 Ji Li1†
Ji Li1† Jing Zhang1†
Jing Zhang1† Sheng-Guang Li1*‡Qian Guo1,2
Sheng-Guang Li1*‡Qian Guo1,2 Jing Xu1,2
Jing Xu1,2 Lina Zhang1Yadan Zou1
Lina Zhang1Yadan Zou1 Ting Long1Ruohan Yu1Yanfeng Zhang1
Ting Long1Ruohan Yu1Yanfeng Zhang1- 1Department of Rheumatology and Immunology, Peking University International Hospital, Beijing, China
- 2Department of Rheumatology and Immunology, Peking University Shougang Hospital, Beijing, China
Background: Long-standing rheumatoid arthritis (RA) complicated by anti–glomerular basement membrane (anti-GBM) disease is exceptionally rare.
Case: A 71-year-old man with long-standing seropositive RA developed a rapidly progressive glomerulonephritis due to anti-GBM disease, without any known drug triggers. Despite plasmapheresis (therapeutic plasma exchange), corticosteroids, and low-dose cyclophosphamide, he remained dialysis-dependent; RA activity was subsequently controlled with tocilizumab. Complications included Stenotrophomonas maltophilia pneumonia, COVID-19 and cytomegalovirus infection, and he died of pneumonia eight months after diagnosis.
Conclusion: This case highlights the need for early serological testing for anti-GBM disease in RA patients with unexplained hematuria/proteinuria and for immunosuppressive therapy mindful of infection risk. Additionally, our literature review identified only ten reported cases of RA with anti-GBM disease, highlighting the rarity of this condition.
Introduction
Rheumatoid arthritis (RA) is a common systemic autoimmune disease that only infrequently causes severe glomerulonephritis. Renal involvement in RA has been documented in approximately 1–5% of patients, usually stemming from medication toxicity (e.g., NSAIDs, gold, penicillamine) or secondary amyloidosis rather than primary vasculitis (1, 2). In contrast, anti-glomerular basement membrane (anti-GBM) disease is an exceptionally rare cause of rapidly progressive glomerulonephritis (annual incidence ~0.6–1.8 cases per million) (3, 4). Without prompt intervention, anti-GBM disease can quickly progress to irreversible end-stage kidney disease (ESKD) and can be potentially fatal. True anti-GBM disease occurring secondary to RA is extraordinarily uncommon—only a handful of cases have been reported—yet when present it poses significant diagnostic and therapeutic challenges.
Here we describe a rare case of anti-GBM disease secondly to a long-standing seropositive RA in a 71-year-old man. Notably, this occurred without typical medication triggers. The presentation was an RPGN that progressed to irreversible kidney failure despite aggressive therapy. We also review the existing literature on RA with anti-GBM disease to explore potential pathogenic links (such as medication-induced immune dysregulation and intrinsic autoimmune propensity) and discuss the complexities of management in this scenario. This case underscores the need for a high index of suspicion for anti-GBM disease in RA patients with unexplained renal signs, as early recognition and intervention are critical for optimizing outcomes.
Case presentation
A 71-year-old male with a 30-year history of poorly controlled seropositive RA was admitted in May 2024 with severe fatigue, anorexia, and oliguria that had progressed over two months. His RA had been characterized by chronic, erosive polyarthritis affecting the hands, wrists, elbows, shoulders, and ankles, leading to significant joint deformities and bilateral elbow contractures.
Past RA treatments included methotrexate (discontinued years prior due to gastrointestinal intolerance), low dose leflunomide (10 mg daily, ongoing), and intermittent use of Tripterygium wilfordii (a Chinese traditional herbal immunosuppressant). He also frequently took NSAIDs for pain management. Despite these therapies, his RA disease activity remained high, with persistent inflammation and deforming joint damage.
Routine laboratory tests five months before admission (December 2023) had already shown early renal abnormalities, specifically: proteinuria (+ on dipstick), gross hematuria (+++), and elevated inflammatory markers (C-reactive protein [CRP] 20.9 mg/L and erythrocyte sedimentation rate [ESR] 79 mm/hour). Unfortunately, these findings were overlooked at the time – they were attributed to a possible urinary tract infection or NSAID use, and no further nephrological evaluation was pursued. The patient did not report any urinary symptoms then, and follow-up was missed amid his ongoing RA issues. This represented a missed opportunity for earlier diagnosis.
By May 2024, the patient’s condition worsened dramatically. He developed marked fatigue, loss of appetite, and his urine output dwindled to <400 mL/day (oliguria). On May 16, 2024, laboratory tests showed a serum creatinine of 1038 µmol/L (baseline ~80 µmol/L in late 2023). Over just a few days, his creatinine spiked further to 1920 µmol/L by May 21. He also exhibited severe uremic symptoms (nausea, confusion) and electrolyte disturbances (potassium 6.3 mmol/L, sodium 125 mmol/L) consistent with AKI. Arterial blood gas revealed metabolic acidosis. He was anemic (hemoglobin 117 g/L, which later dropped further) and had profound hypoalbuminemia (20.9 g/L). Inflammatory markers were extremely elevated (CRP >186 mg/L, ESR >140 mm/hour), and NT-proBNP was >35,000 pg/mL, reflecting fluid overload and possible cardiac strain (Table 1).
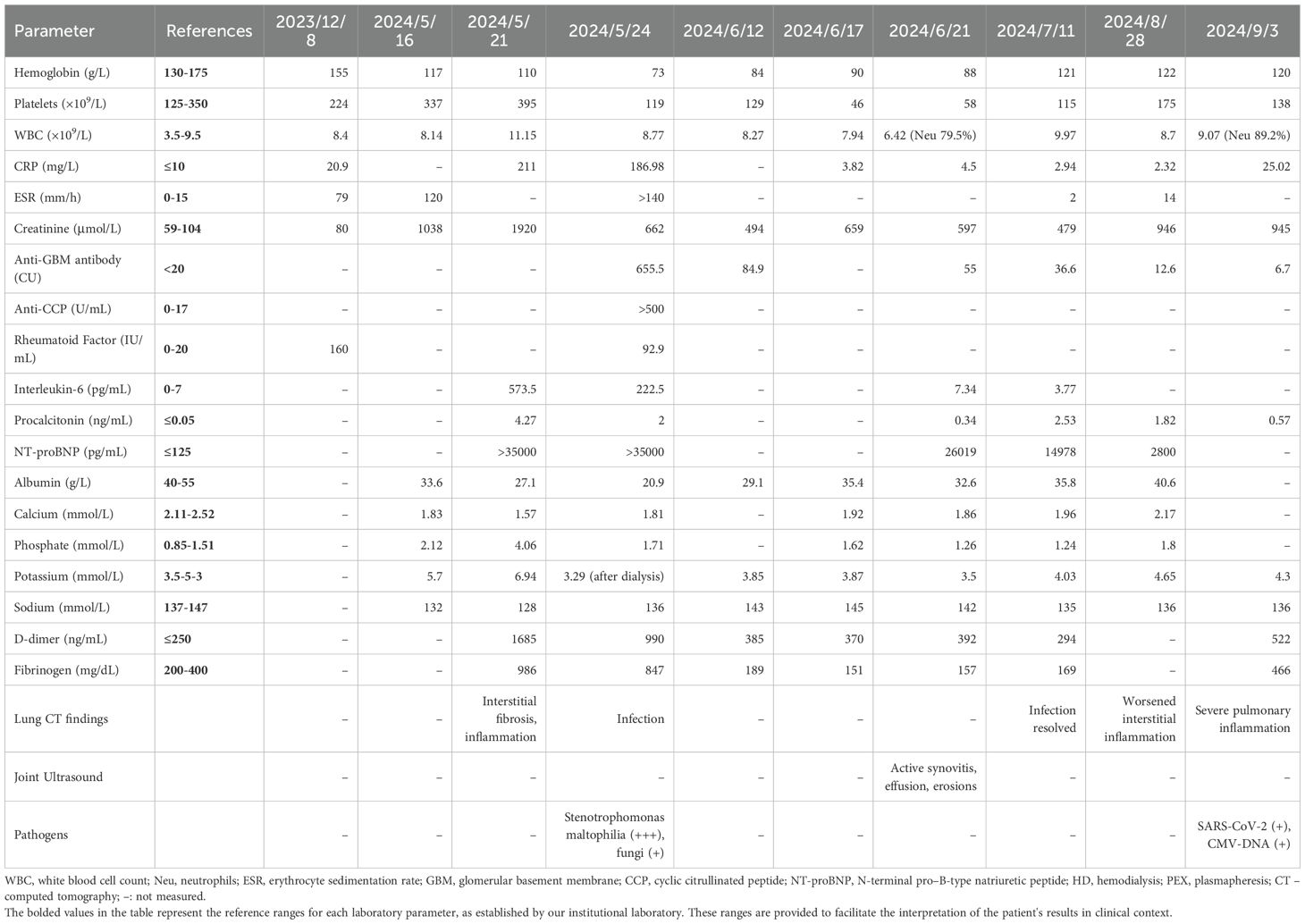
Table 1. Key laboratory and imaging results at critical clinical stages of the present case (RA with anti-GBM disease).
A chest computed tomography (CT) scan on admission showed bilateral interstitial infiltrates and fibrosis, suggestive of RA-associated interstitial lung disease (ILD) versus infection (no discrete lobar consolidation was seen). There were no radiological signs of diffuse alveolar hemorrhage (such as widespread ground-glass opacities), and the patient did not report hemoptysis. Given his critical state, we did not perform bronchoscopy; however, the absence of hemoptysis and the pattern of CT changes made anti-GBM disease-related pulmonary hemorrhage unlikely. The interstitial changes were attributed to chronic RA lung involvement, with superimposed pneumonia also considered. Additionally, a renal ultrasound at admission showed no evidence of obstruction (no hydronephrosis), making a post-renal cause of hematuria unlikely. The urinalysis findings (including red blood cell casts) further supported a glomerular source of bleeding rather than a urologic cause.
On admission, the patient was in critical condition. He was hypotensive (BP 85/50 mmHg) and tachypneic, with signs of fluid overload (marked bilateral leg edema) and uremic encephalopathy (asterixis and confusion). He had basal crackles on lung auscultation (consistent with fluid/inflammatory infiltrates), but no frank pulmonary hemorrhage. Joint exam showed active synovitis in multiple joints, consistent with an ongoing RA flare (he had painful swelling in the wrists, knees, and ankles despite his advanced joint deformities).
Emergency hemodialysis was initiated on admission to manage AKI and metabolic derangements. Empiric broad-spectrum antibiotics were started (meropenem) for possible sepsis. We also obtained extensive immunological tests given the unexplained RPGN. Strikingly, results revealed a very high titer of circulating anti-GBM antibodies (655.5 Chemiluminescence Units; normal <20 CU). Anti-neutrophil cytoplasmic antibodies (ANCA) were negative. RA serologies remained strongly positive (anti-CCP >500 U/mL, rheumatoid factor 92.9 IU/mL), reflecting active disease. Given the combination of oliguric RPGN and a high anti-GBM antibody titer, a diagnosis of anti-GBM disease was made. No renal biopsy was performed – the patient was hemodynamically unstable and coagulopathic, and he (and his family) did not consent to an invasive biopsy. We acknowledge that lacking histology is a limitation; however, the clinical presentation and serology were considered confirmatory in this emergency setting.
Microbiological studies aided in clarifying the lung findings: sputum cultures grew Stenotrophomonas maltophilia (a multi-drug-resistant Gram-negative organism) at heavy growth, and fungal cultures grew Candida species. These results indicated a severe hospital-acquired pneumonia with fungal co-infection. The patient was started on targeted antimicrobial therapy (intravenous trimethoprim-sulfamethoxazole for S. maltophilia, which also served as Pneumocystis jirovecii prophylaxis, and an echinocandin antifungal for Candida coverage). We also implemented prophylactic measures appropriate for his immunosuppressed state – for example, trimethoprim-sulfamethoxazole was continued prophylactically to prevent Pneumocystis pneumonia and other opportunistic infections, and fluconazole was given to prevent fungal thrush. Infection control was a major ongoing concern throughout his treatment.
A multidisciplinary team (nephrology, rheumatology, pulmonology, intensive care) coordinated the next steps. We initiated plasmapheresis on hospital day 2, given the classic indication for anti-GBM disease. He underwent a total of eight plasmapheresis sessions (replacing plasma with albumin) over the next two weeks. Concurrently, we started high-dose corticosteroids (initially IV methylprednisolone 40 mg daily, then tapering to oral prednisone 40 mg/day after one week). For additional immunosuppression, we carefully introduced cyclophosphamide – however, considering the patient’s advanced age and active infections, we used a cautious low-dose regimen (50 mg every other day orally) instead of the standard high-dose protocol. This adjusted dosing was chosen to balance treating the anti-GBM disease with avoiding further immunocompromise.
Remarkably, after about 6 plasmapheresis sessions, the patient’s anti-GBM antibody titer dropped from 655.5 CU to 84.9 CU. After the full 8 plasmapheresis (by early June 2024), the titer further declined to 55 CU (just above the upper limit of normal). Given this near normalization of anti-GBM antibody levels and economic concern, plasmapheresis was concluded after the eighth exchange to minimize additional immunosuppressive exposure and infection risk in this elderly patient. (At follow-up visits in July, August, and September 2024, his anti-GBM antibody titers further decreased to 36.6, 12.6, and 6.7 CU, respectively, as shown in Table 1 and Figure 1). This serological response indicated that our therapy was successful in removing circulating autoantibodies. Clinically, the patient’s pulmonary status improved: follow-up chest imaging in June showed resolution of the pneumonia infiltrates, although the underlying interstitial lung fibrosis had progressed (likely due to RA-ILD). Importantly, he never developed any signs of alveolar hemorrhage during hospitalization, suggesting our timely interventions may have averted the pulmonary manifestations of anti-GBM disease.
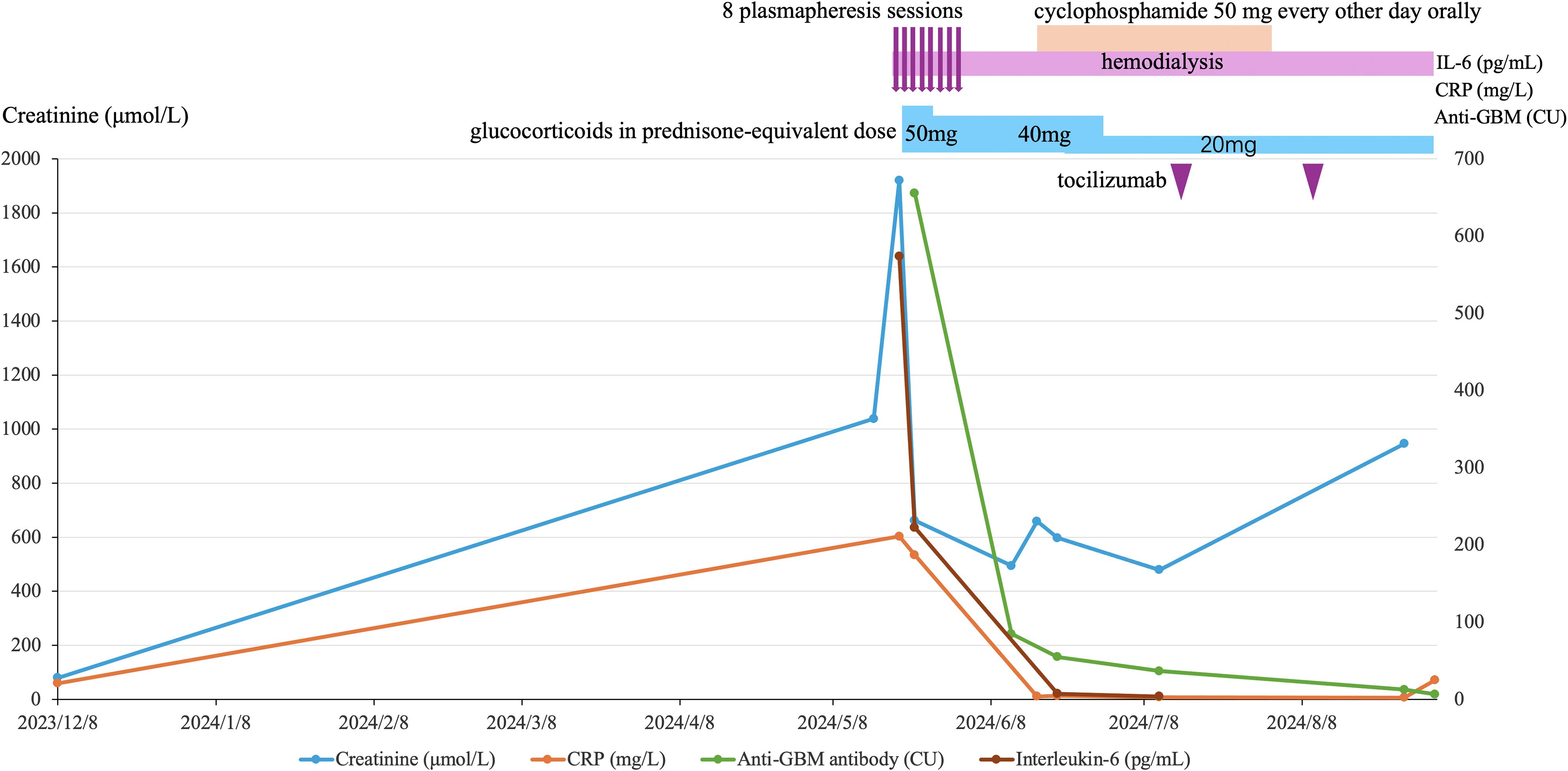
Figure 1. Monthly timeline of key laboratory markers and therapies in anti-GBM disease secondary to long-standing rheumatoid arthritis. From the first evidence of renal injury (Dec 2023) to Aug 2024, the figure plots four markers: serum creatinine (blue, left y-axis), anti-GBM antibody titer (green, right y-axis), CRP (orange, right y-axis), and IL-6 (brown, right y-axis). Treatment overlays above the plot denote 8 plasmapheresis sessions (mid–late May 2024), ongoing hemodialysis, glucocorticoid taper (prednisone-equivalent 50 mg → 40 mg → 20 mg), a short course of oral cyclophosphamide (50 mg every other day), and two tocilizumab infusions. After initiation of plasmapheresis and immunosuppression, anti-GBM, CRP and IL-6 declined sharply, whereas creatinine peaked and then only partially improved, remaining elevated in keeping with dialysis dependence. Curves connect measured time points to illustrate trends.
Despite clearing the anti-GBM antibodies from the circulation, the renal damage was already irreversible at presentation. The patient remained dialysis-dependent; his urine output did not recover (consistent with advanced RPGN and essentially complete ESKD by the time therapy began). We continued intermittent hemodialysis three times per week as chronic renal replacement therapy.
By mid-June, as the acute phase of the anti-GBM disease stabilized, we faced active RA issues. With steroid tapering (we reduced prednisone to 20 mg/day by late June), the patient developed a flare of his arthritis – swelling and pain recurred in multiple joints (particularly knees and shoulders). Considering his refractory RA and contraindications to some other therapies, we started tocilizumab, an IL-6 receptor inhibitor, for its efficacy in RA and relative safety in infection-prone patients. After obtaining clearance from the infectious disease team, tocilizumab 8 mg/kg IV was administered (with doses planned every 4 weeks). This led to a notable improvement in his joint symptoms and inflammatory markers over the next month. We also administered local therapy (intra-articular steroid injections to his shoulders and knees) to provide additional relief during the flare.
During the course of treatment, we were vigilant about prophylaxis and monitoring for opportunistic infections. Despite these efforts, the patient’s recovery was complicated by several serious infections due to his prolonged immunosuppression: in July 2024 he tested positive for COVID-19 (with moderate pneumonia), and shortly thereafter he developed reactivation of cytomegalovirus (CMV) (confirmed by PCR, with CMV viremia and hepatitis). He was treated with supportive care and antivirals (remdesivir for COVID-19, then ganciclovir for CMV). Cyclophosphamide was promptly discontinued by this time (after ~6 weeks of therapy) to help immune recovery, and we managed him with low-dose prednisone plus tocilizumab for RA thereafter. The patient initially recovered from both COVID-19 and CMV infections by August 2024, thanks to aggressive antimicrobial therapy and supportive ICU care.
Unfortunately, on 3 January 2025, approximately eight months after the anti-GBM diagnosis, the patient suffered a new episode of severe pneumonia (unrelated to COVID-19, likely bacterial). Despite broad-spectrum antibiotics and maximal supportive therapy, his condition deteriorated rapidly, and he passed away in the hospital due to respiratory failure. The cause of death was determined to be overwhelming pneumonia on a background of immunosuppression and end-stage renal disease.
This case timeline is visually summarized in Figure 1. It underscores how an initial oversight of renal warning signs led to a cascade of critical illness. Despite timely use of plasmapheresis and immunosuppression once diagnosed (which potentially prevented pulmonary hemorrhage), the delay resulted in permanent dialysis dependence and set the stage for fatal infectious complications. Earlier recognition and intervention might have altered the outcome, highlighting a crucial lesson for clinicians managing complex patients with autoimmune diseases.
Discussion
The coexistence of RA and anti-GBM disease is exceedingly rare. Through our updated review, we identified eight previously reported RA with anti-GBM cases worldwide (six full reports and two conference abstracts). We conducted a search of PubMed, Embase, and Web of Science, together with forward/backward citation tracking and other open resources (e.g., CrossRef/Google Scholar), from inception to September 2025, which identified 292 records (PubMed n = 32; Embase n = 200; Web of Science n = 59; other resources n = 1). After removing 26 duplicates, 266 records were screened; eleven reports underwent full-text assessment, one was excluded because the biopsy confirmed fibrillary GN with negative anti-GBM serology, and ten studies were included, as shown in Figure 2. Each reported case displayed unique features, underscoring substantial heterogeneity in this overlap (5–14). Our patient adds another documented instance to this limited body of evidence and highlights several points that may refine clinical understanding.
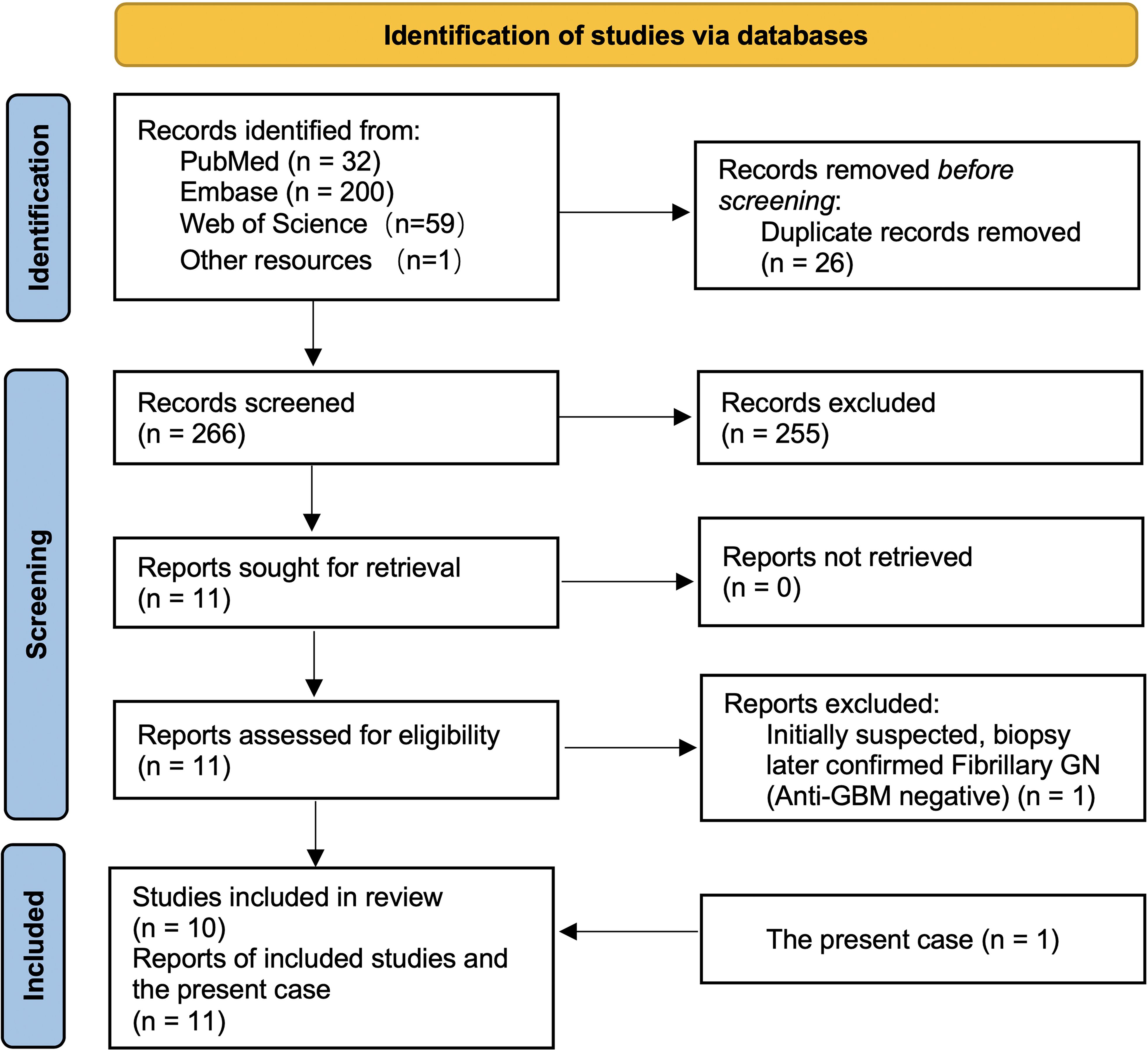
Figure 2. PRISMA flow of study selection for RA complicated by anti-GBM disease. A database search (PubMed = 32, Embase = 200, Web of Science = 59, other sources = 1) identified 292 records. After removing 26 duplicates, 266 records were screened and 255 excluded. Eleven full-text reports were assessed (none unretrieved); one was excluded after biopsy confirmed fibrillary GN with negative anti-GBM. Ten studies were included in the review, and together with the present case (n = 1) yielded eleven reports in total.
Medication triggers vs. intrinsic autoimmunity
Among the ten RA cases, five appear drug-related. One followed D-penicillamine (Peces et al., 1987) (5); one followed leflunomide (Bruyn et al., 2010) (8); and three occurred after adalimumab therapy for RA — reported by Nishimura et al (2012) (9), Sánchez et al (2015) (10), and Heron et al (2020) (11) (see Table 2). Outside RA, Al-Chalabi et al (2021). described an anti-GBM disease in a patient with psoriatic arthritis receiving etanercept (15). Even so, TNF-inhibitor–associated reports remain very few relative to the widespread use of these agents, and experimental models suggest TNF-α blockade may even attenuate anti-GBM nephritis (16–18); therefore, any causal link should be interpreted with caution. By contrast, our patient had no exposure to penicillamine or any TNF inhibitor. He had taken leflunomide and intermittently Tripterygium wilfordii, with occasional NSAIDs; given that leflunomide has been linked to anti-GBM disease in a prior RA case, a drug contribution cannot be excluded but remains unproven.
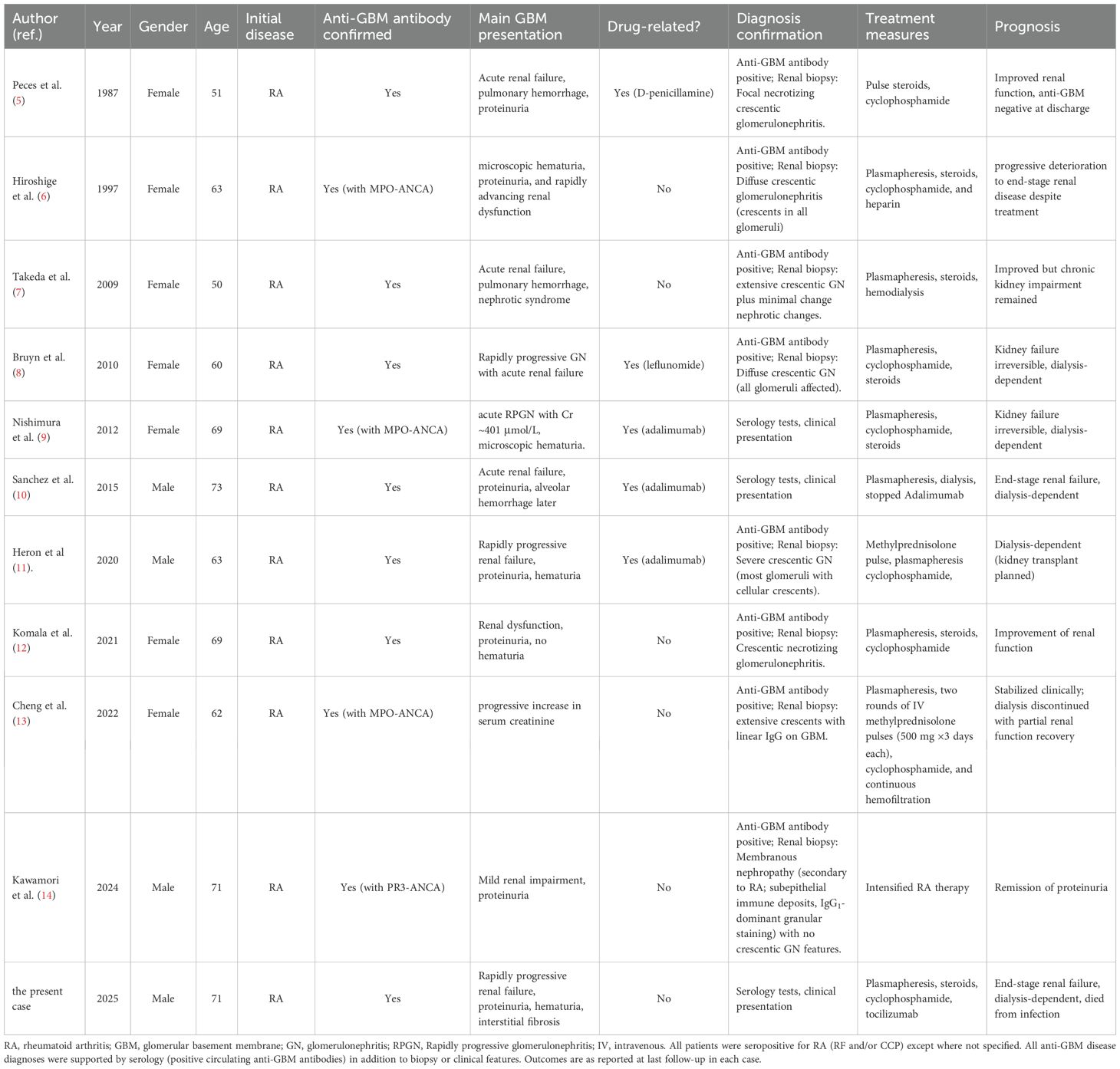
Table 2. Clinical characteristics, diagnosis, treatment, and prognosis of patients with rheumatoid arthritis complicated by anti-GBM disease.
Active RA and renal complications
Active RA itself can cause renal complications (e.g., secondary AA amyloidosis from chronic inflammation), and some RA treatments – notably long-term NSAIDs or certain disease-modifying drugs like gold or penicillamine – are known to have nephrotoxic effects (19, 20). These factors might contribute to mild chronic kidney impairment, but they would not typically provoke the kind of fulminant glomerulonephritis observed in our patient. Indeed, his AKI was far more severe than what NSAIDs or amyloid alone would usually cause, and it correlated with a very high anti-GBM antibody titer, indicating that anti-GBM disease was the primary culprit. This aligns more with other cases where no clear trigger was identified: for example, Takeda et al. (2009) documented idiopathic anti-GBM disease in an RA patient without any drug trigger or ANCA, suggesting that intrinsic immune dysregulation within RA could be enough to generate anti-GBM antibodies (7). Our case supports this idea – that long-standing RA itself, with chronic immune activation, might rarely set the stage for anti-GBM autoimmunity. Potential mechanisms discussed in the literature include genetic predisposition (e.g., certain HLA alleles confer risk for RA and other for anti-GBM) and epitope spreading from prolonged inflammation (where cryptic GBM antigens might be exposed during RA-related tissue damage, inciting a new autoimmune response). While speculative, these mechanisms underscore that RA patients – especially those with uncontrolled inflammation – could harbor an environment conducive to secondary autoimmune phenomena.
Clinical variability and diagnostic challenges
The reported RA–anti-GBM cases have shown variable clinical presentations. Some, like Komala et al. (2021), had atypical features such as absence of hematuria or pulmonary hemorrhage, making diagnosis less straightforward (12). Another case by Kawamori et al. (2024) was double-positive for anti-GBM and ANCA and, interestingly, had a renal biopsy showing membranous nephropathy rather than classic crescentic GN. This underscores that histology can vary and overlap with other pathologies; it also emphasizes that a renal biopsy is invaluable when feasible, to delineate the pathology (something our case regrettably lacked due to the patient’s condition) (14).
Among the 11 cases in our series, 4 patients (36.4%) were ANCA-positive (1 PR3-ANCA and 3 MPO-ANCA), and only 3 cases (27.3%) exhibited pulmonary hemorrhage, whereas the remaining 8 cases (including the present case) had no lung hemorrhage, as summarized in Table 2. This trend is consistent with the meta-analysis by Huang et al., which reported a pulmonary hemorrhage rate of 32.6% in anti-GBM disease (3), indicating that roughly one-third of such patients develop this complication.
Our case presented as a pulmonary-renal syndrome in terms of multi-system involvement, but notably without alveolar hemorrhage. The patient’s respiratory findings were due to infection and RA-ILD rather than any anti-GBM disease–related pulmonary hemorrhage. We cannot overemphasize the importance of thoroughly evaluating such patients for lung hemorrhage: in our patient, careful assessment of imaging and clinical signs (with input from pulmonology) was crucial to confirm that the lungs were “innocent bystanders” apart from infection/RA, allowing us to focus on treating the renal crisis. Epidemiologically, most anti-GBM patients have renal involvement, and approximately two-thirds (≈67%) present without pulmonary hemorrhage, i.e., with a renal-limited course (3).
Diagnostically, our case teaches a cautionary lesson. The patient had early clues (hematuria, proteinuria) that were missed, illustrating how concurrent chronic disease (RA) can distract from recognizing a new, superimposed condition. We reflected on why the warning signs were overlooked: the patient’s mild symptoms in late 2023 did not prompt him to seek extra help, and his providers at the time attributed abnormal labs to benign causes (e.g., a possible UTI or NSAID effect) without pursuing nephrology consult or antibody testing. This represents an instance of anchoring bias – assuming everything is due to the known RA, thereby “anchoring” on that diagnosis and not fully evaluating out-of-character findings.
Pathogenetically, possible links between RA and anti-GBM disease include medication-induced immune dysregulation, genetic susceptibility (HLA alleles), and chronic RA with inflammation possibly exposing cryptic GBM antigens (21). However, detailed genetic studies remain lacking, and further research is needed to clarify the underlying mechanisms definitively.
Prognosis and outcomes
Clinically, prognosis has varied significantly among reported cases, heavily influenced by the severity of initial renal dysfunction, pulmonary involvement, and infectious complications from immunosuppression. Earlier recognition, prompt serological and histological diagnosis, and aggressive plasmapheresis plus immunosuppression significantly influenced outcomes positively in reported cases (as noted in prior literature) (22).
Management challenges – immunosuppression vs. infection
Treating anti-GBM disease requires aggressive immunosuppression (per standard practice, a combination of plasmapheresis, high-dose corticosteroids, and cyclophosphamide). In our patient, this collided with the reality of an elderly host prone to infections and an already immunocompromised state from RA. Our management had to be highly individualized in the absence of any precedent or specific guidelines for this overlap. We followed general principles from KDIGO for the anti-GBM treatment (22), but adjusted: for instance, using a reduced cyclophosphamide dose due to the active infection at presentation. Even with careful dosing and eventually discontinuing cyclophosphamide early, the patient developed severe opportunistic infections. This underlines a key point: there are no established treatment guidelines for RA with anti-GBM disease, so clinicians must balance competing risks on a case-by-case basis. In our discussion, we emphasize that multidisciplinary input (nephrology deciding how aggressively to immunosuppress, rheumatology managing RA therapy, infectious disease overseeing prophylaxis) is essential. The lack of formal guidelines is a gap – all reported cases (including the present case) had different therapeutic approaches and outcomes, which we summarize in Table 2. Until more cases are studied, treatment remains an art of customization. We have highlighted this to illustrate how we tailored our patient’s treatment (for example, plasmapheresis frequency was adjusted based on antibody levels; cyclophosphamide was tapered off early; tocilizumab was chosen for RA control to avoid potential triggers).
Our case also illustrates the delicate timing of introducing RA therapy after the acute anti-GBM illness. We started tocilizumab once the patient’s condition allowed (after initial infections were treated) to address his active RA. This likely improved his quality of life (relieving arthritis) and helped reduce steroid exposure, but it may have contributed to persistent susceptibility to infections (IL-6 inhibition can also impair host defenses). It’s hard to say whether withholding RA therapy would have changed the infection outcome, since uncontrolled RA itself can cause debility and perhaps increase infection risk (through high inflammation and the need for corticosteroids). This scenario highlights the conundrum that managing two active autoimmune diseases simultaneously is a tightrope walk, and decisions must be continuously re-evaluated as the patient’s status evolves.
Prognosis
The prognosis in reported RA–anti-GBM cases has varied widely. Some patients (especially those with milder initial renal impairment or prompt treatment) survived with preserved kidney function. Others, like ours, had poor outcomes with dialysis dependence or death. A common theme is that the severity of initial renal dysfunction and any delays in therapy strongly influence outcomes. In our patient, by the time of diagnosis, his kidneys were virtually non-functional (creatinine ~1900 µmol/L, requiring immediate dialysis). Despite our aggressive measures halting further damage (and likely preventing pulmonary hemorrhage), we could not restore renal function. He remained on dialysis and ultimately succumbed to infection – an outcome that aligns with known observations that worse kidney function at diagnosis portends higher mortality and lower chance of renal recovery.
Mechanistic considerations and immunogenetics
Chronic RA inflammation may facilitate epitope spreading, whereby tissue injury exposes cryptic GBM epitopes and breaks tolerance. Genetically, anti-GBM disease is strongly associated with HLA-DRB115:01 (21), whereas RA is linked to “shared epitope” alleles such as *04:01 and *04:04 (23). These are distinct susceptibility alleles and should not be interpreted as a shared epitope across the two conditions. While a convergent impact on immune activation has been hypothesized, there is no evidence demonstrating a common antigen-presentation overlap between RA and anti-GBM. Complement and IL-6 pathways have been implicated in anti-GBM pathogenesis, supporting consideration of IL-6 blockade for RA control on a case-by-case basis; importantly, no causal link to TNF-α inhibitors has been established.
Infection prevention and monitoring under dual immunosuppression
For patients requiring combined anti-GBM therapy (plasmapheresis + high-dose corticosteroids + cyclophosphamide) and concurrent RA control, evidence-based strategies include: Pneumocystis prophylaxis with trimethoprim–sulfamethoxazole; up-to-date vaccination (influenza, pneumococcal, COVID-19) before or between treatment cycles; baseline screening and periodic PCR monitoring for CMV in high-risk or lymphopenic patients; judicious antifungal use with de-escalation once cultures clear; catheter care and early removal/exchange; growth-factor support and nutritional optimization; and dose/interval tailoring of cytotoxic agents.
Conclusion
In summary, this case underscores several key lessons. Clinicians caring for patients with rheumatoid arthritis should maintain a high level of vigilance for anti-GBM disease when otherwise unexplained hematuria or proteinuria is encountered. Once the diagnosis is established, plasmapheresis and immunosuppression need to be initiated as early as possible to maximize the chance of preserving renal function. At the same time, the coexistence of RA and anti-GBM disease demands an individualized approach that carefully balances the need for effective immunosuppression with the heightened risk of infection. Close collaboration between nephrology, rheumatology, and infectious disease specialists is essential to achieve optimal outcomes in such complex cases.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The requirement of ethical approval was waived by Biomedical Ethics Committee of Peking University International Hospital for the studies involving humans. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
JL: Conceptualization, Project administration, Writing – review & editing, Formal Analysis, Writing – original draft, Data curation, Investigation. JZ: Writing – review & editing, Investigation. S-GL: Methodology, Conceptualization, Writing – original draft, Supervision, Resources, Writing – review & editing, Formal Analysis, Investigation. QG: Writing – review & editing, Resources. JX: Resources, Writing – review & editing. LZ: Investigation, Writing – review & editing. YDZ: Investigation, Writing – review & editing. TL: Investigation, Writing – review & editing. RY: Investigation, Writing – review & editing. YFZ: Investigation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Galesić K, Prkacin I, Tisljar M, and Vergles JM. Renal involvement in patients with rheumatoid arthritis. Reumatizam. (2009) 56:30–5.
2. Vinicki JP, Pellet SC, De Rosa G, Dubinsky D, Laborde HA, Marini A, et al. Analysis of 65 renal biopsies from patients with rheumatoid arthritis (1976-2015): change in treatment strategies decreased frequency and modified histopathological findings. J Clin Rheumatol. (2015) 21:335–40. doi: 10.1097/RHU.0000000000000302
3. Kuang H, Jiang N, Jia XY, Cui Z, and Zhao MH. Epidemiology, clinical features, risk factors, and outcomes in anti-glomerular basement membrane disease: A systematic review and meta-analysis. Autoimmun Rev. (2024) 23:103531. doi: 10.1016/j.autrev.2024.103531
4. Zhang P, Shi KL, Gao CL, Xu F, Jia LL, and Sun JC. Complement in anti-glomerular basement membrane glomerulonephritis. Front Immunol. (2025) 16:3389/fimmu.2025.1442955. doi: 10.3389/fimmu.2025.1442955
5. Peces R, Riera JR, Arboleya LR, López-Larrea C, and Alvarez J. Goodpasture’s syndrome in a patient receiving penicillamine and carbimazole. Nephron. (1987) 45:316–20. doi: 10.1159/000184171
6. Hiroshige K, Soejima M, Yuu K, Muta T, Takasugi M, and Kuroiwa A. Report of a patient with rheumatoid arthritis developing rapidly progressive glomerulonephritis associated with anti-glomerular basement membrane antibody and antineutrophil cytoplasmic antibody. Clin Exp Nephrology. (1997) 1:62–6. doi: 10.1007/BF02480657
7. Takeda Y, Abe A, Toki T, Komaba H, Abe T, Umezu M, et al. A case of Goodpasture syndrome associated with minimal change nephrotic syndrome (MCNS) in a patient with rheumatoid arthritis (RA). Japanese J Nephrology. (2009) 51:897–903.
8. Bruyn GAW, Veenstra RP, Halma C, and Grond J. Anti-glomerular basement membrane antibody-associated renal failure in a patient with leflunomide-treated rheumatoid arthritis. Arthritis Rheumatism. (2010) 48:1164–5. doi: 10.1002/art.10891
9. Nishimura K, Saegusa J, Kawano S, and Morinobu A. Tumor necrosis factor-α inhibitor-induced antiglomerular basement membrane antibody disease in a patient with rheumatoid arthritis. J Rheumatol. (2012) 39:1904. doi: 10.3899/jrheum.120325
10. Sanchez CAC, Cordova HR, and Andujar KZ. Anti-tumor necrosis factor therapy - A trigger for anti-glomerular basement membrane disease. J Am Soc Nephrology. (2015) 26:351A.
11. Heron V, Nicholson M, Wilkinson S, Young A, Govindarajulu S, Stewart A, et al. Anti-glomerular basement membrane-antibody disease in a patient treated with adalimumab for rheumatoid arthritis. J Clin Sci Res. (2020) 9:124 – 7. doi: 10.4103/JCSR.JCSR_15_20
12. Komala MG, Bayly A, and Lin MW. Atypical presentation of anti GBM disease in an elderly woman with no hematuria and subnephrotic proteinuria. J Am Soc Nephrology. (2021) 32:819. doi: 10.1681/ASN.20213210S1819b
13. Cheng T, Zhi H, Liu Y, Zhang S, Song Z, and Li Y. Dual anti-glomerular basement membrane and anti-neutrophil cytoplasmic antibodies-positive rapidly progressive glomerulonephritis with rheumatoid arthritis and sjogren’s syndrome: A case report and literature review. J Clin Med. (2022) 11(22):6793. doi: 10.3390/jcm11226793
14. Kawamori S, Suzuki H, Lee M, Umezawa Y, Sasatsuki Y, Muto M, et al. A case of secondary membranous nephropathy caused by rheumatoid arthritis accompanied by double positive for anti-GBM antibodies and proteinase 3 (PR3)-ANCA. J Am Soc Nephrology. (2024) 35:1307. doi: 10.1681/ASN.20242807ya1e
15. Al-Chalabi S, Wu HHL, Chinnadurai R, and Ponnusamy A. Etanercept-induced anti-glomerular basement membrane disease. Case Rep IN Nephrol AND DIALYSIS. (2021) 11:292–300. doi: 10.1159/000518984
16. Khan SB, Cook HT, Bhangal G, Smith J, Tam FW, and Pusey CD. Antibody blockade of TNF-alpha reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int. (2005) 67:1812–20. doi: 10.1111/j.1523-1755.2005.00279.x
17. Karkar AM, Smith J, and Pusey CD. Prevention and treatment of experimental crescentic glomerulonephritis by blocking tumour necrosis factor-alpha. Nephrol Dial Transplant. (2001) 16:518–24. doi: 10.1093/ndt/16.3.518
18. Le Hir M, Haas C, Marino M, and Ryffel B. Prevention of crescentic glomerulonephritis induced by anti-glomerular membrane antibody in tumor necrosis factor-deficient mice. Lab Invest. (1998) 78:1625–31. doi: 10.1038/labinvest.3780132
19. Icardi A, Araghi P, Ciabattoni M, Romano U, Lazzarini P, and Bianchi G. Kidney involvement in rheumatoid arthritis. Reumatismo. (2003) 55:76–85. doi: 10.4081/reumatismo.2003.76
20. Peng Q, Wang G, and Li J. Commentary: Rheumatoid arthritis and the risk of end-stage renal disease: a nationwide, population-based study. Front Med (Lausanne). (2023) 10:3389/fmed.2023.1194649. doi: 10.3389/fmed.2023.1194649
21. McAdoo SP and Pusey CD. Anti-glomerular basement membrane disease. Clin J Am Soc Nephrol. (2017) 12:1162–72. doi: 10.2215/cjn.01380217
22. Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, et al. Executive summary of the KDIGO 2021 guideline for the management of glomerular diseases. Kidney Int. (2021) 100:753–79. doi: 10.1016/j.kint.2021.05.015
Keywords: rheumatoid arthritis, anti–glomerular basement membrane disease, rapidly progressive glomerulonephritis, autoimmune disease, case report
Citation: Li J, Zhang J, Li S-G, Guo Q, Xu J, Zhang L, Zou Y, Long T, Yu R and Zhang Y (2025) Rapidly progressive anti-GBM disease secondary to long-standing rheumatoid arthritis: a case report and literature review. Front. Immunol. 16:1661117. doi: 10.3389/fimmu.2025.1661117
Received: 07 July 2025; Accepted: 06 October 2025;
Published: 17 October 2025.
Edited by:
Huang Kuang, Peking University First Hospital, ChinaReviewed by:
Min Hui Tan, Hospital Kuala Lumpur, MalaysiaHashim Mohamed Siraj, Ivane Javakhishvili Tbilisi State University Faculty of Medicine, Georgia
Copyright © 2025 Li, Zhang, Li, Guo, Xu, Zhang, Zou, Long, Yu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheng-Guang Li, bGlzaGVuZ2d1YW5nQHZpcC4xNjMuY29t
†These authors have contributed equally to this work
‡ORCID: Sheng-Guang Li, orcid.org/0000-0002-8047-9984