 Niovi Nicolaou
Niovi Nicolaou Maria S. Andreou
Maria S. Andreou Christiana M. Neophytou
Christiana M. Neophytou Panagiotis Papageorgis
Panagiotis Papageorgis- 1Tumor Microenvironment, Metastasis and Experimental Therapeutics Laboratory, Basic and Translational Cancer Research Center, Department of Life Sciences, European University Cyprus, Nicosia, Cyprus
- 2Apoptosis and Cancer Chemoresistance Laboratory, Basic and Translational Cancer Research Center, Department of Life Sciences, European University of Cyprus, Nicosia, Cyprus
Glioblastoma is considered the most common and lethal form of brain cancer. Despite tremendous progress in glioblastoma therapeutics, the profound intra- and inter-tumoral heterogeneity of glioblastoma tumors, the difficulty of agents to cross the blood-brain barrier (BBB), the development of drug resistance as well as the immunosuppressive tumor microenvironment (TME) predominantly account for the failure of existing conventional and targeted therapies. Therefore, there is a growing necessity to decipher the complexity of the TME that promotes immunosuppression and to discover innovative strategies targeting both the tumor and its TME to improve patient treatment outcomes. In this comprehensive review, we present the latest evidence implicating various components of the TME in regulating the efficacy of immunotherapies. We also discuss the current challenges and opportunities of immunotherapy in treating glioblastoma, including ongoing clinical trials using immune checkpoints inhibitors (ICIs), CAR-T cell therapy, vaccines, cytokine therapy and oncolytic viruses.
1 Introduction
Glioblastoma, formerly known as glioblastoma multiforme (GBM), is defined as a WHO Grade 4 adult-type diffuse glioma, indicating that is a fast-growing tumor (1). The term ‘glioblastoma’ is now used for the Isocitrate dehydrogenase (IDH) wildtype tumors only, while the IDH-mutant tumors have been renamed to “Astrocytoma, IDH-mutant, CNS WHO grade 4”. For simplicity, in this review, the term ‘glioblastoma’ will be used throughout for IDH wildtype tumors, unless otherwise specified.
Glioblastoma accounts for around half of all the CNS tumors, making it the most common primary malignancy of the adult brain (2). Although the causes of glioblastoma are not fully understood, evidence suggests that age, obesity, exposure to ionizing radiation and hereditary genetic conditions such as neurofibromatosis (NF), Li-Fraumeni syndrome, tuberous sclerosis (TSC), Turcot Syndrome, and Lynch syndrome, are risk factors for developing the disease (3, 4). The clinical symptoms include cognitive disorder and seizures, slowly progressive impairments of the CNS (motor weakness, sensory and memory loss, visual deficits and speech difficulties), headaches, nausea and vomiting, and changes in personality (5–7). Further to the presentation of these clinical symptoms, glioblastoma is diagnosed through radiological exams, including mostly magnetic resonance imaging (MRI), but also CT (computed tomography) scans, and positron emission tomography (PET) (5, 8–12). Finally, a diagnosis of GMB in adults is made if there is evidence of necrosis, microvascular proliferation, mutation in the telomerase reverse transcriptase (TERT) promoter, gene amplification of the epidermal growth factor receptor (EGFR), or +7/−10 chromosome copy number changes (1). Classification of glioblastoma is based on histological features, with the three main histological variants being giant cell glioblastoma, gliosarcoma and epithelioid glioblastoma, as well as several histological patterns (1).
Glioblastoma is a highly aggressive and lethal tumor, with a median overall survival of 15–18 months, with only 3% of patients having a progression-free survival (PFS) of more than 5 years (2, 13, 14). Current glioblastoma standard of care therapies include surgery, radiation therapy and temozolomide (TMZ) administration (13). However, TMZ’s efficacy is limited due to systemic toxicity and development of resistance resulting in lack of long-term efficacy and cure (15–17). Therefore, alternative therapeutic strategies are being developed to tackle resistance or improve immunotherapy for glioblastoma. Immunotherapy, especially immune checkpoint blockage (ICB) using ICIs, has revolutionized cancer therapy over the last few years, with exceptional success in several cancer types (18, 19). Unfortunately, initial trials in glioblastoma with ICIs have revealed negative results (20, 21). This therapy failure has mainly been attributed to the highly complex and immunosuppressive glioblastoma TME, which comprises glioma and glioma stem cells (GSCs), immune cells, cells of the nervous system, the brain vascular system, and extracellular matrix (ECM) components (22, 23). Novel ICIs and immunotherapeutic approaches aiming to overcome the challenges posed by the limited glioblastoma immunogenicity are currently in development or being evaluated in clinical trials. This review summarizes the current state of immunotherapy in glioblastoma and discusses the underlying mechanisms by which TME components affect its efficacy or can be exploited for the identification of alternative immunotherapeutic targets in the future.
2 Available therapies for glioblastoma
The current standard of care for newly and recurrent diagnosed patients of glioblastoma includes the removal of tumor to the greatest extent that is safe for the patient, followed by radiotherapy with concomitant chemotherapy (13). Other therapeutic options include the alkylating agents Carmustine and Lomustine, the monoclonal antibody Bevacizumab, and Tumor Treating Fields (TTFiels).
2.1 Surgery
As recently reviewed (24), several neurosurgical strategies are available, tailored for each patient according to the tumor volume and location. These include resection of the tumor (supramaximal, gross total, subtotal or near-total) and biopsy. Surgery not only helps to minimize tumor volume and improve patients’ overall survival (OS) but also allows surgeons to biopsy the tumor for classification and to design the appropriate radiotherapy regime. Maximal safe resection is recommended as the initial step in treatment since it alleviates symptoms, enhances OS, and boosts the effectiveness of adjuvant therapies.
2.2 Radiotherapy
Following surgery, radiotherapy is a cornerstone of glioblastoma treatment and has been shown to increase OS (3). Simple 2D and 3D radiotherapy techniques as well as more modern approaches, such as intensity-modulated radiotherapy (IMRT), can maximize the levels of radiation that reach the tumor site, while minimizing off-targeting to non-cancerous areas of the brain, thus reducing neurotoxicity and other adverse side effects. Following radiotherapy, patients with favorable prognostic factors or methylated O6-methylguanine-DNA methyltransferase (MGMT) promoter, typically receive adjuvant chemotherapy, usually with TMZ as discussed below. This combined approach is often referred to as the Stupp regimen and it is considered the standard of care for newly diagnosed glioblastoma (25).
2.3 Approved drugs and implant-based therapy
2.3.1 Temozolomide
TMZ, commercially known as Temodar, has received FDA approval for treating glioblastoma in 2005, based on its improvement in overall OS (26). It belongs to the new class of oral alkylating agents with an imidazole ring, with its chemical designation being 3-methyl-4-oxoimidaz[5,1-d][1,2,3,5]tetrazine-8-carboxamide (27–29). TMZ is a BBB penetrating pro-drug, which gets hydrolyzed under physiological pH to its active drug form, methyltriazen-1yl imidazole-4-carboxamide (MTIC). The drug modifies its targets by adding methyl groups in genomic DNA in guanine and adenine, in N7 and O6, and N3 sites respectively. Methylation causes DNA replication errors and disruption of the mismatch repair (MMR) repair system, which leads to DNA double-strand breaks and eventually programmed cell death. Therefore, the clinical benefit of TMZ is significantly influenced by the methylation of MGMT promoter; patients with methylation on the MGMT promoter show a better response to TMZ. While TMZ is generally well-tolerated, it is accompanied with several side effects including hematological and hepatotoxicity and others (30, 31). Furthermore, glioblastoma has several mechanisms of resistance to TMZ, as reviewed elsewhere (15–17).
2.3.2 Carmustine (BCNU)
BCNU (Bischloroethyl Nitrosourea Carmustine) belongs to the class of N-nitrosoureas [1,3-bis(2-chloroethyl)urea], a monofunctional alkylating agent. It is clinically approved in two forms for glioblastoma treatment. Firstly, intravenous (IV) BCNU was approved in 1977 for treating recurrent glioblastoma, however its use has declined due to the availability of more effective treatments will less toxicity. Secondly, Carmustine Wafers (CWs) marketed as Gliadel®, are biodegradable wafers that are implanted in the surgical site during glioblastoma resection, and they release carmustine locally. They were approved by the FDA for recurrent GBM and malignant glioma in 1996 and 2003 respectively (32). Their use is currently limited and remains a controversial topic among neurosurgeons due to the potential side effect of pulmonary fibrosis, and lack of significant evidence on its impact on the quality of life, infections after surgery and possibility of adjuvant therapy (33). In a recent meta-analysis study, Ricciardi et al. (2022), evaluated the OS and progression-free survival (PFS) in newly high-grade glioma patients that received intraoperative implantation of CWs, and concluded that CWs can significantly improve OS, but patients must be carefully selected based on their age and tumor volume to minimize side effects (34).
2.3.3 Lomustine (CCNU)
Lomustine, also known as CCNU (chloroethyl-cyclohexyl-nitrosourea), is a monofunctional alkylating agent of the nitrosourea family, that alkylates DNA and RNA, which triggers cancer cell death through DNA and RNA cross-linking (35). It is lipid-soluble and thus can successfully cross the BBB. It was FDA-approved for the treatment of brain tumors in 1976, and it is still being widely used for recurrent and progressive glioblastoma, administered orally in 6 to 8 weeks intervals (36, 37). Lomustine is given as monotherapy or in combination with procarbazine and vincristine (PCV regime), and it is considered safe with well-controlled side effects (37, 38). CCNU is increasingly considered as the standard of care option for recurrent glioblastoma, as no other treatment has demonstrated superior outcomes in controlled clinical trials (39).
2.3.4 Bevacizumab
Bevacizumab, also known as Avastin®, is a human recombinant monoclonal antibody to vascular endothelial growth factor (VEGF), a signal protein central to angiogenesis, that has been used for the treatment of several cancer types, both in monotherapy and in combination therapies (40). In 2009, bevacizumab was FDA-approved for recurrent glioblastoma following two Phase II trials, in which it was given in combination with irinotecan, based on its safety and improvement of quality of life (41, 42). In a recent scoping review (43), PFS benefits, and well-controlled side effects were supported for recurrent glioblastoma from bevacizumab, but no benefits in OS were identified. In fact, the European Medicines Agency (EMA) has rejected the use of bevacizumab for treating recurrent GBM, due to the lack of positive benefit-risk (44). It was suggested that combining bevacizumab with other therapies, like TTFields, might improve therapeutic outcome, but more studies are needed (43).
2.3.5 Optune® device
Optune® device, also known as TTFields, made by NovoCure is a portable wearable device, that was initially approved in 2011 for treating patients with recurrent glioblastoma and in 2015 for newly diagnosed glioblastoma (45). TTFields are alternating electric fields that disrupt cancer cell replication both in vitro and in vivo (46). TTFields combined with TMZ have shown significantly improved OS and PFS, however, they have not been adopted as standard care due to several factors such as high cost and inconvenience (47).
3 The glioblastoma immunosuppressive tumor microenviroment
The glioblastoma TME is characterized by significant heterogeneity and complexity, encompassing glioma and GSCs, immune and nervous system cells, ECM components and the brain vascular system. Additionally, TME is highly dynamic, characterized by extensive cell-to-cell communication and regulated by factors such as pH and oxygen levels. Glioblastoma lacks infiltration of immune cells, favoring the development of tumorigenic properties, with the immunosuppressive TME playing a crucial role in cancer cell survival and response to therapy (Figure 1).
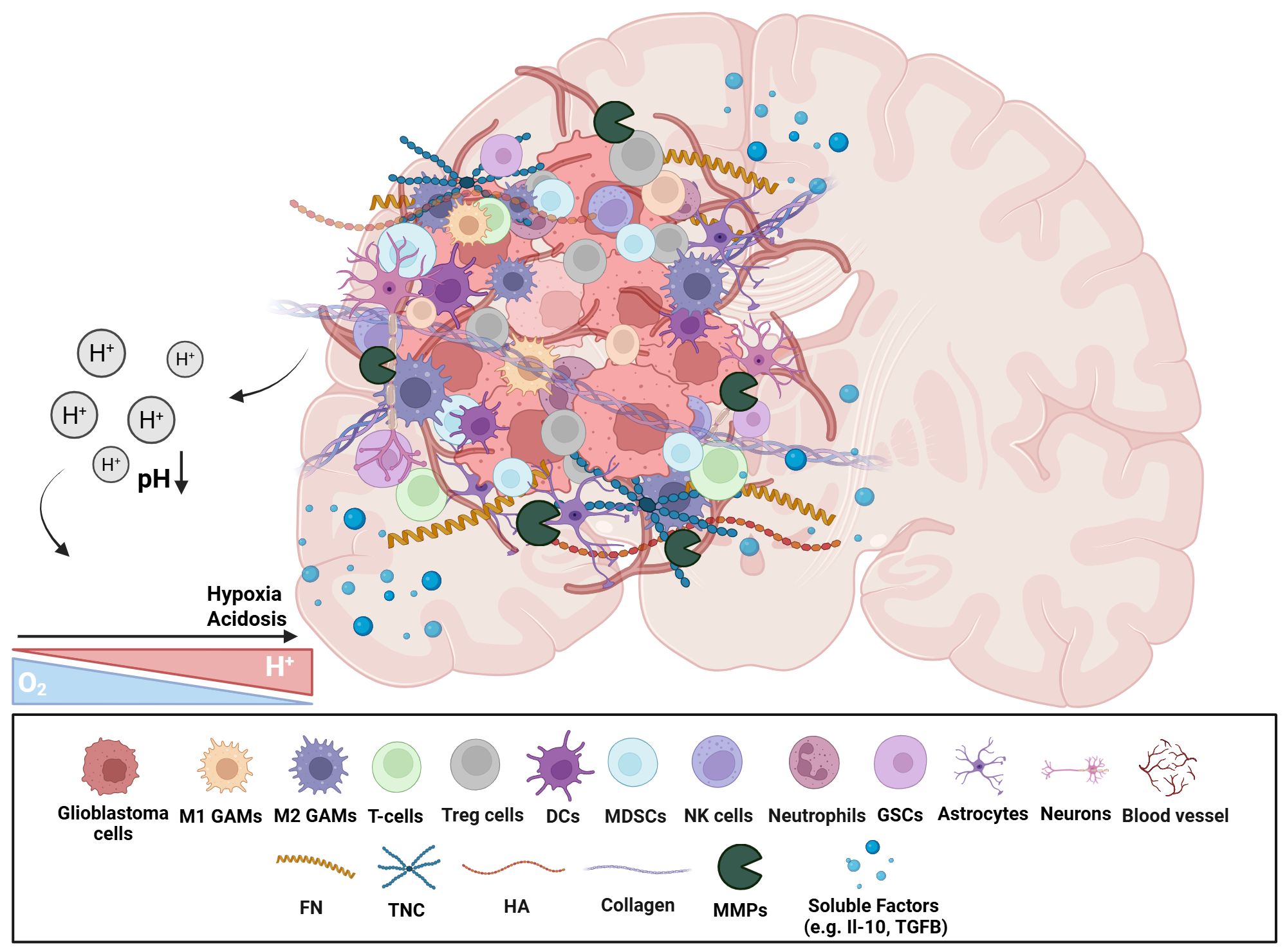
Figure 1. Schematic representation of the immunosuppressive glioblastoma TME. The heterogeneous cellular and non-cellular players of glioblastoma TME are represented. There is an enrichment of immunosuppressive cellular subsets (e.g. Tregs, MDSCs, and GAMs), neural cells, as well as GSCs. Non-cellular aspects include ECM proteins, and soluble factors (e.g. cytokines, growth factors). The hypoxic and acidic environment affects various TME components in numerous ways. This highly complex microenvironment contributes to a strong tumour heterogeneity and immunosuppressive environment, facilitating tumour progression, resistance to therapies, and immune evasion mechanisms. (GAMs, Glioblastoma-associated microglia/macrophages; Tregs, Regulatory T cells; DCs, Dendritic cells; MDSCs, Myeloid-derived suppressor cells; GSCs, Glioblastoma stem cells; FN, Fibronectin; TNC, Tenascin-C; HA, Hyaluronic acid; MMPs, Matrix metalloproteases; ECM, Extracellular matrix). Created in BioRender. Papageorgis, P (2025). https://BioRender.com/qc1ro4l.
3.1 Non-cellular components
3.1.1 ECM
In general, the ECM compromises approximately 20% of the brain mass, and under physiological conditions, it provides structural and biochemical support, as well as regulation of various cellular processes (48). The major components of the ECM, including hyaluronic acid (HA), tenascin-C (TNC), fibronectin (FN), laminin, and collagen, among others, play a vital role in the modulation of invasiveness. In glioblastoma, remodeling, and degrading of the ECM is observed since Matrix metalloproteinases (MMPs), more specifically MMP-2 and MMP-9, are released into the extracellular space. Tenascin-C (TNC), another ECM component, is a matricellular protein (MCP) that is normally expressed at low levels; however, during development and pathological conditions it is highly expressed (49, 50). Glioblastoma cells produce and release TNC and high TNC expression is associated with poor patient survival and disease progression. TNC can interact with multiple proteins (e.g. fibronectin, Toll-like receptors etc.) and thus can promote neovascularization, proliferation, invasiveness, and immunomodulation. Additionally, the presence of TNC stimulates GSCs invasiveness by MMP12 and ADAM metallopeptidase domain 9 (ADAM9) expression and activity, via the c-Jun NH2-terminal kinase pathway. Another ECM component is HA, which activates CD44, a cell surface adhesion protein, stimulating the synthesis and secretion of additional HA, leading to upregulation of MT1-MMP, thus promoting glioblastoma cell infiltration (48). Moreover, FN glycoprotein, also highly expressed in glioblastoma, promotes cell adhesion, differentiation of GSCs, and invasion, and plays a role as a coordinator between ECM and glioblastoma cells (51). Activation of the adhesion kinase/paxillin/Akt signaling pathway is responsible for GSCs adherence and differentiation, while the increase of MMPs’ activity and activation of axis Stat3-ODZ1-RhoA/ROCK, could be responsible for the invasive behavior observed in glioblastoma. Laminin glycoprotein, more specifically laminin-2, -5, and -8, are also found to be highly expressed in glioblastoma patients and they are suspected to have a role in glioblastoma spreading and infiltration (48). Lastly, collagen type I is normally present at low levels in brain tissue, but its expression is slightly elevated in glioblastoma tumors. It is enhanced in the perivascular niche of GSCs, promoting therefore invasiveness via integrin and phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways.
3.1.2 Physicochemical properties
Physiological factors, such as pH and oxygen concentrations, can affect tumor progression, and immunosuppression (48). The intra-tumoral heterogeneity displayed in glioblastoma alters the nutrient supply and availability of oxygen within the tumor, influencing metabolic properties and energy utilization of cancer cells.
3.1.2.1 Hypoxia
Hypoxic conditions are fundamental drivers of oncogenesis. During hypoxic conditions, glioblastoma cells can adapt and persist due to the high expression of hypoxia-inducible factors (HIFs), a family of transcription factors that are stabilized under low oxygen conditions and are regulated by the inhibition of prolyl-4-hydroxylase 2 (PHD2). This accumulation further activates the expression of downstream targets, including proangiogenic and anti-apoptotic genes. The angiogenesis-induced hypoxia in glioblastoma leads to specific features such as necrotic cores and microvascular hyperplasia, that drive tumor growth and invasion. Additionally, under hypoxia, MMP-2 and MMP-9 expression is increased, and the epithelial-to-mesenchymal transition (EMT) process is induced mainly by transcription factors that have hypoxia response elements (HREs) at their gene promoter regions, such as Twist, Snail, and ZEB. Finally, hypoxia can amplify the activities of immunosuppressive cells, including the influx of M2 macrophages and Tregs at the tumor site (52, 53).
3.1.2.2 Acidosis
The TME as well as intrinsic cellular processes of glioblastoma are affected by acidosis (low pH), which facilitates pro-tumorigenic processes including survival, proliferation, migration, and angiogenesis (52, 53). Tumor acidosis further increases the expression of HIF-1α and HIF-2α and alters the interactions among glioblastoma cells and various TME components, significantly impacting the invasion process (53). It reduces the infiltration and activity of effector lymphocytes and NK cells, further promoting glioblastoma persistence. In addition, drug uptake and efficacy are also affected since acidosis neutralizes radiation-induced reactive oxygen species (ROS) formation, inhibiting apoptosis (52, 53). Thereby, acidosis plays a significant role in fostering a highly immunosuppressive TME.
3.2 Cellular components
3.2.1 Immune components
Immune cells found in glioblastoma TME constitute up to 50%; glioblastoma-associated macrophages and microglia (GAMs) are the most prevalent, followed by neutrophils, regulatory T cells (Tregs), dendritic cells (DCs), and myeloid-derived suppressor cells (MDSCs) (54). The immune components present in glioblastoma contribute to the observed immunosuppressive characteristics of the glioblastoma TME.
3.2.1.1 GAMs
GAMs in glioblastoma are composed of macrophages and microglia, which are resident macrophages located in the CNS, both functioning as phagocytic cells. They play a significant and complex role by interacting with glioblastoma cells in various ways that can influence tumor progression (55). While they have the potential to attack glioblastoma cells, they also support tumor growth and invasion. Based on their marker expression and cytokine expression profile, GAMs can be polarized into M1 (anti-tumor) or M2 (pro-tumor) phenotypes. As glioblastoma progresses, however, GAMs tend to be pro-tumorigenic thus favoring an M2 phenotype. The presence of GAMs has been considered to have prognostic value, since higher levels of GAMs are positively correlated with poor prognosis and worsen OS (52). They secrete factors that support tumor growth (e.g. insulin-like growth factor (IGF-1), epidermal growth factor (EGF), platelet-derived growth factor (PDG-F)), as well as anti-inflammatory cytokines (e.g. interleukin-6 (IL-6), IL-10, transforming growth factor beta (TGF-β)), promoting the malignant phenotype of glioblastoma and contributing to the permeability of the BBB (52). More specifically, the proliferation of glioblastoma cells is associated with elevated Ca2+ levels in the tumor, which further stimulate ATP-mediated tumor cells that directly interact with GAMs, leading to their activation. In addition, the secretion of IL-6 by GAMs activates the JAK-STAT3 pathway in endothelial cells (ECs) and downregulates intercellular connexins levels, such as connexin43 (Cx43). This contributes to the disruption of the BBB and its increased permeability (56).
3.2.1.2 Neutrophils
In glioblastoma, neutrophils (CD66b+ and CD16+), a subset of myeloid-derived suppressor cells, upregulate the S100A4 protein, which suppresses the mesenchymal phenotype and facilitates acquired resistance to anti-VEGF therapy (57). They play a role in the oncogenic process of tumor initiation, proliferation, and dissemination via a pro-tumorigenic positive feedback loop. Neutrophils can induce angiogenesis and hinder the functions of DCs, macrophages, and NK cells, thus suppressing the immune system and facilitating in the migration of tumor cells. Neutrophil Extracellular Traps (NETs) secreted by activated neutrophils are extracellular fibrous networks consisting of DNA and proteins (58). Their role in GMB progression is mostly beneficial: they may enhance tumor growth by activating EGFR or TLR4 signaling in tumor cells, they support immune evasion by forming physical barriers for cytotoxic T cells or NK cells and contribute to treatment resistance by activating survival pathways (59).
3.2.1.3 T-cells (CD4+, CD8+, Tregs)
T lymphocytes, a central component of the adaptive immune system, are essential for anti-tumor immunity. In glioblastoma, however, the anti-tumor response is compromised since CD4+/CD8+ T-cells constitute only 2% of infiltrating immune cells. Most of these cells show upregulation of inhibitory receptors/immune checkpoints, which signal anergy, exhaustion, and tolerance, thereby promoting further the immunosuppressive nature of glioblastoma (59). In contrast, regulatory T-cells (Tregs) are found to be enriched in glioblastoma infiltrating immune cells. This enrichment promotes the systemic reduction of CD4+ T-cells, and the inhibition of the cytotoxic responses of CD8+ T-cells, further inducing the effector T-cell anergy and tolerance (60–63). Tregs are suppressor cells, mainly characterized by elevated expression of several transcription factors (e.g. Foxp3, CD25), and cytotoxic T lymphocyte antigen 4 (CTLA-4). Tregs can further inhibit T cell activity by binding to CD80/CD86 on antigen presenting cells (APCs) via CTLA-4. They release immunosuppressive cytokines such as IL-10, IL-4, IL-13, IDO and TGF-β, reducing TNFα and INF-γ levels in effector CD4+ T cells, inhibiting APCs function, and downregulating tumor-specific cytotoxicity within the immune response.
3.2.1.4 NK cells
NK cells (CD56+CD3- cells) mediate antigen-independent immune surveillance as effector lymphoid cells (64). Despite their potential for anti-tumor activity, NK cells are found in low levels or impaired within glioblastoma tumors and thus their cytotoxic effects are suppressed by factors such as TGF-β and IL-10 within the TME.
3.2.1.5 DCs
As a diverse class of professional APCs, DCs are central to the activation and regulation of innate and adaptive immunity (61). In normal conditions, DCs are absent from the brain parenchyma, however in pathological conditions such as glioblastoma, DCs can infiltrate brain tissue via afferent lymphatic vessels or endothelial venules. Although DCs have a pivotal role in antitumor immunity, in the TME of glioblastoma, the overexpression of nuclear factor erythroid-related factor (Nef) in DCs results in their suppression and, consequently, a decrease in the effector T cell activation (61).
3.2.1.6 MDSCs
MDSCs are a heterogeneous population of immature myeloid cells that activate immunosuppressive cells and inhibit the release of inflammatory factors thus mediating anti-tumor immunity. In glioblastoma patients, different MDSC populations are present, with the major population being polymorphonuclear CD15+CD33+HLADR- (PMN-MDSCs) accounting for 82%, followed by lineage-negative (E-MDSCs) at 15% and monocytic (CD14+CD33+HLADR-; M-MDSCs) at 3% (61, 65). Signal transducer and activator of transcription 3 (STAT3) is a hallmark of MDSCs, and its upregulation regulates MDSCs expansion and tolerogenicity. Factors including IL-10, IL-6, VEGF, GM-CSF, PGE-2, and TGF-β2, found upregulated in glioblastoma, also influence MDSCs expansion. MDSCs are key players in glioblastoma immunosuppressive TME, exerting their effects via various mechanisms including amino acid depletion, oxidative stress, decreased DCs maturation, and the indirect induction of Tregs induced by IL-10 and TGF-β (59). PMN-MDSCs can suppress the antigen-presenting capacity of DCs by upregulating myeloperoxidase (MPO) expression, therefore limiting the ability of DCs to cross-present tumor-associated antigens (TAAs). Additional immunosuppressive activities of MDSCs are regulated by CCAAT/enhancer binding protein β (C/EBPβ) which controls the expression of arginase (ARG1) and inducible nitric oxide synthase (iNOS). These expressions can inhibit T cell growth and migration by interfering with the expression of CD3ζ chain and by inducing the nitration of CCL2 chemokine. iNOS further produces nitric oxide (NO) from amino acid L-arginine, which inhibits the IL-2 signaling pathway in an IFN-γ depended-manner, ultimately impairing T-cell proliferation (59). Moreover, MDSCs suppress NK cell cytotoxicity and cytokine release via ROS production. Crosstalk between GAMs and MDSCs also skews GAMs toward a pro-tumor M2 phenotype.
3.2.2 GSCs
GSCs possess the ability to self-renew and differentiate, contributing to intra-tumoral heterogeneity and playing a key role in tumorigenesis and tumor propagation (48, 54, 66). GSCs activate, regulate, and recruit pro-tumor immune cells. They inhibit T-cell proliferation and cytotoxic T-cell activation while suppressing macrophage-mediated tumor-killing by producing cytokines such as IL-10 and TGFβ. Additionally, GSCs express the TGFβ receptor (TGF-βRII) on their surface, and binding of its ligand triggers the secretion of MMP9 by GSCs. GSCs also interact closely with ECs, creating a perivascular niche, and impacting the glioblastoma progression. Subsequently, GSCs express high levels of proangiogenic growth factors such as VEGF, angiopoietin-1 (Ang-1), bradykinin (BK), IL-8, and stromal cell-derived factor-1 I (SDF-1), which induce their differentiation into ECs and pericytes, further enhancing angiogenesis, migratory abilities, and invasiveness.
3.2.3 Neural components
3.2.3.1 Astrocytes
Astrocytes make up about half of the total volume of the human brain and play a key role in brain physiology and disease, as they are integral components of the BBB. Glioblastoma invasiveness is modulated by astrocytes (48). Glioblastoma cells release EVs into the TME, which are internalized by neighboring astrocytes. Astrocytes then become activated and start secreting elevated amounts of chemokines (e.g. IL-6), enhancing glioblastoma cell invasion and tissue infiltration by increased production MMPs, especially MMP-2 and MMP-9. In addition, glioblastoma invasiveness and migration are modulated by the release of factors, such as glial cell line-derived neurotrophic factor (GNDF) and connective tissue growth factor (CTGF) from astrocytes. GNDF promotes glioblastoma invasion by triggering the activation of rearranged during transfection/GNDF family receptor alpha-1 (RET/GFRa1) receptors and pro-tumoral signaling pathways, such as mitogen-activated protein kinases (MAPK) and PIK3/Akt. Whereas CTGF, when bound to integrin β1, it activates the nuclear factor kappa-light-chain-enhancer of the activated B cells (NF-kB) signaling pathway which secretes additional growth factors, such as TGF-β, further facilitating glioblastoma invasiveness. Moreover, glioblastoma-associated astrocytes upregulate the gap junction protein connexin 43 (Cx43), which facilitates direct communication between astrocytes and glioblastoma cells, promoting further tumor invasion and migration.
3.2.3.2 Neurons
Neurons usually interact indirectly with glioblastoma cells in the TME with different mechanisms, including paracrine stimulation, synaptic transmission, and secretion of neurotransmitters. Glioblastoma proliferation, invasion, and resistance to apoptosis are promoted by the binding of TrkB receptors on glioblastoma cells to molecules released from neurons such as brain-derived neurotrophic factor (BDNF) and neuroligin-3 (NLGN3). Additionally, glioblastoma cells form functional synaptic connections with neurons, enabling electrochemical signaling that influences tumor progression. One example is the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) glutamate receptor, which mediates excitatory postsynaptic potentials, enhancing intracellular calcium signaling, further promoting glioblastoma proliferation, survival, and invasion (67).
3.2.3.3 Oligodendrocytes
Oligodendrocytes are responsible for myelinating axons in the CNS, therefore playing a role in brain physiology, regulating neuronal activities, neural plasticity, and metabolic support (52). In glioblastoma, oligodendrocytes are disrupted leading to worsened tumor-induced damage to neural circuits. They are located at the tumor border niches, suggesting their potential influence in both invasion and recurrence. Oligodendrocytes release cytokines such as angiopoietin-2, which enhances glioblastoma cell motility, and are implicated in promoting angiogenesis contributing to the tumor’s vascular support and sustenance (53).
4 Current state of glioblastoma immunotherapy
Cancer immunotherapy works by “re-educating” the patient’s immune system to eliminate tumors and therefore holds great promise for cancer therapy (68). The most widely used strategies, often combined, are ICIs, chimeric antigen receptor (CAR) T cells, vaccines and oncolytic viruses. Other immunotherapeutic strategies include other T-cell based therapeutic approaches, cytokine therapy and other targeted immunomodulatory therapies, including monoclonal antibodies. Currently, 104 interventional clinical trials of immunotherapy in glioblastoma have been identified as active (‘recruiting’, “active not recruiting” and “enrolment with invitation”), in ClinicalTrials.gov, accessed on 01/06/2025 (Tables 1–3, Figures 2, 3).
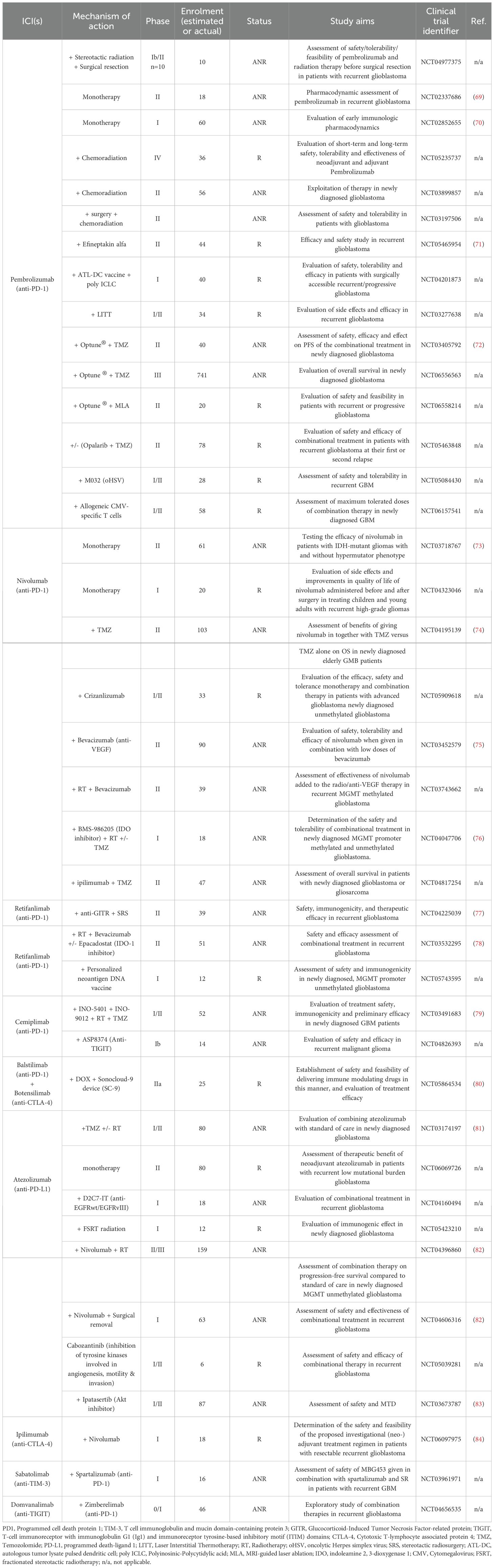
Table 1. ICIs in ongoing interventional clinical trials against glioblastoma - R, “Recruiting”; ANR, “Active, not recruiting”; and EBI, “Enrolling by invitation”.
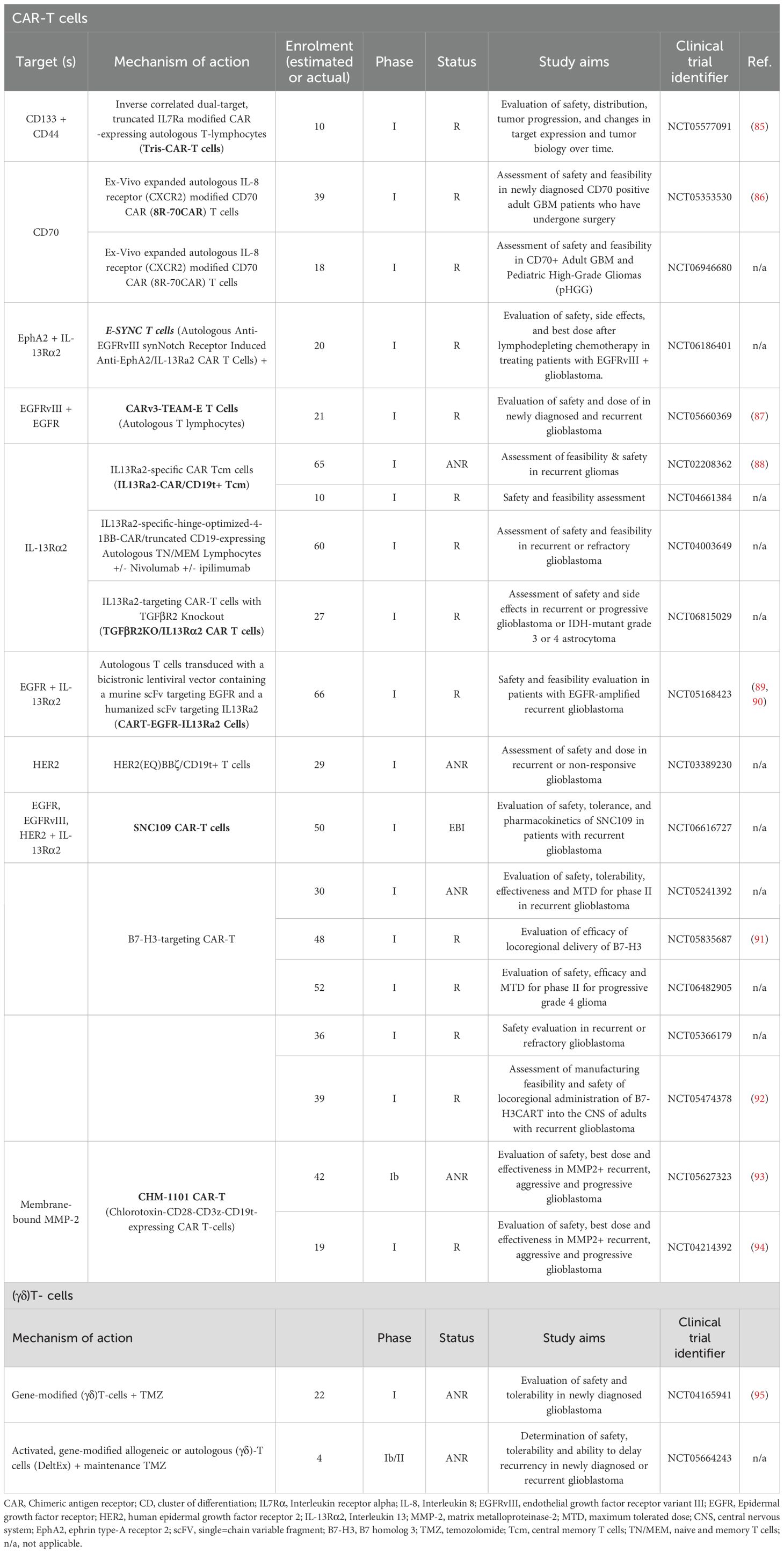
Table 2. T-cell based therapies in ongoing interventional clinical trials against glioblastoma – R, “Recruiting”; ANR, “Active, not recruiting”; and EBI, “Enrolling by invitation”.
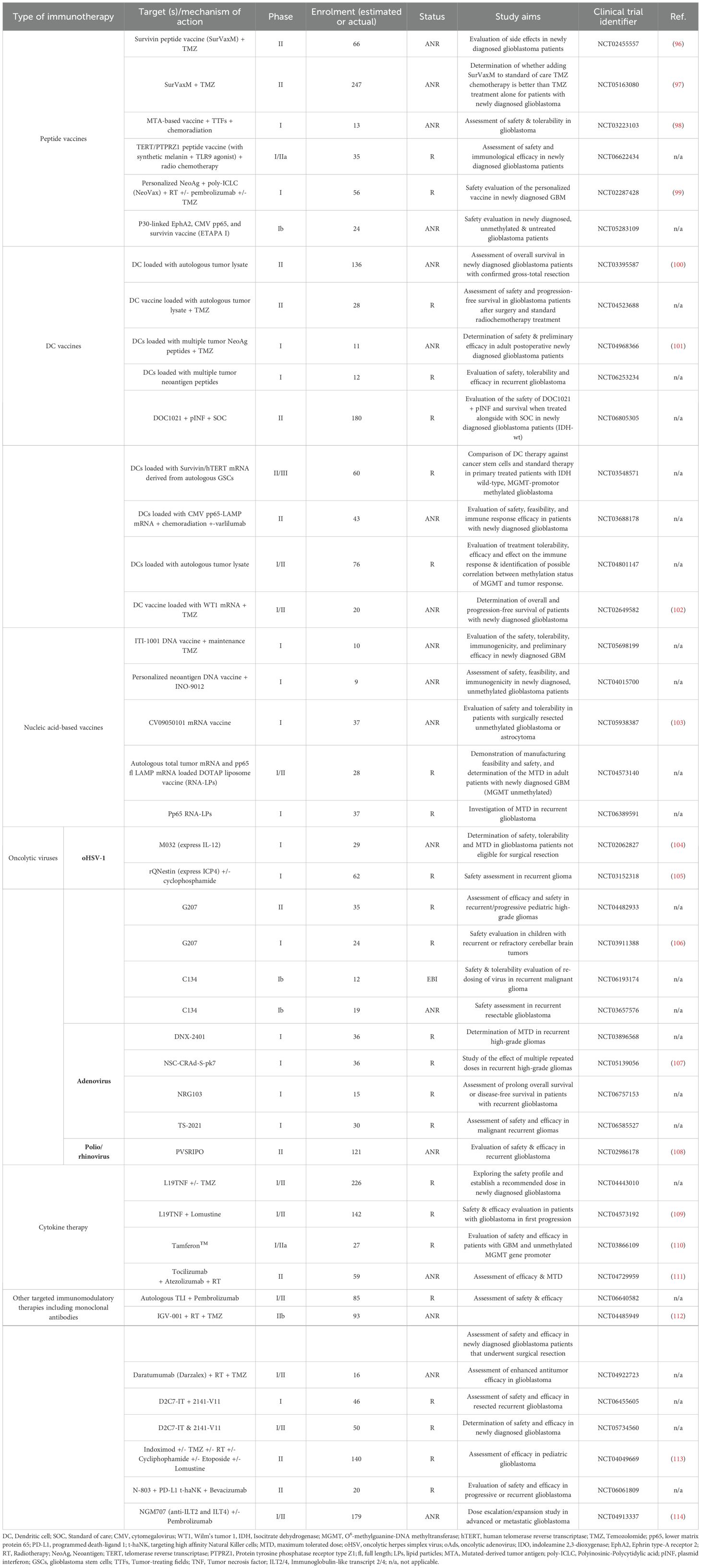
Table 3. Vaccines, oncolytic viruses, cytokine therapy and other targeted immunotherapies in ongoing interventional clinical trials against glioblastoma - R, “Recruiting”; ANR, “Active, not recruiting”; and EBI, “Enrolling by invitation”.
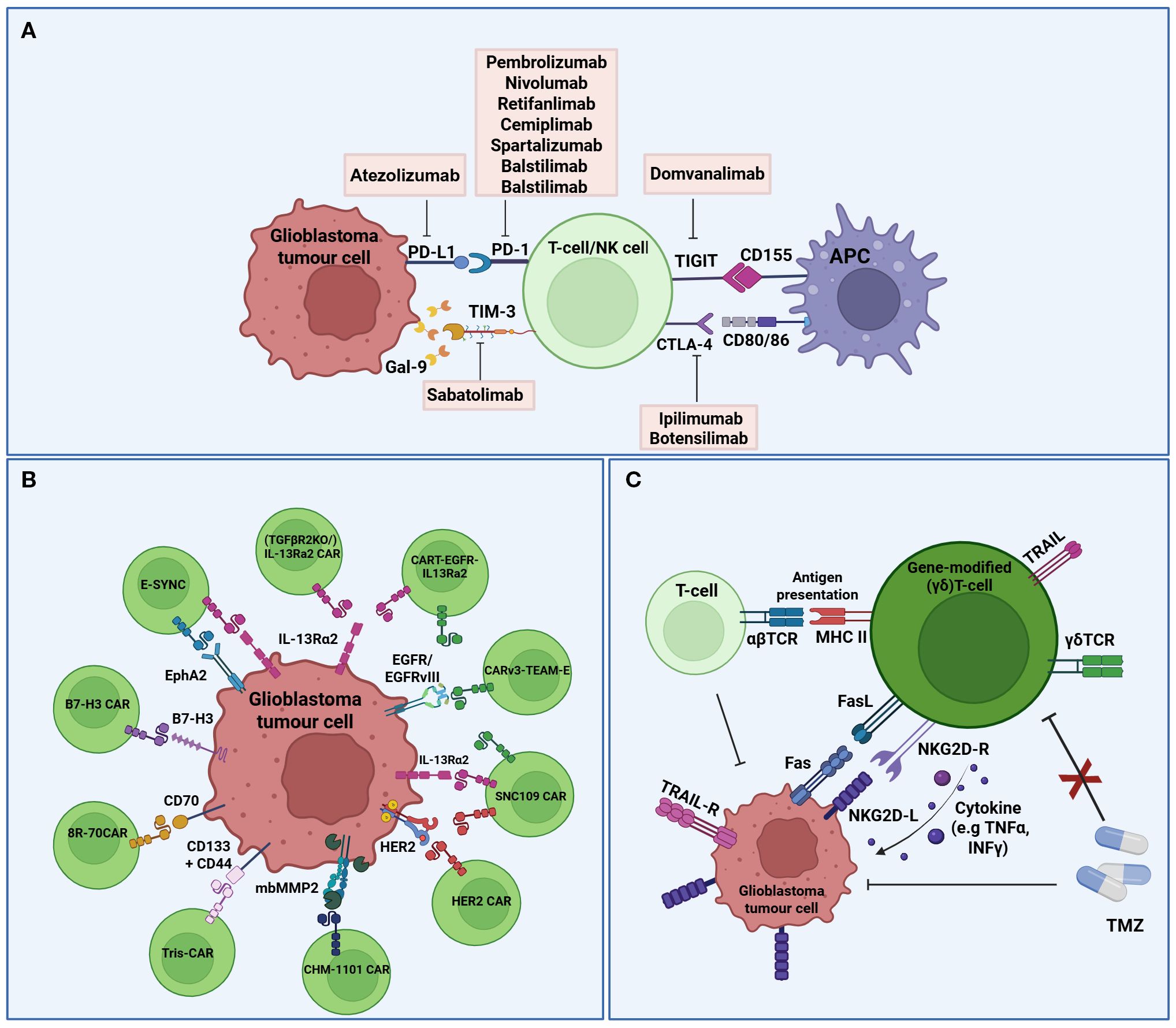
Figure 2. Schematic representation of immune checkpoint inhibitors, CAR-T cells, and (γδ)T-cells in ongoing interventional clinical trials against glioblastoma. (A) Immune Checkpoint Inhibitors; Several ICIs and their targets are being investigated in clinical trials to enhance the anti-tumor response. Exhaustion markers (e.g. PD-1, CTLA-4, TIGIT), which are upregulated on the surface of T cells, interact with immune checkpoint molecules (e.g. PD-L1, CD80/86, CD155) expressed on glioblastoma cells and APCs. (B) CAR-T cells; Several targetable tumour-associated antigens for glioblastoma CAR T-cell therapy. CAR T-cells are engineered to recognize tumor-associated antigens (e.g. IL13Rα2, HER2, B7-H3, etc) via corresponding CAR constructs, enabling selective tumor cell recognition and cytotoxicity. (C) γδT-cells; Gene-modified γδT-cells exhibit direct cytotoxicity against glioblastoma cells and enhance the anti-tumor activity of other immune cells through FasL/TRAIL-mediated apoptosis, antigen presentation, and cytokine secretion. Several antigens and receptors are represented in the same cell for the graphic. (PD1, Programmed cell death protein 1; PD-L1, programmed death-ligand 1; TIM-3, T cell immunoglobulin and mucin domain-containing protein 3; TIGIT, T-cell immunoreceptor with immunoglobulin G1 (Ig1) and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains; CTLA-4, Cytotoxic T-lymphocyte associated protein 4; Gal-9, Galectin-9; MCH, Major histocompatibility complex; APC, Antigen presenting cell; CAR, Chimeric antigen receptor; IL13Rα2, interleukin 13 receptor subunit alpha 2; HER2, human epidermal growth factor receptor 2; mbMMP2, membrane-bound metalloproteinases 2; B7-H3, B7 homolog 3; EphA2, Ephrin type-A receptor 2; EGFR, Epidermal growth factor receptor; TCR, T cell receptor; TMZ, Temozolomide; NKG2D-R, Natural killer group 2 member D receptor; NKG2D-L, NKG2D ligand; TNFα, Tumour necrosis factor 2α; INFγ, Interferon γ; TRAIL, Tumour necrosis factor-related apoptosis-inducing ligand; TRAIL-R, TRAIL receptors). Created in BioRender. Papageorgis, P (2025). https://BioRender.com/mz964iy.
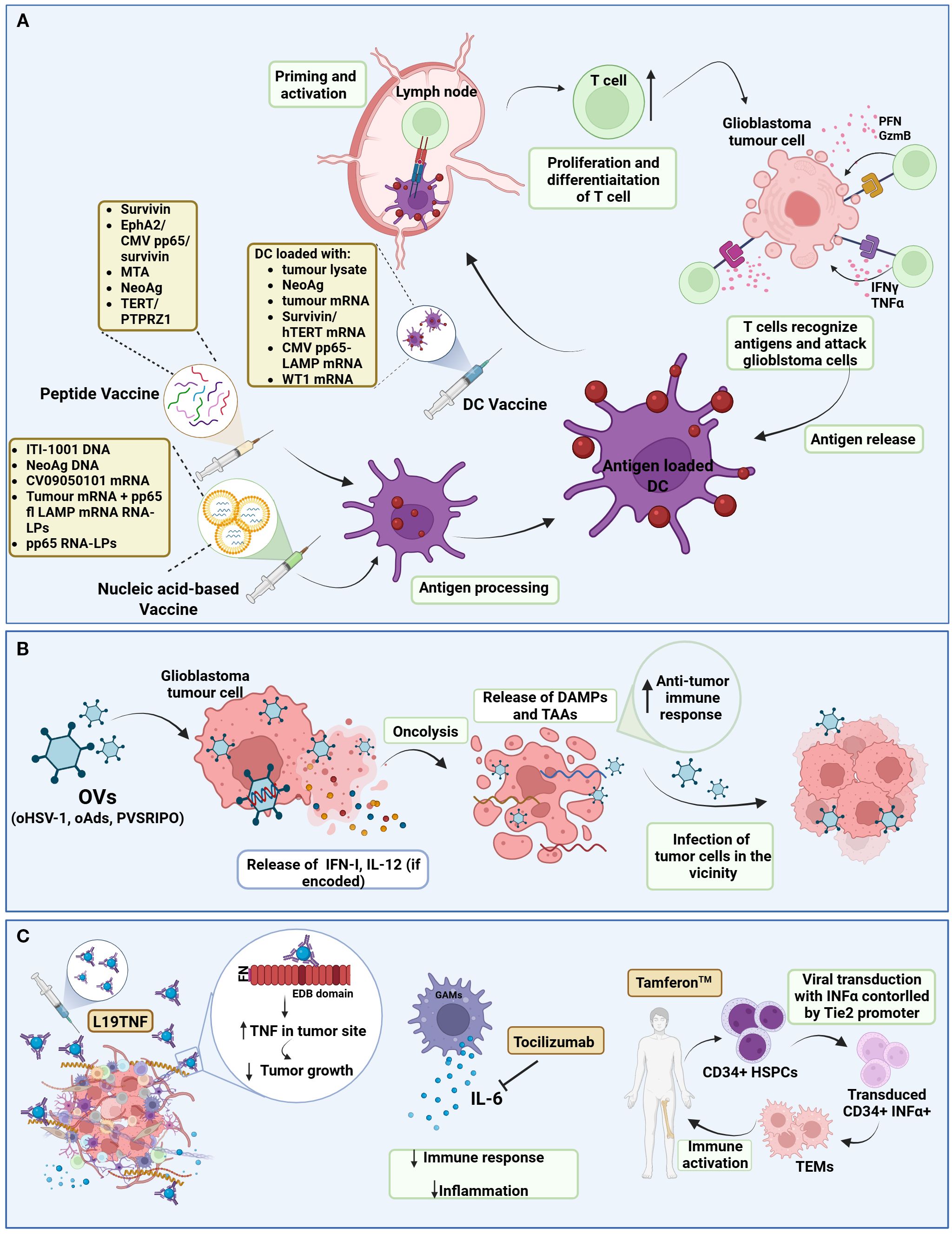
Figure 3. Schematic representation of cancer vaccines, oncolytic viruses, and cytokine therapies in ongoing interventional clinical trials against glioblastoma. (A) Cancer Vaccines; Tumour-immune cycle induced by cancer vaccines. After the administration of cancer vaccines (peptide, nucleic acid-based, DC vaccines), the DCs uptake and process the antigens and then present them to MHC II or MHC I via cross-presentation. Antigen-loaded DCs migrate into lymph nodes to prime and activate T-cells. Activated T-cells, proliferate and differentiate into recognizing tumour antigens and targeting glioblastoma cells. Immunogenic dead glioblastoma cells can release additional antigens, leading to the initiation of a subsequent cycle. (B) Oncolytic viruses; Genetically engineered OVs selectively replicate within glioblastoma cells while sparing normal cells. This process leads to oncolysis, which not only releases virus progeny to infect neighbouring tumour cells but also exposes DAMPs and TAAs, triggering a robust anti-tumour immune response. (C) Cytokine therapies; Cytokine therapies modulate the glioblastoma TME. For example, L19TNF delivers TNF to the TME, reducing tumour growth. Tocilizumab binds to IL-6, decreasing the body’s immune response and inflammation. Tamferon-mediated delivery of INFα via genetically modified CD34+ HSPCs promotes systemic and tumour-localized immune activation. (DC, Dendritic cell; EphA2, Ephrin type-A receptor 2; CMV, Cytomegalovirus; MTA, Mutated-derived tumour antigen; NeoAg, Neoantigen; fl, full length; LPs, lipid particles; TERT, telomerase reverse transcriptase; PTPRZ1, Protein tyrosine phosphatase receptor type Z1; WT1, Wilms; tumour 1; PFN, Perforin; GzmB, Granzyme B; OV, Oncolytic virus; oHSV-1, oncolytic herpes simplex virus-1; oAds, oncolytic adenovirus; INF-I, Interferon Type I; IL-12, Interleukin-12; DAMPs, Damage-associated molecular patterns; TAAs, Tumor-associated antigens; TNF, Tumour-necrosis factor; FN, Fibronectin; EDB, Extra-domain B; GAM, Glioblastoma associated macrophage; IL-6, interleukin 6; TEM, Tie2-expressing macrophages; HSPC, Hematopoietic stem and progenitor cells). Created in BioRender. Papageorgis, P. (2025) https://BioRender.com/n35gcht.
4.1 Immune checkpoint inhibitors
Immune checkpoints are the gatekeepers of the immune system, crucial for preventing autoimmunity under normal physiological conditions and protecting tissues from damage following response of the immune system to pathogens (115). In cancer, the expression of immune-checkpoint proteins is dysregulated and therefore confers immune resistance. Several inhibitory immunoreceptors, referred to as “immune checkpoints”, have been identified and studied in cancer, including, amongst others, programmed death ligand 1 (PD-1), cytotoxic T-lymphocyte associated protein 4 (CTLA-4), lymphocyte activation gene 3 protein (LAG3), T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), and T-cell immunoreceptor with immunoglobulin G1 (Ig1) and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (TIGIT) (116). Blocking their activity with ICIs has revolutionized cancer therapy over the past few years and has been successful for several cancer types (18, 19). Although initial trials with ICIs in glioblastoma revealed disappointing results (20), glioblastoma tumors are still considered a promising candidate for immunotherapy and is therefore being investigated in several clinical trials (Tables 1-3, Figures 2, 3).
4.1.1 Anti-PD-1/PD-L1
PD-1 receptor, expressed on T- and other immune cells, is a dominant negative regulator of T-cell response, when activated by its ligand, PD-L1, which is expressed on tumor cells (117). Anti-PD-1/PDL-1 therapy has been approved for the treatment of several cancer types, including metastatic melanoma, non-small cell lung cancer (NSCLC), head and neck cancers, urothelial carcinoma and others (118). More specifically, there are 6 FDA-approved PD-1/PD-L1 inhibitors, including the PD-1 inhibitors Pembrolizumab (Keytruda), Nivolumab (Opdivo) and Cemiplimab (Libtayo) and the PD-L1 inhibitor Atezolizumab (Tecentriq). PD-1/PDL-1 therapy has been explored in several clinical studies in glioblastoma, and although it is safe, it did not prolong OS (18). Currently, there are no FDA-approved anti-PD-1/PD-L1 inhibitors for glioblastoma (119), but the PD-1 inhibitors Pembrolizumab, Nivolumab, Cemiplimab, Retifanlimab by Zynyz (approved under FDA’s Accelerated Approval Program for Merkel cell carcinoma), and Balstilimab (currently in Phase II clinical trials for several cancers), as well as the PD-L1 inhibitor, Atezolizumab, are being tested in clinical trials for safety, tolerability, feasibility and efficacy in newly diagnosed, recurrent and progressive glioblastoma, both as monotherapy and as part of a therapeutic regime (Table 1).
4.1.2 Anti-CTLA-4
CTLA-4 belongs to the superfamily of CD28–B7 immunoglobulins and it shares its two ligands (B7.1, B7.2) with its co-stimulatory counterpart CD28, and together these molecules are functioning at the tip of the immunological cascade (120). In glioblastoma, CTLA-4 competes with CD28 for binding to costimulatory molecules (CD80 and CD86) on APCs, thereby inhibiting the activation of T cells. Anti-CTLA-4 as a single form of therapy or in combination with other ICIs, enhances endogenous immune responses to immunogenic tumors. Ipilimumab (MDX-010 and Yervoy®) is a humanized monoclonal CTLA-4 antibody that has been approved by the FDA as monotherapy in anti-PD-1 refractory cases or in combination with nivolumab as a first-line treatment of advanced melanoma (121). In glioblastoma clinical trials, ipilimumab is administered in combination with nivolumab (NCT06097975, Table 1). Botensilimab (AGEN1181), is an Fc-enhanced anti-CTLA-4 antibody, which is considered one of the most advanced-next generation ICIs currently in clinical trials, due to its novel FcyR-dependent mechanism to promote superior priming and activation of T cells (121). In glioblastoma, Botensilimab is being tested in a single clinical trial in combination with Balstilimab and chemotherapy, given through a BBB sonication device (NCT05864534, Table 1).
4.1.3 Anti-TIM3
TIM-3 is an immuno-myeloid cell surface marker specific to IFN-γ producing CD4+ and CD8+ T cells, expressed on multiple immune cells and leukemic stem cells (122). In cancer, elevated TIM-3 expression is associated with poor outcome, therefore TIM-3 has become an attractive candidate for immunotherapy. Sabatolimab (MBG453), is a novel high-affinity, humanized, IgG4 antibody targeting the TIM-3 receptor currently under clinical development by Novartis for the treatment of both solid tumors and hematological malignancies (123). Sabatolimab, is tested for safety in a Phase I clinical trial of recurrent glioblastoma, in which is given in combination with spartalizumab (anti-PD-1) (NCT03961971, Table 1).
4.1.4 Anti-TIGIT
TIGIT belongs to the Ig superfamily, and it is expressed on activated CD4+ and CD8+ T cells, as well as NK cells (124–126). TIGIT interacts with its ligand, CD155, which is expressed mostly on DCs and macrophages, and upon TIGIT/CD155 interaction, immune responses are negatively regulated. Specifically, T-cell receptor expression is reduced, resulting in impairment of the function of CD8+ and NK cells, resulting in immunosuppression. Currently, Domvanalimab, an investigational inhibitor, is being evaluated in clinical trials in combination Zimberelimab (anti-PD-1) in recurrent glioblastoma (NCT04656535, Table 1).
Despite extensive research, there is still no ICI approved for the treatment of glioblastoma, since no significant improvement in OS has been observed during clinical trials so far. Despite the disappointing early clinical trial results, the use of ICIs for the treatment of glioblastoma remains under ongoing clinical investigation, aiming to reverse the glioblastoma immunosuppressive TME which is mainly attributed to the upregulation of several immune checkpoint molecules, such as PD-1, PD-L1, CTLA-4, LAG-3, TIM-3 and TIGIT. A variety of ICIs are currently being investigated in clinical trials in glioblastoma (early, aggressive or recurrent), for their safety and effectiveness, often in combination with other therapies to overcome the limitations of monotherapy and improve therapeutic outcomes. The use of novel delivery systems in several trials, like the Optune® device, suggests a commitment in overcoming delivery limitations especially the BBB. Furthermore, combining ICIs with personalized neoantigen vaccines aims to enhance T-cell activation and specificity. In addition, targeting myeloid cells aims to enhance antigen presentation. Importantly, discovery of new biomarkers to predict response to ICI therapy is essential in GMB; for example, in other types of cancer, patients with low PDL-1 levels also benefit from anti-PD-1 therapy, suggesting other mechanisms are involved in their action (127). The lack of reliable biomarkers for the use of ICIs in GMB is discussed in recent reviews (128, 129). The future of ICIs in glioblastoma, depends on the delivery systems, the drug combination strategies, the tackling of the cold glioblastoma immune microenvironment, and the identification of personalized biomarkers based on molecular signatures or immune profiles that could help tailor ICI therapies to responsive subpopulations of patients. Another important factor is optimizing the timing of ICI administration. For instance, recent findings suggest that administering combination ICI in the neoadjuvant setting can stimulate the infiltration, activation, and proliferation of tumor-specific T cells in patients with newly diagnosed glioblastoma (130). In fact, a clinical trial, based on this, is already ongoing (NCT06816927).
4.2 T-cell based therapies
4.2.1 CAR T-cells
CAR-T cell therapy is at the forefront of T-cell based therapies, providing a powerful tool for cancer treatment (131–133). It involves the inducible expression of a chimeric antigen receptor (CAR), engineered to target a specific antigen of interest on autologous T cells, hence bypassing the need for antigen presentation by Major Histocompatibility Complex (MHC) otherwise required for the activation of endogenous T cells (131–134). Since 2017, six CAR-T cell products have been approved for the treatment of several cancers, including acute lymphoblastic leukemia (ALL), diffuse large B-cell lymphoma (DLBCL), refractory follicular lymphoma (FL), recurrent or refractory mantle cell lymphoma (MCL), and relapsed or refractory multiple myeloma (133). Currently there are no FDA-approved CAR-T cell therapies for glioblastoma, but several clinical trials (mostly in Phase I) are evaluating CAR-T cell therapy in glioblastoma (Table 2; Figure 2B), including one that is combining CAR-T cell therapy with ICIs (NCT04003649). The glioblastoma antigens currently targeted in the clinic, often in combination, are Cluster of Differentiation proteins (CD133, CD44 & CD70), IL receptors (IL-13Ra2), EGFR and EGFRvIII, ephrin receptor A2 (EphA2), human epidermal growth factor receptor 2 (HER2) and B7-H3 (Table 2).
IL-7 is a hematopoietic cytokine that promotes the activation, differentiation and homeostasis of naïve T-cells, as well as the survival, expansion and proliferation of memory T-cells (135). It has been shown that engineered T-cells constitutively expressing the IL-7 receptor alpha (IL-7Rα) have great antitumor efficacies in both breast cancer (136) and glioblastoma models (137), therefore, IL-7 and its receptor are great candidates for immunotherapy. For the clinical trial NCT05577091 (Table 2), autologous T-cells have been genetically modified to express a CAR targeting CD133 and CD44, and a truncated form of the IL-7Rα. These CAR-T cells are believed to have immunostimulating and anti-neoplastic activities since they target CD133 and CD44, two markers of GSCs, associated with the proliferative or invasive state of glioblastoma cells (138). Also, the IL-7Rα-Tris-CAR-T cells induce selective toxicity to tumor cells, and the IL-7/IL-7Ra-mediated signaling promotes the proliferation and survival of T cells.
IL-8 is another important chemokine, that coupled with its receptor, IL-8R, play a role in tumor invasion, proliferation, survival and angiogenesis, as well as in the promotion of the malignant properties of the glioblastoma stem cells (139–142). CD70, is an antigen that is overexpressed in glioblastoma and is associated with poor survival (143). It was also hypothesized that it correlates with a mesenchymal phenotype and immunosuppression via recruitment of macrophages and CD8+ T-cell death. This information on IL-8 and CD70, let to the generation of CD70-targeting CAR with a modified IL-8 receptor (called 8R-70CAR) that let to complete tumor regression of advance cancers in pre-clinical studies, including glioblastoma (144). (NCT05353530 & NCT06946680, Table 2).
IL-13Rα2 is a high affinity membrane receptor that is overexpressed in glioblastoma, and is associated with poor outcome, mesenchymal gene profile, immunity, and the tumor microenvironment, which make it an important therapeutic target (145, 146). First-generation of IL-13Rα2-targeted CAR-T cells showed evidence of antitumor efficacy, but limited persistence of T-cells (147), therefore new approaches were explored in the design and production of IL-13Rα2-targeted CAR-T cells. Brown et al. (2018) (148), designed a second-generation of IL-13Rα2-targeted CAR, by engineering memory-enriched T cells to express IL-13Rα2 and 41BB-constimulatory CAR, a member of the tumor necrosis factor (TNF) receptor superfamily that enhances CAR-T survival and persistence (149, 150). Furthermore, the engineered T-cells expressed a truncated form of CD19, a pan B-cell marker (151) that has been targeted for the treatment of several hematological malignancies (152), and was shown to be highly expressed in brain and endothelial cells causing increased BBB permeability and neurotoxicity after targeted CAR-T immunotherapy (153, 154). These second-generation CAR-T cells not only improved anti-tumor activity but also T-cell persistence. Furthermore, it was shown that when delivered intracranially, they had a greater anti-tumor effect compared to intravenous delivery in orthotopic glioblastoma mouse models. This study, led to the protocol design for the NCT02208362 trial (Table 2), in which multiple intracranial infusions of these CAR-T cells, were not only proven to be safe, but also let to the regression of glioblastoma, increased levels of immune cells and cytokines, and persistence of response for up to 7.5 months after treatment initiation (155). Results from this trial, demonstrated that locoregional therapy with IL-13Rα2-targeted CAR-T is safe with promising clinical activity in a subpopulation of patients (88). Later, Chang Xu et al. (2022) (156), developed the first humanized third-generation CAR-targeting IL-13Rα2 that showed great anti-tumor efficacy and reduced expression of immunosuppressive cytokines such as IL-6 (88, 157) Like second and third generation IL-13Rα2-targeted CAR-T cells, HER2-specific, 41BB-costimulatory CAR with a truncated CD19 have been engineered and are being assessed in recurrent or non-responsive glioblastoma (NCT03389230, Table 2). HER2 is another important target, since it was found to be overexpressed in glioblastoma and is associated with poor prognosis (158).
EGFRs are transmembrane receptors, part of the ErbB family of receptor tyrosine kinases (RTKs), activated by several ligands (e.g. TGFα) binding to the extracellular domain (ECD) (159). Ligand-receptor formation of homo- or hetero-dimers, leads to the activation of several downstream pathways (e.g. MAPK, STAT3 and PI3K) that regulate cell survival, proliferation, angiogenesis and migration. When EGFRs are overexpressed or mutated, they stay constitutively active, which leads to uncontrolled cell proliferation and therefore tumor progression. EGFR is overexpressed in ~60% of primary glioblastoma and 10% of secondary glioblastomas (160). Furthermore, several mutants have been found in glioblastoma, with the most common being EGRvIII (159, 160). Overexpression and mutations in EGFR lead to a more aggressive glioblastoma phenotype and increased tumor heterogeneity, therefore several strategies have been developed to target EGFR (Table 2) (87, 89, 161, 162) B7-H3 (also known as CD276) is a type I transmembrane protein, that expressed in >70% of glioblastoma patients (163, 164), and is associated with progression, metastasis, poor outcome and immune evasion (165), as it is an immune checkpoint molecule expressed on antigen-presenting cells. The antitumor efficacy of B7-H3 CAR-T cells in glioblastoma in vitro and in vivo models was first reported in Xing Tang et al. (2019) (166). Several clinical trials, currently in recruiting phase, in refractory or recurrent glioblastoma, are evaluating the safety, feasibility, dose and efficacy of B7-H3 CAR-T cells (NCT05835687, NCT06482905, NCT05366179 & NCT05474378 Table 2). In NCT05241392 (Table 2), the preliminary data were recently published and showed that the use of B7-H3 CAR-T cells is safe and tolerated and holds great promise toward improving patients’ overall survival (167). Regarding the pharmacokinetic profile of B7-H3 CAR-T cells, a study reported that local delivery led to cerebrospinal fluid (CSF) persistence and localized immune activation rather than systemic effects (168). It is worth noting, that in a recent pre-clinical study, B7-H3 CAR-T cells consisting of IL-7Rα, have been shown to suppress tumor growth and prolong overall survival in glioblastoma mouse models (169), suggesting the potential implication of B7-H3-IL-7Rα CAR-T cells in the clinic.
Finally, chlorotoxin (CLTX) is a small 36-amino-acid peptide purified from the venom of the scorpion Leiurus quinquestriatus (170). CLTX was initially characterized based on its inhibition of glioma-specific chloride ion channels (GCC), however recent studies identified MMP-2 as the principal receptor for CLTX on the surface of glioblastoma cells. As mentioned above, MMP-2 expression is increased in glioblastoma TME, contributing significantly to the tumor invasiveness. CLTX has demonstrated specific and selective binding to membrane-bound MMP-2 and minimal binding to normal brain tissue, therefore CAR-T cells engineered to incorporate CTLX as an antigen recognition domain are considered a promising approach for MMP-2 positive glioblastoma treatment, redirecting cytotoxic T cells towards glioblastoma cells (171).
4.2.2 (γδ)T-cells
(γδ)T-cells comprise a unique type of innate immune T cells that express a γδ T cell receptor (TCR), and are found abundant in several tissues, including lymphocytes that infiltrate solid tumors (172, 173). They can directly kill tumor cells through (a) cytokine-mediated cytotoxicity (e.g. TNFα and IFNγ), (b) perforin (PFN) & granzyme (GzmB) and FasL & TRAILR mediated target cell apoptosis, (c) antibody-dependent cell-mediated cytotoxicity, and (d) antigen processing and presentation. They can also have indirect anti-tumor effects through interaction with immune cells (e.g. NK cells and αβ T-cells). In addition, they have no autologous limitations, can be derived from healthy donors, and can be easily expanded. These properties make them a powerful tool in immunotherapy. The first therapeutic mechanism involves the selective amplification of (γδ)T-cells in vivo using antibodies or bisphosphonate antigens. In the second mechanism, which is adoptive cell therapy, tumors are treated with allogeneic (γδ)T-cells (natural or genetically engineered) or (γδ)T-cells that have been expanded in vitro. The first-in-human Phase I clinical trial (NCT04165941, Table 2) involves the intracranial infusion of TMZ-resistant (γδ)T-cells in newly diagnosed glioblastoma patients. During TMZ treatment, the natural killer group 2, member D (NKG2D) receptor ligands (NDG2DL), the master activators of NK cells (174), are upregulated, but this immune response is impaired due to lymphodepletion (172, 175). Therefore, genetic modification of (γδ)T-cells, naturally expressing the NKG2D receptor, were genetically modified and expanded ex vivo with an MGMT-expressing lentivector that provided resistance to TMZ, allowing therefore the simultaneous infusion of (γδ)T-cell with chemotherapy, and targeting therefore the tumor when NKG2DL are maximally expressed (Figure 2C). Modifying the cells to be resistant to TMZ is crucial, as they need to be able to resist the cytotoxic effects of TMZ and be able to survive and function on the presence of chemotherapy. In another trial, gene modified allogeneic or autologous (γδ)T-Cells (DeltEx), again conferring TMZ resistance, are evaluated for safety, tolerance and ability to delay recurrence in newly or recurrent glioblastoma (NCT05664243, Table 2; Figure 2C).
The use of CAR-T cells in glioblastoma is based on targeting the tumor-specific or tumor-enriched antigens like IL-13Rα2, EGFR/EGFRvIII, HER2, CD70, B7-H3, and CD133/CD44. Apart from antigen-specificity, the use of innovative constructs in active clinical trials, such as next-generation CAR-T that integrate co-stimulatory domains, can improve T-cell persistence, activation, and tumor selectivity. In some trials, locoregional delivery of CAR-T cells through intracranial administration aims to enhance the efficacy and reduce systemic toxicity, although is more invasive and complex. Since glioblastoma is characterized by profound heterogeneity that limits the durability of response due to downregulation of target antigens, CAR-T cells that target multiple antigens have been developed that aim to reduce escape via antigen heterogeneity. (γδ)T-cells use a different strategy from CAR-T, exploiting innate-like immune response. Based on their recognition of ligands independently of MHC, they are ideal for glioblastoma that is immune-evasive. Another advantage is that they are multifunctional, since use different mechanisms for killing tumor cells. On the other hand, the safety and efficacy of (γδ)T-cells is still not well established and they also require complex genetic engineering. Although highly innovative, both CAR-T and (γδ)T-cells need to overcome antigen escape, and the suppressive microenvironment. Moving forward, further clinical exploration is needed, and their success will depend on effectively reshaping the TME.
4.3 Vaccines
As recently reviewed, cancer vaccines are another promising form of immunotherapy in glioblastoma (176). Briefly, anti-cancer vaccines aim to provoke an immune response within the body against tumor-specific antigen(s) and are generally composed of the antigen (neoantigen, tumor-associated antigen or pathogen derived-antigen) and the platform/carrier type (DC vaccine, peptide vaccine, nucleic acid vaccine or viral vector vaccine). Several vaccines are in active clinical trials in glioblastoma (Table 2, Figure 3A).
4.3.1 Peptide vaccines
Peptide vaccines are composed of 8–30 amino acid including tumor-specific or tumor-associated antigens that elicit an anti-tumor T cell response (177, 178). As they are not highly immunogenic, they can be combined with other forms of immunotherapy or chemoradiation. They can be individualized, single-targeted or multi-targeted. One target of peptide vaccines is the survivin protein; a member of the inhibitor of apoptosis (IAPs) family, that is overexpressed in glioblastoma and is associated with poor prognosis (179). The survivin peptide vaccine, SurVaxM, is currently being evaluated in two clinical trials in combination with TMZ (NCT02455557 & NCT05163080, Table 3). Preliminary results showed that SurVaxM is safe and well-tolerated and its combination with TMZ is very promising (96). pp65 and survivin are also being targeted together with EphA2 through a multi-peptide vaccine (ETAPA I) in HLA-A*0201 positive patients with a newly diagnosed, unmethylated, and untreated glioblastoma (NCT05283109, Table 3). pp65 is overexpressed in high grade gliomas and medulloblastomas, but not in adjacent brain, and plays a significant role in glioblastoma progression. When tested in children and young adults, it was proven to be well-tolerated and promoted antigen-specific immune responses (180). Other targets include Telomerase Reverse Transcriptase (TERT), Protein tyrosine phosphatase receptor type Z1 (PTPRZ1) and Toll-like receptors (TLRs), that are being targeted in combination in NCT06622434 (Table 3). PTPRZ1 is a clinically relevant antibody in glioblastoma associated with stemness (181), and telomerase (TRT) is a major oncogene, whose promoter is mutated in approximately 80% of glioblastoma patients and is associated with tumor progression (96, 182, 183). TLRs are ubiquitously expressed receptors that recognize pathogens and lie at the first line of defense in the innate immune system (184). In several tumor types, upon ligand recognition, TLRs activate downstream intracellular signaling pathways either supporting or suppressing tumor growth, thus they are a great candidate for immunotherapy. Finally, several autologous or allogeneic multipartite vaccines, designed to induce a variety of neoantigen-specific immune responses, are tested in clinical trials in combination with other therapies, such as ICIs and nucleic acid vaccines (NCT03223103 & NCT02287428, Table 3).
4.3.2 DC vaccines
DC are the most superior APC cells of the immune system, thus playing a vital role in presenting antigens in the lymph nodes eliciting T-cell priming and distant anti-tumor response (185, 186). DC vaccines are generated by culturing hematopoietic progenitor cells or monocytes ex vivo in the presentation of a cytokine cocktail to induce their maturation. Following maturation, DCs are loaded into tumor antigens and subsequently injected into patients. Not only they can achieve priming of CD4+ T cells by peptide-MHCII complex, but also, they elicit CD8+ T-cell antitumor responses. Several clinical trials are underway in glioblastoma investigating the safety and efficacy of DC vaccines in newly diagnosed or recurrent glioblastoma (Table 3). Some are loaded with autologous tumor lysates (NCT03395587, NCT04523688 & NCT04801147, Table 3), and even “double-loaded” (NCT06805305), and others are loaded with multiple tumor neoantigens (NCT04968366 & NCT06253234, Table 3). They are administered either alone or in combination with chemoradiation or ICIs. Finally, other DC vaccines are loaded with mRNAs for specific proteins, including Survivin/hTERT derived from autologous GSCs (NCT03548571, Table 3), the tumor-associated antigen Wilm’s tumor 1 (WT1) (NCT02649582, Table 3) and the human CMV immunodominant protein pp65-LAMP (NCT03688178, Table 3).
4.3.3 Nucleic acid-based vaccines
Other than the DC and peptide vaccines, a few other vaccines are being exploited in glioblastoma immunotherapy trials based on the delivery of nucleic acids (Table 3). Nucleic acid-based vaccines introduce a segment of DNA or RNA that encodes a specific or multiple tumor antigens, to elicit and immune response (176). The current vaccines clinical trials include DNA vaccines (NCT05698199, & NCT04015700), an mRNA vaccine (NCT05938387), and two vaccines that use lipid nanoparticle technology (187) to deliver RNA (NCT04573140 & NCT06389591). Apart from the mRNA vaccine that encodes several GBM peptides (188), all the others are targeting the immunogenic and viral antigens of CMV.
Cancer vaccines are a growing field in glioblastoma immunotherapy, and they hold a great promise, since they aim to evoke tumor-specific immune response through tumor associated or neoantigen presentation. In addition, they have shown great tolerability and minimal toxicity in early-phase trials (e.g. SurVaxM). Furthermore, they allow personalized and/or multi-antigen targeting to address the heterogeneity present in glioblastoma. Especially DC vaccines, that are loaded with autologous tumor lysates, offer T-cell priming, overcoming the limitations of peptide vaccines (priming of weak immune response). The efficacy of vaccines might be limited the high infiltration of Tregs, and antigenic heterogeneity, therefore combining vaccines with other immunotherapies (CAR-T cells, ICIs and oncolytic viruses) may further improve immunogenicity and efficacy. Future progress hinges on optimizing vaccine formulations for better delivery, refining antigen targets, and leveraging synergies with other immunotherapies.
4.4 Oncolytic viruses
The development, use and clinical relevance of oncolytic virus in cancer immunotherapy have been extensively reviewed elsewhere (189–192). Briefly, oncolytic viruses (Ovs) are genetically engineered viruses that selectively attack and lyse tumor cells without disrupting normal cells via different biological mechanisms. They are manipulated in such way to enhance tumor selectivity, promote replication competence, limit pathogenicity and increase immunogenic cell death (ICD). In glioblastomas, treatment with OVs is very suitable due to their alignment to the brain environment, the fact that they do not form distant metastases, and finally that they are fast growing tumor cells that attract virus replication. OVs are classified into two major groups: (1) replication-competent OVs that selectively replicate in cancer cells e.g. Herpes Simplex Virus (oHSV) and adenoviruses (oAds) and (2) replication-deficient viral vectors used as vehicles for other therapeutic genes e.g. polioviruses.
Most ongoing clinical trials in glioblastoma are using oHSVs (Table 3, Figure 3B) (193–197).
Apart from oHSVs, trials utilizing oAds are currently ongoing in glioblastoma (Table 3, Figure 3B). DNX-2401 oAds has two stable genetic changes the dsDNA adenovirus genome that allows the selective and efficient replication in current cells (197). When combined with pembrolizumab, DNX-2401 not only was proven safe, but it also induced durable cell death by direct oncolytic activity and immune response in high-grade glioma patients. The drug is now in Phase I clinical trials in patients with recurrent glioblastoma (NCT03896568, Table 3). However, DNX-2401 is not intended for systemic BBB penetration due to its viral nature and size (195). NSC-CRAd-S-pk7, is another very promising adenovirus developed against glioblastoma (198). It has several features including: (1) a survivin promoter for enhancing specific replication in tumor cells, since survivin is found to be overexpressed in glioblastoma, (2) a modified Ad5 protein through insertion of polylysine sequence (pk7), which binds to heparin sulfate proteoglycans also overexpressed in glioblastoma and (3) neural stem cells (NSCs) as its carrier, also contributing to selectivity. In Phase I trials, NSC-CRAd-S-pk7 was shown to be safe, improve OS and result in an increase of cytotoxic T-cells (105). A clinical trial against recurrent high-grade glioma is currently underway (NCT05139056, Table 3). NRG-103 is a novel gene therapy agent engineered to enhance the tumor-specific recognition and cytolytic activity of an oncolytic virus, while simultaneously eliciting a robust anti-tumor immune response. It leverages in situ transdifferentiation technology, incorporating multiple engineered mutations within the adenoviral genome. Notably, NRG-103 expresses two transcription factors capable of efficiently reprogramming residual glioblastoma cells into neuronal-like cells, thereby aiming to delay tumor recurrence and improve long-term survival. In preclinical models it exhibited significant anti-tumor activity, and its overall or disease-free survival is being evaluated in patients with recurrent glioblastoma (NCT06757153, Table 3). Lastly, the safety and efficacy of the oncolytic virus TS-2021, is evaluated in the clinical trial NCT06585527 (Table 3). TS-2021 is a third-generation oncolytic adenovirus that can efficiently target glioblastoma cells overexpressing Ki67 (proliferation marker) and TGF-β2 and can inhibit invasiveness through targeting of the MKK4/JNK/MMP3 pathway (199).
Finally, PVSRIPO is under clinical investigation for safety and efficacy in recurrent glioblastoma (NCT02986178, Table 3). PVSRIPO is a type 1 (Sabin) live-attenuated poliovirus vaccine that carries a heterologous internal ribosomal entry site (IRES) of human rhinovirus type 2 (HRV2) (200). Its synthesis allows selective expression in tumor cells after coupled with its receptor, CD155, expressed in several tumors including glioblastoma, exerting antitumor effects.
Ovs are an innovative class of immunotherapies that offer direct tumor cells lysis as well as indirect stimulation of anti-tumor immune response. They exhibit several advantages including their selective replication in tumor cells minimizing the damage to normal brain tissue (e.g. oHSV G207 and C134 are designed with deletions in neurovirulence genes to limit replication in healthy neural tissue while enhancing tumor-specific lysis) (201). They can induce ICD, reprogram the immunosuppressive microenvironment (e.g. CAN-3110) and improve T-cell activation (e.g. NSC-CRAd-S-pk7). On the other hand, glioblastoma heterogeneity and pre-existing immunity against viral vectors, can limit OV spread and propagation. Furthermore, and tumor cells can develop resistance through interferon signaling or downregulation of viral receptors (e.g., CD155 for PVSRIPO). Even though promising results have been demonstrated in early-phase trials, their benefit on PFS and OS must be demonstrated in Phase II/III trials. Ongoing advances in vector engineering, biomarker-guided patient selection, and combinations with other immunotherapies (e.g. with ICIs) are expected to shape the next generation of OV-based therapies.
4.5 Cytokine therapies
Another form of immunotherapy are the cytokine-based therapies that are very promising, as they can potentially alter the immunosuppressive microenvironment and trigger anti-tumor immunity (195). However, the systemic administration of therapeutic doses of pro-inflammatory cytokines can cause toxicity. To tackle this systemic toxicity, L19TNF was generated. Composed of TNF fused to the L19 antibody, L19TNF is a fully human antibody–cytokine fusion designed to selectively deliver TNF to tumors by binding a fibronectin epitope uniquely expressed in the tumor extracellular matrix, where it induces cell death, and at the same time the reduction of toxicity to healthy organs (202) (Figure 3C). Ongoing trials are investigating the safety, efficacy and recommended dose of intravenous administration of L19TNF in newly diagnosed (NCT04443010, Table 3) and recurrent glioblastoma (NCT04573192, Table 3), in combination with TMZ-based chemoradiation or lomustine accordingly. In another ongoing clinical trial in recurrent glioblastoma (NCT04729957, Table 3), the efficacy and MTD of tocilizumab in combination with atezolizumab and stereotactic RT is being investigated. Tocilizumab is an anti-IL-6 monoclonal antibody that reduces the body’s immune response and inflammation (Figure 3C). Therefore, it suppresses the inhibitory effect of immune cells surrounding glioblastoma and consequently allow atezolizumab, an anti-PD-L1 treatment, to activate the immune response against glioblastoma. Finally, Tamferon™, was designed to increase the production of IFNα to cause immune activation (203). More specific, CD34+ HSPCS are isolated from the patient and are transduced ex-vivo with a lentivirus expressing IFNα downstream a Tie2 promoter, so that IFNα expression is confined to Tie-2 expressing macrophages (TEMs) (Figure 3C). The safety and efficacy of Tamferon™, is evaluated in patients with MGMT-unmethylated glioblastoma (NCT03866109, Table 3).
Cytokine therapy is a highly promising but still evolving frontier. It has an immunomodulation potential, since it aims to reprogram the TME through cytokines such as TNF and IFNα, enabling a more effective anti-tumor response. Innovations like L19TNF and Tamferon™ help to overcome the systemic toxicity by directing cytokine action to the tumor site. Furthermore, the latter utilizes patient-specific, gene-modified immune cells, showing a shift towards personalized immunotherapy. Cytokine therapy is often combined with other therapies, including the standard of care, to address other resistance mechanisms. Like other immunotherapeutic approaches, immune evasion and tumor heterogeneity are still a concern. Furthermore, delivery is still an issue, especially since cytokines have a limited therapeutic window, and local overexpression can cause neurotoxicity or inflammation. The issues need to be addressed, and success in clinical trials remains to be proven.
4.6 Other targeted immunomodulatory therapies
Finally, a few other strategies that do not explicitly fall into the categories already discussed, have been developed to induce immune-specific responses and are under clinical evaluation in glioblastoma (Table 3). For example, Tumor-infiltrating lymphocytes (TILs) therapy, an innovative form of adoptive cell therapy that utilizes the patient’s own immune cells to target and destroy cancer cells, is given in combination with pembrolizumab in patients with advanced gliomas (NCT06640582, Table 3). In addition, IGV-001 that is being evaluated in NCT04485949 (Table 3), is a first-in-class autologous immunotherapeutic product from Goldspire™, that combines personalized whole tumor-derived cells with an insulin-like growth factor receptor 1 (IGF-1R) antisense oligonucleotide (IMV-001) in an implantable biodiffusion chambers (204). It has been reported that inhibiting IGF-1R can effectively suppress the growth of GBM cells directly or indirectly through suppression of cell proliferation or angiogenesis respectively (205–208). In addition, the N-803 from Anktiva, a modified IL-15-based fusion protein (IL-15Rα-Fc), functions as an immunostimulatory agent that drives the expansion and activation of NK cells and CD8+ T lymphocytes (209), is being evaluated in progressive or recurrent glioblastoma (NCT06061809). It is given in combination with PD-L1 targeting high-affinity NK (t-haNK) cells (210) and the anti-VEGF antibody, Bevacizumab. While N-803 generally demonstrate improved tolerability, long-term exposure may still carry immunological risks. Sustained stimulation of NK and CD8+ T cells can lead to chronic immune activation, increasing the risk of cytokine release syndrome (CRS), immune-mediated tissue damage and inflammatory toxicities such as fever, and hypotension. Finally, the drug NGM707 is being evaluated as monotherapy and in combination with pembrolizumab in advanced or metastatic glioblastoma in NCT04913337 (Table 3). NGM707, is a dual humanized monoclonal antibody that targets Immunoglobulin-like transcript (ILT)2 and ILT4 resulting in early efficacy and biomarker signals in advanced or metastatic solid tumors (113), probably through the generation of immune niche and immune-checkpoint blockage (211).
These novel immunomodulatory therapies for glioblastoma are conceptually sound and mechanistically diverse, representing a hopeful direction. TILs and the IGV-001 personalized vaccine provide tumor-specific cytotoxicity since the former are isolated from patient’s tumor and the latter combines patient-specific tumor cells with IGF-1R antisense oligonucleotide. Combination of TLIs with pembrolizumab maximizes activity by preventing T-cell exhaustion. Daratumumab and other monoclonal antibodies are designed against tumor-specific antigens, aiming to enhance DC priming and T-cell activation, boosting anti-tumor immunity. Indoximod and N-803 both enhance NK function, and finally NGM707 aims checkpoint inhibition beyond PD-1/PD-L1, as ILT2/ILT4 target innate immune checkpoints, potentially reviving exhausted myeloid cells and enhancing antigen presentation. However, despite their innovative approach, common hurdles persist including tumor heterogeneity leading to immune escape, impaired delivery due to the BBB and complex trial designs. Most evidence remains preclinical or Phase I, making long-term benefit speculative.
5 Conclusions & future prospectives
Glioblastoma remains among the most aggressive and therapeutically challenging malignancies due to its complex, heterogeneous, and profoundly immunosuppressive tumor microenvironment (TME). Despite the significant progress in cancer immunotherapy in some tumor types, mainly using ICIs and CAR-T cell therapies, these approaches have not yet demonstrated substantial clinical benefits in glioblastoma patients, primarily due to intrinsic resistance mechanisms facilitated by the glioblastoma TME. The etiology of this phenomenon is clearly multifactorial and is largely attributed to the highly immunosuppressive nature and heterogeneity of GBM tumors. Immunosuppression in the TME is mediated via a multitude of underlying mechanisms, including the secretion of immunosuppressive cytokines, the abundance of Tregs, MDSCs and GAMs in the TME, the insufficient infiltration and elimination of antigen-specific T cells, the sequestration of T-cells in the bone marrow leading to their dysfunction, T-cell exhaustion, antigen escape as well as upregulation of multiple immune checkpoint molecules. In addition, the low TMB present in the majority of GBM tumors leads to limited number of produced neoantigens, which are needed to elicit durable T-cell responses, and contributes to the limited efficacy of immunotherapy. Moreover, physiological barriers like the blood-brain barrier (BBB), along with hypoxic and acidic conditions within the TME, significantly hinder therapeutic efficacy and immune response.
This review provides a comprehensive description of immunotherapy clinical trials. However, interpretations based on these trials are limited due to mixed treatment regimens (monotherapy vs. combinations) and ethnic bias, as many cited studies were conducted in predominantly Japanese cohorts, reducing generalizability across broader populations. To enhance the success of glioblastoma immunotherapies, future strategies must involve a comprehensive and functional understanding of distinct components of the TME at the single-cell level, such as tumor profiling using spatial transcriptomics and proteomics as well as patient-specific neoantigen identification, that will enable personalized and precision targeting to improve immunotherapy efficacy and patient outcomes. In addition, advancing glioblastoma therapy requires overcoming simultaneously both the biological and physical barriers that influence the therapeutic efficacy, such as the immunosuppressive TME, ECM composition and BBB permeability. Therefore, technological advancements in BBB penetration and targeted drug delivery (e.g., advanced nanomedicine, ultrasound-mediated BBB disruption) hold substantial promise and should be further explored.
In addition, combining immunotherapeutic agents with strategies that modulate the non-cellular components of the TME, such as agents targeting hypoxia, acidosis, and ECM constituents, could also enhance therapeutic outcomes. Further exploration of combination therapies integrating ICIs, CAR-T cells, vaccines, and oncolytic viruses with standard therapies (chemotherapy, radiotherapy) and novel targeted treatments is critical. However, mitigating short- and long-term side-effects in patients, especially in combination treatments likely remains one of the major challenges that need to be addressed.
To advance the field of glioblastoma immunotherapy, future efforts should move beyond incremental improvements. One promising direction is the application of artificial intelligence (AI) and machine learning (ML) to optimize clinical trial design, enabling real-time patient stratification and prediction of therapeutic response based on multi-omics data. Subsequently, spatial mapping of the TME through spatial transcriptomics and multiplexed imaging can uncover regional immune niches and patterns of immune suppression within glioblastoma, guiding the localization of targeted therapies. Coupling these insights with biomarker-driven patient stratification could personalize immunotherapy approaches and increase clinical efficacy.
Finally, integrating novel biomarkers and robust pre-clinical models using state-of-the art humanized patient-derived xenograft models into clinical trial designs will facilitate better patient stratification, treatment personalization, and evaluation of immunotherapy efficacy. Future interdisciplinary research efforts must focus on refining patient selection criteria and developing multimodal therapies that target both the tumor and its immunosuppressive milieu. These will be crucial in overcoming existing barriers, emphasizing on exploiting glioblastoma-specific vulnerabilities to ultimately transform the treatment landscape for glioblastoma. Despite numerous challenges, immunotherapy remains one of the most promising treatment strategies for glioblastoma.
Author contributions
NN: Conceptualization, Writing – original draft, Writing – review & editing. MA: Writing – original draft, Writing – review & editing. CN: Writing – original draft, Writing – review & editing. PP: Supervision, Writing – review & editing, Writing – original draft, Conceptualization.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23:1231. doi: 10.1093/NEUONC/NOAB106
2. Grochans S, Cybulska AM, Simińska D, Korbecki J, Kojder K, Chlubek D, et al. Epidemiology of glioblastoma multiforme–literature review. Cancers (Basel). (2022) 14:2412. doi: 10.3390/CANCERS14102412
3. Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. (2020) 18:3. doi: 10.1038/s41571-020-00447-z
4. Oronsky B, Reid TR, Oronsky A, Sandhu N, and Knox SJ. A review of newly diagnosed glioblastoma. Front Oncol. (2021) 10:574012. doi: 10.3389/fonc.2020.574012
5. Posti JP, Bori M, Kauko T, Sankinen M, Nordberg J, Rahi M, et al. Presenting symptoms of glioma in adults. Acta Neurol Scand. (2015) 131:88–93. doi: 10.1111/ANE.12285
6. McKinnon C, Nandhabalan M, Murray SA, and Plaha P. Glioblastoma: clinical presentation, diagnosis, and management. BMJ. (2021) 374:374. doi: 10.1136/BMJ.N1560
7. Yuile P, Dent O, Cook R, Biggs M, and Little N. Survival of glioblastoma patients related to presenting symptoms, brain site and treatment variables. J Clin Neurosci. (2006) 13:747–51. doi: 10.1016/j.jocn.2005.10.011
8. Shukla G, Alexander GS, Bakas S, Nikam R, Talekar K, Authors Gaurav Shukla A, et al. Advanced magnetic resonance imaging in glioblastoma: A review. JHN J. (2018) 13:5. doi: 10.29046/JHNJ.013.1.005
9. Peeken JC, Goldberg T, Pyka T, Bernhofer M, Wiestler B, Kessel KA, et al. Combining multimodal imaging and treatment features improves machine learning-based prognostic assessment in patients with glioblastoma multiforme. Cancer Med. (2019) 8:128–36. doi: 10.1002/CAM4.1908
10. Abd-Elghany AA, Naji AA, Alonazi B, Aldosary H, Alsufayan MA, Alnasser M, et al. Radiological characteristics of glioblastoma multiforme using CT and MRI examination. J Radiat Res Appl Sci. (2019) 12:289–93. doi: 10.1080/16878507.2019.1655864
11. Dumba M, Fry A, Shelton J, Booth TC, Jones B, Shuaib H, et al. Imaging in patients with glioblastoma: A national cohort study. Neurooncol Pract. (2022) 9:487–95. doi: 10.1093/NOP/NPAC048
12. Drake LR, Hillmer AT, and Cai Z. Approaches to PET imaging of glioblastoma. Molecules. (2020) 25:568. doi: 10.3390/MOLECULES25030568
13. Kotecha R, Odia Y, Khosla AA, and Ahluwalia MS. Key clinical principles in the management of glioblastoma. Wolters Kluwer Health. (2023) 19:180–9. doi: 10.1200/OP.22.00476
14. Ntafoulis I, Koolen SLW, Leenstra S, and Lamfers MLM. Drug repurposing, a fast-track approach to develop effective treatments for glioblastoma. Cancers. (2022) 14:3705. doi: 10.3390/CANCERS14153705
15. Teraiya M, Perreault H, and Chen VC. An overview of glioblastoma multiforme and temozolomide resistance: can LC-MS-based proteomics reveal the fundamental mechanism of temozolomide resistance? Front Oncol. (2023) 13:1166207/BIBTEX. doi: 10.3389/FONC.2023.1166207/BIBTEX
16. Almeida Lima K, Osawa IYA, Ramalho MCC, de Souza I, Guedes CB, de Souza Filho CHD, et al. Temozolomide resistance in glioblastoma by NRF2: protecting the evil. Biomedicines. (2023) 11:1081. doi: 10.3390/BIOMEDICINES11041081
17. Chien CH, Hsueh WT, Chuang JY, and Chang KY. Dissecting the mechanism of temozolomide resistance and its association with the regulatory roles of intracellular reactive oxygen species in glioblastoma. J Biomed Sci. (2021) 28:1. doi: 10.1186/S12929-021-00717-7
18. Shiravand Y, Khodadadi F, Mohammad S, Kashani A, Hosseini-Fard R, Hosseini S, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol. (2022) 29:3044–60. doi: 10.3390/curroncol29050247
19. Meng L, Wu H, Wu J, An Ding P’, He J, Sang M, et al. Mechanisms of immune checkpoint inhibitors: insights into the regulation of circular RNAS involved in cancer hallmarks. Cell Death Dis. (2024) 15:3. doi: 10.1038/s41419-023-06389-5
20. Omuro A. Immune-checkpoint inhibitors for glioblastoma: what have we learned? Arq Neuropsiquiatr. (2022) 80:266. doi: 10.1590/0004-282X-ANP-2022-S129
21. Arrieta VA, Dmello C, McGrail DJ, Brat DJ, Lee-Chang C, Heimberger AB, et al. Immune checkpoint blockade in glioblastoma: from tumor heterogeneity to personalized treatment. J Clin Invest. (2023) 133:133. doi: 10.1172/JCI163447
22. Sharma P, Aaroe A, Liang J, and Puduvalli VK. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol Adv. (2023) 5:1–16. doi: 10.1093/NOAJNL/VDAD009
23. Zhang X, Zhao L, Zhang H, Zhang Y, Ju H, Wang X, et al. The immunosuppressive microenvironment and immunotherapy in human glioblastoma. .Front Immunol (2022) 13:1003651. doi: 10.3389/fimmu.2022.1003651
24. Rončević A, Koruga N, Soldo Koruga A, Rončević R, Rotim T, Šimundić T, et al. Personalized treatment of glioblastoma: current state and future perspective. Biomedicines. (2023) 11:1579. doi: 10.3390/BIOMEDICINES11061579
25. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMOA043330/SUPPL_FILE/987SA1.PDF
26. Stupp R, Taillibert S, Kanner A, Read W, Steinberg DM, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. (2017) 318:2306–16. doi: 10.1001/JAMA.2017.18718
27. Tentori L, Vernole P, Lacal PM, Madaio R, Portarena I, Levati L, et al. Cytotoxic and clastogenic effects of a DNA minor groove binding methyl sulfonate ester in mismatch repair deficient leukemic cells. Leukemia. (2000) 14:1451–9. doi: 10.1038/sj.leu.2401842
28. Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,l-on-l,2,3,5-tetrazin-4(37/)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine1. Cancer Res. (1987) 47:5846–52.
29. Koumarianou A, Kaltsas G, Kulke MH, Oberg K, Strosberg JR, Spada F, et al. Temozolomide in advanced neuroendocrine neoplasms: pharmacological and clinical aspects. Neuroendocrinology. (2015) 101:274–88. doi: 10.1159/000430816
30. He Q, Liu J, Liang J, Liu X, Li W, Liu Z, et al. Towards improvements for penetrating the blood-brain barrier-recent progress from a material and pharmaceutical perspective. Cells. (2018) 7:24. doi: 10.3390/CELLS7040024
31. Ostermann S, Csajkag C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in Malignant glioma patients. Clin Cancer Res. (2004) 10:3728–36. doi: 10.1158/1078-0432.CCR-03-0807
32. Ray DB. BCNU (bischloroethyl nitrosourea) induced toxicity. Encyclopedia Toxicol. (2024) 1:923–6. doi: 10.1016/B978-0-12-824315-2.00308-0
33. Roux A, Peeters S, Zanello M, Nassif RB, Lahoud GA, Edouard D, et al. Extent of resection and Carmustine wafer implantation safely improve survival in patients with a newly diagnosed glioblastoma: a single center experience of the current practice. J Neurooncol. (2017) 3:83–92. doi: 10.1007/s11060-017-2551-4
34. Ricciardi L, Manini I, Cesselli D, Trungu S, Piazza A, Mangraviti A, et al. Carmustine wafers implantation in patients with newly diagnosed high grade glioma: is it still an option? Front Neurol. (2022) 13:884158/BIBTEX. doi: 10.3389/FNEUR.2022.884158/BIBTEX
35. Nikolova T, Roos WP, Krämer OH, Strik HM, and Kaina B. Chloroethylating nitrosoureas in cancer therapy: DNA damage, repair and cell death signaling. Biochim Biophys Acta (BBA) - Rev Cancer. (2017) 1868:29–39. doi: 10.1016/J.BBCAN.2017.01.004
36. Hochberg FH, Wolfson L, Linggood R, Baker WH, and Kornblith P. Quality and duration of survival in glioblastoma multiforme: combined surgical, radiation, and lomustine therapy. JAMA. (1979) 241:1016–8. doi: 10.1001/JAMA.1979.03290360032023
37. Weller M and Le Rhun E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat Rev. (2020) 87:102029. doi: 10.1016/j.ctrv.2020.102029
38. Vaz-Salgado MA, Villamayor M, Albarrán V, Alía V, Sotoca P, Chamorro J, et al. Recurrent glioblastoma: A review of the treatment options. Cancers. (2023) 15:4279. doi: 10.3390/CANCERS15174279
39. Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. (2017) 18:e315–29. doi: 10.1016/S1470-2045(17)30194-8/ASSET/444F9D58-D351-460F-AA38-E632D79873A9/MAIN.ASSETS/GR3.SML
40. Kazazi-Hyseni F, Beijnen JH, and Schellens JHM. Bevacizumab. Oncologist. (2010) 15:819. doi: 10.1634/THEONCOLOGIST.2009-0317
41. Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. Am Soc Clin Oncol. (2009) 27:4733–40. doi: 10.1200/JCO.2008.19.8721
42. Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. (2009) 27:740. doi: 10.1200/JCO.2008.16.3055
43. Fu M, Zhou Z, Huang X, Chen Z, Zhang L, Zhang J, et al. Use of Bevacizumab in recurrent glioblastoma: a scoping review and evidence map. BMC Cancer. (2023) 23:1–23. doi: 10.1186/S12885-023-11043-6/TABLES/5
44. Yu Z, Zhao G, Zhang Z, Li Y, Chen Y, Wang N, et al. Efficacy and safety of bevacizumab for the treatment of glioblastoma. Exp Ther Med. (2016) 11:371. doi: 10.3892/ETM.2015.2947
45. Rominiyi O, Vanderlinden A, Clenton SJ, Bridgewater C, Al-Tamimi Y, and Collis SJ. Tumour treating fields therapy for glioblastoma: current advances and future directions. Br J Cancer. (2020) 124:4. doi: 10.1038/s41416-020-01136-5
46. Kirson ED, Gurvich Z, Schneiderman R, Dekel E, Itzhaki A, Wasserman Y, et al. Disruption of cancer cell replication by alternating electric fields. Cancer Res. (2004) 64:3288–95. doi: 10.1158/0008-5472.CAN-04-0083
47. Fisher JP and Adamson DC. Current FDA-approved therapies for high-grade Malignant gliomas. Biomedicines. (2021) 9:324. doi: 10.3390/BIOMEDICINES9030324
48. Erices JI, Bizama C, Niechi I, Uribe D, Rosales A, Fabres K, et al. Glioblastoma microenvironment and invasiveness: new insights and therapeutic targets. Int J Mol Sci. (2023) 24:7047. doi: 10.3390/IJMS24087047
49. Fu Z, Zhu G, Luo C, Chen Z, Dou Z, Chen Y, et al. Matricellular protein tenascin C: Implications in glioma progression, gliomagenesis, and treatment. Front Oncol. (2022) 12:971462. doi: 10.3389/FONC.2022.971462
50. Xia S, Lal B, Tung B, Wang S, Goodwin CR, and Laterra J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. (2016) 18:507. doi: 10.1093/NEUONC/NOV171
51. Han Z and Lu ZR. Targeting fibronectin for cancer imaging and therapy. J materials Chem B Materials Biol Med. (2017) 5:639. doi: 10.1039/C6TB02008A
52. White J, White MPJ, Wickremesekera A, Peng L, and Gray C. The tumour microenvironment, treatment resistance and recurrence in glioblastoma. J Trans Med. (2024) 22:1. doi: 10.1186/S12967-024-05301-9
53. Giles B, Nakhjavani M, Wiesa A, Knight T, Shigdar S, and Samarasinghe RM. Unravelling the glioblastoma tumour microenvironment: can aptamer targeted delivery become successful in treating brain cancers? Cancers. (2023) 15:4376. doi: 10.3390/CANCERS15174376
54. Silver A, Feier D, Ghosh T, Rahman M, Huang J, Sarkisian MR, et al. Heterogeneity of glioblastoma stem cells in the context of the immune microenvironment and geospatial organization. Front Oncol. (2022) 12:1022716/BIBTEX. doi: 10.3389/FONC.2022.1022716/BIBTEX
55. Losurdo A, Di Muzio A, Cianciotti BC, Dipasquale A, Persico P, Barigazzi C, et al. T cell features in glioblastoma may guide therapeutic strategies to overcome microenvironment immunosuppression. Cancers. (2024) 16:603. doi: 10.3390/CANCERS16030603
56. Lin H, Liu C, Hu A, Zhang D, Yang H, and Mao Y. REVIEW Open Access Understanding the immunosuppressive microenvironment of glioma: mechanistic insights and clinical perspectives. J Hematol Oncol. (2024) 17:31. doi: 10.1186/s13045-024-01544-7
57. Wang X, Li X, Wu Y, Hong J, and Zhang M. The prognostic significance of tumor-associated neutrophils and circulating neutrophils in glioblastoma (WHO CNS5 classification). BMC Cancer. (2023) 23:1–15. doi: 10.1186/S12885-022-10492-9/FIGURES/6
58. Zhao J and Jin J. Neutrophil extracellular traps: New players in cancer research. Front Immunol. (2022) 13:937565/XML. doi: 10.3389/FIMMU.2022.937565/XML
59. Inocencio JF, Mitrasinovic S, Asad M, Parney IF, Zang X, and Himes BT. Immune checkpoint pathways in glioblastoma: a diverse and evolving landscape. Front Immunol. (2024) 15:1424396. doi: 10.3389/FIMMU.2024.1424396
60. Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell. (2020) 181:1626–42.e20. doi: 10.1016/J.CELL.2020.04.055/ASSET/CB899E09-3258-4E0B-A1BB-4A93235E667F/MAIN.ASSETS/GR7.JPG
61. De Leo A, Ugolini A, and Veglia F. Myeloid cells in glioblastoma microenvironment. Cells. (2020) 10:18. doi: 10.3390/CELLS10010018
62. Yin B, Cai Y, Chen L, Li Z, and Li X. Immunosuppressive MDSC and Treg signatures predict prognosis and therapeutic response in glioma. Int Immunopharmacol. (2024) 141:112922. doi: 10.1016/J.INTIMP.2024.112922
63. Galvez-Cancino F, Navarrete M, Beattie G, Puccio S, Conde-Gallastegi E, Foster K, et al. Regulatory T cell depletion promotes myeloid cell activation and glioblastoma response to anti-PD1 and tumor-targeting antibodies. Immunity. (2025) 58:1236–53.e8. doi: 10.1016/J.IMMUNI.2025.03.021/ASSET/EDBEF7E0-F40A-4AD1-A306-6CD8EEA81ADA/MAIN.ASSETS/FX1.JPG
64. Tripathy DK, Panda LP, Biswal S, and Barhwal K. Insights into the glioblastoma tumor microenvironment: current and emerging therapeutic approaches. Front Pharmacol. (2024) 15:1355242/BIBTEX. doi: 10.3389/FPHAR.2024.1355242/BIBTEX
65. Mi Y, Guo N, Luan J, Cheng J, Hu Z, Jiang P, et al. The emerging role of myeloid-derived suppressor cells in the glioma immune suppressive microenvironment. Front Immunol. (2020) 11:737. doi: 10.3389/FIMMU.2020.00737
66. Xu X, Zheng Y, Luo L, You Z, Chen H, Wang J, et al. Glioblastoma stem cells deliver ABCB4 transcribed by ATF3 via exosomes conferring glioblastoma resistance to temozolomide. Cell Death Dis. (2024) 15:5. doi: 10.1038/s41419-024-06695-6
67. Geng Z, Zhang Z, Wang M, Yu Z, Wang S, Lu J, et al. Targeting stromal cells in tumor microenvironment as a novel treatment strategy for glioma. Cancer Cell Int. (2025) 25:1–16. doi: 10.1186/S12935-025-03692-3/FIGURES/3
68. Bausart M, Préat V, and Malfanti A. Immunotherapy for glioblastoma: the promise of combination strategies. J Exp Clin Cancer Res. (2021) 41:35. doi: 10.1186/s13046-022-02251-2
69. De Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. (2019) 22:539. doi: 10.1093/NEUONC/NOZ185
70. McFaline-Figueroa JR, Sun L, Youssef GC, Huang R, Li G, Kim J, et al. Neoadjuvant anti-PD1 immunotherapy for surgically accessible recurrent glioblastoma: clinical and molecular outcomes of a stage 2 single-arm expansion cohort. Nature Communications. Nat Res. (2024) 15:10757. doi: 10.1038/S41467-024-54326-7
71. Webb M, Burns TC, Twohy E, Sener U, Kizilbash SH, Ruff MW, et al. Efficacy and safety study of neoadjuvant efineptakin alfa (NT-I7) and pembrolizumab in recurrent glioblastoma. J Clin Oncol. (2023) 41:TPS2085–TPS2085. doi: 10.1200/JCO.2023.41.16_SUPPL.TPS2085
72. Chen D, Le SB, Ghiaseddin AP, Manektalia H, Li M, O’Dell A, et al. Efficacy and safety of adjuvant TTFields plus pembrolizumab and temozolomide in newly diagnosed glioblastoma: A phase 2 study. Med. (2025) 6:100708. doi: 10.1016/j.medj.2025.100708
73. Wu J, Yuan Y, Mentges K, Reyes J, Vera E, Mendoza T, et al. Phase II trial evaluating nivolumab in patients with recurrent IDH-mutant gliomas with and without hypermutation phenotype. J Clin Oncol. (2023) 41:e14043–3. doi: 10.1200/JCO.2023.41.16_SUPPL.E14043
74. Sim HW, Wachsmuth L, Barnes EH, Yip S, Koh ES, Hall M, et al. NUTMEG: A randomized phase II study of nivolumab and temozolomide versus temozolomide alone in newly diagnosed older patients with glioblastoma. Neurooncol Adv. (2023) 5:vdad124. doi: 10.1093/NOAJNL/VDAD124
75. Ahluwalia MS, Peereboom DM, Ozair A, Khosla AA, Rauf Y, Nayak L, et al. A randomized, controlled, phase 2 trial of nivolumab plus standard-dose or low-dose bevacizumab for recurrent glioblastoma (NAVAL). J Clin Oncol. (2024) 42:2072–2. doi: 10.1200/JCO.2024.42.16_SUPPL.2072
76. Lukas R, Sachdev S, Kumthekar P, Dixit K, Grimm S, Gondi V, et al. CTIM-12. A phase 1 trial of immunoradiotherapy with the IDO enzyme inhibitor (BMS-986205) and nivolumab in patients with newly diagnosed mgmt promoter unmethylated IDHwt GLIOBLASTOMA. Neuro Oncol. (2021) 23:vi51. doi: 10.1093/NEUONC/NOAB196.204
77. Bagley SJ, Mathew D, Desai AS, Chen K, Long Q, Shabason JE, et al. PD1 inhibition and GITR agonism in combination with fractionated stereotactic radiotherapy for the treatment of recurrent glioblastoma: A phase 2, multi-arm study. J Clin Oncol. (2023) 41:2004–4. doi: 10.1200/JCO.2023.41.16_SUPPL.2004
78. Campian JL, Butt OH, Huang J, Luo J, Tao Y, Strowd RE, et al. Preliminary results of a phase II study of retifanlimab (PD-1 inhibitor) plus or minus epacadostat (IDO1 inhibitor) in combination with bevacizumab and hypofractionated radiotherapy for recurrent glioblastoma: NCT03532295. J Clin Oncol. (2022) 40:2058–8. doi: 10.1200/JCO.2022.40.16_SUPPL.2058
79. Reardon DA, Brem S, Desai AS, Bagley SJ, Kurz SC, Dela Fuente MI, et al. Intramuscular (IM) INO-5401 + INO-9012 with electroporation (EP) in combination with cemiplimab (REGN2810) in newly diagnosed glioblastoma. J Clin Oncol. (2022) 40:2004–4. doi: 10.1200/JCO.2022.40.16_SUPPL.2004
80. Kim KS, Habashy K, Gould A, Zhao J, Najem H, Amidei C, et al. Fc-enhanced anti-CTLA-4, anti-PD-1, doxorubicin, and ultrasound-mediated blood-brain barrier opening: A novel combinatorial immunotherapy regimen for gliomas. Neuro Oncol. (2024) 26:2044–60. doi: 10.1093/NEUONC/NOAE135
81. Weathers SP, Li X, Zhu H, Damania AV, Knafl M, McKinley B, et al. Improved overall survival in an anti-PD-L1 treated cohort of newly diagnosed glioblastoma patients is associated with distinct immune, mutation, and gut microbiome features: a single arm prospective phase I/II trial. Nat Commun. (2025) 16:3950. doi: 10.1038/S41467-025-56930-7
82. Markus L, Sun L, Hugo W, Van den Abbeele AD, Wen P, and Prins R. IMMU-38. intratumoral correlation of multiplex immunofluorescence AND89ZR-CREFMIRLIMAB berdoxam immunopet in recurrent gbm patients treated with neoadjuvant ANTI-PD-1 +/- ANTI-CTLA-4 therapy. Neuro Oncol. (2023) 25:v150. doi: 10.1093/NEUONC/NOAD179.0570
83. Tiu C, Yau WH, Welsh LC, Jones TL, Zachariou A, Prout T, et al. Abstract CT093: Preliminary evidence of antitumor activity of Ipatasertib (Ipat) and Atezolizumab (A) in glioblastoma (GBM) patients (pts) with PTEN loss in the Phase 1 Ice-CAP trial (NCT03673787). Cancer Res. (2023) 83:CT093–3. doi: 10.1158/1538-7445.AM2023-CT093
84. Dirven I, Lescrauwaet L, Bertels C, Stevens L, Geeraerts X, Vaeyens F, et al. A phase I clinical trial on combined (neo-)adjuvant intravenous plus intracranial administration of ipilimumab and nivolumab in recurrent glioblastoma (NEO-GLITIPNI). J Clin Oncol. (2025) 43:2049–9. doi: 10.1200/JCO.2025.43.16_SUPPL.2049
85. Shmatko A, Jin D, Patel A, Rahmanzade R, Wick W, Krieg S, et al. PL02.1.A locoregional BI-specific CAR-T cells targeting CD44 and CD133 in recurrent glioblastoma: the interim results of the first in-human clinical trial. Neuro Oncol. (2024) 26:v1–2. doi: 10.1093/NEUONC/NOAE144.003
86. Ghiaseddin A, Tao H, Deleyrolle P, Rahman M, Rinonos SZ, Kresak J, et al. CTIM-21. First-in-human phase I clinical trial using 8R-70CAR T cells for newly diagnosed adult glioblastoma. Neuro Oncol. (2024) 26:viii90–0. doi: 10.1093/NEUONC/NOAE165.0354
87. Choi BD, Gerstner ER, Frigault MJ, Leick MB, Mount CW, Balaj L, et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. New Engl J Med. (2024) 390:1290–8. doi: 10.1056/NEJMOA2314390/SUPPL_FILE/NEJMOA2314390_DATA-SHARING.PDF
88. Brown CE, Hibbard JC, Alizadeh D, Blanchard MS, Natri HM, Wang D, et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med Nat Research. (2024) 30:1001–12. doi: 10.1038/S41591-024-02875-1;SUBJMETA=1059,1922,2325,250,251,631,67;KWRD=CANCER+IMMUNOTHERAPY,CNS+CANCER,IMMUNOTHERAPY
89. Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ, et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med. (2024) 30:1320–9. doi: 10.1038/s41591-024-02893-z
90. Bagley SJ, Binder ZA, Fraietta JA, Desai AS, Nabavizadeh A, Marshall A, et al. A phase 1 study of intracerebroventricular (ICV) delivery of bivalent chimeric antigen receptor (CAR) T-cells targeting EGFR and IL13Ra2 in patients with recurrent glioblastoma (rGBM). J Clin Oncol. (2025) 43:102–2. doi: 10.1200/JCO.2025.43.16_SUPPL.102
91. Bertrand KC, Kostecka A, Smith K, Han K, Chiang J, Pinto S, et al. CTIM-15. Locoregional administration OF B7-H3-car T cells expressing 41BB ligand for pediatric patients with primary CNS tumors: preliminary results from a phase i study. Neuro Oncol. (2024) 26:viii88–8. doi: 10.1093/NEUONC/NOAE165.0348
92. Li G, Stocksdale BR, Song K-W, Mahdi J, Tanner K, Bertrand S, et al. A phase 1 study of B7H3 CAR-T cells administered intracranially in recurrent glioblastoma. J Clin Oncol. (2025) 43:2018–8. doi: 10.1200/JCO.2025.43.16_SUPPL.2018
93. Litten JB, Ramakrishnan A, Astrow SH, Harrison C, Aliki A, and Badie B. Phase 1b multicenter study to evaluate CHM 1101 in patients with recurrent or progressive glioblastoma. J Clin Oncol. (2023) 41:TPS2086–TPS2086. doi: 10.1200/JCO.2023.41.16_SUPPL.TPS2086
94. Badie B, Barish ME, Chaudhry A, D’Apuzzo M, Forman SJ, Portnow J, et al. A phase 1 study to evaluate chimeric antigen receptor (CAR) T cells incorporating a chlorotoxin tumor-targeting domain for patients with MMP2+ Recurrent or progressive glioblastoma (NCT04214392). J Clin Oncol. (2021) 39:TPS2662–TPS2662. doi: 10.1200/JCO.2021.39.15_SUPPL.TPS2662
95. Nabors LB, Lamb LS, Beelen MJ, Pillay T, ter Haak M, Youngblood S, et al. Phase 1 trial of drug resistant immunotherapy: A first-in-class combination of MGMT-modified γδ t cells and temozolomide chemotherapy in newly diagnosed glioblastoma. J Clin Oncol. (2021) 39:2057–7. doi: 10.1200/JCO.2021.39.15_SUPPL.2057
96. Ahluwalia MS, Reardon DA, Abad AP, Curry WT, Wong ET, Figel SA, et al. Phase IIa study of surVaxM plus adjuvant temozolomide for newly diagnosed glioblastoma. J Clin Oncol. (2022) 41:1453–65. doi: 10.1200/JCO.22
97. Ahluwalia MS, Ciesielski MJ, Reardon DA, Butowski NA, Aiken R, Venur V, et al. A multicenter, randomized controlled phase 2b trial of survivin vaccine SurVaxM plus adjuvant temozolomide for newly diagnosed glioblastoma (SURVIVE). J Clin Oncol. (2024) 42:TPS2099–TPS2099. doi: 10.1200/JCO.2024.42.16_SUPPL.TPS2099
98. Tabatabai G, Freres P, Glas M, Hau P, Von Baumgarten L, Van Den Bent M, et al. CTIM-10. phase 1 trial of personalized neoantigen vaccines in combination with standard care to treat glioblastoma. Neuro Oncol. (2024) 26:viii86–7. doi: 10.1093/NEUONC/NOAE165.0343
99. Miller A, Kosaloglu-Yalcin Z, Westernberg L, Montero L, Bahmanof M, Frentzen A, et al. A phase 1b study of personalized neoantigen vaccine plus pembrolizumab in adults with advanced cancer. J Clin Oncol. (2021) 39:2615–5. doi: 10.1200/JCO.2021.39.15_SUPPL.2615
100. Rapp M, Grauer OM, Kamp M, Sevens N, Zotz N, Sabel M, et al. A randomized controlled phase II trial of vaccination with lysate-loaded, mature dendritic cells integrated into standard radiochemotherapy of newly diagnosed glioblastoma (GlioVax): Study protocol for a randomized controlled trial. Trials. (2018) 19:293. doi: 10.1186/S13063-018-2659-7
101. Zhang Y, Chi X, Cheng X, Zhang Y, Zhang K, Huang N, et al. Personalized neoantigen-pulsed autologous dendritic cells vaccine as an adjuvant treatment for patients with newly-diagnosed glioblastoma multiforme: A phase I trial. J Clin Oncol. (2024) 42:e14025–5. doi: 10.1200/JCO.2024.42.16_SUPPL.E14025
102. Van Genechten T, De Laere M, Van Den Bossche J, Stein B, De Rycke K, Deschepper C, et al. Adjuvant Wilms’ tumour 1-specific dendritic cell immunotherapy complementing conventional therapy for paediatric patients with high-grade glioma and diffuse intrinsic pontine glioma: protocol of a monocentric phase I/II clinical trial in Belgium. BMJ Open. (2024) 14:e077613. doi: 10.1136/BMJOPEN-2023-077613
103. Von Baumgarten L, Van Den Bent MJ, Burger MC, Freres P, Glas M, Hau P, et al. Phase 1 dose-finding study to evaluate safety and tolerability of CVGBM in patients with newly diagnosed and surgically resected MGMT-unmethylated glioblastoma. J Clin Oncol. (2024) 42:TPS2095–TPS2095. doi: 10.1200/JCO.2024.42.16_SUPPL.TPS2095
104. Estevez-Ordonez D, Stein J, Maleknia P, Gallegos C, Atchley T, Laskay N, et al. CTIM-13. Phase I clinical trial of oncolytic HSV-1 M032, a second-generation virus armed to expressed IL-12, for the treatment of adult patients with recurrent or progressive Malignant glioma. Neuro Oncol. (2023) 25:64–v64. doi: 10.1093/NEUONC/NOAD179.0253
105. Chiocca EA, Solomon I, Nakashima H, Lawler SE, Triggs D, Zhang A, et al. First-in-human CAN-3110 (ICP-34.5 expressing HSV-1 oncolytic virus) in patients with recurrent high-grade glioma. J Clin Oncol. (2021) 39:2009–9. doi: 10.1200/JCO.2021.39.15_SUPPL.2009
106. Bernstock JD, Bag AK, Fiveash J, Kachurak K, Elsayed G, Chagoya G, et al. Design and rationale for first-in-human Phase 1 Immunovirotherapy clinical trial of oncolytic HSV G207 to treat Malignant pediatric cerebellar brain tumors. Hum Gene Ther. (2020) 31:1132. doi: 10.1089/HUM.2020.101
107. Portnow J, Badie B, Blanchard S, D’Apuzzo M, Nath A, Synold TW, et al. Phase 1 study of multiple intracerebral doses of a neural stem cell-based oncolytic virotherapy for treatment of recurrent high-grade gliomas. J Clin Oncol. (2024) 42:TPS2102–TPS2102. doi: 10.1200/JCO.2024.42.16_SUPPL.TPS2102
108. Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. New Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMOA1716435/SUPPL_FILE/NEJMOA1716435_DISCLOSURES.PDF
109. Weiss T, Puca E, Covelli A, Gramatzki D, Roth P, Neri D, et al. CTIM-22. Phase I study results of the antibody-cytokine fusion protein L19TNF in combination with lomustine for patients with recurrent glioblastoma reveal promising safety and durable tumor responses. Neuro Oncol. (2023) 25:v66. doi: 10.1093/NEUONC/NOAD179.0262
110. Eoli M, Gentner B, Anghileri E, Farina F, Capotondo A, D’Alessandris G, et al. P11.19. A macrophages-based immunotherapy of solid tumors microenvironment: the TEM-GBM study (NCT03866109). Neuro Oncol. (2023) 25:ii76. doi: 10.1093/NEUONC/NOAD137.253
111. Bagley S, Polley M-Y, Kotecha R, Brem S, Tolakanahalli R, Iwamoto F, et al. CTIM-21. NRG-BN010: a safety run-in and phase II study evaluating the combination of tocilizumab, atezolizumab, and fractionated stereotactic radiotherapy in recurrent glioblastoma – trial in progress. Neuro Oncol. (2022) 24:vii64–4. doi: 10.1093/NEUONC/NOAC209.253
112. Wong ET, Cielo D, Svokos K, Doberstein C, Sampath P, Donahue JE, et al. IGV-001 cellular immunotherapy for newly diagnosed glioblastoma: overcoming the logistic challenge. Front Oncol. (2025) 15:1556450/PDF. doi: 10.3389/FONC.2025.1556450/PDF
113. Johnson TS, Pacholczyk R, Ring EK, Aguilera D, Castellino RC, Fangusaro JR, et al. IMMU-09. Phase 2 trial using indoximod-based chemo-immunotherapy for patients with childhood brain cancer: interim analysis of the GCC1949 study (NCT04049669). Neuro Oncol. (2024) 26:0. doi: 10.1093/NEUONC/NOAE064.380
114. Wang JS, Sommerhalder D, Sharma M, Edenfield WJ, Sehgal K, Oh D-Y, et al. Phase 1/2 study of NGM707, an ILT2/ILT4 dual antagonist antibody, in advanced solid tumors: Interim results from dose-escalation. J Clin Oncol. (2024) 42:2582–2. doi: 10.1200/JCO.2024.42.16_SUPPL.2582
115. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012). doi: 10.1038/nrc3239
116. He X and Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. (2020). 30:660–9. doi: 10.1038/s41422-020-0343-4
117. Hudson K, Cross N, Jordan-Mahy N, and Leyland R. The extrinsic and intrinsic roles of PD-L1 and its receptor PD-1: implications for immunotherapy treatment. Front Immunol. (2020) 11:568931. doi: 10.3389/FIMMU.2020.568931
118. Marin-Acevedo JA, Kimbrough EMO, and Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol. (2021) 14:1. doi: 10.1186/S13045-021-01056-8
119. Wang H, Yang J, Li X, and Zhao H. Current state of immune checkpoints therapy for glioblastoma. Heliyon. (2024) 10:e24729. doi: 10.1016/J.HELIYON.2024.E24729
120. Peggs KS, Quezada SA, Korman AJ, and Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol Elsevier Curr Trends. (2006) 18:206–13. doi: 10.1016/J.COI.2006.01.011
121. Ziogas DC, Theocharopoulos C, Lialios PP, Foteinou D, Koumprentziotis IA, Xynos G, et al. Beyond CTLA-4 and PD-1 inhibition: novel immune checkpoint molecules for melanoma treatment. Cancers (Basel). (2023) 15:2718. doi: 10.3390/CANCERS15102718
122. Acharya N, Sabatos-Peyton C, and Carrizosa Anderson A. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. (2020) 8:911. doi: 10.1136/jitc-2020-000911
123. Xu S, Zhang N, Rinne ML, Sun H, and Stein AM. Sabatolimab (MBG453) model-informed drug development for dose selection in patients with myelodysplastic syndrome/acute myeloid leukemia and solid tumors. CPT Pharmacometrics Syst Pharmacol. (2023) 12:1653–65. doi: 10.1002/PSP4.12962
124. Rousseau A, Parisi C, and Barlesi F. Anti-TIGIT therapies for solid tumors: a systematic review. ESMO Open. (2023) 8:101184. doi: 10.1016/J.ESMOOP.2023.101184
125. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
126. Ge Z, Peppelenbosch MP, Sprengers D, and Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol. (2021) 12:699895. doi: 10.3389/FIMMU.2021.699895
127. Wojas-Krawczyk K, Kalinka E, Grenda A, Krawczyk P, and Milanowski J. Beyond PD-L1 markers for lung cancer immunotherapy. Int J Mol Sci. (2019) 20:1915. doi: 10.3390/IJMS20081915
128. Senhaji N, Houssaini AS, Lamrabet S, Louati S, and Bennis S. Molecular and circulating biomarkers in patients with glioblastoma. Int J Mol Sci. (2022) 23:7474. doi: 10.3390/IJMS23137474
129. Seyhan AA. Circulating liquid biopsy biomarkers in glioblastoma: advances and challenges. Int J Mol Sci. (2024) 25:7974. doi: 10.3390/IJMS25147974
130. Long GV, Shklovskaya E, Satgunaseelan L, Mao Y, da Silva IP, Perry KA, et al. Neoadjuvant triplet immune checkpoint blockade in newly diagnosed glioblastoma. Nat Med. (2025) 31:1557–66. doi: 10.1038/S41591-025-03512-1;SUBJMETA=1922,580,631,67;KWRD=CNS+CANCER,TUMOUR+IMMUNOLOGY
131. Utkarsh K, Srivastava N, Kumar S, Khan A, Dagar G, Kumar M, et al. CAR-T cell therapy: a game-changer in cancer treatment and beyond. Clin Trans Oncol. (2024) 26:6. doi: 10.1007/S12094-023-03368-2
132. Luksik AS, Yazigi E, Shah P, and Jackson CM. CAR T cell therapy in glioblastoma: overcoming challenges related to antigen expression. Cancers. (2023) 15:663. doi: 10.3390/cancers15051414
133. Chen YJ, Abila B, and Mostafa Kamel Y. CAR-T: what is next? Cancers (Basel). (2023) 15:663. doi: 10.3390/CANCERS15030663
134. Chmielewski M, Hombach AA, and Abken H. Antigen-specific T-cell activation independently of the MHC: chimeric antigen receptor-redirected T cells. Front Immunol. (2013) 4:371. doi: 10.3389/FIMMU.2013.00371
135. Ma K and Hu P. Chimeric antigen receptor T-cell therapy for glioblastoma. Cancers (Basel). (2023) 15:371. doi: 10.3390/CANCERS15235652
136. Zhao Z, Li Y, Liu W, and Li X. Engineered IL-7 receptor enhances the therapeutic effect of AXL-CAR-T cells on triple-negative breast cancer. BioMed Res Int. (2020) 2020:4795171. doi: 10.1155/2020/4795171
137. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
138. Brown DV, Filiz G, Daniel PM, Hollande F, Dworkin S, Amiridis S, et al. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PloS One. (2017) 12:e0172791. doi: 10.1371/JOURNAL.PONE.0172791
139. Infanger DW, Cho Y, Lopez BS, Mohanan S, Liu SC, Gursel D, et al. Tumor and stem cell biology glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. (2013) 73:7079. doi: 10.1158/0008-5472.CAN-13-1355
140. Brat DJ, Bellail AC, and Van Meir EG. Neuro-Oncology The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis 1. Neuro Oncol. (2005) 7:4–106. doi: 10.1215/S1152851704001061
141. Dwyer J, Hebda JK, Guelte L, Galan-Moya A, and Smith E-MS. Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PLoS One. (2012) 7:45562. doi: 10.1371/journal.pone.0045562
142. Hasan T, Caragher SP, Shireman JM, Park CH, Atashi F, Baisiwala S, et al. Interleukin-8/CXCR2 signaling regulates therapy-induced plasticity and enhances tumorigenicity in glioblastoma. Cell Death Dis. (2019) 10:4. doi: 10.1038/s41419-019-1387-6
143. Goutnik M, Iakovidis A, Still MEH, Moor RSF, Melnick K, Yan S, et al. Advancements in chimeric antigen receptor-expressing T-cell therapy for glioblastoma multiforme: Literature review and future directions. Neurooncol Adv. (2024) 6:vdae025. doi: 10.1093/NOAJNL/VDAE025
144. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1-or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
145. Zeng J, Zhang J, Yang YZ, Wang F, Jiang H, Chen HD, et al. IL13RA2 is overexpressed in Malignant gliomas and related to clinical outcome of patients. Am J Transl Res. (2020) 12:4702.
146. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One. (2013) 8:e77769. doi: 10.1371/JOURNAL.PONE.0077769
147. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, and Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev. (2019) 290:60–84. doi: 10.1111/IMR.12773
148. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther. (2018) 26:31. doi: 10.1016/J.YMTHE.2017.10.002
149. Vinay DS and Kwon BS. 4-1BB (CD137), an inducible costimulatory receptor, as a specific target for cancer therapy. BMB Rep. (2014) 47:122. doi: 10.5483/BMBREP.2014.47.3.283
150. Philipson BI, O’Connor RS, May MJ, June CH, Albelda SM, and Milone MC. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci Signal. (2020) 13:eaay8248. doi: 10.1126/SCISIGNAL.AAY8248
151. Hammer O. CD19 as an attractive target for antibody-based therapy. MAbs. (2012) 4:571. doi: 10.4161/MABS.21338
152. Park JH, Geyer MB, and Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic Malignancies: interpreting clinical outcomes to date. Blood. (2016) 127:3312–20. doi: 10.1182/BLOOD-2016-02-629063
153. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404. doi: 10.1158/2159-8290.CD-17-0698
154. Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell. (2020) 183:126. doi: 10.1016/J.CELL.2020.08.022
155. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMOA1610497
156. Xu C, Bai Y, An Z, Hu Y, Zhang C, and Zhong X. IL-13Rα2 humanized scFv-based CAR-T cells exhibit therapeutic activity against glioblastoma. Mol Ther Oncolytics. (2022) 24:443–51. doi: 10.1016/J.OMTO.2022.01.002
157. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. (2020) 5:e133977. doi: 10.1172/JCI.INSIGHT.133977
158. Ramezani M, Siami S, Rezaei M, Khazaei S, and Sadeghi M. An immunohistochemical study of HER2 expression in primary brain tumors. Biomedicine (Taipei. (2020) 10:21. doi: 10.37796/2211-8039.1001
159. Ezzati S, Salib S, Balasubramaniam M, and Aboud O. Epidermal growth factor receptor inhibitors in glioblastoma: current status and future possibilities. Int J Mol Sci. (2024) 25:2316. doi: 10.3390/IJMS25042316
160. Xu H, Zong H, Ma C, Ming X, Shang M, Li K, et al. Epidermal growth factor receptor in glioblastoma. Oncol Lett. (2017) 14:512. doi: 10.3892/OL.2017.6221
161. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. (2021) 13:eabe7378. doi: 10.1126/SCITRANSLMED.ABE7378
162. Wykosky J, Gibo DM, Stanton C, and Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. (2005) 3:541–51. doi: 10.1158/1541-7786.MCR-05-0056
163. Zhang C, Zhang Z, Li F, Shen Z, Qiao Y, Li L, et al. Large-scale analysis reveals the specific clinical and immune features of B7-H3 in glioma. Oncoimmunology. (2018) 7:e1461304. doi: 10.1080/2162402X.2018.1461304
164. Picarda E, Ohaegbulam KC, and Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. (2016) 22:3425–31. doi: 10.1158/1078-0432.CCR-15-2428
165. Li S, Zhang M, Wang M, Wang H, Wu H, Mao L, et al. B7-H3 specific CAR-T cells exhibit potent activity against prostate cancer. Cell Death Discov. (2023) 9:147. doi: 10.1038/s41420-023-01453-7
166. Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics. (2019) 14:279–87. doi: 10.1016/J.OMTO.2019.07.002
167. Zhang Y, Feng R, Chi X, Xian N, Chen X, Huang N, et al. Safety and efficacy of B7-H3 targeting CAR-T cell therapy for patients with recurrent GBM. Am Soc Clin Oncol. (2024) 42:2062–2. doi: 10.1200/JCO20244216_suppl2062
168. Vitanza NA, Wilson AL, Huang W, Seidel K, Brown C, Gustafson JA, et al. Intraventricular B7-H3 CAR T cells for diffuse intrinsic pontine glioma: preliminary first-in-human bioactivity and safety. Cancer Discov. (2023) 13:114–31. doi: 10.1158/2159-8290.CD-22-0750/709786/AM/INTRAVENTRICULAR-B7-H3-CAR-T-CELLS-FOR-DIFFUSE
169. Sakunrangsit N, Khuisangeam N, Inthanachai T, Yodsurang V, Taechawattananant P, Suppipat K, et al. Incorporating IL7 receptor alpha signaling in the endodomain of B7H3-targeting chimeric antigen receptor T cells mediates antitumor activity in glioblastoma. Cancer Immunol Immunother. (2024) 73:98. doi: 10.1007/s00262-024-03685-7
170. Lyons SA, O’Neal J, and Sontheimer H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia. (2002) 39:162–73.
171. Deshane J, Garner CC, and Sontheimer H. Chlorotoxin Inhibits Glioma Cell Invasion via Matrix Metalloproteinase-2. J Biol Chem. (2002) 278:4135–44. doi: 10.1074/jbc.M205662200
172. Zhao Y, Dong P, He W, Zhang J, and Chen H. γδ T cells: Major advances in basic and clinical research in tumor immunotherapy. Chin Med J (Engl). (2024) 137:21. doi: 10.1097/CM9.0000000000002781
173. Chen Y, Li J, Zeng X, Yuan W, and Xu Y. γδ T cells and their roles in immunotherapy: a narrative review. Ann Blood. (2022) 7. doi: 10.21037/AOB-21-33/COIF
174. Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer. (2019) 18 :29. doi: 10.1186/s12943-019-0956-8
175. Lobbous M, Goswami T, Lamb LS, Rochlin K, Pillay T, Youngblood S, et al. INB-200 phase I study of gene modified autologous gamma-delta (γδ) T cells in patients with newly diagnosed glioblastoma multiforme (GBM) receiving maintenance temozolomide (TMZ). J Clin Neurosci. (2023) 42:2007. doi: 10.1200/JCO.2023.41.16_SUPPL.2007
176. Xiong Z, Raphael I, Olin M, Okada H, Li X, and Kohanbash G. Glioblastoma vaccines: past, present, and opportunities. EBioMedicine. (2024) 100. doi: 10.1016/J.EBIOM.2023.104963/ASSET/92417CDD-1178-4475-9C46-B46EE72AC983/MAIN.ASSETS/GR1.JPG
177. Squalli Houssaini A, Lamrabet S, Nshizirungu JP, Senhaji N, Sekal M, Karkouri M, et al. Glioblastoma vaccines as promising immune-therapeutics: challenges and current status. Vaccines. (2024) 12:655. doi: 10.3390/VACCINES12060655
178. Yang T, Shi Y, Liang T, Xing H, Ma W, Michael Li Y, et al. Peptide vaccine against glioblastoma: from bench to bedside. Holistic Integr Oncol. (2022) 1:21. doi: 10.1007/s44178-022-00021-w
179. Tong X, Yang P, Wang K, Liu Y, Liu X, Shan X, et al. Survivin is a prognostic indicator in glioblastoma and may be a target of microRNA-218. Oncol Lett. (2019) 18:359. doi: 10.3892/OL.2019.10335
180. Thompson E, Ashley DM, Ayasoufi K, Norberg P, Archer GE, Buckley E, et al. Outcomes and immune response after peptide vaccination targeting human cytomegalovirus antigen pp65 in children and young adults with recurrent high-grade glioma and medulloblastoma. J Clin Oncol. (2024) 42:2039–9. doi: 10.1200/JCO.2024.42.16_SUPPL.2039
181. Chih Y-C, Dietsch AC, Koopmann P, Ma X, Agardy DA, Zhao B, et al. Vaccine-induced T cell receptor T cell therapy targeting a glioblastoma stemness antigen. Nat Commun. (2025) 16:1262. doi: 10.1038/S41467-025-56547-W
182. Aquilanti E, Kageler L, Wen PY, and Meyerson M. Telomerase as a therapeutic target in glioblastoma. Neuro Oncol. (2021) 23:2004. doi: 10.1093/NEUONC/NOAB203
183. Olympios N, Gilard V, Marguet F, Clatot F, Di Fiore F, and Fontanilles M. TERT promoter alterations in glioblastoma: A systematic review. Cancers (2021) 13:1147. doi: 10.3390/cancers13051147
184. Xun Y, Yang H, Kaminska B, and You H. Toll-like receptors and toll-like receptor-targeted immunotherapy against glioma. J Hematol Oncol. (2021) 14:176. doi: 10.1186/S13045-021-01191-2
185. Van Gool SW, Makalowski J, Kampers LFC, Van de Vliet P, Sprenger T, Schirrmacher V, et al. Dendritic cell vaccination for glioblastoma multiforme patients: has a new milestone been reached? Transl Cancer Res. AME Publishing Company. (2023) 12:2224–8. doi: 10.21037/TCR-23-603
186. Hoffman J, Beutler B, Palucka K, and Banchereau J. Innate immune system Cancer immunotherapy via dendritic cells. Nat Rev Cancer. (2012) 12:265–277. doi: 10.1038/nrc3258
187. Dai S, Wang H, and Deng F. Advances and challenges in enveloped virus-like particle (VLP)-based vaccines. J Immunol Sci. (2018) 2:36–41. doi: 10.29245/2578-3009/2018/2.1118
188. Deyhimfar R, Izady M, Shoghi M, Kazazi MH, Ghazvini ZF, Nazari H, et al. The clinical impact of mRNA therapeutics in the treatment of cancers, infections, genetic disorders, and autoimmune diseases. Heliyon. (2024) 10:e26971. doi: 10.1016/J.HELIYON.2024.E26971
189. Gunasegaran B, Ashley CL, Marsh-Wakefield F, Guillemin GJ, and Heng B. Viruses in glioblastoma: an update on evidence and clinical trials. BJC Rep. (2024) 2:33. doi: 10.1038/s44276-024-00051-z
190. Shalhout SZ, Miller DM, Emerick KS, and Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. (2023) 20:3. doi: 10.1038/s41571-022-00719-w
191. Zolaly MA, Mahallawi W, Khawaji ZY, and Alahmadi MA. The clinical advances of oncolytic viruses in cancer immunotherapy. Cureus. (2023) 15. doi: 10.7759/CUREUS.40742
192. Hamad A, Yusubalieva GM, Baklaushev VP, Chumakov PM, and Lipatova AV. Recent developments in glioblastoma therapy: oncolytic viruses and emerging future strategies. Viruses. (2023) 15. doi: 10.3390/V15020547
193. Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I, et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. New Engl J Med. (2021) 384:1613–22. doi: 10.1056/NEJMOA2024947/SUPPL_FILE/NEJMOA2024947_DATA-SHARING.PDF
194. Cassady KA. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol. (2005) 79:8707–15. doi: 10.1128/JVI.79.14.8707-8715.2005
195. Webb MJ, Sener U, and Vile RG. Current status and challenges of oncolytic virotherapy for the treatment of glioblastoma. Pharmaceuticals. (2023) 16:793. doi: 10.3390/PH16060793
196. Ling AL, Solomon IH, Landivar AM, Nakashima H, Woods JK, Santos A, et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature. (2023) 623:157–66. doi: 10.1038/S41586-023-06623-2
197. Nassiri F, Patil V, Yefet LS, Singh O, Liu J, Dang RMA, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. (2023) 29:6. doi: 10.1038/s41591-023-02347-y
198. Fares J, Ahmed AU, Ulasov IV, Sonabend AM, Miska J, Lee-Chang C, et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed Malignant glioma: a first-in-human, phase 1, dose-escalation trial. Lancet Oncol. (2021) 22:1103–14. doi: 10.1016/S1470-2045(21)00245-X
199. Wang J, Zhang J, Zhang Q, Zhang W, Zhang Q, Jin G, et al. TS-2021, a third-generation oncolytic adenovirus that carried Ki67 promoter, TGF-β2 5′UTR, and IL–15 against experimental glioblastoma. J Med Virol. (2024) 96:e29335. doi: 10.1002/JMV.29335
200. Holl EK, Brown MC, Boczkowski D, McNamara MA, George DJ, Bigner DD, et al. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget. (2016) 7:79828. doi: 10.18632/ONCOTARGET.12975
201. Cassady KA, Bauer DF, Roth J, Chambers MR, Shoeb T, Coleman J, et al. Pre-clinical assessment of C134, a chimeric oncolytic herpes simplex virus, in mice and non-human primates. Mol Ther Oncolytics. (2017) 5:1. doi: 10.1016/J.OMTO.2017.02.001
202. Song K. Current development status of cytokines for cancer immunotherapy. Biomol Ther (Seoul). (2024) 32:13. doi: 10.4062/BIOMOLTHER.2023.196
203. Bunse L, Kratzmann M, Chih Y, Sanghvi K, De Roia A, Harbottle R, et al. P06.08.A clinical outcome and translational insights from a phase I/II trial with the tumor-targeting antibody-cytokine fusion protein l19TNF for patients with recurrent glioblastoma. Neuro Oncol. (2023) 25:ii46–7. doi: 10.1093/NEUONC/NOAD137.148
204. Finocchiaro G, Gentner B, Farina F, Capotondo A, Eoli M, Anghileri E, et al. A phase I-IIa study of genetically modified Tie-2 expressing monocytes in patients with glioblastoma multiforme (TEM-GBM Study). J Clin Oncol. (2021) 39:2532–2. doi: 10.1200/JCO.2021.39.15_SUPPL.2532
205. Lee IY, Hanft S, Schulder M, Judy KD, Wong ET, Elder JB, et al. Autologous cell immunotherapy (IGV-001) with IGF-1R antisense oligonucleotide in newly diagnosed glioblastoma patients. Future Oncol Future Oncol. (2024) 20:579–91. doi: 10.2217/FON-2023-0702
206. Zamykal M, Martens T, Matschke J, Gunther HS, Kathagen A, Schulte A, et al. Inhibition of intracerebral glioblastoma growth by targeting the insulin-like growth factor 1 receptor involves different context-dependent mechanisms. Neuro Oncol. (2015) 17:1076–85. doi: 10.1093/NEUONC/NOU344
207. Lerouge L, Ruch A, Pierson J, Thomas N, and Barberi-Heyob M. Non-targeted effects of radiation therapy for glioblastoma. Heliyon. (2024) 10. doi: 10.1016/J.HELIYON.2024.E30813
208. Fox E, Oliver T, Rowe M, Thomas S, Zakharia Y, Gilman PB, et al. Indoximod: an immunometabolic adjuvant that empowers T cell activity in cancer. Front Oncol. (2018) 8:370. doi: 10.3389/FONC.2018.00370
209. Chamie K, Chang SS, Gonzalgo M, Kramolowsky EV, Sexton WJ, Bhar P, et al. Final clinical results of pivotal trial of IL-15RαFc superagonist N-803 with BCG in BCG-unresponsive CIS and papillary nonmuscle-invasive bladder cancer (NMIBC). J Clin Oncol. (2022) 40:4508–8. doi: 10.1200/JCO.2022.40.16_SUPPL.4508
210. Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J Immunother Cancer. (2020) 8:e000450. doi: 10.1136/JITC-2019-000450
Keywords: immunotherapy, glioblastoma, immunosuppressive tumor microenvironment, drug resistance, immune checkpoint inhibitors
Citation: Nicolaou N, Andreou MS, Neophytou CM and Papageorgis P (2025) Emerging insights into the immunosuppressive tumor microenvironment and its implications for glioblastoma immunotherapy. Front. Immunol. 16:1665742. doi: 10.3389/fimmu.2025.1665742
Received: 14 July 2025; Accepted: 18 September 2025;
Published: 24 October 2025.
Edited by:
Raul Silva García, Mexican Social Security Institute, MexicoReviewed by:
Angeliki Datsi, University Hospital of Düsseldorf, GermanyKecheng Huang, Huazhong University of Science and Technology, China
Copyright © 2025 Nicolaou, Andreou, Neophytou and Papageorgis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis Papageorgis, cC5wYXBhZ2Vvcmdpc0BldWMuYWMuY3k=
†These authors have contributed equally to this work