 Junyi Zhang1†Liyuan Cui2,3,4†Xinhang Meng2,3,4Yujie Luo2,3,4Jingmin Ou1*
Junyi Zhang1†Liyuan Cui2,3,4†Xinhang Meng2,3,4Yujie Luo2,3,4Jingmin Ou1* Songcun Wang2,3,4*
Songcun Wang2,3,4* Mingke Qiu1*
Mingke Qiu1*- 1Department of Interventional Vascular Surgery, Xinhua Hospital, Shanghai JiaoTong University, School of Medicine, Shanghai, China
- 2Laboratory for Reproductive Immunology, Obstetrics & Gynecology Hospital of Fudan University, Shanghai, China
- 3Shanghai Key Lab of Reproduction and Development, Obstetrics & Gynecology Hospital of Fudan University, Shanghai, China
- 4Shanghai Key Lab of Female Reproductive Endocrine Related Diseases, Obstetrics & Gynecology Hospital of Fudan University, Shanghai, China
Arteriosclerosis obliterans (ASO) is a chronic vascular disease characterized by narrowing or occlusion of the vascular lumen. Its pathogenesis is complex and closely associated with lipid metabolism disorders and chronic inflammation. Although notable progress has been made in the treatment of ASO, it still remains a cause of surgical limb loss globally. In recent years, immune checkpoints have been identified as critical regulators of the immune microenvironment that play a significant role in ASO. Furthermore, immune checkpoints can affect lipid metabolism by regulating the metabolic pathways of immune cells, thereby indirectly modulating lipid metabolic processes, such as lipid absorption, transport, and degradation, which are crucial in the development and progression of atherosclerosis. Here, we summarized and discussed progress in studies related to lipid metabolism and immune checkpoints during ASO, and highlighted how immune checkpoints regulate lipid metabolism to affect ASO. Further exploration of the interactions between lipid metabolism regulators and immune checkpoints may uncover novel potential therapeutic targets for ASO management.
1 Introduction
Arteriosclerosis obliterans (ASO) is a chronic occlusive vascular disease caused by atherosclerosis, mainly affecting the arteries of the lower extremities (1). Due to the progression of atherosclerosis within an arterial lumen, accumulation of atherosclerotic plaques leads to narrowing or even occlusion of the arterial lumen, which further triggers a series of symptoms and signs in the affected limb such as ulcers, gangrene, and even amputation (2, 3). Notable progress has been made in ASO treatment, including surgical techniques, endovascular interventions, and pharmacological treatments. However, restenosis usually relapses within 1 – 2 years after therapy, and ASO remains a cause of surgical limb loss globally (4, 5). Moreover, most patients with early-stage ASOs exhibit no obvious clinical symptoms, leading to delayed treatment. Therefore, early diagnostic markers and new therapeutic approaches for ASO are needed.
Dysregulation of lipid metabolism is a key factor in the pathophysiology of ASO because it promotes lipid deposition, triggers inflammatory responses, and impairs endothelial function. Collectively, these processes drive the development and progression of atherosclerosis (6, 7). Immune checkpoints, the key regulatory molecules of immune activation, can influence plaque formation and vascular function by regulating lipid metabolism and inflammation, and are potentially involved in the occurrence and development of ASO (8–10).
This review summarized the general diagnosis and pathological changes in ASO, and highlighted how immune checkpoints regulate lipid metabolism to cause ASO. By integrating the latest study progress on immune checkpoints and lipid metabolism regulation, novel immune-metabolic combination therapies may be explored to achieve a precise ASO treatment.
2 Arteriosclerosis obliterans
ASO is a subtype of peripheral artery disease with increasing global incidence (11, 12). Given its profound impact on patients’ quality of life, early detection, effective prevention, and timely intervention are of paramount importance (13, 14). The diagnosis and treatment of ASO are closely related to atherosclerotic plaques (15, 16). The primary treatment goal is to identify and eliminate arterial plaques, alleviate symptoms, improve quality of life, and reduce the risk of amputation.
2.1 Pathology of arteriosclerosis obliterans
Formation of arterial plaques and subsequent vascular narrowing play crucial roles in ASO development (17). The accumulation of atherosclerotic material, coupled with secondary thrombosis and vascular endothelial dysfunction, contributes to the thickening of the intima in lower extremity arteries. This results in the narrowing of arterial lumen and complete occlusion in severe cases (18). These changes lead to a range of clinical manifestations and symptoms in affected limbs. The disruption of lipid metabolism plays an important role in the earliest lesions in ASO (19). The core mechanism involves lipid deposition and chain reactions. Low-density lipoproteins (LDL) penetrate the intima through vascular endothelial cells and undergo local oxidation to form oxidized LDL (ox-LDL). Ox-LDL induces monocytes to adhere to endothelial cells, migrates into the intima, and transforms into macrophages (20, 21). After engulfing ox-LDL, macrophages form foam cells that promote arterial plaques (22). Lipid deposition triggers local inflammatory responses, stimulating the activation of surrounding vascular smooth muscle cells (VSMCs) and fibroblasts (23). Continuous secretion of inflammatory mediators is understood to be a self-amplifying inflammatory cascade that ultimately promotes an unstable plaque phenotype, plaque erosion and rupture, and the formation of occlusive arterial thrombi that restrict blood flow and cause critical tissue ischemia (24–26).
2.2 High-risk factors of arteriosclerosis obliterans
Many risk factors, including smoking, age, sex, genetics, diabetes, hypertension, and hyperlipidemia, can lead to ASO, and these factors are often associated with lipid metabolism disorders and inflammation (17, 27).
Smoking is a significant risk factor for vascular diseases, especially those affecting lipids and cytokines, which contribute to vascular damage and ASO. Smokers may have higher concentrations of serum total cholesterol and LDL than non-smokers, increasing their risk of atherosclerosis and coronary artery disease (28, 29). Nicotine and its primary metabolite, cotinine, activate nuclear factor kappa-B (NF-κB) transcription factor, thereby driving tissue factor expression in endothelial cells (ECs) and VSMCs (30).
With aging, vascular walls gradually lose their elasticity and endothelial cell function deteriorates, leading to a reduced capacity for vascular repair following injury (31, 32). Aging is accompanied by chronic inflammation and alterations in lipid metabolism, which further accelerate arteriosclerosis progression (33, 34). Hypertension, hyperlipidemia, and diabetes can also disrupt lipid metabolism and trigger inflammation, all of which are high-risk factors for ASO (35–38). Genetic conditions, such as familial hypercholesterolemia or familial mixed hyperlipidemia, directly affect lipid metabolism pathways, leading to LDL accumulation (39, 40).
Therefore, smoking cessation is critical for the prevention and treatment of ASO. Attention should also be paid to regulating lipid metabolism, such as adopting a healthy diet, controlling blood lipid concentrations, and using lipid-lowering medications (e.g., statins), as part of a comprehensive approach. Moreover, the inflammation triggered by various factors cannot be ignored in ASO.
2.3 Lipid metabolism and arteriosclerosis obliterans
Lipid metabolism refers to the entire process of digestion, absorption, transportation, synthesis, breakdown, and utilization of lipid substances in the body (41). Lipid metabolism affects arterial plaques and vascular function through lipid accumulation, fatty acid metabolism, cholesterol transport, and inflammation, thereby contributing to ASO development (42, 43). For example, LDL accumulation in a vessel wall, which is converted into foam cells, and the effect of fatty acids on phenotypic changes in macrophages can exacerbate ASO (44, 45). Additionally, lipid metabolic products, such as oxidized cholesterol derivatives, can activate inflammatory pathways and damage vascular endothelium (46). By activating receptors (such as CD36) on macrophages and VSMCs, ox-LDL triggers an inflammatory response that leads to the progression of atherosclerotic plaques (47, 48). Numerous molecules, such as sterol regulatory element-binding proteins (SREBPs), adenosine monophosphate-activated protein kinase (AMPK), and liver X receptors (LXR) may play pivotal roles in these processes (49–51). Here, we focused on lipid metabolism-related molecules that contribute to ASO.
2.3.1 Sterol regulatory element-binding proteins
SREBPs are a class of transcription factors that play key roles in lipid metabolism, cholesterol synthesis, and fatty acid synthesis (50). SREBP - 1 regulates the transcription of acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN) genes to promote lipogenesis, which relies on protein kinase B (Akt)/mammalian target of rapamycin complex 1 (mTORC1) signaling (52, 53). SREBP - 2 overactivation enhance cholesterol synthesis and LDL uptake, leading to hypercholesterolemia (54). SREBP - 2 can directly bind to protein phosphatase 2A or be activated by molecules, such as Erb-B2 receptor tyrosine kinase 4, thereby promoting LDL uptake (55, 56). The SREBP - 2 signaling pathway could be interfered with by histone deacetylase inhibitors (such as butyrate), resulting in a cholesterol-lowering effect (57). Additionally, SREBP - 1c contributes to fatty acid synthesis and lipid accumulation, which exacerbates lipotoxicity and vascular inflammation, and accelerates plaque development (58). In addition to lipid accumulation, cholesterol synthesis also contributes to vascular inflammation, including NOD-like receptor protein 3 inflammasome activation, oxidative stress, and endothelial dysfunction, all of which are key events in atherosclerosis (59, 60). This implies that SREBPs regulate cholesterol metabolism to affect vascular function, further affecting ASO.
2.3.2 Adenosine monophosphate-activated protein kinase
AMPK is a heterotrimeric complex that is activated under conditions of energy stress such as decrease intracellular adenosine triphosphate (ATP) concentrations (61, 62). It reduces lipid accumulation and inflammation by regulating metabolic balance (63). AMPK phosphorylates and inhibits ACC, a key enzyme in fatty acid synthesis that promotes fatty acid oxidation (FAO) (64, 65). Lepropre et al. demonstrated that the AMPK-ACC signaling pathway modulates platelet phospholipid content, thereby regulating arachidonic acid production (62). This process affects thromboxane generation and granule release during platelet activation, ultimately playing a critical role in the regulation of thrombosis formation (66). AMPK also promotes FAO by relieving the inhibitory effect on CPT1A (67). Moreover, AMPK plays a pivotal role in regulating cholesterol concentrations by upregulating ATP-binding cassette (ABC) transporters (e.g., ABCA1 and ABCG1), downregulating the expression of cholesterol synthesis gene 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), inhibiting SREBP - 1, suppressing the mTORC1 pathway, and mitigating plaque formation (68–71). AMPK activation not only modulates lipid metabolism to mitigate arterial plaque formation but also effectively reduces vascular inflammation. Studies have shown that AMPK activation can suppress the release of pro-inflammatory cytokines, such as interleukin-6 (IL - 6) and tumor necrosis factor-α (TNF-α), which contribute significantly to plaque instability (72, 73). Thus, AMPK reduces foam cell formation, plaque instability, and vascular inflammation, making it a promising therapeutic target for ASO.
2.3.3 Peroxisome proliferator-activated receptors
PPARs are ligand-activated transcription factors belonging to the nuclear receptor superfamily. They regulate the transcription of target genes by forming dimers and binding to specific DNA regions, thereby participating in various physiological processes. There are three main subtypes of PPARs: PPAR-α, PPAR-γ, and PPAR-δ/β (74).
PPARα’s central function in FAO is to regulate downstream genes, promoting the uptake and activation of long-chain fatty acids (75). Activated PPAR-α bound to the retinoid X receptor to form a heterodimer, which initiates the transcription of target genes such as fatty acid transport (FAT) and carnitine palmitoyltransferase-1 (CPT-1), thereby reducing lipid accumulation (67, 76). Moreover, PPARα could upregulate lipoprotein lipase and inhibit apolipoprotein C-III expression, thereby reducing triglyceride concentrations in blood (67, 77). In addition to breaking down triglycerides, PPAR-α agonist like LY518674 might promote high-density lipoprotein (HDL) production and reverse cholesterol transport, effectively clearing cholesterol from the vascular walls (78–80). Cholesterol efflux is promoted by the activation of ABCA1 and scavenger receptor class B type I (SR-BI). Notably, a study revealed that SR-BI regulates transcription factor EB expression by enhancing PPAR-α activation (81). This finding identifies SR-BI as a potential new therapeutic target for atherosclerosis (81).
PPAR-γ depends on phosphatidylinositol-3-kinase (PI3K)/Akt/mTOR signal regulation as a key regulator of adipocyte differentiation (82). It promotes the uptake and storage of free fatty acids in adipocytes by inducing the expression of genes, such as fatty acid-binding protein 4 (FABP4), thereby reducing the concentration of fatty acids in the bloodstream (83). PPAR-γ upregulates the expression of antioxidant-related genes, such as glutathione peroxidase (GPx) and superoxide dismutase (SOD), thereby reducing ox-LDL production (84). PPAR-γ might reduce vascular endothelial damage through a reduction of oxidative stress responses (85). At the same time, PPAR-γ decreases ox-LDL uptake by macrophages in vascular walls (86).
PPAR-δ/β activates key downstream genes, such as CPT-1 and acyl-CoA oxidase 1 through ligand binding, thereby promoting β-oxidation (87). Additionally, PPAR-δ/β regulates the expression of FASN and FABP, which were involved in fat synthesis and storage, thereby reducing fat accumulation (67, 88). Furthermore, PPAR-δ/β facilitates lipolysis by activating genes such as adipose triglyceride lipase (89).
PPARs not only regulates lipid metabolism but also play a crucial role in inhibiting inflammation. PPAR-α suppresses the transcription of inflammatory genes by interfering with the NF-κB signaling pathway (90). It also downregulates chemokines and intercellular adhesion molecule-1, thereby reducing monocyte infiltration into the arterial wall (91). PPAR-γ regulates macrophage phenotypes, thereby promoting transition from pro-inflammatory M1 to anti-inflammatory M2 (92). PPAR-δ/β might inhibit the expression of chemokines, such as chemokine ligand 2 and CXC-chemokine ligand-8, reducing the accumulation of inflammatory cells in local tissues (93, 94). In contrast, PPAR-δ/β activates antioxidant genes like heme oxygenase-1 and quinone oxidoreductase-1, enhancing cellular antioxidant defense (95). Furthermore, PPAR-δ/β modulates the production of pro-inflammatory factors (such as vascular cell adhesion molecule-1) in ECs, thereby alleviating inflammatory response in a vascular wall (96). Thus, PPARs have broad application prospects in the prevention and treatment of ASO, as they regulate lipid metabolism, reduce inflammatory responses, and improve vascular function.
2.3.4 Liver X receptors
LXR is a nuclear receptor transcription factor that exists as two main subtypes: LXRα and LXRβ (51). It serves as a critical regulator of lipid metabolism, cholesterol transport, and anti-inflammatory responses by modulating target gene expression. LXR promotes the efflux of cholesterol from macrophages and foam cells to HDL by activating the downstream genes ABCA1 and ABCG1, thereby reducing intracellular cholesterol accumulation (97). LXRα can be upregulated by PPARγ to promote ABCA1 expression and enhance cholesterol efflux (97). Kim et al. found that LXR activation can inhibit toll-like receptor signaling and reduce the expression of inflammatory genes by inducing changes in membrane lipid composition mediated by ABCA1 (98). Additionally, LXR could activate the cholesterol 7 alpha-hydroxylase gene, thereby promoting cholesterol conversion to bile acids and accelerating cholesterol clearance (99). Moreover, LXR plays a role in upregulating SREBP - 1c expression, interaction with AMPK, and collaboration with PPAR-γ to regulate lipid metabolism, further contributing to lipid regulation (100). Therefore, as a crucial factor in lipid metabolism that influences ASO, in-depth studies on the bidirectional regulatory effects of LXR on lipid metabolism and inflammatory responses is essential.
3 Immune checkpoints and arteriosclerosis obliterans
Immune checkpoints are molecules that regulate immune responses. In most cases, they prevent immune system overactivation, thereby protecting normal cells and healthy tissues from harm (101). Common immune checkpoints include programmed cell death protein 1 (PD - 1), cytotoxic T lymphocyte-associated protein 4 (CTLA - 4), T cell immunoglobulin and mucin-domain containing-3 (Tim-3), etc. These checkpoints play various roles in immune cell activation, differentiation, and immune tolerance (102, 103). Atherosclerosis is closely associated with immune system dysregulation, particularly during endothelial injury, inflammation, and immune cell infiltration (104, 105). Inflammation serves as a core driver of atherosclerosis, connecting traditional risk factors (such as LDL and hypertension) with alterations in vascular wall biology (106). The immune system promotes plaque formation by initiating an inflammatory response (107). By modulating immune cell function and either promoting or inhibiting anti-inflammatory responses, immune checkpoints may influence the critical stages of atherosclerosis, thus playing vital roles in ASO development (Figure 1).
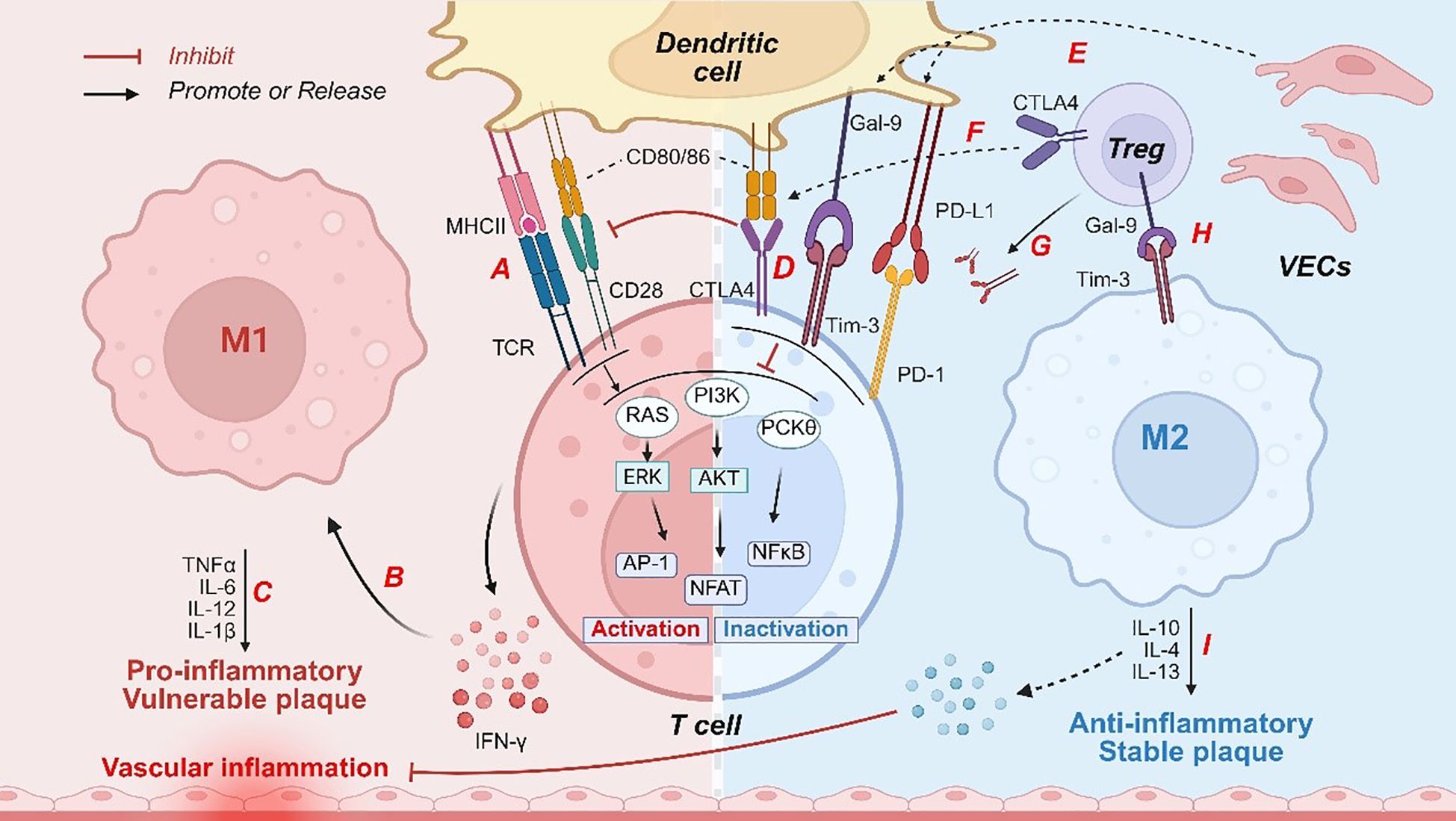
Figure 1. Immune checkpoints regulate immune responses to influence vascular inflammation and plaque stability. Pathogenic factors, such as ox-LDL, induce activation of DCs, characterized by upregulation of (A) MHC II molecules and costimulatory markers (CD80/CD86). These activated dendritic cells engage T-cell receptors, initiating downstream signaling pathways including RAS, PI3K, and PKCθ. Subsequently, (B) IFN-γ secretion by T cells promotes M1 macrophage polarization. M1-derived (C) pro-inflammatory cytokines directly compromise vascular endothelial integrity, exacerbating vascular inflammation and destabilizing plaques. Under inflammatory conditions, (D, E) DCs or VECs express ligands for immune checkpoint molecules, such as PD-L1 and Gal-9, which bind to immune checkpoint molecules on T cells (with CTLA4 competitively inhibiting CD80/CD86 binding to CD28), thereby suppressing the signaling pathways associated with T cell activation. Moreover, (F, G) CTLA - 4 expressed on Tregs depleted CD80/CD86 and released free PD-L1. (H) The Gal-9 secreted by Tregs interacts with Tim-3 receptors on macrophages, driving the polarization of macrophages toward the M2 phenotype. Subsequently, (I) M2 macrophages secrete anti-inflammatory mediators that inhibit inflammation and enhance the stability of atherosclerotic plaques. PI3K, phosphatidylinositol-3-kinase; DCs, dendritic cells; PD - 1, programmed cell death protein -1; CTLA - 4, cytotoxic T lymphocyte associated protein 4; Tim-3, T cell immunoglobulin and mucin-domain containing-3; MHC, major histocompatibility complex; TCR, T-cell receptor; Tregs, regulatory T cells; PD-L1, programmed death ligand-1; Gal-9, galectin-9; TGF-β, transforming growth factor β; ox-LDL, oxidized LDL; NF-κB, nuclear factor kappa-B; ECs, endothelial cells; RAS, rat sarcoma; Akt, protein kinase B; AP - 1, activator protein 1; ERK, extracellular signal-regulated kinase; NFAT, nuclear factor of activated T cells; TNF-α, tumor necrosis factor-α; IFN-γ, interferon γ; IL, interleukin.
3.1 Immune checkpoints and immune responses in arteriosclerosis obliterans
After macrophages and dendritic cells (DCs) phagocytose ox-LDL, antigens are presented via major histocompatibility complex (MHC) molecules on the surfaces of antigen-presenting cells (APCs). Subsequently, MHC binding to T-cell receptor (TCR) activates signaling pathways such as rat sarcoma, PI3K, and PKCθ, thereby initiating T cell activation (108–110). However, full activation of T cells requires co-stimulatory signals such as the interaction between CD28 on the T cell surface and CD80/CD86 ligands on the surface of APCs (111).
CTLA-4 is a structural homolog of CD28 and is primarily expressed in activated T cells and regulatory T cells (Tregs). CTLA - 4 binds to CD80/CD86 on the APC surface with high affinity, thereby directly competitively blocking CD28 signaling and inhibiting TCR signal transduction (112). Studies have shown that the overexpression of CTLA - 4 significantly reduces the area of atherosclerotic lesions and decreases the infiltration of macrophages and CD4+ T cells within plaques via mechanisms involving the inhibition of CD4+ T cell proliferation, downregulation of CD80/CD86 expression, and suppression of T cell activation (113). In contrast, CTLA - 4 inhibition promotes CD4+ T cell differentiation to T-helper type 1 (Th1) cells, ultimately exacerbating atherosclerosis (114). Furthermore, CTLA - 4 is a key molecule through which Tregs exert their immunosuppressive functions (115). CTLA - 4 expression on Tregs enhances their immunosuppressive effects by inhibiting the activation of effector T cells and reducing inflammatory responses (116). Tekguc et al. found that Treg-expressing CTLA - 4 depleted CD80/CD86 and released free programmed death ligand-1(PD-L1) on APCs, exerting dual suppressive effects on T-cell immune responses (117).
PD-1 is an inhibitory co-receptor broadly expressed on the surface of activated T cells. Upon binding to its ligand PD-L1/PD-L2, PD - 1 delivers a negative regulatory signal to the cell. In the atherosclerotic environment, PD - 1 suppresses excessive T cell activation and limited Th1 differentiation, thereby reducing the release of pro-inflammatory cytokines such as interferon γ (IFN-γ) and TNF-α (9). A study revealed that in PD - 1 agonist-treated mice, atherogenic IFN-γ-producing splenic CD4+T cells and cytotoxic CD8+T cells were reduced, while atheroprotective IL - 10-producing CD4+T cells were increased. Additionally, the levels of regulatory B cells, B1 cells, and atheroprotective circulating ox-LDL-specific IgM were significantly elevated (118). In PD - 1 and LDL receptor-deficient mice, predominant activation of pro-inflammatory T cells leads to dyslipidemia, vascular inflammation, and atherosclerosis (119).
Tim-3 is an inhibitory receptor expressed on activated T cells, Tregs, macrophages, and DCs, and its primary ligand is galectin-9 (Gal-9) (120). Tim-3 signaling directly induces the apoptosis of pro-inflammatory T cells (such as Th1 cells) (121). It can also regulate inflammatory response by inhibiting NF-κB activation (122). Therefore, the Tim-3 pathway suppresses immune inflammatory responses and exerts atheroprotective effects. Animal experiments demonstrated that administration of anti-Tim-3 antibodies significantly increased the area of lipid streaks and mature plaques, accompanied by an increase in macrophages and CD4+ T cells, while the proportion of Treg cells decrease (123). Moreover, our study group found that, at an early stage of atherosclerosis, the proportion of PD - 1+ Tim-3+ CD8+ T cells increased in peripheral or arterial blood of patients. Dual blockade of these two immune checkpoints had led to elevated TNF-α and IFN-γ levels, along with decreased IL - 10 and IL - 4 levels (124). At the higher stage of atherosclerosis, the proportion of PD - 1+ Tim-3+ CD4+ T cells was higher in peripheral or arterial blood of patients. Furthermore, simultaneous blockade of the Tim-3 and PD - 1 signaling pathways exacerbates the pro-atherogenic Th1 response in lower extremity ASO (125). Moreover, Tim-3 signaling drives macrophages toward an anti-inflammatory phenotype. In a glioma study, Gal-9 was shown to activate Tim-3 and its downstream pathways to promote M2 macrophage polarization. Enhanced Tim-3 expression predicts poor prognosis in patients with cancer Conversely, blocking Tim-3 signaling inhibits M2 polarization of macrophages and suppresses tumor growth (126). In the transforming growth factor β (TGF-β)-activated tumor microenvironment, Tim-3 expression was significantly correlated with M2 macrophage polarization. In vitro experiments confirmed that TGF-β induced Tim-3 expression in monocytes and M2 macrophages (127). In the context of atherosclerosis, the relationship between Tim-3 and macrophage polarization remains unknown. However, the specific mechanisms by which Tim-3 regulates macrophage inflammatory responses and plaque formation require further exploration.
PD-L1 is expressed in ECs and detected in atherosclerotic plaques. Blocking PD-L1 signaling resulted in a marked increase in IFN-γ+ CD8+ T cells within plaques, promoting inflammation and worsening atherosclerotic burden (128). Additionally, ECs produce and secrete Gal-9, and plasma Gal-9 levels are elevated in patients with peripheral arterial disease. However, in high-fat diet–fed mice, genetic deletion of Gal-9 led to a significant increase in atherosclerotic plaque formation (129). Although the expression of these EC-derived ligands is upregulated in atherosclerosis, their participation in protecting endothelial cell function and the specific mechanisms remain to be elucidated.
Notably, an increasing number of studies have shown that immune checkpoint inhibitors can precipitate atherosclerotic cardiovascular events (130, 131). For example, preclinical studies have suggested that immune checkpoint inhibitors may exacerbate inflammatory responses in atherosclerosis and promote plaque progression, whereas retrospective studies have further confirmed that immune checkpoint inhibitors could increase the risk of atherosclerotic vascular events (132). Clinical and imaging studies have shown that treatment with immune checkpoint inhibitors is associated with an increased risk of atherosclerotic cardiovascular disease (133). This further confirms the protective role of checkpoint molecules in atherosclerosis and suggests that close attention should be paid to ASO development when using these inhibitors.
4 Immune checkpoints in lipid metabolism regulation during arteriosclerosis obliterans
Immune checkpoints affect lipid metabolism by regulating the metabolic pathways of immune cells (47). The activation, differentiation, and functions of immune cells are critically dependent on the dynamic homeostasis of lipid metabolism (134). In immune cells, Tim-3, PD - 1, and CTLA - 4 collaboratively inhibit the glycolytic pathway and enhance PPAR/AMPK-dependent FAO, and also reduce lipid biosynthesis by suppressing PI3K/Akt/mTORC1 signaling (135–137). These regulations may drive T cells and macrophages to favor lipids as their energy source, thereby lowering lipid accumulation within arterial plaques. For example, Tim-3 inhibition has been shown to increase plaque area in mice fed with high-fat diet (123). Here, we discussed how immune checkpoints regulate lipid metabolism and affect ASO from specific immune checkpoint molecules (Table 1, Figure 2).
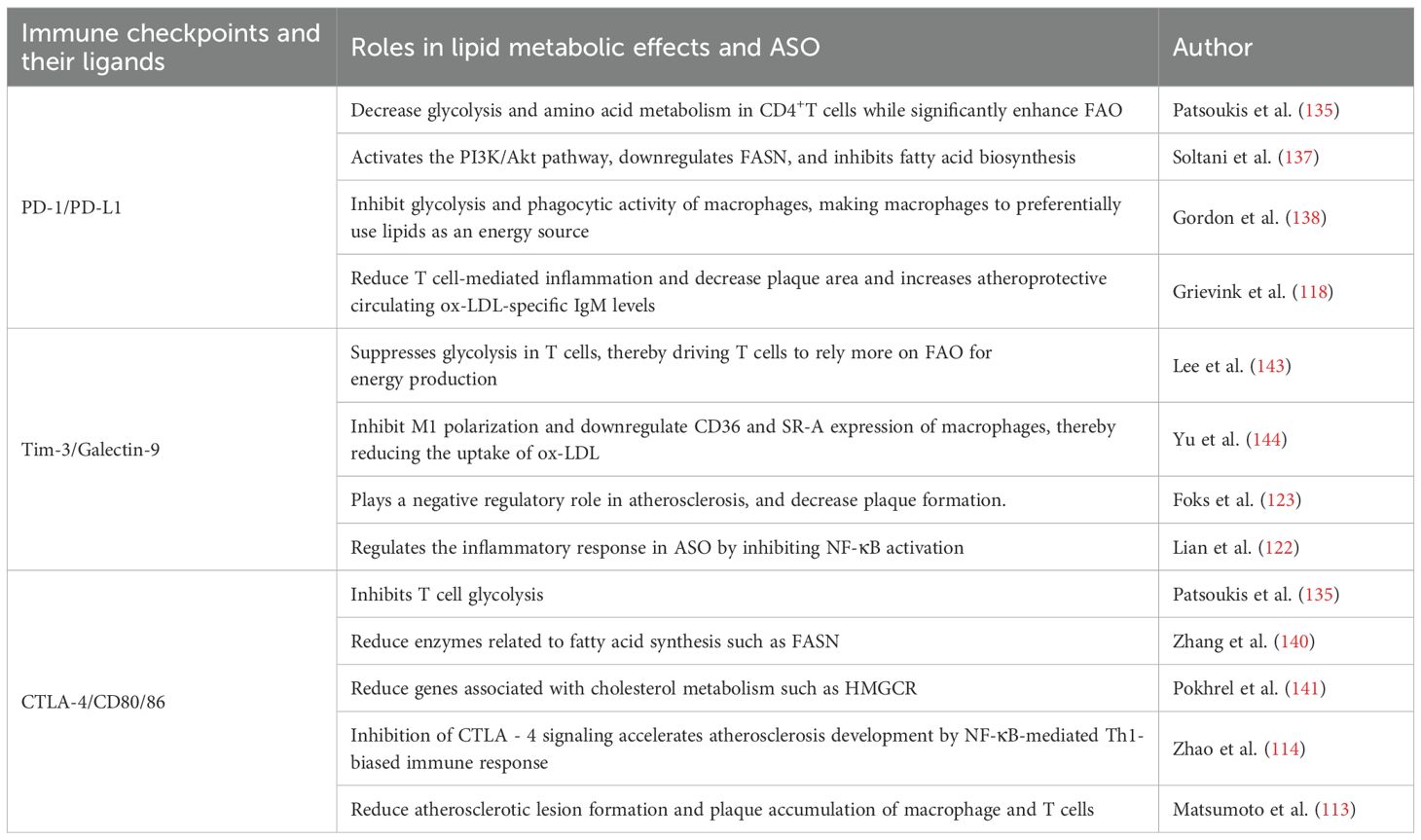
Table 1. Functional interaction of the major immune checkpoints and lipid metabolism during arteriosclerosis obliterans.

Figure 2. Immune checkpoints regulate lipid metabolism to cause ASO. After binding to its ligand, immune checkpoint molecules cause downregulation of the PI3K/Akt/mTOR signaling pathway, (A) thereby upregulating of PPAR-γ and its downstream gene like SOD, leads to a reduction in ox-LDL concentrations, (B) leading to a reduction in SREBP concentrations and subsequent downregulation of its downstream target genes, such as FASN and ACC, ultimately resulting in decreased lipogenesis. Following the inhibition of the PI3K/Akt/mTOR signaling pathway, (C) the metabolic profile shifts toward the predominant FAO and activates the AMPK pathway. Once activated, (D) AMPK induces HMGCR downregulation, resulting in decreased cholesterol synthesis. Moreover, (E) AMPK upregulates ABCA1 and its related targets, thereby increasing cholesterol efflux, and simultaneously (F) upregulates CPT1A, which further enhances FAO and promotes lipolysis. Collectively, these effects lead to reduced lipid accumulation in the endothelial cells and blood vessels, thereby mitigating ASO. ox-LDL, oxidized LDL; SREBPs, sterol regulatory element-binding proteins; AMPK, AMP-activated protein kinase; ACC, acetyl-CoA carboxylase; FAO, fatty acid oxidation; ABC, ATP-binding cassette; mTOR, mammalian target of rapamycin; FAT, fatty acid transport; CPT - 1, carnitine palmitoyltransferase-1; FABP4, fatty acid binding protein 4; SR-BI, scavenger receptor class B type I; GPx, glutathione peroxidase; SOD, superoxide dismutase; FASN, fatty acid synthase; PI3K, phosphatidylinositol-3-kinase; PD - 1, programmed cell death protein -1; CTLA - 4, cytotoxic T lymphocyte associated protein 4; Tim-3, T cell immunoglobulin and mucin-domain containing-3; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; CEH, cholesterol ester hydrolase; PPAR, peroxisome proliferator–activated receptors.
4.1 PD - 1/PD-L1
PD-1 is an important regulator of the immune system and essential for regulating lipid metabolism. Upon PD-L1 binding to PD - 1 and subsequent activation, T cells are unable to carry out glycolysis and amino acid metabolism normally, yet endogenous FAO is enhanced as PD - 1 upregulates the lipases CPT1A and ATGL, thereby promoting endogenous lipolysis and FAO (135). Moreover, PD - 1 exerts metabolic regulatory effects on macrophages. Recent studies have shown that PD - 1 signaling markedly inhibits glycolysis and phagocytic activity in tumor-associated macrophages (138). These metabolic changes may cause macrophages to preferentially use lipids as an energy source, thereby reducing lipid accumulation in arterial plaques and slowing the progression of atherosclerosis. However, the precise mechanism through which PD - 1 regulates lipid metabolism requires further investigation. We hypothesized that a similar mechanism might operate in ASO to reduce plaque formation. Additionally, patients treated with PD - 1/PD-L1 inhibitors for tumors exhibit an increased risk of cardiovascular events, including the aggravation of atherosclerotic occlusive disease (139). Thus, the PD - 1/PD-L1 axis offers a new perspective for ASO treatment. Furthermore, caution should be exercised when using PD - 1 inhibitors in cancer patients with coexisting ASO.
4.2 CTLA - 4
Studies have indicated that CTLA - 4 inhibits glucose uptake in Treg cells; however, unlike PD - 1, it does not significantly enhance FAO in T cells (135). In other words, CTLA - 4 primarily maintains T cells in a metabolically suppressed or homeostatic state rather than actively triggering lipid metabolic pathways. Moreover, in a tumor microenvironment, the CTLA - 4 signaling pathway can affect blood lipid concentrations by regulating enzymes related to fatty acid synthesis (such as FASN) and genes associated with cholesterol metabolism such as HMGCR (140, 141). In mice subjected to antibody-mediated CTLA - 4 blockade, cholesterol synthesis and LDL uptake significantly increased, exacerbating atherosclerotic lesions (142). Although multiple lines of evidence have shown that CTLA - 4 is closely associated with lipid metabolism and plays a role in slowing atherosclerosis, further studies are needed to investigate how CTLA - 4 maintains T cell homeostasis to reduce plaque formation during ASO.
4.3 Tim-3
Experiments showed that in Tim-3-overexpressing Jurkat T cell lines, glucose uptake, lactate production, and glucose transporter-1 concentrations were downregulated, whereas Tim-3 knockout exhibited opposite effects (143). This indicates that Tim-3 signaling suppresses glycolysis in T cells, potentially driving T cells to rely more on FAO for energy production. Moreover, Tim-3 also influences lipid metabolism in macrophages. CD36 and SR-A are primarily responsible for the uptake of lipoprotein-derived cholesterol by macrophages, and are predominantly expressed in M1 macrophages (144). Tim-3 may inhibit M1 polarization, thereby downregulating the expression of CD36 and SRA. Thus, Tim-3 may reduce ox-LDL uptake, decrease foam cell formation, and slow ASO progression. However, studies also found that in human monocyte-derived macrophages, Tim-3 overexpression suppresses miR-155-induced cholesterol ester hydrolase (CEH) expression. Furthermore, miR-155 normally promotes macrophage cholesterol efflux and reduces cholesterol ester accumulation by upregulating CEH expression, thereby inhibiting foam cell formation and atherosclerosis development (145). This implies that Tim-3 accelerates atherosclerosis progression by inhibiting the miR-155-CEH axis. Thus, in-depth studies of the regulatory mechanisms of Tim-3 may provide new directions and targets for ASO treatment.
5 Conclusion and prospects
ASO is a complex chronic vascular disease characterized by disruption of lipid metabolism and chronic inflammation. Lipid accumulation, foam cell formation, and inflammatory responses are critical in ASO development, while immune checkpoints, such as PD - 1, CTLA - 4, and Tim-3, serve as key regulatory elements in the interplay between lipid metabolism and immune activity. These immune checkpoints may affect the metabolic and inflammatory environments of ASOs, thereby influencing lipid uptake, FAO, cholesterol efflux, and macrophage polarization. Therefore, immune checkpoint molecules are potential biomarkers for early ASO diagnosis. Drugs targeting immune checkpoints could be developed to delay ASO progression and prevent postoperative recurrence. Further exploration of the interactions between lipid metabolism regulators (such as AMPK, PPARs, and LXR) and immune checkpoints may reveal novel pathways and potential therapeutic targets for ASO management.
Although immune checkpoint inhibitors can enhance immune responses to tumors by blocking checkpoints and have become a breakthrough in cancer therapy (146), studies on immune checkpoint inhibitors have demonstrated their potential effect on cardiovascular diseases, including heart failure and arteriosclerosis (125, 147). Many studies have reported adverse events related to atherosclerosis caused by immune checkpoint inhibitors (130, 148). Therefore, enhanced monitoring of ASO and cardiovascular risk in patients undergoing immune checkpoint inhibitor therapy is recommended.
Author contributions
JZ: Writing – original draft. LC: Conceptualization, Writing – review & editing. XM: Writing – review & editing, Data curation, Investigation. YL: Investigation, Writing – review & editing, Software. JO: Writing – review & editing, Funding acquisition, Supervision. SW: Funding acquisition, Writing – review & editing, Conceptualization. MQ: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by grants from the National Nature Science Foundation of China (NSFC) (32470992, 82470427, 82201852, and 82370402). Special Youth Project for Clinical Research in Health Industry of the Shanghai Municipal Health Commission (20224Y0005).
Acknowledgments
We thank BioRender.com for its expert assistance in pattern drawing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ASO, arteriosclerosis obliterans; LDL, low-density lipoproteins; ox-LDL, oxidized LDL; VSMCs, vascular smooth muscle cells; NF-κB, nuclear factor kappa-B; ECs, endothelial cells; SREBPs, sterol regulatory element-binding proteins; AMPK, AMP-activated protein kinase; LXR, liver X receptors; ATP, adenosine triphosphate; ACC, acetyl-CoA carboxylase; FAO, fatty acid oxidation; ABC, ATP-binding cassette; mTORC1, mammalian target of rapamycin complex 1; DCs, dendritic cells; IL - 6, interleukin-6; TNF-α, tumor necrosis factor-α; IFN-γ, interferon γ; FAT, fatty acid transport; CPT - 1, carnitine palmitoyltransferase-1; Akt, protein kinase B; HDL, high-density lipoprotein; FABP4, fatty acid binding protein 4; SR-BI, scavenger receptor class B type I; GPx, glutathione peroxidase; SOD, superoxide dismutase; FASN, fatty acid synthase; PI3K, phosphatidylinositol-3-kinase; PD - 1, programmed cell death protein -1; CTLA - 4, cytotoxic T lymphocyte associated protein 4; Tim-3, T cell immunoglobulin and mucin-domain containing-3; MHC, major histocompatibility complex; APCs, antigen-presenting cells; TCR, T-cell receptor; Tregs, regulatory T cells; PD-L1, programmed death ligand-1; Th1, T-helper type 1; Gal-9, galectin-9; TGF-β, transforming growth factor β; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; CEH, cholesterol ester hydrolase; PPAR, peroxisome proliferator–activated receptors.
References
1. Bai J, Wang F, Wang X, Mutu E, Duan C, Qi Y, et al. Expression and clinical significance of HSP27 and its phosphorylation in lower extremity arteriosclerosis obliterans. PeerJ. (2020) 8:e9305. doi: 10.7717/peerj.9305
3. Diehm C, Allenberg JR, Pittrow D, Mahn M, Tepohl G, Haberl RL, et al. Mortality and vascular morbidity in older adults with asymptomatic versus symptomatic peripheral artery disease. Circulation. (2009) 120:2053–61. doi: 10.1161/circulationaha.109.865600
4. Wooten C, Hayat M, du Plessis M, Cesmebasi A, Koesterer M, Daly KP, et al. Anatomical significance in aortoiliac occlusive disease. Clin Anat (New York NY). (2014) 27:1264–74. doi: 10.1002/ca.22444
5. Setacci C, Castelli P, Chiesa R, Grego F, Simoni GA, Stella A, et al. Restenosis: a challenge for vascular surgeon. J Cardiovasc surgery. (2012) 53:735–46.
6. Bäck M, Yurdagul A Jr., Tabas I, Öörni K, and Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
7. Gisterå A and Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. (2017) 13:368–80. doi: 10.1038/nrneph.2017.51
8. Vuong JT, Stein-Merlob AF, Nayeri A, Sallam T, Neilan TG, and Yang EH. Immune checkpoint therapies and atherosclerosis: mechanisms and clinical implications: JACC state-of-the-art review. J Am Coll Cardiol. (2022) 79:577–93. doi: 10.1016/j.jacc.2021.11.048
9. Yousif LI, Tanja AA, de Boer RA, Teske AJ, and Meijers WC. The role of immune checkpoints in cardiovascular disease. Front Pharmacol. (2022) 13:989431. doi: 10.3389/fphar.2022.989431
10. Kusters PJH, Lutgens E, and Seijkens TTP. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc Res. (2018) 114:368–77. doi: 10.1093/cvr/cvx248
11. Adou C, Magne J, Gazere N, Aouida M, Chastaingt L, and Aboyans V. Global epidemiology of lower extremity artery disease in the 21st century (2000 - 21): a systematic review and meta-analysis. Eur J Prev Cardiol. (2024) 31:803–11. doi: 10.1093/eurjpc/zwad381
12. Criqui MH, Matsushita K, Aboyans V, Hess CN, Hicks CW, Kwan TW, et al. Lower extremity peripheral artery disease: contemporary epidemiology, management gaps, and future directions: A scientific statement from the American heart association. Circulation. (2021) 144:e171–e91. doi: 10.1161/cir.0000000000001005
13. Aboyans V, Ricco JB, Bartelink MEL, Björck M, Brodmann M, Cohnert T, et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS): Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteriesEndorsed by: the European Stroke Organization (ESO)The Task Force for the Diagnosis and Treatment of Peripheral Arterial Diseases of the European Society of Cardiology (ESC) and of the European Society for Vascular Surgery (ESVS). Eur Heart J. (2018) 39:763–816. doi: 10.1093/eurheartj/ehx095
14. Hirsch AT, Criqui MH, Treat-Jacobson D, Regensteiner JG, Creager MA, Olin JW, et al. Peripheral arterial disease detection, awareness, and treatment in primary care. Jama. (2001) 286:1317–24. doi: 10.1001/jama.286.11.1317
15. Shah AJ, Pavlatos N, and Kalra DK. Preventive therapies in peripheral arterial disease. Biomedicines. (2023) 11:3157. doi: 10.3390/biomedicines11123157
16. Franzone A, Ferrone M, Carotenuto G, Carbone A, Scudiero L, Serino F, et al. The role of atherectomy in the treatment of lower extremity peripheral artery disease. BMC surgery. (2012) 12 Suppl 1:S13. doi: 10.1186/1471-2482-12-s1-s13
17. Della Corte V, Todaro F, Cataldi M, and Tuttolomondo A. Atherosclerosis and its related laboratory biomarkers. Int J Mol Sci. (2023) 24:15546. doi: 10.3390/ijms242115546
18. Freedman JE and Loscalzo J. Endothelial dysfunction and atherothrombotic occlusive disease. Drugs. (1997) 54 Suppl 3:41–9. doi: 10.2165/00003495-199700543-00007
19. Ferreira HB, Trindade F, Nogueira-Ferreira R, Leite-Moreira A, Ferreira R, Dias-Neto M, et al. Lipidomic insights on abdominal aortic aneurysm and peripheral arterial disease. J Mol Med. (2025). doi: 10.1007/s00109-025-02524-1
20. Cao H, Jia Q, Yan L, Chen C, Xing S, and Shen D. Quercetin suppresses the progression of atherosclerosis by regulating MST1-mediated autophagy in ox-LDL-induced RAW264.7 macrophage foam cells. Int J Mol Sci. (2019) 20:6093. doi: 10.3390/ijms20236093
21. Wang Y, Zou Y, Jiang Q, Li W, Chai X, Zhao T, et al. Ox-LDL-induced CD80(+) macrophages expand pro-atherosclerotic NKT cells via CD1d in atherosclerotic mice and hyperlipidemic patients. Am J Physiol Cell Physiol. (2024) 326:C1563–c72. doi: 10.1152/ajpcell.00043.2024
22. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, and Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med. (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8
23. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, and Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
24. Khan A, Roy P, and Ley K. Breaking tolerance: the autoimmune aspect of atherosclerosis. Nat Rev Immunol. (2024) 24:670–9. doi: 10.1038/s41577-024-01010-y
25. Marchini T, Hansen S, and Wolf D. ApoB-specific CD4(+) T cells in mouse and human atherosclerosis. Cells. (2021) 10:446. doi: 10.3390/cells10020446
26. Bentzon JF, Otsuka F, Virmani R, and Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114:1852–66. doi: 10.1161/circresaha.114.302721
27. Firnhaber JM and Powell CS. Lower extremity peripheral artery disease: diagnosis and treatment. Am Family physician. (2019) 99:362–9.
28. Miller ER 3rd, Appel LJ, Jiang L, and Risby TH. Association between cigarette smoking and lipid peroxidation in a controlled feeding study. Circulation. (1997) 96:1097–101. doi: 10.1161/01.cir.96.4.1097
29. Gellert C, Schöttker B, Müller H, Holleczek B, and Brenner H. Impact of smoking and quitting on cardiovascular outcomes and risk advancement periods among older adults. Eur J Epidemiol. (2013) 28:649–58. doi: 10.1007/s10654-013-9776-0
30. Cirillo P, DER S, Pacileo M, Gargiulo A, Leonardi A, Angri V, et al. Nicotine induces tissue factor expression in cultured endothelial and smooth muscle cells. J Thromb haemostasis: JTH. (2006) 4:453–8. doi: 10.1111/j.1538-7836.2006.01741.x
31. Hwang HJ, Kim N, Herman AB, Gorospe M, and Lee JS. Factors and pathways modulating endothelial cell senescence in vascular aging. Int J Mol Sci. (2022) 23:10135. doi: 10.3390/ijms231710135
32. Tian XL and Li Y. Endothelial cell senescence and age-related vascular diseases. J Genet Genomics = Yi Chuan xue bao. (2014) 41:485–95. doi: 10.1016/j.jgg.2014.08.001
33. Soysal P, Arik F, Smith L, Jackson SE, and Isik AT. Inflammation, frailty and cardiovascular disease. Adv Exp Med Biol. (2020) 1216:55–64. doi: 10.1007/978-3-030-33330-0_7
34. Zeng Q, Gong Y, Zhu N, Shi Y, Zhang C, and Qin L. Lipids and lipid metabolism in cellular senescence: Emerging targets for age-related diseases. Ageing Res Rev. (2024) 97:102294. doi: 10.1016/j.arr.2024.102294
35. Ma J, Li Y, Yang X, Liu K, Zhang X, Zuo X, et al. Signaling pathways in vascular function and hypertension: molecular mechanisms and therapeutic interventions. Signal transduction targeted Ther. (2023) 8:168. doi: 10.1038/s41392-023-01430-7
36. Yuan T, Yang T, Chen H, Fu D, Hu Y, Wang J, et al. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. (2019) 20:247–60. doi: 10.1016/j.redox.2018.09.025
37. Giacco F and Brownlee M. Oxidative stress and diabetic complications. Circ Res. (2010) 107:1058–70. doi: 10.1161/circresaha.110.223545
38. Norton L, Shannon C, Gastaldelli A, and DeFronzo RA. Insulin: The master regulator of glucose metabolism. Metabolism: Clin experimental. (2022) 129:155142. doi: 10.1016/j.metabol.2022.155142
39. Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, and Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis primers. (2017) 3:17093. doi: 10.1038/nrdp.2017.93
40. González-Lleó AM, Sánchez-Hernández RM, Boronat M, and Wägner AM. Diabetes and familial hypercholesterolemia: interplay between lipid and glucose metabolism. Nutrients. (2022) 14:1503. doi: 10.3390/nu14071503
41. DeBose-Boyd RA. Significance and regulation of lipid metabolism. Semin Cell Dev Biol. (2018) 81:97. doi: 10.1016/j.semcdb.2017.12.003
42. Bauersachs R and Zannad F. Rivaroxaban: A new treatment paradigm in the setting of vascular protection? Thromb haemostasis. (2018) 118:S12–s22. doi: 10.1055/s-0038-1636530
43. Yu XH, Zhang DW, Zheng XL, and Tang CK. Cholesterol transport system: An integrated cholesterol transport model involved in atherosclerosis. Prog Lipid Res. (2019) 73:65–91. doi: 10.1016/j.plipres.2018.12.002
44. Ference BA, Braunwald E, and Catapano AL. The LDL cumulative exposure hypothesis: evidence and practical applications. Nat Rev Cardiol. (2024) 21:701–16. doi: 10.1038/s41569-024-01039-5
45. Kotlyarov S and Kotlyarova A. Involvement of fatty acids and their metabolites in the development of inflammation in atherosclerosis. Int J Mol Sci. (2022) 23:1308. doi: 10.3390/ijms23031308
46. Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, and Orekhov AN. The diabetes mellitus-atherosclerosis connection: the role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci. (2020) 21:1835. doi: 10.3390/ijms21051835
47. Adachi H and Tsujimoto M. Endothelial scavenger receptors. Prog Lipid Res. (2006) 45:379–404. doi: 10.1016/j.plipres.2006.03.002
48. Luo Y, Duan H, Qian Y, Feng L, Wu Z, Wang F, et al. Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell Res. (2017) 27:352–72. doi: 10.1038/cr.2017.8
49. Wang Q, Liu S, Zhai A, Zhang B, and Tian G. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol Pharm bulletin. (2018) 41:985–93. doi: 10.1248/bpb.b17-00724
50. Shen S, Shen M, Kuang L, Yang K, Wu S, Liu X, et al. SIRT1/SREBPs-mediated regulation of lipid metabolism. Pharmacol Res. (2024) 199:107037. doi: 10.1016/j.phrs.2023.107037
51. Wang B and Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol. (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
52. Laplante M and Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr biol: CB. (2009) 19:R1046–52. doi: 10.1016/j.cub.2009.09.058
53. Zhou Y, Tao J, Calvisi DF, and Chen X. Role of lipogenesis rewiring in hepatocellular carcinoma. Semin liver disease. (2022) 42:77–86. doi: 10.1055/s-0041-1731709
54. Musso G, Gambino R, and Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res. (2013) 52:175–91. doi: 10.1016/j.plipres.2012.11.002
55. Haskins JW, Zhang S, Means RE, Kelleher JK, Cline GW, Canfran-Duque A, et al. Neuregulin-activated ERBB4 induces the SREBP - 2 cholesterol biosynthetic pathway and increases low-density lipoprotein uptake. Sci Signal. (2015) 8:ra111. doi: 10.1126/scisignal.aac5124
56. Rice LM, Donigan M, Yang M, Liu W, Pandya D, Joseph BK, et al. Protein phosphatase 2A (PP2A) regulates low density lipoprotein uptake through regulating sterol response element-binding protein-2 (SREBP - 2) DNA binding. J Biol Chem. (2014) 289:17268–79. doi: 10.1074/jbc.M114.570390
57. Bridgeman S, Woo HC, Newsholme P, and Mamotte C. Butyrate lowers cellular cholesterol through HDAC inhibition and impaired SREBP - 2 signalling. Int J Mol Sci. (2022) 23:15506. doi: 10.3390/ijms232415506
58. Ferre P, Phan F, and Foufelle F. SREBP - 1c and lipogenesis in the liver: an update1. Biochem J. (2021) 478:3723–39. doi: 10.1042/BCJ20210071
59. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
60. Yang DR, Wang MY, Zhang CL, and Wang Y. Endothelial dysfunction in vascular complications of diabetes: a comprehensive review of mechanisms and implications. Front endocrinol. (2024) 15:1359255. doi: 10.3389/fendo.2024.1359255
61. Herzig S and Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
62. Trefts E and Shaw RJ. AMPK: restoring metabolic homeostasis over space and time. Mol Cell. (2021) 81:3677–90. doi: 10.1016/j.molcel.2021.08.015
63. Garcia D and Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. (2017) 66:789–800. doi: 10.1016/j.molcel.2017.05.032
64. Randriamboavonjy V, Isaak J, Frömel T, Viollet B, Fisslthaler B, Preissner KT, et al. AMPK α2 subunit is involved in platelet signaling, clot retraction, and thrombus stability. Blood. (2010) 116:2134–40. doi: 10.1182/blood-2010-04-279612
65. Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, and Wakil SJ. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci United States America. (2000) 97:1444–9. doi: 10.1073/pnas.97.4.1444
66. Lepropre S, Kautbally S, Octave M, Ginion A, Onselaer MB, Steinberg GR, et al. AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood. (2018) 132:1180–92. doi: 10.1182/blood-2018-02-831503
67. Nakamura MT, Yudell BE, and Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res. (2014) 53:124–44. doi: 10.1016/j.plipres.2013.12.001
68. Hu HJ, Wang XH, Zhang TQ, Liu Y, Chen ZR, Zhang ZZ, et al. PLK1 promotes cholesterol efflux and alleviates atherosclerosis by up-regulating ABCA1 and ABCG1 expression via the AMPK/PPARγ/LXRα pathway. Biochim Biophys Acta Mol Cell Biol lipids. (2022) 1867:159221. doi: 10.1016/j.bbalip.2022.159221
69. An T, Zhang X, Li H, Dou L, Huang X, Man Y, et al. GPR120 facilitates cholesterol efflux in macrophages through activation of AMPK signaling pathway. FEBS J. (2020) 287:5080–95. doi: 10.1111/febs.15310
70. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. (2011) 13:376–88. doi: 10.1016/j.cmet.2011.03.009
71. Dai X, Jiang C, Jiang Q, Fang L, Yu H, Guo J, et al. AMPK-dependent phosphorylation of the GATOR2 component WDR24 suppresses glucose-mediated mTORC1 activation. Nat Metab. (2023) 5:265–76. doi: 10.1038/s42255-022-00732-4
72. Nerstedt A, Johansson A, Andersson CX, Cansby E, Smith U, and Mahlapuu M. AMP-activated protein kinase inhibits IL - 6-stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3). Diabetologia. (2010) 53:2406–16. doi: 10.1007/s00125-010-1856-z
73. Ji G, Zhang Y, Yang Q, Cheng S, Hao J, Zhao X, et al. Genistein suppresses LPS-induced inflammatory response through inhibiting NF-κB following AMP kinase activation in RAW 264.7 macrophages. PloS One. (2012) 7:e53101. doi: 10.1371/journal.pone.0053101
74. Wagner N and Wagner KD. The role of PPARs in disease. Cells. (2020) 9:2367. doi: 10.3390/cells9112367
75. Chung KW, Lee EK, Lee MK, Oh GT, Yu BP, and Chung HY. Impairment of PPARα and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J Am Soc Nephrol: JASN. (2018) 29:1223–37. doi: 10.1681/asn.2017070802
76. Foster DW. The role of the carnitine system in human metabolism. Ann New York Acad Sci. (2004) 1033:1–16. doi: 10.1196/annals.1320.001
77. Ruby MA, Goldenson B, Orasanu G, Johnston TP, Plutzky J, and Krauss RM. VLDL hydrolysis by LPL activates PPAR-alpha through generation of unbound fatty acids. J Lipid Res. (2010) 51:2275–81. doi: 10.1194/jlr.M005561
78. Nissen SE, Nicholls SJ, Wolski K, Howey DC, McErlean E, Wang MD, et al. Effects of a potent and selective PPAR-alpha agonist in patients with atherogenic dyslipidemia or hypercholesterolemia: two randomized controlled trials. Jama. (2007) 297:1362–73. doi: 10.1001/jama.297.12.1362
79. Millar JS, Duffy D, Gadi R, Bloedon LT, Dunbar RL, Wolfe ML, et al. Potent and selective PPAR-alpha agonist LY518674 upregulates both ApoA-I production and catabolism in human subjects with the metabolic syndrome. Arteriosclerosis thrombosis Vasc Biol. (2009) 29:140–6. doi: 10.1161/atvbaha.108.171223
80. Dai J, Li Y, Kametani F, Cui X, Igarashi Y, Huo J, et al. Curcumin promotes AApoAII amyloidosis and peroxisome proliferation in mice by activating the PPARα signaling pathway. eLife. (2021) 10:e63538. doi: 10.7554/eLife.63538
81. Tao H, Yancey PG, Blakemore JL, Zhang Y, Ding L, Jerome WG, et al. Macrophage SR-BI modulates autophagy via VPS34 complex and PPARα transcription of Tfeb in atherosclerosis. J Clin Invest. (2021) 131:e94229. doi: 10.1172/jci94229
82. Sun H, Zhu X, Cai W, and Qiu L. Hypaphorine attenuates lipopolysaccharide-induced endothelial inflammation via regulation of TLR4 and PPAR-γ Dependent on PI3K/akt/mTOR signal pathway. Int J Mol Sci. (2017) 18:844. doi: 10.3390/ijms18040844
83. Moseti D, Regassa A, and Kim WK. Molecular regulation of adipogenesis and potential anti-adipogenic bioactive molecules. Int J Mol Sci. (2016) 17:124. doi: 10.3390/ijms17010124
84. Reddy AT, Lakshmi SP, Banno A, and Reddy RC. Role of GPx3 in PPARγ-induced protection against COPD-associated oxidative stress. Free Radical Biol Med. (2018) 126:350–7. doi: 10.1016/j.freeradbiomed.2018.08.014
85. Mukohda M, Stump M, Ketsawatsomkron P, Hu C, Quelle FW, and Sigmund CD. Endothelial PPAR-γ provides vascular protection from IL - 1β-induced oxidative stress. Am J Physiol Heart Circulatory Physiol. (2016) 310:H39–48. doi: 10.1152/ajpheart.00490.2015
86. Nicholson AC and Hajjar DP. CD36, oxidized LDL and PPAR gamma: pathological interactions in macrophages and atherosclerosis. Vasc Pharmacol. (2004) 41:139–46. doi: 10.1016/j.vph.2004.08.003
87. Toral M, Romero M, Jiménez R, Mahmoud AM, Barroso E, Gómez-Guzmán M, et al. Carnitine palmitoyltransferase-1 up-regulation by PPAR-β/δ prevents lipid-induced endothelial dysfunction. Clin Sci (London England: 1979). (2015) 129:823–37. doi: 10.1042/cs20150111
88. Jahansouz C, Xu H, Hertzel AV, Kizy S, Steen KA, Foncea R, et al. Partitioning of adipose lipid metabolism by altered expression and function of PPAR isoforms after bariatric surgery. Int J Obes (2005). (2018) 42:139–46. doi: 10.1038/ijo.2017.197
89. Tang T, Abbott MJ, Ahmadian M, Lopes AB, Wang Y, and Sul HS. Desnutrin/ATGL activates PPARδ to promote mitochondrial function for insulin secretion in islet β cells. Cell Metab. (2013) 18:883–95. doi: 10.1016/j.cmet.2013.10.012
90. Tang Y, Yang LJ, Liu H, Song YJ, Yang QQ, Liu Y, et al. Exosomal miR-27b-3p secreted by visceral adipocytes contributes to endothelial inflammation and atherogenesis. Cell Rep. (2023) 42:111948. doi: 10.1016/j.celrep.2022.111948
91. Naidenow J, Hrgovic I, Doll M, Hailemariam-Jahn T, Lang V, Kleemann J, et al. Peroxisome proliferator-activated receptor (PPAR) α and δ activators induce ICAM - 1 expression in quiescent non stimulated endothelial cells. J Inflammation (London England). (2016) 13:27. doi: 10.1186/s12950-016-0135-2
92. He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. (2021) 37:109955. doi: 10.1016/j.celrep.2021.109955
93. Kharroubi I, Lee CH, Hekerman P, Darville MI, Evans RM, Eizirik DL, et al. BCL - 6: a possible missing link for anti-inflammatory PPAR-delta signalling in pancreatic beta cells. Diabetologia. (2006) 49:2350–8. doi: 10.1007/s00125-006-0366-5
94. Planavila A, Rodríguez-Calvo R, Jové M, Michalik L, Wahli W, Laguna JC, et al. Peroxisome proliferator-activated receptor beta/delta activation inhibits hypertrophy in neonatal rat cardiomyocytes. Cardiovasc Res. (2005) 65:832–41. doi: 10.1016/j.cardiores.2004.11.011
95. Jimenez R, Toral M, Gómez-Guzmán M, Romero M, Sanchez M, Mahmoud AM, et al. The role of nrf2 signaling in PPARβ/δ-mediated vascular protection against hyperglycemia-induced oxidative stress. Oxid Med Cell longevity. (2018) 2018:5852706. doi: 10.1155/2018/5852706
96. Luz-Crawford P, Ipseiz N, Espinosa-Carrasco G, Caicedo A, Tejedor G, Toupet K, et al. PPARβ/δ directs the therapeutic potential of mesenchymal stem cells in arthritis. Ann rheumatic diseases. (2016) 75:2166–74. doi: 10.1136/annrheumdis-2015-208696
97. Soumian S, Albrecht C, Davies AH, and Gibbs RG. ABCA1 and atherosclerosis. Vasc Med (London England). (2005) 10:109–19. doi: 10.1191/1358863x05vm593ra
98. Kim J, Kim JY, Byeon HE, Kim JW, Kim HA, Suh CH, et al. Inhibition of toll-like receptors alters macrophage cholesterol efflux and foam cell formation. Int J Mol Sci. (2024) 25:6808. doi: 10.3390/ijms25126808
99. Gupta S, Pandak WM, and Hylemon PB. LXR alpha is the dominant regulator of CYP7A1 transcription. Biochem Biophys Res Commun. (2002) 293:338–43. doi: 10.1016/s0006-291x(02)00229-2
100. Su F and Koeberle A. Regulation and targeting of SREBP - 1 in hepatocellular carcinoma. Cancer metastasis Rev. (2024) 43:673–708. doi: 10.1007/s10555-023-10156-5
101. Nirschl CJ and Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. (2013) 19:4917–24. doi: 10.1158/1078-0432.Ccr-12-1972
102. Burke KP, Patterson DG, Liang D, and Sharpe AH. Immune checkpoint receptors in autoimmunity. Curr Opin Immunol. (2023) 80:102283. doi: 10.1016/j.coi.2023.102283
103. Saleh R, Toor SM, Sasidharan Nair V, and Elkord E. Role of epigenetic modifications in inhibitory immune checkpoints in cancer development and progression. Front Immunol. (2020) 11:1469. doi: 10.3389/fimmu.2020.01469
104. Wang R, Wang M, Ye J, Sun G, and Sun X. Mechanism overview and target mining of atherosclerosis: Endothelial cell injury in atherosclerosis is regulated by glycolysis (Review). Int J Mol Med. (2021) 47:65–76. doi: 10.3892/ijmm.2020.4798
105. Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ, and Han M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal transduction targeted Ther. (2022) 7:131. doi: 10.1038/s41392-022-00955-7
106. Libby P. Inflammation in atherosclerosis. Arteriosclerosis thrombosis Vasc Biol. (2012) 32:2045–51. doi: 10.1161/atvbaha.108.179705
107. Laera N, Malerba P, Vacanti G, Nardin S, Pagnesi M, and Nardin M. Impact of immunity on coronary artery disease: an updated pathogenic interplay and potential therapeutic strategies. Life (Basel Switzerland). (2023) 13:2128. doi: 10.3390/life13112128
108. Kortum RL, Rouquette-Jazdanian AK, and Samelson LE. Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol. (2013) 34:259–68. doi: 10.1016/j.it.2013.02.004
109. Johansen KH, Golec DP, Thomsen JH, Schwartzberg PL, and Okkenhaug K. PI3K in T cell adhesion and trafficking. Front Immunol. (2021) 12:708908. doi: 10.3389/fimmu.2021.708908
110. Brezar V, Tu WJ, and Seddiki N. PKC-theta in regulatory and effector T-cell functions. Front Immunol. (2015) 6:530. doi: 10.3389/fimmu.2015.00530
111. Burke KP, Chaudhri A, Freeman GJ, and Sharpe AH. The B7:CD28 family and friends: Unraveling coinhibitory interactions. Immunity. (2024) 57:223–44. doi: 10.1016/j.immuni.2024.01.013
112. Slavik JM, Hutchcroft JE, and Bierer BE. CD28/CTLA-4 and CD80/CD86 families: signaling and function. Immunologic Res. (1999) 19:1–24. doi: 10.1007/bf02786473
113. Matsumoto T, Sasaki N, Yamashita T, Emoto T, Kasahara K, Mizoguchi T, et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arteriosclerosis thrombosis Vasc Biol. (2016) 36:1141–51. doi: 10.1161/atvbaha.115.306848
114. Zhao ML, Liang C, Jiang WW, Zhang M, Guan H, Hong Z, et al. Inhibition of CTLA - 4 accelerates atherosclerosis in hyperlipidemic mice by modulating the Th1/Th2 balance via the NF-κB signaling pathway. Heliyon. (2024) 10:e37278. doi: 10.1016/j.heliyon.2024.e37278
115. Zong Y, Deng K, and Chong WP. Regulation of Treg cells by cytokine signaling and co-stimulatory molecules. Front Immunol. (2024) 15:1387975. doi: 10.3389/fimmu.2024.1387975
116. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA - 4 control over Foxp3+ regulatory T cell function. Sci (New York NY). (2008) 322:271–5. doi: 10.1126/science.1160062
117. Tekguc M, Wing JB, Osaki M, Long J, and Sakaguchi S. Treg-expressed CTLA - 4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci United States America. (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
118. Grievink HW, Smit V, Verwilligen RAF, Bernabé Kleijn MNA, Smeets D, Binder CJ, et al. Stimulation of the PD - 1 pathway decreases atherosclerotic lesion development in ldlr deficient mice. Front Cardiovasc Med. (2021) 8:740531. doi: 10.3389/fcvm.2021.740531
119. Cochain C, Chaudhari SM, Koch M, Wiendl H, Eckstein HH, and Zernecke A. Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T cells in atherosclerosis-prone mice. PloS One. (2014) 9:e93280. doi: 10.1371/journal.pone.0093280
120. Zhao L, Cheng S, Fan L, Zhang B, and Xu S. TIM - 3: An update on immunotherapy. Int immunopharmacol. (2021) 99:107933. doi: 10.1016/j.intimp.2021.107933
121. Zhu C, Anderson AC, and Kuchroo VK. TIM - 3 and its regulatory role in immune responses. Curr topics Microbiol Immunol. (2011) 350:1–15. doi: 10.1007/82_2010_84
122. Lian C, Wang Z, Qiu J, Jiang B, Lv J, He R, et al. TIM−3 inhibits PDGF−BB−induced atherogenic responses in human artery vascular smooth muscle cells. Mol Med Rep. (2020) 22:886–94. doi: 10.3892/mmr.2020.11167
123. Foks AC, Ran IA, Wasserman L, Frodermann V, Ter Borg MN, de Jager SC, et al. T-cell immunoglobulin and mucin domain 3 acts as a negative regulator of atherosclerosis. Arteriosclerosis thrombosis Vasc Biol. (2013) 33:2558–65. doi: 10.1161/atvbaha.113.301879
124. Qiu MK, Wang SC, Dai YX, Wang SQ, Ou JM, and Quan ZW. PD - 1 and tim-3 pathways regulate CD8+ T cells function in atherosclerosis. PloS One. (2015) 10:e0128523. doi: 10.1371/journal.pone.0128523
125. Cui L, Chen L, Dai Y, Ou J, Qiu M, and Wang S. Increased level of tim-3(+)PD-1(+)CD4(+)T cells with altered function might be associated with lower extremity arteriosclerosis obliterans. Front Immunol. (2022) 13:871362. doi: 10.3389/fimmu.2022.871362
126. Ni X, Wu W, Sun X, Ma J, Yu Z, He X, et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci Adv. (2022) 8:eabl5165. doi: 10.1126/sciadv.abl5165
127. Katagata M, Okayama H, Nakajima S, Saito K, Sato T, Sakuma M, et al. TIM - 3 expression and M2 polarization of macrophages in the TGFβ-activated tumor microenvironment in colorectal cancer. Cancers. (2023) 15:4943. doi: 10.3390/cancers15204943
128. Li Q, Wei S, Li Y, Wu F, Qin X, Li Z, et al. Blocking of programmed cell death-ligand 1 (PD-L1) expressed on endothelial cells promoted the recruitment of CD8(+)IFN-γ(+) T cells in atherosclerosis. Inflammation Res. (2023) 72:783–96. doi: 10.1007/s00011-023-01703-5
129. Krautter F, Hussain MT, Zhi Z, Lezama DR, Manning JE, Brown E, et al. Galectin-9: A novel promoter of atherosclerosis progression. Atherosclerosis. (2022) 363:57–68. doi: 10.1016/j.atherosclerosis.2022.11.014
130. Drobni ZD, Alvi RM, Taron J, Zafar A, Murphy SP, Rambarat PK, et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation. (2020) 142:2299–311. doi: 10.1161/circulationaha.120.049981
131. Thuny F, Naidoo J, and Neilan TG. Cardiovascular complications of immune checkpoint inhibitors for cancer. Eur Heart J. (2022) 43:4458–68. doi: 10.1093/eurheartj/ehac456
132. Inno A, Chiampan A, Lanzoni L, Verzè M, Molon G, and Gori S. Immune checkpoint inhibitors and atherosclerotic vascular events in cancer patients. Front Cardiovasc Med. (2021) 8:652186. doi: 10.3389/fcvm.2021.652186
133. Poels K, Neppelenbroek SIM, Kersten MJ, Antoni ML, Lutgens E, and Seijkens TTP. Immune checkpoint inhibitor treatment and atherosclerotic cardiovascular disease: an emerging clinical problem. J immunother Cancer. (2021) 9:e002916. doi: 10.1136/jitc-2021-002916
134. Dang Q, Li B, Jin B, Ye Z, Lou X, Wang T, et al. Cancer immunometabolism: advent, challenges, and perspective. Mol cancer. (2024) 23:72. doi: 10.1186/s12943-024-01981-5
135. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD - 1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. (2015) 6:6692. doi: 10.1038/ncomms7692
136. Bakan I and Laplante M. Connecting mTORC1 signaling to SREBP - 1 activation. Curr Opin lipidol. (2012) 23:226–34. doi: 10.1097/MOL.0b013e328352dd03
137. Soltani M, Ghanadian M, Ghezelbash B, Shokouhi A, Zamyatnin AA Jr., Bazhin AV, et al. PD-L1 stimulation can promote proliferation and survival of leukemic cells by influencing glucose and fatty acid metabolism in acute myeloid leukemia. BMC cancer. (2023) 23:447. doi: 10.1186/s12885-023-10947-7
138. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD - 1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
139. Abulizi A, Yan G, Xu Q, Muhetaer R, Wu S, Abudukelimu K, et al. Cardiovascular adverse events and immune-related adverse events associated with PD - 1/PD-L1 inhibitors for head and neck squamous cell carcinoma (HNSCC). Sci Rep. (2024) 14:25919. doi: 10.1038/s41598-024-75099-5
140. Zhang M, Yu L, Sun Y, Hao L, Bai J, Yuan X, et al. Comprehensive analysis of FASN in tumor immune infiltration and prognostic value for immunotherapy and promoter DNA methylation. Int J Mol Sci. (2022) 23:15603. doi: 10.3390/ijms232415603
141. Pokhrel RH, Acharya S, Ahn JH, Gu Y, Pandit M, Kim JO, et al. AMPK promotes antitumor immunity by downregulating PD - 1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol cancer. (2021) 20:133. doi: 10.1186/s12943-021-01420-9
142. Poels K, van Leent MMT, Reiche ME, Kusters PJH, Huveneers S, de Winther MPJ, et al. Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells. (2020) 9:1987. doi: 10.3390/cells9091987
143. Lee MJ, Yun SJ, Lee B, Jeong E, Yoon G, Kim K, et al. Association of TIM - 3 expression with glucose metabolism in Jurkat T cells. BMC Immunol. (2020) 21:48. doi: 10.1186/s12865-020-00377-6
144. Yu XH, Fu YC, Zhang DW, Yin K, and Tang CK. Foam cells in atherosclerosis. Clinica chimica acta; Int J Clin Chem. (2013) 424:245–52. doi: 10.1016/j.cca.2013.06.006
145. Zhang F, Zhao J, Sun D, and Wei N. MiR-155 inhibits transformation of macrophages into foam cells via regulating CEH expression. Biomed pharmacother = Biomed pharmacotherapie. (2018) 104:645–51. doi: 10.1016/j.biopha.2018.05.068
146. Abril-Rodriguez G and Ribas A. SnapShot: immune checkpoint inhibitors. Cancer Cell. (2017) 31:848–.e1. doi: 10.1016/j.ccell.2017.05.010
147. Screever EM, Yousif LIE, Moslehi JJ, Salem JE, Voors AA, Silljé HHW, et al. Circulating immune checkpoints predict heart failure outcomes. ESC Heart failure. (2023) 10:2330–7. doi: 10.1002/ehf2.14304
Keywords: arteriosclerosis obliterans, lipid metabolism, immune checkpoints, vascular inflammatory responses, endothelial function
Citation: Zhang J, Cui L, Meng X, Luo Y, Ou J, Wang S and Qiu M (2025) Functional interaction between immune checkpoints and lipid metabolism in the development of arteriosclerosis obliterans. Front. Immunol. 16:1665454. doi: 10.3389/fimmu.2025.1665454
Received: 14 July 2025; Accepted: 18 August 2025;
Published: 01 September 2025.
Edited by:
Yang Zhang, Brigham and Women’s Hospital and Harvard Medical School, United StatesReviewed by:
Katarzyna Napiórkowska-Baran, Nicolaus Copernicus University in Toruń, PolandNicola Laera, University of Brescia and ASST-Spedali Civili di Brescia, Italy
Copyright © 2025 Zhang, Cui, Meng, Luo, Ou, Wang and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingke Qiu, cWl1bWluZ2tlQHhpbmh1YW1lZC5jb20uY24=; Songcun Wang, c29uZ2N1bndhbmdAZnVkYW4uZWR1LmNu; Jingmin Ou, amluZ21pbm91QDE2My5jb20=
†These authors have contributed equally to this work