 Abdullah Jabri1
Abdullah Jabri1 Abdulrahman Elsalti2
Abdulrahman Elsalti2 Mohamed Alsharif1
Mohamed Alsharif1 Raghad Alsharif3
Raghad Alsharif3 Tasnim Abbad1
Tasnim Abbad1 Dania Sibai1
Dania Sibai1 Bader Taftafa1
Bader Taftafa1 Abdulaziz Mhannayeh1
Abdulaziz Mhannayeh1 Mohammad Imran Khan1,4
Mohammad Imran Khan1,4 Ahmed Yaqinuddin1*
Ahmed Yaqinuddin1*- 1College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 2International School of Medicine, Istanbul Medipol University, Istanbul, Türkiye
- 3College of Medicine, Almaarefa University, Riyadh, Saudi Arabia
- 4King Faisal Specialist Hospital and Research Center, Jeddah, Saudi Arabia
β-cell destruction by autoreactive T cells is a hey hallmark of type 1 diabetes mellitus (T1D). Epigenetic mechanisms—including DNA methylation, histone modifications, chromatin remodeling, and non-coding RNAs—play critical roles in regulating T-cell development, activation, and tolerance. Disruption of these processes contributes to immune imbalance and the onset of T1D. This review summarizes current insights into how epigenetic regulation shapes T-cell function and highlights emerging evidence linking these changes to environmental influences such as gut microbiota, diet, and viral infections. Exploring the interaction between genetic susceptibility and environmental triggers through an epigenetic framework not only advances our understanding of T1D pathogenesis but also provides opportunities for biomarker discovery and the development of targeted epigenetic therapies. With further research, these advances hold promise for improving precision medicine strategies in T1D.
1 Introduction
Type 1 diabetes (T1D) is an autoimmune disease in which the immune system mistakenly attacks and destroys pancreatic β-cells, eventually leading to insulin shortage (1). This β-cell loss primarily happens via autoreactive T-cell mechanisms in genetically predisposed individuals, often initiated or modulated by environmental factors (2). Epigenetic regulation—through mechanisms such as DNA methylation, histone modifications, non-coding RNAs, and chromatin remodeling—has emerged as a critical mediator linking genetic susceptibility to environmental influences in T1D (3). These heritable yet reversible modifications govern T-cell development, activation, and tolerance, thereby shaping immune balance and disease risk (4).
The disease progresses through three clinically and biologically distinct, largely silent stages (5, 6). In Stage 1, individuals have detectable autoantibodies directed against pancreatic β-cell antigens, indicating an active immune attack on the islets, but glucose metabolism remains within the normal range and there are no symptoms. In Stage 2, ongoing immune-mediated β-cell injury produces measurable impairment of glucose regulation, for example abnormal responses on glucose tolerance testing or a rising A1c although fasting glucose and symptom status may still be non-diabetic. In Stage 3, β-cell loss reaches a threshold at which persistent hyperglycemia develops, meeting diagnostic criteria for diabetes and often accompanied by typical symptoms such as polyuria, polydipsia, and weight loss. CD4+ and CD8+ T-cells are essential factors in the progression of T1D and significant elements of the islet infiltration. Initially, autoreactive T cells are stimulated by β-cell antigens shown by antigen-presenting cells (APCs) (7). The activated CD4+ T-cells invade the pancreas and are believed to aid in β-cell damage through the activation of macrophages and CD8+ T-cells. These in turn are directly responsible for the destruction of β-cells through their interaction with major histocompatibility complex (MHC) class I molecules and by the secretion of perforin and granzyme (8). Usually, regulatory T cells (Tregs), the main regulators of inflammatory responses, are responsible for immune tolerance and homeostasis (9, 10). The lack of Tregs may become one of the reasons for the development of human autoimmune diseases like T1D, whereas an excess of Tregs may lead to the weakening of the immune response to cancer or infections (11). Established T1D risk genes include the human leukocyte antigen (HLA) region, insulin (INS), protein tyrosine phosphatase non-receptor type 22 (PTPN22), interleukin-2 receptor alpha (IL2RA), and cytotoxic T-lymphocyte associated protein 4 (CTLA4), among others (12). Importantly, their expression is strongly influenced by epigenetic mechanisms, which may explain how environmental exposures—such as viral infections, microbiota alterations, or dietary factors—trigger or accelerate disease onset. In this review, we aimed to explore the role of epigenetic regulation of T cells in the pathogenesis of T1D, with a particular focus on how mechanisms such as DNA methylation, histone modifications, non-coding RNAs, and chromatin remodeling influence T-cell development, activation, and tolerance. By summarizing current findings on epigenetic dysregulation in both CD4+ and CD8+ T-cell subsets, and examining the interplay between environmental triggers and genetic susceptibility, we highlight the growing importance of epigenetic biomarkers for diagnosis and the therapeutic potential of epigenome-targeting strategies.
2 Overview of epigenetic modifications
2.1 DNA methylation: a versatile and targeted regulator
DNA methylation is a key regulatory process of the addition of a methyl group to cytosine bases within Cytosine-phosphate-Guanine (CpG) dinucleotides and is mediated by DNA methyltransferases (DNMTs). In general, promoter methylation is silencing and demethylation is activation (13). For example, the transcription factor FOXP3, essential for Treg development, is silenced when its regulatory regions are hypermethylated and activated when hypomethylated (14). This helps to maintain immune tolerance and Treg lineage fidelity.
Region specific hypomethylation also activates immune related genes. Genes such as HLA-DQB1 and GAD2 have lower methylation at their promoters and enhancers under immunostimulatory conditions which in turn enhance antigen presentation and cytokine responsiveness (15). These are not stochastic but occur at defined regulatory loci, so it’s a tightly controlled system of gene activation and silencing.
Methylation variability refers to the differences in DNA methylation patterns observed across individuals, tissues, developmental stages, or environmental conditions (16). It has been noticed even between genetically identical monozygotic twins. High resolution methylome studies show differential methylation at loci including INS-IGF2, SH2B3 and MEG3 (17). This inter-individual variation gives insight into how genetically similar individuals can have different immunological outcomes. Abnormal methylation variability is often associated with pathological conditions, including cancer, autoimmune disorders, and neurological diseases, making it a valuable biomarker for disease risk and progression.
2.2 Histone modifications: balancing activation and Repression
Histone modifications are post-translational changes to the histone tails that wrap DNA into chromatin (15). These changes control chromatin accessibility and help recruit transcription factors. For example, H3K9 acetylation (H3KAc) is associated with open chromatin and active transcription, especially at immune genes like HLA-DRB1/DQB1 in APCs (13). H3K9 demethylation (H3K9me2) at loci like CTLA4 is linked to a repressive chromatin state, preserving immune checkpoints and preventing autoreactivity.
Histone acetylation patterns are sensitive to environmental cues. Microbial metabolites like butyrate, a short chain fatty acid produced by commensal Clostridium species (clusters IV/XIVa) inhibit histone deacetylases (HDACs) and promote acetylation at immune regulatory loci like FOXP3 (18). This leads to enhanced Treg differentiation and immune homeostasis. Epigenetic integration of microbiota derived signals is key to immune tolerance.
Metabolic factors also affect histone modification patterns. For example, hyperglycemia decreases the activity of NAD+-dependent deacetylases SIRT2 and SIRT6, leading to persistent acetylation at histone residues H3K9, H3K14 and H3K27 (19). These modifications impair β-cell function, alter stress response gene expression and may contribute to long term metabolic complications. Histone modifications are epigenetic sensors of both microbial and metabolic environments.
2.3 Non-coding RNAs: epigenetic regulators in health and disease
Both post-transcriptional and chromatin levels of gene expression are controlled by non-coding RNAs (ncRNAs), which include microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) (20). miRNAs usually attach to the 3′ untranslated regions of target mRNAs to prevent translation or degradation. Treg migration, cytokine signaling and immunological homeostasis are controlled by miRNAs like miR-125a-5p and miR-342 in immune cells. Changes in immune response and disease susceptibility are linked to polymorphisms in regulatory miRNAs like miR-146a and miR-155 (15).
Regulatory miRNAs like miR-375 are present in pancreatic β-cells where they have a role in insulin secretion and β-cell survival. miR-375 is upregulated in normal conditions resulting in the suppression of insulin secretion by targeting exocytosis-related genes (e.g. Myotrophin, PDK1), but chronic high glucose downregulates miR-375 and leads to dysregulated insulin release and β-cell stress (13, 15, 21). miRNAs maintain endocrine cell identity while fine-tuning immunological responses.
lncRNAs are more than 200 nucleotides long and act through various mechanisms, including chromatin looping, transcriptional interference and enhancer modulation. HI-LNC25 (LINC01370) regulates the transcription factor GLIS3 which is critical for β-cell survival and differentiation, while PLUTO promotes the expression of PDX1 a master regulator of insulin production (15). These lncRNAs have been shown to be tissue-specific epigenetic regulators. Importantly, recent evidence suggests that lncRNAs may contribute to disease susceptibility by interacting with non-coding genomic regions. More than 90% of T1D-associated single nucleotide polymorphism (SNP) are in non-coding regions, and lncRNAs are implicated in the development of autoimmune risk. One example is the SNP of NONHSAG044354 lncRNA within the BACH2 locus, a gene involved in immunoregulation and tolerance (15, 22). lncRNAs also maintain epigenetic memory by stabilizing transcriptional activity at inflammatory loci even after cytokine signaling has ceased, and thus preserve cellular identity over time (21).
2.4 Chromatin remodeling: organizing the accessible genome
Chromatin remodeling is the repositioning of nucleosomes by ATP-dependent complexes like SWI/SNF which control DNA accessibility to transcription factors (23). This is important during T-cell lineage differentiation, β-cell specification and enhancer activation (14).
Recent single-nucleus assay for transposase-accessible chromatin using sequencing (snATAC-seq) studies on over 130,000 nuclei have shown that many autoimmune risk variants map to cis-regulatory elements (cCREs) in memory CD8+ T cells and Tregs (23). These elements are required for gene accessibility of CTLA4 and FOXP3 which are central to immune regulation (22). Chromatin accessibility at these sites is controlled by transcription factor binding and is disrupted by disease associated variants.
Genome organizers such as Special AT-rich Sequence-Binding Protein 1 (SATB1) control long range enhancer-promoter interactions to shape chromatin (14). SATB1 promotes thymic growth and peripheral function in Tregs by opening chromatin at super-enhancers near FOXP3 and CTLA4 (24). Regulatory programs during T cell activation and differentiation rely on these remodeling activities. Inflammatory cytokines like IL-1β and IFN-γ also dynamically control chromatin accessibility. These signals open up closed chromatin regions enriched for IRF, STAT and NF-κB motifs (21, 22). This plasticity allows for rapid transcriptional responses in immune and endocrine cells. Furthermore, HLA class II haplotypes, DR3/DQ2 regulate allele specific chromatin remodeling (25). For example they control HLA-DRB5 in dendritic cells and immunological tolerance and antigen presentation.
3 Epigenetic dysregulation in T1D-associated T-cells
3.1 CD4+ T-Cells (Th1, Th17, Tregs)
Tregs in T1D undergo epigenetic changes that can disable them. Alterations in FOXP3 methylation have been reported in subsets of autoimmune diabetes. Examples include FOXP3 promoter/Treg-specific demethylated region (TSDR) hypermethylation and reduced FOXP3 expression in CD4+ T cells from Latent Autoimmune Diabetes in Adults (LADA) and fulminant T1D patients, and enrichment of TSDR-methylated FOXP3+IFN-γ+ cells in T1D cohorts (26–28). This epigenetic silencing can be exacerbated by environmental factors; reduced butyrate from gut dysbiosis decreases histone acetylation at the FOXP3 enhancer and further destabilizes Treg function (18). IL2RA (CD25) promoter hypermethylation limits IL-2 signaling, necessary for Treg survival and suppressive capacity (29). These changes present early in disease progression, thus may contribute to breakdown of immune tolerance before clinical onset (17).
Unlike Treg dysfunction, effector CD4+ subsets (Th1 and Th17 cells) in T1D display activating epigenetic modifications at pro-inflammatory cytokine loci. Studies of Th1/Th17 lineage-specific chromatin have shown that enhancers of cytokine genes such as IFN-γ and IL-17 are marked by activating histone modifications, including H3K27ac, which facilitates transcriptional upregulation (22, 30). Moreover, single-cell chromatin accessibility analyses suggest that T1D risk variants are enriched in Th1/Th17-specific regulatory elements, potentially altering transcription factor binding and cytokine expression (23).
It remains unclear whether Th1 and Th17 markers arise from the same cells (reflecting cellular plasticity) or from distinct subsets, as studies report both scenarios (31). Similarly, checkpoint receptor changes such as CTLA4 and PD-1 may not be uniform across all T1D patients; some studies report reduced PD-1 expression on Tregs, whereas others find normal levels. Frequencies and suppressive function of Tregs also show conflicting results across cohorts. Comparative commentary indicates that while some studies report increased Th17 cells in T1D, others find no change in IL-17–producing cells under baseline conditions. These inconsistencies underscore heterogeneity among patients and highlight the need for careful interpretation of immune signatures. Together, Treg dysfunction and Th1/Th17 hyperactivity may create a self-reinforcing cycle of autoimmunity (Figure 1).
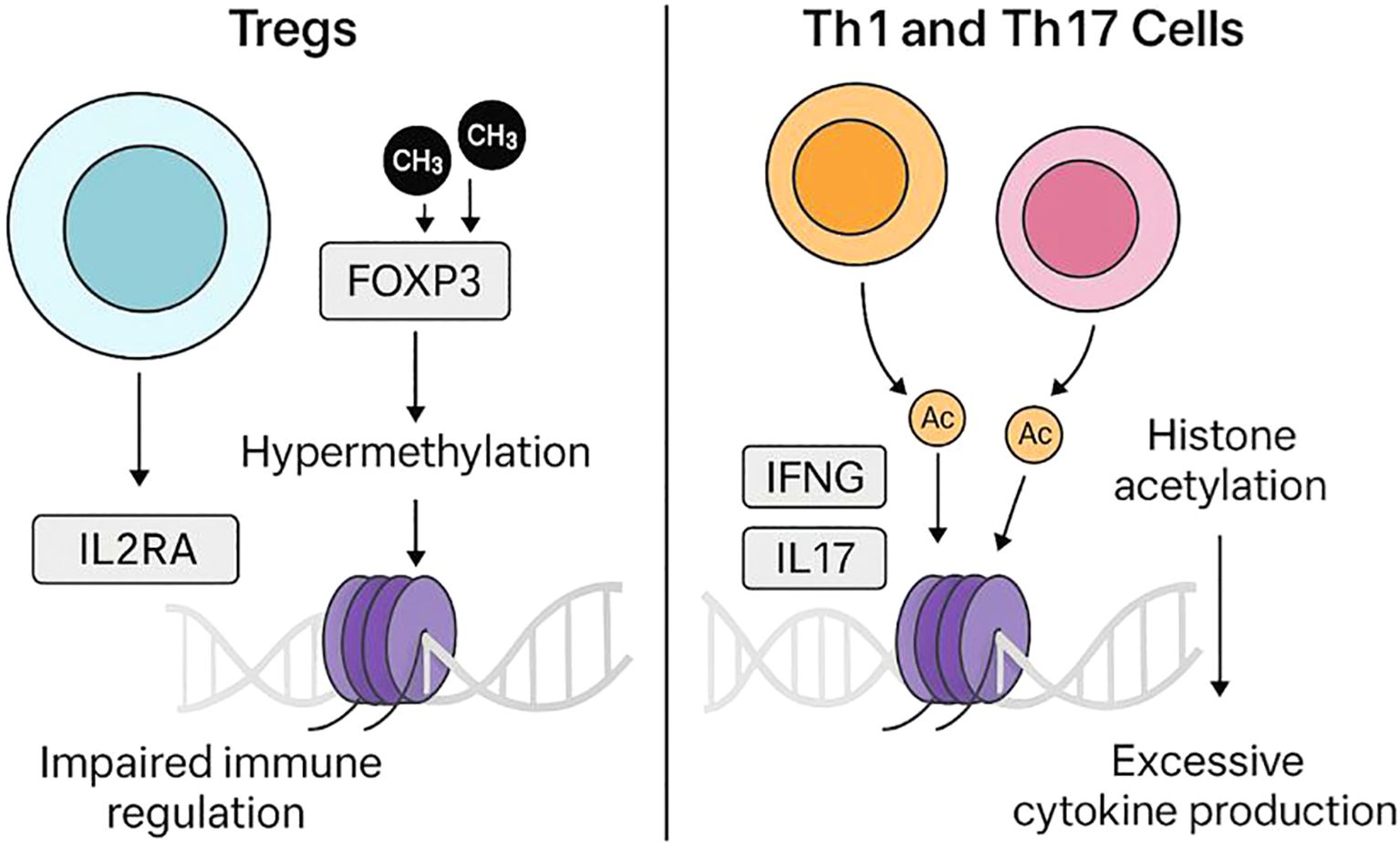
Figure 1. Epigenetic dysregulation of T-cell subsets in T1D. On the left, regulatory T cells (Tregs) exhibit altered epigenetic regulation at immune tolerance–related loci, including FOXP3 and IL2RA, which may impair their suppressive function. On the right, effector T cells (Th1 and Th17) display activating histone acetylation (e.g., H3K27ac) at cytokine gene loci (IFNG, IL17A), increasing pro-inflammatory cytokine production. Together, these opposing epigenetic changes weaken tolerance and promote autoimmunity in T1D.
3.2 CD8+ T-cells (cytotoxic T-cells)
CD8+ T-cells are primed for autoreactivity in T1D through epigenetic changes that make them more reactive to β-cell antigens. Epigenetic variation in immune cells, such as altered methylation at loci including INS and IL2RA, has been associated with T1D risk. In parallel, molecular mimicry between β-cell autoantigens (e.g., insulin, GAD65) and microbial peptides may promote activation of autoreactive T cells (16, 18). Enhancers near genes involved in cytotoxic function, such as SOCS1 (cytokine signaling) and STXBP1 (vesicle fusion) are commonly disrupted in T1D patients according to chromatin accessibility profiling (30). CD8+ T-cells are more cytotoxic in T1D patients due to these epigenetic changes.
The T1D microenvironment activates CD8+ T-cells. IFN-γ induces MHC class I on β-cells, making them more visible to cytotoxic T-cells (32). miR-23b, miR-590-5p dysregulate CD8+ T-cell survival by suppressing TRAIL and FAS (33). Notably, these epigenetic changes occur early in disease progression, as seen by hypomethylation at the LDHC locus in children who later develop autoantibodies (25). Thus, these mechanisms create a self-reinforcing cycle where epigenetic priming activates CD8+ T-cells, which in turn destroy more β-cells and release more antigen.
3.3 Dysregulation of immune tolerance
One mechanism of tolerance breakdown in T1D is epigenetic silencing of immunological checkpoint molecules. T1D patients have hypermethylation at the CTLA4 and PD-1 loci which reduces expression of these inhibitory receptors (30, 34). Variants in the CTLA4 enhancer region can make this worse by disrupting chromatin architecture and transcription factor binding (IRF1) in Tregs (24). These epigenetic changes lead to autoimmune β-cell death,compromised checkpoint function and uncontrolled T cell activation. Table 1. summarizes several epigenetic changes in association with T1D.
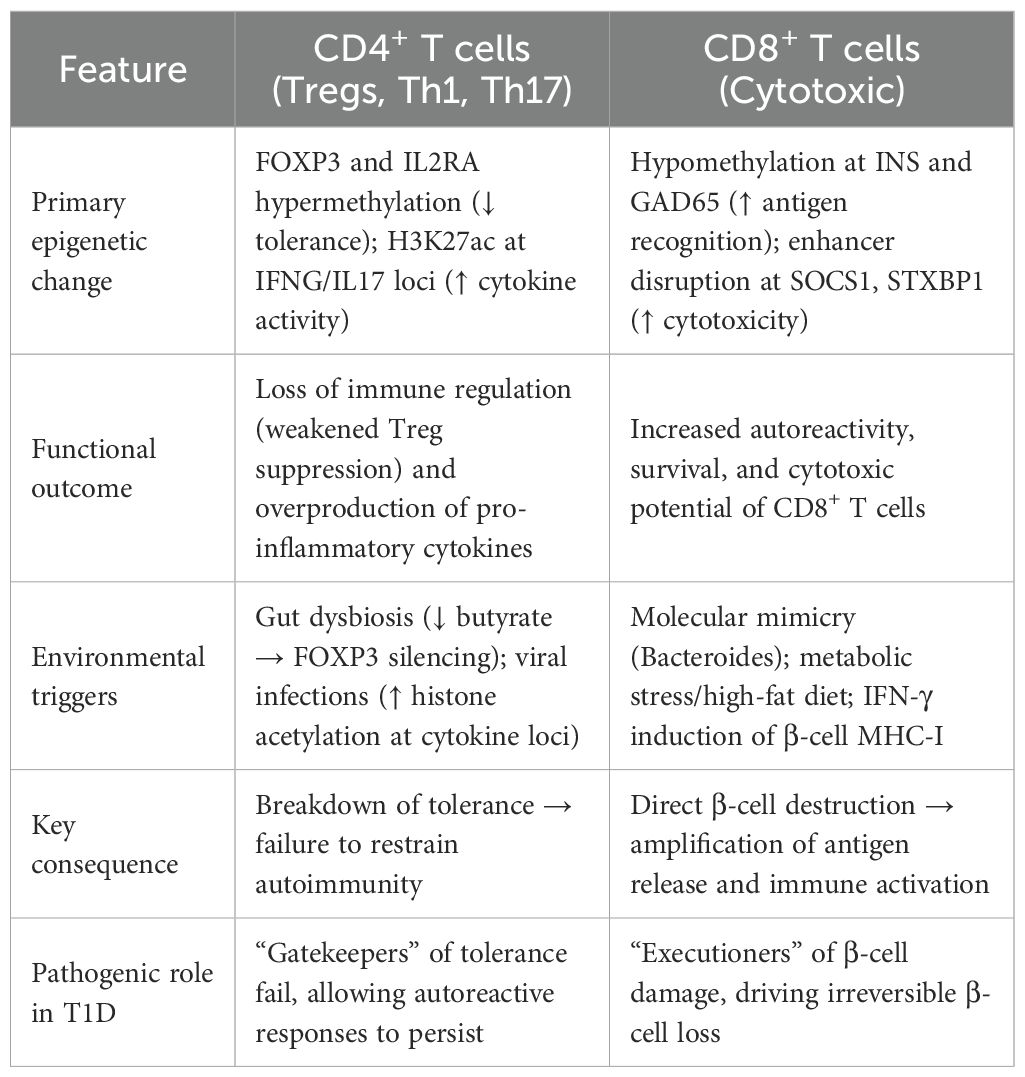
Table 1. Contrasting epigenetic regulation of CD4+ vs CD8+ T cells in T1D.
3.4 Long non-coding RNAs in T-cell regulation
lncRNAs shape T-cell fate and effector programs by scaffolding chromatin modifiers, guiding transcription-factor recruitment, and modulating enhancer–promoter communication. In Tregs, Flicr (Foxp3 long intergenic non-coding RNA) acts as a negative tuner of FOXP3, altering chromatin accessibility at Foxp3 regulatory elements; genetic ablation increases FOXP3 and improves tolerance in autoimmune-prone backgrounds, highlighting Flicr as a rheostat of Treg stability (35).
In Th1 cells, the antisense lncRNA NeST (also known as Tmevpg1/Ifng-AS1) is induced in a T-bet/STAT4–dependent manner and promotes IFNG transcription by recruiting WDR5/MLL to deposit H3K4 methylation at the Ifng locus; NeST thus reinforces Th1 polarization and IFN-γ output (36–38).
For Th2 programs, lincR-Ccr2-5′AS cooperates with GATA-3 to regulate a chemokine-receptor cluster (CCR1/2/3/5), and its knockdown impairs Th2 migration in vivo, illustrating how lncRNAs coordinate lineage-specific trafficking with gene programs (39).
4 Gene–environment–epigenome interactions
4.1 Genetic susceptibility
T1D susceptibility is strongly influenced by genetics, with 78 risk loci now identified by large GWAS and fine-mapping studies (23, 40–52). Many of these risk variants fall in regulatory elements active in immune and pancreatic cell types, suggesting functional effects on gene expression (23). Recent research indicates that epigenetic mechanisms may be involved in the development of T1D due to genetic risk variations. SNPs in INS (rs689) and IL2RA (rs12722495) in particular were linked to altered DNA methylation at immune cell promoter CpG sites. Higher methylation in CD8+ T cells was associated with the risk allele rs689, but lower methylation was found in B cells with rs12722495. These methylation alterations specific to a genotype may affect immunological functioning and increase the risk of developing the disease (53). Table 2 summarizes key genes and their associated SNPs linked to immune function, β-cell regulation, and T1D risk (49). However, many of these genes, such as CCR5, IL10, IL27, and GPX7, have been variably reported in association with T1D. For instance, CCR5 has shown associations in some populations but not others, and IL10/IL27 findings have been inconsistent across cohorts. These discrepancies suggest that some of these loci may have modest effect sizes or population-specific effects. Therefore, while these genes are candidates for contributing to T1D susceptibility, many associations remain tentative, and their precise functional roles in disease pathogenesis are still uncertain.
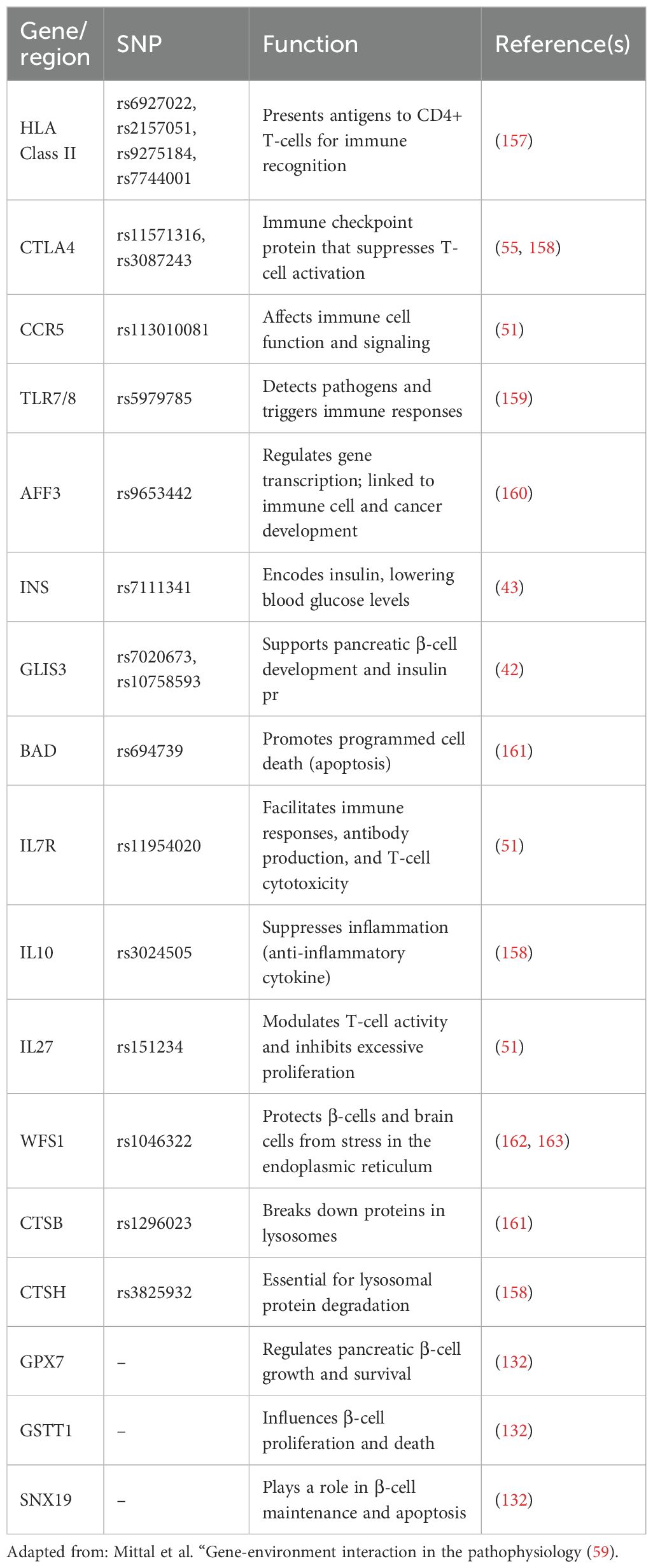
Table 2. Summary of key genes and their associated SNPs linked to immune function, β-cell regulation, and T1D risk.
4.1.1 Human leukocyte antigen
About 50% of the lifetime risk of T1D is attributed to mutations in the HLA class II genes on chromosome 6, which increases the chance of acquiring the disease (47, 54). Specifically, the DR4-DQ8 (DQA1*03:01 – DQB1*03:02) or DR3-DQ2 (DQA1*05:01 – DQB1*02:01) haplotypes are present in 90% of children with T1D. The largest risk factor for contracting the disease is the combination of these two haplotypes in a person’s genotype (55). Numerous studies have examined the connection between T1D risk and variations in the HLA gene. These genetic correlations have implications for disease prediction and means of prevention in addition to aiding in our understanding of the pathophysiology of T1D. HLA typing, for instance, is utilized in T1D prevention trials to identify people who might benefit from early interventions and to stratify risk (56).
4.1.2 Cathepsin H
Other gene loci, including the susceptibility locus of cathepsin H (CTSH), have also been linked to the development of T1D in addition to HLA. CTSH has been linked to a higher incidence of T1D by genome-wide association studies (GWAS) (51). Using integrated data from quantitative trait locus (eQTL) with GWAS, a study identified the possible pathogenic pathways of the CTSH gene in T1D (57). Single cell RNA sequencing (scRNA) revealed that the pancreas of T1D patients had a significant upregulation of the CTSH gene in acinar cells as compared to the control group. Additionally, a group of genes co-expressed with CTSH that had a substantial positive connection with T1D were found using single-cell weighted gene co-expression network analysis (WGCNA). The CTSH gene in the exocrine pancreas was thought to enhance the antiviral response based on functional enrichment analysis. An inflammatory milieu is produced as a result of this amplification, which also raises the expression of pro-inflammatory cytokines. T1D is likely to develop as a result of this process, which is likely to harm β-cells. High CTSH expression, which is influenced by other environmental factors such post-translational modifications and epigenetics, was found to connect with the risk of T1D in another study (58). When combined, these studies demonstrate how CTSH contributes to a higher risk of T1D development.
4.1.3 Other genes
It has been demonstrated that additional potential genes, including INS, GLIS3, CCR5, BAD, GPX7, GSTT1, and SNX19, increase vulnerability to T1D (23, 40–51). A few of these genes have a direct impact on pancreatic β-cell growth and death. Table 3 provides a detailed list of all the genes linked to a higher risk of T1D along with an explanation of their roles.
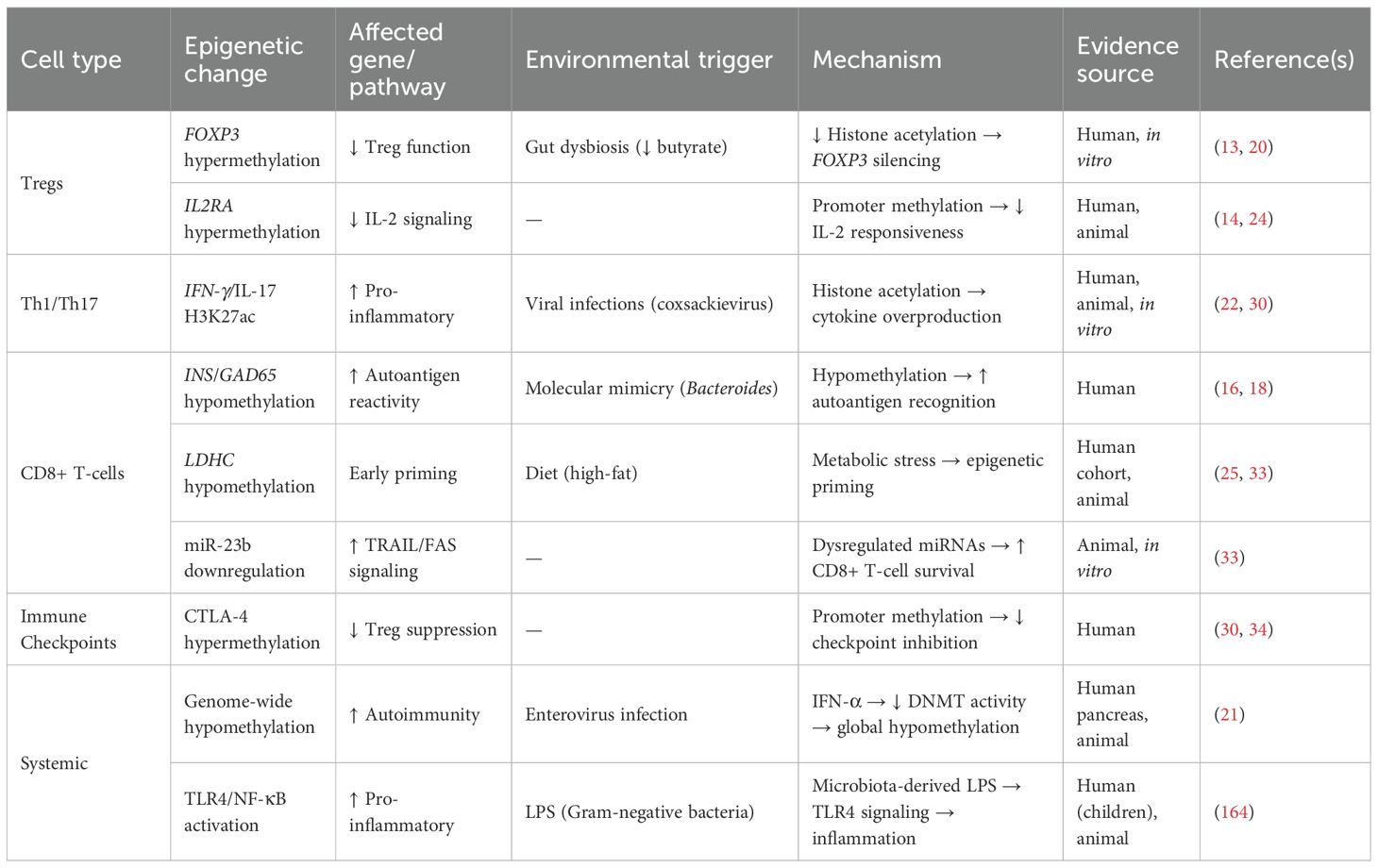
Table 3. Epigenetic dysregulation in T1D-associated T-cells.
Recent research have demonstrated that the pathophysiology of T1D is complex, despite the fact that genetics has been found to play a significant influence in the disease. Identical twin studies have revealed that if one twin has T1D, the other twin may not be at all susceptible to the condition, indicating that genetic factors by themselves are insufficient to fully explain how T1D develops (59).
4.2 Environmental triggers
In addition to genetics, environmental factors have been linked to the development of T1D independently. These include viral infections, pesticide exposure, lifestyle and eating habits, and vitamin D deficiency (60–62).
4.2.1 Viral infections
Viral infection-induced autoimmunity may be a significant factor in the development of T1D (Figure 2) (63). Enteroviruses have been linked to the etiopathogenesis of T1D on several levels, including infecting pancreatic β-cells and triggering autoimmunity against them (64). Most commonly, T1D incidence has been linked to Coxsackie B viruses (65–67). Enterovirus proteins have been detected in the pancreas during the outset of illness in people with T1D (68). It has been demonstrated that several enterovirus species can infect and impair the function of pancreatic β-cells since these cells also contain many receptors that enteroviruses employ to entry into cells. Interferons, which are produced in response to these viral infections, drive gene transcription; newly diagnosed T1D patients have been found to exhibit this IFN-stimulated gene expression. The later emergence of autoantibodies against pancreatic β-cells has also been linked to this gene transcription. Given that viremia was missing in children with quick onset T1D in the TEDDY research, it is possible that infections could cause autoimmunity gradually over time as opposed to suddenly (69). Moreover, pancreatic β-cell antigens and certain viruses, like enteroviruses, have structural similarities. This similarity may result in a condition called molecular mimicry, in which the body’s own cells, including β-cells that produce insulin, are mistakenly attacked by the immune system, which is triggered to combat the virus, causing T1D (60). In pancreatic β-cells, enteroviruses have been demonstrated to interfere with the miRNA-mediated inhibition of pro-inflammatory pathways, whereas related Picornaviridae viruses, like rhinovirus, can modify the expression of cytokine genes by altering DNA methylation (70–73). The offspring may be primed for autoimmune reactions and have a higher chance of developing T1D later in life if the mother’s enteroviral infection during pregnancy causes long-lasting epigenetic changes in the fetal immune-related genes (74–77).
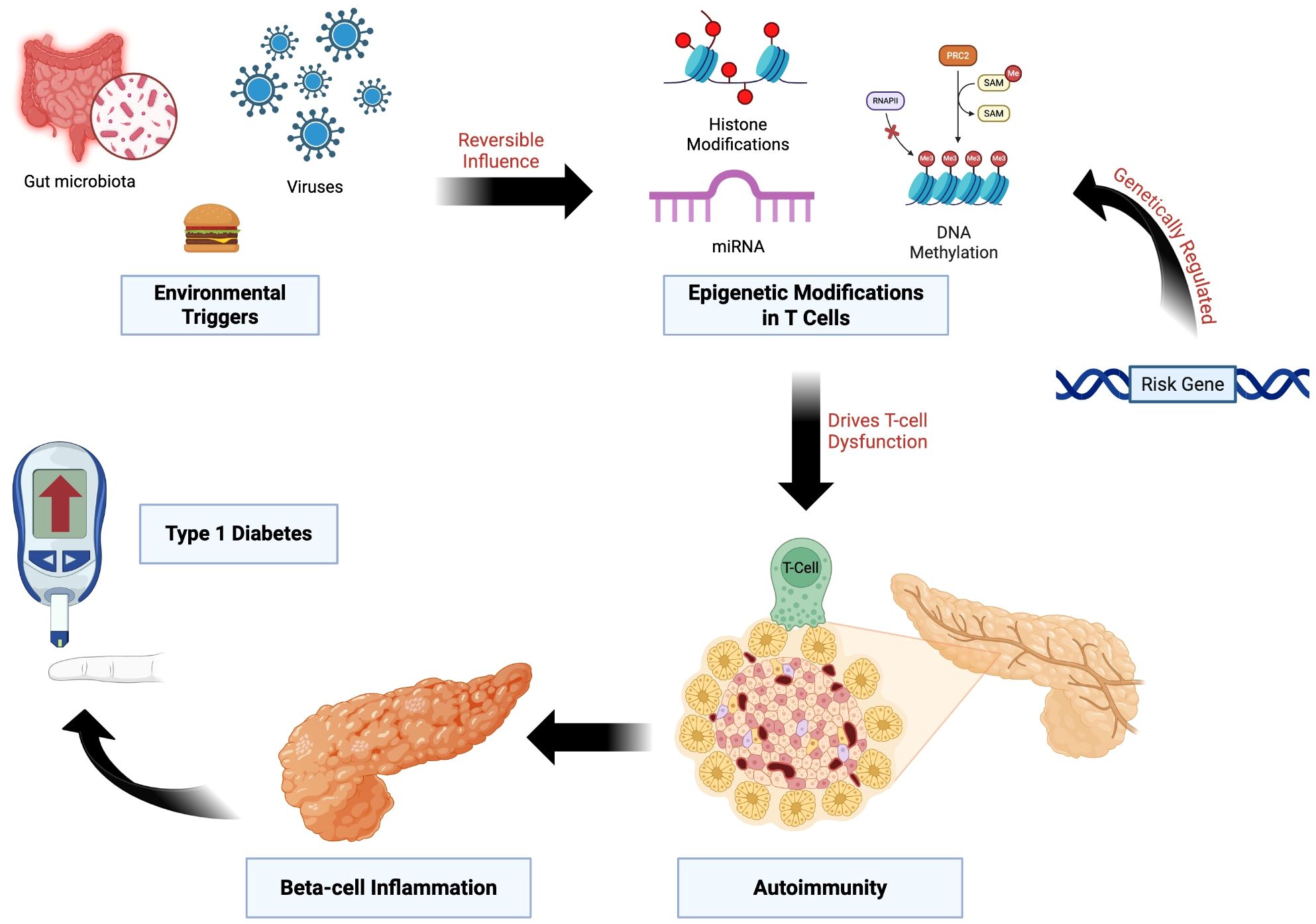
Figure 2. Epigenetic Dysregulation Linking Genetic Risk and Environmental Triggers to T1D. Genetic susceptibility loci influence epigenetic regulation in T cells through mechanisms including DNA methylation, histone modifications, and non-coding RNAs. Environmental exposures, such as viral infections, altered gut microbiota, and dietary factors, exert reversible effects on these pathways. The resulting epigenetic dysregulation promotes loss of immune tolerance, β-cell inflammation, and progression to T1D.
4.2.2 Pesticide exposure
T1D development has been linked to pesticide exposure. Chemicals called pesticides are used extensively in agriculture to control pests, but there have been worries about their possible effects on human health. Studies have examined the link between pesticide exposure and T1D, although research in this field is still ongoing and results are inconclusive (78). Epidemiological studies suggest that environmental toxins such as pesticides may interact with genetic susceptibility to influence disease onset. There may be a connection between pesticide exposure and T1D, according to epidemiological research. Even at low concentrations, pesticide exposure has been linked to the occurrence of T1D and prediabetes, also known as aberrant glucose regulation (79). Men and women had different causal relationships between pesticide exposure and impaired glucose control; in men, a U-shaped dose-response relationship was more pronounced.
Pesticides may also cause or hasten the autoimmune reaction that destroys β-cells in the pancreas, according to some theories. Although the exact processes behind this possible link are unknown, they might have to do with oxidative stress induction or immune function disturbance (59). These findings illustrate how environmental exposures beyond viral infections can contribute to T1D development, setting the stage to examine lifestyle and dietary influences.
4.2.3 Lifestyle and diet
Numerous studies have examined the relationship between dietary and lifestyle factors and the onset of T1D, identifying a number of connections and possible mechanisms (80). Diet and lifestyle represent modifiable environmental factors that may mediate T1D risk, in part through their effects on gut microbiota and immune function. It is clear that dietary practices that alter the composition of the gut microbiota may be a major factor in the development of T1D. Up to now, the most convincing evidence for a causal link between intestinal microbiome and the disease comes from well-controlled intervention studies in murine models (81). Although not fully understood, a complicated relationship between gut permeability, the immune system, and intestinal microbiota has previously been discovered (82). The gut barrier, which is made up of enterocytes, mucus, gut microbiota, tight junction (TJ) proteins, and the innate and adaptive immune cells that make up the gut-associated lymphoid tissue, regulates gut permeability (83). Intestinal permeability and the passage of microbial antigens, products, or microbes themselves can result from the breakdown of TJ and the compromise of the intestinal barrier. The expression of TJ proteins, which include claudin-2, occludin, cingulin, and zonula occludens (ZO) proteins, controls the TJ of the intestinal barrier. According to some research, intestinal permeability is dependent on elevated zonulin levels, which are impacted by bacterial colonization (84, 85). It is also known that zonulin modulates TJ to reversibly modify intestinal permeability (86–88). It is interesting to note that elevated blood zonulin levels occur prior to the development of clinically noticeable T1D (89). However, subsequent studies have raised concerns regarding the specificity of zonulin assays and the generalizability of these findings. Critical reviews indicate that while zonulin represents a potentially important modulator of intestinal permeability, its measurement can be affected by cross-reactivity and methodological variability, and not all individuals with T1D show elevated levels. Therefore, interpreting zonulin data requires caution, and it should be considered alongside other markers and functional assessments of intestinal barrier integrity. Furthermore, an increase in intestinal paracellular permeability has been found in T1D patients, supporting the concept of barrier dysfunction as a feature of disease pathogenesis (90–93).
Intestinal permeability was higher in children with multiple islet autoantibodies (≥2 IA) who developed T1D than in those who did not, indicating a role for intestinal permeability in the pathophysiology of T1D (94, 95). The intestinal barrier’s permeability is modulated by a variety of gut commensals (96). The data that certain gut bacteria create gamma-aminobutyric acid and express GAD supports a theory. By acting as an antigen to activate submucosal T-cells, the GAD produced from bacteria as a result of gut bacterial death (e.g., by viral or antibiotic-mediated mechanisms) may miseducate the host immune system and result in the development of T1D (97, 98).
Some of the bacteria can carry peptide sequences that resemble insulin, which could cause auto-immunity, according to bioinformatics research (99). Remarkably, T-cell clones that are directed against preproinsulin peptides have demonstrated a high degree of cross-reactivity with peptides from Clostridium and Bacteroides species (100). A peptide generated by Parabacteroides distasonis that resembles the β-chain of insulin has been found in a NOD mouse model (101). T-cells are able to identify this peptide, which triggers an immunological reaction to this insulin chain.
The gnotobiotic zebrafish model has shown that the intestinal microbiota is necessary for the normal growth of the pancreatic β-cell population during early larval development. This is due to the action of a bacterial protein called β-cell expansion factor A (BefA), which is produced by gut microbes (102). These results raise the possibility that the gut microbiota plays a part in the formation of early pancreatic β-cells and point to a connection between juvenile fecal microbiota composition and an elevated risk of diabetes.
Studies have repeatedly shown that T1D is linked to notable changes in the makeup of the gut microbiota. In comparison to healthy controls, children who subsequently developed T1D had different microbial patterns, including lower levels of Lactococcus lactis and Streptococcus thermophilus and greater levels of Bifidobacterium spp., according to the seminal TEDDY study (103). Bacteroides species are more prevalent in both established T1D patients and at-risk individuals, according to several independent studies (104–106). Certain strains, such as B. dorei and B. vulgatus, are particularly enriched in high-risk Finnish children (107), while B. stercoris, B. intestinalis, B. cellulosilyticus, and B. fragilis are found in Italian patients (108).
However, results across cohorts have not always been consistent, indicating that microbial signatures of T1D risk are still unclear. For example, while the TEDDY longitudinal analysis found differences in early microbiota, other cohorts such as Diabimmune and DIPP report different taxa changes (103). Recent reviews and meta-analyses emphasize that microbial findings vary by geography and study population, and some studies fail to replicate specific “diabetogenic” bacteria (109).
Two trends that stand out in functional investigations of the gut microbiome in T1D are the significant drop in butyrate-producing bacteria from Clostridium clusters IV and XIVa and the decreased number of species that break down mucin, such as Prevotella and Akkermansia (110, 111). Studies using metagenomic and metabolomic techniques have found common microbial traits between T1D patients and their siblings, such as higher Clostridiales and Dorea with concomitant reductions in Dialister and Akkermansia (111) These alterations seem to be clinically significant.
One especially noteworthy observation is the reduction of butyrate-producing bacteria, which has been linked in several studies to greater intestinal permeability and an increased risk of T1D (110, 112–114). Intervention studies that demonstrate that butyrate supplementation can enhance metabolic parameters and cause disease remission in NOD mice models further reinforce this relationship (115, 116). The exact molecular pathways are still unclear, highlighting a crucial field for further investigation, even though these findings collectively strongly link gut microbiota dysbiosis to T1D development, especially through processes involving barrier function and immune modulation.
Systemic immunological responses are significantly shaped by short-chain fatty acids (SCFAs) generated from the microbiota, especially butyrate and propionate (117). The pancreas and lymph nodes are among the distal tissues that these compounds affect after diffusing through the intestinal epithelium. Through processes including HDACs inhibition and free fatty acid receptor (FFAR) activation, SCFAs control T-cell activity. This results in epigenetic remodeling that promotes the formation of regulatory T cells and lowers inflammation. However, host-specific variables including nutrition, metabolic status, and microbiome makeup affect SCFA effects, which are highly context-dependent. In order to completely comprehend and utilize SCFA-driven immune regulation in the setting of T1D, it may be necessary to integrate metagenomic, metabolomic, and epigenetic techniques.
Vitamin D represents another dietary factor that may influence T1D risk through immune modulation. The onset of T1D has been linked to low vitamin D levels (118–121). This correlation is believed to result from vitamin D’s possible ability to influence immune system modulation, which may have an effect on the autoimmune processes implicated in T1D. Other research, however, has found no link between low vitamin D levels and increased incidence of T1D (122, 123). Furthermore, interventional studies investigating vitamin D supplementation for T1D prevention have produced mixed results. For example, a 2021 meta-analysis reported limited and inconsistent evidence that vitamin D supplementation reduces T1D risk, highlighting variability in study design, population, and dosing regimens. These findings indicate that, while vitamin D may have immunomodulatory effects, supplementation alone has not been conclusively shown to prevent T1D (124).
Vitamin D affects immune function through genetic pathways involving the vitamin D receptor (VDR), in addition to its traditional role in maintaining mineral homeostasis (125). VDR binds to vitamin D response elements (VDREs) and forms a heterodimer with retinoid X receptor (RXR) upon binding 1,25(OH)D (126). This heterodimer regulates transcription differently depending on the cell type. Vitamin D’s specific immunomodulatory effects on T cells and other immunological subsets implicated in T1D may be explained by this. Furthermore, complexes of vitamin D and VDR can disrupt transcription factors like CREB, altering gene expression without the involvement of RXR and pointing to different epigenetic processes of immune control (112).
4.2.4 Antibiotic use
Research has indicated a link between the use of antibiotics and a higher risk of developing T1D (80, 127, 128). Depending on the route of delivery, using broad-spectrum antibiotics during the first two years of life has been linked to an increased risk of developing T1D (129). Interestingly, only infants born via cesarean section showed a correlation between broad-spectrum antibiotics and T1D, but kids born vaginally did not. Other research, however, finds no connection between T1D and antibiotic use (130, 131). To determine the role of antibiotic use and delivery method in the development of T1D, more research is necessary.
4.3 Integrated mechanisms
It is currently unclear what causes pancreatic β cell loss and the development of T1D in certain people, despite the distinct roles of environmental risk factors and genetic vulnerability. There is a growing theory that the pathophysiology of T1D is significantly influenced by the interplay between genetic predisposition and environmental variables (Figure 3). The impact of gene variations that cause autoimmunity and result in T1D clinical symptoms may be amplified by environmental variables (59).
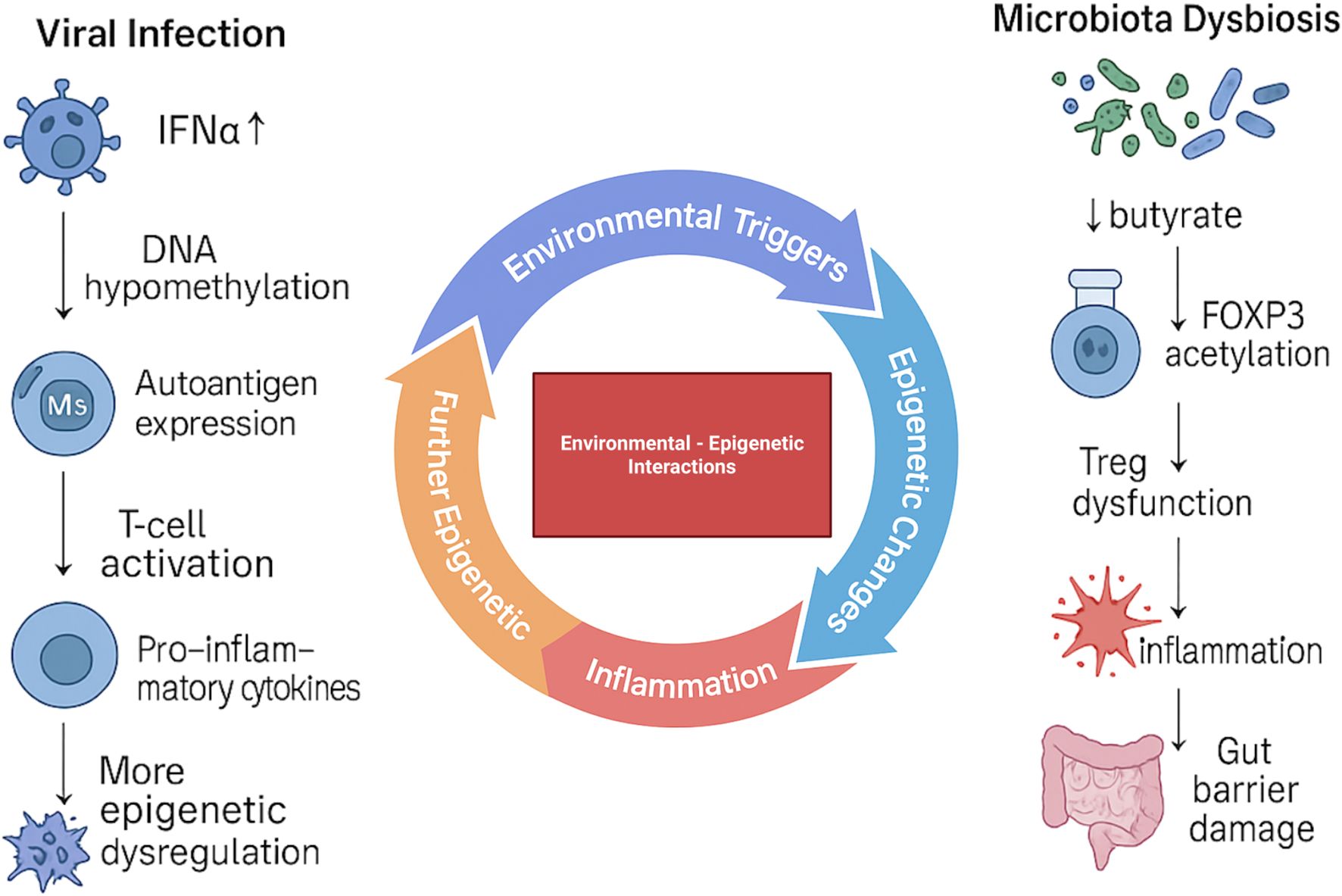
Figure 3. Environmental triggers and their epigenetic impact on T1D pathogenesis. Viral infections promote DNA hypomethylation, autoantigen expression, T-cell activation, and pro-inflammatory cytokine release, leading to progressive epigenetic dysregulation. In parallel, microbiota dysbiosis reduces butyrate availability, decreases FOXP3 acetylation, and impairs Treg function, driving inflammation and gut barrier damage. Together, these environmental–epigenetic interactions amplify immune dysregulation and contribute to the development of T1D.
Together with environmental variables, epigenetic modulators have become important regulators of gene expression and cellular phenotype (34, 132–136). One of the main molecular pathways through which gene-environment interactions may heighten vulnerability to T1D is thought to be epigenetics (12, 133, 137). Investigations into epigenetic mechanisms, such as changes in DNA methylation, have revealed abnormal patterns in genes related to insulin control and immune function in people with T1D (138–141) Laajala et al. did not observe differences in DNA methylation between cases and controls in cord blood samples (142). By contrast, Johnson et al. reported DNA methylation changes that preceded seroconversion, indicating that methylation alterations can occur before the appearance of islet autoantibodies. They analyzed multiple pre-disease peripheral-blood samples and identified longitudinal differences in the rate of age-related methylation change at 10 genomic regions. Several of these differences were detectable as early as birth and in samples taken before onset of islet autoimmunity (143). Additionally, in the setting of T1D, histone alterations have demonstrated their impact on immune response gene dysregulation (144). Another aspect of epigenetics that has been highlighted is the function of miRNAs, specifically in regulating inflammatory and immunological responses in T1D (133, 145–148). In addition to their implications for biomarker discovery, epigenetic changes linked to T1D risk also pave the way for precision medicine approaches in T1D diagnosis, risk assessment, and treatment (59).
5 Epigenetic biomarkers and therapeutic potential
Epigenetic biomarkers are emerging as valuable tools that can be used in understanding, diagnosing, and potentially treating various diseases including T1D. Longitudinal studies show that specific methylation changes occur before clinical disease onset. In T1D, early demethylation events at immune and β-cell genes can be detected months to years before diagnosis (20). For instance, hypomethylation at the INS promoter, a hallmark of active insulin transcription, correlates with β-cell function and can be detected in circulating cell-free DNA (15, 17, 29).
Similarly, circulating miRNA pose as a promising biomarker and predictor of T1D progression. miR-25 has been found to be negatively associated with residual β-cell function, and positively associated with glycemic control 3 months after onset (149). This suggests that miR-25 may have a role in cell proliferation of pancreatic endocrine cells, thus making it of benefit in evaluating T1D progression and management. Another study suggested the usage of hsa-miR-1-3p in monitoring T1D progression and associated cardiovascular complications (146). Assessing for such epigenetic changes may aid in early detection, diagnosis, and prognosis of T1D among other diseases as well.
Epigenetic mechanisms are being explored as therapeutic targets. One promising strategy involves the use of small molecule inhibitors that target enzymes involved in epigenetic modifications including HDACs and DNMTs. HDAC inhibitors can promote a more permissive chromatin state, thereby enhancing the expression of genes involved in immune regulation and tolerance (150, 151). DNMT inhibitors, on the other hand, may reverse aberrant DNA hypermethylation and restore the expression of silenced checkpoint inhibitors such as PD-1 or CTLA-4 (152). Early preclinical models suggest that modulating these enzymes in T-cells may reduce autoreactivity and promote immune tolerance in autoimmune settings, although translation to humans is still in early stages (153).
Another emerging area of therapeutic research involves miRNA-based therapies. Since specific miRNAs contribute to the dysregulation of T-cell function in T1D, strategies that restore the balance of miRNA expression may help re-establish immune homeostasis (154). This could involve the use of miRNA mimics to restore deficient regulatory miRNAs or antagomirs to inhibit pro-inflammatory miRNAs. While these approaches offer a degree of precision not seen with conventional immunosuppressive therapies and may reduce off-target effects, challenges remain regarding delivery methods, tissue specificity, and potential immune responses. CRISPR/dCas9-mediated epigenetic editing is an emerging approach that enables precise, reversible control of gene expression without altering the DNA sequence. Unlike conventional CRISPR, the catalytically inactive “dead” Cas9 (dCas9) is fused to epigenetic modifiers such as p300 (a histone acetyltransferase) or TET1 (a DNA demethylase) and guided to specific genomic loci by custom-designed guide RNAs. This system allows researchers to modulate the epigenetic landscape of immune-regulatory genes in T-cells with high specificity. For example, targeting dCas9-p300 to the promoter of the FOXP3 gene in mouse primary T-cells significantly enhanced and stabilized FOXP3 expression, promoting a regulatory T-cell phenotype even under inflammatory conditions (155). Similarly, in human T-cell models, dCas9-TET1 systems have been used to reduce methylation at FOXP3 enhancer regions and induce functional suppressive Treg-like cells (156). These findings highlight the potential of epigenetic editing to reprogram autoreactive T-cells, restore immune tolerance, and ultimately serve as a targeted, gene-specific immunotherapy for autoimmune diseases such as T1D; however, clinical translation is limited, and challenges such as efficient in vivo delivery, immune cell targeting, and off-target effects remain to be addressed.
6 Conclusion
The immunological landscape of T1D is significantly shaped by epigenetic mechanisms, particularly in regulating T-cell growth, activation, and tolerance. These pathways operate at the intersection of genetic susceptibility and environmental exposures, offering a more integrated view of T1D pathogenesis. Recent findings on DNA methylation, histone remodeling, and non-coding RNAs shed light on why immune tolerance fails in some individuals but not others. Crucially, the reversibility of epigenetic modifications enables the possibility of therapeutic immune cell reprogramming. At the same time, epigenetic signatures hold promise as biomarkers for early risk stratification, prediction of disease progression, and monitoring of therapeutic response. Despite this potential, major challenges remain, including limited understanding of the causal hierarchy among epigenetic changes, variability across patient populations, and difficulty distinguishing disease-driving modifications from secondary changes. As tools such as CRISPR-based editing and single-cell epigenomics advance, integrating biomarker discovery with mechanistic insights will be essential for translating epigenetic research into durable and precise strategies for preventing or delaying T1D onset.
Author contributions
AJ: Conceptualization, Data curation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. AE: Conceptualization, Data curation, Methodology, Visualization, Writing – original draft, Writing – review & editing. MA: Data curation, Methodology, Visualization, Writing – original draft, Writing – review & editing. RA: Data curation, Methodology, Visualization, Writing – original draft, Writing – review & editing. TA: Data curation, Methodology, Visualization, Writing – original draft, Writing – review & editing. DA: Data curation, Methodology, Visualization, Writing – original draft, Writing – review & editing. BT: Data curation, Methodology, Visualization, Writing – original draft, Writing – review & editing. AM: Data curation, Methodology, Visualization, Writing – original draft, Writing – review & editing. MK: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing. AY: Conceptualization, Data curation, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lucier J and Mathias PM. Type 1 Diabetes. In: StatPearls. Treasure Island, FL, USA: StatPearls Publishing (2025).
2. Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. (2017) 3:17016. doi: 10.1038/nrdp.2017.16
3. Wang Y, Hou C, Wisler J, Singh K, Wu C, Xie Z, et al. Elevated histone H3 acetylation is associated with genes involved in T lymphocyte activation and glutamate decarboxylase antibody production in patients with type 1 diabetes. J Diabetes Investig. (2019) 10:51–61. doi: 10.1111/jdi.12867
4. Cerna M. Epigenetic regulation in etiology of type 1 diabetes mellitus. Int J Mol Sci. (2019) 21. doi: 10.3390/ijms21010036
5. Mauvais F-X and van Endert PM. Type 1 diabetes: A guide to autoimmune mechanisms for clinicians. Diabetes Obes Metab. (2025) 27:40–56. doi: 10.1111/dom.16460
6. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the endocrine society, and the american diabetes association. Diabetes Care. (2015) 38:1964–74. doi: 10.2337/dc15-1419
7. Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, and Unanue ERA. Minor subset of batf3-dependent antigen-presenting cells in islets of langerhans is essential for the development of autoimmune diabetes. Immunity. (2014) 41:657–69. doi: 10.1016/j.immuni.2014.09.012
8. White TD, Almutairi A, Gai-Tusing Y, Stephenson DJ, Stephenson BD, Chalfant CE, et al. Differential lipid signaling from CD4+ and CD8+ T cells contributes to type 1 diabetes development. Front Immunol. (2024) 15:1444639. doi: 10.3389/fimmu.2024.1444639
9. Hull CM, Peakman M, and Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia. (2017) 60:1839–50. doi: 10.1007/s00125-017-4377-1
10. Syed Khaja AS, Binsaleh NK, Qanash H, Alshetaiwi H, Ginawi IAM, and Saleem M. Dysregulation and therapeutic prospects of regulatory T cells in type 1 diabetes. Acta Diabetol. (2025) 62:785–800. doi: 10.1007/s00592-025-02478-3
11. ElEssawy B and Li XC. Type 1 diabetes and T regulatory cells. Pharmacol Res. (2015) 98:22–30. doi: 10.1016/j.phrs.2015.04.009
12. Zhang J, Chen L-M, Zou Y, Zhang S, Xiong F, and Wang C-Y. Implication of epigenetic factors in the pathogenesis of type 1 diabetes. Chin Med J (Engl). (2021) 134:1031–42. doi: 10.1097/CM9.0000000000001450
13. Akil A-SA-S, Jerman LF, Yassin E, Padmajeya SS, Al-Kurbi A, and Fakhro KA. Reading between the (Genetic) lines: how epigenetics is unlocking novel therapies for type 1 diabetes. Cells. (2020) 9. doi: 10.3390/cells9112403
14. Ohkura N and Sakaguchi S. Transcriptional and epigenetic basis of treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res. (2020) 30:465–74. doi: 10.1038/s41422-020-0324-7
15. Klak M, Gomółka M, Kowalska P, Cichoń J, Ambrożkiewicz F, Serwańska-Świętek M, et al. Type 1 diabetes: genes associated with disease development. Cent Eur J Immunol. (2020) 45:439–53. doi: 10.5114/ceji.2020.103386
16. Giwa AM, Ahmed R, Omidian Z, Majety N, Karakus KE, Omer SM, et al. Current understandings of the pathogenesis of type 1 diabetes: genetics to environment. World J Diabetes. (2020) 11:13–25. doi: 10.4239/wjd.v11.i1.13
17. Zhang H and Pollin TI. Epigenetics variation and pathogenesis in diabetes. Curr Diabetes Rep. (2018) 18:121. doi: 10.1007/s11892-018-1091-4
18. Del Chierico F, Rapini N, Deodati A, Matteoli MC, Cianfarani S, and Putignani L. Pathophysiology of type 1 diabetes and gut microbiota role. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232314650
19. Wang G, Shen W-B, Chen AW, Reece EA, and Yang P. Diabetes and early development: epigenetics, biological stress, and aging. Am J Perinatol. (2025) 42:977–87. doi: 10.1055/a-2405-1493
20. Zajec A, Trebušak Podkrajšek K, Tesovnik T, Šket R, Čugalj Kern B, Jenko Bizjan B, et al. Pathogenesis of type 1 diabetes: established facts and new insights. Genes (Basel). (2022) 13. doi: 10.3390/genes13040706
21. Akhbari P, Richardson SJ, and Morgan NG. Type 1 diabetes: interferons and the aftermath of pancreatic beta-cell enteroviral infection. Microorganisms. (2020) 8. doi: 10.3390/microorganisms8091419
22. Benaglio P, Zhu H, Okino M-L, Yan J, Elgamal R, Nariai N, et al. Type 1 diabetes risk genes mediate pancreatic beta cell survival in response to proinflammatory cytokines. Cell Genomics. (2022) 2:100214. doi: 10.1016/j.xgen.2022.100214
23. Chiou J, Geusz RJ, Okino M-L, Han JY, Miller M, Melton R, et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature. (2021) 594:398–402. doi: 10.1038/s41586-021-03552-w
24. Bettini M and Bettini ML. Function, failure, and the future potential of tregs in type 1 diabetes. Diabetes. (2021) 70:1211–9. doi: 10.2337/dbi18-0058
25. Kindt ASD, Fuerst RW, Knoop J, Laimighofer M, Telieps T, Hippich M, et al. Allele-specific methylation of type 1 diabetes susceptibility genes. J Autoimmun. (2018) 89:63–74. doi: 10.1016/j.jaut.2017.11.008
26. Li Y, Zhao M, Hou C, Liang G, Yang L, Tan Y, et al. Abnormal DNA methylation in CD4+ T cells from people with latent autoimmune diabetes in adults. Diabetes Res Clin Pract. (2011) 94:242–8. doi: 10.1016/j.diabres.2011.07.027
27. Wang Z, Zheng Y, Hou C, Yang L, Li X, Lin J, et al. DNA methylation impairs TLR9 induced foxp3 expression by attenuating IRF-7 binding activity in fulminant type 1 diabetes. J Autoimmun. (2013) 41:50–9. doi: 10.1016/j.jaut.2013.01.009
28. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol. (2011) 186:3918–26. doi: 10.4049/jimmunol.1003099
29. Belot M-P, Fradin D, Mai N, Le Fur S, Zélénika D, Kerr-Conte J, et al. CpG methylation changes within the IL2RA promoter in type 1 diabetes of childhood onset. PloS One. (2013) 8:e68093. doi: 10.1371/journal.pone.0068093
30. Minniakhmetov I, Yalaev B, Khusainova R, Bondarenko E, Melnichenko G, Dedov I, et al. Genetic and epigenetic aspects of type 1 diabetes mellitus: modern view on the problem. Biomedicines. (2024) 12. doi: 10.3390/biomedicines12020399
31. Qin Y, Li Y, Wang Y, Wei Q, Dai L, Huang M, et al. Plasticity deficits of tregs remodeling toward th1-like and th17-like tregs in individuals with type 1 diabetes. J Endocrinol Invest. (2025) 48:1495–509. doi: 10.1007/s40618-025-02557-w
32. Marroqui L, Perez-Serna AA, Babiloni-Chust I, and Dos Santos RS. Type I interferons as key players in pancreatic β-cell dysfunction in type 1 diabetes. Int Rev Cell Mol Biol. (2021) 359:1–80. doi: 10.1016/bs.ircmb.2021.02.011
33. Kohil A, Al-Asmakh M, Al-Shafai M, and Terranegra A. The interplay between diet and the epigenome in the pathogenesis of type-1 diabetes. Front Nutr. (2020) 7:612115. doi: 10.3389/fnut.2020.612115
34. Čugalj Kern B, Trebušak Podkrajšek K, Kovač J, Šket R, Jenko Bizjan B, Tesovnik T, et al. The role of epigenetic modifications in late complications in type 1 diabetes. Genes (Basel). (2022) 13. doi: 10.3390/genes13040705
35. Zemmour D, Pratama A, Loughhead SM, Mathis D, and Benoist C. Flicr, a long noncoding RNA, modulates foxp3 expression and autoimmunity. Proc Natl Acad Sci U.S.A. (2017) 114:E3472–80. doi: 10.1073/pnas.1700946114
36. Collier SP, Collins PL, Williams CL, Boothby MR, and Aune TM. Cutting edge: influence of tmevpg1, a long intergenic noncoding RNA, on the expression of ifng by th1 cells. J Immunol. (2012) 189:2084–8. doi: 10.4049/jimmunol.1200774
37. Collier SP, Henderson MA, Tossberg JT, and Aune TM. Regulation of the th1 genomic locus from ifng through tmevpg1 by T-bet. J Immunol. (2014) 193:3959–65. doi: 10.4049/jimmunol.1401099
38. Gomez JA, Wapinski OL, Yang YW, Bureau J-F, Gopinath S, Monack DM, et al. The neST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ Locus. Cell. (2013) 152:743–54. doi: 10.1016/j.cell.2013.01.015
39. Hu G, Tang Q, Sharma S, Yu F, Escobar TM, Muljo SA, et al. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat Immunol. (2013) 14:1190–8. doi: 10.1038/ni.2712
40. Hehenkamp P, Hoffmann M, Kummer S, Reinauer C, Döing C, Förtsch K, et al. Interleukin-7-dependent nonclassical monocytes and CD40 expression are affected in children with type 1 diabetes. Eur J Immunol. (2021) 51:3214–27. doi: 10.1002/eji.202149229
41. Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA, et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein bim. PloS Genet. (2013) 9:e1003532. doi: 10.1371/journal.pgen.1003532
42. Duarte GCK, Assmann TS, Dieter C, de Souza BM, and Crispim D. GLIS3 rs7020673 and rs10758593 polymorphisms interact in the susceptibility for type 1 diabetes mellitus. Acta Diabetol. (2017) 54:813–21. doi: 10.1007/s00592-017-1009-7
43. Törn C, Hadley D, Lee H-S, Hagopian W, Lernmark Å, Simell O, et al. Role of type 1 diabetes-associated SNPs on risk of autoantibody positivity in the TEDDY study. Diabetes. (2015) 64:1818–29. doi: 10.2337/db14-1497
44. Sandholm N, Rubio García A, Pekalski ML, Inshaw JRJ, Cutler AJ, and Todd JA. Thymocyte regulatory variant alters transcription factor binding and protects from type 1 diabetes in infants. Sci Rep. (2022) 12:14137. doi: 10.1038/s41598-022-18296-4
45. Rich SS. Genetics and its potential to improve type 1 diabetes care. Curr Opin Endocrinol Diabetes Obes. (2017) 24:279–84. doi: 10.1097/MED.0000000000000347
46. Shapiro MR, Thirawatananond P, Peters L, Sharp RC, Ogundare S, Posgai AL, et al. De-coding genetic risk variants in type 1 diabetes. Immunol Cell Biol. (2021) 99:496–508. doi: 10.1111/imcb.12438
47. Noble JA and Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diabetes Rep. (2011) 11:533–42. doi: 10.1007/s11892-011-0223-x
48. Pociot F and Lernmark Å. Genetic risk factors for type 1 diabetes. Lancet. (2016) 387:2331–9. doi: 10.1016/S0140-6736(16)30582-7
49. Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Wellcome trust case control consortium genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447:661–78. doi: 10.1038/nature05911
50. Inshaw JRJ, Cutler AJ, Crouch DJM, Wicker LS, and Todd JA. Genetic variants predisposing most strongly to type 1 diabetes diagnosed under age 7 years lie near candidate genes that function in the immune system and in pancreatic β-cells. Diabetes Care. (2020) 43:169–77. doi: 10.2337/dc19-0803
51. Onengut-Gumuscu S, Chen W-M, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet. (2015) 47:381–6. doi: 10.1038/ng.3245
52. Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen W-M, Santa Cruz DF, Yang H, et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet. (2021) 53:962–71. doi: 10.1038/s41588-021-00880-5
53. Pahkuri S, Ekman I, Vandamme C, Näntö-Salonen K, Toppari J, Veijola R, et al. DNA methylation differences within INS, PTPN22 and IL2RA promoters in lymphocyte subsets in children with type 1 diabetes and controls. Autoimmunity. (2023) 56. doi: 10.1080/08916934.2023.2259118
54. Noble JA, Valdes AM, Varney MD, Carlson JA, Moonsamy P, Fear AL, et al. HLA class I and genetic susceptibility to type 1 diabetes: results from the type 1 diabetes genetics consortium. Diabetes. (2010) 59:2972–9. doi: 10.2337/db10-0699
55. Bradfield JP, Qu H-Q, Wang K, Zhang H, Sleiman PM, Kim CE, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PloS Genet. (2011) 7:e1002293. doi: 10.1371/journal.pgen.1002293
56. Nguyen C, Varney MD, Harrison LC, and Morahan G. Definition of high-risk type 1 diabetes HLA-DR and HLA-DQ types using only three single nucleotide polymorphisms. Diabetes. (2013) 62:2135–40. doi: 10.2337/db12-1398
57. Song Z, Li S, Shang Z, Lv W, Cheng X, Meng X, et al. Integrating multi-omics data to analyze the potential pathogenic mechanism of CTSH gene involved in type 1 diabetes in the exocrine pancreas. Brief Funct Genomics. (2024) 23:406–17. doi: 10.1093/bfgp/elad052
58. Ye J, Stefan-Lifshitz M, and Tomer Y. Genetic and environmental factors regulate the type 1 diabetes gene CTSH via differential DNA methylation. J Biol Chem. (2021) 296:100774. doi: 10.1016/j.jbc.2021.100774
59. Mittal R, Camick N, Lemos JRN, and Hirani K. Gene-environment interaction in the pathophysiology of type 1 diabetes. Front Endocrinol (Lausanne). (2024) 15:1335435. doi: 10.3389/fendo.2024.1335435
60. Houeiss P, Luce S, and Boitard C. Environmental triggering of type 1 diabetes autoimmunity. Front Endocrinol (Lausanne). (2022) 13:933965. doi: 10.3389/fendo.2022.933965
61. Wei Y, Wang L, and Liu J. The diabetogenic effects of pesticides: evidence based on epidemiological and toxicological studies. Environ pollut. (2023) 331:121927. doi: 10.1016/j.envpol.2023.121927
62. Rewers M and Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet. (2016) 387:2340–8. doi: 10.1016/S0140-6736(16)30507-4
63. Elsalti A and Mahroum N. Viral Infections and Type 1 Diabetes Mellitus – Guilty Viruses in the Court of Autoimmunity. In: Infection and Autoimmunity. Amsterdam, Netherlands: Elsevier (2024). p. 271–83.
64. Isaacs SR, Roy A, Dance B, Ward EJ, Foskett DB, Maxwell AJ, et al. Enteroviruses and risk of islet autoimmunity or type 1 diabetes: systematic review and meta-analysis of controlled observational studies detecting viral nucleic acids and proteins. Lancet Diabetes Endocrinol. (2023) 11:578–92. doi: 10.1016/S2213-8587(23)00122-5
65. Carré A, Vecchio F, Flodström-Tullberg M, You S, and Mallone R. Coxsackievirus and type 1 diabetes: diabetogenic mechanisms and implications for prevention. Endocr Rev. (2023) 44:737–51. doi: 10.1210/endrev/bnad007
66. Filippi C and von Herrath M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell Immunol. (2005) 233:125–32. doi: 10.1016/j.cellimm.2005.04.009
67. Nekoua MP, Alidjinou EK, and Hober D. Persistent coxsackievirus B infection and pathogenesis of type 1 diabetes mellitus. Nat Rev Endocrinol. (2022) 18:503–16. doi: 10.1038/s41574-022-00688-1
68. Blanter M, Sork H, Tuomela S, and Flodström-Tullberg M. Genetic and environmental interaction in type 1 diabetes: A relationship between genetic risk alleles and molecular traits of enterovirus infection? Curr Diabetes Rep. (2019) 19:82. doi: 10.1007/s11892-019-1192-8
69. TEDDY Study Group. The environmental determinants of diabetes in the young (TEDDY) study. Ann N Y Acad Sci. (2008) 1150:1–13. doi: 10.1196/annals.1447.062
70. Kim KW, Ho A, Alshabee-Akil A, Hardikar AA, Kay TWH, Rawlinson WD, et al. Coxsackievirus B5 infection induces dysregulation of microRNAs predicted to target known type 1 diabetes risk genes in human pancreatic islets. Diabetes. (2016) 65:996–1003. doi: 10.2337/db15-0956
71. Alidjinou EK, Engelmann I, Bossu J, Villenet C, Figeac M, Romond M-B, et al. Persistence of coxsackievirus B4 in pancreatic ductal-like cells results in cellular and viral changes. Virulence. (2017) 8:1229–44. doi: 10.1080/21505594.2017.1284735
72. Engelmann I, Alidjinou EK, Bertin A, Bossu J, Villenet C, Figeac M, et al. Persistent coxsackievirus B4 infection induces microRNA dysregulation in human pancreatic cells. Cell Mol Life Sci. (2017) 74:3851–61. doi: 10.1007/s00018-017-2567-0
73. McErlean P, Favoreto S, Costa FF, Shen J, Quraishi J, Biyasheva A, et al. Human rhinovirus infection causes different DNA methylation changes in nasal epithelial cells from healthy and asthmatic subjects. BMC Med Genomics. (2014) 7:37. doi: 10.1186/1755-8794-7-37
74. Dahlquist GG, Ivarsson S, Lindberg B, and Forsgren M. Maternal enteroviral infection during pregnancy as a risk factor for childhood IDDM. A population-based case-control study. Diabetes. (1995) 44:408–13. doi: 10.2337/diab.44.4.408
75. Dahlquist GG. Viruses and other perinatal exposures as initiating events for beta-cell destruction. Ann Med. (1997) 29:413–7. doi: 10.3109/07853899708999371
76. Viskari HR, Roivainen M, Reunanen A, Pitkäniemi J, Sadeharju K, Koskela P, et al. Maternal first-trimester enterovirus infection and future risk of type 1 diabetes in the exposed fetus. Diabetes. (2002) 51:2568–71. doi: 10.2337/diabetes.51.8.2568
77. Dahlquist GG, Forsberg J, Hagenfeldt L, Boman J, and Juto P. Increased prevalence of enteroviral RNA in blood spots from newborn children who later developed type 1 diabetes: A population-based case-control study. Diabetes Care. (2004) 27:285–6. doi: 10.2337/diacare.27.1.285
78. Xu Z-R, Yuan X-X, Chen R-M, Wei H-Y, Chen L-Q, Du H-W, et al. Association between new onset type 1 diabetes and real-world antibiotics and neonicotinoids’ Exposure-related gut microbiota perturbation. World J Pediatr. (2022) 18:671–9. doi: 10.1007/s12519-022-00589-3
79. Kim S-K, Oh H-J, Oh S-S, and Koh S-B. Pesticide exposure in relation to the incidence of abnormal glucose regulation: A retrospective cohort study. Int J Environ Res Public Health. (2022) 19. doi: 10.3390/ijerph19127550
80. Quinn LM, Wong FS, and Narendran P. Environmental determinants of type 1 diabetes: from association to proving causality. Front Immunol. (2021) 12:737964. doi: 10.3389/fimmu.2021.737964
81. Zhou H, Sun L, Zhang S, Zhao X, Gang X, and Wang G. Evaluating the causal role of gut microbiota in type 1 diabetes and its possible pathogenic mechanisms. Front Endocrinol (Lausanne). (2020) 11:125. doi: 10.3389/fendo.2020.00125
82. Vaarala O, Atkinson MA, and Neu J. The “Perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes. (2008) 57:2555–62. doi: 10.2337/db08-0331
83. Bibbò S, Dore MP, Pes GM, Delitala G, and Delitala AP. Is there a role for gut microbiota in type 1 diabetes pathogenesis? Ann Med. (2017) 49:11–22. doi: 10.1080/07853890.2016.1222449
84. Wang W, Uzzau S, Goldblum SE, and Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. (2000) 113:4435–40. doi: 10.1242/jcs.113.24.4435
85. Asmar R, Gosse P, Topouchian J, N’tela G, Dudley A, and Shepherd GL. Effects of telmisartan on arterial stiffness in type 2 diabetes patients with essential hypertension. J Renin Angiotensin Aldosterone Syst. (2002) 3:176–80. doi: 10.3317/jraas.2002.038
86. Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci U.S.A. (2005) 102:2916–21. doi: 10.1073/pnas.0500178102
87. Fasano A. Intestinal permeability and its regulation by zonulin: diagnostic and therapeutic implications. Clin Gastroenterol Hepatol. (2012) 10:1096–100. doi: 10.1016/j.cgh.2012.08.012
88. Kelly CP, Green PHR, Murray JA, Dimarino A, Colatrella A, Leffler DA, et al. Larazotide acetate in patients with coeliac disease undergoing a gluten challenge: A randomised placebo-controlled study. Aliment Pharmacol Ther. (2013) 37:252–62. doi: 10.1111/apt.12147
89. Sapone A, de Magistris L, Pietzak M, Clemente MG, Tripathi A, Cucca F, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. (2006) 55:1443–9. doi: 10.2337/db05-1593
90. Mønsted MØ, Falck ND, Pedersen K, Buschard K, Holm LJ, and Haupt-Jorgensen M. Intestinal permeability in type 1 diabetes: an updated comprehensive overview. J Autoimmun. (2021) 122:102674. doi: 10.1016/j.jaut.2021.102674
91. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia. (2006) 49:2824–7. doi: 10.1007/s00125-006-0465-3
92. Secondulfo M, Iafusco D, Carratù R, deMagistris L, Sapone A, Generoso M, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig Liver Dis. (2004) 36:35–45. doi: 10.1016/j.dld.2003.09.016
93. Paroni R, Fermo I, Molteni L, Folini L, Pastore MR, Mosca A, et al. Lactulose and mannitol intestinal permeability detected by capillary electrophoresis. J Chromatogr B Analyt Technol BioMed Life Sci. (2006) 834:183–7. doi: 10.1016/j.jchromb.2006.02.050
94. Maffeis C, Martina A, Corradi M, Quarella S, Nori N, Torriani S, et al. Association between intestinal permeability and faecal microbiota composition in italian children with beta cell autoimmunity at risk for type 1 diabetes. Diabetes Metab Res Rev. (2016) 32:700–9. doi: 10.1002/dmrr.2790
95. Harbison JE, Roth-Schulze AJ, Giles LC, Tran CD, Ngui KM, Penno MA, et al. Gut microbiome dysbiosis and increased intestinal permeability in children with islet autoimmunity and type 1 diabetes: A prospective cohort study. Pediatr Diabetes. (2019) 20:574–83. doi: 10.1111/pedi.12865
96. Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, and Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr. (2011) 141:769–76. doi: 10.3945/jn.110.135657
97. Bedi S, Richardson TM, Jia B, Saab H, Brinkman FSL, and Westley M. Similarities between bacterial GAD and human GAD65: implications in gut mediated autoimmune type 1 diabetes. PloS One. (2022) 17:e0261103. doi: 10.1371/journal.pone.0261103
98. Jamshidi P, Hasanzadeh S, Tahvildari A, Farsi Y, Arbabi M, Mota JF, et al. Is there any association between gut microbiota and type 1 diabetes? A systematic review. Gut Pathog. (2019) 11:49. doi: 10.1186/s13099-019-0332-7
99. Altindis E, Vomund AN, Chow I-T, Damasio M, Kwok W, Unanue ER, et al. Identification of cross reactive insulin immunogenic epitopes from commensal gut microbes. Diabetes. (2018) 67. doi: 10.2337/db18-95-OR
100. Cole DK, Bulek AM, Dolton G, Schauenberg AJ, Szomolay B, Rittase W, et al. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J Clin Invest. (2016) 126:2191–204. doi: 10.1172/JCI85679
101. Girdhar K, Huang Q, Chow I-T, Vatanen T, Brady C, Raisingani A, et al. A gut microbial peptide and molecular mimicry in the pathogenesis of type 1 diabetes. Proc Natl Acad Sci U.S.A. (2022) 119:e2120028119. doi: 10.1073/pnas.2120028119
102. Hill JH, Franzosa EA, Huttenhower C, and Guillemin KA. Conserved bacterial protein induces pancreatic beta cell expansion during zebrafish development. Elife. (2016) 5. doi: 10.7554/eLife.20145
103. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature. (2018) 562:589–94. doi: 10.1038/s41586-018-0620-2
104. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. (2011) 5:82–91. doi: 10.1038/ismej.2010.92
105. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: A case-control study. BMC Med. (2013) 11:46. doi: 10.1186/1741-7015-11-46
106. Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes. (2015) 64:3510–20. doi: 10.2337/db14-1847
107. Davis-Richardson AG and Triplett EW. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia. (2015) 58:1386–93. doi: 10.1007/s00125-015-3614-8
108. Biassoni R, Di Marco E, Squillario M, Barla A, Piccolo G, Ugolotti E, et al. Gut microbiota in T1DM-onset pediatric patients: machine-learning algorithms to classify microorganisms as disease linked. J Clin Endocrinol Metab. (2020) 105:e3114-e3126. doi: 10.1210/clinem/dgaa407
109. Rampanelli E and Nieuwdorp M. Gut microbiome in type 1 diabetes: the immunological perspective. Expert Rev Clin Immunol. (2023) 19:93–109. doi: 10.1080/1744666X.2023.2150612
110. de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. (2014) 57:1569–77. doi: 10.1007/s00125-014-3274-0
111. Del Chierico F, Conta G, Matteoli MC, Fierabracci A, Reddel S, Macari G, et al. Gut microbiota functional traits, blood PH, and anti-GAD antibodies concur in the clinical characterization of T1D at onset. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms231810256
112. de Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes. (2013) 62:1238–44. doi: 10.2337/db12-0526
113. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PloS One. (2011) 6:e25792. doi: 10.1371/journal.pone.0025792
114. de Groot PF, Belzer C, Aydin Ö, Levin E, Levels JH, Aalvink S, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PloS One. (2017) 12:e0188475. doi: 10.1371/journal.pone.0188475
115. Gonzalez A, Krieg R, Massey HD, Carl D, Ghosh S, Gehr TWB, et al. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol Dial Transplant. (2019) 34:783–94. doi: 10.1093/ndt/gfy238
116. Jacob N, Jaiswal S, Maheshwari D, Nallabelli N, Khatri N, Bhatia A, et al. Butyrate induced tregs are capable of migration from the GALT to the pancreas to restore immunological tolerance during type-1 diabetes. Sci Rep. (2020) 10:19120. doi: 10.1038/s41598-020-76109-y
117. Feng C, Jin C, Liu K, and Yang Z. Microbiota-derived short chain fatty acids: their role and mechanisms in viral infections. Biomedicine Pharmacotherapy. (2023) 160:114414. doi: 10.1016/j.biopha.2023.114414
118. Berridge MJ. Vitamin D deficiency and diabetes. Biochem J. (2017) 474:1321–32. doi: 10.1042/BCJ20170042
119. Pozzilli P, Manfrini S, Crinò A, Picardi A, Leomanni C, Cherubini V, et al. IMDIAB group low levels of 25-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 in patients with newly diagnosed type 1 diabetes. Horm Metab Res. (2005) 37:680–3. doi: 10.1055/s-2005-870578
120. Yu J, Sharma P, Girgis CM, and Gunton JE. Vitamin D and beta cells in type 1 diabetes: A systematic review. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232214434
121. Bener A, Alsaied A, Al-Ali M, Al-Kubaisi A, Basha B, Abraham A, et al. High prevalence of vitamin D deficiency in type 1 diabetes mellitus and healthy children. Acta Diabetol. (2009) 46:183–9. doi: 10.1007/s00592-008-0071-6
122. Manousaki D, Harroud A, Mitchell RE, Ross S, Forgetta V, Timpson NJ, et al. Vitamin D levels and risk of type 1 diabetes: A mendelian randomization study. PloS Med. (2021) 18:e1003536. doi: 10.1371/journal.pmed.1003536
123. Reinert-Hartwall L, Honkanen J, Härkönen T, Ilonen J, Simell O, Peet A, et al. No association between vitamin D and β-cell autoimmunity in finnish and Estonian children. Diabetes Metab Res Rev. (2014) 30:749–60. doi: 10.1002/dmrr.2550
124. Zipitis CS and Akobeng AK. Vitamin D supplementation in early childhood and risk of type 1 diabetes: A systematic review and meta-analysis. Arch Dis Child. (2008) 93:512–7. doi: 10.1136/adc.2007.128579
125. Voltan G, Cannito M, Ferrarese M, Ceccato F, and Camozzi V. Vitamin D: an overview of gene regulation, ranging from metabolism to genomic effects. Genes (Basel). (2023) 14. doi: 10.3390/genes14091691
126. Zella LA, Meyer MB, Nerenz RD, Lee SM, Martowicz ML, and Pike JW. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol Endocrinol. (2010) 24:128–47. doi: 10.1210/me.2009-0140
127. Kilkkinen A, Virtanen SM, Klaukka T, Kenward MG, Salkinoja-Salonen M, Gissler M, et al. Use of antimicrobials and risk of type 1 diabetes in a population-based mother-child cohort. Diabetologia. (2006) 49:66–70. doi: 10.1007/s00125-005-0078-2
128. Boursi B, Mamtani R, Haynes K, and Yang Y-X. The effect of past antibiotic exposure on diabetes risk. Eur J Endocrinol. (2015) 172:639–48. doi: 10.1530/EJE-14-1163
129. Clausen TD, Bergholt T, Bouaziz O, Arpi M, Eriksson F, Rasmussen S, et al. Broad-spectrum antibiotic treatment and subsequent childhood type 1 diabetes: A nationwide danish cohort study. PloS One. (2016) 11:e0161654. doi: 10.1371/journal.pone.0161654
130. Kemppainen KM, Vehik K, Lynch KF, Larsson HE, Canepa RJ, Simell V, et al. Association between early-life antibiotic use and the risk of islet or celiac disease autoimmunity. JAMA Pediatr. (2017) 171:1217–25. doi: 10.1001/jamapediatrics.2017.2905
131. Tapia G, Størdal K, Mårild K, Kahrs CR, Skrivarhaug T, Njølstad PR, et al. Antibiotics, acetaminophen and infections during prenatal and early life in relation to type 1 diabetes. Int J Epidemiol. (2018) 47:1538–48. doi: 10.1093/ije/dyy092
132. Olsson AH, Volkov P, Bacos K, Dayeh T, Hall E, Nilsson EA, et al. Genome-wide associations between genetic and epigenetic variation influence MRNA expression and insulin secretion in human pancreatic islets. PloS Genet. (2014) 10:e1004735. doi: 10.1371/journal.pgen.1004735
133. Xie Z, Chang C, Huang G, and Zhou Z. The role of epigenetics in type 1 diabetes. Adv Exp Med Biol. (2020) 1253:223–57. doi: 10.1007/978-981-15-3449-2_9
134. Fu W, Farache J, Clardy SM, Hattori K, Mander P, Lee K, et al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and β Cells. Elife. (2014) 3:e04631. doi: 10.7554/eLife.04631
135. Syreeni A, El-Osta A, Forsblom C, Sandholm N, Parkkonen M, Tarnow L, et al. Genetic examination of SETD7 and SUV39H1/H2 methyltransferases and the risk of diabetes complications in patients with type 1 diabetes. Diabetes. (2011) 60:3073–80. doi: 10.2337/db11-0073
136. Rosen ED, Kaestner KH, Natarajan R, Patti M-E, Sallari R, Sander M, et al. Epigenetics and epigenomics: implications for diabetes and obesity. Diabetes. (2018) 67:1923–31. doi: 10.2337/db18-0537
137. Jerram ST, Dang MN, and Leslie RD. The role of epigenetics in type 1 diabetes. Curr Diabetes Rep. (2017) 17:89. doi: 10.1007/s11892-017-0916-x
138. Stefan M, Zhang W, Concepcion E, Yi Z, and Tomer Y. DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J Autoimmun. (2014) 50:33–7. doi: 10.1016/j.jaut.2013.10.001
139. Dashti M, Nizam R, Hebbar P, Jacob S, John SE, Channanath A, et al. Differentially methylated and expressed genes in familial type 1 diabetes. Sci Rep. (2022) 12:11045. doi: 10.1038/s41598-022-15304-5
140. Starskaia I, Laajala E, Grönroos T, Härkönen T, Junttila S, Kattelus R, et al. Early DNA methylation changes in children developing beta cell autoimmunity at a young age. Diabetologia. (2022) 65:844–60. doi: 10.1007/s00125-022-05657-x
141. Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PloS Genet. (2011) 7:e1002300. doi: 10.1371/journal.pgen.1002300
142. Laajala E, Kalim UU, Grönroos T, Rasool O, Halla-aho V, Konki M, et al. Umbilical cord blood DNA methylation in children who later develop type 1 diabetes. Diabetologia. (2022) 65:1534–40. doi: 10.1007/s00125-022-05726-1
143. Johnson RK, Vanderlinden LA, Dong F, Carry PM, Seifert J, Waugh K, et al. Longitudinal DNA methylation differences precede type 1 diabetes. Sci Rep. (2020) 10:3721. doi: 10.1038/s41598-020-60758-0
144. Miao F, Chen Z, Zhang L, Liu Z, Wu X, Yuan Y-C, et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J Biol Chem. (2012) 287:16335–45. doi: 10.1074/jbc.M111.330373
145. Mostahfezian M, Azhir Z, Dehghanian F, and Hojati Z. Expression pattern of microRNAs, miR-21, miR-155 and miR-338 in patients with type 1 diabetes. Arch Med Res. (2019) 50:79–85. doi: 10.1016/j.arcmed.2019.07.002
146. Morales-Sánchez P, Lambert C, Ares-Blanco J, Suárez-Gutiérrez L, Villa-Fernández E, Garcia AV, et al. Circulating miRNA expression in long-standing type 1 diabetes mellitus. Sci Rep. (2023) 13:8611. doi: 10.1038/s41598-023-35836-8
147. Al-Nakhle H, Mohsen I, Elnaem B, Alharbi A, Alnakhli I, Almoarfi S, et al. Altered expression of vitamin D metabolism genes and circulating microRNAs in PBMCs of patients with type 1 diabetes: their association with vitamin D status and ongoing islet autoimmunity. Noncoding RNA. (2023) 9. doi: 10.3390/ncrna9050060
148. Bahreini F, Rayzan E, and Rezaei N. MicroRNAs and diabetes mellitus type 1. Curr Diabetes Rev. (2022) 18:e021421191398. doi: 10.2174/1573399817666210215111201
149. Nielsen LB, Wang C, Sørensen K, Bang-Berthelsen CH, Hansen L, Andersen M-LM, et al. Circulating levels of microRNA from children with newly diagnosed type 1 diabetes and healthy controls: evidence that miR-25 associates to residual beta-cell function and glycaemic control during disease progression. Exp Diabetes Res. (2012) 2012:896362. doi: 10.1155/2012/896362
150. Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, and Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. (2015) 3:1375–85. doi: 10.1158/2326-6066.CIR-15-0077-T
151. Borcoman E, Kamal M, Marret G, Dupain C, Castel-Ajgal Z, and Le Tourneau C. HDAC inhibition to prime immune checkpoint inhibitors. Cancers (Basel). (2021) 14. doi: 10.3390/cancers14010066
152. Saleh MH, Wang L, and Goldberg MS. Improving cancer immunotherapy with DNA methyltransferase inhibitors. Cancer Immunol Immunother. (2016) 65:787–96. doi: 10.1007/s00262-015-1776-3
153. Gomez S, Tabernacki T, Kobyra J, Roberts P, and Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin Cancer Biol. (2020) 65:99–113. doi: 10.1016/j.semcancer.2019.12.019
154. Scherm MG and Daniel C. MiRNA-mediated immune regulation in islet autoimmunity and type 1 diabetes. Front Endocrinol (Lausanne). (2020) 11:606322. doi: 10.3389/fendo.2020.606322
155. Okada M, Kanamori M, Someya K, Nakatsukasa H, and Yoshimura A. Stabilization of foxp3 expression by CRISPR-DCas9-based epigenome editing in mouse primary T cells. Epigenet Chromatin. (2017) 10:24. doi: 10.1186/s13072-017-0129-1
156. Huang S, Lau C-H, Tin C, and Lam RHW. Extended replicative lifespan of primary resting T cells by CRISPR/DCas9-based epigenetic modifiers and transcriptional activators. Cell Mol Life Sci. (2024) 81:407. doi: 10.1007/s00018-024-05415-9
157. McKinnon E, Morahan G, Nolan D, and James I. Diabetes genetics consortium association of MHC SNP genotype with susceptibility to type 1 diabetes: A modified survival approach. Diabetes Obes Metab. (2009) 11 Suppl 1:92–100. doi: 10.1111/j.1463-1326.2008.01009.x
158. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. (2009) 41:703–7. doi: 10.1038/ng.381
159. Cooper JD, Walker NM, Smyth DJ, Downes K, Healy BC, and Todd JA. Type I diabetes genetics consortium follow-up of 1715 SNPs from the wellcome trust case control consortium genome-wide association study in type I diabetes families. Genes Immun. (2009) 10 Suppl 1:S85–94. doi: 10.1038/gene.2009.97
160. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. (2007) 39:857–64. doi: 10.1038/ng2068
161. Evangelou M, Smyth DJ, Fortune MD, Burren OS, Walker NM, Guo H, et al. A method for gene-based pathway analysis using genomewide association study summary statistics reveals nine new type 1 diabetes associations. Genet Epidemiol. (2014) 38:661–70. doi: 10.1002/gepi.21853
162. Eiberg H, Hansen L, Kjer B, Hansen T, Pedersen O, Bille M, et al. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet. (2006) 43:435–40. doi: 10.1136/jmg.2005.034892
163. Rigoli L, Bramanti P, Di Bella C, and De Luca F. Genetic and clinical aspects of wolfram syndrome 1, a severe neurodegenerative disease. Pediatr Res. (2018) 83:921–9. doi: 10.1038/pr.2018.17
Keywords: type 1 diabetes, epigenetics, T-cell dysfunction, autoimmunity, DNA methyaltion
Citation: Jabri A, Elsalti A, Alsharif M, Alsharif R, Abbad T, Sibai D, Taftafa B, Mhannayeh A, Khan MI and Yaqinuddin A (2025) Linking epigenetic mechanisms of T cell dysfunction with pathophysiology of type 1 diabetes mellitus. Front. Immunol. 16:1664255. doi: 10.3389/fimmu.2025.1664255
Received: 11 July 2025; Accepted: 15 October 2025;
Published: 29 October 2025.
Edited by:
Ubaid Ullah Kalim, University of Turku, FinlandReviewed by:
Aquib Ehtram, La Jolla Institute for Immunology (LJI), United StatesCaio Santos Bonilha, University of Glasgow, United Kingdom
Inna Starskaia, University of Turku, Finland
Copyright © 2025 Jabri, Elsalti, Alsharif, Alsharif, Abbad, Sibai, Taftafa, Mhannayeh, Khan and Yaqinuddin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Yaqinuddin, YXlhcWludWRkaW5AYWxmYWlzYWwuZWR1