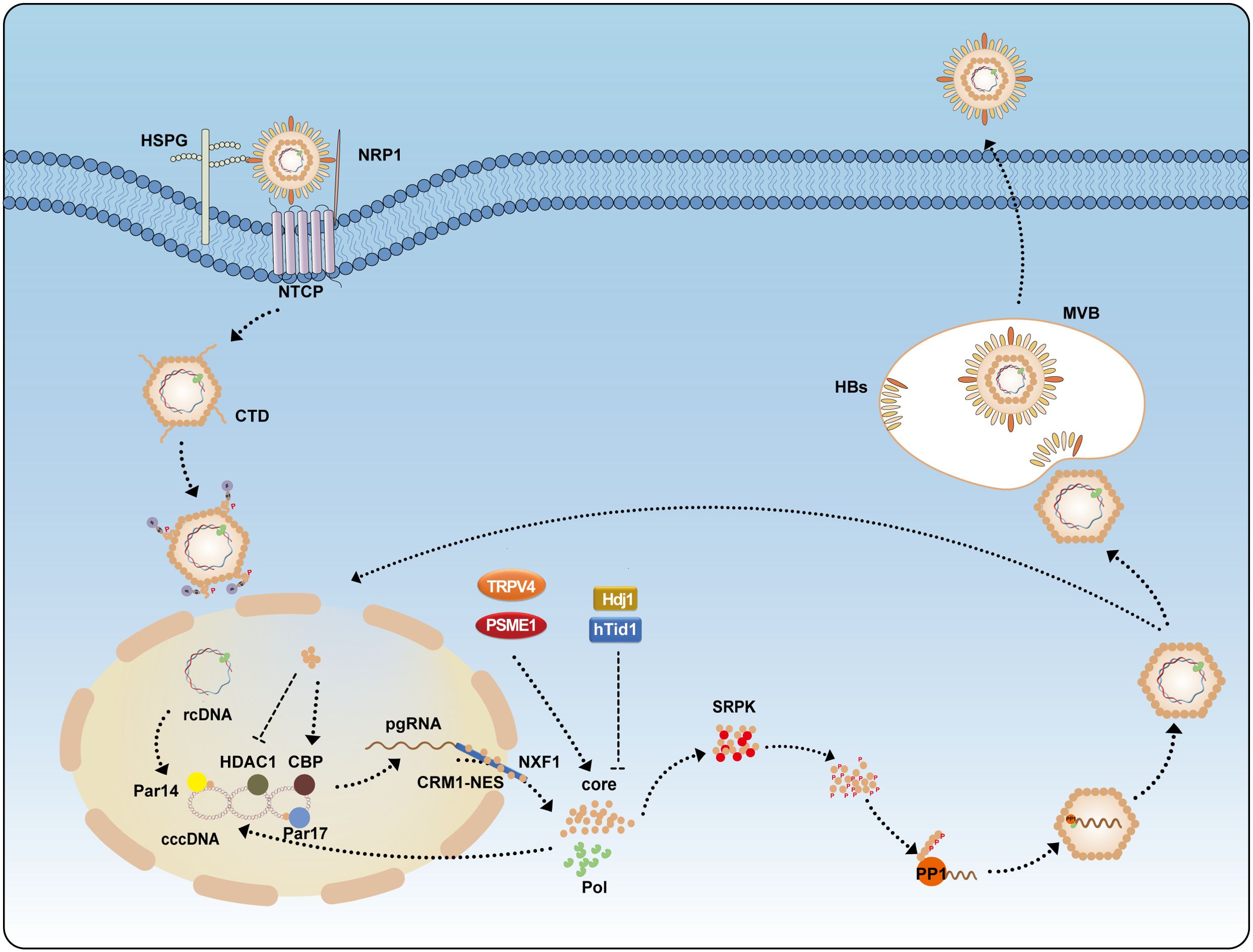
Figure 2. The life cycle of HBV. HBV enters hepatocytes through the receptor NTCP, assisted by HSPG and NRP1. Following entry, CTD is phosphorylated to facilitate its nuclear import. Inside the nucleus, HBc modulates the activity of Par14, Par17, CBP, and HDAC1 on cccDNA to enhance transcription. Newly synthesized HBc, translated from pgRNA, can be exported to the cytoplasm via the CRM1-NES and NXF1 pathways. Its expression is promoted by TRPV4 and PSME1 but inhibited by Hdj1 and hTid1. SRPK binds the unphosphorylated CTD to inhibit capsid assembly; CTD phosphorylation triggers the release of SRPK, allowing PP1 to dephosphorylate the CTD and facilitate nucleocapsid packaging. Mature nucleocapsids are either recycled to the nucleus to replenish the cccDNA pool or enveloped by HBs in MVBs and secreted as infectious virions.CBP, CREB-binding protein; HDAC1, histone deacetylase 1; HSPG, heparan sulfate proteoglycans; HBs, hepatitis B surface antigen; MVBs, multivesicular bodies; NRP1, neuropilin-1; NTCP, sodium taurocholate cotransporting polypeptide; NXF1, nuclear RNA export factor 1; PP1, protein phosphatase 1; PSME1, proteasome activator subunit 1; SRPK, serine-arginine protein kinase; TRPV4, transient receptor potential vanilloid 4.
Without the HBV capsid, Pol cannot reverse transcribe pgRNA into rcDNA. The core CTD contains 16 arginine residues, with 14 of them grouped into four clusters of three or four. These arginine clusters contribute to the reverse transcription process, including negative strand DNA synthesis extension, primer translocation, cyclization, and positive strand DNA extension (51). The phosphorylation state of the duck hepatitis B virus (DHBV) nucleocapsid dynamically changes during reverse transcription. Immature nucleocapsids (containing pgRNA), mature nucleocapsids (containing dsDNA), and secreted nucleocapsids were isolated from DHBV virus, and it was found that HBc was phosphorylated in immature nucleocapsids, but dephosphorylated in mature and secreted nucleocapsids (52). Phosphorylated HBc promotes negative strand DNA synthesis and the initiation of positive strand DNA extension, and the extension of these strands leads to HBc dephosphorylation. Dephosphorylated HBc, in turn, promotes further maturation of the positive strand DNA and stabilizes the mature nucleocapsid (53). It is hypothesized that the DNA synthesis process is associated with the dephosphorylation of CTD, but studies have shown that dephosphorylation of Ser155, Ser162, and Ser170 decreases the efficiency of negative strand DNA extension, primer translocation, cyclization, and positive strand DNA extension (54). While phosphorylation of Ser155, Ser162, and Ser170 also reduces these steps’ efficiency, the reduction is less pronounced, indicating that phosphorylation is required for rcDNA synthesis (55).
3.3 Regulation of cccDNA formation
It has proposed that HBc may negatively regulate cccDNA formation through its involvement in nucleocapsid envelopment and virion secretion by interacting with the viral envelope proteins. This hypothesis is supported by studies showing that mutations in either HBc or the L surface protein, which impair nucleocapsid envelopment and virion secretion, lead to increased cccDNA formation via intracellular amplification (56–61). However, there is no direct evidence to support the notion that HBc itself directly inhibits cccDNA formation. The cleavage of the nucleocapsid is regulated by the dynamic phosphorylation and dephosphorylation of HBc, controlling the release of the rcDNA genome and its conversion into cccDNA. In addition to CTD being phosphorylated, the NTD also has two conserved phosphorylation sites, Ser44 and Ser49. Substituting alanine to mimic the dephosphorylated state did not affect HBc expression, capsid assembly, pgRNA packaging, DNA synthesis, or complete virion secretion, but it inhibited cccDNA synthesis. Substituting glutamic acid to simulate the phosphorylated state partially impaired pgRNA packaging but destabilized the mature nucleocapsid, promoting cccDNA synthesis and enhancing CTD phosphorylation. These results suggest that phosphorylation of the NTD sites should not occur during the early stages of virus assembly but should occur after nucleocapsid maturation to facilitate the next stage of replication, such as during infection or nuclear entry, when nucleocapsid uncoating occurs to promote cccDNA formation (62).
Furthermore, HBc regulates the nuclear import of rcDNA to control cccDNA formation. During nucleocapsid maturation, an increase in negative charges from rcDNA synthesis may induce the exposure of CTD inside the immature nucleocapsid to the surface of the mature nucleocapsid, allowing the nuclear localization signal (NLS) in CTD to function and transport rcDNA into the nucleus for cccDNA formation (63). A study has found that there are two highly conserved lysine residues on HBc, namely K7 and K96, which are not required for HBV replication. However, the codon encoding K7 constitutes part of the RNA polyadenylation signal and is indispensable for cccDNA transcription (64). HBc also participates in cccDNA transcription by binding to cccDNA, reducing the nucleosomal spacing of cccDNA-histone complexes, altering the chromatin structure of cccDNA, and promoting its transcription (65). HBc prefers to bind to regions of cccDNA that are rich in CpG dinucleotide sequences. HBc promotes the binding of the acetyltransferase CREB-binding protein (CBP) to cccDNA and inhibits the binding of histone deacetylase 1 (HDAC1) to cccDNA, promoting histone acetylation and enhancing cccDNA transcription (66). However, studies have also shown that HBc does not participate in cccDNA transcription, neither overexpression nor deletion of HBc affects HBV RNA levels (67, 68).
3.4 Immunogenicity and application potential of HBc
HBc has significant immunogenicity during HBV infection, stimulating the immune system to produce specific antibodies. HBc can induce T cell responses, and patients with high frequencies of HBc and Pol-specific T cells can control HBV well after discontinuation of nucleos(t)ide analog (NAs) therapy. Patients who successfully clear the virus typically exhibit strong HBc-specific CTL responses (69, 70). Studies have found that both thymus-dependent and thymus-independent IgM and IgG antibodies can be detected in mice, suggesting that HBc can directly activate B cells to produce antibodies without T cell involvement, although thymus-dependent mice generate more antibodies, facilitating a more effective immune response. This indicates that HBc is both a T cell-dependent and T cell-independent antigen (71). Studies have shown that HBc can specifically bind to the immunoglobulin (mIg) antigen receptor on the surface of resting mouse B cells, and B cells can present HBc to naïve Th cells in vivo and to T cell hybridomas in vitro, promoting T cell proliferation (72, 73). HBV-specific CD8+ T cells have been shown to play a crucial role in suppressing HBV replication, and C64–72 may be the immunodominant epitope of HBV core antigen, which binds with high affinity to HLA-A*0201 and induces specific CTL responses (74). The protein transduction domain (PTD) of the human immunodeficiency virus type 1 (HIV-1) Tat protein can cross the lipid bilayer of cells, either alone or as a fusion protein. The PTD- HBc fusion protein increases IFN-γ+ CD8+ T cells, enhances CTL responses, and suppresses HBV replication (75).
4 Interaction between HBV core protein and host
HBc is a key protein in the viral replication process, and the host can influence viral replication through its interaction with HBc. In hepatocytes, HBc promotes HBV replication by enhancing the binding ability of the nuclear factor κB (NF-κB) dimer p50/p65 to DNA, thereby activating the enhancer II/core promoter (EnII/Cp) (76). Peptidyl-prolyl cis-trans isomerases Par14 and Par17 interact with HBc through the 133RP134 motif, bind to HBc, and are also incorporated into core particles. Par14/Par17 enhance the stability of HBc and core particles and recruit HBc to cccDNA, promoting HBV replication (77). Members of the Hsp40/DnaJ protein family, Hdj1 and hTid1, bind to HBc and promote its degradation, thereby inhibiting capsid formation and HBV replication (78).
The proteasome pathway is the major route for intracellular protein degradation. Transient receptor potential vanilloid 4 (TRPV4) inhibits HBc degradation through a ubiquitin-dependent proteasome pathway, thereby increasing HBc stability, promoting capsid assembly, and enhancing HBV replication. Additionally, TRPV4 can increase H3K4 methylation, further promoting cccDNA-dependent transcription (79). Similarly, proteasome activator subunit 1 (PSME1) interacts with HBc to reduce its binding to the 26S proteasome, inhibiting ubiquitination and enhancing its stability. Moreover, PSME1 increases H3K27 acetylation levels, thereby promoting the transcriptional activity of cccDNA (80) (Figure 2).
In summary, HBc not only facilitates intracellular transport of the viral genome and modulates host immune responses to promote viral replication, but it is also recognized by the host. While this recognition can activate antiviral immunity to suppress the virus, it may also lead to excessive immune activation, potentially mediating immunopathological damage to liver tissue.
4.1 Intracellular Transport and Localization
During infection, HBc shuttles between the nucleolus and cytoplasm of hepatocytes. The nucleocapsid, with a diameter of 32/36 nm, is close to the size limit for transport through nuclear pores but can still enter the nucleus in its intact form (81). The nucleocapsid is directionally transported to the nuclear pore via the microtubule system (82). Four subcellular localization signals have been identified in the arginine-rich domain (ARD) of HBc. ARD-I and ARD-III synergistically function as nuclear localization signals (NLS), while ARD-II and ARD-IV independently serve as nuclear export signals (NES), both of which are critical for the bidirectional trafficking of HBc or capsids (83–85). The NLS is exposed on the capsid surface through a pore at a quasi-sixfold vertex of the icosahedral structure. importin α1 binds to the NLS, exposing the importin β-binding (IBB) sequence of importin α, which then binds to importin β to form a trimeric complex. Importin β mediates the docking of the complex at the nuclear pore complex (NPC) and facilitates its translocation through the nuclear pore to the nuclear basket. Only intact nuclear capsids can release the genome into the nucleus (86–90).
Phosphorylation of the CTD (C-terminal domain) is an essential step for transporting the viral genome into the nucleus. Studies have shown that phosphorylation at Ser155, Ser162, and Ser170 promotes CTD compaction, facilitates the externalization of CTD, and exposes the NLS on the capsid without affecting its affinity for importin α1 (89, 91).
The nuclear export of HBc and capsids depends on two different pathways. The ARD of HBc interacts with NXF1 (nuclear export factor 1), mediating the nuclear export of mRNA and proteins (85). Each tip region of the HBc particle contains two CRM1 (chromosome region maintenance protein 1)-dependent nuclear export signals (NES^CRM1), and there are 480 NES^CRM1 signals on the surface of a capsid. This high density of NES^CRM1 signals enables efficient binding of HBc particles to CRM1, facilitating nuclear export of the capsids. Additionally, CRM1 facilitates the secretion of mature nucleocapsids from the nuclear pores to the endoplasmic reticulum/Golgi apparatus for viral particle formation by coupling with the microtubule system (92, 93).
HBc monomers depend on the NXF1 pathway to be transported from the nucleus to the cytoplasm, participating in pgRNA packaging. Nucleocapsids rely on the CRM1 pathway to be exported from the nucleus to the cytoplasm for reverse transcription and viral assembly.
4.2 Regulation of host immune responses
The innate immune response serves as the first line of defense against invading pathogens. Pattern recognition receptors (PRRs) detect pathogens and induce the production of antiviral factors to control HBV infection. Moreover, the innate immune response is crucial for activating the adaptive immune response, as it mediates the recruitment of adaptive immune cells. T cells and B cells recognize and eliminate HBV-infected hepatocytes. However, persistent exposure to viral antigens can lead to immune exhaustion, which eventually establishes chronic infection by dampening host responses (94–96).
The capsid, via its arginine-rich domain, binds to heparan sulfate on macrophage surfaces and induces the production of cytokines such as TNF-α, IL-6, and IL-12p40. This process involves activation of NF-κB, ERK-1/2, and p38 MAPK signaling pathways, thereby enhancing immune responses and activating adaptive immunity (97). HBV infection can trigger the production of interferons (IFNs) and induce interferon-stimulated genes (ISGs) to exert antiviral effects (98). IFN-β can induce the expression of galectin-9 (GAL9), which, mediated by another ISG-encoded protein, viperin, interacts with HBc and promotes HBc accumulation in the cytoplasm. Interestingly, the autophagosome marker LC3 colocalizes with HBc. Further studies revealed that GAL9 enhances the auto-ubiquitination of E3 ubiquitin ligase RNF13, promoting p62 recruitment and interaction with LC3 to form autophagosomes, leading to selective autophagy of HBc (99) (Figure 3).
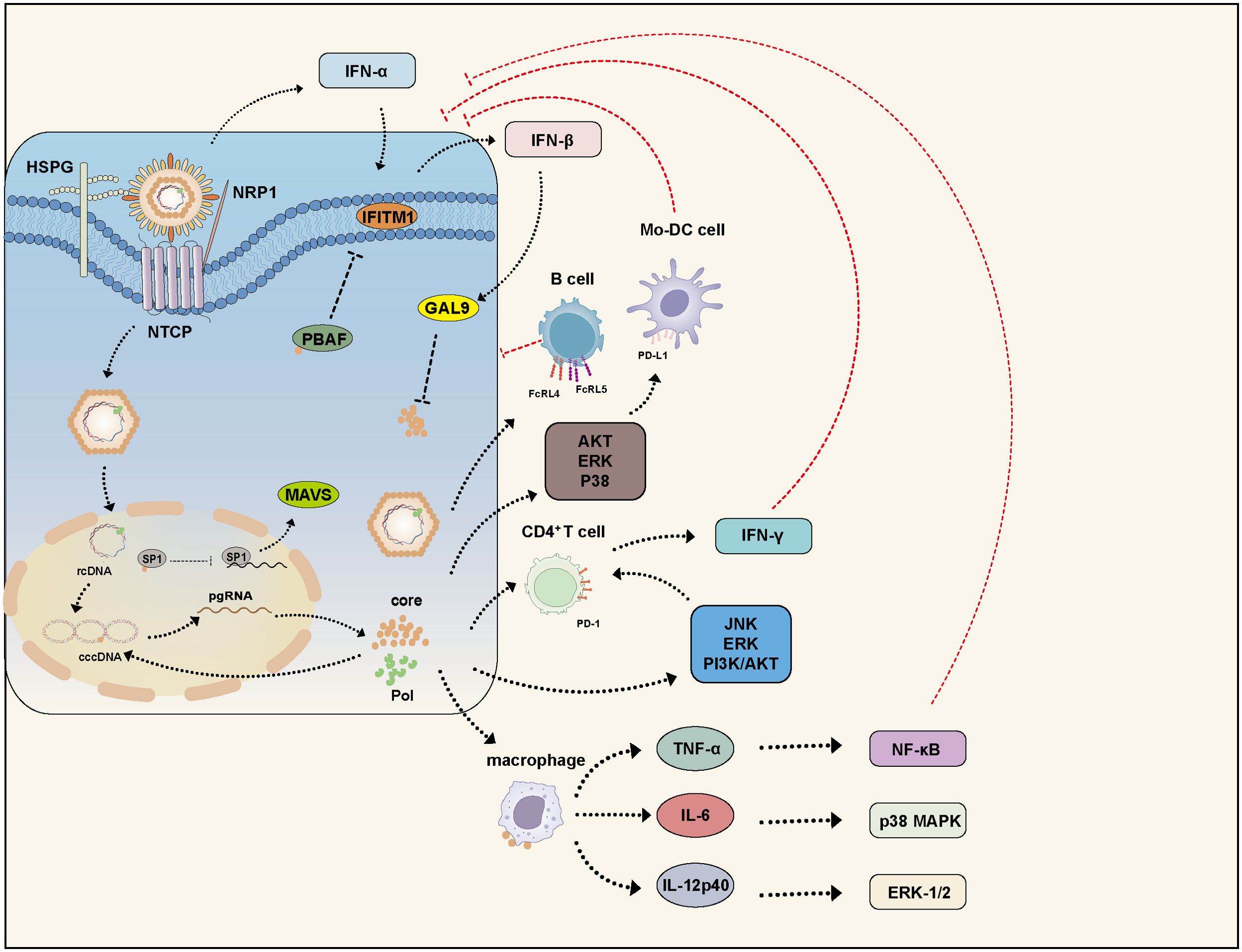
Figure 3. HBc modulates the host immune response. HBV infection activates multiple IFN signaling pathways. IFN-α induces the expression of IFITM1, while IFN-β upregulates GAL9 to inhibit HBV replication. However, HBc counteracts these defenses by disrupting the stability of PBAF, thereby suppressing IFITM1 expression. Additionally, HBc interacts with the transcription factor SP1 to suppress MAVS expression, leading to inhibition of the IFN-β signaling pathway. Furthermore, HBc participates in various signaling pathways and impairs the functions of B cells, Mo-DC, CD4+ T cells, and macrophages, thereby facilitating HBV persistence. GAL9, Galectin-9; PBAF, polybromo-associated BAF; IFITM, interferon-induced transmembrane protein; MAV, mitochondrial antiviral-signaling protein.
HBc also suppresses MAVS (mitochondrial antiviral-signaling protein) expression by interacting with the transcription factor SP1 and inhibiting its binding to the MAVS promoter. MAVS activation can suppress HBV replication via IFN-β induction, and combined use of MAVS and IFN-α has been shown to enhance antiviral effects both in vitro and in vivo. Our group has identified several ISGs that inhibit HBV by targeting the HBx protein, However, IFN-α treatment alone is not fully effective against HBV infection, as IFN-α also induces the RNA-editing enzyme ADAR1, which targets the 3’UTR of MAVS mRNA to suppress its expression (100–108). Interestingly, research found that IFN-induced IFIT3 significantly promotes HBV replication, suggesting that IFN may also activate certain ISGs that enhance viral replication (109). Early studies found that during acute HBV infection in chimpanzees, there was no induction of genes associated with innate immune responses. HBV remains undetected and spreads before the adaptive immune response initiates, effectively evading recognition by the innate immune system during early infection (110, 111).
The BRG1-associated factor (BAF) and polybromo-associated BAF (PBAF) are mammalian SWI/SNF chromatin remodeling complexes. They regulate the expression of various interferon-inducible genes via ATP-dependent chromatin remodeling. BAF200 is a specific subunit of PBAF. HBc disrupts PBAF stability by interacting with BAF200, thereby suppressing the expression of interferon-induced transmembrane proteins (IFITMs) and partially restoring HBV replication despite IFN-α-induced immune responses (112) (Figure 3).
Phosphorylation of serine residues within the arginine-rich domain of HBc inhibits the binding of capsids to macrophages and the secretion of cytokines. HBc remains dephosphorylated during asymptomatic infection but is phosphorylated during viral replication in hepatocytes, indicating that HBc can escape immune responses via phosphorylation modifications (97, 113). In HBV-infected patients (especially those with acute hepatitis), HBc can promote the proliferation of CD4+ T cells and stimulate the production of IFN-γ and lymphotoxin, thereby regulating immune responses and facilitating HBV clearance (114). Deletion of HBc leads to high levels of HBsAg in mice, with 93.3% of mice remaining HBsAg positive after 12 weeks. A C-terminal deletion of 10 amino acids in HBc abolishes effective HBc-specific IFN-γ responses, promoting HBV persistence (115).
Programmed death receptor 1 (PD-1) overexpression is a hallmark of exhausted T cells. HBc induces PD-1 expression on CD4+ T cells via JNK, ERK, and PI3K/AKT signaling pathways. PD-1 expression correlates with HBV DNA levels during the immune clearance phase, influencing viral clearance and contributing to HBV persistence (116). Programmed death ligand 1 (PD-L1, also known as B7-H1) binds to PD-1, transmitting co-inhibitory signals to T cells and regulating their activation and tolerance. HBc upregulates B7-H1 expression in monocyte-derived dendritic cells (Mo-DCs) through activation of AKT, ERK, and p38 pathways, leading to Mo-DC apoptosis and impaired HBV DNA clearance (117). Fc receptor-like proteins 4 (FcRL4) and 5 (FcRL5) are inhibitory receptors on B cells that regulate immune responses by suppressing B cell activation and proliferation. HBc binds specifically to B cells, inducing high expression of FcRL4 and FcRL5, thereby impairing B cell function (118) (Figure 3).
Overall, HBc modulates both innate and adaptive immune responses, enabling immune evasion and contributing to viral persistence.
4.3 HBc and host cell cytokines
The immune system acts as a double-edged sword: while it clears infected cells, it also triggers inflammation and necrosis in liver tissue, leading to the destruction of infected hepatocytes. Prolonged chronic inflammation can result in liver fibrosis, cirrhosis, and even hepatocellular carcinoma. Many cytokines are associated with adverse outcomes in hepatitis B. HBc, which is highly immunogenic, can induce the production of various cytokines, contributing to disease progression in hepatitis B.
HBc promotes the expression of NLRP3 (NOD-like receptor family pyrin domain-containing 3), a component of the NLRP3 inflammasome, and activates caspase-1, leading to the secretion of IL-1β and IL-18 through the NLRP3 inflammasome pathway, thereby triggering liver inflammation (119). HBc also enhances the expression and secretion of IL-6 by activating the ERK, p38 MAPK, and NF-κB signaling pathways (120).
In the presence of IL-6, transforming growth factor-β (TGF-β) promotes the differentiation of naïve T lymphocytes into Th17 cells, thereby fostering autoimmunity and inflammation (121–124). Cytokines secreted by Th17 cells, particularly IL-17, are associated with the progression of liver fibrosis and cirrhosis. In patients with hepatitis B, IL-17 is mainly found in fibrotic areas, and its levels closely correlate with the severity of fibrosis (125).
IL-17 activates hepatic stellate cells (HSCs) by interacting with IL-17 receptors (IL-17R) on their surface. Additionally, IL-17 can activate HSCs via the p38 and ERK1/2 signaling pathways. It also stabilizes TGF-β receptor II in a JNK-dependent manner, thereby activating the Smad2/3 pathway to further stimulate HSC activation and enhance liver fibrosis (126, 127).
5 HBc clinical relevance
5.1 Clinical applications of anti-HBc
Anti-HBc is a classical serological marker. Once infected with HBV, Anti-HBc usually persists for a long time, often for life. Therefore, Anti-HBc is the most useful marker for determining past exposure to or infection with HBV (128). Anti-HBc IgM is an early marker of HBV infection, typically appearing 6 to 8 weeks after infection and peaking during the acute phase. However, it gradually disappears within 6 months after infection. Anti-HBc IgM levels can also rise during acute exacerbations of chronic hepatitis, though the titers are lower compared to those during acute hepatitis, allowing differentiation between acute and chronic infections (129).
Anti-HBc IgG follows Anti-HBc IgM, usually appearing 4 to 8 months after acute infection and persisting for a long time. A positive Anti-HBc IgG result indicates past HBV infection, which could either be a resolved past infection or a chronic infection (130, 131). If both HBsAg and Anti-HBs are negative while Anti-HBc is positive, several scenarios must be considered, such as false positivity, the window period of acute infection, a recovered past infection with declining antibody levels, occult infection, co-infection with other hepatitis viruses, or mutations in HBsAg (132, 133). Therefore, in clinical diagnostics, Anti-HBc testing is typically combined with other markers (such as HBsAg, HBeAg, and HBV DNA) to improve diagnostic accuracy and predictive power. Anti-HBc IgG is a surrogate indicator of HBV-specific activation of adaptive immune response (134). Anti-HBc IgG includes four subtypes: IgG1, IgG2, IgG3, and IgG4. Studies have reported that in chronic carriers, IgG1 > IgG3, whereas in recovered individuals, IgG3 > IgG1 (135, 136). HBeAg and HBc are translated from different start codons within the C ORF and share an identical 149-amino-acid core domain (137). HBeAg is a serum marker, which is closely related to the number of infectious hepatitis B virus particles in the serum (138).HBeAg suppresses T-helper (Th) cell function, blocks the IgM-to-IgG class transition, and significantly reduces the production of anti-HBc IgG. It also induces T-cell tolerance to diminish the immune response against intracellular HBc, thereby promoting persistent viral infection (139).
The hepatitis B vaccine, based on recombinant DNA technology to express HBsAg, protects against HBV infection by stimulating humoral immune responses; thus, vaccinated individuals develop only anti-HBs antibodies (140). For patients who are solely Anti-HBc positive, vaccination can still elicit a good immune response. Moreover, their response to vaccination may provide additional diagnostic information: patients who respond to the vaccine may have had a false-positive Anti-HBc, while non-responders may harbor occult HBV infection (133, 141).
Quantitative Anti-HBc (qAnti-HBc) levels vary significantly across different stages of HBV infection and are determined by the host’s immune status. qAnti-HBc can more accurately reflect hepatic inflammation and is associated with the degree of liver fibrosis, exacerbation during chronic infection, and the presence of occult infection. The qAnti-HBc level can serve as a predictive marker for spontaneous or treatment-induced HBeAg and HBsAg seroclearance, relapse after treatment cessation, reinfection post-liver transplantation, and viral reactivation during immunosuppression (142, 143).
Anti-HBc and qAnti-HBc each have distinct clinical focuses: Anti-HBc is mainly used for preliminary screening and long-term monitoring of HBV infection, whereas qAnti-HBc offers significant advantages in assessing hepatic inflammation, predicting treatment responses, and estimating relapse risk.
5.2 HBc variants and drug resistance
Nucleoside analogs (NAs) inhibit viral replication by suppressing the function of the Pol, but the rate of sustained virological response is very low, requiring long-term treatment. Long-term NA therapy increases the risk of antiviral drug resistance. Resistance mutations primarily occur in the Pol, but studies have shown that HBc mutations can compensate for the replication defects of drug-resistant HBV, enhancing the virus’s survival ability under drug pressure.
HBc mutations (P5T, S35T, P79Q, E83D, S87G, I97L, and Q177K) were found in patients with lamivudine (LMV) resistance, promoting the packaging of pgRNA and thereby enhancing HBV DNA replication, with the P5T mutation playing a significant role (144). In patients undergoing NA therapy, an increased frequency of the P130L mutation has been observed. Researchers speculate that the P130L mutation may alter the secondary structure of HBc, reducing its antigenicity and preventing immune recognition, leading to immune escape. However, this hypothesis requires further experimental validation (145) (Table 1).
In summary, nucleotide analog resistance mutations do not occur solely in the Pol; compensatory mutations in HBc can also promote HBV replication.
5.3 HBc as a therapeutic target
HBc plays an indispensable role in multiple steps of the HBV life cycle. Therefore, several drugs aim to target HBc to disrupt various stages of the replication cycle. Capsid assembly modulators (CAMs) are antiviral agents that interfere with the dynamics and morphology of the capsid (Figure 4). Based on their functions, CAMs are classified into two types:
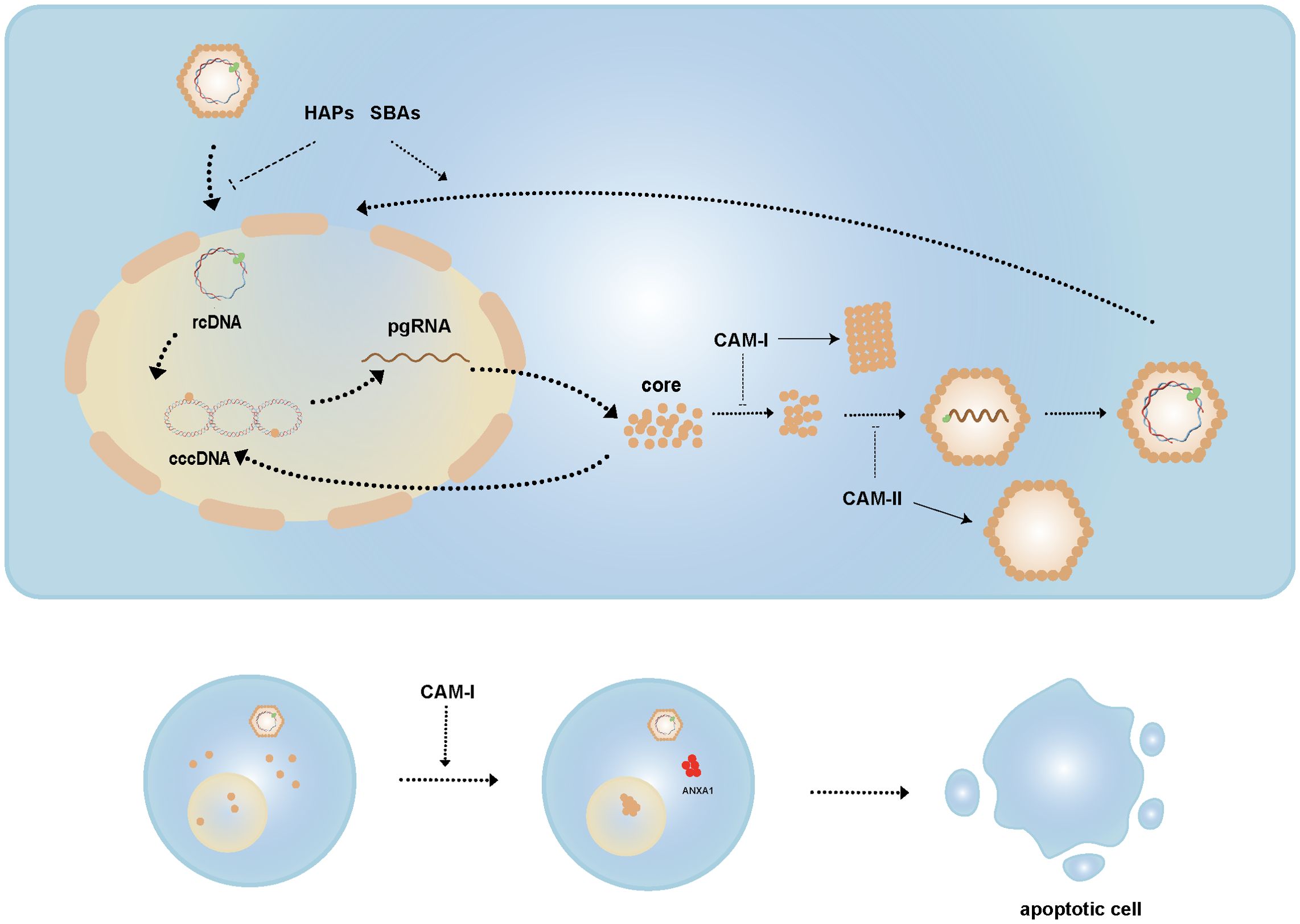
Figure 4. Mechanisms of CAMs. CAM-I acts by altering capsid conformation, whereas CAM-II interferes with pgRNA packaging to generate empty capsids. HAPs and SBAs affect nucleocapsid disassembly, inhibiting this process during infection but inducing it in mature nucleocapsids. CAM-I induces nuclear aggregation of HBc and promotes ANXA1 expression, ultimately leading to apoptosis of HBV-infected cells.
● CAM-A, such as heteroaryl pyrimidines (HAPs), induce the misassembly of HBc dimers, leading to aberrant capsid structures and HBc aggregation.
● CAM-E, such as sulfonylbenzamide (SBAs), phenylpropenamide (PPA), pyrazolyl thiazole (PT), ethoxamide-pyrrolamide (GPA), and dibenzothiazepine-2-one (DPT), stabilize dimer-dimer interactions to form intact capsids that lack the viral genome (146, 147).
CAMs disrupt HBc assembly by binding to a hydrophobic pocket at the dimer interface, enhancing dimer-dimer interactions. CAM-A induce HBc phosphorylation, altering the capsid’s conformation and affecting its stability, whereas CAM-E interfere with the packaging of pgRNA, resulting in the formation of empty capsids (35, 148).
CAMs not only affect capsid assembly but also play dual roles during HBV replication. Studies have shown that HAPs and SBAs not only impair capsid assembly but also induce the disassembly of viral particles and cytoplasmic mature nucleocapsids, thereby regulating the formation of cccDNA. Specifically, by triggering the disassembly of nucleocapsids in viral particles, CAMs prevent rcDNA from entering the nucleus and inhibit cccDNA synthesis during new infections. Conversely, by inducing the disassembly of mature cytoplasmic nucleocapsids, they accelerate rcDNA nuclear import and conversion to cccDNA (149).
Additionally, research has found that high concentrations of HAP12, in collaboration with importin β, tend to promote the disassembly of HBV empty capsids (150). Some studies suggest that CAMs induce the aggregation of HBc within the nucleus, promoting the expression of apoptosis-related gene ANXA1 and activating the interferon signaling pathway, thereby helping to clear HBV-infected cells. However, CAM-induced cell death was not observed in HBV-infected primary human hepatocytes (PHHs), likely because HBc aggregates form a complex with the STIP1 homology and U box-containing protein 1 (STUB1) and heat shock protein 70 (Hsp70), recruit Bcl-2 associated athanogene 3 (BAG3) to transport them to the perinuclear region, and are subsequently degraded via the ubiquitin-binding protein p62-mediated autophagosome formation (147, 151–153).
Due to the inability of existing drugs to eliminate cccDNA and the emergence of drug resistance, CAMs, by intervening at multiple steps of the viral life cycle, may offer a more durable antiviral effect and reduce the risk of resistance, making them a focus of current drug development efforts. In recent years, several CAMs have entered clinical development stages.
RO7049389, a HAP compound, induces aberrant capsid assembly leading to degradation of the viral core protein, thereby inhibiting pgRNA packaging and HBV DNA replication (154). It is currently in Phase 2 clinical trials. Results from Phase 1 trials showed that after 4 weeks of treatment in patients with chronic hepatitis B (CHB), RO7049389 led to a reduction in HBV DNA with infrequent and mild adverse events, comparable to those in the NAs control group (155). Additionally, given the high prevalence of HBV in Asia, the safety of RO7049389 was validated in healthy Chinese volunteers during Phase 1 trials (156).
Studies have shown that ABI-H2158 promotes abnormal capsid assembly, blocks pgRNA encapsidation, inhibits HBV DNA replication, and prevents cccDNA formation by inducing premature disassembly of capsids during infection. Although ABI-H2158 had entered Phase 2 clinical trials, development was halted due to hepatotoxicity (157).
JNJ-6379, similar in mechanism to ABI-H2158, is a CAM-E evaluated for safety, pharmacokinetics, and antiviral activity in Phase 1 clinical studies. Data indicated that JNJ-6379 was well tolerated with mostly mild to moderate adverse events. HBV DNA and RNA levels decreased in a dose-dependent manner, reaching levels below the lower limit of quantification at the highest doses. However, there was no significant effect on HBsAg and HBeAg levels, and viral rebound was observed after treatment cessation (158).
Phase 2 studies found that virologic breakthrough could occur when JNJ-6379 was used as monotherapy, but this was not observed when combined with NAs. The combination significantly enhanced the reduction of HBV DNA and HBV RNA levels, though the impact on HBsAg and HBeAg remained limited (159).
JNJ-3989, a small interfering RNA (siRNA), reduces levels of all HBV proteins and pregenomic RNA by targeting and cleaving HBV RNA transcripts. In Phase 2 studies, the combination of JNJ-6379, JNJ-3989, and NAs not only maintained their respective functions but also significantly reduced HBsAg levels and minimized HBV DNA rebound after treatment cessation (160).
6 Future research directions and challenges
The ultimate goal in treating hepatitis B virus (HBV) infection is the complete eradication of the virus, ideally through the elimination of covalently closed circular DNA (cccDNA). However, current therapies struggle to effectively clear cccDNA. At present, the most achievable treatment goal is considered to be a functional cure, characterized by the sustained absence of detectable HBsAg and HBV DNA in serum (with or without seroconversion of HBsAg), improvement in liver inflammation and fibrosis, and long-term maintenance of these outcomes after cessation of therapy.
Clinical studies have demonstrated that CAMs can significantly reduce HBV DNA levels, but they exert limited effects on the established cccDNA pool. CAMs function by binding to the hydrophobic pocket at the HBc dimer-dimer interface; thus, mutations within HBc could potentially impact CAM efficacy. JNJ-6379 has shown good antiviral activity against clinical isolates from HBV genotypes A–H, with the exception of one genotype D strain, which exhibited substitution at position 33 of HBc—a site located within the CAM binding pocket (161).
Residue T109, located within the CAM binding pocket, is one of the most frequently mutated sites in HBc. Mutations T109I and T109M promote normal capsid assembly, rendering them relatively resistant to CAMs, while the T109S mutation impairs capsid assembly and increases sensitivity to CAMs (162, 163). Mutations at residues D29G, T33N/S, and Y118F within the CAM binding pocket significantly reduce the antiviral activity of BAY41-4109, whereas mutations at S106T, T128I, and L140I enhance its antiviral efficacy (Table 1). These mutations alter the characteristics of the binding pocket, thereby affecting drug binding and antiviral potency. Similarly, other studies have confirmed that numerous amino acid mutations within the CAM binding pocket impact CAM activity (161, 164).
The hydrophobic nature of the amino acids within the CAM binding pocket is crucial for maintaining drug functionality. Interestingly, research has identified a second binding pocket on the HBV capsid, located at the spike apex. This pocket, located at the tip of the capsid spikes and targeted by the peptide dimers, can induce HBc aggregation, thereby interfering with capsid formation and viral assembly (165). This secondary pocket also represents a potential target for future drug development.
7 Conclusion
HBc is an indispensable and multifunctional protein within the HBV life cycle, playing critical roles in viral replication, nucleocapsid formation, and persistent infection through complex interactions with host cellular pathways. These interactions not only facilitate viral survival but also contribute to immune evasion and hepatocarcinogenesis, positioning HBc as a pivotal therapeutic target.
Despite these advancements, several research challenges remain. One major obstacle is the limited impact of current CAMs on pre-existing intrahepatic cccDNA pools, which are central to HBV persistence. Additionally, HBc mutations within the CAM-binding hydrophobic pocket can significantly alter drug susceptibility, leading to variable therapeutic responses and potential resistance. The heterogeneity of HBV genotypes further complicates drug development, as genotype-specific mutations (e.g., at positions T109 and T33) may influence HBc structure and CAM efficacy.
Future research should focus on three main directions: (1) the development of next-generation CAMs with broader genotype coverage and improved potency against mutant HBc variants; (2) deeper mechanistic insights into HBc’s role in cccDNA maintenance and host epigenetic modulation, aiming to discover novel therapeutic leverage points; and (3) the integration of CAMs into rational combination regimens that include immune-based therapies, with the goal of achieving a complete cure, defined as the elimination or permanent silencing of cccDNA and integrated HBV DNA.
Ultimately, while functional cure remains a realistic near-term objective, the complete eradication of HBV will require innovative strategies to overcome the resilience of cccDNA, understand host-virus dynamics at a deeper level, and preempt or overcome resistance pathways. HBc-targeted research remains central to this endeavor.
Author contributions
GT: Writing – original draft, Writing – review & editing, Validation, Funding acquisition. YZ: Writing – original draft, Writing – review & editing, Validation. HS: Validation, Writing – review & editing. FX: Writing – review & editing, Validation. MH: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (grant no. 82371765 to GT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jeng WJ, Papatheodoridis GV, and Lok ASF. Hepatitis B. Lancet. (2023) 401:1039–52. doi: 10.1016/S0140-6736(22)01468-4
2. Liang TJ. Hepatitis B: the virus and disease. Hepatology. (2009) 49:S13–21. doi: 10.1002/hep.22881
3. Chen Y and Tian Z. HBV-induced immune imbalance in the development of HCC. Front Immunol. (2019) 10:2048. doi: 10.3389/fimmu.2019.02048
4. Chuang YC, Tsai KN, and Ou JJ. Pathogenicity and virulence of Hepatitis B virus. Virulence. (2022) 13:258–96. doi: 10.1080/21505594.2022.2028483
5. Cao J, Zhang J, Lu Y, Luo S, Zhang J, Zhu P, et al. Cryo-EM structure of native spherical subviral particles isolated from HBV carriers. Virus Res. (2019) 259:90–6. doi: 10.1016/j.virusres.2018.10.015
6. Lei B, Song H, Xu F, Wei Q, Wang F, Tan G, et al. When does hepatitis B virus meet long-stranded noncoding RNAs? Front Microbiol. (2022) 13:962186. doi: 10.3389/fmicb.2022.962186
7. Schäfer G, Blumenthal MJ, and Katz AA. Interaction of human tumor viruses with host cell surface receptors and cell entry. Viruses. (2015) 7:2592–617. doi: 10.3390/v7052592
8. Watashi K. HBV slow maturation process leads to infection. Trends Microbiol. (2016) 24:597–9. doi: 10.1016/j.tim.2016.06.007
9. Asami J, Terakado Kimura K, Fujita-Fujiharu Y, Ishida H, Zhang Z, Nomura Y, et al. Structure of the bile acid transporter and HBV receptor NTCP. Nature. (2022) 606:1021–6. doi: 10.1038/s41586-022-04845-4
10. Fukano K, Tsukuda S, Oshima M, Suzuki R, Aizaki H, Ohki M, et al. Troglitazone impedes the oligomerization of sodium taurocholate cotransporting polypeptide and entry of hepatitis B virus into hepatocytes. Front Microbiol. (2018) 9:3257. doi: 10.3389/fmicb.2018.03257
11. Yu H, Ren J, Deng H, Li L, Zhang Z, Cheng S, et al. Neuropilin-1 is a novel host factor modulating the entry of hepatitis B virus. J Hepatol. (2025) 82:37–50. doi: 10.1016/j.jhep.2024.06.032
12. Wei L and Ploss A. Mechanism of hepatitis B virus cccDNA formation. Viruses. (2021) 13(8):1463. doi: 10.3390/v13081463
13. Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. (2015) 64:1972–84. doi: 10.1136/gutjnl-2015-309809
14. Iannacone M and Guidotti LG. Immunobiology and pathogenesis of hepatitis B virus infection. Nat Rev Immunol. (2022) 22:19–32. doi: 10.1038/s41577-021-00549-4
15. Selzer L and Zlotnick A. Assembly and release of hepatitis B virus. Cold Spring Harb Perspect Med. (2015) 5(12):a021394. doi: 10.1101/cshperspect.a021394
16. Beck J and Nassal M. Hepatitis B virus replication. World J Gastroenterol. (2007) 13:48–64. doi: 10.3748/wjg.v13.i1.48
17. Wu Q, Glitscher M, Tonnemacher S, Schollmeier A, Raupach J, Zahn T, et al. Presence of intact hepatitis B virions in exosomes. Cell Mol Gastroenterol Hepatol. (2023) 15:237–59. doi: 10.1016/j.jcmgh.2022.09.012
18. Liu K, Luckenbaugh L, Ning X, Ji X, and Hu J. Multiple roles of core protein linker in hepatitis B virus replication. PloS Pathog. (2018) 14:e1007085. doi: 10.1371/journal.ppat.1007085
19. Wynne SA, Crowther RA, and Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. (1999) 3:771–80. doi: 10.1016/S1097-2765(01)80009-5
20. Packianathan C, Katen SP, Dann C, and Zlotnick A. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. . J Virol. (2010) 84:1607–15. doi: 10.1128/JVI.02033-09
21. Liu C, Fan G, Wang Z, Chen H-S, and Yin C-C. Allosteric conformational changes of human HBV core protein transform its assembly. Sci Rep. (2017) 7:1404. doi: 10.1038/s41598-017-01568-9
22. Alexander CG, Jürgens MC, Shepherd DA, Freund SMV, Ashcroft AE, Ferguson N, et al. Thermodynamic origins of protein folding, allostery, and capsid formation in the human hepatitis B virus core protein. Proc Natl Acad Sci U.S.A. (2013) 110:E2782–91. doi: 10.1073/pnas.1308846110
23. Yu X, Jin L, Jih J, Shih C, and Zhou ZH. 3.5Å cryoEM structure of hepatitis B virus core assembled from full-length core protein. PloS One. (2013) 8:e69729. doi: 10.1371/journal.pone.0069729
24. Chotiyaputta W and Lok AS. Hepatitis B virus variants. Nat Rev Gastroenterol Hepatol. (2009) 6:453–62. doi: 10.1038/nrgastro.2009.107
25. Zhu Y, Jin Y, Cai X, Bai X, Chen M, Chen T, et al. Hepatitis B virus core protein variations differ in tumor and adjacent nontumor tissues from patients with hepatocellular carcinoma. Intervirology. (2012) 55:29–35. doi: 10.1159/000322832
26. Kefalakes H, Budeus B, Walker A, Jochum C, Hilgard G, Heinold A, et al. Adaptation of the hepatitis B virus core protein to CD8(+) T-cell selection pressure. Hepatology. (2015) 62:47–56. doi: 10.1002/hep.27771
27. Le Pogam S, Yuan TTT, Sahu GK, Chatterjee S, and Shih C. Low-level secretion of human hepatitis B virus virions caused by two independent, naturally occurring mutations (P5T and L60V) in the capsid protein. J Virol. (2000) 74:9099–105. doi: 10.1128/JVI.74.19.9099-9105.2000
28. Zhang Y, Ren Y, Wu Y, Zhao B, Qiu L, Li X, et al. The L60V variation in hepatitis B virus core protein elicits new epitope-specific cytotoxic T lymphocytes and enhances viral replication. J Virol. (2013) 87:8075–84. doi: 10.1128/JVI.00577-13
29. Roseman AM, Berriman JA, Wynne SA, Butler PJG, and Crowther RA. A structural model for maturation of the hepatitis B virus core. Proc Natl Acad Sci U.S.A. (2005) 102:15821–6. doi: 10.1073/pnas.0504874102
30. Böttcher B and Nassal M. Structure of mutant hepatitis B core protein capsids with premature secretion phenotype. J Mol Biol. (2018) 430:4941–54. doi: 10.1016/j.jmb.2018.10.018
31. Shih C, Wu SY, Chou SF, and Yuan TT. Virion secretion of hepatitis B virus naturally occurring core antigen variants. Cells. (2020) 10(1):43. doi: 10.3390/cells10010043
32. Ceres P, Stray SJ, and Zlotnick A. Hepatitis B virus capsid assembly is enhanced by naturally occurring mutation F97L. J Virol. (2004) 78:9538–43. doi: 10.1128/JVI.78.17.9538-9543.2004
33. Yuan TT and Shih C. A frequent, naturally occurring mutation (P130T) of human hepatitis B virus core antigen is compensatory for immature secretion phenotype of another frequent variant (I97L). J Virol. (2000) 74:4929–32. doi: 10.1128/jvi.74.10.4929-4932.2000
34. de Rocquigny H, Rat V, Pastor F, Darlix JL, Hourioux C, Roingeard P, et al. Phosphorylation of the arginine-rich C-terminal domains of the hepatitis B virus (HBV) core protein as a fine regulator of the interaction between HBc and nucleic acid. Viruses. (2020) 12(7):738. doi: 10.3390/v12070738
35. Viswanathan U, Mani N, Hu Z, Ban H, Du Y, Hu J, et al. Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antiviral Res. (2020) 182:104917. doi: 10.1016/j.antiviral.2020.104917
36. Zheng C-L, Fu Y-M, Xu Z-X, Zou Y, and Deng K. Hepatitis B virus core protein dimer−dimer interface is critical for viral replication. Mol Med Rep. (2019) 19:262–70. doi: 10.3892/mmr.2018.9620
37. Birnbaum F and Nassal M. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J Virol. (1990) 64:3319–30. doi: 10.1128/jvi.64.7.3319-3330.1990
38. Ludgate L, Liu K, Luckenbaugh L, Streck N, Eng S, Voitenleitner C, et al. Cell-free hepatitis B virus capsid assembly dependent on the core protein C-terminal domain and regulated by phosphorylation. J Virol. (2016) 90:5830–44. doi: 10.1128/JVI.00394-16
39. Watts NR, Conway JF, Cheng N, Stahl SJ, Belnap DM, Steven AC, et al. The morphogenic linker peptide of HBV capsid protein forms a mobile array on the interior surface. EMBO J. (2002) 21:876–84. doi: 10.1093/emboj/21.5.876
40. Shim HY, Quan X, Yi YS, and Jung G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology. (2011) 410:161–9. doi: 10.1016/j.virol.2010.11.005
41. Nassal M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol. (1992) 66:4107–16. doi: 10.1128/jvi.66.7.4107-4116.1992
42. Jung J, Hwang SG, Chwae Y-J, Park S, Shin H-J, Kim K, et al. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J Virol. (2014) 88:8754–67. doi: 10.1128/JVI.01343-14
43. Chen C, Wang JC, and Zlotnick A. A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PloS Pathog. (2011) 7:e1002388. doi: 10.1371/journal.ppat.1002388
44. Yip RPH, Kwok DCY, Lai LTF, Ho S-M, Wong ICK, Chan C-P, et al. SRPK2 mediates HBV core protein phosphorylation and capsid assembly via docking interaction. PloS Pathog. (2024) 20:e1011978. doi: 10.1371/journal.ppat.1011978
45. Porterfield JZ, Dhason MS, Loeb DD, Nassal M, Stray SJ, Zlotnick A, et al. Full-length hepatitis B virus core protein packages viral and heterologous RNA with similarly high levels of cooperativity. J Virol. (2010) 84:7174–84. doi: 10.1128/JVI.00586-10
46. Ning X, Basagoudanavar SH, Liu K, Luckenbaugh L, Wei D, Wang C, et al. Capsid phosphorylation state and hepadnavirus virion secretion. J Virol. (2017) 91(9):e00092–17. doi: 10.1128/JVI.00092-17
47. Gazina EV, Fielding JE, Lin B, and Anderson DA. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol. (2000) 74:4721–8. doi: 10.1128/jvi.74.10.4721-4728.2000
48. Zhao Q, Hu Z, Cheng J, Wu S, Luo Y, Chang J, et al. Hepatitis B virus core protein dephosphorylation occurs during pregenomic RNA encapsidation. J Virol. (2018) 92(13):e02139–17. doi: 10.1128/JVI.02139-17
49. Hu Z, Ban H, Zheng H, Liu M, Chang J, Guo J-T, et al. Protein phosphatase 1 catalyzes HBV core protein dephosphorylation and is co-packaged with viral pregenomic RNA into nucleocapsids. PloS Pathog. (2020) 16:e1008669. doi: 10.1371/journal.ppat.1008669
50. Hsieh CL, Chang LY, Chen PJ, and Yeh SH. HBV polymerase recruits the phosphatase PP1 to dephosphorylate HBc-Ser170 to complete encapsidation. PloS Pathog. (2025) 21:e1012905. doi: 10.1371/journal.ppat.1012905
51. Lewellyn EB and Loeb DD. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J Virol. (2011) 85:1298–309. doi: 10.1128/JVI.01957-10
52. Perlman DH, Berg EA, O'Connor PB, Costello CE, and Hu J. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U.S.A. (2005) 102:9020–5. doi: 10.1073/pnas.0502138102
53. Basagoudanavar SH, Perlman DH, and Hu J. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J Virol. (2007) 81:1641–9. doi: 10.1128/JVI.01671-06
54. Niklasch M, Zimmermann P, and Nassal M. The hepatitis B virus nucleocapsid-dynamic compartment for infectious virus production and new antiviral target. Biomedicines. (2021) 9(11):1577. doi: 10.3390/biomedicines9111577
55. Lewellyn EB and Loeb DD. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PloS One. (2011) 6:e17202. doi: 10.1371/journal.pone.0017202
56. Mendenhall MA, Hong X, and Hu J. Hepatitis B virus capsid: the core in productive entry and covalently closed circular DNA formation. Viruses. (2023) 15(3):642. doi: 10.3390/v15030642
57. Xi J, Luckenbaugh L, and Hu J. Multiple roles of PP2A binding motif in hepatitis B virus core linker and PP2A in regulating core phosphorylation state and viral replication. PloS Pathog. (2021) 17:e1009230. doi: 10.1371/journal.ppat.1009230
58. Gao W and Hu J. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol. (2007) 81:6164–74. doi: 10.1128/JVI.02721-06
59. Summers J, Smith PM, and Horwich AL. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol. (1990) 64:2819–24. doi: 10.1128/jvi.64.6.2819-2824.1990
60. Cui X, Luckenbaugh L, Bruss V, and Hu J. Alteration of mature nucleocapsid and enhancement of covalently closed circular DNA formation by hepatitis B virus core mutants defective in complete-virion formation. J Virol. (2015) 89:10064–72. doi: 10.1128/JVI.01481-15
61. Hong X, Menne S, and Hu J. Constrained evolution of overlapping genes in viral host adaptation: Acquisition of glycosylation motifs in hepadnaviral precore/core genes. PloS Pathog. (2022) 18:e1010739. doi: 10.1371/journal.ppat.1010739
62. Luo J, Xi J, Gao L, and Hu J. Role of Hepatitis B virus capsid phosphorylation in nucleocapsid disassembly and covalently closed circular DNA formation. PloS Pathog. (2020) 16:e1008459. doi: 10.1371/journal.ppat.1008459
63. Liu H, Xi J, and Hu J. Regulation of hepatitis B virus replication by cyclin docking motifs in core protein. J Virol. (2021) 95(12):e00230–21. doi: 10.1128/JVI.00230-21
64. Hong X and Hu J. Conserved lysine residues of hepatitis B virus core protein are not required for covalently closed circular DNA formation. J Virol. (2022) 96:e0071822. doi: 10.1128/jvi.00718-22
65. Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, et al. Structural organization of the hepatitis B virus minichromosome. J Mol Biol. (2001) 307:183–96. doi: 10.1006/jmbi.2000.4481
66. Guo Y-H, Li Y-N, Zhao J-R, Zhang J, and Yan Z. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics. (2011) 6:720–6. doi: 10.4161/epi.6.6.15815
67. Zhong Y, Wu C, Xu Z, Teng Y, Zhao L, Zhao K, et al. Hepatitis B virus core protein is not required for covalently closed circular DNA transcriptional regulation. J Virol. (2022) 96:e0136222. doi: 10.1128/jvi.01362-22
68. Tu T, Zehnder B, Qu B, and Urban S. D e novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. (2021) 3:100195. doi: 10.1016/j.jhepr.2020.100195
69. Hoofnagle JH, Gerety RJ, and Barker LF. Antibody to hepatitis-B-virus core in man. Lancet. (1973) 2:869–73. doi: 10.1016/S0140-6736(73)92004-7
70. Rivino L, Le Bert N, Gill US, Kunasegaran K, Cheng Y, Tan DZ, et al. Hepatitis B virus-specific T cells associate with viral control upon nucleos(t)ide-analogue therapy discontinuation. J Clin Invest. (2018) 128:668–81. doi: 10.1172/JCI92812
71. Milich DR and McLachlan A. The nucleocapsid of hepatitis B virus is both a T-cell-independent and a T-cell-dependent antigen. Science. (1986) 234:1398–401. doi: 10.1126/science.3491425
72. Milich DR, Chen M, Schödel F, Peterson DL, Jones JE, Hughes JL, et al. Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci U.S.A. (1997) 94:14648–53. doi: 10.1073/pnas.94.26.14648
73. Ye G, Chen C, Zhou Y, Tang L, Cai J, Huang Y, et al. Anti-HBc mirrors the activation of HBV-specific CD8(+) T cell immune response and exhibits a direct effect on HBV control. Antiviral Res. (2024) 230:105975. doi: 10.1016/j.antiviral.2024.105975
74. Liu Q, Zheng Y, Yu Y, Tan Q, and Huang X. Identification of HLA-A*0201-restricted CD8+ T-cell epitope C64-72 from hepatitis B virus core protein. Int Immunopharmacol. (2012) 13:141–7. doi: 10.1016/j.intimp.2012.03.018
75. Chen X, Lai J, Pan Q, Tang Z, Yu Y, Zang G, et al. The delivery of HBcAg via Tat-PTD enhances specific immune response and inhibits Hepatitis B virus replication in transgenic mice. Vaccine. (2010) 28:3913–9. doi: 10.1016/j.vaccine.2010.03.070
76. Kwon JA and Rho HM. Hepatitis B viral core protein activates the hepatitis B viral enhancer II/pregenomic promoter through the nuclear factor kappaB binding site. Biochem Cell Biol. (2002) 80:445–55. doi: 10.1139/o02-133
77. Saeed U, Piracha ZZ, Kwon H, Kim J, Kalsoom F, Chwae Y-J, et al. The HBV core protein and core particle both bind to the PPiase par14 and par17 to enhance their stabilities and HBV replication. Front Microbiol. (2021) 12:795047. doi: 10.3389/fmicb.2021.795047
78. Sohn SY, Kim SB, Kim J, and Ahn BY. Negative regulation of hepatitis B virus replication by cellular Hsp40/DnaJ proteins through destabilization of viral core and X proteins. J. Gen Virol. (2006) 87:1883–91. doi: 10.1099/vir.0.81684-0
79. Zhang Y, Yuan X, Wang J, Han M, Lu H, Wang Y, et al. TRPV4 promotes HBV replication and capsid assembly via methylation modification of H3K4 and HBc ubiquitin. J Med Virol. (2024) 96:e29510. doi: 10.1002/jmv.29510
80. Liu Y, Yang J, Wang Y, Zeng Q, Fan Y, Huang A, et al. The proteasome activator subunit PSME1 promotes HBV replication by inhibiting the degradation of HBV core protein. Genes Dis. (2024) 11:101142. doi: 10.1016/j.gendis.2023.101142
81. Panté N and Kann M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol Biol Cell. (2002) 13:425–34. doi: 10.1091/mbc.01-06-0308
82. Rabe B, Glebe D, and Kann M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J Virol. (2006) 80:5465–73. doi: 10.1128/JVI.02303-05
83. Yeh CT, Liaw YF, and Ou JH. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J Virol. (1990) 64:6141–7. doi: 10.1128/jvi.64.12.6141-6147.1990
84. Li H-C, Huang E-Y, Su P-Y, Wu S-Y, Yang C-C, Lin Y-S, et al. Nuclear export and import of human hepatitis B virus capsid protein and particles. PloS Pathog. (2010) 6:e1001162. doi: 10.1371/journal.ppat.1001162
85. Yang C-C, Huang E-Y, Li H-C, Su P-Y, and Shih C. Nuclear export of human hepatitis B virus core protein and pregenomic RNA depends on the cellular NXF1-p15 machinery. PloS One. (2014) 9:e106683. doi: 10.1371/journal.pone.0106683
86. Görlich D and Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. (1999) 15:607–60. doi: 10.1146/annurev.cellbio.15.1.607
87. Goldfarb DS, Corbett AH, Mason DA, Harreman MT, and Adam SA. Importin alpha: a multipurpose nuclear-transport receptor. Trends Cell Biol. (2004) 14:505–14. doi: 10.1016/j.tcb.2004.07.016
88. Jiang B and Hildt E. Intracellular trafficking of HBV particles. Cells. (2020) 9(9):2023. doi: 10.3390/cells9092023
89. Yang R, Ko Y-H, Li F, Lokareddy RK, Hou C-FD, Kim C, et al. Structural basis for nuclear import of hepatitis B virus (HBV) nucleocapsid core. Sci Adv. (2024) 10:eadi7606. doi: 10.1126/sciadv.adi7606
90. Rabe B, Vlachou A, Panté N, Helenius A, and Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U.S.A. (2003) 100:9849–54. doi: 10.1073/pnas.1730940100
91. Kann M, Sodeik B, Vlachou A, Gerlich WH, and Helenius A. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol. (1999) 145:45–55. doi: 10.1083/jcb.145.1.45
92. Su P-Y, Yen S-CB, Yang C-C, Chang C-H, Lin W-C, Shih C, et al. Hepatitis B virus virion secretion is a CRM1-spike-mediated late event. J BioMed Sci. (2022) 29:44. doi: 10.1186/s12929-022-00827-w
93. Yang C-C, Chang C-H, Chen H-L, Chou M-C, Yang C-J, Jhou R-S, et al. CRM1-spike-mediated nuclear export of hepatitis B virus encapsidated viral RNA. Cell Rep. (2022) 38:110472. doi: 10.1016/j.celrep.2022.110472
94. Khanam A, Chua JV, and Kottilil S. Immunopathology of chronic hepatitis B infection: role of innate and adaptive immune response in disease progression. Int J Mol Sci. (2021) 22(11):5497. doi: 10.3390/ijms22115497
95. Zheng JR, Wang ZL, and Feng B. Hepatitis B functional cure and immune response. Front Immunol. (2022) 13:1075916. doi: 10.3389/fimmu.2022.1075916
96. Tsai HT, Tsai TH, Lu TM, and Yang CC. Immunopathology of hepatitis B virus infection. Int Rev Immunol. (2008) 27:427–46. doi: 10.1080/08830180802295757
97. Cooper A, Tal G, Lider O, and Shaul Y. Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J Immunol. (2005) 175:3165–76. doi: 10.4049/jimmunol.175.5.3165
98. Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. (2015) 42:123–32. doi: 10.1016/j.immuni.2014.12.016
99. Miyakawa K, Nishi M, Ogawa M, Matsunaga S, Sugiyama M, Nishitsuji H, et al. Galectin-9 restricts hepatitis B virus replication via p62/SQSTM1-mediated selective autophagy of viral core proteins. Nat Commun. (2022) 13:531. doi: 10.1038/s41467-022-28171-5
100. Wei Q, Song H, Gao Y, Xu F, Xiao Q, Wang F, et al. Dual-role of cholesterol-25-hydroxylase in regulating hepatitis B virus infection and replication. mBio. (2022) p:e0067722. doi: 10.1128/mbio.00677-22
101. Xu F, Song H, Xiao Q, Wei Q, Pang X, Gao Y, et al. Type-III interferon stimulated gene TRIM31 mutation in an HBV patient blocks its ability in promoting HBx degradation. Virus Res. (2022) 308:198650. doi: 10.1016/j.virusres.2021.198650
102. Xu F, Song H, Xiao Q, Li N, Zhang H, Cheng G, et al. Type III interferon-induced CBFbeta inhibits HBV replication by hijacking HBx. Cell Mol Immunol. (2019) 16:357–66. doi: 10.1038/s41423-018-0006-2
103. Tan G, Yi Z, Song H, Xu F, Li F, Aliyari R, et al. Type-I-IFN-stimulated gene TRIM5gamma inhibits HBV replication by promoting HBx degradation. Cell Rep. (2019) 29:3551–3563 e3. doi: 10.1016/j.celrep.2019.11.041
104. Tan G, Xu F, Song H, Yuan Y, Xiao Q, Ma F, et al. Identification of TRIM14 as a type I IFN-stimulated gene controlling hepatitis B virus replication by targeting HBx. Front Immunol. (2018) 9:1872. doi: 10.3389/fimmu.2018.01872
105. Song H, Xu F, Pang X, Xiao Q, Wei Q, Lei B, et al. STAT3-dependent gene TRIM5gamma interacts with HBx through a zinc binding site on the BBox domain. Front Microbiol. (2021) 12:663534. doi: 10.3389/fmicb.2021.663534
106. Tan G, Xiao Q, Song H, Ma F, Xu F, Peng D, et al. Type I IFN augments IL-27-dependent TRIM25 expression to inhibit HBV replication. Cell Mol Immunol. (2018) 15:272–81. doi: 10.1038/cmi.2016.67
107. Song H. TRIM25 inhibits HBV replication by promoting HBx degradation and the RIG-I-mediated pgRNA recognition. (Engl). (2023) 136:799–806. doi: 10.1097/CM9.0000000000002617
108. Li T, Yang X, Li W, Song J, Li Z, Zhu X, et al. ADAR1 stimulation by IFN-α Downregulates the expression of MAVS via RNA editing to regulate the anti-HBV response. Mol Ther. (2021) 29:1335–48. doi: 10.1016/j.ymthe.2020.11.031
109. Xu F, Song H, An B, Xiao Q, Cheng G, Tan G, et al. NF-κB-dependent IFIT3 induction by HBx promotes hepatitis B virus replication. Front Microbiol. (2019) 10:2382. doi: 10.3389/fmicb.2019.02382
110. Busca A and Kumar A. Innate immune responses in hepatitis B virus (HBV) infection. Virol J. (2014) 11:22. doi: 10.1186/1743-422X-11-22
111. Wieland S, Thimme R, Purcell RH, and Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U.S.A. (2004) 101:6669–74. doi: 10.1073/pnas.0401771101
112. Li T, Ke Z, Liu W, Xiong Y, Zhu Y, Liu Y, et al. Human hepatitis B virus core protein inhibits IFNα-induced IFITM1 expression by interacting with BAF200. Viruses. (2019) 11(5):427. doi: 10.3390/v11050427
113. Yeh CT, Wong SW, Fung YK, and Ou JH. Cell cycle regulation of nuclear localization of hepatitis B virus core protein. Proc Natl Acad Sci U.S.A. (1993) 90:6459–63. doi: 10.1073/pnas.90.14.6459
114. Jung MC, Spengler U, Schraut W, Hoffmann R, Zachoval R, Eisenburg J, et al. Hepatitis B virus antigen-specific T-cell activation in patients with acute and chronic hepatitis B. J Hepatol. (1991) 13:310–7. doi: 10.1016/0168-8278(91)90074-L
115. Lin Y-J, Huang L-R, Yang H-C, Tzeng H-T, Hsu P-N, Wu H-L, et al. Hepatitis B virus core antigen determines viral persistence in a C57BL/6 mouse model. Proc Natl Acad Sci U.S.A. (2010) 107:9340–5. doi: 10.1073/pnas.1004762107
116. Li M, Sun X-H, Zhu X-J, Jin S-G, Zeng Z-J, Zhou Z-H, et al. HBcAg induces PD-1 upregulation on CD4+T cells through activation of JNK, ERK and PI3K/AKT pathways in chronic hepatitis-B-infected patients. Lab Invest. (2012) 92:295–304. doi: 10.1038/labinvest.2011.157
117. Li M, Zhou Z-H, Sun X-H, Zhang X, Zhu X-J, Jin S-G, et al. Hepatitis B core antigen upregulates B7-H1 on dendritic cells by activating the AKT/ERK/P38 pathway: a possible mechanism of hepatitis B virus persistence. Lab Invest. (2016) 96:1156–64. doi: 10.1038/labinvest.2016.96
118. Poonia B, Ayithan N, Nandi M, Masur H, and Kottilil S. HBV induces inhibitory FcRL receptor on B cells and dysregulates B cell-T follicular helper cell axis. Sci Rep. (2018) 8:15296. doi: 10.1038/s41598-018-33719-x
119. Ding X, Lei Q, Li T, Li L, and Qin B. Hepatitis B core antigen can regulate NLRP3 inflammasome pathway in HepG2 cells. J Med Virol. (2019) 91:1528–36. doi: 10.1002/jmv.25490
120. Chen Z, Li Y-X, Fu H-J, Ren Y-L, Zou L, Shen S-Z, et al. Hepatitis B Virus Core Antigen Stimulates IL-6 Expression via p38, ERK and NF-κB Pathways in Hepatocytes. Cell Physiol Biochem. (2017) 41:91–100. doi: 10.1159/000455954
121. Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PloS One. (2011) 6:e18909. doi: 10.1371/journal.pone.0018909
122. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
123. Ge J, Wang K, Meng Q-H, Qi Z-X, Meng F-L, Fan Y-C, et al. Implication of Th17 and Th1 cells in patients with chronic active hepatitis B. J Clin Immunol. (2010) 30:60–7. doi: 10.1007/s10875-009-9328-2
124. Zhang F, Yao S, Yuan J, Zhang M, He Q, Yang G, et al. Elevated IL-6 receptor expression on CD4+ T cells contributes to the increased Th17 responses in patients with chronic hepatitis B. Virol J. (2011) 8:270. doi: 10.1186/1743-422X-8-270
125. Wang L, Chen S, and Xu K. IL-17 expression is correlated with hepatitis B−related liver diseases and fibrosis. Int J Mol Med. (2011) 27:385–92. doi: 10.3892/ijmm.2011.594
126. Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. (2013) 191:1835–44. doi: 10.4049/jimmunol.1203013
127. Fabre T, Kared H, Friedman SL, and Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol. (2014) 193:3925–33. doi: 10.4049/jimmunol.1400861
128. Jackson K, Locarnini S, and Gish R. Diagnostics of hepatitis B virus: standard of care and investigational. Clin Liver Dis (Hoboken). (2018) 12:5–11. doi: 10.1002/cld.729
129. Zamor PJ and Lane AM. Interpretation of HBV serologies. Clin Liver Dis. (2021) 25:689–709. doi: 10.1016/j.cld.2021.06.012
130. Song JE and Kim DY. Diagnosis of hepatitis B. Ann Transl Med. (2016) 4:338. doi: 10.21037/atm.2016.09.11
131. Guvenir M and Arikan A. Hepatitis B virus: from diagnosis to treatment. Pol J Microbiol. (2020) 69:391–9. doi: 10.33073/pjm-2020-044
132. Pondé RA, Cardoso DD, and Ferro MO. The underlying mechanisms for the ‘anti-HBc alone’ serological profile. Arch Virol. (2010) 155:149–58. doi: 10.1007/s00705-009-0559-6
133. Wang Q, Klenerman P, and Semmo N. Significance of anti-HBc alone serological status in clinical practice. Lancet Gastroenterol Hepatol. (2017) 2:123–34. doi: 10.1016/S2468-1253(16)30076-0
134. Kim S, Yoo S, Lee JI, Kim S, Chang HY, Kim D, et al. Anti-HBc igG levels: A predictor of HBsAg seroclearance in chronic hepatitis B patients with nucleos(t)ide analogue-induced HBeAg seroclearance. Dig Dis Sci. (2022) 67:321–8. doi: 10.1007/s10620-021-06845-2
135. Yang Y-Y, Huang C-F, Wei JCC, Ho M-S, Wang L-N, Lin S-J, et al. The study of IgG subclass profiles of anti-hbc in populations with different status of HBV infection. Cell Mol Immunol. (2005) 2:393–8.
136. Huang C-F, Lin S-S, Ho Y-C, Chen F-L, and Yang C-C. The immune response induced by hepatitis B virus principal antigens. Cell Mol Immunol. (2006) 3:97–106.
137. Venkatakrishnan B and Zlotnick A. The structural biology of hepatitis B virus: form and function. Annu Rev Virol. (2016) 3:429–51. doi: 10.1146/annurev-virology-110615-042238
138. You SL, Yang HI, and Chen CJ. Seropositivity of hepatitis B e antigen and hepatocellular carcinoma. Ann Med. (2004) 36:215–24. doi: 10.1080/07853890310021580
139. Chen MT, Billaud J-N, Sällberg M, Guidotti LG, Chisari FV, Jones J, et al. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U.S.A. (2004) 101:14913–8. doi: 10.1073/pnas.0406282101
140. Pattyn J, Hendrickx G, Vorsters A, and Van Damme P. Hepatitis B vaccines. J Infect Dis. (2021) 224:S343–s351. doi: 10.1093/infdis/jiaa668
141. Ren W, Li J, Cheng R, Wu Z, Liu Y, Qiu Y, et al. Long-term persistence of anti-HBs after hepatitis B vaccination among isolated anti-HBc positive adults in China: 8-years results. Hum Vaccin Immunother. (2021) 17:1190–5. doi: 10.1080/21645515.2020.1806672
142. Song L-W, Liu P-G, Liu C-J, Zhang T-Y, Cheng X-D, Wu H-L, et al. Quantitative hepatitis B core antibody levels in the natural history of hepatitis B virus infection. Clin Microbiol Infect. (2015) 21:197–203. doi: 10.1016/j.cmi.2014.10.002
143. Lazarevic I, Banko A, Miljanovic D, and Cupic M. Clinical utility of quantitative HBV core antibodies for solving diagnostic dilemmas. Viruses. (2023) 15(2):373. doi: 10.3390/v15020373
144. Zhang Y, Zhang H, Zhang J, Zhang J, and Guo H. Naturally occurring core protein mutations compensate for the reduced replication fitness of a lamivudine-resistant HBV isolate. Antiviral Res. (2019) 165:47–54. doi: 10.1016/j.antiviral.2019.03.006
145. Liang Y, Yano Y, Putri WA, Mardian Y, Okada R, Tanahashi T, et al. Early changes in quasispecies variant after antiviral therapy for chronic hepatitis B. Mol Med Rep. (2018) 17:5528–37. doi: 10.3892/mmr.2018.8551
146. Taverniti V, Ligat G, Debing Y, Buh Kum D, Baumert TF, Verrier ER, et al. Capsid assembly modulators as antiviral agents against HBV: molecular mechanisms and clinical perspectives. J Clin Med. (2022) 11(5):1349. doi: 10.3390/jcm11051349
147. Taverniti V, Meiss-Heydmann L, Gadenne C, Vanrusselt H, Buh Kum D, Giannone F, et al. CAM-A-dependent HBV core aggregation induces apoptosis through ANXA1. JHEP Rep. (2024) 6:101134. doi: 10.1016/j.jhepr.2024.101134
148. Lecoq L, Brigandat L, Huber R, Fogeron M-L, Wang S, Dujardin M, et al. Molecular elucidation of drug-induced abnormal assemblies of the hepatitis B virus capsid protein by solid-state NMR. Nat Commun. (2023) 14:471. doi: 10.1038/s41467-023-36219-3
149. Guo F, Zhao Q, Sheraz M, Cheng J, Qi Y, Su Q, et al. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PloS Pathog. (2017) 13:e1006658. doi: 10.1371/journal.ppat.1006658
150. Kim C, Barnes LF, Schlicksup CJ, Patterson AJ, Bothner B, Jarrold MF, et al. Core protein-directed antivirals and importin β Can synergistically disrupt hepatitis B virus capsids. J Virol. (2022) 96:e0139521. doi: 10.1128/JVI.01395-21
151. Kornyeyev D, Song Z, Eng S, Soulette C, Ramirez R, Tang J, et al. Selective depletion of HBV-infected hepatocytes by class A capsid assembly modulators requires high levels of intrahepatic HBV core protein. Antimicrob Agents Chemother. (2024) 68:e0042024. doi: 10.1128/aac.00420-24
152. Buh Kum D, Vanrusselt H, Acosta Sanchez A, Taverniti V, Verrier ER, Baumert TF, et al. Class A capsid assembly modulator RG7907 clears HBV-infected hepatocytes through core-dependent hepatocyte death and proliferation. Hepatology. (2023) 78:1252–65. doi: 10.1097/HEP.0000000000000428
153. Lin J, Yin L, Xu X-Z, Sun H-C, Huang Z-H, Ni X-Y, et al. Bay41-4109-induced aberrant polymers of hepatitis b capsid proteins are removed via STUB1-promoted p62-mediated macroautophagy. PloS Pathog. (2022) 18:e1010204. doi: 10.1371/journal.ppat.1010204
154. Hou J, Gane E, Balabanska R, Zhang W, Zhang J, Lim TH, et al. Efficacy, safety, and pharmacokinetics of capsid assembly modulator linvencorvir plus standard of care in chronic hepatitis B patients. Clin Mol Hepatol. (2024) 30:191–205. doi: 10.3350/cmh.2023.0422
155. Yuen M-F, Zhou X, Gane E, Schwabe C, Tanwandee T, Feng S, et al. Safety, pharmacokinetics, and antiviral activity of RO7049389, a core protein allosteric modulator, in patients with chronic hepatitis B virus infection: a multicentre, randomised, placebo-controlled, phase 1 trial. Lancet Gastroenterol Hepatol. (2021) 6:723–32. doi: 10.1016/S2468-1253(21)00176-X
156. Wu X, Feng S, Zhang J, Zhang W, Zhang Y, Zhu M, et al. Evaluation of the safety, tolerability, and pharmacokinetics of RO7049389 in healthy Chinese volunteers. Clin Transl Sci. (2022) 15:195–203. doi: 10.1111/cts.13134
157. Yan R, Cai D, Zong Y, Guo L, Zhou Y, Tang A, et al. Preclinical characterization of ABI-H2158, an HBV core inhibitor with dual mechanisms of action. Antiviral Res. (2023) 209:105485. doi: 10.1016/j.antiviral.2022.105485
158. Zoulim F, Lenz O, Vandenbossche JJ, Talloen W, Verbinnen T, Moscalu I, et al. JNJ-56136379, an HBV capsid assembly modulator, is well-tolerated and has antiviral activity in a phase 1 study of patients with chronic infection. Gastroenterology. (2020) 159:521–533.e9. doi: 10.1053/j.gastro.2020.04.036
159. Janssen HLA, Hou J, Asselah T, Chan HLY, Zoulim F, Tanaka Y, et al. Randomised phase 2 study (JADE) of the HBV capsid assembly modulator JNJ-56136379 with or without a nucleos(t)ide analogue in patients with chronic hepatitis B infection. Gut. (2023) 72:1385–98. doi: 10.1136/gutjnl-2022-328041
160. Agarwal K, Buti M, van Bömmel F, Lampertico P, Janczewska E, Bourliere M, et al. JNJ-73763989 and bersacapavir treatment in snucleos(t)ide analogue-suppressed patients with chronic hepatitis B: REEF-2. J Hepatol. (2024) 81:404–14. doi: 10.1016/j.jhep.2024.03.046
161. Verbinnen T, Tan Y, Wang G, Dehertogh P, Vergauwen K, Neefs J-M, et al. Anti-HBV activity of the HBV capsid assembly modulator JNJ-56136379 across full-length genotype A-H clinical isolates and core site-directed mutants in vitro. J Antimicrob Chemother. (2020) 75:2526–34. doi: 10.1093/jac/dkaa179
162. Ruan L, Hadden JA, and Zlotnick A. Assembly properties of hepatitis B virus core protein mutants correlate with their resistance to assembly-directed antivirals. J Virol. (2018) 92(20):e01082–18. doi: 10.1128/JVI.01082-18
163. Klumpp K, Lam AM, Lukacs C, Vogel R, Ren S, Espiritu C, et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U.S.A. (2015) 112:15196–201. doi: 10.1073/pnas.1513803112
164. Berke JM, Tan Y, Verbinnen T, Dehertogh P, Vergauwen K, Vos A, et al. Antiviral profiling of the capsid assembly modulator BAY41–4109 on full-length HBV genotype A-H clinical isolates and core site-directed mutants in vitro. Antiviral Res. (2017) 144:205–15. doi: 10.1016/j.antiviral.2017.06.016
Keywords: hepatitis B core protein (HBc), HBV replication, immune evasion, capsidassembly modulators (CAMs), therapeutic target
Citation: Zhu Y, Song H, Xu F, Huang M and Tan G (2025) HBc: the multifunctional architect of HBV replication, immune evasion, and therapeutic innovation. Front. Immunol. 16:1657920. doi: 10.3389/fimmu.2025.1657920
Received: 02 July 2025; Accepted: 28 October 2025;
Published: 17 November 2025.
Edited by:
Gennady Bocharov, Marchuk Institute of Numerical Mathematics (RAS), RussiaReviewed by:
H. Syed Iqbal, YR Gaitonde Centre for AIDS Research and Education, IndiaJiacheng Lin, Fujian Medical University, China
Copyright © 2025 Zhu, Song, Xu, Huang and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangyun Tan, dGd5MDQyNUBqbHUuZWR1LmNu