 Shahinur Acter
Shahinur Acter Qing Lin*
Qing Lin*- Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
The S100 superfamily of proteins consists of Ca2+-binding proteins characterized by the EF-hand motif. Certain members of this protein family, such as S100A8, S100A9, and S100A12, have been effectively utilized as biomarkers for the detection and evaluation of prognosis in immunological diseases. These proteins are also identified as damage-associated molecular pattern (DAMP) molecules, which exhibit significant upregulation in various autoimmune disorders, cancers, and neurodegenerative diseases. Following tissue injury, necrotic or immune cells release or secrete DAMPs to initiate inflammatory responses. This signaling further creates autocrine and paracrine positive feedback loops that amplify and sustain the inflammatory response. The NLRP3 inflammasome pathway is a pivotal component in these DAMP-induced immune regulatory mechanisms. This review summarizes the regulatory roles of S100 protein family in NLRP3 inflammasome signaling and their functions in innate and adaptive immunity, with an emphasis on pulmonary hypertension. Moreover, we examine the interactive feedback mechanisms among NLRP3 inflammasome, S100A8/A9, and Gasdermin D, exploring their implications in autoimmune diseases.
1 Introduction
An inflammasome is a protein complex consisting of three parts: a sensor protein, an adaptor, and pro-caspase-1 (1). It is an important part of the innate immune system, also known as the body’s defense against pathogens and tissue damage (2). In recent years, the NOD-like receptors (NLR) family has been instrumental in inflammatory responses and has attracted enormous research attention from scientists in the field of immunology research (3). NLR family are subdivided into four subfamilies: NLRA, NLRB, NLRC, and NLRP, These subfamilies have been found to promote inflammasome assembly and inflammatory response by activating multiple downstream signals (4). Among these, NLRP3 (NOD-like receptor protein 3) has gained extra research attention given its in-depth link to the innate immune system; in particular, it is expressed predominantly in macrophages, neutrophils, and dendritic cells as a component of the inflammasome (4). It is an important innate sensor of structurally diverse metabolic damage-associated molecular pattern molecules (DAMPs) (1). This NLRP3 complex protein mediates caspase-1 activation, followed by the secretion of proinflammatory cytokines IL-1/IL-18 in response to threats such as microbial infection and cellular damage. It is also known as apoptosis-associated speck-like protein (5). Regulating the NLPR3 inflammasome is essential due to its crucial role in the immune response. Increasing evidence suggests that the dysregulated condition of NLPR3 inflammasome can result in excessive inflammation, which contributes to the development of chronic inflammatory diseases, including autoimmune disorders, metabolic diseases, cardiovascular diseases, and cancer (6, 7). Therefore, a clear understanding of the mechanism behind NLPR3 response is crucial in order to maintain desirable immune function and tissue homeostasis.
Growing evidence suggests that S100 protein family proteins play a significant role as mediators in the initiation and maintenance of inflammation (8, 9). These proteins play a dual role within inflammatory networks: they act as inflammation-responsive proteins but can also amplify the inflammatory signal which produced them in the first place (8). As a family of calcium-binding cytosolic proteins with multifunction properties, S100s have an extensive range of intracellular and extracellular functions through regulating calcium balance. The intracellular functions of this protein family involve interaction with intracellular receptors (8, 9). This protein family plays regulatory roles in multiple cellular processes, including apoptosis, proliferation, differentiation, migration and invasion, energy metabolism, calcium homeostasis, and protein phosphorylation (shown in Figure 1) in a number of cells types, including monocytes, macrophages, neutrophils, lymphocytes, myoblast, epithelial cells, endothelial cells, smooth muscle cells, neurons, and fibroblasts. Moreover, S100 proteins have demonstrated their significant role in governing immune homeostasis, post-traumatic injury, and inflammation (10). There are some members of these S100 proteins that function as DAMP molecules that could be released from the cells upon stress or activation of phagocytes such as neutrophils and macrophages (10). DAMPs are a series of intracellular molecules that are linked with apoptosis and tissue damage via instigating a rapid inflammatory response (11). Studies suggest that by binding to the pattern receptors such as Toll-like receptors (TLRs) and receptors for advanced glycation end products (RAGE), S100 proteins actualized as threat signals leading to the activation of immune cells and endothelial cells (12). S100A8 is a calcium-binding protein belonging to the S100 protein family. The S100A8 protein plays a key role in stimulating the NLPR3 inflammasome, which has been identified as a pro-inflammatory mediator, by triggering the activation of this immune complex, resulting in the release of inflammatory cytokines like pro-interleukin-1β (IL-1β) (13, 14).
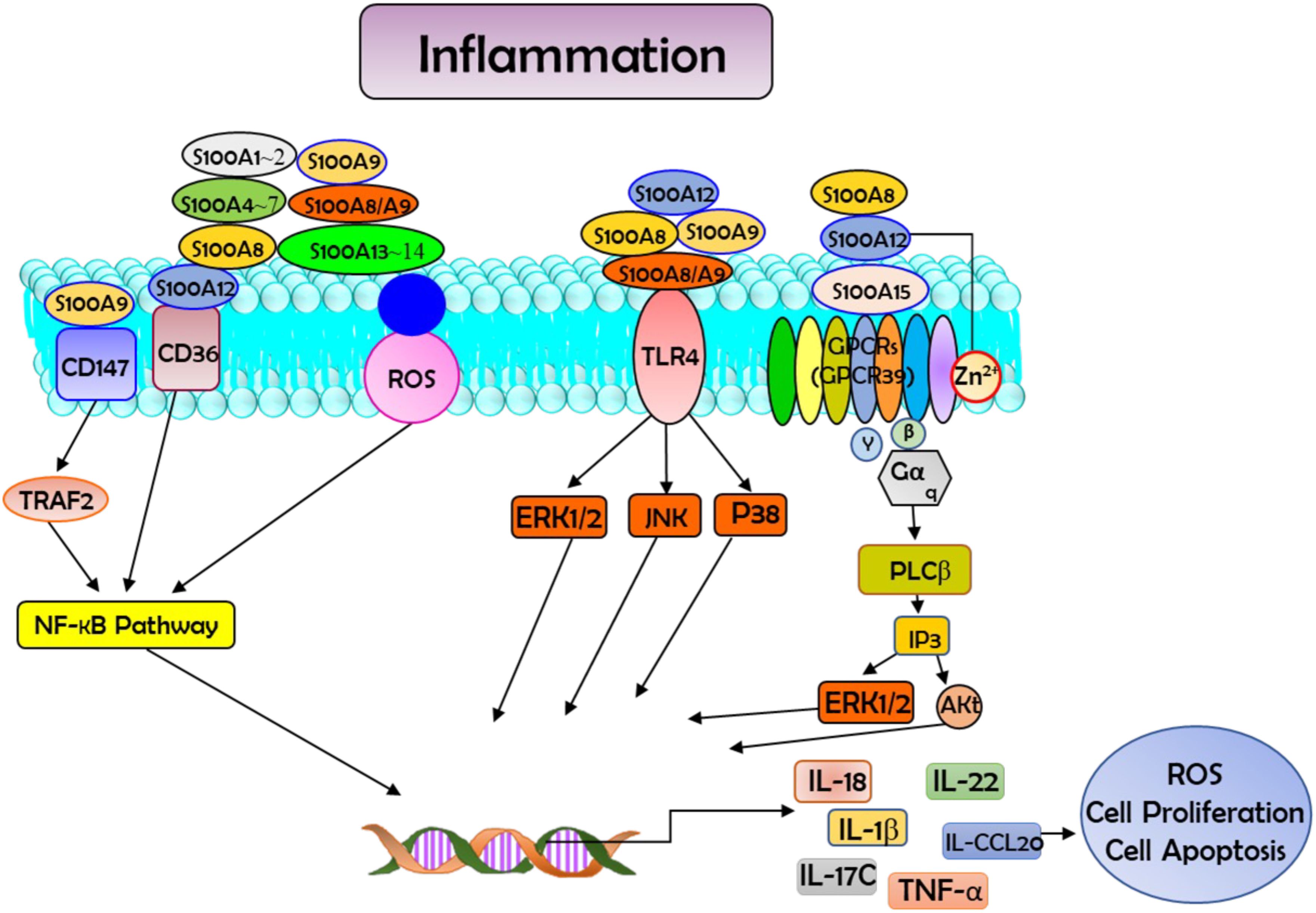
Figure 1. Schematic diagram showing the interaction between S100 proteins and their receptors in the cells. Such interactions activate the downstream signal transductions and regulate multiple cellular processes, such as reactive oxygen species (ROS) production, apoptosis, and cell proliferation. In this process they participate in the modulation of inflammation and have vital roles in combating microbial infection through zinc-mediated nutritional immunity. Akt=protein kinase B; CCL20, ERK1/2=fibroblast growth factor receptors; GPCRs, G protein coupled receptors (GPR39 is used as an example); IL-18, interleukin-18; IP3, Inositol Trisphosphate; NF-κB, nuclear factor kappa B; P38, p38 MAP-Kinases; PI3K, phosphatidylinositol 3-kinase; PLCβ, Phospholipase C β; RAGE, receptor for advanced glycation end products; STAT, Signal Transducer and Activator of Transcription; TLR4, Toll-like receptor 4; TNF-α, tumor necrosis factor-α; TRAF2, tumor necrosis factor receptor-associated factor 2.
In this review article, we provide a comprehensive and detailed overview of the NLPR3 inflammasome and S100 protein family and the biological functions of DAMP molecules (S100A8/A9) and highlight their multifunctional role during immune response, with a major focus on their role in inflammatory conditions.
2 S100A protein family and their functions
2.1 S100 protein family proteins function as immune markers
The S100A subfamily is a group of the superfamily of Ca2+-binding proteins, which is characterized by the specific Ca2+-binding motif, the elongation factor hand (EF-hand) (15). In 1965, S100 proteins S100A1 and S100B were first extracted as neural proteins from the bovine brain and were named S100 due to their solubility in 100% saturated ammonium sulfate (16). There are at least 24 different small acidic proteins (10-12k Da) in this protein family, and they each share 25-65% similarity in their amino acid sequence (16). Among the S100 protein family, S100A8 (MRP8, calgranulin A) and S100A9 (MRP14, calgranulin B) are calcium-binding proteins expressed in cells of myeloid origin (12). This protein family is found as anti-parallel homo- and heterodimers, with each monomer consisting of two helix-loop-helix EF-hands (EF-1 and EF-2) that are connected by a hinge region and flanked by conserved hydrophobic residues at the C- and N-terminal ends (17). Over the years, 3D structures of S100 proteins have been revealed in three different forms that include bound to Ca2+, bound to its target protein, or in its apo (Ca2+free) form (18). Studies suggest that certain S100 members have been described to bind to the same target molecules (19). For example, S100A1, S100A6, and S100B interact with annexin A6, while S100A1, S100A2, S100A4, and S100B bind to the tumor p53 (19). Studies on the structure of the S100-target complexes have revealed that S100 protein family members apply different mechanisms for target recognition despite the similar conformational change induced by Ca2+ binding in all S100 protein family members (20). Moreover, the region exposed upon Ca2+ binding comprises the most variable portions of the S100 sequences (hinge and C-terminal regions) that are found enough to discriminate against different target proteins (18, 20). Additionally, the distribution of hydrophobic and charged residues, together with differences in surface configurations, contributes to the specific target binding patterns described among all the members of the S100 protein family (18, 20). The roles of the S100 protein family are summarized in Figure 2.
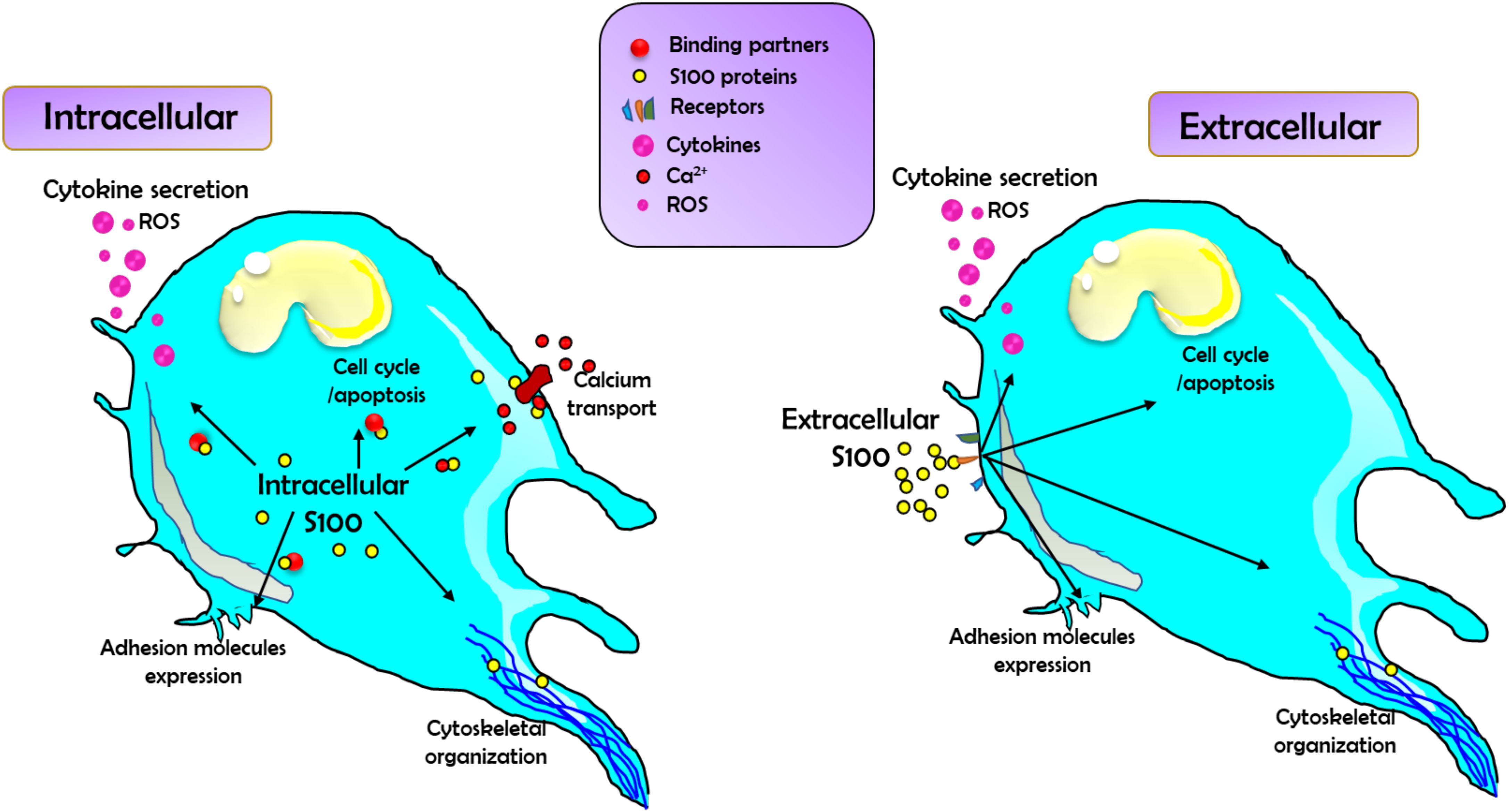
Figure 2. The schematic diagram demonstrates the summary of the general roles of S100 family proteins in regulating cell function. Left: S100 family proteins act intracellularly by binding to and modulating the function of a wide range of binding partners, including those involved in regulating cytokine and reactive oxygen species (ROS) production and release, calcium transport (both through directly binding Ca2+ channels and Ca2+ regulators), cell cycle, apoptosis, and adhesion molecule expression, as well binding to cytoskeletal components. Right: Secreted S100 proteins can also act as DAMPs to signal via an array of cell surface receptors. This binding will activate signaling pathways to affect changes in cytokine/ROS production, cell cycle, apoptosis, adhesion molecule expression, and changes in cytoskeletal reorganization and migration.
Studies show that extracellular S100 proteins are involved in the activation of G protein-coupled receptors, such as heparan sulfate proteoglycans or N-Glycans and scavenger receptors in autocrine and paracrine manners (15). S100A proteins can be detected in biological fluids, including urine, cerebrospinal fluid, serum, sputum, and feces. Therefore, extracellular S100 proteins are considered biomarkers that are associated with certain diseases (9, 15). It has been demonstrated that certain members of the S100 proteins family, including S100A12, S100A8/A9, and S100B, are linked to specific diseases like autoinflammatory diseases, stroke, and trauma (9). Oesterle et al. have successfully detected an increased amount of S100A12 in the blood of patients with diabetes, and they found that this increase correlated with a higher risk of the development of cardiovascular disease (21). Another research group, Bogdanova et al., detected an increased amount of serum concentration of S100A12 and other acute-phase inflammatory markers after investigating 35 patients with periodic disease (PD), and they found that the level of S100A12 in PD was significantly higher in comparison to other familial periodic fevers (22). S100A12 was more sensitive to the subclinical activity of autoinflammatory diseases when compared to other inflammatory biomarkers, such as neutrophil counts, fibrinogen, C-reactive protein (CRP), and erythrocyte sedimentation rate (22). A study done by another group has demonstrated similar findings, where upregulated concentrations of S100A12 in serum were observed in patients with Familial Mediterranean fever in comparison to controls, which suggests this protein is a novel biomarker (23).
In various studies, S100A8 and S100A9 have been identified as novel diagnostic markers to aid in differential diagnosis (24, 25). Recently, it has been shown that the expression of S100A8/A9 was high in human atherosclerotic lesions, and the blood levels were also on the rise among the patients with coronary artery diseases (CAD), suggesting implied S100A8/A9 might act as a biomarker for cardiovascular events (26). Similar findings were demonstrated by Xia et al., where serum S100A8/A9 levels were elevated in 178 CAD patients with unstable angina pectoris or acute myocardial infarction, and the level of S100A8/A9 was significantly positively linked with CRP (27). These studies represent S100A8/A9 as a novel biomarker for the CAD (28). The serum levels of S100A8/A9 were dramatically increased in IL-1Ra−/− mice, contributing to bone erosion, cartilage damage, and synovial inflammation (21). Therefore, S100A8/A9 is considered a systemic or local biomarker to evaluate the extent of inflammation and inflammatory joint destruction in seronegative arthritis (21). The S100A8/A9 protein complex plays a crucial role as a mediator in the initiation and maintenance of inflammation and has been applied as a valuable clinical biomarker for therapeutic response monitoring (29). Additionally, it was identified as an interesting biomarker to monitor disease activity in chronic inflammatory disorders, including inflammatory bowel disease and rheumatoid arthritis (29).
Increasing evidence suggests that S100 proteins are secreted from cells and exert cytokine-like functions through the binding and activation of cell surface receptors such as the RAGE, TLR-4, ErbB4 receptor, the dopamine D2 receptor, amyloid-β, and annexins (29). It has been demonstrated that various S100 proteins contribute to leukocyte migration (9). For example, S100A8/A9 has been reported to induce pro-inflammatory cytokine production in macrophages and nucleophiles through the activation of the nuclear factor-kB (NF-κB) and p38 mitogen-activated protein kinase pathways and mediate immune cell migration (9, 30). Moreover, the evidence shows that S100A12 has an impact in inducing the production of pro-inflammatory cytokines interleukin (IL) -6 and -8 through RAGE-dependent NF-κB activation, which results in the recruitment of monocytes (9). Additionally, S100A10 protein has been reported to recruit macrophages to tumor sites, while S100A8/S100A9 have been shown to signal through RAGE to mediate the effect of tumor necrosis factor (TNF)-α on the differentiation of myeloid-derived suppressor cells, which demonstrates their regulatory involvement in immune responses (9, 31). The roles and associations of S100A proteins with NLRP3 inflammasome signaling are complex, involving various cellular activation processes observed across multiple studies. Table 1 summarizes the effects of different S100 family proteins on NLRP3 regulation and related diseases (32–35).

Table 1. Summary of the roles of S100 family proteins in NLRP3 regulation, their immunoregulatory mechanisms, and associated diseases.
2.2 Expression and distribution of S100A8/S100A9 proteins
Recent studies show that among the S100 protein family, S100A8 and S100A9 are specifically linked to innate immune function through their expression in immune cells of the myeloid lineage (36, 37). S100A8/A9 proteins are mainly derived from myeloid cells such as monocytes, neutrophils, and macrophages, with constitutive expression believed to be limited to neutrophils and monocytes (29). S100A8/A9 proteins comprise 93 and 113 amino acids with molecular weights of 10.8 and 13.2 kDa, respectively (29, 38). S100A8/A9 proteins comprise approximately 45% of the cytoplasmic proteins in neutrophils, and only 5% are constitutively expressed in monocytes (29, 38). These proteins are found in various structures, including homodimers, heterodimers, and tetramers (29, 39). The homodimer is less stable, leading to a preference for the formation of noncovalently bonded complexes, and this is the predominant form of S100A8/A9 in physiological settings (40). After reaching Ca2+ concentration at a certain threshold, S100A8/A9 heterodimers form (termed S100A8/A9, or calprotectin) tetramers, a configuration that is vital for their biological activity (36). These proteins are mainly secreted by immune cells such as dendritic cells, neutrophils, monocytes, and activated macrophages (41).
These proteins are predominantly localized in the cytoplasm and relocate to the cytoskeleton and plasma membrane to increase intracellular calcium levels. Studies suggest that S100A8/A9 expression is downregulated during the maturation of monocytes to macrophages; its expression is also observed in macrophages present in inflamed tissues (36). Moreover, S100A8/A9 expression is seen in activated epithelial cells, endothelial cells, and keratinocytes. In both dendritic and myeloid-derived suppressor cells, S100A8/A9 protein expression is observed, with noticeably upregulated expression seen in dendritic cells treated with IL-10 (35). Fibrocytes are another population of S100A8/A9-expressing cells that are derived from myeloid lineage cells, which have been found to play a strong role in tissue repair and fibrosis. Upon exposure to the gram-negative bacterium Porphyromonas gingivalis, the S100A8/A9 protein complex can be induced in vascular smooth muscle cells, suggesting that this bacterium can trigger an inflammatory response within the blood vessel walls by activating S100A8/A9 in these cells (36).
2.3 Damage-associated molecular patterns
DAMPs are molecules within cells that are components of the innate immune response. They are released during cellular damage or cell apoptosis and tissue damage caused by trauma or pathogen infection (42). DAMPs cannot be recognized by the immune system under normal physiological conditions (43, 44). DAMP was first described by Seong and Matzinger in 2004 (45). DAMPs largely depend on the type of cells, such as epithelial or mesenchymal and injured tissue; however, DAMPs all share the common feature of stimulating an innate immune response within an organism (46). DAMPs serve as crucial mediators that link sterile inflammation to end-organ damage and life-threatening disease through the modulation of the innate immune response (11, 46).
Many of these DAMPs are intracellular proteins such as S100 proteins and HMGB1. Studies suggest that the HMGB1 protein has dual functions as a nonhistone nucleoprotein and an extracellular inflammatory cytokine. Intracellular HMGB1 is extensively bound to DNA and involved in transcriptional regulation, DNA replication and repair, telomere maintenance, and nucleosome assembly (47, 48). HMGB1 is prone to bind other proinflammatory molecules including DNA, RNA, histones, nucleosomes, lipopolysaccharide (LPS), SDF-1, IL-1α, IL-1β, and additional factors. These complexes act in synergy via cognate receptors to the HMGB1-partner molecules (47, 48). During injury or pathogenetic infection, HMGB1 is released and promotes inflammation. During this process, HMGB1 is passively released by necrotic but not apoptotic death of normal cells and actively secreted by a variety of activated immune and nonimmune cells (36, 47, 48).
However, some studies have demonstrated that HMGB1 is not a pro-inflammatory cytokine per se (47–49). HMGB1 by itself has little or no pro-inflammatory activity, but it binds to mediators of inflammation such as LPS, DNA, or IL-1β and induces signaling pathways leading to NF-κB activation, thereby potentiating inflammatory responses (50). Although the signaling pathways elicited by HMGB1 are not fully defined, there are studies suggesting that the triggering occurs via several receptors including the multiligand RAGE, TLR2, and TLR4 (51).
Likely, S100 proteins act both as intracellular mediators and as extracellular signaling proteins, which are able to regulate activities of the target cells in either a paracrine or autocrine manner. As intracellular calcium-binding molecules, S100A8/A9 have a role in migration and cytoskeletal metabolism. Cell damage or activation of phagocytes triggers their release into the extracellular space where they become danger signals that activate immune cells and vascular endothelium. S100A8/A9 seem to interact with RAGE4 and TLRs (52). Another member of S100 protein family, S100A12, is also found at high concentrations in inflamed tissue, where neutrophils and monocytes belong to the most abundant cell types. Even though S100A12 binds to RAGE, at least part of the proinflammatory effects of the S100A8/A9 complex depend upon interaction with other receptors (12).
DAMPs are associated with various diseases, including osteoarthritis (OA), neurodegenerative diseases, cancer, autoimmune diseases, and cardiovascular diseases (53). OA has been regarded as a degenerative joint disease that is characterized by the destruction of cartilage. Physical trauma and obesity are considered risk factors for OA (54). However, there is evidence suggesting that DAMPs-induced inflammation plays a crucial role, where S100 proteins are involved in the pathogenesis of OA (54). S100A8/A9 protein expression was elevated in the synovium of a collagenase-induced OA mouse model (54). Although S100A12 expression was unchanged in the serum between the OA patients and the healthy controls, the S100A12 level in the synovial fluid of OA patients was significantly increased in comparison to the healthy controls (55). Additionally, S100A12 escalated the secretion of MMP-13 and vascular endothelial growth factor in human OA chondrocytes, which suggests that S100A12 has a role in the progression of OA (56).
DAMPs are known to be associated with neuroinflammation in neurodegenerative disorders, including Alzheimer’s disease (AD) and Parkinson’s disease (PD) (57, 58). In AD patients, the levels of HMGB1 and soluble RAGE are significantly increased, which correlates with the levels of amyloid beta (59). A recent report demonstrated that HMGB1 and thrombin are triggers of inflammation and dysfunction of the blood-brain barrier (59). In AD patients, the serum levels of S100B were intimately related to the severity of the disease (60). The administration of pentamidine, an S100B inhibitor, reduced the levels of S100B and RAGE, thereby inhibiting neuroinflammation in the brain of an AD mouse model. Moreover, the role of the HMGB1-TLR4 axis is essential in the pathogenesis of PD. It has been demonstrated that the serum HMGB1 and TLR4 protein levels were significantly elevated in PD patients and correlated with the PD stages (61). Additionally, the S100B protein level was increased in the substantia nigra and cerebrospinal fluid of PD patients, and S100B was increased as well in the ventral midbrain of a mouse model treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (62).
The role of DAMPs in the pathogenesis of cancer is controversial. However, research suggests that DAMPs may mediate tumor progression by inducing chronic inflammation, which is a compound risk factor for tumor progression (63–65). Various studies demonstrated that DAMPs such as HMGB1 and S100 proteins activate inflammatory pathways and release promoters of carcinogenesis such as IL-1, IL-6, lymphotoxin (LT) -β, IFN-γ, TNF, and transforming growth factor (TGF)-β, which suggests DAMPs has a role in the development of early-stage carcinogenesis (63, 64, 66).
The involvement of DAMPs with rheumatoid arthritis has been demonstrated in various studies (67, 68). These studies suggest that upregulated S100A8/A9 and S100A11/A12 proteins were observed in the synovial tissue, synovial fluid, or serum of rheumatoid arthritis patients (69, 70). In addition, the expression of HMGB1 was elevated in the serum and synovial fluid of rheumatoid arthritis patients in a number of studies (47, 71).
Involvement of S100 proteins in the pathogenesis of atherosclerosis indicating a significant role of DAMPs in cardiovascular diseases (72). Atherosclerosis is an inflammatory disease of the arterial wall, in which the vessels narrow due to accumulating plaques of inflammatory cells and lipids (73). It has been demonstrated that S100A8/A9 exist in plaques, and they increase atherogenesis by activating neutrophils and monocytes in arterial lesions (72). Moreover, S100A8/A9 and S100A12 have a crucial role in the mediation of inflammation and increase atherosclerosis in human and rodent models by interacting with RAGE, which plays an important role in endothelial dysfunction and inflammation (21). In addition, it has been reported that, among conventional risk factors, S100A12 showed the strongest association with the risk of coronary heart disease (74). DAMPs may also have a role in pulmonary hypertension, which is characterized by perivascular infiltration of inflammatory cells and pulmonary vascular remodeling, ultimately resulting in the right heart failure and premature death (75).
During cellular stress or tissue damage, released DAMPs can be detected by multiple pattern recognition receptors. NLRP3 inflammasome is a cytosolic pattern recognition receptor that is commonly associated with the immune response to bacteria, viruses, fungi, and parasites (5). In most cases, the recognition of pathogens in the immune response is indirect. TLRs recognize the particular components of the invader and then induce the NLRP3 inflammasome components to be transcribed and assembled (76). Upon activation by cellular damage or tissue stress, NLRP3 triggers the formation of the inflammasome, also known as a multiprotein signaling complex, that activates caspase-1, leads to the secretion of IL-1β and IL-18, and induces pyroptosis, a form of cell death that is a major pathway of inflammation (5). Studies demonstrate a close association to various inflammatory diseases, including type 2 diabetes, gout, neurodegenerative diseases, and pulmonary hypertension (77).
Growing evidence suggests that two DAMPs, S100A8 and S100A9, play a significant role in activating the NLRP3 inflammasome (13, 29). They are involved in inflammation caused by stress, injury, and microorganisms. After being affected, neutrophils, macrophages, and monocytes intensely express and secrete S100A8/A9 to modulate inflammatory processes with the induction of inflammatory cytokines, ROS, and nitric oxide (13, 38, 39). The released S100A8/A9 proteins can promote IL-1β secretion in neutrophils and macrophages in an autocrine or paracrine fashion, as well as prime the NLRP3 inflammasome (29, 78). Studies have shown that S100A8/A9 proteins activate NLPR3 inflammasome signaling to promote the pathogenesis of several diseases, including myelodysplastic syndromes and airway obstructive diseases (13, 41). In one study, a close correlation between S100A8 and pyroptosis was observed, and a direct effect of S100A8 on macrophage pyroptosis was identified (13). This study also demonstrated that S100A8 interacts with the TLR4 and then activates downstream Nm NF-κB with transcriptional upregulation of NLRP3, pro-IL-1β, and pro-IL-18 (13). ROS has been reported as the second signal for NLRP3 activation, playing a crucial role in fibrotic progression (13, 20, 79). A number of studies suggest that S100A8/A9 have an immunoregulatory role as intracellular differentiation markers. After secretion to the extracellular compartment, they also function as innate amplifiers of inflammation in various inflammatory diseases via two receptors, TLR4 and RAGE, which leads to increased production of pro-inflammatory cytokines like IL-1β and contributes to various inflammatory diseases including pulmonary hypertension (20, 79, 80).
3 Regulatory role of S100A8/A9 in NLRP3 inflammasome
During inflammation, S100A8/A9 is released vigorously and exerts a critical role in modulating the inflammatory response by activating NF-κB1 through ROS-dependent activation (80). The calcium binding proteins S100A8/A9 serve as a candidate biomarker for diagnosis and follow-up, and they have a role as a predictive indicator of therapeutic responses to inflammation-associated diseases (80).
3.1 Function of NLRP3 inflammasome
NLRP3 is an intracellular receptor that senses foreign pathogens and self-danger signals, leading to the formation and activation of the NLRP3 inflammasome (29). NLRP3 inflammasome is an important component of the innate immune system’s response to pathogens, which consists of a set of cytoplasmic multiprotein complexes (5). Different inflammasomes have distinct stimulatory signals but have very conserved downstream effects, especially in the activation of caspase-1, which in turn triggers the three key substances: IL-1β, pro-IL-18, and gasdermin D (GSDMD) (3, 4).
The NLRP3 protein belongs to the family of nucleotide-binding oligomerization domain-like receptors (NLRs), and NLRP3 protein contains a leucine-rich repeat domain at the carboxyl terminus, a pyrin domain at the amino terminus, and a nucleotide-binding domain in the center domain (5). It has been reported that the NLRP3 inflammasome can rapidly and effectively eliminate microbial infection and repair damaged tissue (3, 5). In a number of studies, DAMP molecules S100A8/A9 were shown to be actively involved in the inflammatory response by regulating neutrophil functions (9). S100A8/A9 induce the secretion of several pro-inflammatory cytokines in monocytes, including IL-6, TNFα, and IL-1β, through stimulating the production of ROS (9, 72). This phenomenon activates the transcription factor NF-kB, which leads to cytokine secretion and expression and results in the activation of the NLRP3 inflammasome (29, 81). NLRP3 activation and function is shown in Figure 3.

Figure 3. A schematic diagram for NLR-related protein 3 (NLRP3) inflammasome activation. A number of stimuli trigger the activation of NLRP3, which secretes interleukin (IL)-1β and IL-18. Diverse pathogen-associated molecular patterns (PAMP) and/or DAMP stimulation potentiate two signals that activate the NLRP3 inflammasome. Signal 1 activation leads to the expression of the pro-IL-1β gene and the production of the pro-IL-1β protein through the toll-like receptor (TLR)-MyD88-NFκB signaling pathway. Signal 2 is a critical step in inflammasome activation. These signals or agonists trigger the assembly of a large macromolecular complex through the recruitment of the apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain adaptor protein and pro-caspase-1 to NLRP3. Several mechanisms have been suggested for NLRP3 inflammasome activation, including pore formation through P2X7 receptor and K+ efflux, mitochondrial reactive oxygen species generation, phagocytic pathway activation by particulate or crystalline structures (e.g., monosodium urate crystals, aluminum potassium sulfate, or silica nanoparticles), and lysosome rupture. The molecular mechanisms by which NLRP3 inflammasome activation occurs are not yet fully understood. ATP, adenosine triphosphate.
3.2 S100A8/A9 in the priming and activation of the NLRP3 inflammasome
The NLRP3 inflammasome is a multiprotein complex that is a critical component of the innate immune system. NLRP3 inflammasome consists of a sensor NLRP3; it is an NLR family protein containing a pyrin domain 3, an adaptor, apoptosis-associated speck-like protein, which contains a caspase activation and recruitment domain and an effector called caspase-1 (82). This tripartite protein comprises three domains: an amino-terminal pyrin domain, a central NACHT domain (domain present in NAIP, CIITA, HETE, and TP1), and a carboxy-terminal leucine-rich repeat domain (82, 83). It has been demonstrated that the activation process of NLRP3 inflammasome depends upon two signals: a priming signal that is required for the upregulation of NLRP3 and pro-IL-1β and a second signal that triggers assembly into the NLRP3 inflammasome complex shown in Figure 4 (82). These two signals have been proposed to underlie NLRP3 activation: priming and activation (Figure 4).
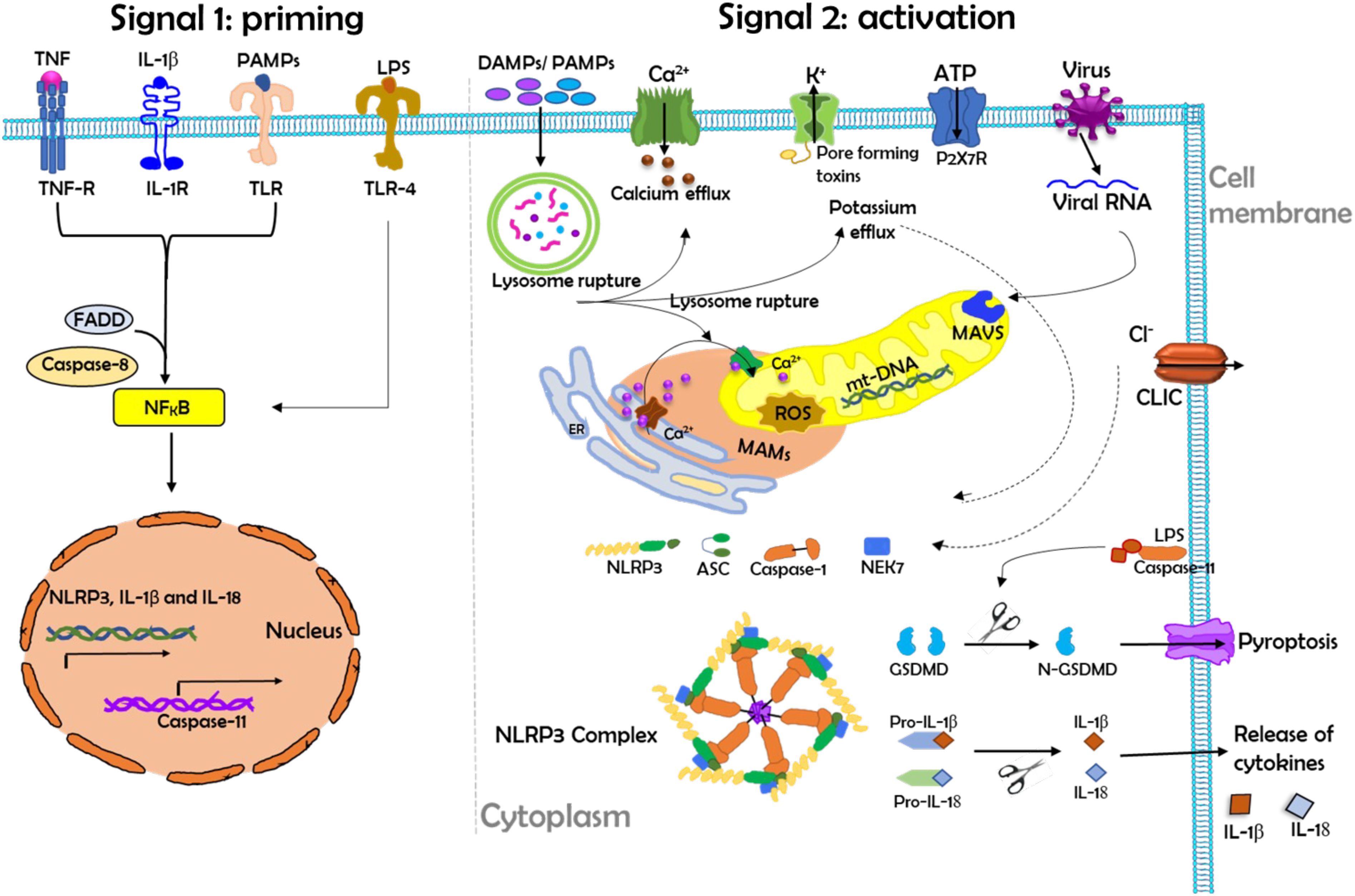
Figure 4. The schematic diagram demonstrates the priming and activation of the NLRP3 inflammasome. Herein, the activation process of the NLRP3 inflammasome requires two main signals. Left: Signal 1 (priming) leads to the activation of the transcription factor NF-κB and the subsequent transcription of canonical and noncanonical NLRP3 inflammasome components. Priming is provided by exposure to pathogen-associated molecular patterns (PAMPs) such as LPS or by endogenous cytokines that activate receptors at the cell membrane. During priming, the induction of NLRP3 expression is controlled by FAS-associated death domain protein (FADD) and caspase-8. Right: Signal 2 (activation) is responsible for NLRP3 complex assembly and the subsequent release of inflammatory cytokines (IL-1β and IL-18). NLRP3 activation is provided by a plethora of stimuli, such as PAMPs or DAMPs, ATP, and viral RNA. NLRP3 activation in turn triggers downstream signaling events such as mitochondrial damage, mitochondrial ROS production, lysosomal disruption, and ion (K+ and Ca2+) efflux. During activation, mitochondrial antiviral signaling protein (MAVS) mediates the NLRP3 activation induced by RNA viruses, where excessive Ca2+ released from the ER causes mitochondrial dysfunction and is implicated in NLRP3 inflammasome activation. Chloride intracellular channel protein (CLIC)-mediated Cl- efflux promotes the NEK7-NLRP3 interaction and subsequent NLRP3 inflammasome assembly. Herein, LPS can directly activate TLR4 to induce the transcription and activation of caspase-11, which in turn cleaves the pore-forming protein gasdermin D (GSDMD), which can induce pyroptosis. IL-18, interleukin-18; IL-1β, interleukin-1beta; IL-1R, interleukin 1 receptor; mt-DNA, mitochondrial DNA; ROS, reactive oxygen species; TLR, Toll-like receptor.
The priming signal is triggered by the engagement of TLRs by their ligands or endogenous molecules, such as TNF or IL-β, which then leads to the transcription of NLRP3 and pro-IL-1β via the regulation of NF-κB (84). Recently, it has been shown that the induction of NLRP3 expression during priming is also controlled by FAS-associated death domain protein and caspase-8 (83). During the activation signal, NLRP3 responds to many stimuli for activation of the inflammasome. A variety of pathogen-associated molecular patterns (PAMPs) and DAMPs provide the activation signal for NLRP3. The PAMPs and DAMPs originated from numerous pathogens, a large number of pore-forming toxins, adenosine triphosphate, and particulate crystals and aggregates (76, 82, 85).
Recent studies have demonstrated that cell stress in NLRP3 inflammasome-associated autoinflammatory disease enhanced ATP release and maintained high levels of IL-1β and IL-18 in blood monocytes (82). Moreover, ATP-induced NLRP3 inflammasome activation is differentially regulated between dendritic cells and macrophages (82). As observed in the dendritic cells, stimulation with TLR ligands in the absence of ATP was sufficient to produce mature IL-1β (86). Increasing evidence suggests a regulatory role of S100A8/A9 in the NLRP3 inflammasome. For example, S100A8/A9 are capable of promoting the expression of pro-inflammatory mediators, including TNF-a, IL-1β, IL-6, and IL-8, through NF-kB activation in THP-1 cell line and human macrophages (87). Additionally, S100A8/A9 have a regulatory role in NLRP3 inflammasome, as these proteins influence the redox balance in human monocytes (87). The mechanism of enhanced ROS production is still unclear. However, evidence suggested mediating effects on ROS production via TLR4 activation (87).
It has been proposed that intracellular signals induced by S100A8/A9 are likely to be similar to LPS (87). LPS is known to trigger ROS production, which is crucial for the expression of inflammatory cytokines through the activation of redox-sensitive transcription factors (87). LPS stimulates the NF-kB-driven protein synthesis that primes the NLRP3 inflammasome, which greatly potentiates the release of mature IL-1β (87). However, several studies have shown that prolonged stimulation of monocytes without a co-signal like ATP leads to a slow activation of caspase-1 and the release of IL-1β (87–90). On their own, S100A8/A9 induce IL-1β secretion after 24 h of stimulation; however, the timeframe is greatly reduced by the addition of ATP (87). One study reported that 4 h of priming with S100A8/A9 and 30 min of stimulation with ATP were sufficient to observe caspase-1 cleavage and IL-1β release (87). It demonstrates the ROS- and NF-κB-mediated NLRP3 inflammasome priming by S100A8/A9 in human peripheral blood mononuclear cells, suggesting that S100A8/A9 plays a crucial role in redox-sensitive biological responses (87). In another study, Prunenster et al. demonstrated a governing role of S100A8/A9 in NLRP3 inflammasome induced by E-selection (13). This cell adhesion molecule is induced in endothelial cells by inflammatory stimulation, injury, or other factors. Recently, Liu Yan et al. reported a role of S100A8 as a stimulator NLRP3 inflammasome dependent pyroptosis in macrophages via activating TLR4/NF-κB signaling and inducing ROS abundance, which leads to the progression of liver fibrosis (13). Additionally, a review article by David A. Sallman et al. described how S100A9 trigger the pyroptosis through the generation of reactive oxygen species, which leads to assembly and activation of the redox-sensitive NLRP3 inflammasome, assuring propagation of the myelodysplastic syndromes (91).
3.3 DAMP-NLPR3 inflammasome signaling axis in autoimmune diseases
Multiple studies have shown that overactivation of the NLRP3 inflammasome results in excessive inflammation and tissue damage, leading to pathological conditions that contribute to autoimmune diseases (92). Mechanistically, the NLRP3 inflammasome serves as a key checkpoint in both innate and adaptive immunity during the development of autoimmune diseases. Early immune responses can be triggered by immune dysfunction, often due to the erroneous targeting of self-antigens, which is frequently associated with heightened immune activity (92, 93). Damaged tissues and cells release DAMP molecules, which act as alarm signals (94). Interestingly, direct binding of stimuli to NLRP3 is rarely observed (92), suggesting that NLRP3 detects common upstream signals induced by inflammasome activators. This mode of indirect activation is fundamental to understanding how NLRP3 functions at a molecular level. The interaction between NLRP3 inflammasome and DAMP signaling is central to elucidating this mechanism. DAMP signaling can initiate priming via NF-κB, leading to the increased expression of inflammasome components, including NLRP3, pro-caspase-1, pro-IL-18, and pro-IL-1β. After priming, NLRP3 assembly results in full inflammasome activation, culminating in caspase-1-dependent cleavage and activation of IL-1β and IL-18 (95). As a DAMP player, HMGB1 has been identified as a regulator of the NLRP3 inflammasome, indicating that NLRP3 inflammasome interacts with S100A8 and S100A9, as these two DAMP molecules share the same receptors as HMGB1, including RAGE and TLR4 (96).
DAMP molecules amplify and sustain injury-induced inflammatory responses via autocrine and paracrine mechanisms, primarily through RAGE-dependent pathways (97). This process also facilitates post-inflammatory fibrotic repair and vascular remodeling in injured tissues (98, 99). Specifically, S100A8 and S100A9 regulate the NLRP3 inflammasome through RAGE, TLR4, and NF-κB signaling pathways (87). These two S100 molecules are crucial neutrophil-derived proteins that are highly conserved between humans and mice (100–102). They have been identified as potent inducers of the pro-inflammatory phenotype in macrophages (36, 103). Consequently, through regulation of innate immune cells—particularly neutrophils and macrophages—activation of DAMP-inflammasome signaling acts as a pivotal component of the innate immune response to various stimuli, including self-antigens, thereby contributing to the onset and progression of autoimmune diseases.
Adaptive immunity extends from innate immunity (5, 93). Innate immune responses initiate the adaptive immune process, enabling the host to develop long-lasting, effective defenses (5, 93). DAMP-inflammasome signaling would play a crucial role in regulating adaptive immune responses in autoimmune diseases (93). Cytokines produced by the NLRP3 inflammasome, specifically IL-1β and IL-18 (93), promote the differentiation of naive T cells into effector and memory T cells, thereby activating adaptive immunity (3, 92). By driving both innate and adaptive immune responses, the NLRP3 inflammasome is linked to the pathogenesis of various autoimmune diseases, including type 1 diabetes, cystic fibrosis, rheumatoid arthritis, autoimmune colitis, psoriasis, systemic lupus erythematosus, and systemic sclerosis (4, 93). Elevated levels of S100A8 and S100A9 proteins, released from activated phagocytes in patients with the above-listed autoimmune disorders, serve as biomarkers for diagnosis and prognosis (104). Notably, diseases like psoriasis and arthritis exhibit patchy inflammation despite systemic immune dysregulation (93), indicating that localized DAMPs are essential for inflammatory manifestations at the sites. Conversely, systemic autoimmune conditions such as systemic sclerosis can progress to organ-specific diseases like pulmonary hypertension (105). Studies have shown that activation of DAMP signaling pathways, particularly HMGB1-RAGE, triggers macrophage activation, pulmonary endothelial cell apoptosis, and vascular smooth muscle cell proliferation, mechanisms central to pulmonary vascular remodeling post-injury (98, 99, 106, 107). As RAGE ligands, S100A8/A9 would be critical in regulating inflammasome pathways in pulmonary hypertension, a topic further elaborated below. Prior research indicates positive feedback loops mediated by DAMP signaling including TLRs, RAGE, and NF-κB (97–99, 106, 107), which are crucial for transitioning from acute to chronic immune responses as well as for switching immune cells from a pro-inflammatory to a pro-proliferative phenotype (99). These mechanisms facilitate the progression from innate to adaptive immunity, contribute to inflammation-driven fibrosis, and highlight the pivotal role of the DAMP-inflammasome signaling axis in orchestrating this transition.
3.4 Contribution of 100A8/A9 and NLRP3 in pulmonary hypertension
Pulmonary hypertension remains one of the most challenging entities to diagnose and treat due to the subtlety and nonspecificity of its symptoms and signs, the lack of availability of sensitive, noninvasive, accurate diagnostic tests (108). There is increasing evidence suggesting the association of systemic autoimmune diseases with pulmonary hypertension (108). Various studies have demonstrated the role of S100A8/A9 in promoting inflammation, fibroblast growth, and collagen production in various lung diseases including pulmonary hypertension. S100A8/A9 is a proinflammatory factor that activates the complement factor system to amplify the inflammatory response. For example, Guo et al. have demonstrated a correlation between S100A8/A9 and sepsis-induced lung damage (109). They have reported that S100A8/A9 expression significantly increased in the lungs of cecal ligation and puncture (CLP) operated mice (109). The knockout (KO) of S100A8/A9 noticeably mitigated pulmonary inflammation, vascular leakage, and acute lung injury, resulting in improved survival outcomes in septic mice (109). In another study, Du et al. demonstrated a role of S100A8/A9 as an initial proinflammatory factor that triggers cardiac fibroblasts activation, thus amplifying the inflammatory response and initiating cardiac damage (110).
NLPR3 inflammasomes are multi-protein complexes involved in sensing both endogenous and exogenous cellular stress. They are triggered by DAPMs and PAMPs based on the identity of pattern recognition receptor within the complex (2). The formation of the NLRP3 inflammasome complex is beneficial to the proximity-induced autocatalytic activation of pro-caspase-1 to cleaved caspase-1. In this process, the role of caspase-1 downstream is the cleavage of cytokines such as pro-IL-1β and pro-IL-18 to their biologically active forms. Herein, caspase-1 additionally cleaves GSDMD (111). GSDMD is a protein in which the N-terminal subunits assemble into a multi-unit complex, forming a pore in the plasma membrane that acts as a key executioner in the release of active IL-1β and induction of pyroptosis (112). A number of studies have demonstrated the association of NLRP3 inflammasome and GSDMD in pulmonary hypertension. Both NLRP3 and GSDMD have been found playing a role in pulmonary hypertension by activating the proinflammatory cytokines and releasing cytosolic contents into the extracellular space (111, 113). In particular, GSDMD mediated pyroptosis plays major role in endothelial dysfunction, which leading to pulmonary hypertension (112).
Our ongoing research has also indicated the relation of DAMP molecules and NLRP3 inflammasome in inducing pulmonary hypertension, which is consistent with other studies (85, 111). DAMP molecules regulate NLRP3 inflammasome through the receptors RAGE and TLR4 (87), which then triggers a signaling cascade involving NF-κB transcription factor, ultimately leading to the activation of the NLRP3 inflammasome complex and the secretion of GSDMD, causing cell death through pyroptosis (114). DAMPs such as S100A8/A9 are reported as crucial components for inducing NLRP3 inflammasome priming. S100A8/A9 are proinflammatory mediators released by myeloid cells during many acute and chronic inflammatory disorders, suggesting a regulatory role in NLRP3 inflammasome (13, 29, 83). However, the contribution of NLRP3 inflammasome, S100A8/A9, and GSDMD as a team to pulmonary hypertension is unclear. Understanding the interactive and feedback mechanism between NLRP3 inflammasome, S100A8/A9, and GSDMD is necessary to explore their role in pulmonary hypertension. Moreover, finding the precise mechanism of how to maintain regulated NLRP3 activation might be the key to control various autoimmune diseases including pulmonary hypertension, which has yet to be investigated.
4 Conclusion
In this review, we summarized the immunoregulatory mechanisms by which S100 family proteins regulate the inflammasome signaling axis. A fundamental commonality among S100 family proteins is their engagement of the DAMP–NLRP3 pathway. Many members—including S100A8/A9, S100A12, and S100B—interact with DAMP receptors such as RAGE and TLR4, leading to NF-κB activation, ROS generation, and subsequent inflammasome priming (12, 20, 29, 87, 95). These shared pathways enable S100 family proteins to amplify inflammatory responses across diverse pathological contexts.
Within this family, S100A8 and S100A9 possess distinctive regulatory features. They are among the most abundantly expressed DAMPs in neutrophils and monocytes (29, 38), constituting up to 40% of neutrophil cytosolic protein and approximately 5% in monocytes (39, 115). With such high expression and potent immunoregulatory properties, they can influence both the priming and activation phases of NLRP3 inflammasome assembly (29, 87, 95). Notably, S100A8/A9 can synergize with secondary signals such as ATP to accelerate IL-1β maturation (29, 87), promote pyroptosis via GSDMD activation (13, 91), and sustain inflammatory loops in chronic diseases. The dual role of S100A8/A9 as potent inflammatory mediators, DAMP molecules, and sensitive biomarkers, combined with their strong translational relevance in conditions such as pulmonary hypertension and autoimmune diseases (87, 98, 99, 105, 106), underpins our emphasis on these proteins as the primary focus of this review. At the same time, we recognize the important contributions of other S100 family members, which are summarized in Table 1, and highlight the need for further studies to delineate their individual and overlapping roles, as well as the interactive mechanisms between different S100 proteins in NLRP3 inflammasome regulation.
S100A8 and S100A9 levels are markedly increased in a wide range of inflammatory conditions, including autoimmune diseases, cancers, and neurodegenerative disorders, and they may serve as effective biomarkers for disease detection and prognosis (116). As key DAMPs, these proteins are crucial in host anti-infective immunity. Excessive activation of the NLRP3 inflammasome is linked to the development of various autoimmune diseases, underscoring the importance of maintaining its activity within a regulated range. Further investigation is needed to elucidate the mechanistic relationships between NLRP3 inflammasome signaling and DAMPs in diverse autoimmune diseases. In particular, dissecting the feedback processes between the NLRP3 inflammasome, S100A8/A9, and GSDMD could be a crucial step in understanding their roles in inflammatory pathology. Such insights may ultimately facilitate the development of strategies to modulate the DAMP–NLRP3 axis and maintain balanced inflammasome activation, aiding in the diagnosis and treatment of autoimmune and inflammatory diseases, including pulmonary hypertension, which remains one of the most challenging conditions to diagnose and manage today.
Author contributions
QL: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. SA: Visualization, Writing – original draft, Investigation, Software.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Institutes of Health grant R01HL166717 (to QL, MPI), the American Heart Association grant #938614/2022 (to QL), and the Johns Hopkins University Stimulating and Advancing ACCM Research (StAAR) grant (to QL).
Acknowledgments
We thank Brittni Delmaine for editing this article in manuscript form.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dai Y, Zhou J, and Shi C. Inflammasome: structure, biological functions, and therapeutic targets. MedComm (2020). (2023) 4:e391. doi: 10.1002/mco2.391
2. de Zoete MR, Palm NW, Zhu S, and Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. (2014) 6:a016287. doi: 10.1101/cshperspect.a016287
3. Kim YK, Shin JS, and Nahm MH. Nod-like receptors in infection, immunity, and diseases. Yonsei Med J. (2016) 57:5–14. doi: 10.3349/ymj.2016.57.1.5
4. Chen Y, Ye X, Escames G, Lei W, Zhang X, Li M, et al. The nlrp3 inflammasome: contributions to inflammation-related diseases. Cell Mol Biol Lett. (2023) 28:51. doi: 10.1186/s11658-023-00462-9
5. Kelley N, Jeltema D, Duan Y, and He Y. The Nlrp3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
6. Sharma BR and Kanneganti TD. Nlrp3 inflammasome in cancer and metabolic diseases. Nat Immunol. (2021) 22:550–9. doi: 10.1038/s41590-021-00886-5
7. Toldo S, Mezzaroma E, Buckley LF, Potere N, Di Nisio M, Biondi-Zoccai G, et al. Targeting the Nlrp3 inflammasome in cardiovascular diseases. Pharmacol Ther. (2022) 236:108053. doi: 10.1016/j.pharmthera.2021.108053
8. Sreejit G, Flynn MC, Patil M, Krishnamurthy P, Murphy AJ, and Nagareddy PR. S100 family proteins in inflammation and beyond. Adv Clin Chem. (2020) 98:173–231. doi: 10.1016/bs.acc.2020.02.006
9. Xia C, Braunstein Z, Toomey AC, Zhong J, and Rao X. S100 proteins as an important regulator of macrophage inflammation. Front Immunol. (2017) 8:1908. doi: 10.3389/fimmu.2017.01908
10. Xia P, Ji X, Yan L, Lian S, Chen Z, and Luo Y. Roles of S100a8, S100a9 and S100a12 in infection, inflammation and immunity. Immunology. (2024) 171:365–76. doi: 10.1111/imm.13722
11. Land WG. The role of damage-associated molecular patterns in human diseases: part I - promoting inflammation and immunity. Sultan Qaboos Univ Med J. (2015) 15:e9–e21. doi: 10.18295/2075-0528.1648
12. Foell D, Wittkowski H, Vogl T, and Roth J. S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J Leukoc Biol. (2007) 81:28–37. doi: 10.1189/jlb.0306170
13. Liu Y, Kong X, You Y, Xiang L, Zhang Y, Wu R, et al. S100a8-mediated nlrp3 inflammasome-dependent pyroptosis in macrophages facilitates liver fibrosis progression. Cells. (2022) 11:3579. doi: 10.3390/cells11223579
14. Cai Z, Xie Q, Hu T, Yao Q, Zhao J, Wu Q, et al. S100a8/A9 in myocardial infarction: A promising biomarker and therapeutic target. Front Cell Dev Biol. (2020) 8:603902. doi: 10.3389/fcell.2020.603902
15. Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med. (2013) 13:24–57. doi: 10.2174/156652413804486214
16. Sreejit G, Flynn MC, Patil M, Krishnamurthy P, Murphy AJ, and Nagareddy PR. S100 family proteins in inflammation and beyond. In: Makowski GS, editor. Adv Clin Chem. (2020) 98:173–231. doi: 10.1016/bs.acc.2020.02.006
17. Liang H, Li J, and Zhang K. Pathogenic role of S100 proteins in psoriasis. Front Immunol. (2023) 14:1191645. doi: 10.3389/fimmu.2023.1191645
18. Bresnick AR, Weber DJ, and Zimmer DB. S100 proteins in cancer. Nat Rev Cancer. (2015) 15:96–109. doi: 10.1038/nrc3893
19. Santamaria-Kisiel L, Rintala-Dempsey AC, and Shaw GS. Calcium-dependent and -independent interactions of the S100 protein family. Biochem J. (2006) 396:201–14. doi: 10.1042/bj20060195
20. Gonzalez LL, Garrie K, and Turner MD. Role of S100 proteins in health and disease. Biochim Biophys Acta (BBA) - Mol Cell Res. (2020) 1867:118677. doi: 10.1016/j.bbamcr.2020.118677
21. Oesterle A and Bowman MA. S100a12 and the S100/calgranulins: emerging biomarkers for atherosclerosis and possibly therapeutic targets. Arterioscler Thromb Vasc Biol. (2015) 35:2496–507. doi: 10.1161/atvbaha.115.302072
22. Bogdanova MV, Rameev VV, Kozlovskaya LV, Fedorov ES, and Salugina SO. Serum calgranulin C is a highly sensitive autoinflammation activity indicator in patients with familial periodic fevers. Ter Arkh. (2016) 88:58–64. doi: 10.17116/terarkh201688658-64
23. Kallinich T, Wittkowski H, Keitzer R, Roth J, and Foell D. Neutrophil-derived S100a12 as novel biomarker of inflammation in familial mediterranean fever. Ann Rheum Dis. (2010) 69:677–82. doi: 10.1136/ard.2009.114363
24. Kim H-J, Kang HJ, Lee H, Lee S-T, Yu M-H, Kim H, et al. Identification of S100a8 and S100a9 as serological markers for colorectal cancer. J Proteome Res. (2009) 8:1368–79. doi: 10.1021/pr8007573
25. Huang X, Liu J, and Huang W. Identification of S100a8 as a common diagnostic biomarkers and exploring potential pathogenesis for osteoarthritis and metabolic syndrome. Front Immunol. (2023) 14:1185275. doi: 10.3389/fimmu.2023.1185275
26. Ionita MG, Vink A, Dijke IE, Laman JD, Peeters W, van der Kraak PH, et al. High levels of myeloid-related protein 14 in human atherosclerotic plaques correlate with the characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol. (2009) 29:1220–7. doi: 10.1161/atvbaha.109.190314
27. Xia GL, Wang YK, and Huang ZQ. The correlation of serum myeloid-related protein-8/14 and eosinophil cationic protein in patients with coronary artery disease. BioMed Res Int. (2016) 2016:4980251. doi: 10.1155/2016/4980251
28. Geven EJ, van den Bosch MH, Di Ceglie I, Ascone G, Abdollahi-Roodsaz S, Sloetjes AW, et al. S100a8/A9, a potent serum and molecular imaging biomarker for synovial inflammation and joint destruction in seronegative experimental arthritis. Arthritis Res Ther. (2016) 18:247. doi: 10.1186/s13075-016-1121-z
29. Wang S, Song R, Wang Z, Jing Z, Wang S, and Ma J. S100a8/A9 in inflammation. Front Immunol. (2018) 9:1298. doi: 10.3389/fimmu.2018.01298
30. Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H, et al. The S100a8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and P38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. (2006) 8:R69. doi: 10.1186/ar1939
31. Paik S, Kim JK, Silwal P, Sasakawa C, and Jo EK. An update on the regulatory mechanisms of Nlrp3 inflammasome activation. Cell Mol Immunol. (2021) 18:1141–60. doi: 10.1038/s41423-021-00670-3
32. D’Ambrosi N, Milani M, and Apolloni S. S100a4 in the physiology and pathology of the central and peripheral nervous system. Cells. (2021) 10:798. doi: 10.3390/cells10040798
33. Pruenster M, Immler R, Roth J, Kuchler T, Bromberger T, Napoli M, et al. E-selectin-mediated rapid nlrp3 inflammasome activation regulates S100a8/S100a9 release from neutrophils via transient gasdermin D pore formation. Nat Immunol. (2023) 24:2021–31. doi: 10.1038/s41590-023-01656-1
34. Yang Z, Yan WX, Cai H, Tedla N, Armishaw C, Di Girolamo N, et al. S100a12 provokes mast cell activation: A potential amplification pathway in asthma and innate immunity. J Allergy Clin Immunol. (2007) 119:106–14. doi: 10.1016/j.jaci.2006.08.021
35. Zhou H, Zhao C, Shao R, Xu Y, and Zhao W. The functions and regulatory pathways of S100a8/A9 and its receptors in cancers. Front Pharmacol. (2023) 14:1187741. doi: 10.3389/fphar.2023.1187741
36. Averill MM, Kerkhoff C, and Bornfeldt KE. S100a8 and S100a9 in cardiovascular biology and disease. Arteriosclerosis Thrombosis Vasc Biol. (2012) 32:223–9. doi: 10.1161/ATVBAHA.111.236927
37. Lee J, Kim H, Kim M, Yoon S, and Lee S. Role of lymphoid lineage cells aberrantly expressing alarmins S100a8/A9 in determining the severity of Covid-19. Genes Genomics. (2023) 45:337–46. doi: 10.1007/s13258-022-01285-2
38. Xu Z, Cheng C, Kong R, Liu Y, Wang S, Ma Y, et al. S100a8 and S100a9, both transcriptionally regulated by pu.1, promote epithelial-mesenchymal transformation (Emt) and invasive growth of dermal keratinocytes during scar formation post burn. Aging (Albany NY). (2021) 13:15523–37. doi: 10.18632/aging.203112
39. Vogl T, Gharibyan AL, and Morozova-Roche LA. Pro-inflammatory S100a8 and S100a9 proteins: self-assembly into multifunctional native and amyloid complexes. Int J Mol Sci. (2012) 13:2893–917. doi: 10.3390/ijms13032893
40. Polakowska M, Steczkiewicz K, Szczepanowski RH, and Wysłouch-Cieszyńska A. Toward an understanding of the conformational plasticity of S100a8 and S100a9 Ca(2+)-binding proteins. J Biol Chem. (2023) 299:102952. doi: 10.1016/j.jbc.2023.102952
41. Cremers NAJ, van den Bosch MHJ, van Dalen S, Di Ceglie I, Ascone G, van de Loo F, et al. S100a8/A9 increases the mobilization of pro-inflammatory ly6chigh monocytes to the synovium during experimental osteoarthritis. Arthritis Res Ther. (2017) 19:217. doi: 10.1186/s13075-017-1426-6
42. Ma M, Jiang W, and Zhou R. Damps and damp-sensing receptors in inflammation and diseases. Immunity. (2024) 57:752–71. doi: 10.1016/j.immuni.2024.03.002
43. Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest. (2012) 122:2711–9. doi: 10.1172/jci62423
44. Newton K and Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. (2012) 4:a006049. doi: 10.1101/cshperspect.a006049
45. Seong S-Y and Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. (2004) 4:469–78. doi: 10.1038/nri1372
46. Tang D, Kang R, Coyne CB, Zeh HJ, and Lotze MT. Pamps and damps: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
47. Pisetsky DS, Erlandsson-Harris H, and Andersson U. High-mobility group box protein 1 (Hmgb1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther. (2008) 10:209. doi: 10.1186/ar2440
48. Yang H, Antoine DJ, Andersson U, and Tracey KJ. The many faces of hmgb1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. (2013) 93:865–73. doi: 10.1189/jlb.1212662
49. Lin Q, Fang J, Fang D, Li B, Zhou H, and Su SB. Production of recombinant human hmgb1 and anti-hmgb1 rabbit serum. Int Immunopharmacol. (2011) 11:646–51. doi: 10.1016/j.intimp.2011.01.005
50. Ge Y, Huang M, and Yao YM. The effect and regulatory mechanism of high mobility group box-1 protein on immune cells in inflammatory diseases. Cells. (2021) 10:1044. doi: 10.3390/cells10051044
51. Klune JR, Dhupar R, Cardinal J, Billiar TR, and Tsung A. Hmgb1: endogenous danger signaling. Mol Med. (2008) 14:476–84. doi: 10.2119/2008-00034.Klune
52. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. (2007) 13:1042–9. doi: 10.1038/nm1638
53. Roh JS and Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
54. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
55. Wang LC, Zhang HY, Shao L, Chen L, Liu ZH, He X, et al. S100a12 levels in synovial fluid may reflect clinical severity in patients with primary knee osteoarthritis. Biomarkers. (2013) 18:216–20. doi: 10.3109/1354750x.2013.766262
56. Nakashima M, Sakai T, Hiraiwa H, Hamada T, Omachi T, Ono Y, et al. Role of S100a12 in the pathogenesis of osteoarthritis. Biochem Biophys Res Commun. (2012) 422:508–14. doi: 10.1016/j.bbrc.2012.05.036
57. Venegas C and Heneka MT. Danger-associated molecular patterns in Alzheimer’s disease. J Leukoc Biol. (2017) 101:87–98. doi: 10.1189/jlb.3MR0416-204R
58. Herrero MT, Estrada C, Maatouk L, and Vyas S. Inflammation in Parkinson’s disease: role of glucocorticoids. Front Neuroanat. (2015) 9:32. doi: 10.3389/fnana.2015.00032
59. Festoff BW, Sajja RK, van Dreden P, and Cucullo L. Hmgb1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J Neuroinflamm. (2016) 13:194. doi: 10.1186/s12974-016-0670-z
60. Chaves ML, Camozzato AL, Ferreira ED, Piazenski I, Kochhann R, Dall’Igna O, et al. Serum levels of S100b and Nse proteins in Alzheimer’s disease patients. J Neuroinflamm. (2010) 7:6. doi: 10.1186/1742-2094-7-6
61. Yang Y, Han C, Guo L, and Guan Q. High expression of the Hmgb1–Tlr4 axis and its downstream signaling factors in patients with Parkinson’s disease and the relationship of pathological staging. Brain Behav. (2018) 8:e00948. doi: 10.1002/brb3.948
62. Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al. S100b Is Increased in Parkinson’s Disease and Ablation Protects against Mptp-Induced Toxicity through the Rage and Tnf-A Pathway. Brain. (2012) 135:3336–47. doi: 10.1093/brain/aws250
63. Hernandez C, Huebener P, and Schwabe RF. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene. (2016) 35:5931–41. doi: 10.1038/onc.2016.104
64. Grivennikov SI, Greten FR, and Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
65. Lin Q, Jin S, Han M, Zheng W, Liu J, and Wei X. Inflammation in the tumor microenvironment. J Immunol Res. (2018) 2018:1965847. doi: 10.1155/2018/1965847
66. Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. (2009) 16:295–308. doi: 10.1016/j.ccr.2009.08.021
67. McInnes IB and Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
68. van Boekel MA, Vossenaar ER, van den Hoogen FH, and van Venrooij WJ. Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. (2002) 4:87–93. doi: 10.1186/ar395
69. Baillet A, Trocmé C, Berthier S, Arlotto M, Grange L, Chenau J, et al. Synovial fluid proteomic fingerprint: S100a8, S100a9 and S100a12 proteins discriminate rheumatoid arthritis from other inflammatory joint diseases. Rheumatology. (2010) 49:671–82. doi: 10.1093/rheumatology/kep452
70. Andrés Cerezo L, Šumová B, Prajzlerová K, Veigl D, Damgaard D, Nielsen CH, et al. Calgizzarin (S100a11): A novel inflammatory mediator associated with disease activity of rheumatoid arthritis. Arthritis Res Ther. (2017) 19:79. doi: 10.1186/s13075-017-1288-y
71. Goldstein RS, Bruchfeld A, Yang L, Qureshi AR, Gallowitsch-Puerta M, Patel NB, et al. Cholinergic anti-inflammatory pathway activity and high mobility group box-1 (Hmgb1) serum levels in patients with rheumatoid arthritis. Mol Med. (2007) 13:210–5. doi: 10.2119/2006–00108.Goldstein
72. Schiopu A and Cotoi OS. S100a8 and S100a9: damps at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflammation. (2013) 2013:828354. doi: 10.1155/2013/828354
73. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352:1685–95. doi: 10.1056/NEJMra043430
74. Ligthart S, Sedaghat S, Ikram MA, Hofman A, Franco OH, and Dehghan A. En-rage: A novel inflammatory marker for incident coronary heart disease. Arterioscler Thromb Vasc Biol. (2014) 34:2695–9. doi: 10.1161/atvbaha.114.304306
75. Xiao G, Zhuang W, Wang T, Lian G, Luo L, Ye C, et al. Transcriptomic analysis identifies toll-like and nod-like pathways and necroptosis in pulmonary arterial hypertension. J Cell Mol Med. (2020) 24:11409–21. doi: 10.1111/jcmm.15745
76. Franchi L, Muñoz-Planillo R, and Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012) 13:325–32. doi: 10.1038/ni.2231
77. Jin M, Yang F, Yang I, Yin Y, Luo JJ, Wang H, et al. Uric acid, hyperuricemia and vascular diseases. Front Biosci (Landmark Ed). (2012) 17:656–69. doi: 10.2741/3950
78. Kligman D and Hilt DC. The S100 protein family. Trends Biochem Sci. (1988) 13:437–43. doi: 10.1016/0968-0004(88)90218-6
79. Zhou Y, Hann J, Schenten V, Plançon S, Bueb JL, Tolle F, et al. Role of S100a8/A9 for cytokine secretion, revealed in neutrophils derived from Er-Hoxb8 progenitors. Int J Mol Sci. (2021) 22:8845. doi: 10.3390/ijms22168845
80. Singh P and Ali SA. Multifunctional role of S100 protein family in the immune system: an update. Cells. (2022) 11:2274. doi: 10.3390/cells11152274
81. Yang CS, Shin DM, and Jo EK. The role of Nlr-related protein 3 inflammasome in host defense and inflammatory diseases. Int Neurourol J. (2012) 16:2–12. doi: 10.5213/inj.2012.16.1.2
82. Jo EK, Kim JK, Shin DM, and Sasakawa C. Molecular mechanisms regulating Nlrp3 inflammasome activation. Cell Mol Immunol. (2016) 13:148–59. doi: 10.1038/cmi.2015.95
83. Missiroli S, Perrone M, Boncompagni C, Borghi C, Campagnaro A, Marchetti F, et al. Targeting the Nlrp3 inflammasome as a new therapeutic option for overcoming cancer. Cancers. (2021) 13:2297. doi: 10.3390/cancers13102297
84. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: Nf-Kappab activating pattern recognition and cytokine receptors license Nlrp3 inflammasome activation by regulating Nlrp3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
85. Koizumi Y, Toma C, Higa N, Nohara T, Nakasone N, and Suzuki T. Inflammasome activation via intracellular Nlrs triggered by bacterial infection. Cell Microbiol. (2012) 14:149–54. doi: 10.1111/j.1462-5822.2011.01707.x
86. He Y, Franchi L, and Núñez G. Tlr agonists stimulate Nlrp3-dependent Il-1β Production independently of the purinergic P2x7 receptor in dendritic cells and in vivo. J Immunol. (2013) 190:334–9. doi: 10.4049/jimmunol.1202737
87. Simard JC, Cesaro A, Chapeton-Montes J, Tardif M, Antoine F, Girard D, et al. S100a8 and S100a9 induce cytokine expression and regulate the Nlrp3 inflammasome via Ros-dependent activation of Nf-Kb(1.). PloS One. (2013) 8:e72138. doi: 10.1371/journal.pone.0072138
88. Cheneval D, Ramage P, Kastelic T, Szelestenyi T, Niggli H, Hemmig R, et al. Increased mature interleukin-1beta (Il-1beta) secretion from Thp-1 cells induced by Nigericin is a result of activation of P45 Il-1beta-converting enzyme processing. J Biol Chem. (1998) 273:17846–51. doi: 10.1074/jbc.273.28.17846
89. Schumann RR, Belka C, Reuter D, Lamping N, Kirschning CJ, Weber JR, et al. Lipopolysaccharide activates caspase-1 (Interleukin-1-converting enzyme) in cultured monocytic and endothelial cells. Blood. (1998) 91:577–84. doi: 10.1182/blood.V91.2.577
90. Grahames CB, Michel AD, Chessell IP, and Humphrey PP. Pharmacological characterization of Atp- and Lps-induced Il-1beta release in human monocytes. Br J Pharmacol. (1999) 127:1915–21. doi: 10.1038/sj.bjp.0702732
91. Sallman DA, Cluzeau T, Basiorka AA, and List A. Unraveling the pathogenesis of mds: the Nlrp3 inflammasome and pyroptosis drive the Mds phenotype. Front Oncol. (2016) 6:151. doi: 10.3389/fonc.2016.00151
92. Zhang Y, Yang W, Li W, and Zhao Y. Nlrp3 inflammasome: checkpoint connecting innate and adaptive immunity in autoimmune diseases. Front Immunol. (2021) 12:732933. doi: 10.3389/fimmu.2021.732933
93. Li Z, Guo J, and Bi L. Role of the Nlrp3 inflammasome in autoimmune diseases. Biomedicine Pharmacotherapy. (2020) 130:110542. doi: 10.1016/j.biopha.2020.110542
94. Lin Q, Yang XP, Fang D, Ren X, Zhou H, Fang J, et al. High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler Thromb Vasc Biol. (2011) 31:1024–32. doi: 10.1161/ATVBAHA.111.224048
95. Swanson KV, Deng M, and Ting JP. The Nlrp3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
96. Lin Q, Li M, Fang D, Fang J, and Su SB. The essential roles of toll-like receptor signaling pathways in sterile inflammatory diseases. Int Immunopharmacol. (2011) 11:1422–32. doi: 10.1016/j.intimp.2011.04.026
97. van Beijnum JR, Buurman WA, and Griffioen AW. Convergence and amplification of toll-like receptor (Tlr) and receptor for advanced glycation end products (Rage) signaling pathways via high mobility group B1 (Hmgb1). Angiogenesis. (2008) 11:91–9. doi: 10.1007/s10456-008-9093-5
98. Lin Q, Fan C, Gomez-Arroyo J, Van Raemdonck K, Meuchel LW, Skinner JT, et al. Himf (Hypoxia-induced mitogenic factor) signaling mediates the Hmgb1 (High mobility group box 1)-dependent endothelial and smooth muscle cell crosstalk in pulmonary hypertension. Arterioscler Thromb Vasc Biol. (2019) 39:2505–19. doi: 10.1161/ATVBAHA.119.312907
99. Lin Q, Fan C, Skinner JT, Hunter EN, Macdonald AA, Illei PB, et al. Relmalpha licenses macrophages for damage-associated molecular pattern activation to instigate pulmonary vascular remodeling. J Immunol. (2019) 203:2862–71. doi: 10.4049/jimmunol.1900535
100. Stackowicz J, Jonsson F, and Reber LL. Mouse models and tools for the in vivo study of neutrophils. Front Immunol. (2019) 10:3130. doi: 10.3389/fimmu.2019.03130
101. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. (2019) 50:1317–34.e10. doi: 10.1016/j.immuni.2019.03.009
102. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, and Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. (2020) 20:485–503. doi: 10.1038/s41568-020-0281-y
103. Ganta VC, Choi M, Farber CR, and Annex BH. Antiangiogenic vegf(165)B regulates macrophage polarization via S100a8/S100a9 in peripheral artery disease. Circulation. (2019) 139:226–42. doi: 10.1161/CIRCULATIONAHA.118.034165
104. Kerkhoff C, Voss A, Scholzen TE, Averill MM, Zanker KS, and Bornfeldt KE. Novel insights into the role of S100a8/A9 in skin biology. Exp Dermatol. (2012) 21:822–6. doi: 10.1111/j.1600-0625.2012.01571.x
105. Lin Q and Johns RA. Resistin family proteins in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. (2020) 319:L422–L34. doi: 10.1152/ajplung.00040.2020
106. Lin Q, Kumar S, Kariyawasam U, Yang X, Yang W, Skinner JT, et al. Human resistin induces cardiac dysfunction in pulmonary hypertension. J Am Heart Assoc. (2023) 12:e027621. doi: 10.1161/JAHA.122.027621
107. Lin Q, Price SA, Skinner JT, Hu B, Fan C, Yamaji-Kegan K, et al. Systemic evaluation and localization of resistin expression in normal human tissues by a newly developed monoclonal antibody. PloS One. (2020) 15:e0235546. doi: 10.1371/journal.pone.0235546
108. Gurubhagavatula I and Palevsky HI. Pulmonary hypertension in systemic autoimmune disease. Rheumatic Dis Clinics North America. (1997) 23:365–94. doi: 10.1016/S0889-857X(05)70335-5
109. Yu J, Zhao B, Pi Q, Zhou G, Cheng Z, Qu C, et al. Deficiency of S100a8/A9 attenuates pulmonary microvascular leakage in septic mice. Respir Res. (2023) 24:288. doi: 10.1186/s12931-023-02594-0
110. Wu Y, Li Y, Zhang C, A X, Wang Y, Cui W, et al. S100a8/A9 released by cd11b+Gr1+ Neutrophils activates cardiac fibroblasts to initiate angiotensin ii–induced cardiac inflammation and injury. Hypertension. (2014) 63:1241–50. doi: 10.1161/HYPERTENSIONAHA.113.02843
111. Foley A, Steinberg BE, and Goldenberg NM. Inflammasome activation in pulmonary arterial hypertension. Front Med (Lausanne). (2021) 8:826557. doi: 10.3389/fmed.2021.826557
112. Mulla J, Katti R, and Scott MJ. The role of gasdermin-D-mediated pyroptosis in organ injury and its therapeutic implications. Organogenesis. (2023) 19:2177484. doi: 10.1080/15476278.2023.2177484
113. Al-Qazazi R, Lima PDA, Prisco SZ, Potus F, Dasgupta A, Chen KH, et al. Macrophage-Nlrp3 activation promotes right ventricle failure in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2022) 206:608–24. doi: 10.1164/rccm.202110-2274OC
114. Kesavardhana S, Malireddi RKS, and Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol. (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
115. Hessian PA, Edgeworth J, and Hogg N. Mrp-8 and mrp-14, two abundant Ca(2+)-binding proteins of neutrophils and monocytes. J Leukoc Biol. (1993) 53:197–204. doi: 10.1002/jlb.53.2.197
Keywords: S100A8, S100A9, DAMP, inflammasome, gasdermin D, pulmonary hypertension
Citation: Acter S and Lin Q (2025) S100 proteins as a key immunoregulatory mechanism for NLRP3 inflammasome. Front. Immunol. 16:1663547. doi: 10.3389/fimmu.2025.1663547
Received: 10 July 2025; Accepted: 14 August 2025;
Published: 28 August 2025.
Edited by:
Soohyun Kim, Konkuk University, Republic of KoreaReviewed by:
Marcos Edgar Herkenhoff, Santa Catarina State University, BrazilMengyuan Li, Ningxia Medical College, China
Copyright © 2025 Acter and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Lin, cWxpbjJAamhtaS5lZHU=