 Grace Bernard1,2
Grace Bernard1,2 Laura Evgin1,2,3*
Laura Evgin1,2,3*- 1Interdisciplinary Oncology Program, University of British Columbia, Vancouver, BC, Canada
- 2Basic and Translational Research Department, BC Cancer Research Institute, Vancouver, BC, Canada
- 3Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada
The chimeric antigen receptor (CAR) is a synthetic and modular molecule composed of both signaling and non-signaling domains that allows a T cell to recognize cell surface antigens and trigger cytolytic functionality. It is appreciated that the non-signaling structural components, including the linker, hinge, and transmembrane domains, can dramatically alter how the CAR molecule interacts with itself and other endogenous molecules in the immune synapse. Herein, we describe the current understanding of how the structural domains can alter CAR T cell therapeutic efficacy and highlight how knowledge of the target antigen characteristics can inform CAR design choices.
1 Introduction
Chimeric antigen receptor (CAR) modified T cells have provided a paradigm shift in the management of hematological cancers, and there are seven Food and Drug Administration (FDA) approved therapies for relapsed/refractory acute lymphoblastic leukemia (ALL) (1–3), aggressive B cell lymphoma (4–6), mantle cell lymphoma (7), indolent B cell lymphoma (8, 9), and multiple myeloma (10–12). CAR T cells targeting CD19 and BMCA have induced prolonged remissions in patients with advanced malignancies with minimal long-term toxicities, and this success has been facilitated by the lineage-restricted and uniform expression of these antigens (13). CD19 and BCMA-specific CAR T cells are also being repurposed for the treatment of autoimmune diseases, where autoreactive cells of the B cell lineage (B cells themselves, plasmablasts, and plasma cells) are central mediators of disease pathology (14, 15).
2 Overview of the architecture of a CAR
The synthetic CAR molecule brings together the recognition capabilities of an antibody with the signaling properties of the T cell receptor (TCR) complex to redirect T cell function in a TCR-major histocompatibility complex (MHC) independent manner (Figure 1). The ectodomain contains a binding region responsible for recognizing the target cell antigen, which is frequently derived from a single-chain variable fragment (scFv) or a camelid nanobody (16, 17). However, specificity can also be conferred by a variety of natural ligands (18, 19), receptors (20), short peptides (21), or even fully synthetic binders, such as the D-domain (22). This recognition domain is fused to the hinge and transmembrane domains which provide flexibility and embed the protein in the membrane, respectively (23). The TCR α and β subunits do not have signaling properties but rather associate with CD3 chains. Thus, to mimic the TCR signal, the cytosolic portion of CD3ζ is fused at the distal end of the CAR molecule, where three (or fewer) (24, 25) ITAMs are involved in an LCK-mediated phosphorylation cascade involving LAT, SLP-76, and PLCγ; ultimately resulting in CAR T cell activation (26). Since effective T cell responses require both a primary signal from the TCR, as well as a secondary signal from a costimulatory molecule, the endo domain also includes the cytosolic region of one or more costimulatory molecules, membrane-proximal to the CD3ζ domain. While clinically approved CAR T cells incorporate CD28 or 4-1BB, many alternatives have been explored, including ICOS (27), OX40 (28), CD2 (29), CD27 (30), and IL-2RB in combination with a STAT3 binding motif (31), and shuffling these costimulatory domains alters the kinetic, differentiation, persistence, and cytolytic properties of CAR T cells (32, 33).
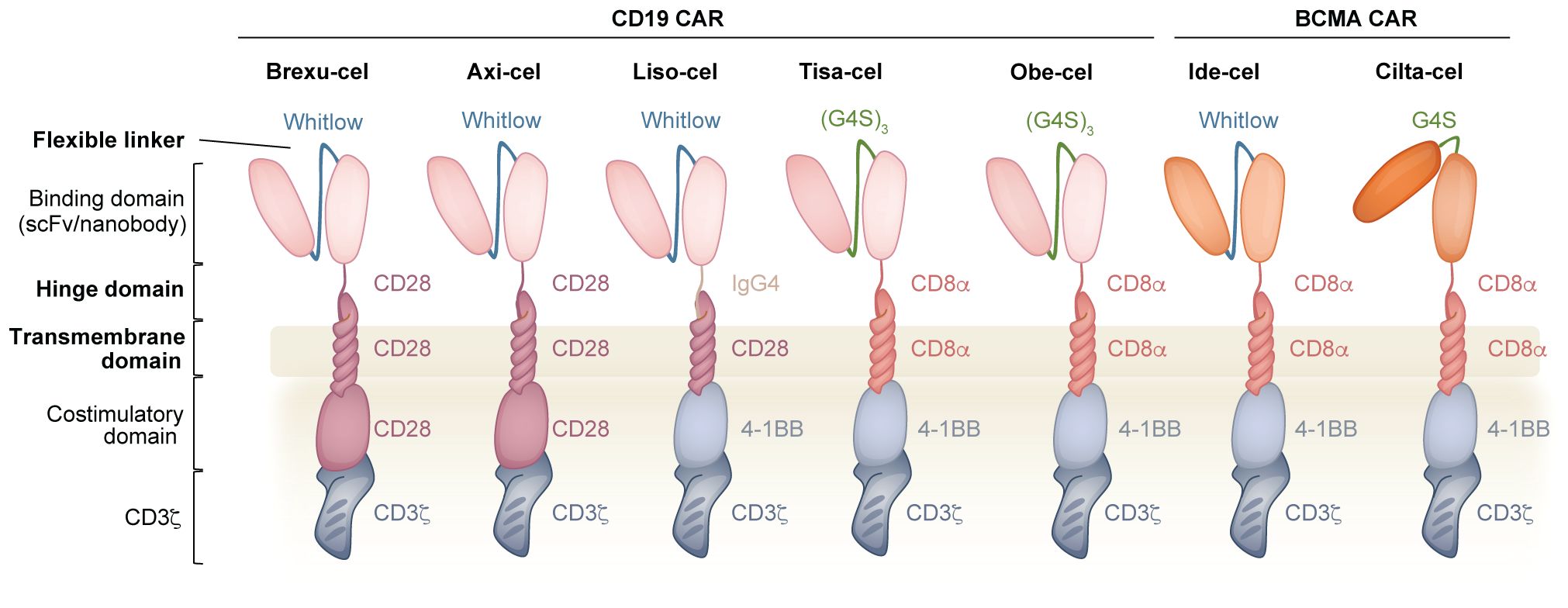
Figure 1. Overview of the CAR and the components that are used in clinically approved CD19 and BCMA CAR T cell products. The binding domain of the CAR comprises an scFv (or nanobody) where the VH and VL (or two VH domains) are connected via a flexible linker (Whitlow or (G4S)n). The hinge domain (derived CD28 or CD8α or IgG4) connects the binding domain to the transmembrane domain (derived CD28 or CD8α). In the intracellular region, the costimulatory domain (derived from CD28 or 4-1BB) is connected to CD3ζ. Bolded domains are described in the text.
3 Non-signaling components in the CAR
Beyond the hematological targets CD19 and BCMA, the modularity of the CAR system allows for applications in solid tumors, infectious and autoimmune diseases, aging, and fibrosis, etc., by a simple substitution of domains (34–38). Whether designing a novel CAR, or optimizing the sensitivity or function of an existing CAR, many possible permutations exist given the library of previously described domains. While the recognition and signaling domains have previously been extensively reviewed (39–41), herein, we describe the origin, nature and functional characteristics imparted by the non-signaling components of the CAR, including the linker, hinge and transmembrane domains. Moreover, we explore how some structure-activity relationships may be generalized, whereas others are construct, antigen, or epitope-specific. Finally, we propose open questions to make informed receptor design choices and guide future work. While these domains are scaffold components of the CAR, they profoundly shape CAR T cell functional properties and thus therapeutic outcomes. Finally, our review focuses on the design of CARs for expression in T cells, but we acknowledge that these synthetic molecules can redirect other cell types, including NK cells and macrophages, and the optimal structural elements for these cellular substrates differ. For example, in NK cells, a CAR containing the NKG2D transmembrane domain displayed superior functionality to the CD28 transmembrane domain (42).
3.1 Linker region
In CARs that incorporate an scFv as the mode of recognition, the linker is a synthetic peptide that covalently joins the variable heavy (VH) and light (VL) chains. One commonly used linker is composed of a pentapeptide series of four glycine residues followed by serine and is repeated three or four times as a 15-mer (G4S)3 or 20-mer (G4S)4 (43). The small size of these residues provides flexibility, and the polar nature of serine increases linker solubility, limiting interference with the variable region folding and target binding (44). Alternatively, the 218 linker, commonly known as the Whitlow linker (GSTSGSGKPGSGEGSTKG), is also frequently used and was designed to have reduced susceptibility to aggregation and proteolysis compared to the earlier 202 linker (45–47). Both linkers are used in clinically approved products: Whitlow (Brexu-cel, Liso-cel, Axi-cel and Ide-cel); G4S (Tisa-cel, Obe-cel and Cilta-cel) (Figure 1) (48, 49). Antibodies with specificity to these linkers facilitate both the detection of surface expression (48, 50, 51) and the purification (52) of CAR-expressing cells.
3.1.1 Identity of the linker
As they are similar in length, it is not clear whether the choice of the G4S or Whitlow linker imparts significant differences on CAR T cell functional outcome, nor has this been extensively reported on. Kouro et al. compared the use of the Whitlow and (G4S)4 linkers with an scFv with a propensity for tonic signaling in the context of both 4-1BB and CD28 co-stimulation. No differences were observed in surface aggregation, NFAT and NFκB signaling intensity, or in vivo tumor control in a xenograft model. However, the Whitlow-linked CAR did produce higher levels of IL4, TNFα, GM-CSF and IL10 in a costimulatory domain-specific manner (53).
3.1.2 Length of the linker
The properties conferred on an scFv by the length of the linker have been investigated in detail (54). An scFv with a linker longer than 12 amino acids exists as a monomer whereas a linker of 3 to 12 residues promotes association with a second scFv molecule to produce a “diabody” (55–58). In turn, short linker-based multivalent scFvs have greater binding valency to target molecules on the cell surface and reduced off-rates compared to monovalent scFvs (59, 60).
In the context of a CAR, Singh et al. examined the efficacy of two CD22-targeting CARs that differed only in the length of their linker: a CD22 long linker consisting of four G4S repeats (CD22-L) or a short linker composed of a single G4S (CD22-S) (61). This investigation was prompted by discrepant clinical outcomes using CAR T cells incorporating the same VH and VL chains, albeit connected via linkers of different lengths (62). The CD22-S CAR molecule was found to aggregate and drive low-level antigen-independent tonic signaling, as measured by activation of the phosphotidyl-inositol-3 kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling pathways. Compared to the CD22-L CAR T cells, the CD22-S CAR T cells also remained in contact longer with target cells, consistent with the slower off-rate of scFv multimers, producing more effector cytokines and demonstrating superior killing both in vitro and in vivo. Although not experimentally tested in this study, the authors suggest that shortening the linker to enhance affinity could be a useful design strategy for targeting antigens expressed at low density. These advantages were observed with the 4-1BB but not CD28 costimulatory domain-based CAR, in line with previous reports that the CD28 costimulatory domain exacerbates tonic signaling-associated dysfunction (63, 64). The enhanced tonic signaling and antigen-dependent activation conferred by the short hinge were also observed with CD33 but not CD19-based CARs. Therefore, while shortening the linker may promote clustering for some VH and VL domain pairings, this does not represent a universal strategy to enhance CAR T cell activity, as susceptibility to clustering and tonic signaling is also determined by biochemical properties of the framework region (65).
3.2 Hinge domain
The hinge domain, also known as the spacer region, connects the binding domain to the transmembrane domain, and provides the flexibility for the CAR to interact with a specific antigen of interest. The most commonly used hinges are derived from CD8α and CD28, however, IgG molecules have also been used preclinically and clinically (Figure 1) (13). Nerve growth factor receptor (NGFR) and CD34, two molecules normally absent from mature T cells, have also been used as alternative hinge domains to facilitate the detection of the CAR and the immunomagnetic sorting of CAR T cells (66, 67). The properties of the hinge, including its identity and length, shape how the CAR responds to antigen density and epitope position, ultimately affecting sensitivity and signaling strength.
3.2.1 Identity of the hinge
The hinge domain retains features of the native molecule, which affect the functional properties of the CAR. For example, CD28 typically exists as a homodimer due to an interdomain disulfide bond (68, 69), and the critical cysteine at position 123 is incorporated into the CAR hinge domain. A series of CD19 FMC63-based CARs incorporating various hinge and transmembrane domains identified that the cysteine in the CD28 hinge domain can stabilize a heterodimer of the CAR and endogenous CD28 (further described in the transmembrane section below) (70). Similarly, CD8α is expressed on T cells as a mixture of CD8αα homo- and CD8αβ heterodimers, with dimerization mediated by Ig domain interactions in the ectodomain as well as disulfide bonds formed by cysteine residues in the hinge and transmembrane regions (71, 72). The CD8α-derived hinge region incorporated into the CAR is known to be intrinsically disordered and dynamically transitions between conformation states involving proline cis–trans isomerization. A CD8α hinge-based CAR targeting CD22 was found to outperform a CD28 hinge-based CAR in vitro against low antigen density leukemia, and it was suggested that the flexibility of the CD8α hinge, driven by cis-trans isomerization, in combination with disulfide bridging between dimeric molecules, enhanced the signal transmission and sensitivity of the CAR (72). In contrast to the disorder of the CD8α hinge, the structural rigidity of the IgG4 hinge, in combination with two embedded cysteine residues, was found to promote homodimerization and the interaction of camelid VH domains in a GPC1 CAR to amplify T cell signaling (73). Finally, IgG4 or IgG1-derived hinges containing the CH2-CH3 domain retain the ability to interact with the Fc receptor, and while these CAR T cells perform well in vitro, in vivo, they lacked persistence and therapeutic activity, likely due to interaction with Fc-receptor-bearing myeloid cells. However, T cells equipped with a CAR bearing either a deletion of the CH2 domain, which interacts with the Fc receptor, or a specific mutation of the involved residues (L235E, N297Q) within the CH2 region, exhibited improved persistence and elicited more potent anti-tumor efficacy in mice (74–76).
3.2.2 Length of the hinge
The length of the hinge contributes to the overall functionality of a CAR in an antigen- and epitope-specific manner. The “kinetic segregation” model was originally proposed to describe peptide-MHC-TCR activation, where exclusion of the CD45 tyrosine phosphatase from the TCR signalosome shifts the equilibrium in favor of Lck-mediated TCR complex phosphorylation (77–79). In an analogous manner, Xiao et al. proposed the “size exclusion” model for CAR triggering where antigen binding narrows the intermembrane space and segregates the bulky CD45 phosphatase from the CAR zone, thus facilitating phosphorylation (Figure 2A) (80). Although described with different terminology, both models invoke the same principle where binding creates spatial constraints that exclude CD45 and enable phosphorylation. Using a CD19-targeting CAR with various hinge lengths, the size of the extracellular domain was determined to be inversely proportional to CD45 exclusion from the immune synapse, CD3ζ and ERK phosphorylation, cytokine production, and in vitro killing. This size-dependent activation also held true in vivo, where the shorter hinge CAR constructs provided superior tumor control in xenograft mouse models. To contextualize these findings, the CD8α hinge was used as the base construct, and additional Ig domains were added to increase the length by 4–16 nm. Consistent with this model where a shorter intermembrane distance elicits stronger exclusion of CD45, using CD22- and CEA-specific CARs, it was also shown that membrane-proximal epitopes stimulate CAR T cell activation better than distal epitopes (80). This new framework also helps explain previous reports showing that CARs with scFvs targeting membrane-proximal epitopes exhibit superior functional properties compared to those targeting distal epitopes (Figure 2B) (81, 82).
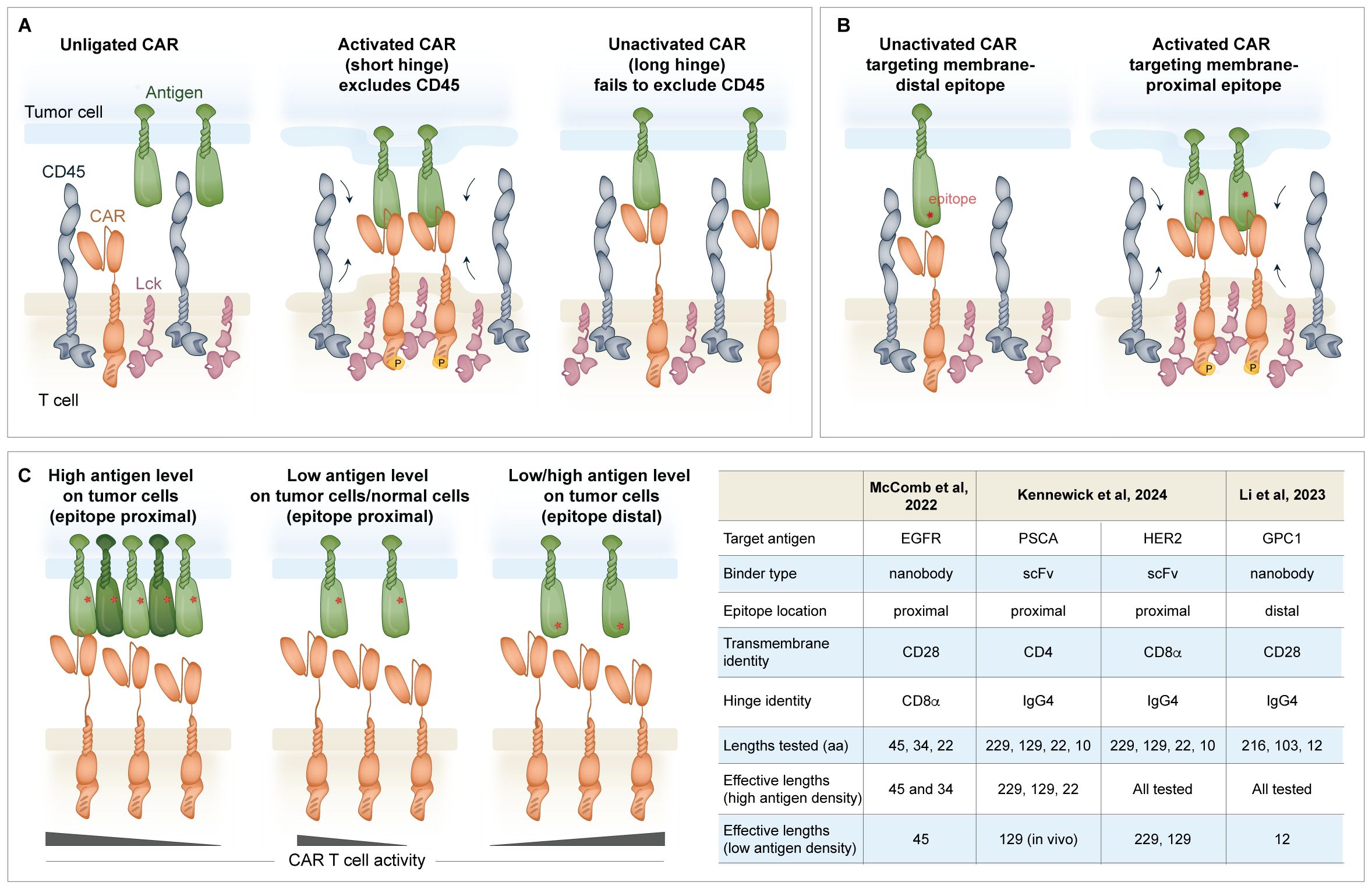
Figure 2. Regulation of the immune synapse formation between the CAR and the antigen of interest. (A) Effective activation of a CAR T cell is described by the “size exclusion model” where antigen binding by the CAR narrows the intermembrane space and excludes the CD45 phosphatase from the CAR zone, favoring Lck phosphorylation of the ITAMs in CD3ζ. If the hinge is too long, CD45 is ineffectively excluded (80). (B) The location of the epitope on the antigen of interest is important. scFvs targeting proximal epitopes may be better suited than those targeting distal epitopes to trigger the immune synapse. (C) The hinge can be shortened to increase the selectivity of the CAR for cells with high antigen density. This is useful for CARs that recognize shared antigens (i.e. HER2 and others) that are expressed at a lower level on normal cells. The hinge may also be shortened to accommodate binders that target membrane distal epitopes to promote effective formation of the immune synapse.
Given these implications on activation, together with knowledge of the target epitope location and antigen density, the length of the hinge can be manipulated to fine-tune CAR sensitivity (Figure 2C). The selection of shorter hinges can promote antigen-driven activation of CARs targeting membrane distal epitopes (73, 83). Conversely, shortening the hinge length is a useful method to attenuate the activation of a CAR targeting a membrane-proximal epitope of an overexpressed but not cancer-specific antigen, such as epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), or prostate stem cell antigen (PSCA) (84, 85). While tumor cells with high antigen density elicited the activation of HER2 CAR T cells with a conventional 45 amino acid CD8α hinge as well as the 34 amino acid truncated hinge, tumor cells with low antigen density selectively activated the longer hinge bearing CAR (84). Similarly, PSCA-targeted CAR T cells incorporating a 229 or 129-amino acid IgG4-derived hinge activated against high and low antigen targets, whereas a CAR containing a 22-amino acid long IgG4 hinge only displayed activity against high antigen targets (85). This mechanism of selectivity is likely independent of the size exclusion model, as CARs with the longest hinge are still capable of activation (i.e., all CARs can exclude CD45). Instead, other structural or steric factors, such as restricted flexibility or suboptimal scFv orientation when the hinge is too short, may influence activation. Although this approach tunes the safety and selectivity profile of the CAR, it may also have the unwanted effect of selecting for tumor variants with even lower antigen density that can escape CAR T cell activation.
Taken together, these results underscore that there is not a one-size-fits-all approach to hinge engineering and that optimization is required for each CAR. Nonetheless, understanding the properties of the scFv (i.e. affinity, oligomerization propensity), the position of the target epitope on the antigen and its proximity to the membrane, as well as overall antigen density may help to predict a ‘goldilocks’ hinge identity and length.
3.3 Transmembrane domain
The transmembrane domain links the extracellular region to the intracellular signaling domains and anchors the CAR to the surface of the cell. The transmembrane domain from CD8α and CD28 are the most popular choice preclinically and clinically (Figure 1), however, CD3ζ and CD4 transmembrane domains have also been described (86, 87). Although in some cases the hinge and transmembrane domains are studied separately, they are often used en bloc, thereby making it difficult to distinguish the functional contributions of each domain.
3.3.1 Surface expression
Fujiwara et al. investigated the roles of the hinge and transmembrane domains in regulating the surface expression of the CAR using domains from CD4, CD8α, and CD28 (87). T cells transduced with a VEGFR2-specific CAR with a hinge and transmembrane domain from CD8α or CD28 showed higher surface expression than CARs with a hinge and transmembrane domain derived from CD3ζ or CD4, suggesting these domains play an important role in the stability of surface presentation (87). Similar findings were also observed with an NKp30 CAR where CD8α or CD28 transmembrane domains provided superior surface expression compared to a CD3ζ transmembrane domain (88).
3.3.2 Association with endogenous molecules
Not only are transmembrane domains actively involved in regulating surface expression, but they are also capable of mediating interactions with endogenous proteins. A first-generation carcinoembryonic antigen (CEA)-specific CAR containing a CD3ζ-derived transmembrane domain was found to dimerize and form complexes with endogenous TCRs (86). As noted in the section on the hinge domain, CARs with a CD28 transmembrane domain, but not those with a CD8α transmembrane domain, were shown to heterodimerize with endogenous CD28 (70). This interaction was demonstrated by co-immunoprecipitation studies, as well as stimulation with anti-CD28, which elicited CAR-dependent proliferation. This phenomenon was attributed to four polar amino acids found in the CD28 transmembrane domain, and disruption of these amino acids abrogated the interaction (70).
3.3.3 Regulation of functional sensitivity
To address the issue of CAR T cell evasion by antigen-low tumor variants, Majzner et al. explored how the CD8α or CD28 hinge and transmembrane domains affect the cytolytic properties of an FMC63-based CD19 CAR (89). Although functionally equivalent in a high antigen density setting, CARs containing the CD8α hinge/transmembrane displayed reduced killing and cytokine production in an antigen low setting compared to the CD28 hinge/transmembrane CAR. The T cells with the CD28 hinge/transmembrane CAR killed their targets more quickly post-engagement, and this was attributed to their ability to form microclusters, followed by supramolecular activation clusters (cSMACs), and recruit ZAP70. In this way, the hinge/transmembrane domain can tune the threshold for antigen recognition to enhance efficacy against antigen low targets (89).
3.3.4 Toxicity implications of hinge/transmembrane choices
Both anti-CD28 stimulation, as well as cell-based expression of the natural ligands of CD28, CD80 and CD86, have been shown to activate T cells expressing high levels of CD28 transmembrane-based CARs, raising the concern of off-target activation (70). Although co-stimulation blockade with CTLA4-Ig was shown to block this effect in vitro, and may also provide a therapeutic solution, further investigation is warranted to understand the implications of this phenomenon. CAR T cell–mediated tumor killing may activate antigen-presenting cells (APCs), such as macrophages, to upregulate CD80/86, creating a positive feedback loop that drives antigen-independent proliferation and function of CAR T cells containing the CD28 hinge and transmembrane domains, thereby contributing to cytokine release syndrome (CRS). Similar mechanisms may arise in other clinical settings, such as infectious complications, where innate immune activation induces CD80/86 expression on APCs, potentially stimulating these CAR T cells. Because this effect was most pronounced when CAR surface expression was very high, engineering strategies such as targeted CAR insertion into the TRAC locus may help limit expression levels and reduce this risk (90).
Alabanza et al. showed that for two CD19 targeting CARs (FMC63 and a humanized scFv), those incorporating CD8α hinge and transmembrane domains produced lower levels of cytokines and exhibited less activation-induced cell death in vitro versus CARs incorporating the CD28 hinge and transmembrane domains (91). Because cytokine-mediated toxicity is a key factor in managing patient care after CAR T cell therapy, the authors propose that selecting CARs with reduced cytokine release profiles may represent a favorable design strategy. Indeed, clinical testing also demonstrated that the CD8α hinge and transmembrane domain containing CAR (Hu19-CD828ζ) exhibited fewer neurologic toxicities and serum cytokine levels than historical use of the CD28 hinge and transmembrane containing CAR (FMC63–28ζ) (92).
4 Open questions
● What is the degree of generalizability of the above-mentioned findings? How does the integration of the sum of the components attenuate or exacerbate trends? For example, shortening the hinge has been shown to increase selectivity for highly expressed antigen targets with proximal binding epitopes (Figure 2C). However, precisely how long or short the hinge should be differs between studies, suggesting that the other features of the CAR and the epitope also determine the threshold.
○ McComb et al. consider the full-length 45 amino acid CD8α hinge to be long, whereas Li et al. consider the 226 amino acid IgG4 hinge to be long (73, 84). In the McComb et al. study, functionality fell off in the low antigen setting between 34 and 22 amino acids, and in the Li et al. study, functionality was limited at 22 amino acids. In the low antigen setting where the CAR targets a proximal antigen, a variety of “longer” hinges may be tolerated, but an activation cut-off may converge across constructs in the range of 30 amino acids.
● When setting out to develop a new CAR, it is not possible to try all permutations of linker, hinge and transmembrane domains, so what combination of pieces might mitigate risk to develop a highly sensitive CAR? Should binder campaigns prioritize validation of scFvs targeting membrane-proximal epitopes? Should membrane-distal parts of the antigen not be part of the bait molecule used to identify binders? Should the CD28 hinge/transmembrane be tested first, as it provides the greatest sensitivity?
● Is shortening the linker a general strategy to increase the functional avidity of the CAR, particularly for antigenic targets that are not abundantly expressed? Singh et al. might suggest that only some scFv/epitope combinations are amenable to this approach. Depending on the antigen binding site, the orientation may or may not be amenable to a CAR dimer binding multiple targets. Although a 4-1BB costimulatory domain may be required to avoid dysfunction, are other engineering approaches also essential to elicit synthetic T cell states to accommodate low-level tonic signaling (93)?
● How does the hinge length and flexibility affect the ability of a CAR to bind in cis and exert either protective effects where the target is expressed on the T cell (94), or epitope masking on tumor cells (95)? This flexibility and masking property is likely to be similarly determined by the size of the antigen and the epitope location.
● Will these properties established in ex vivo engineered T cells be translatable, or do accommodations need to be made for in vivo engineered CARs where the molecule is transiently expressed by RNA? A CD28 hinge/transmembrane configuration was preferred over the CD8α-derived domain in a study testing mRNA/lipid nanoparticle (LNP) in vivo engineering, although this comparison was not investigated in depth (96).
● CARs are also being expressed in other cell types, including macrophages and NK cells. How the hinge and transmembrane domain can be tailored for these settings is only beginning to be explored.
5 Discussion
Seemingly minor structural changes to a CAR can impart strong functional characteristics. To design an efficient receptor, a deep understanding of the individual CAR modular elements, the characteristics of the antigen of interest, and the intersection of these properties is required. Decisions about which structural components to select can have profound consequences regarding the susceptibility to tonic signaling, interaction with or exclusion of endogenous molecules from the signalosome, and can tune the signal strength and threshold of required antigen density for activity. However, as the biochemical design rules are being uncovered, the degree to which they are broadly applicable or unique to an experimental context needs to be recognized.
Not all antigens are created equal. They may have a wide or narrow window of tumor selectivity. They may be densely or sparsely expressed and have variable propensities to reduce expression in response to CAR selective pressure. The specific location of the binding epitope on the antigen of interest is also a key variable. All these factors can be experimentally considered in the construction of a CAR. For example, low antigen density has been addressed by inducing tonic signaling with short linkers to prime the CAR T cells to respond when needed (61) and by the incorporation of the CD28 hinge and transmembrane domains, which have superior sensitivity to coordinate an organized immune synapse. Finally, scFvs targeting membrane distal epitopes can be accommodated by shortening the length of the hinge region (73) to tighten the intermembrane space and exclude the CD45 phosphatase (80).
Taken together, the synthetic CAR molecule delivered as a cell therapy has revolutionized the way we think about disease treatment and immunotherapy. CARs are celebrated for their modular nature, and the choice of each component represents an opportunity to maximize function and tailor the therapy to the unique context.
Author contributions
GB: Conceptualization, Writing – original draft, Writing – review & editing. LE: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. GB is supported by a scholarship from the Canadian Institute for Health Research. LE is a Michael Smith Health Research BC Scholar (SCH-2021-1570) and a Tier 2 Canada Research Chair in Cancer Immunotherapy (CRC-2023-00133), and is further supported by BC Cancer and the BC Cancer Foundation, a Terry Fox Research Institute New Investigator Award (1131), and the Canadian Institute for Health Research (PJT-183637).
Conflict of interest
LE reports SAB and consulting activities for NanoVation Therapeutics and receiving in-kind reagents.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
2. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukemia: phase 2 results of the single-arm, open-label, multicenter ZUMA-3 study. Lancet. (2021) 398:491–502. doi: 10.1016/S0140-6736(21)01222-8
3. Roddie C, Sandhu KS, Tholouli E, Logan AC, Shaughnessy P, Barba P, et al. Obecabtagene autoleucel in adults with B-cell acute lymphoblastic leukemia. N Engl J Med. (2024) 391:2219–30. doi: 10.1056/NEJMoa2406526
4. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
5. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicenter seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
6. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
7. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
8. Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med. (2022) 28:325–32. doi: 10.1038/s41591-021-01622-0
9. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicenter, phase 2 trial. Lancet Oncol. (2022) 23:91–103. doi: 10.1016/S1470-2045(21)00591-X
10. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
11. Martin T, Usmani SZ, Berdeja JG, Agha M, Cohen AD, Hari P, et al. Ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol. (2023) 41:1265–74. doi: 10.1200/JCO.22.00842
12. Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
13. Cappell KM and Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
14. Muller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Volkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - A case series with follow-up. N Engl J Med. (2024) 390(8):687–700. doi: 10.1056/NEJMoa2308917
15. Muller F, Wirsching A, Hagen M, Volkl S, Tur C, Raimondo MG, et al. BCMA CAR T cells in a patient with relapsing idiopathic inflammatory myositis after initial and repeat therapy with CD19 CAR T cells. Nat Med. (2025) 31:1793–7. doi: 10.1038/s41591-025-03718-3
16. Sidana S, Patel KK, Peres LC, Bansal R, Kocoglu MH, Shune L, et al. Safety and efficacy of standard-of-care ciltacabtagene autoleucel for relapsed/refractory multiple myeloma. Blood. (2025) 145:85–97. doi: 10.1182/blood.2024025945
17. Li D, Wang R, Liang T, Ren H, Park C, Tai CH, et al. Camel nanobody-based B7-H3 CAR-T cells show high efficacy against large solid tumors. Nat Commun. (2023) 14:5920. doi: 10.1038/s41467-023-41631-w
18. Schmidts A, Ormhoj M, Choi BD, Taylor AO, Bouffard AA, Scarfo I, et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. (2019) 3:3248–60. doi: 10.1182/bloodadvances.2019000703
19. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
20. Zhang T, Lemoi BA, and Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. (2005) 106:1544–51. doi: 10.1182/blood-2004-11-4365
21. Hebbar N, Epperly R, Vaidya A, Thanekar U, Moore SE, Umeda M, et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun. (2022) 13:587. doi: 10.1038/s41467-022-28243-6
22. Frigault MJ, Bishop MR, Rosenblatt J, O’Donnell EK, Raje N, Cook D, et al. Phase 1 study of CART-ddBCMA for the treatment of subjects with relapsed and refractory multiple myeloma. Blood Adv. (2023) 7:768–77. doi: 10.1182/bloodadvances.2022007210
23. Sadelain M, Brentjens R, and Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
24. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. (2019) 25:82–8. doi: 10.1038/s41591-018-0290-5
25. Park JH, Palomba ML, Perica K, Devlin SM, Shah G, Dahi PB, et al. Results from first-in-human phase I study of a novel CD19-1XX chimeric antigen receptor with calibrated signaling in large B-cell lymphoma. J Clin Oncol. (2025) 43(21):2418–28. doi: 10.1200/JCO-24-02424
26. Lindner SE, Johnson SM, Brown CE, and Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv. (2020) 6:eaaz3223. doi: 10.1126/sciadv.aaz3223
27. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. (2014) 124:1070–80. doi: 10.1182/blood-2013-10-535245
28. Hombach AA, Heiders J, Foppe M, Chmielewski M, and Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology. (2012) 1(4):458–66. doi: 10.4161/onci.19855
29. Posey AD Jr. and June CH. 211. CD2, the first identified T cell co-stimulator, demonstrates more effective chimeric antigen receptor activity over CD28 and 4-1BB. Mol Ther. (2015) 23:S83. doi: 10.1016/S1525-0016(16)33816-3
30. Song DG, Ye Q, Poussin M, Harms GM, Figini M, and Powell DJ Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. (2012) 119:696–706. doi: 10.1182/blood-2011-03-344275
31. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. (2018) 24:352–9. doi: 10.1038/nm.4478
32. Gordon KS, Kyung T, Perez CR, Holec PV, Ramos A, Zhang AQ, et al. Screening for CD19-specific chimeric antigen receptors with enhanced signaling via a barcoded library of intracellular domains. Nat BioMed Eng. (2022) 6:855–66. doi: 10.1038/s41551-022-00896-0
33. Castellanos-Rueda R, Di Roberto RB, Bieberich F, Schlatter FS, Palianina D, Nguyen OTP, et al. speedingCARs: accelerating the engineering of CAR T cells by signaling domain shuffling and single-cell sequencing. Nat Commun. (2022) 13:6555. doi: 10.1038/s41467-022-34141-8
34. Liu B, Zhang W, Xia B, Jing S, Du Y, Zou F, et al. Broadly neutralizing antibody-derived CAR T cells reduce viral reservoir in individuals infected with HIV-1. J Clin Invest. (2021) 131(19):e150211. doi: 10.1172/JCI150211
35. Schett G, Muller F, Taubmann J, Mackensen A, Wang W, Furie RA, et al. Advancements and challenges in CAR T cell therapy in autoimmune diseases. Nat Rev Rheumatol. (2024) 20:531–44. doi: 10.1038/s41584-024-01139-z
36. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
37. Yang D, Sun B, Li S, Wei W, Liu X, Cui X, et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci Transl Med. (2023) 15:eadd1951. doi: 10.1126/scitranslmed.add1951
38. Du B, Qin J, Lin B, Zhang J, Li D, and Liu M. CAR-T therapy in solid tumors. Cancer Cell. (2025) 43:665–79. doi: 10.1016/j.ccell.2025.03.019
39. Nix MA and Wiita AP. Alternative target recognition elements for chimeric antigen receptor (CAR) T cells: beyond standard antibody fragments. Cytotherapy. (2024) 26:729–38. doi: 10.1016/j.jcyt.2024.02.024
40. Cappell KM and Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. (2021) 18:715–27. doi: 10.1038/s41571-021-00530-z
41. Rafiq S, Hackett CS, and Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
42. Li Y, Hermanson DL, Moriarity BS, and Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92 e5. doi: 10.1016/j.stem.2018.06.002
43. Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A. (1988) 85:5879–83. doi: 10.1073/pnas.85.16.5879
44. van Rosmalen M, Krom M, and Merkx M. Tuning the flexibility of glycine-serine linkers to allow rational design of multidomain proteins. Biochemistry. (2017) 56:6565–74. doi: 10.1021/acs.biochem.7b00902
45. Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. (1993) 6:989–95. doi: 10.1093/protein/6.8.989
46. Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, et al. Single-chain antigen-binding proteins. Science. (1988) 242:423–6. doi: 10.1126/science.3140379
47. Chen X, Zaro JL, and Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Delivery Rev. (2013) 65:1357–69. doi: 10.1016/j.addr.2012.09.039
48. Grahnert A, Seiffert S, Wenk K, Schmiedel D, Boldt A, Vucinic V, et al. Evaluation of Anti-CAR Linker mAbs for CAR T Monitoring after BiTEs/bsAbs and CAR T-Cell Pretreatment. Biomedicines. (2024) 12(8):1641. doi: 10.3390/biomedicines12081641
49. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
50. Singh A, L’Heureux S, Sinapius R, Kvorjak M, Wade E, Lohmueller J, et al. Abstract 898: Generation and validation of anti-linker monoclonal antibodies for the detection of surface expressed scFv-based CARs. Cancer Res. (2023) 83:898–. doi: 10.1158/1538-7445.AM2023-898
51. Schindler K, Ruppel KE, Muller C, Koehl U, Fricke S, and Schmiedel D. Linker-specific monoclonal antibodies present a simple and reliable detection method for scFv-based CAR NK cells. Mol Ther Methods Clin Dev. (2024) 32:101328. doi: 10.1016/j.omtm.2024.101328
52. Harrer DC, Li SS, Kaljanac M, Bezler V, Barden M, Pan H, et al. Magnetic CAR T cell purification using an anti-G4S linker antibody. J Immunol Methods. (2024) 528:113667. doi: 10.1016/j.jim.2024.113667
53. Kouro T, Hoshino D, Mano Y, Tsuji S, Himuro H, Imai K, et al. Improvement in the function of self-activating chimeric antigen receptor by replacing the linker sequence. Front Immunol. (2025) 16:1502607. doi: 10.3389/fimmu.2025.1502607
54. Hudson PJ and Kortt AA. High avidity scFv multimers; diabodies and triabodies. J Immunol Methods. (1999) 231:177–89. doi: 10.1016/S0022-1759(99)00157-X
55. Holliger P, Prospero T, and Winter G. Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A. (1993) 90:6444–8. doi: 10.1073/pnas.90.14.6444
56. Perisic O, Webb PA, Holliger P, Winter G, and Williams RL. Crystal structure of a diabody, a bivalent antibody fragment. Structure. (1994) 2:1217–26. doi: 10.1016/S0969-2126(94)00123-5
57. Atwell JL, Breheney KA, Lawrence LJ, McCoy AJ, Kortt AA, and Hudson PJ. scFv multimers of the anti-neuraminidase antibody NC10: length of the linker between VH and VL domains dictates precisely the transition between diabodies and triabodies. Protein Eng. (1999) 12:597–604. doi: 10.1093/protein/12.7.597
58. Dolezal O, Pearce LA, Lawrence LJ, McCoy AJ, Hudson PJ, and Kortt AA. ScFv multimers of the anti-neuraminidase antibody NC10: shortening of the linker in single-chain Fv fragment assembled in V(L) to V(H) orientation drives the formation of dimers, trimers, tetramers and higher molecular mass multimers. Protein Eng. (2000) 13:565–74. doi: 10.1093/protein/13.8.565
59. Kortt AA, Lah M, Oddie GW, Gruen CL, Burns JE, Pearce LA, et al. Single-chain Fv fragments of anti-neuraminidase antibody NC10 containing five- and ten-residue linkers form dimers and with zero-residue linker a trimer. Protein Eng. (1997) 10:423–33. doi: 10.1093/protein/10.4.423
60. Pluckthun A and Pack P. New protein engineering approaches to multivalent and bispecific antibody fragments. Immunotechnology. (1997) 3:83–105. doi: 10.1016/S1380-2933(97)00067-5
61. Singh N, Frey NV, Engels B, Barrett DM, Shestova O, Ravikumar P, et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat Med. (2021) 27:842–50. doi: 10.1038/s41591-021-01326-5
62. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
63. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. (2015) 3:356–67. doi: 10.1158/2326-6066.CIR-14-0186
64. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
65. Landoni E, Fuca G, Wang J, Chirasani VR, Yao Z, Dukhovlinova E, et al. Modifications to the framework regions eliminate chimeric antigen receptor tonic signaling. Cancer Immunol Res. (2021) 9:441–53. doi: 10.1158/2326-6066.CIR-20-0451
66. Bister A, Ibach T, Haist C, Smorra D, Roellecke K, Wagenmann M, et al. A novel CD34-derived hinge for rapid and efficient detection and enrichment of CAR T cells. Mol Ther Oncolytics. (2021) 23:534–46. doi: 10.1016/j.omto.2021.11.003
67. Bister A, Ibach T, Haist C, Gerhorst G, Smorra D, Soldierer M, et al. Optimized NGFR-derived hinges for rapid and efficient enrichment and detection of CAR T cells in vitro and in vivo. Mol Ther Oncolytics. (2022) 26:120–34. doi: 10.1016/j.omto.2022.05.012
68. Evans EJ, Esnouf RM, Manso-Sancho R, Gilbert RJ, James JR, Yu C, et al. Crystal structure of a soluble CD28-Fab complex. Nat Immunol. (2005) 6:271–9. doi: 10.1038/ni1170
69. Lazar-Molnar E, Almo SC, and Nathenson SG. The interchain disulfide linkage is not a prerequisite but enhances CD28 costimulatory function. Cell Immunol. (2006) 244:125–9. doi: 10.1016/j.cellimm.2007.02.014
70. Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front Immunol. (2021) 12:639818. doi: 10.3389/fimmu.2021.639818
71. Srinivasan S, Zhu C, and McShan AC. Structure, function, and immunomodulation of the CD8 co-receptor. Front Immunol. (2024) 15:1412513. doi: 10.3389/fimmu.2024.1412513
72. Chen X, Mirazee JM, Skorupka KA, Matsuo H, Youkharibache P, Taylor N, et al. The CD8alpha hinge is intrinsically disordered with a dynamic exchange that includes proline cis-trans isomerization. J Magn Reson. (2022) 340:107234. doi: 10.1016/j.jmr.2022.107234
73. Li N, Quan A, Li D, Pan J, Ren H, Hoeltzel G, et al. The IgG4 hinge with CD28 transmembrane domain improves V(H)H-based CAR T cells targeting a membrane-distal epitope of GPC1 in pancreatic cancer. Nat Commun. (2023) 14:1986. doi: 10.1038/s41467-023-37616-4
74. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
75. Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. (2015) 23:757–68. doi: 10.1038/mt.2014.208
76. Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. (2016) 5:e1253656. doi: 10.1080/2162402X.2016.1253656
77. Davis SJ and van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol. (2006) 7:803–9. doi: 10.1038/ni1369
78. Xu X, Li H, and Xu C. Structural understanding of T cell receptor triggering. Cell Mol Immunol. (2020) 17:193–202. doi: 10.1038/s41423-020-0367-1
79. Furlan G, Minowa T, Hanagata N, Kataoka-Hamai C, and Kaizuka Y. Phosphatase CD45 both positively and negatively regulates T cell receptor phosphorylation in reconstituted membrane protein clusters. J Biol Chem. (2014) 289:28514–25. doi: 10.1074/jbc.M114.574319
80. Xiao Q, Zhang X, Tu L, Cao J, Hinrichs CS, and Su X. Size-dependent activation of CAR-T cells. Sci Immunol. (2022) 7:eabl3995. doi: 10.1126/sciimmunol.abl3995
81. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. (2008) 180:7028–38. doi: 10.4049/jimmunol.180.10.7028
82. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. (2013) 121:1165–74. doi: 10.1182/blood-2012-06-438002
83. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. (2013) 19:3153–64. doi: 10.1158/1078-0432.CCR-13-0330
84. McComb S, Nguyen T, Shepherd A, Henry KA, Bloemberg D, Marcil A, et al. Programmable attenuation of antigenic sensitivity for a nanobody-based EGFR chimeric antigen receptor through hinge domain truncation. Front Immunol. (2022) 13:864868. doi: 10.3389/fimmu.2022.864868
85. Kennewick KT, Yamaguchi Y, Gibson J, Gerdts EA, Jeang B, Tilakawardane D, et al. Nonsignaling extracellular spacer regulates tumor antigen selectivity of CAR T cells. Mol Ther Oncol. (2024) 32:200789. doi: 10.1016/j.omton.2024.200789
86. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, and Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. (2010) 184:6938–49. doi: 10.4049/jimmunol.0901766
87. Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M, and Okada N. Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells. (2020) 9(5):1182. doi: 10.3390/cells9051182
88. Zhang T, Wu MR, and Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. (2012) 189:2290–9. doi: 10.4049/jimmunol.1103495
89. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. (2020) 10:702–23. doi: 10.1158/2159-8290.CD-19-0945
90. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumor rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
91. Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. (2017) 25:2452–65. doi: 10.1016/j.ymthe.2017.07.013
92. Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. (2020) 26:270–80. doi: 10.1038/s41591-019-0737-3
93. Tien-Ching Chang AH, Lattin J, Barrett A, Landmann J, Warrington J, Tenzin Y, et al. BACH2 regulates T cell lineage states to overcome dysfunction driven by tonic CAR signaling. doi: 10.21203/rs.3.rs-5845875/v1
94. Quach DH, Ganesh HR, Briones YD, Nouraee N, Ma A, Hadidi YF, et al. Rejection resistant CD30.CAR-modified Epstein-Barr virus-specific T cells as an off-the-shelf platform for CD30(+) lymphoma. Mol Ther Oncol. (2024) 32:200814. doi: 10.1016/j.omton.2024.200814
95. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. (2018) 24:1499–503. doi: 10.1038/s41591-018-0201-9
Keywords: chimeric antigen receptor (CAR), T cell, transmembrane domain (TMD), hinge domain, linker region
Citation: Bernard G and Evgin L (2025) Non-signaling but all important: how the linker, hinge, and transmembrane domains in the CAR hold it all together. Front. Immunol. 16:1664403. doi: 10.3389/fimmu.2025.1664403
Received: 11 July 2025; Accepted: 06 October 2025;
Published: 27 October 2025.
Edited by:
Mathieu Crupi, Ottawa Hospital Research Institute (OHRI), CanadaReviewed by:
Kaushik Choudhuri, The University of Utah, United StatesHao Sun, Dana–Farber Cancer Institute, United States
Copyright © 2025 Bernard and Evgin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Evgin, bGV2Z2luQGJjZ3NjLmNh