 Blanka Wolszczak-Biedrzycka1*
Blanka Wolszczak-Biedrzycka1* Beata Cieślikiewicz1Filip Studniarz2Łukasz Dąbrowski3Mateusz Fąs2Krystyna Matyszkiewicz–Suchodolska2Monika Harasimowicz3
Beata Cieślikiewicz1Filip Studniarz2Łukasz Dąbrowski3Mateusz Fąs2Krystyna Matyszkiewicz–Suchodolska2Monika Harasimowicz3 Justyna Dorf4
Justyna Dorf4- 1Department of Psychology and Sociology of Health and Public Health, University of Warmia and Mazury in Olsztyn, Olsztyn, Poland
- 2The Oncology Center of the Region of Warmia and Mazury in Olsztyn, Clinical Hospital of the Ministry of the Interior and Administration, Olsztyn, Poland
- 3Medical Laboratory, “Diagnostyka” Olsztyn, Olsztyn, Poland
- 4Department of Clinical Laboratory Diagnostics, Medical University of Bialystok, Bialystok, Poland
Since the beginning of the COVID-19 pandemic, research has been ongoing to find the best diagnostic parameters to identify patients with a high risk of severe infection. Numerous studies have examined chemokine biomarkers in COVID-19 as a biomarker for high risk patients. The four main structural proteins of the SARS-CoV-2, spike protein, membrane protein, envelope protein and nucleocapsid protein enable the virus to penetrate host cells and stimulate the immune system. SARS-CoV-2 enters host cells via ACE2 in upper respiratory tract the virus entries by binding to the spike protein. Uncontrolled activation and enhancement of the immune response leads to massive release of cytokines and chemokines known as cytokine storm (CS). Chemokines are described as important cytokines in COVID-19 with a potential role as prognostic factor particularly for the severity of the infection and the risk of death from complications, to identify high-risk patients. Our review contains chemokines (CCL2, CCL3, CCL5, CXCL8, CXCL10), which level is significantly higher in patients with COVID-19 infection vs control individuals.
Introduction
From the beginning of the pandemic until July 2024, more than 775 million people worldwide have been diagnosed with COVID-19, and the virus claimed 7 million lives. In Poland, there were 6,517,494 confirmed cases of COVID-19 and 119,622 fatalities (1). The high incidence of severe infections and the high fatality rate prompted research into the pathogenesis and significance of the cytokine storm in COVID-19.
Overproduction of pro-inflammatory cytokines and chemokines contributes to pneumonia and, in extreme cases, acute respiratory distress syndrome (ARDS) (2). Research has shown that the levels of pro- and anti-inflammatory cytokines and chemokines are correlated with the severity of SARS-CoV-2 infection and the risk of death from complications (3). Normal or reduced white blood counts (WBC) and lymphocytopenia are observed in most patients with mild and moderate SARS-CoV-2 infection. However, in patients with severe COVID-19, the levels of circulating neutrophils, plasma D-dimer levels and serum urea levels are elevated, whereas lymphocyte counts are reduced (4). These patients are also characterized by higher levels of cytokines and chemokines, mainly interleukin 6 (IL-6), interleukin 10 (IL- 10), and tumor necrosis factor alpha (TNF-α) (5, 6). An increase in the levels of interleukin 2 (IL-2), interleukin 7 (IL-7), macrophage colony-stimulating factor (M-CSF), granulocyte colony-stimulating factor (G -CSF), granulocyte-macrophage colony-stimulating factor (GM- CSF), interferon gamma-induced protein 10 (IP-10), monocyte chemoattractant protein-1 (MCP-1), and macrophage inflammatory protein-1 alpha (MIP 1-α) has also been observed in patients with severe symptoms of infection who require hospitalization in an intensive care unit (ICU) (7–11). Last researches shown that the binding of CCR1 and CXCR6 to monocytes and CD8 T lymphocytes influence on the course of COVID-19. An elevated expression of genetics-risk genes, increase in inflammatory cytokines and chemokines (12–15).
Many COVID-19 patients still require hospitalization and die from complications, which is why new biomarkers that are potentially useful for diagnosing the disease in early stages of progression, monitoring treatment, and identifying patients at high risk of severe infection and death should be sought. Such biomarkers would enable clinicians do develop more detailed diagnostic protocols, more effective treatments and measures to prevent infection in patients at the highest risk of severe COVID-19.
This literature review summarizes the current knowledge on the diagnostic utility of chemokines as tools for monitoring the progression of COVID-19 and the health status of patients infected with SARS-CoV-2. Chemokine concentrations as inflammatory biomarkers could be used to more rapidly identify patients at high risk of severe infection and death.
Systematic review
The literature review was conducted in the PubMed database, and it involved articles published between 2020 and January 2024. The following keywords were used: COVID-19 and chemokines, SARS-CoV-2 virus and chemokines, COVID-19 and CCl2, COVID-19 and CCl3, COVID-19 and CCl5, COVID-19 and CXCl8, COVID-19 and CXCl10. The inclusion and exclusion criteria are presented in Table 1. The systematic review yielded 310 research articles in the Medline database (PubMed). Of those, 220 were rejected due to misleading titles. 90 abstracts were read, 22 were assessed for eligibility based on the adopted inclusion/exclusion criteria. 7 articles were unrelated to the studied topic. Ultimately, only 15 research articles were included in the study (Figure 1). The search and selection process are according to the PRISMA (16).

Table 1. Inclusion and exclusion criteria in the systematic review.
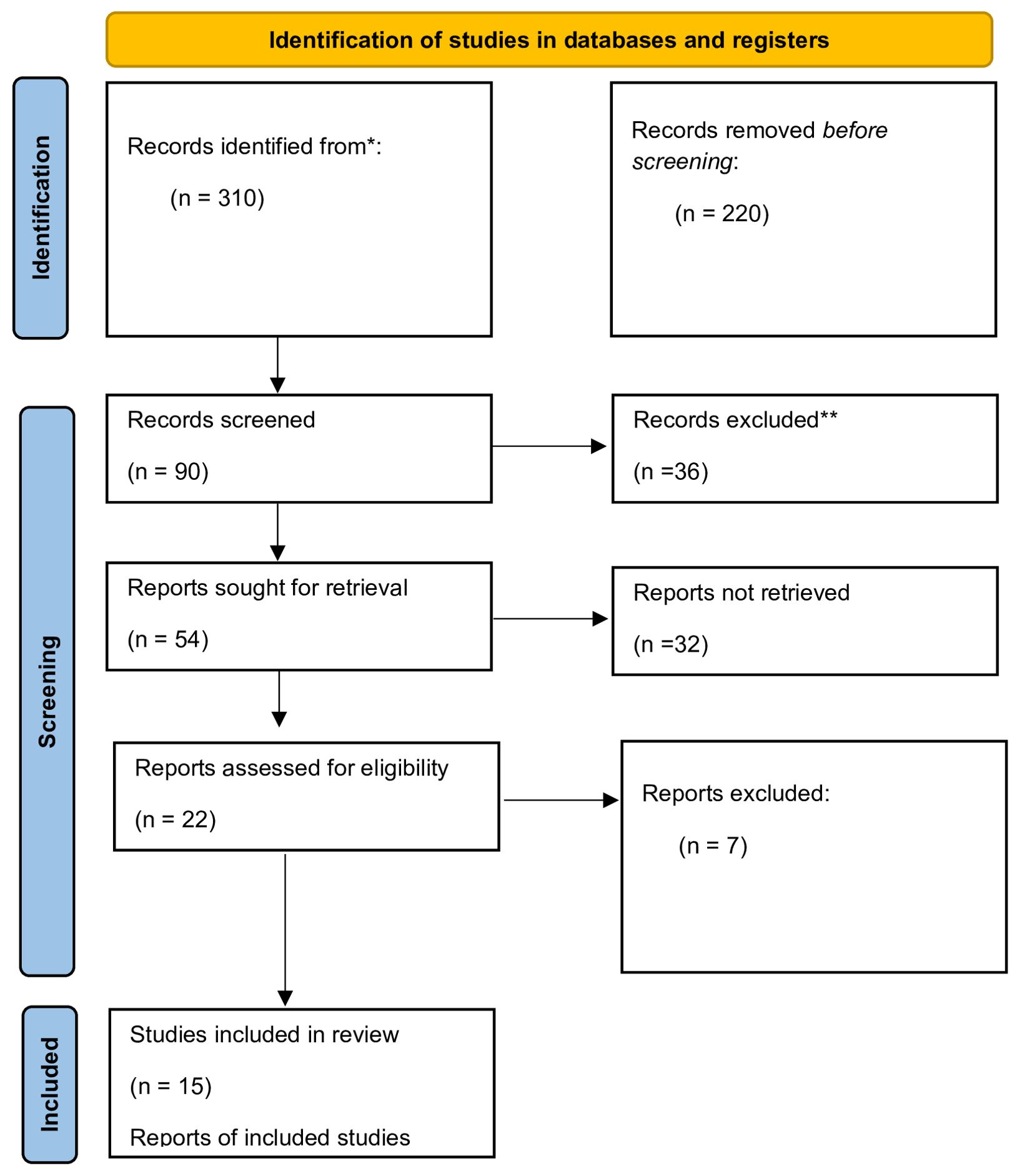
Figure 1. Flowchart of the systematic review process (Prisma).
Structure of SARS-CoV-2
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) belongs to a large and diverse family of Coronaviruses (Coronaviridae, CoV), which cause infections in animals - birds and mammals, including humans. SARS-CoV-2 is a β-coronavirus which, similarly to SARS-CoV and the Middle East Respiratory Syndrome Coronavirus (MERS-CoV), infects mammals. It is currently regarded as the most pathogenic virus in this group (17). SARS-CoV-2 and other Coronaviruses are spherical or oval particles with a lipid envelope containing proteins that enable the virus to penetrate host cells. Viral particles have a diameter of around 80–120 nm (18). The members of the Coronavirus family owe their name to the presence of spiculated surface glycoproteins that resemble a solar corona (19). SARS-CoV-2 is an enveloped, positive-sense ssRNA virus with a genome of ~29.9 kb. Its genetic material is composed of a positive-sense single-stranded RNA (ssRNA (+)) with an estimated length of 60–140 nm. The RNA contains around 30,000 nucleotides (20). The nucleotides are arranged into 14 open reading frames (ORFs) that represent two-thirds of information encoded within the virus’ genetic material and are responsible for the production of 16 non-structural proteins (NSPs). NSPs participate in the replication and transcription of new RNA molecules (21). The remaining one third of the genome encodes four major structural proteins (Figure 2) that play important roles in the pathogenesis of COVID-19, as well as accessory proteins that participate in virion-host cell interactions (22). The four major structural proteins that bind to host cells and elicit an immune response are the spike (S) protein - mediates receptor binding and membrane fusion, membrane (M) protein - shapes the envelope and assembly, envelope (E) protein - viroporin involved in assembly/release, and nucleocapsid (N) protein - packages the RNA genome (21). The S protein is a type I transmembrane glycoprotein that enables the virus to bind to cell membrane receptors and penetrate host cells. The S protein is composed of two functional subunits, S1 and S2. Subunit S1 binds to receptors on the surface of host cells, whereas S2 mediates the fusion of the viral envelope and the host cell membrane (Figure 1) (21). In mature SARS-CoV-2 particles, the S protein is assembled as a trimer with three binding domains that attach to the angiotensin-converting enzyme (ACE2) (23). Cryogenic electron tomography revealed the presence of around 30–40 trimeric S proteins on the surface of each viral particle in a pre-fusion state (24). The M protein is responsible for the shape of the viral envelope. Together with S and N proteins, the M protein interacts with the endoplasmic reticulum and the Golgi apparatus, where newly created virions are folded and released (25). The E protein is a viroporin, a small glycoprotein that speeds up the infectious process in the host organism. Protein-lipid pores are formed in the membranes of infected cells, and they can act as ion channels that increase membrane permeability and promote the release of progeny virions (26). The N protein binds, protects, and packages viral RNA to form ribonucleoprotein structures enclosed within the viral capsid (11).
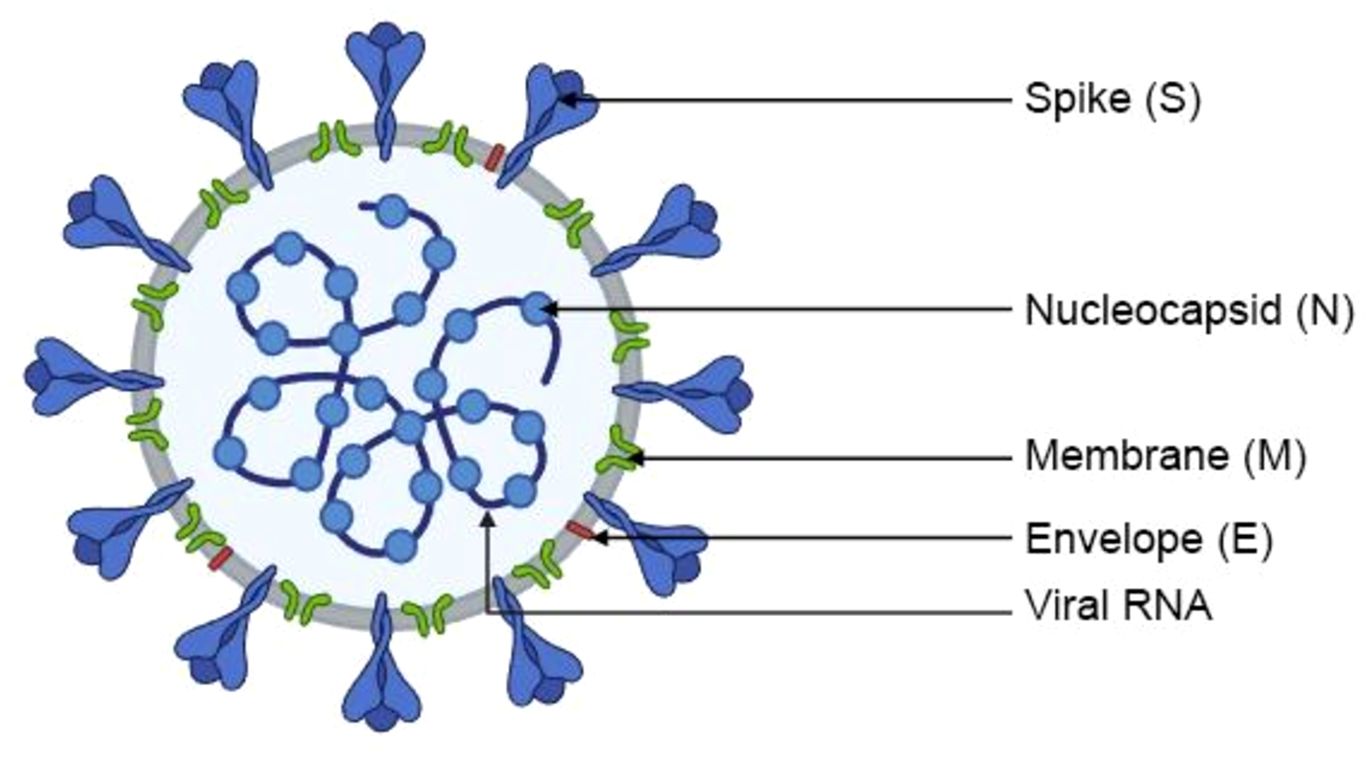
Figure 2. Structure of SARS-CoV-2.
Cell entry mechanism and life cycle of SARS-CoV-2
SARS-CoV-2 easily enters host cells, which contributes to its high infectivity and is a key element in the pathogenesis of COVID-19 (Figure 3) (25). The pathogen uses human ACE2 as the entry receptor. ACE2 catalyzes the conversion of angiotensin II into angiotensin 1–7 which has vasodilating, anti-inflammatory, anti-thrombotic, and anti-proliferative properties. Under physiological conditions, ACE2 is expressed on type I and type II pneumocytes (26). According to research, the binding affinity of SARS-CoV-2 to ACE2 is around 10–20 stronger in comparison with other coronaviruses. The ACE2 receptor is localized on epithelial cells in the upper respiratory tract and lungs, enterocytes, gallbladder and bile duct cells, renal tubule cells, skeletal muscle cells, myocardial cells, and vascular endothelial cells (27). The S protein undergoes conformational changes when it binds to the ACE2 receptor, which increases its sensitivity to the host’s proteolytic enzymes (28). In the first stage of viral entry into host cells, the S protein is cleaved into subunits S1 and S2, and the proteolytic cleavage process is mediated by enzymes such as transmembrane protease serine 2 (TMPRSS2) and 4 (TMPRSS4), cathepsin L, trypsin, furin, and human airway trypsin-like protease. The infectivity of SARS- CoV-2 is determined by the S protein’s ability to bind with ACE2 and undergo cleavage because these processes promote endocytosis and facilitate the fusion of viral and endosomal membranes. This mechanism enables the virion to enter the host cell. The pH inside endosomes is low, which additionally promotes the fusion of the viral envelope with the endosomal membrane (29).
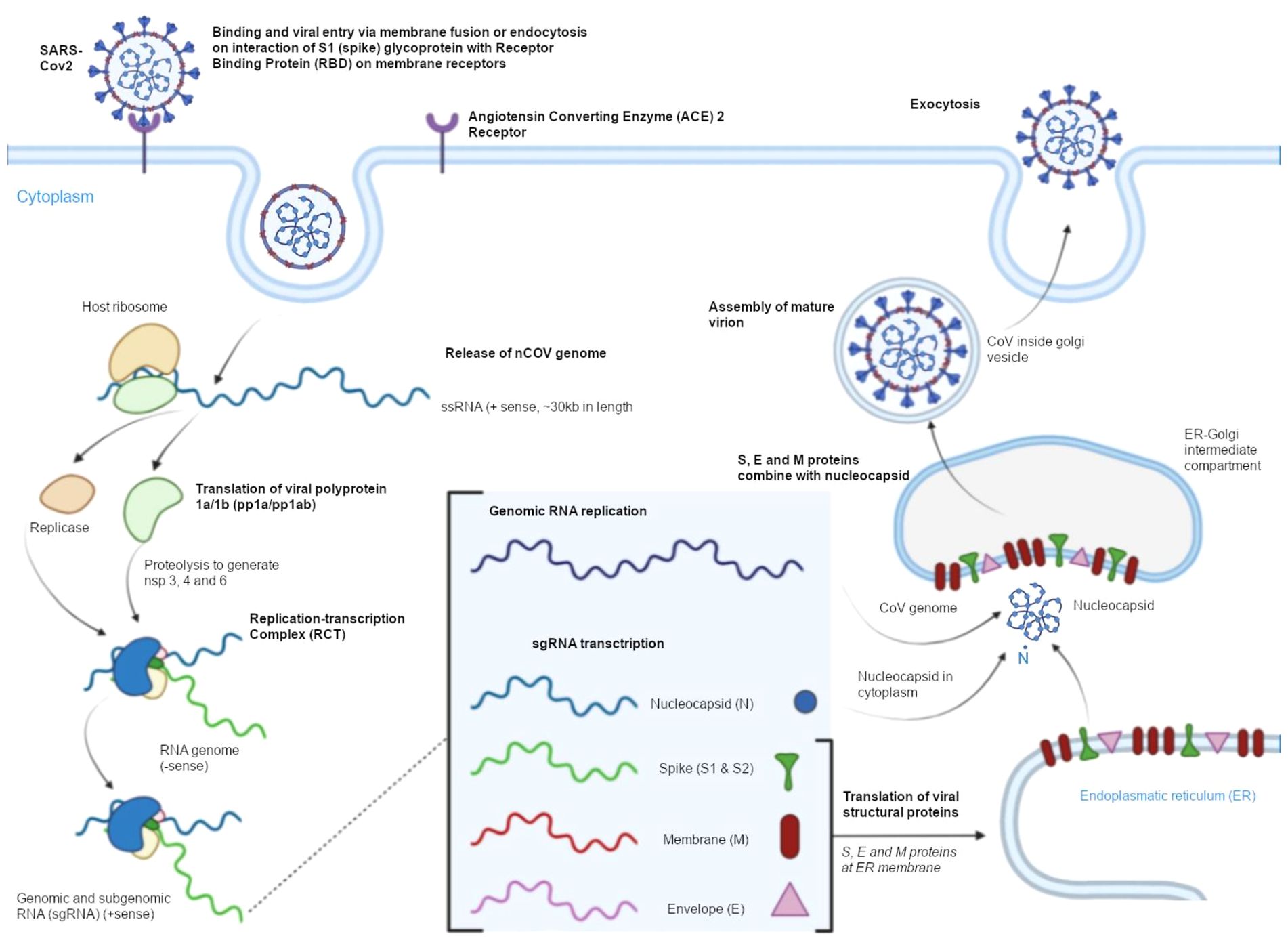
Figure 3. Cell entry mechanism and the life cycle of SARS-CoV-2.
In the following stage, viral RNA is released and directly incorporated into host cell ribosomes. The SARS-CoV-2 replicase is translated, the virus’ genetic information is transcribed and replicated, and structural proteins are translated. In the final stage, new virions are folded in the endoplasmic reticulum – Golgi intermediate compartment (ERGIC). Newly formed, mature SARS-CoV-2 particles are transported by Golgi-derived vesicles to the plasma membrane and released from the infected cell into extracellular space in a process known as exocytosis (30–32).
Inflammation caused by SARS-CoV-2
After entering host cells, viruses induce an inflammatory response by activating the immune system which is responsible for protecting the host organism and eliminating the pathogen. In viral infections, the innate (nonspecific) immune response is the first line of defense, which does not require prior contact with the pathogen (2).
Pattern/pathogen recognition receptors (PRRs) detect viruses in the human body. These receptors are found on the surface of cells or in the cytoplasm. PRRs are molecular alarms that sense pathogens in the host organism. They are present on the cells of the innate immune system, including neutrophils, monocytes, macrophages, dendritic cells, natural killer (NK) cells, endothelial cells, epithelial cells, and keratinocytes (33). These receptors recognize unknown viral protein patterns, activate immune cells, and trigger the release of pro- and anti- inflammatory cytokines (including IL-1, IL-6, IL-7, and TNF-α) and chemokines (including IP- 10). As a result, immune cells are rapidly recruited to the site of infection (3). PRRs have been divided into two types: receptors that are associated with the cell membrane, including toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic receptors, including NOD-like receptors (NLRs) and RIG-I-like receptors (RLRs) (34). The viral elements recognized by PRRs are known as pathogen-associated molecular patterns (PAMPs) which belong to a family of evolutionary conserved structural elements that are pathogen-specific and essential for the survival of pathogens. SARS-CoV-2 produces PAMPs such as ssRNA (detected by intracellular RLRs), the S protein and other envelope proteins (detected by TLRs). When SARS-CoV-2 is sensed, PRRs secrete interferons (IFNs), antiviral cytokines that play a key role in the immune response to viral infections. Type I-III IFNs participate in the primary (innate) immune response (35, 36). This group of cytokines includes IFN-α and IFN-β. Type I IFN binds to interferon-α/β receptors (INFAR) that are present on the surface of most cells. Transcription factors such as signal transducer and activator of transcription 1 (STAT1) are phosphorylated, which leads to the activation of interferon-stimulated genes (ISGs). ISGs participate in the inflammatory response and inhibit viral replication and transmission by slowing down cell metabolism, enhancing the secretion of pro-inflammatory cytokines, and increasing PRR expression (37, 38).
Viral entry and the resulting damage to host cells leads to the release of damage- associated molecular patterns (DAMPs) which are also recognized by PRRs. Adenosine triphosphate (ATP) and nucleic acids are the best-known DAMPs. Alarmins regulate the migration of macrophages, monocytes, and T cells to the site of infection and intensify the local inflammatory response by releasing pro-inflammatory cytokines. Excessive recruitment of immune cells causes tissue damage. In patients with severe COVID-19, the above can lead to lung inflammation observed in chest X-rays (39, 40).
SARS-CoV-2 has defense mechanisms, which prevent immune cells from sensing the virus and disrupt the host immune response. As a result, the virus can replicate and infect successive cells. Research has shown that SARS-CoV-2 inhibits IFN release, and in patients with severe COVID-19, IFN levels are lower than in patients with mild or moderate symptoms of the disease. SARS-CoV-2 also inhibits other antiviral innate immune responses by influencing PRRs and their signaling pathways (41, 42).
Cytokine storm
Patients with SARS-CoV-2 infection can experience a wide range of clinical manifestations. Some infections present with mild symptoms or no symptoms and do not require treatment. However, severe infections can be life-threatening and may require hospitalization (43). Severe symptoms of COVID-19 are caused by both local and systemic inflammatory responses and the overproduction of pro-inflammatory factors, a process that is known as the cytokine storm or the cytokine release syndrome (CRS) (44). The cytokine storm is caused by the uncontrolled activation and enhancement of the host’s immune system (45), which leads to the massive release of cytokines and chemokines (46).Neutrophils, macrophages, NK cells, Th1, Th2, Th9 and Th12 lymphocytes, cytotoxic lymphocytes (Tc) (47), and cytokines, including IL-1, IL-6, IL-12, IL-18, TNF and IFN-γ are implicated in the pathogenesis of the cytokine storm (48, 49). Neutrophils promote clotting and produce cytokines. Macrophages present antigens, participate in phagocytosis, and induce cytokine production, whereas NK cells kill target cells and participate in immunoregulatory processes. In turn, IL-1 and IFN-γ produced by Th1 lymphocytes recruit macrophages and dendritic cells, and IL-4, IL-5 and IL-13 secreted by Th2 lymphocytes recruit eosinophils and basophils. IL-9 and IL-1 produced by Th9 lymphocytes activate mast cells, whereas IL-17, IL- 21 and IL-22 secreted by Th17 lymphocytes regulate neutrophil production. Tc lymphocytes produce IFN-γ, perforins and granzymes that exert cytotoxic effects on cells infected with SARS-CoV-2 (50, 51). IFN-γ and macrophages trigger a strong inflammatory response which leads to the cytokine storm. IFN-γ, IL-1, IL-6, IL-18, and TNF play the key role during CRS and trigger symptoms such as fever, headache, dizziness, and fatigue. The activation of the complement system is regulated by cytokines, which can lead to an increase or a decrease in cytokine production. The clinical symptoms of CRS include high fever, lymphadenopathy, hepatosplenomegaly, cytopenia, hyperferritinemia, and disorders of the central nervous system (CNS) (Figure 4) (52).
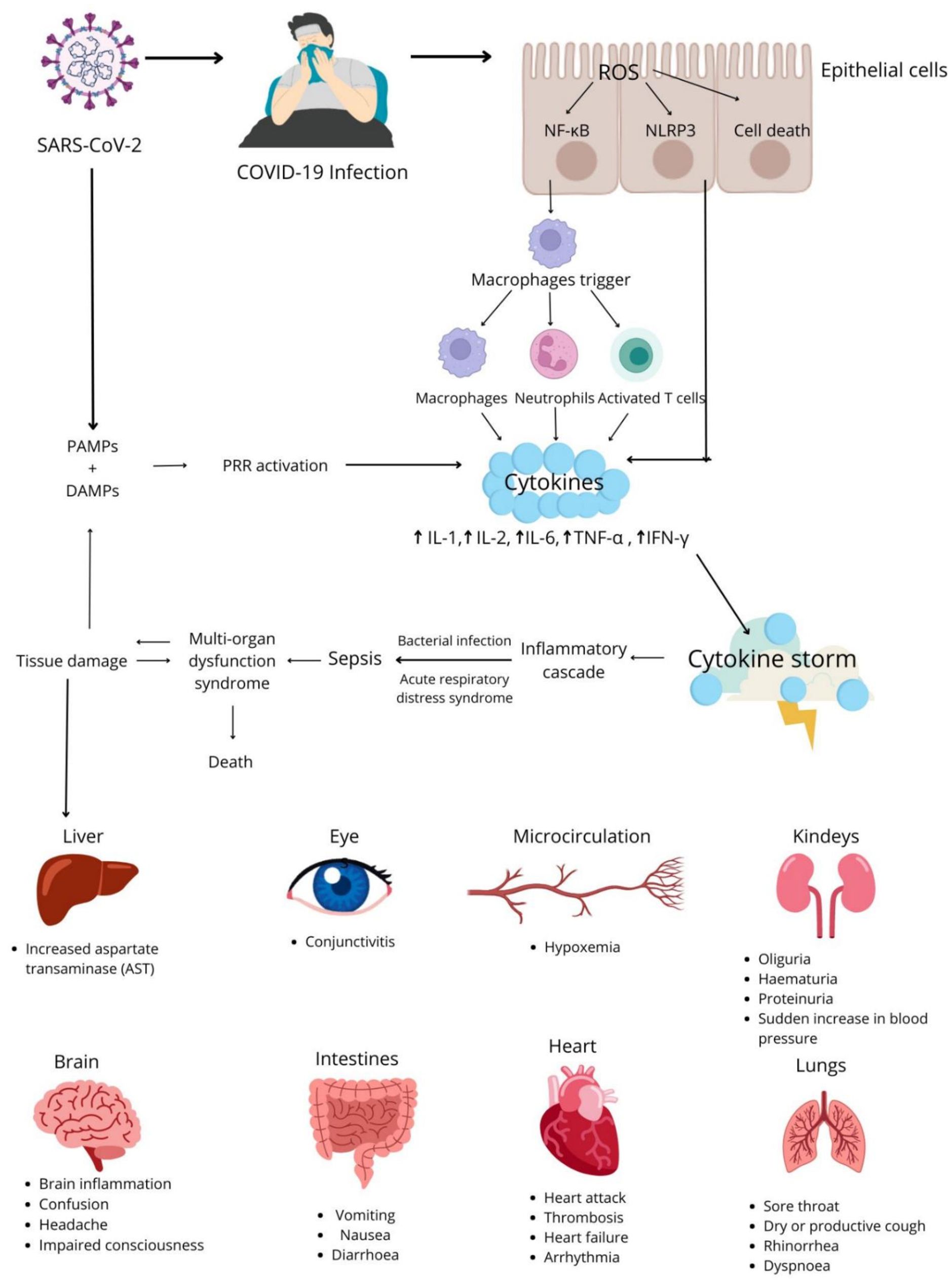
Figure 4. Pathomechanism of the cytokine storm and its consequences in COVID-19 patients.
Chemokines, in particular monocyte chemoattractant protein-1 (MCP-1/CCL2), macrophage inflammatory protein-1α (MIP-1α/CCL3), MIP-1β/CCL4, the regulated on activation, normal T-cell expressed and secreted (RANTES/CCL5) chemokine, monokine induced by γ-interferon (MIG/CXCL9), and IFN-gamma-inducible protein (IP-10/CXCL10), also play important roles in COVID-19 (53, 54). These proteins are secreted by monocytes, macrophages, dendritic cells, blood platelets, fibroblasts, granulocytes, and epithelial cells in response to the identified pathogens, including SARS-CoV-2 (55). Chemokines are expressed in inflamed tissues, and they influence the maturation and differentiation of immune cells, including T cells, neutrophils, eosinophils, and macrophages. They also stimulate the migration of immune cells to the inflammation site to prevent acute respiratory failure (56). Chemokines fight inflammation and participate in tissue healing in COVID-19 patients (57).
Potential complications of COVID-19 include pneumonia and, in severe cases, ARDS, which decreases blood oxygen levels and leads to multiorgan failure and septic shock. ARDS, caused among others by the cytokine storm, is one of the most common causes of death in COVID-19 patients (27, 58). Enhanced inflammatory responses in COVID-19 can also cause damage to many other organs and tissues. The substances released during the cytokine storm are filtered by renal tubules, which can lead to acute kidney injury (AKI) (59). In COVID-19 patients, CRS also contributes to liver and bile duct fibrosis, and it can cause damage to the nervous system by increasing the risk of encephalopathy and stroke. Research studies examining the impact of SARS-CoV-2 on the CNS produced ambiguous results. It is assumed that the virus can cause hypoxemia, the cytokine storm can exert an indirect effect of on the brain, or the virus can directly damage the brain provided that it is able to cross the blood-brain barrier (60).
Chemokines
Chemokines, or chemotactic cytokines, are a large group of proteins that are structurally homologous to cytokines, stimulate the movement of leukocytes and control their migration from the bloodstream to tissues. Chemokines participate in inflammatory processes and in the pathogenesis of many diseases, including COVID-19 (61). These polypeptides are composed of 66 to 111 amino acids with a tertiary structure stabilized by disulfide bonds between cysteines (62). Chemokines are functionally divided into constitutive chemokines and inducible chemokines that are formed under inflammatory conditions. Constitutive chemokines are produced in lymphoid tissues, including the bone marrow and the thymus, and they control basic migration processes and the development of immune system cells. Pro-inflammatory chemokines are secreted by various tissues and leukocytes in response to toxins and pro- inflammatory cytokines IL-1, TNFα, and IFNγ (63, 64). The chemokines described in this article belong to the group of inducible chemokines, and changes in their concentrations play an important role in different stages of SARS-CoV-2 infection (Supplementary Table 1).
CCL2 (monocyte chemoattractant protein-1 – MCP1)
CCL2 is a small chemokine of the CC chemokine family. It is encoded by a gene on chromosome 17q12 (65). CCL2 recruits monocytes and basophils to the site of infection. It stimulates basophils and mast cells to release granules into extracellular space. This chemokine regulates the anti-cancer activity of monocytes and participates in the formation of granulomas (66, 67). CCL2 also recruits mast cells in the progenitor stage and increases neutrophil accumulation. This chemokine stimulates fibroblasts to produce procollagen (68).
During SARS-CoV-2 infection, CCL2 is overproduced by macrophage-derived vesicles, T cells, and lung endothelial cells. Monocytes, the main immune system cells, are activated, and they infiltrate lungs and elicit a strong inflammatory response. Acute inflammation damages the lung endothelium and may lead to diffuse alveolar hemorrhage and fibroproliferative disorders in patients with ARDS. Increased expression of CCL2 is associated with an acute inflammatory response in early stages of infection, and with prolonged stay in the ICU (53).
CCL3 (macrophage inflammatory protein-1 alpha – MIP-1α)
CCL3, a chemokine of the CC family, is encoded by the CCL3 gene on chromosome 17q12 (69). This chemokine is secreted by macrophages and monocytes in response to the stimulation with bacterial endotoxin, as well as by the pro-inflammatory cytokine IL-1β. CCL3 can be also expressed by most hematopoietic cells, fibroblasts, endothelial cells, vascular smooth muscle cells, and activated blood platelets. This chemokine also acts as a ligand for receptors CCR1, CCR4, and CCR5 (70).
CCL3 contributes to inflammation through chemotaxis by directing the migration of inflammatory cells to tissues. It affects mainly monocytes, T cells, dendrocytes, NK cells, and blood platelets, and it activates granulocytes (65). This chemokine also induces the synthesis and release of other pro-inflammatory cytokines, including IL-1, IL-6, and TNF-α, from fibroblasts and macrophages (70).
Due to its functional properties, CCL3 plays an important role in viral infections, including COVID-19. A rapid and significant increase in CCL3 expression was observed in the blood serum of patients infected with SARS-CoV-2. The concentration of this chemokine was found to be higher in patients with moderate or severe COVID-19 than in asymptomatic patients, which suggests that CCL3 could be a useful biomarker for monitoring patients infected with SARS-CoV-2. It should also be noted that the pro-inflammatory effects of CCL3 could contribute to various pathological processes. This chemokine is implicated in the recruitment and activation/degranulation of granulocytes (in particular eosinophils), which can lead to an acute neutrophil-mediated inflammatory response and severe tissue damage (71).
CCL5 (RANTES)
CCL5 is a pro-inflammatory chemokine of the CC family, and it is encoded by the CCL5 gene localized on chromosome 17q12 (72). CCL5 is chemotactic for T cells, eosinophils, basophils, monocytes, NK cells, dendrocytes, and mast cells (73, 74). Under the influence of IL-2 and IFN-γ secreted by T cells, CCL5 induces the proliferation and activation of NK cells known as CC-Chemokine-activated killer (CHAK) cells. This chemokine also induces the expression of extracellular matrix metalloproteinases (MMPs) which regulate the migration of immune cells to the site of inflammation (75). CCL5 is expressed late (3–5 days) after T cell activation via TCR, and this unusual kinetic profile is important for the maintenance of inflammation.
The role of CCL5 in COVID-19 has not been fully elucidated to date, and research studies analyzing this chemokine have produced contradictory results. It is generally believed that CD8+ T cells specific for SARS-CoV-2 are the main source of CCL5 in early stages of infection. In addition, the CCL5/CCR5 axis inhibits the apoptosis of macrophages that play a key role in the pathophysiology of COVID-19. However, elevated levels of CCL5 can also cause acute kidney and liver failure. These contradictory findings could be attributed to differences in the study populations and the duration and methods of measurement. Therefore, CCL5 cannot be univocally regarded as a reliable prognostic marker in COVID-19, and further research is needed to elucidate its role in SARS-CoV-2 infection (54).
CXCL8 (interleukin 8 – IL-8)
CXCL8 is a pro-inflammatory chemokine which, similarly to other chemokines of the CXC family, is encoded by the CXCL8 gene on chromosome 4q13.3 (76). Macrophages and monocytes are the main sources of CXCL8, but this chemokine is also produced by other cells with toll-like receptors that participate in the innate immune response, as well as epithelial cells, airway smooth muscle cells, and endothelial cells. CXCL8 secretion is induced by factors IL- 17A and IL-17F which are produced by IL-6 and Th17 cells and whose levels are elevated in the peripheral blood of patients with SARS-CoV-2 infection (54). This chemokine binds mainly to receptors CXCR1 and CXCR2, and it shows higher affinity for CXCR1.
CXCL8 is implicated in the recruitment, activation, and accumulation of neutrophils. It induces chemotaxis, mainly in neutrophils, but also in other granulocytes. This chemokine also stimulates phagocytosis in the recruited cells. CXCL8 induces various physiological processes that stimulate adhesion to endothelial cells and migration to target tissues, including histamine release, increased expression of LFA-1 integrins on leukocyte membranes, increased concentration of intracellular Ca2+ ions, and the respiratory burst, a process in which reactive oxygen species (ROS) and digestive enzymes are activated to facilitate leukocyte migration to tissues (77). The activity of CXCL8 is strictly dependent on transcription factor AP-1 and associated with SARS-CoV-2 spike and nucleocapsid proteins. CXCL8 directly inhibits IFN induction by viral proteins, thus minimizing the anti-viral activity of IFN, in particular in early stages of infection (78). This chemokine also induces the production of neutrophil extracellular traps (NETs), strongly immunogenic and toxic networks of fibers that induce an inflammatory response and damage epithelial and endothelial cells. CXCL8 achieves these effects by stimulating exocytosis and the respiratory burst in neutrophils. In turn, NETs enhance the release of CXCL8, which increases the recruitment of neutrophils and prevents their apoptosis (54). Significantly elevated levels of CXCL8 were noted in patients who died from COVID-19 complications, which suggests that this chemokine can be used as a prognostic marker of mortality in patients infected with SARS-CoV-2 (51). IL-8 levels were found to be significantly higher in COVID-19 patients than in healthy subjects. In SARS-CoV-2 infection, uncontrolled production of IL-8, IL-2, IL-6, and TNF-α leads to the cytokine storm, disrupts immune system activity, and causes acute lung injury, ARDS (79–81), and multiorgan failure. Research has demonstrated that the risk of death from COVID-19 complications was positively correlated with an increase in IL-8 levels (82).
CXCL10 (interferon gamma-induced protein 10 – IP-10)
CXCL10, a chemokine of the CXC family, is encoded by the CXCL10 gene on chromosome 4q.21.1 It is secreted by various cells, including monocytes, endothelial cells, and fibroblasts, in response to interferon gamma (83–85). CXCL10 binds mainly to receptor CXCR3 (86–89). CXCL10 participates in the recruitment of monocytes, macrophages, T cells, NK cells, and dendritic cells, and increases T cell adhesion to endothelial cells. CXCL10 modulates anti- cancer immune responses and promotes the inhibition of angiogenesis (90).
CXC10L is the key chemokine that participates in the TRL4-TRIF signaling pathway which is implicated in the pathogenesis of lung injury during viral infections. This chemokine contributes to the apoptosis of T cells and, consequently, lymphopenia in SARS-CoV-2 infection. Despite the fact that CXCL10 lacks the ELR sequence, it plays an important role in pulmonary neutrophil infiltration, which intensifies its production. The CXCL10/CXCR3 axis affects the recruited neutrophils and causes the respiratory burst, which contributes to inflammation and ARDS. CXCL10 is a reliable prognostic marker for risk assessment in patients infected with SARS-CoV-2 (86, 87).
Conclusions
This literature review indicates that the immune response elicited by SARS-CoV-2 leads to the activation of various chemokines. The observed changes in chemokine levels are associated mainly with severe COVID-19, a poor prognosis, and a higher risk of mortality due to complications. The most recent research findings concerning chemokines’ role in the pathogenesis and progression of SARS-CoV-2 infection were summarized.
Author contributions
BW-B: Writing – review & editing, Supervision, Conceptualization, Validation, Project administration. BC: Writing – original draft. FS: Writing – original draft. ŁD: Writing – original draft. MF: Writing – original draft. KM–S: Writing – original draft, Software. MH: Writing – original draft. JD: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1662643/full#supplementary-material
Glossary
ACE2: angiotensin-converting enzyme
AKI: Acute kidney injury
ARDS: Acute respiratory distress syndrome
ATP: Adenosine triphosphate
CHAK: CC-Chemokine-activated killer
CLRs: C-type lectin receptors
CNS: Central nervous system
CoV: Coronaviruses (Coronaviridae)
CRS: Cytokine release syndrome
CSF: Cerebral spine fluid
DAMPs: Damage-associated molecular patterns
ERGIC: Endoplasmic reticulum – Golgi intermediate compartment
G -CSF: granulocyte colony-stimulating factor
GM-CSF: granulocyte-macrophage colony-stimulating factor
ICU: Intensive care unit
IFN-γ: Interferon gamma
IFNs: Interferons
IL-1: Interleukin 1
IL-2: Interleukin 2
IL-4: Interleukin 4
IL-5: Interleukin 5
IL-6: interleukin 6
IL-7: interleukin 7
IL-9: interleukin 9
IL-10: Interleukin 10
IL-12: Interleukin 12
IL-13: Interleukin 13
IL-17: Interleukin 17
IL-17A: Interleukin 17A
IL-17F: Interleukin 17F
IL-18: Interleukin 18
IL-21: Interleukin 21
IL-22: Interleukin 22
INFAR : Interferon-α/β receptors
IP-10: Interferon gamma-induced protein 10, CXCL10
ISGs: Interferon-stimulated genes
M-CSF: Macrophage colony-stimulating factor
MCP-1: Monocyte chemoattractant protein-1, CCL2
MERS-CoV: Middle East Respiratory Syndrome Coronavirus
MIP-1α: Macrophage inflammatory protein-1α, CCL3
MIP-1β: Macrophage inflammatory protein-1β, CCL4
MMPs: Extracellular matrix metalloproteinases
NETs: Neutrophil extracellular traps
NK: Natural killer (cells)
NLRs: NOD-like receptors
NSPs: Non-structural proteins
ORFs: Open reading frames
PAMPs: Pathogen-associated molecular patterns
PRRs: Pattern/pathogen Pecognition receptors
RF: Respiratory failure
RLRs: RIG-I-like receptors
RNA: Ribonucleid acid
ROS: Reactive oxygen species
SARS-CoV-2: Severe Acute Respiratory Syndrome Coronavirus 2
SOFA: Sequential organ failure assessment
ssRNA: Single-stranded RNA
STAT1: Signal transducer and activator of transcription 1
Tc: Cytotoxic lymphocytes
Th1: T helper1 lymphocyte
Th2: T helper2 lymphocyte
Th9: T helper9 lymphocyte
Th12: T helper12 lymphocyte
TLRs: Toll-like receptors
TMPRSS2: Transmembrane protease serine 2
TMPRSS4: Transmembrane protease serine 4
TNF-α: Tumor necrosis factor alpha
WBC: White blood counts
References
1. COVID-19 Data Explorer - Our World in Data . Available online at: https://ourworldindata.org/explorers/coronavirusdata-explorer (Accessed July 17, 2024).
2. Kunnumakkara AB, Rana V, Parama D, Banik K, Girisa S, Henamayee S, et al. COVID-19, cytokines, inflammation, and spices: How are they related? Life Sci. (2021) 284. doi: 10.1016/j.lfs.2021.119201
3. Pum A, Ennemoser M, Adage T, and Kungl AJ. Cytokines and chemokines in SARS-CoV-2 infections— therapeutic strategies targeting cytokine storm. Biomolecules. (2021) 11. doi: 10.3390/biom11010091
4. Toori KU, Qureshi MA, and Chaudhry A. Lymphopenia: A useful predictor of COVID-19 disease severity and mortality. Pak J Med Sci. (2021) 37:1984. doi: 10.12669/PJMS.37.7.4085
5. Wolszczak-Biedrzycka B. Changes in chemokine and growth factor levels may be useful biomarkers for monitoring disease severity in COVID-19 patients; a pilot study. Front Immunol. (2024) 14:1320362. doi: 10.3389/fimmu.2023.1320362
6. Wolszczak-Biedrzycka B, Dorf J, Wojewódzka-żelezniakowicz M, Żendzian-Piotrowska M, Dymicka-Piekarska VJ, Matowicka-Karna J, et al. Unveiling COVID-19 secrets: harnessing cytokines as powerful biomarkers for diagnosis and predicting severity. J Inflammation Res. (2023) 16:6055–70. doi: 10.2147/JIR.S439217
7. Dymicka-Piekarska V, Dorf J, Milewska A, Łukaszyk M, Kosidło JW, Kamińska J, et al. Neutrophil/lymphocyte ratio (NLR) and lymphocyte/monocyte ratio (LMR)– risk of death inflammatory biomarkers in patients with COVID-19. J Inflammation Res. (2023) 16. doi: 10.2147/jir.s409871
8. Wolszczak-Biedrzycka B, Dorf J, Milewska A, Łukaszyk M, Naumnik W, Kosidło JW, et al. The diagnostic value of inflammatory markers (CRP, IL6, CRP/IL6, CRP/L, LCR) for assessing the severity of COVID-19 symptoms based on the MEWS and predicting the risk of mortality. J Inflammation Res. (2023) 16. doi: 10.2147/jir.s406658
9. Kosidło JW, Wolszczak-Biedrzycka B, Dymicka-Piekarska V, Dorf J, and Matowicka-Karna J. Clinical significance and diagnostic utility of NLR, LMR, PLR and SII in the course of COVID-19: A literature review. J Inflammation Res. (2023) 16:539–62. doi: 10.2147/JIR.S395331
10. Eissa M, Shaarawy S, and Abdellateif MS. The role of different inflammatory indices in the diagnosis of COVID-19. Int J Gen Med. (2021) 14:7843–53. doi: 10.2147/IJGM.S337488
11. Pedersen SF and Ho YC. SARS-CoV-2: A storm is raging. J Clin Invest. (2020) 130. doi: 10.1172/JCI137647
12. Yunlong M, Fei Q, and Chunyu D. Integrating single-cell sequencing data with GWAS summary statistics reveals CD16+monocytes and memory CD8+T cells involved in severe COVID-19. Genome Med. (2022) 14:16. doi: 10.1186/s13073-022-01021-1
13. Yunlong M, Chunyu D, and Yijun Z. Polygenic regression uncovers trait-relevant cellular contexts through pathway activation transformation of single-cell RNA sequencing data. Cell Genom. (2023) 3:100383. doi: 10.1016/j.xgen.2023.100383
14. Stephenson E, Reynolds G, and Botting RA. Single-cell multi-omics analysis of the immune response in COVID-19. Nat Med. (2021) 27:904–16. doi: 10.1038/s41591-021-01329-2
15. Ji-Yuan Z, Xiang-Ming W, and Xing X. Single-cell landscape of immunological responses in patients with COVID-19. Nat Immunol. (2020) 21:1107–18. doi: 10.1038/s41590-020-0762-x
16. Shamseer L, Moher D, and Clarke M. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ. (2015) 7647. doi: 10.1136/bmj.g7647
17. Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, et al. Molecular architecture of the SARS-coV-2 virus. Cell. (2020) 183. doi: 10.1016/j.cell.2020.09.018
18. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. (2021) 19:409–24. doi: 10.1038/s41579-021-00573-0
19. Bakhshandeh B, Jahanafrooz Z, Abbasi A, Goli MB, Sadeghi M, Mottaqi MS, et al. Mutations in SARS-CoV-2; Consequences in structure, function, and pathogenicity of the virus. Microb Pathog. (2021) 154. doi: 10.1016/j.micpath.2021.104831
20. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, and Veesler D. Structure, function, and antigenicity of the SARS-coV-2 spike glycoprotein. Cell. (2020) 181. doi: 10.1016/j.cell.2020.02.058
21. Harrison AG, Lin T, and Wang P. Mechanisms of SARS-coV-2 transmission and pathogenesis. Trends Immunol. (2020) 41. doi: 10.1016/j.it.2020.10.004
22. Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the Origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. (2020) 7. doi: 10.1093/nsr/nwaa036
23. Klein S, Cortese M, Winter SL, Wachsmuth-Melm M, Neufeldt CJ, Cerikan B, et al. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat Commun. (2020) 11. doi: 10.1038/s41467-020-19619-7
24. Pawlik L, Śpiołek E, Fichna J, and Aleksandra Tarasiuk I. Charakterystyka wirusa SARS-CoV-2 i potencjalne farmakologiczne sposoby leczenia. Postepy Biochem. (2020) 66:83–90. doi: 10.18388/PB.2020_321
25. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. (2020) 34:327–31. doi: 10.23812/CONTI-E
26. Zheng M. ACE2 and COVID-19 susceptibility and severity. Aging Dis. (2022) 13. doi: 10.14336/AD.2021.0805
27. Zhang X, Li S, and Niu S. ACE2 and COVID-19 and the resulting ARDS. Postgrad Med J. (2020) 96. doi: 10.1136/postgradmedj-2020-137935
28. Takeshita H and Yamamoto K. Tryptophan metabolism and COVID-19-induced skeletal muscle damage: is ACE2 a key regulator? Front Nutr. (2022) 9:868845. doi: 10.3389/fnut.2022.868845
29. Piva F, Sabanovic B, Cecati M, and Giulietti M. Expression and co-expression analyses of TMPRSS2, a key element in COVID-19. Eur J Clin Microbiol Infect Dis. (2021) 40. doi: 10.1007/s10096-020-04089-y
30. Wang Y and Gandy S. The Golgi apparatus: Site for convergence of COVID-19 brain fog and Alzheimer’s disease? Mol Neurodegener. (2022) 17. doi: 10.1186/s13024-022-00568-2
31. Scherer KM, Mascheroni L, Carnell GW, Wunderlich LCS, Makarchuk S, Brockhoff M, et al. SARS-CoV-2 nucleocapsid protein adheres to replication organelles before viral assembly at the Golgi/ERGIC and lysosome-mediated egress. Sci Adv. (2022) 8. doi: 10.1126/sciadv.abl4895
32. Lundstrom K, Hromić-Jahjefendić A, Bilajac E, Aljabali AAA, Baralić K, Sabri NA, et al. COVID-19 signalome: Pathways for SARS-CoV-2 infection and impact on COVID-19 associated comorbidity. Cell Signal. (2023) 101. doi: 10.1016/j.cellsig.2022.110495
33. Yamada T and Takaoka A. Innate immune recognition against SARS-CoV-2. Inflammation Regener. (2023) 43. doi: 10.1186/s41232-023-00259-5
34. Manes NP and Nita-Lazar A. Molecular mechanisms of the toll-like receptor, STING, MAVS, inflammasome, and interferon pathways. MSystems. (2021) 6. doi: 10.1128/msystems.00336-21
35. Wijeratne T, Gillard Crewther S, Sales C, and Karimi L. COVID-19 pathophysiology predicts that ischemic stroke occurrence is an expectation, not an exception—A systematic review. Front Neurol. (2021) 11:607221. doi: 10.3389/fneur.2020.607221
36. Biedrzycki G, Wolszczak-Biedrzycka B, Dorf J, and Maciejczyk M. The antioxidant barrier, oxidative/nitrosative stress, and protein glycation in allergy: from basic research to clinical practice. Front Immunol. (2024) 1-1440313. doi: 10.3389/fimmu.2024.1440313
37. Zheng Z. Establishment and Validation of an Interferon-Stimulated Genes (ISGs) Prognostic Signature in Pan-cancer Patients: A Multicenter, Real-world Study. Int. J. Biol. Sci. (2022) 18(9):3762–76. doi: 10.7150/ijbs.71385
38. Ailioaie LM, Ailioaie C, and Litscher G. Implications of SARS-coV-2 infection in systemic juvenile idiopathic arthritis. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23084268
39. Zanin N, Viaris de Lesegno C, Podkalicka J, Meyer T, Gonzalez Troncoso P, Bun P, et al. STAM and Hrs interact sequentially with IFN-α Receptor to control spatiotemporal JAK–STAT endosomal activation. Nat Cell Biol. (2023) 25. doi: 10.1038/s41556-022-01085-6
40. Zhou H, Zhao J, and Perlman S. Autocrine interferon priming in macrophages but not dendritic cells results in enhanced cytokine and chemokine production after coronavirus infection. MBio. (2010) 1. doi: 10.1128/mBio.00219-10
41. Sun BL, Sun X, Kempf CL, Song JH, Casanova NG, Camp SM, et al. Involvement of eNAMPT/TLR4 inflammatory signaling in progression of non-alcoholic fatty liver disease, steatohepatitis, and fibrosis. FASEB J. (2023) 37. doi: 10.1096/fj.202201972RR
42. Bermudez T, Sammani S, Song JH, Hernon VR, Kempf CL, Garcia AN, et al. eNAMPT neutralization reduces preclinical ARDS severity via rectified NFkB and Akt/mTORC2 signaling. Sci Rep. (2022) 12. doi: 10.1038/s41598-021-04444-9
43. Flisiak R, Parczewski M, Horban A, Jaroszewicz J, Kozielewicz D, Pawłowska M, et al. Management of SARS-CoV-2 infection: Recommendations of the polish association of epidemiologists and infectiologists. Annex no. 2 as of October 13, 2020. Pol Arch Intern Med. (2020) 130:915–8. doi: 10.20452/PAMW.15658
44. Gupta A, Jayakumar MN, Saleh MA, Kannan M, Halwani R, Qaisar R, et al. SARS-CoV-2 infection- induced growth factors play differential roles in COVID-19 pathogenesis. Life Sci. (2022) 304. doi: 10.1016/j.lfs.2022.120703
45. Hasanvand A. COVID-19 and the role of cytokines in this disease. Inflammopharmacology. (2022) 30. doi: 10.1007/s10787-022-00992-2
46. Alturaiki W, Alkadi H, Alamri S, Awadalla ME, Alfaez A, Mubarak A, et al. . doi: 10.1016/j.heliyon.2022.e12653
47. Rokni M, Hamblin MR, and Rezaei N. Cytokines and COVID-19: friends or foes? Hum Vaccin Immunother. (2020) 16. doi: 10.1080/21645515.2020.1799669
48. Arsentieva NA, Liubimova NE, Batsunov OK, Korobova ZR, Kuznetsova RN, Rubinstein AA, et al. PREDICTIVE VALUE OF SPECIFIC CYTOKINES FOR LETHAL COVID-19 OUTCOME. Russian J Infect Immun. (2022) 12. doi: 10.15789/2220-7619-PVO-2043
49. Zhao Z, Wei Y, and Tao C. An enlightening role for cytokine storm in coronavirus infection. Clin Immunol. (2021) 222. doi: 10.1016/j.clim.2020.108615
50. Guo Y, Hu K, Li Y, Lu C, Ling K, Cai C, et al. Targeting TNF-α for COVID-19: recent advanced and controversies. Front Public Health. (2022) 10:833967. doi: 10.3389/fpubh.2022.833967
51. Smail SW, Babaei E, Amin K, and Abdulahad WH. Serum IL-23, IL-10, and TNF-α predict in-hospital mortality in COVID-19 patients. Front Immunol. (2023) 14:1145840/BIBTEX. doi: 10.3389/FIMMU.2023.1145840/BIBTEX
52. Fishchuk L, Rossokha Z, Pokhylko V, Cherniavska Y, Tsvirenko S, Kovtun S, et al. Modifying effects of TNF-α, IL-6 and VDR genes on the development risk and the course of COVID-19. Pilot study. Drug Metab Pers Ther. (2022) 37. doi: 10.1515/dmpt-2021-0127
53. Vieira SM, Lemos HP, Grespan R, Napimoga MH, Dal-Secco D, Freitas A, et al. A crucial role for TNF-α in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5: RESEARCH PAPER. Br J Pharmacol. (2009) 158:779–89. doi: 10.1111/j.1476-5381.2009.00367.x
54. Smail SW, Babaei E, and Amin K. Ct, IL-18 polymorphism, and laboratory biomarkers for predicting Chemosensory dysfunctions and mortality in COVID-19. Future Sci OA. (2023) 9. doi: 10.2144/FSOA-2022-0082/ASSET/IMAGES/LARGE/FIGURE4.JPEG
55. Coperchini F, Chiovato L, Croce L, Magri F, and Rotondi M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. (2020) 53. doi: 10.1016/j.cytogfr.2020.05.003
56. Khalil BA, Elemam NM, and Maghazachi AA. Chemokines and chemokine receptors during COVID-19 infection. Comput Struct Biotechnol J. (2021) 19. doi: 10.1016/j.csbj.2021.01.034
57. Olivarria G and Lane TE. Evaluating the role of chemokines and chemokine receptors involved in Coronavirus infection. Expert Rev Clin Immunol. (2022) 18. doi: 10.1080/1744666X.2022.2017282
58. Cavalcanti NV, Torres LC, da Matta MC, Lindoso CD, Carvalho LNA, Duarte MCMB, et al. Chemokine patterns in children with acute bacterial infections. Scand J Immunol. (2016) 84. doi: 10.1111/sji.12492
59. Viejo-Borbolla A, Martinez-Martín N, Nel HJ, Rueda P, Martín R, Blanco S, et al. Enhancement of chemokine function as an immunomodulatory strategy employed by human herpesviruses. PloS Pathog. (2012) 8. doi: 10.1371/journal.ppat.1002497
60. NCT04569877, GM-CSF inhalation to prevent ARDS in COVID-19Pneumonia (2020). Available online at: https://Clinicaltrials.Gov/Show/NCT04569877 (Accessed September 10, 2025).
61. Bendib I, Beldi-Ferchiou A, Schlemmer F, Surenaud M, Maitre B, Plonquet A, et al. Alveolar compartmentalization of inflammatory and immune cell biomarkers in pneumonia-related ARDS. Crit Care. (2021) 25. doi: 10.1186/s13054-020-03427-y
62. Yu H, Sun T, and Feng J. Complications and pathophysiology of COVID-19 in the nervous system. Front Neurol. (2020) 11:573421. doi: 10.3389/fneur.2020.573421
63. Grodecka M and Waśniowska K. Interceptors:–”silent” chemokine receptors. Postepy Hig Med Dosw. (2007) 61. doi: 10.1186/1476-4598-13-198
64. Murdoch C and Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. (2000) 95. doi: 10.1182/blood.v95.10.3032.010k17_3032_3043
65. Sallusto F and Baggiolini M. Chemokines and leukocyte traffic. Nat Immunol. (2008) 9. doi: 10.1038/ni.f.214
66. Komolafe K and Pacurari M. CXC chemokines in the pathogenesis of pulmonary disease and pharmacological relevance. Int J Inflam. (2022) 2022). doi: 10.1155/2022/4558159
67. Mehrabian M, Sparkes RS, Mohandas T, Fogelman AM, and Lusis AJ. Localization of monocyte chemotactic protein-1 gene (SCYA2) to human chromosome 17q11.2-q21.1. Genomics. (1991) 9. doi: 10.1016/0888-7543(91)90239-B
68. Bischoff SC, Krieger M, Brunner T, and Dahinden CA. Monocyte chemotactic protein 1 is a potent activator of human basophils. J Exp Med. (1992) 175. doi: 10.1084/jem.175.5.1271
69. Conti P, Boucher W, Letourneau R, Feliciani C, Reale M, Barbacane RC, et al. Monocyte chemotactic protein-1 provokes mast cell aggregation and [3H]5HT release. Immunology. (1995) 86.
70. Irving SG, Zipfel PF, Balke J, Mcbride OW, Morton CC, Burd PR, et al. Two inflammatory mediator cytokine genes are closely linked and variably amplified on chromosome 17q. Nucleic Acids Res. (1990) 18. doi: 10.1093/nar/18.11.3261
71. Lee J, Kim BY, Son Y, Giang DH, Lee D, Eo SK, et al. 4’-O-Methylalpinumisoflavone inhibits the activation of monocytes/macrophages to an immunostimulatory phenotype induced by 27-hydroxycholesterol. Int J Mol Med. (2019) 43. doi: 10.3892/ijmm.2019.4135
72. Bhavsar I, Miller CS, and Al-Sabbagh M. Macrophage Inflammatory Protein-1 Alpha (MIP-1 alpha)/CCL3: As a biomarker. In: General methods in Biomarker Research and Their Applications. Springer, Dordrecht (2015). doi: 10.1007/978-94-007-7696-8_27
73. Schaller TH, Batich KA, Suryadevara CM, Desai R, and Sampson JH. Chemokines as adjuvants for immunotherapy: implications for immune activation with CCL3. Expert Rev Clin Immunol. (2017) 13. doi: 10.1080/1744666X.2017.1384313
74. Donlon TA, Krensky AM, Wallace MR, Collins FS, Lovett M, and Clayberger C. Localization of a human T-cell-specific gene, RANTES (D17S136E), to chromosome 17q11.2-q12. Genomics. (1990) 6. doi: 10.1016/0888-7543(90)90485-D
75. Zeng Z, Lan T, Wei Y, and Wei X. CCL5/CCR5 axis in human diseases and related treatments. Genes Dis. (2022) 9. doi: 10.1016/j.gendis.2021.08.004
76. Appay V and Rowland-Jones SL. RANTES: A versatile and controversial chemokine. Trends Immunol. (2001) 22. doi: 10.1016/S1471-4906(00)01812-3
77. Maghazachi AA, Al-Aoukaty A, and Schall TJ. CC chemokines induce the generation of killer cells from CD56+ cells. Eur J Immunol. (1996) 26. doi: 10.1002/eji.1830260207
78. Krensky AM and Ahn YT. Mechanisms of disease: Regulation of RANTES (CCL5) in renal disease. Nat Clin Pract Nephrol. (2007) 3. doi: 10.1038/ncpneph0418
79. Almeida RS, Ferreira MLB, Sonon P, Cordeiro MT, Sadissou I, Diniz GTN, et al. Cytokines and soluble HLA-G levels in the acute and recovery phases of arbovirus-infected Brazilian patients exhibiting neurological complications. Front Immunol. (2021) 12:582935. doi: 10.3389/fimmu.2021.582935
80. Modi WS, Dean M, Seuanez HN, Mukaida N, Matsushima K, and O’Brien SJ. Monocyte-derived neutrophil chemotactic factor (MDNCF/IL-8) resides in a gene cluster along with several other members of the platelet factor 4 gene superfamily. Hum Genet. (1990) 84. doi: 10.1007/BF00208938
81. Dixit N and Simon SI. Chemokines, selectins and intracellular calcium flux: Temporal and spatial cues for leukocyte arrest. Front Immunol. (2012) 3:188. doi: 10.3389/fimmu.2012.00188
82. Vilotić A, Nacka-Aleksić M, Pirković A, Bojić-Trbojević Ž, Dekanski D, and Jovanović Krivokuća M. IL-6 and IL-8: an overview of their roles in healthy and pathological pregnancies. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232314574
83. Gonzalez-Aparicio M and Alfaro C. Significance of the IL-8 pathway for immunotherapy. Hum Vaccin Immunother. (2020) 16. doi: 10.1080/21645515.2019.1696075
84. Chang Y, Bai M, and You Q. Associations between serum interleukins (IL-1 β, IL-2, IL-4, IL-6, IL-8, and IL-10) and disease severity of COVID-19: A systematic review and meta-analysis. BioMed Res Int. (2022) 2022. doi: 10.1155/2022/2755246
85. Bergantini L, d’Alessandro M, Cameli P, Otranto A, Luzzi S, Bianchi F, et al. Cytokine profiles in the detection of severe lung involvement in hospitalized patients with COVID-19: The IL-8/IL-32 axis. Cytokine. (2022) 151. doi: 10.1016/j.cyto.2022.155804
86. Luster AD, Unkeless JC, and Ravetch JV. γ-Interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. (1985) 315. doi: 10.1038/315672a0
87. Booth V, Keizer DW, Kamphuis MB, Clark-Lewis I, and Sykes BD. The CXCR3 binding chemokine IP-10/CXCL10: Structure and receptor interactions. Biochemistry. (2002) 41. doi: 10.1021/bi026020q
88. Campanella GSV, Lee EMJ, Sun J, and Luster AD. CXCR3 and heparin binding sites of the chemokine IP-10 (CXCL10). J Biol Chem. (2003) 278. doi: 10.1074/jbc.M212077200
89. Gudowska-Sawczuk M and Mroczko B. What is currently known about the role of CXCL10 in SARS-coV-2 infection? Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23073673
Keywords: COVID-19, cytokine storm, chemokines, SARS-CoV-2, biomarkers
Citation: Wolszczak-Biedrzycka B, Cieślikiewicz B, Studniarz F, Dąbrowski Ł, Fąs M, Matyszkiewicz–Suchodolska K, Harasimowicz M and Dorf J (2025) Chemokines as potential biomarkers for predicting the course of COVID-19 – a review of the literature. Front. Immunol. 16:1662643. doi: 10.3389/fimmu.2025.1662643
Received: 09 July 2025; Accepted: 25 August 2025;
Published: 23 September 2025.
Edited by:
Danielle Aparecida Sousa Rodrigues, Federal University of Rio de Janeiro, BrazilReviewed by:
Yunlong Ma, University of Pennsylvania, United StatesZahra’a Abdul Al-Aziz Yousif, Ibn Sina University for Medical and Pharmaceutical Sciences, Iraq
Copyright © 2025 Wolszczak-Biedrzycka, Cieślikiewicz, Studniarz, Dąbrowski, Fąs, Matyszkiewicz–Suchodolska, Harasimowicz and Dorf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Blanka Wolszczak-Biedrzycka, YmxhbmthLndvbHN6Y3pha0B1d20uZWR1LnBs