 Yaning Guan1,2†Qian Li1,2†Yongjing Liu1,2Pingping Zhang1,2Maolin Huang1,2Yimin Guo1,2Jing Chen1,2Yan Chen1,2*
Yaning Guan1,2†Qian Li1,2†Yongjing Liu1,2Pingping Zhang1,2Maolin Huang1,2Yimin Guo1,2Jing Chen1,2Yan Chen1,2* Zuochen Du1,2*
Zuochen Du1,2* Pei Huang1,3*
Pei Huang1,3*- 1Department of Pediatrics, Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 2Guizhou Children’s Hospital, Zunyi, China
- 3Laboratory of Hematology and Immunology, Guizhou Children’s Hospital, Zunyi, China
Introduction: Netherton syndrome (NS) is a rare autosomal recessive disorder caused by mutations in the SPINK5 gene, which encodes the serine protease inhibitor LEKTI. It is characterized by congenital ichthyosis, hair shaft abnormalities, and atopic manifestations. Previous reports suggest that intravenous immunoglobulin (IVIG) may provide partial clinical benefit in NS. Here, we report the clinical and immunological characterization of an infant with NS effectively treated with IVIG therapy.
Methods: Clinical information was collected and reviewed from a 1-year-6-month-old boy presenting with NS. Hair shaft abnormalities were examined by scanning electron microscopy. Pathogenic variants in SPINK5 were identified using whole-exome and Sanger sequencing. Protein expression was assessed by Western blotting and ELISA. Peripheral lymphocyte subsets were analyzed by flow cytometry, and cytokine levels were evaluated with the Olink® Target 48 Cytokine panel.
Results: The patient presented with typical clinical manifestations of NS. Genetic analysis identified a novel heterozygous deletion spanning the SPINK5 gene (chr5:147,443,561-147,719,327), together with the c.1258A>G (p.K420E) polymorphism. LEKTI expression was markedly decreased, consistent with the genetic findings. Immune profiling revealed markedly reduced unswitched memory and marginal zone-like B cells, increased naïve B cells, and elevated γδ T cells compared with healthy controls. Cytokine analysis showed significantly increased levels of multiple pro-inflammatory cytokines, including TGFA, IL17 family members, CXCL8, CCL2, TNF, CCL19, and IL18. Following IVIG therapy, the patient demonstrated significant clinical improvement, with recovery of skin manifestations, and partial normalization of lymphocyte subsets and cytokine levels, indicating restoration of immune regulation.
Discussion: This study reports a novel compound heterozygous SPINK5 mutation in an infant with NS, comprising a large deletion and c.1258A>G polymorphism, resulting in LEKTI deficiency and immune dysregulation. IVIG therapy effectively alleviated clinical symptoms and restored immune balance, highlighting its potential as a therapeutic option for NS and related immunodeficiency disorders.
Introduction
Netherton syndrome (NS) is a rare genetic disorder characterized clinically by a triad of ichthyosis, hair shaft abnormalities, and atopic manifestations (1). This condition results from mutations in the serine peptidase inhibitor Kazal type 5 (SPINK5) gene, encoding the serine protease inhibitor LEKTI (Lympho-Epithelial Kazal-type-related Inhibitor) (2). LEKTI is essential for maintaining skin barrier integrity and modulating immune responses and physiological desquamation by specifically inhibiting serine proteases kallikrein-related peptidase 5 (KLK5) and KLK7, among others (3). Mutations in SPINK5 cause LEKTI deficiency, compromising the skin barrier function and increasing susceptibility to allergens and infections (4, 5). Clinically, NS typically manifests from birth with severe dermatological symptoms, including ichthyosis linearis circumflexa, erythroderma, and atopic-like dermatitis. These symptoms are often complicated by recurrent infections, growth retardation, and a variety of immunological abnormalities (6). Clinical management of NS is challenging, given the significant discomfort and diminished quality of life due to persistent dermatologic symptoms and related complications (7).
Recent studies have documented a diverse spectrum of SPINK5 gene mutations associated with NS, including point mutations, small insertions, deletions, and splice-site mutations. Such mutations typically impair LEKTI function, producing the characteristic clinical phenotype of the syndrome (8). Although a number of specific mutation sites have been reported, such as nonsense mutations (e.g., p.R790*, p.R218*) and missense mutations (e.g., p.T808I, p.D106N), polymorphisms and large deletions in NS are less commonly documented (8–10).
Beyond affecting skin barrier integrity, SPINK5 mutations significantly impact immune function. Patients harboring these mutations frequently exhibit compromised humoral immunity, characterized by reduced populations of specific B cell subsets, such as memory B cells, transitional B cells, and plasmablasts. Such immunodeficiency contributes to increased susceptibility to recurrent infections and inflammatory conditions (11, 12). Furthermore, elevated immunoglobulin E (IgE) and eosinophil counts commonly observed in these patients indicate a heightened allergic response (13). Dysfunctional LEKTI also disrupts the regulation of cytokine production, leading to an imbalance between pro-inflammatory and anti-inflammatory cytokines. Notably, elevated levels of interleukin-17 (IL-17) family cytokines and tumor necrosis factor-alpha (TNF-α) are frequently reported in patients with SPINK5 mutations, fueling chronic inflammation and immune dysregulation (14, 15). A comprehensive understanding of these immune alterations is essential for the development of targeted therapeutic strategies aimed at modulating immune responses and improving clinical outcomes in NS.
Currently, NS remains incurable, and existing treatments primarily offer symptomatic relief. Systemic therapies, including topical calcineurin inhibitors (16, 17) and biological agents such as ustekinumab (18), have demonstrated variable efficacy. Nevertheless, there remains a significant gap in effective and targeted treatments for this challenging disease.
In this study, we conducted a comprehensive evaluation of the clinical, genetic, and immunological profiles of a 1-year-6-month-old patient presenting with severe NS. We assessed the extensive flaking affecting the patient’s entire body from birth, confirmed the presence and characteristics of genetic mutations and deletions through genetic testing, and measured LEKTI and KLK5 expression to elucidate the pathological implications of these genetic alterations. We comprehensively examined the changes in lymphocyte subpopulations, fine immune typing and various cytokine levels before and after the treatment to assess the modulation of disease immune function by intravenous immunoglobulin (IVIG). Through these evaluations, this study aims to deepen the understanding of the molecular and immunological mechanisms underpinning NS and highlight the therapeutic potential of IVIG in managing this complex and debilitating disorder.
Materials and methods
Patient recruitment and ethical considerations
This study involved a pediatric patient, a 1-year-6-month-old East Asian boy. Clinical data and blood specimens were collected after obtaining informed consent from his legal guardians during the patient’s initial hospitalization. Additionally, fourteen demographically matched (by age, race, and ethnicity) healthy controls were recruited. The study was conducted in strict adherence to the ethical principles outlined in the Declaration of Helsinki and was approved by the Ethics Committee of the Affiliated Hospital of Zunyi Medical University.
Genetic analysis
To identify the genetic basis of the patient’s condition, previous genetic sequencing results were reviewed, which had been performed by the Center for Genetic Diagnosis of Rare and inherited skin diseases (Guangdong, China).
To confirm the SPINK5 gene variant, genomic DNA was extracted from peripheral blood samples using a commercial DNA extraction kit (DP304-02, TIANGEN, China). PCR amplification was then performed using primers flanking the mutation site (19) (forward primer: 5’-CAGGGTTAGGCACATCACATTC-3’; reverse primer: 5’-TAAGGAATGCACGTGTTCCCTG-3’) (Sangon Biotech, China), and the resulting PCR products were subsequently validated by Sanger sequencing (Sangon Biotech, China).
Cytokine analysis
Changes in multiple cytokines before and after treatment were analyzed using the Olink Target 48 Cytokine panel (Olink, China). Blood samples were collected from both the patient and healthy controls, and plasma was isolated through centrifugation of whole blood samples. The separated plasma samples were then submitted to Olink for cytokine profiling according to the manufacturer’s protocols.
Scanning electron microscopy of hair
Hair samples obtained from the patient were sent to LiLai Biomedicine (Chengdu, China) for scanning electron microscopy analysis to characterize hair shaft abnormalities.
Isolation of PBMCs
Peripheral blood samples were collected into vacutainers containing sodium heparin. The samples were first centrifuged at 2,500 rpm for 5 minutes at room temperature to separate plasma, which was collected and stored for subsequent analyses. The remaining blood cells were gently diluted with PBS and carefully layered onto Ficoll Paque PREMIUM (LTS1077, TBDscience, China). PBMCs were isolated by centrifugation at 500 × g for 20 minutes at room temperature, after which the PBMC fraction was carefully collected for further experimental procedures.
Flow cytometry
Peripheral lymphocyte profiles were analyzed in a single experiment using 50 μL of whole blood, as previously described (20). The following antibodies, all purchased from BioLegend (USA), were used for cell staining: PE–anti-CD21 (354904), Brilliant Violet 421–anti-IgM (314516), PerCP–anti-CD38 (303520), Brilliant Violet 510–anti-IgD (348220), PE/Cy7–anti-CD27 (302838), APC–anti-CD19 (302212), APC/Cy7–anti-CD31 (303120), AF488–anti-CD24 (311108), Brilliant Violet 711–anti-IgG (410740), APC/Cy7–anti-CD23 (338520), Percp–anti-CD3 (300326), FITC–anti-CD4 (300506), APC–anti-CXCR3 (353708), Brilliant Violet 510–anti-CD8a (301048), PE–anti-CD127 (351304), PE/Cy7–anti-CD45RA (304126), Brilliant Violet 421–anti-CD185 (356920), PB–anti-CD38 (356628), Brilliant Violet 605–anti-CD27 (302830), PE/Fire640–anti-CD196 (353449), Brilliant Violet 711–anti-CD45RO (304236), Brilliant Violet 785–anti-PD-1 (329930), PE/Dazzle594–anti-CD57 (359620), AF660–anti-TCR γδ (331240), Brilliant Violet 650–anti-CD25 (302634), APC/Fire810–anti-HLA-DR (307674), PE–anti-TCR aβ (306708), Brilliant Violet 421–anti-TCR γδ (331218), PerCP–anti-CD3 (300326), FITC–anti-CD4 (300506), APC–anti-CD27 (356410), APC–anti-CD19 (302212), PE–anti-CD24 (311106), AF488–anti-IgD (348216), and PB–anti-CD27 (302822), APC–anti-CD20 (302310) and Zombie NIR™ Fixable Viability Kit (423106). Samples were analyzed on a FACS Canto Plus flow cytometer (BD Biosciences, USA) or Cytek® Aurora/Northern Lights™ (Cytek, USA), and data analysis was performed using FlowJo software. The results were summarized and compared to age-matched healthy controls.
Western blotting
PBMCs were lysed in RIPA buffer supplemented with protease inhibitors (Servicebio, China). Protein extracts mixed with loading buffer were separated by 10% SDS-PAGE gel electrophoresis and subsequently transferred onto PVDF membranes (Millipore, Germany). Membranes were blocked in 5% skim milk and incubated overnight at 4°C with primary antibodies against LEKTI (29808-1-AP, Proteintech, USA), antibodies against KLK5 (38528, SAB, USA) and HRP-conjugated β-actin recombinant rabbit monoclonal antibody (ET1702-67, HUABIO, China). After incubation with HRP-linked secondary antibodies (RGAR001, Proteintech, China), membranes were washed in TBST and visualized using an enhanced chemiluminescence detection system (BIO-RAD, USA).
Enzyme-linked immunosorbent assay
Plasma was collected from peripheral blood samples, and the level of KLK5 was measured using a Human KLK5 ELISA Kit (JM-6832H2, JINGMEI, China). The assay was performed according to the manufacturer’s instructions, and KLK5 concentrations were calculated by interpolation from the standard calibration curve.
Statistical analysis
Statistical analyses were performed using SPSS 29.0 (IBM, USA) and GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA, USA). Comparisons between groups were assessed using the unpaired two-tailed Student’s t-test. Statistical significance was defined as a P-value less than 0.05.
Results
Clinical presentation and medical history
The patient, a boy aged 1 year and 6 months, is the first child of healthy, non-consanguineous parents, born following an uncomplicated pregnancy. Immediately after birth, he exhibited widespread dermatological symptoms, including ichthyosis, erythroderma, and atopic-like dermatitis, characterized by severe scaling, erythema, and pruritus. These skin manifestations significantly affected his comfort and quality of life. The patient also experienced recurrent respiratory infections and persistent diarrhea, necessitating multiple hospital admissions and frequent antibiotic therapy. Additionally, he exhibited failure to thrive.
Routine blood examinations revealed that white blood cell (WBC) counts were generally within normal ranges, though elevations occurred during episodes of infection (Figures 1A, B). Consistently elevated eosinophil counts were noted (Figure 1C), whereas hemoglobin, platelet (PLT), and neutrophil levels remained within normal limits (Figures 1D–F). Scanning Electron Microscopy examination of hair shafts identified “bamboo hair” deformities (Figure 1G).
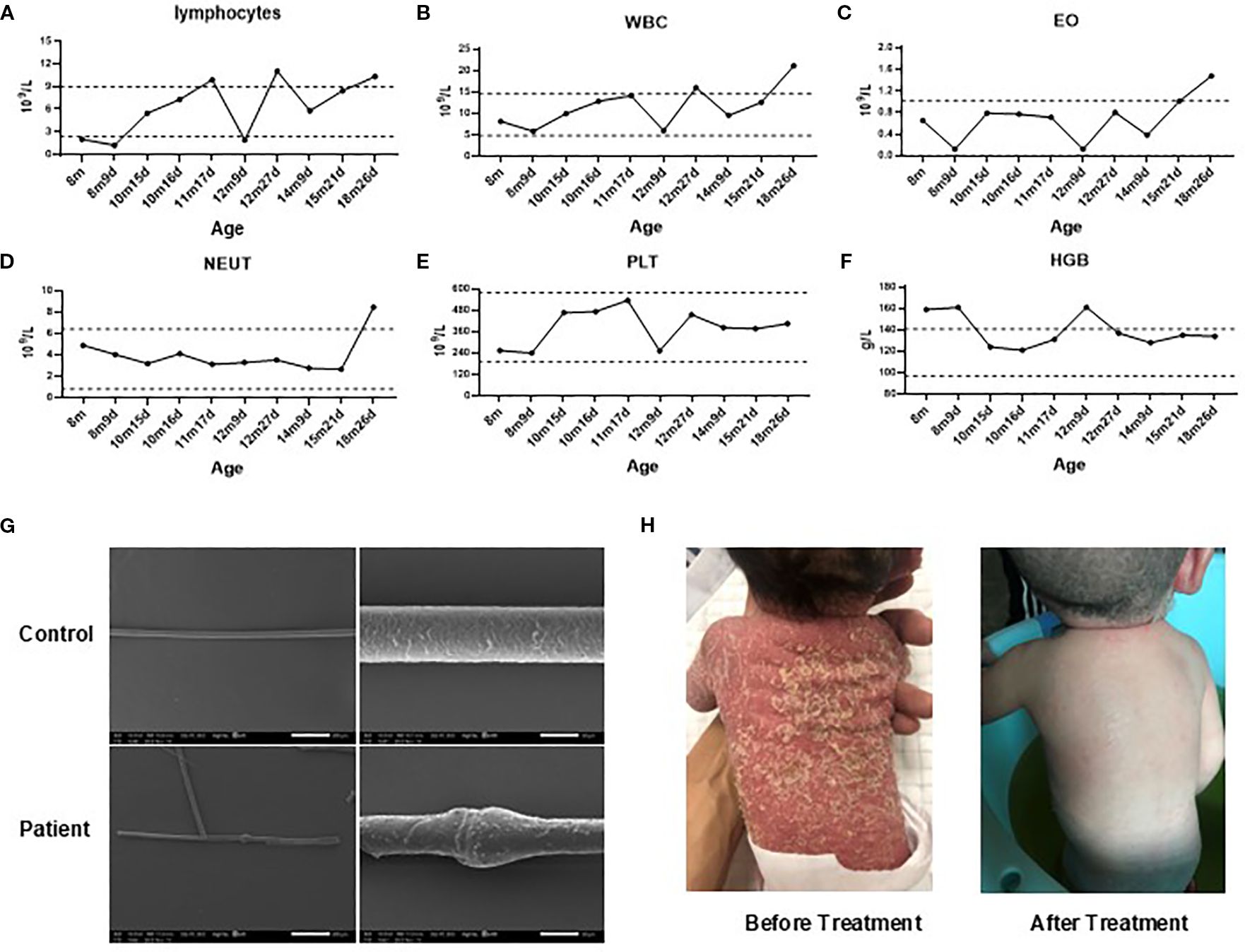
Figure 1. Clinical findings. (A–F). Changes in the patient’s lymphocytes (A), white blood cell counts (WBC, B), eosinophil counts (EO, C), neutrophils (NETU, D), platelet (PLT, E) counts and hemoglobin levels (HGB, F) over time. (G). Scanning electron microscopy of hair from the patient and his mother. (H) Skin desquamation before and after IVIG treatment. Gray dashed lines represent reference ranges. m, months; d, days.
Since birth, the patient had received symptomatic skin care, including topical corticosteroids and emollients, along with specialized hypoallergenic formula feeding to manage potential food allergies. Despite these interventions, recurrent skin rashes and growth delays persisted. Regular IVIG therapy (500 mg/kg, once monthly) was initiated at 8 months of age. After 11 months of continuous treatment, the patient exhibited notable clinical improvement, including significant regression of skin lesions (Figure 1H), relief of gastrointestinal symptoms, and reduced frequency of respiratory infections. However, the family declined further therapy, which subsequently led to disease relapse.
Genetic analysis and protein expression
We reviewed and validated the patient’s previous genetic testing results. Genetic analyses identified the presence of a G1258A polymorphism (NM_006846; exon14 c.1258A>G, p.K420E) and a heterozygous deletion of approximately 257.8 kb involving the SPINK5 gene locus (Figure 2A). Sanger sequencing subsequently confirmed the G1258A polymorphism in SPINK5 (Figure 2B).
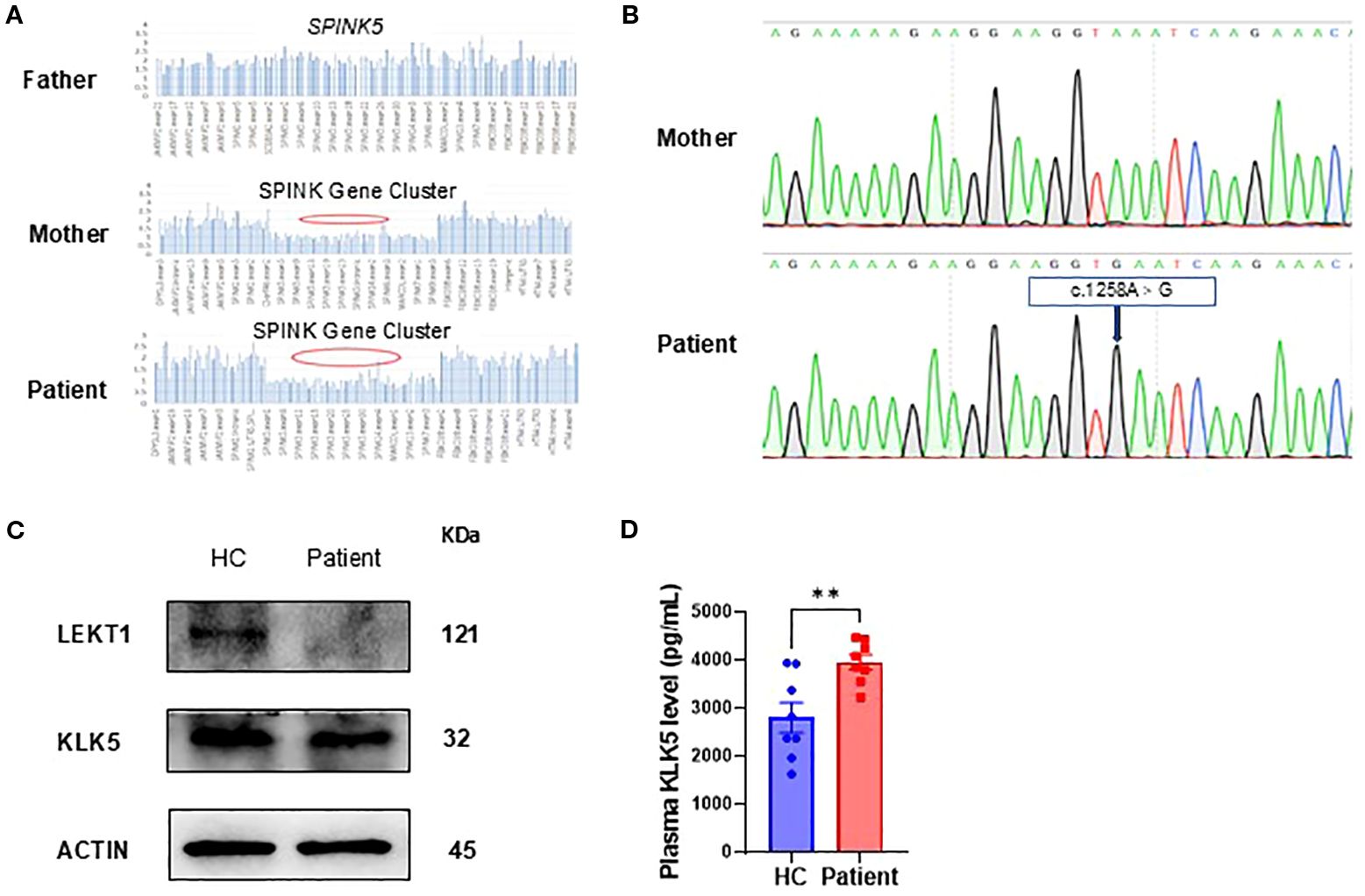
Figure 2. Genetic and protein expression analysis. (A) Whole-exome sequencing identified a heterozygous deletion spanning the SPINK gene cluster, including SPINK5. (B) Sanger sequencing of the patient and his mother revealed a polymorphism in SPINK5 (NM_006846; exon 14, c.1258A>G, p.K420E). (C) LEKTI and KLK5 expression in peripheral blood mononuclear cells (PBMCs) from the patient compared to healthy controls (HC, n=3). (D) KLK5 level in the patient’s plasma (seven time points from initial hospitalization) compared with that in healthy controls (HC, n = 8).
To evaluate the functional consequences of these genetic alterations, we assessed LEKTI and KLK5 protein expression in PBMCs. The analysis revealed markedly decreased LEKTI and relatively low KLK5 expression in the patient compared with healthy controls, consistent with the identified genetic defects (Figure 2C). In contrast, we found a significant increase in plasma KLK5 levels (Figure 2D).
Immunological assessment by multiparametric flow cytometry
We further investigated the patient’s immune function using multiparametric flow cytometry before and after IVIG therapy. Prior to IVIG administration, the patient’s B-cell compartments, including unswitched memory B cells, transitional B cells, and plasmablasts, were markedly reduced. Following IVIG therapy, these B-cell subsets significantly increased, suggesting an enhanced immune response and improved B-cell maturation. Although initial assessments of T-cell populations showed no notable abnormalities, post-treatment evaluations revealed substantial increases in total T cells, including CD8+ T cells, CD4+ T cells, CD4+ TEMRA cells, CD8+ TEMRA cells, CD4+ naive, and CD8+ naive T cells. These results indicate a substantial improvement in T-cell-mediated immunity following IVIG therapy (Table 1).
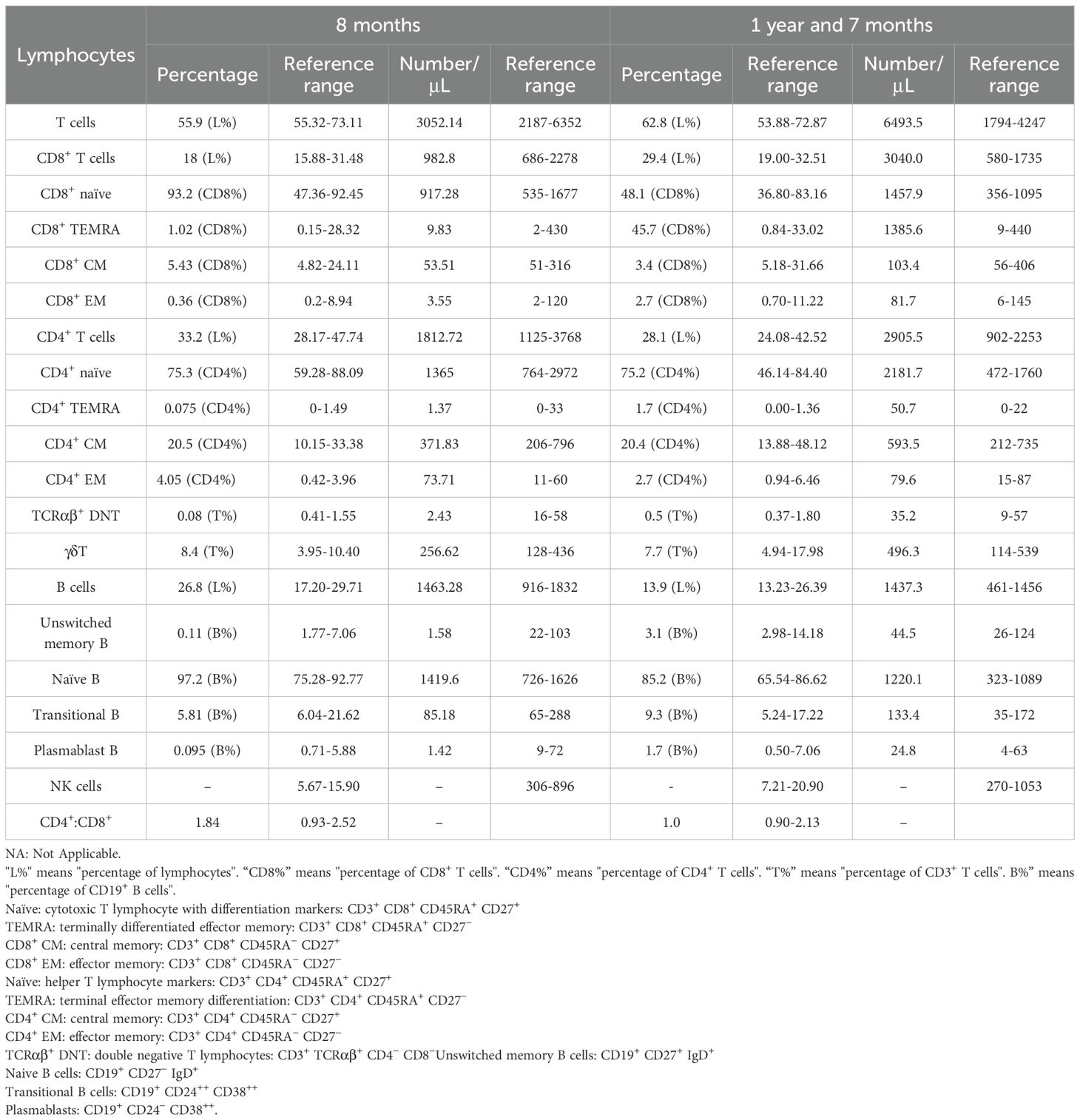
Table 1. Patient immunological profile.
Lymphocyte subset assessment
To obtain a comprehensive view of the patient’s immunological status, we compared lymphocyte subsets before and after IVIG therapy with demographically matched healthy controls (HC). The patient exhibited significantly decreased proportions of switched memory B cells and CD27+IgD+ marginal zone-like (MZ-like) B cells compared with controls. Conversely, naive B cell populations were elevated in the patient, and this subset did not show significant changes following IVIG treatment (Figures 3A, B). Additionally, the patient showed markedly elevated proportions of γδ T cells compared to healthy controls; these were notably reduced following IVIG therapy (Figure 3C). The proportions of follicular regulatory T cells (cTfr), Th1/17 cells, and activated CD8+ T cells were significantly higher in the patient (Figures 3D–F). Post-IVIG therapy, these T cell subsets demonstrated a downward trend towards normalization.
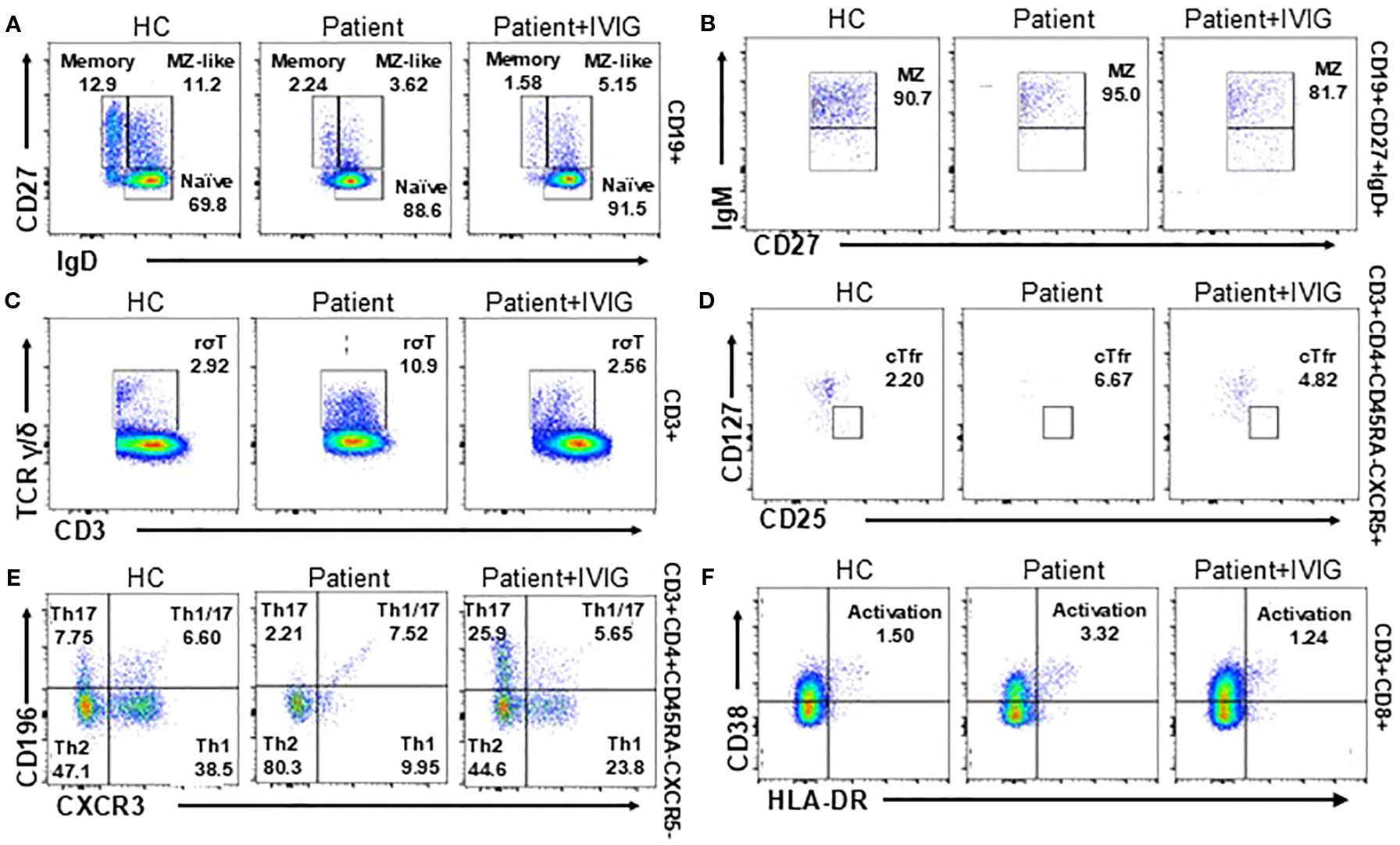
Figure 3. T and B cell subset phenotyping before and after treatment and comparison with healthy controls (HC, n=3). (A, B) Flow cytometry analysis of B cell subsets, including switched memory B cells, naive B cells (A) and CD27+IgD+ marginal zone-like (MZ-like) cells (B). (C-F) Flow cytometry of T cell subsets, including γδ T cells (C), cTfr cells (D), Th cells (E) and activated CD8+ cells (F) in the flow cytometry.
Cytokine profile analysis
To evaluate the immunological effects of the identified SPINK5 mutation, a comprehensive multiplex cytokine profile analysis was performed. Our results revealed significant alterations in multiple key cytokines. Specifically, the patient exhibited notably elevated levels of TGFA, IL17C, IL17A, IL17F, CXCL8, CCL2, TNF, CCL19, and IL18 prior to IVIG therapy (Figure 4A). Following IVIG treatment, substantial reductions in the levels of CCL2, TNF, CCL19, IL18, IL17F, TGFA, and IL17A were observed (Figure 4B), indicating significant immune modulation by IVIG. These cytokines alterations likely contribute to the clinical improvements observed in the patient, such as reduced inflammation and enhanced immune regulation. This detailed cytokine profiling thus provides critical insights into the underlying therapeutic mechanisms of IVIG.
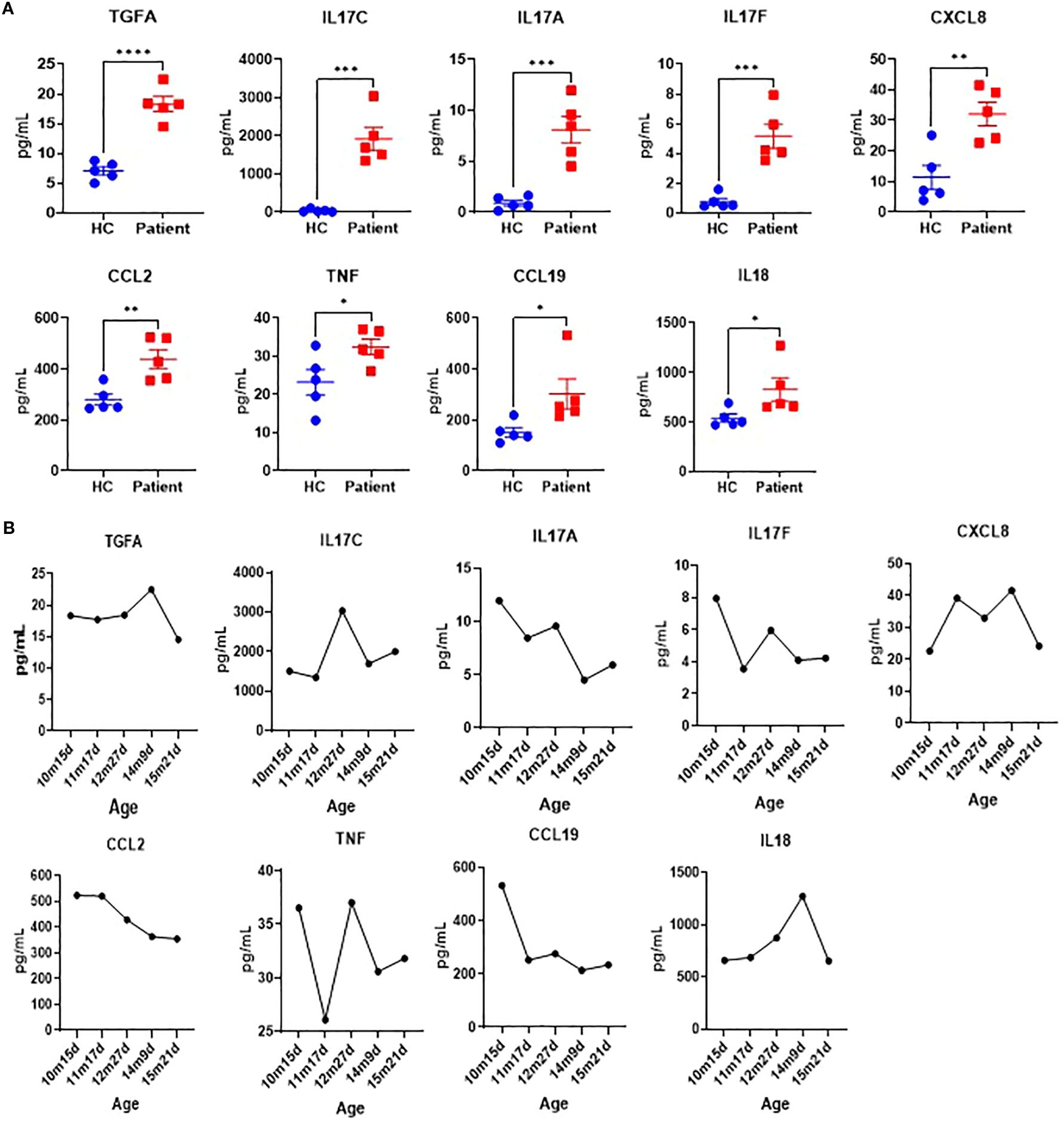
Figure 4. Cytokine profile analysis. (A) Cytokine profiles of the patient (measured monthly over five time points from initial hospitalization) and healthy control (HC, n=5). (B) Cytokine levels change before (10m15d) and after IVIG treatment over time.
Discussion
In this study, we conducted a comprehensive evaluation of a 1-year and 6-month-old pediatric patient with a confirmed diagnosis of NS, who presented with severe dermatological manifestations, immunological deficiencies, and growth retardation associated with mutations in the SPINK5 gene. From birth, the patient exhibited hallmark clinical features, including ichthyosis, erythroderma, and atopic dermatitis, along with recurrent respiratory infections and gastrointestinal disturbances. Genetic analysis revealed a rare combination of a G1258A polymorphism and a heterozygous deletion spanning approximately 257.8 kb that encompasses the SPINK5 gene. To date, only two cases involving large genomic deletions including the SPINK5 gene have been reported worldwide (9, 21). SPINK5 polymorphisms have been shown to affect LEKTI function and contribute to the clinical variability of NS (22, 23). To the best of our knowledge, this is the first reported case of a compound mutation comprising both a large fragment deletion and the c.1258A>G polymorphism in SPINK5, suggesting a potentially unique pathogenic mechanism and further emphasizing the functional heterogeneity of LEKTI-related deficiency. This mutation profile was associated with markedly reduced expression of LEKTI protein, confirming its pathogenic impact. Immunological assessments demonstrated a significant reduction in specific B-cell subsets, while T-cell populations remained within the normal range. Following IVIG therapy, substantial improvement in both B-cell and T-cell subsets was observed, indicating a restoration of immune functionality. In addition, multiplex cytokine profiling revealed notable modulation of inflammatory mediators after treatment, supporting the therapeutic efficacy of IVIG in modulating immune responses in NS.
Pathogenic variants of SPINK5 associated with NS have been identified across all functional domains of the gene. To date, more than 80 distinct variants have been reported, including point mutations, deletions, and polymorphisms (8). Among these, point mutations are the most frequently documented, while reports of large deletions and polymorphisms remain relatively rare (9, 21). In our case, we identified a G1258A polymorphism in combination with a heterozygous deletion spanning approximately 257.8 kb that encompasses the entire SPINK5 gene. To our knowledge, this combined large-fragment deletion and G1258A polymorphism represents a novel genetic alteration, further expanding the mutational spectrum of SPINK5 and providing new insights into the genetic pathogenesis of NS. In NS patients, epidermal homeostasis is severely disrupted (with characteristic structural defects), accompanied by dysregulated expression of KLK5, a critical epidermal protease. This contrasts paradoxically with the well-established essential roles of KLK5/KLK7 in murine NS models (24, 25), suggesting gaps in understanding human NS pathogenesis-particularly given inconsistent reports on KLK5/KLK7 activity in clinical cases (26, 27). Our study demonstrates significantly decreased LEKTI expression in patient PBMCs, alongside relatively low KLK5 expression but elevated plasma KLK5 levels, consistent with previously reported findings (28). These findings reinforce the LEKTI-KLK5-axis role in disease pathogenesis and highlight the need for further investigation into the precise roles of these proteases in human NS pathophysiology.
Our detailed analysis of the patient’s lymphocyte subsets revealed several noteworthy findings, some of which align with existing literature on SPINK5 mutations, while others provide novel insights. Prior to IVIG therapy, the patient exhibited markedly reduced levels of key B-cell subsets, including memory B cells, transitional B cells, and plasmablasts. These findings are consistent with previous reports of impaired humoral immunity in SPINK5-related immunodeficiency (12, 29). Notably, we also observed an increased proportion of naive B cells, which remained elevated even after IVIG treatment. This persistent elevation contrasts with prior studies, which have not emphasized this feature (12), suggesting a potential marker of disrupted B-cell maturation that may be resistant to IVIG modulation. Additionally, our study identified significantly elevated γδ T cells before treatment, which decreased after IVIG administration. This dynamic response of γδ T cells is rarely reported in the context of SPINK5 mutations and highlights a previously underrecognized component of the T-cell response to immunoglobulin therapy. Moreover, the patient demonstrated elevated levels of cTfr, Th1/17 cells, and activated CD8+ T cells, populations associated with immune activation and inflammation. These subsets were substantially reduced following IVIG treatment, consistent with its known immunomodulatory effects and capacity to dampen pro-inflammatory responses (11). Importantly, the detailed profiling of these specific lymphocyte subsets provides a deeper understanding of the immune dysregulation associated with SPINK5 mutations and the immune-restorative potential of IVIG. While prior studies have broadly characterized immune abnormalities in Netherton syndrome, our findings contribute novel perspectives by delineating how distinct B and T cell subsets respond to treatment. These insights not only reinforce existing knowledge but also suggest new avenues for immunophenotypic monitoring and tailored therapeutic strategies in patients with SPINK5-related immunodeficiencies.
Our comprehensive multiplex cytokine analysis revealed significant alterations in multiple cytokines following IVIG therapy, extending beyond previous studies that primarily focused on elevated IL-17 levels in patients with SPINK5 mutations. While the increased expression of IL-17A, IL-17C, and IL-17F is consistent with existing literature (30, 31), our study identified additional cytokines with notable changes, including TGFA, CXCL8, CCL2, TNF, CCL19, and TNFSF12. The elevated levels of CXCL8 and CCL2, chemokines critical for recruiting immune cells to sites of inflammation (32, 33), along with TNF, a key pro-inflammatory cytokine (34), reflect a heightened state of immune activation. Furthermore, the upregulation of TNFSF12, involved in immune regulation (35), and CCL19, which plays a role in lymphoid tissue organization (36), provides new insight into the complex immune dysregulation observed in SPINK5 mutation-associated disease. Following IVIG therapy, the observed decline in these cytokines suggests a robust immunomodulatory effect, likely contributing to the clinical improvements noted in our patient. This expanded cytokine profile not only deepens our understanding of the inflammatory landscape in SPINK5-related disorders but also introduces novel candidate biomarkers for monitoring disease activity and treatment efficacy. These findings support the use of cytokine profiling as a valuable tool in guiding therapeutic strategies and advancing personalized care for patients with SPINK5 mutations.
In conclusion, this study underscores the considerable clinical and immunological challenges associated with SPINK5 mutations and highlights the therapeutic potential of IVIG in improving immune function and alleviating disease manifestations. The identification of a G1258A polymorphism in combination with a heterozygous 257.8 kb deletion encompassing the SPINK5 gene represents a novel genetic finding. Furthermore, the comprehensive profiling of cytokine dynamics and lymphocyte subset alterations provides new insights into the immunopathogenesis of NS. Although this study is limited to a single case, the findings emphasize the importance of further research involving larger patient cohorts to validate and expand upon these observations, as well as long-term efficacy evaluations to better understand the sustained therapeutic impact of IVIG in NS. Ultimately, such efforts will advance the diagnosis, treatment, and overall management of SPINK5-related disorders, contributing to improved outcomes and quality of life for affected individuals.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Ethics Committee of Affiliated Hospital of Zunyi Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
YaG: Data curation, Formal analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. QL: Data curation, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. YL: Formal analysis, Investigation, Validation, Writing – review & editing. PZ: Investigation, Validation, Writing – review & editing. MH: Investigation, Validation, Writing – original draft. YiG: Investigation, Data curation, Writing – review & editing. JC: Writing – review & editing. YC: Conceptualization, Funding acquisition, Resources, Writing – review & editing. ZD: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. PH: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (32360184), the China Postdoctoral Science Foundation (2024MD754045), the Guizhou Provincial Program on Commercialization of Scientific and Technological Achievements (QKHCG(2024)ZD012), and the Medical Research Union Fund for High-quality Health Development of Guizhou Province (2024GZYXKYJJXM0062, 2024GZYXKYJJXM0061), Guizhou Association for Science and Technology Youth Talent Lifting Project (ZD), and the Key Advantageous Discipline Construction Project of Guizhou Provincial Health Commission in 2023.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. The authors confirm the responsible use of generative artificial intelligence in the preparation of this manuscript and assume full responsibility for its application. AI-assisted technology was employed solely for non-substantive tasks including language polishing (grammar refinement and syntax optimization) and reference formatting standardization. All critical academic content—including research conception, data interpretation, and scientific conclusions—was exclusively developed by the authors. A rigorous manual review process ensured complete consistency between AI-generated content and original research data. The authors retain ultimate accountability for all scholarly claims and manuscript integrity.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barbati F, Giovannini M, Oranges T, Lodi L, Barni S, Novembre E, et al. Netherton syndrome in children: management and future perspectives. Front Pediatr. (2021) 9:645259. doi: 10.3389/fped.2021.645259
2. Sprecher E, Chavanas S, DiGiovanna JJ, Amin S, Nielsen K, Prendiville JS, et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: implications for mutation detection and first case of prenatal diagnosis. J Invest Dermatol. (2001) 117:179–87. doi: 10.1046/j.1523-1747.2001.01389.x
3. Flora A and Smith A. Netherton’s syndrome: A case of two male siblings diagnosed in adulthood. Case Rep Dermatol. (2020) 12:64–9. doi: 10.1159/000507359
4. Herz-Ruelas ME, Chavez-Alvarez S, Garza-Chapa JI, Ocampo-Candiani J, Cab-Morales VA, and Kubelis-López DE. Netherton syndrome: case report and review of the literature. Skin Appendage Disord. (2021) 7:346–50. doi: 10.1159/000514699
5. Wang Y, Song H, Yu L, Wu N, Zheng X, Liang B, et al. A novel mutation in SPINK5 gene underlies a case of atypical Netherton syndrome. Front Genet. (2022) 13:943264. doi: 10.3389/fgene.2022.943264
6. DiGiovanna JJ and Robinson-Bostom L. Ichthyosis: etiology, diagnosis, and management. Am J Clin Dermatol. (2003) 4:81–95. doi: 10.2165/00128071-200304020-00002
7. Nouwen AEM, Schappin R, Nguyen NT, Ragamin A, Bygum A, Bodemer C, et al. Outcomes of systemic treatment in children and adults with netherton syndrome: A systematic review. Front Immunol. (2022) 13:864449. doi: 10.3389/fimmu.2022.864449
8. Sarri CA, Roussaki-Schulze A, Vasilopoulos Y, Zafiriou E, Patsatsi A, Stamatis C, et al. Netherton syndrome: A genotype-phenotype review. Mol Diagn Ther. (2017) 21:137–52. doi: 10.1007/s40291-016-0243-y
9. Wang Q, Qiu F, Wu H, and Fan YM. New compound heterozygous SPINK5 mutations in a Chinese infant with Netherton syndrome. J Eur Acad Dermatol Venereol. (2021) 35:e782–4. doi: 10.1111/jdv.17457
10. Moltrasio C, Romagnuolo M, Riva D, Colavito D, Ferrucci SM, Marzano AV, et al. Netherton syndrome caused by heterozygous frameshift mutation combined with homozygous c.1258A>G polymorphism in SPINK5 gene. Genes (Basel). (2023) 14(5):1080. doi: 10.3390/genes14051080
11. Renner ED, Hartl D, Rylaarsdam S, Young ML, Monaco-Shawver L, Kleiner G, et al. Comèl-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol. (2009) 124:536–43. doi: 10.1016/j.jaci.2009.06.009
12. Eränkö E, Ilander M, Tuomiranta M, Mäkitie A, Lassila T, Kreutzman A, et al. Immune cell phenotype and functional defects in Netherton syndrome. Orphanet J Rare Dis. (2018) 13:213. doi: 10.1186/s13023-018-0956-6
13. Smith DL, Smith JG, Wong SW, and deShazo RD. Netherton’s syndrome: a syndrome of elevated IgE and characteristic skin and hair findings. J Allergy Clin Immunol. (1995) 95:116–23. doi: 10.1016/S0091-6749(95)70159-1
14. Paller AS, Renert-Yuval Y, Suprun M, Esaki H, Oliva M, Huynh TN, et al. An IL-17-dominant immune profile is shared across the major orphan forms of ichthyosis. J Allergy Clin Immunol. (2017) 139:152–65. doi: 10.1016/j.jaci.2016.07.019
15. Czarnowicki T, He H, Leonard A, Malik K, Magidi S, Rangel S, et al. The major orphan forms of ichthyosis are characterized by systemic T-cell activation and Th-17/Tc-17/Th-22/Tc-22 polarization in blood. J Invest Dermatol. (2018) 138:2157–67. doi: 10.1016/j.jid.2018.03.1523
16. Cury Martins J, Martins C, Aoki V, Gois AF, Ishii HA, and da Silva EM. Topical tacrolimus for atopic dermatitis. Cochrane Database Syst Rev. (2015) 2015:Cd009864. doi: 10.1002/14651858.CD009864.pub2
17. Bin Saif G and Al-Khenaizan S. Netherton syndrome: successful use of topical tacrolimus and pimecrolimus in four siblings. Int J Dermatol. (2007) 46:290–4. doi: 10.1111/j.1365-4632.2006.02956.x
18. Volc S, Maier L, Gritsch A, Aichelburg MC, and Volc-Platzer B. Successful treatment of Netherton syndrome with ustekinumab in a 15-year-old girl. Br J Dermatol. (2020) 183:165–7. doi: 10.1111/bjd.18892
19. Morizane S, Ouchida M, Sunagawa K, Sugimoto S, Kobashi M, Sugihara S, et al. Analysis of all 34 exons of the SPINK5 gene in Japanese atopic dermatitis patients. Acta Med Okayama. (2018) 72:275–82. doi: 10.18926/AMO/56073
20. Ding Y, Zhou L, Xia Y, Wang W, Wang Y, Li L, et al. Reference values for peripheral blood lymphocyte subsets of healthy children in China. J Allergy Clin Immunol. (2018) 142:970–973.e978. doi: 10.1016/j.jaci.2018.04.022
21. Zhang Z, Pan C, Wei R, Li H, Yang Y, Chen J, et al. Netherton syndrome caused by compound heterozygous mutation, c.80A>G mutation in SPINK5 and large-sized genomic deletion mutation, and successful treatment of intravenous immunoglobulin. Mol Genet Genomic Med. (2021) 9:e1600. doi: 10.1002/mgg3.1600
22. Walley AJ, Chavanas S, Moffatt MF, Esnouf RM, Ubhi B, Lawrence R, et al. Gene polymorphism in Netherton and common atopic disease. Nat Genet. (2001) 29:175–8. doi: 10.1038/ng728
23. Di WL, Hennekam RC, Callard RE, and Harper JI. A heterozygous null mutation combined with the G1258A polymorphism of SPINK5 causes impaired LEKTI function and abnormal expression of skin barrier proteins. Br J Dermatol. (2009) 161:404–12. doi: 10.1111/j.1365-2133.2009.09231.x
24. Furio L, Pampalakis G, Michael IP, Nagy A, Sotiropoulou G, and Hovnanian A. KLK5 inactivation reverses cutaneous hallmarks of netherton syndrome. PloS Genet. (2015) 11:e1005389. doi: 10.1371/journal.pgen.1005389
25. Cork MJ, Danby SG, Vasilopoulos Y, Hadgraft J, Lane ME, Moustafa M, et al. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol. (2009) 129:1892–908. doi: 10.1038/jid.2009.133
26. Descargues P, Deraison C, Prost C, Fraitag S, Mazereeuw-Hautier J, D'Alessio M, et al. Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J Invest Dermatol. (2006) 126:1622–32. doi: 10.1038/sj.jid.5700284
27. Blunder S, Hermann-Kleiter N, Mahmuti R, Hermann M, Ortner D, Reider D, et al. Blocking of IL-4/IL-13 signalling with dupilumab results in restoration of serum and cutaneous abnormalities in netherton syndrome. Exp Dermatol. (2025) 34:e70113. doi: 10.1111/exd.70113
28. Komatsu N, Saijoh K, Jayakumar A, Clayman GL, Tohyama M, Suga Y, et al. Correlation between SPINK5 gene mutations and clinical manifestations in Netherton syndrome patients. J Invest Dermatol. (2008) 128:1148–59. doi: 10.1038/sj.jid.5701153
29. Hannula-Jouppi K, Laasanen SL, Ilander M, Furio L, Tuomiranta M, Marttila R, et al. Intrafamily and interfamilial phenotype variation and immature immunity in patients with netherton syndrome and finnish SPINK5 founder mutation. JAMA Dermatol. (2016) 152:435–42. doi: 10.1001/jamadermatol.2015.5827
30. Petrova E, López-Gay JM, Fahrner M, Leturcq F, Villartay JD, Barbieux C, et al. Comparative analyses of Netherton syndrome patients and Spink5 conditional knock-out mice uncover disease-relevant pathways. Commun Biol. (2024) 7:152. doi: 10.1038/s42003-024-05780-y
31. Mahajan R, Bakshi S, Kumar A, De D, and Handa S. Case report: Interleukin-17 targeted biological therapy in netherton syndrome. Front Pediatr. (2023) 11:1297658. doi: 10.3389/fped.2023.1297658
32. Yalcin AD. A case of netherton syndrome: successful treatment with omalizumab and pulse prednisolone and its effects on cytokines and immunoglobulin levels. Immunopharmacol Immunotoxicol. (2016) 38:162–6. doi: 10.3109/08923973.2015.1115518
33. Semple BD, Kossmann T, and Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab. (2010) 30:459–73. doi: 10.1038/jcbfm.2009.240
34. Kalliolias GD and Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
35. Zhang Y, Zeng W, and Xia Y. TWEAK/Fn14 axis is an important player in fibrosis. J Cell Physiol. (2021) 236:3304–16. doi: 10.1002/jcp.30089
Keywords: SPINK5, Netherton syndrome, IVIG, immune modulation, immunophenotyping
Citation: Guan Y, Li Q, Liu Y, Zhang P, Huang M, Guo Y, Chen J, Chen Y, Du Z and Huang P (2025) Clinical and immunological characterization of a Netherton syndrome infant with a large SPINK gene cluster deletion and a c.1258A>G polymorphism in SPINK5. Front. Immunol. 16:1658444. doi: 10.3389/fimmu.2025.1658444
Received: 02 July 2025; Accepted: 29 August 2025;
Published: 25 September 2025.
Edited by:
Suneel Kumar, The State University of New Jersey, United StatesReviewed by:
Satomi Igawa, Asahikawa Medical University, JapanPeiguang Wang, Anhui Medical University, China
Copyright © 2025 Guan, Li, Liu, Zhang, Huang, Guo, Chen, Chen, Du and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Chen, Y3l6NjAwQDE2My5jb20=; Zuochen Du, ZHpjOTAzNkAxMjYuY29t; Pei Huang, ZmVuZ2xpbjQ2MjBAMTYzLmNvbQ==
†These authors have contributed equally to this work