 Andrea Spinazzola1
Andrea Spinazzola1 Giovanni Maria Iannantuono1
Giovanni Maria Iannantuono1 James L. Gulley2Elena Giudice3
James L. Gulley2Elena Giudice3 Marco Filetti1Stefano Sganga1Francesca Lo Bianco1
Marco Filetti1Stefano Sganga1Francesca Lo Bianco1 Charalampos S. Floudas2
Charalampos S. Floudas2 Gennaro Daniele1*
Gennaro Daniele1*- 1Phase 1 Unit, Fondazione Policlinico Universitario A. Gemelli, Istituto di ricovero e cura a carattere scientifico (IRCCS), Rome, Italy
- 2Center for Immuno-Oncology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
- 3Division of Gynecologic Oncology, Humanitas San Pio X, Milan, Italy
T-cell engagers (TCEs) are an emerging class of immunotherapeutic agents designed to harness the immune system to target and eliminate cancer cells. These molecules bridge T lymphocytes with tumor cells, generating an immunologic synapse that leads to potent immune-mediated tumor destruction. Although the clinical activity of TCEs in patients with solid tumors remains insufficient, recent technological advancements have led to the development of several candidates in early-phase clinical trials, with some showing encouraging signs of efficacy. This review examines the current landscape of TCEs in early clinical development for the treatment of solid tumors, describing their mechanism, clinical progress, efficacy, and challenges.
1 Introduction
Immunotherapies have revolutionized the approach to cancer treatment, with numerous strategies pursued over the last two decades. The core concept of immunotherapies is harnessing, through various means, the immune system of the host to eradicate the tumor. To date, the primary focus of cancer immunotherapy has been on T lymphocytes, which belong to the adaptive immune system and possess direct cytotoxicity against tumor cells (1–3).
A critical aspect of an efficient T cell-mediated response against cancer is the presence of tumor-associated antigens (TAAs) that T lymphocytes can recognize and react to. These typically include neoantigens, which are newly formed antigens that derive from mutations in the DNA. Other relevant types of TAAs encompass proteins whose expression is physiologically restricted to fetal development (known as oncofetal antigens), proteins that in the adult are expressed almost exclusively in the testes (known as cancer-testis antigens: CTAs), and proteins that are physiologically expressed by adult tissues but overexpressed by tumor cells (4, 5).
The most successful applications of immunotherapies involve monoclonal antibodies that block immunomodulatory molecules known as immune checkpoints, as well as chimeric antigen receptor-T (CAR-T) cells, which consist of engineered autologous T cells expressing a synthetic receptor against a TAA. While CAR-T cells offer a targeted therapeutic approach against cancer, immune checkpoint inhibitors (ICIs) work in part by inducing a robust generalized activation of T cells. Additional strategies that have been comprehensively explored, with mixed results, include vaccines targeting TAAs and the use of tumor-infiltrating lymphocytes (TILs) (1). Although immunotherapies have proven highly effective in prolonging the survival of patients with cancer, this benefit is generally restricted to only a subset of patients and to specific cancer types, and the extent of survival gain varies depending on the indication.
T-cell engagers (TCEs) function by simultaneously binding one or more TAAs and the CD3ϵ subunit of the T-cell receptor (TCR) complex expressed by T lymphocytes. This engagement redirects T cells to cancer cells, with TCEs bridging an effective immune synapse independently of the epitope specificity of the lymphocyte. As a result, T cells are activated and promoted to proliferate, produce cytokines, and selectively kill tumor cells through the release of perforin, which induces the formation of pores in the plasma membrane, along with granzymes, a family of serine proteases that cleave various intracellular proteins to induce cell death through apoptosis (6) (Figure 1A). A key feature of this process is that, unlike the antitumor immune response that occurs naturally or after treatment with ICIs, cancer vaccines, or TILs, it does not depend on antigen recognition on major histocompatibility complex (MHC) molecules. This enables T cells to attack cancer cells that do not express MHC molecules or whose TAAs are not efficiently mounted on MHC molecules, both of which are mechanisms that contribute to tumor immune evasion and resistance to immunotherapies (7, 8). Additionally, in contrast to ICIs and cancer vaccines, TCEs induce T cell-mediated lysis of tumor cells without requiring activation by antigen-presenting cells or the engagement of costimulatory molecules, and their activity does not depend on the epitope specificity of the TCR. Accordingly, TCEs present several advantages over traditional immunotherapies, offering a specific, effective, and immediate approach to enhance antitumor immunity that bypasses different steps of the complex process of T-cell activation against cancer cells.
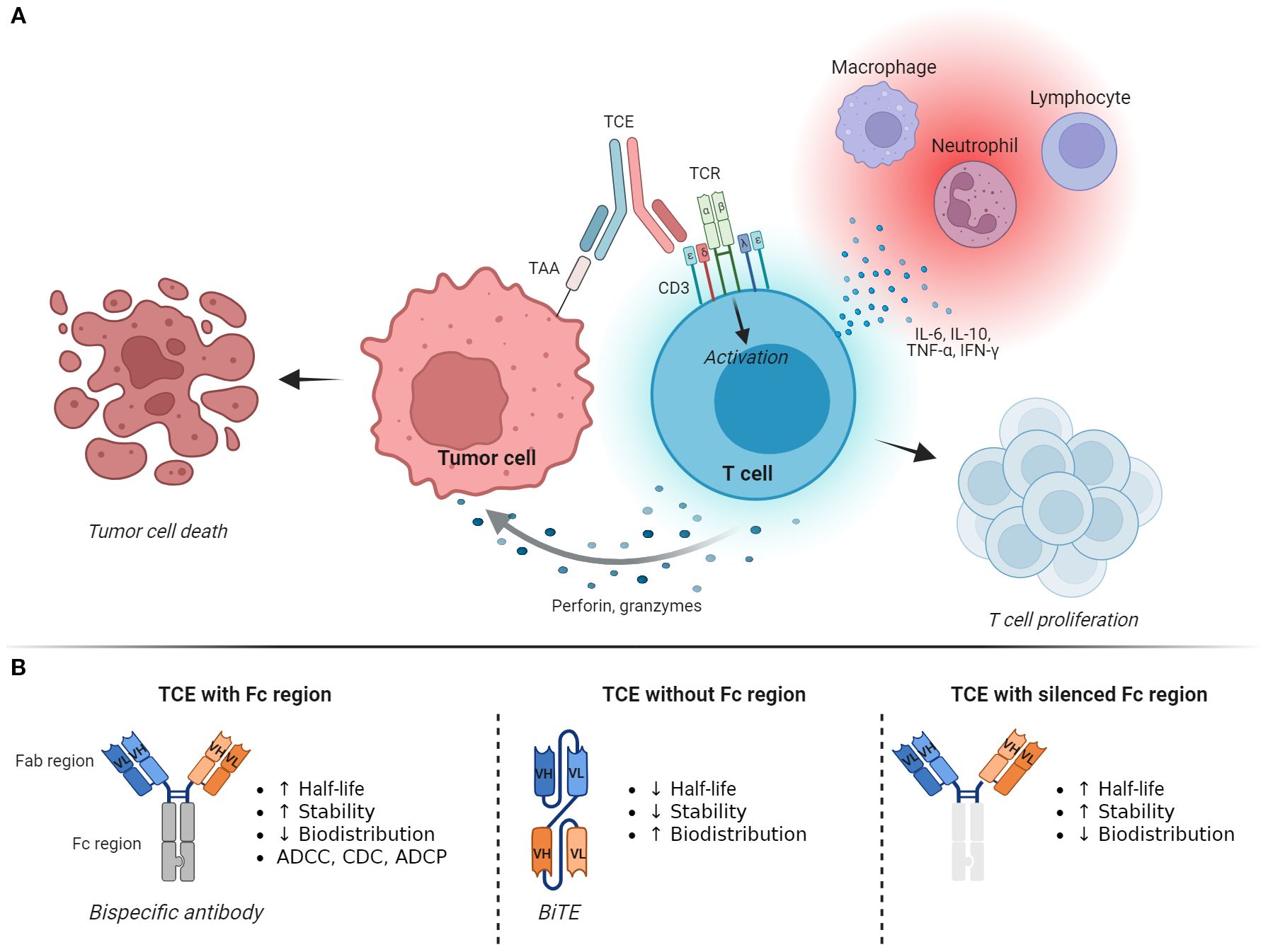
Figure 1. Mechanism of action and structure of T-cell engagers. (A) T-cell engagers (TCEs) generate an immunological synapse between tumor cells and T lymphocytes by binding a tumor-associated antigen (TAA) and the CD3ϵ subunit of the T-cell receptor (TCR) complex. This leads to T-cell activation, proliferation, and killing of the tumor cell through the release of perforin and granzymes. Activated T cells also produce and secrete various cytokines that induce inflammation and recruitment of other immune cells. (B) The basic structure of a TCE is represented by a bispecific antibody, which can be designed as an IgG-like full-length antibody that includes the Fc domain, as a fragment-based subtype that lacks the Fc region, or as a molecule with a silenced/effectorless Fc region. These changes confer different functional characteristics to the TCE, affecting its half-life, biodistribution, toxicity profile, and potentially its antitumor activity. Image created with Biorender.com. ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cell-mediated phagocytosis; BiTE, bispecific T-cell engager; CD3, cluster of differentiation 3; CDC, complement-dependent cytotoxicity; Fab region, fragment antigen-binding region; Fc region, fragment crystallizable region; IFN-γ, interferon-gamma; IL-6, interleukin-6; IL-10, interleukin-10; TAA, tumor-associated antigens; TCE, T-cell engager; TCR, T-cell receptor complex; TNF-α, tumor necrosis factor-alpha.
The field of TCEs is rapidly advancing, with numerous new molecules entering clinical trials. However, in solid tumors, the development of many promising agents of this class is often discontinued due to the lack of meaningful activity, leading to a situation where most TCEs do not proceed to the late stages of clinical development (9). Accordingly, we provide an updated description of the current landscape of TCEs undergoing early-phase clinical evaluation in solid tumors, with a focus on efficacy data. We identified relevant phase 1 and 2 clinical trials on ClinicalTrials.gov and PubMed. Clinical data were obtained from published articles, abstract presentations at international conferences, and press releases from the sponsors, with a cutoff date of April 30, 2025. We prioritized TCEs that are currently undergoing clinical development or whose clinical trial results have been published within the last 5 years. For ease of narrative, we grouped the different TCEs based on the specific category of the TAAs they target.
2 TCEs under clinical evaluation in solid tumors
2.1 Structural and functional aspects
The basic structure of a TCE consists of a bispecific antibody (BsAb), an engineered artificial antibody that simultaneously binds two different antigens. As such, TCEs can be designed as IgG-like full-length antibodies that include the Fc domain or as fragment-based subtypes that lack an Fc region, such as bispecific T-cell engagers (BiTEs), nanobodies, and diabodies (6, 10, 11).
Functionally, these changes confer different characteristics that can be harnessed to optimize the design of the drug (Figure 1B). The presence of the Fc fragment confers a longer half-life, higher stability, and the potential for interactions with complement proteins and Fc receptors on innate immune cells. These interactions may enhance the antitumor effect of the drug through antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cell-mediated phagocytosis (ADCP). However, this design also carries disadvantages, such as its large size, which may limit tissue penetration and distribution, and possible adverse events (AEs) due to TAA-independent T-cell activation and ADCC and CDC toward the T cells (12, 13). On the other hand, TCEs that do not contain the Fc region exhibit better tissue penetration at the cost of lower stability and shorter plasma half-life. They also lack the potential antitumor effect related to ADCC, CDC, and ADCP. Molecules with silenced Fc fragments have been developed to abolish Fc-receptor binding and complement activation while retaining stability and extended half-life.
Additional strategies and innovative designs are being explored to improve patient safety and enhance antitumor activity. For instance, certain TCEs are administered as prodrugs that require proteolytic cleavage to unmask one or both of their binding domains (14). This technology leverages the high level of proteases typically found in the tumor microenvironment (TME), localizing the treatment in the tumor while remaining inactive in healthy tissues. Multispecific TCEs can bind to multiple TAAs or different epitopes of the same TAA, thereby increasing tumor selectivity and sensitivity. Alternatively, they can contain domains that engage ligands and receptors involved in T-cell activation, further stimulating antitumor immunity (15). Finally, TCEs may contain an additional binding domain for human serum albumin (HSA), which extends their half-life in the bloodstream (16).
The majority of available TCEs target TAAs that are localized on the cell membrane. This limits their applications and efficacy in solid tumors for two reasons. First, most TAAs are expressed as intracellular proteins, which can be exposed on the surface only as peptide fragments bound to MHC molecules. Secondly, many cell membrane-bound TAAs are also expressed by normal cells, which can result in on-target off-tumor toxicity (6, 17). To overcome these obstacles, various strategies have been pursued, most notably the development of molecules that combine a soluble TCR-targeting domain with an anti-CD3 effector domain, such as ImmTAC and TCER biologics. In contrast to classical TCEs, these agents redirect T cells toward tumor cells expressing a peptide fragment of the antigen presented by specific MHC class I molecules (18, 19).
Table 1 provides an overview of the TCEs currently undergoing clinical development in solid tumors. Figure 2 displays these agents based on their clinical indication, while Figure 3 illustrates the TAAs that have been attempted as targets.
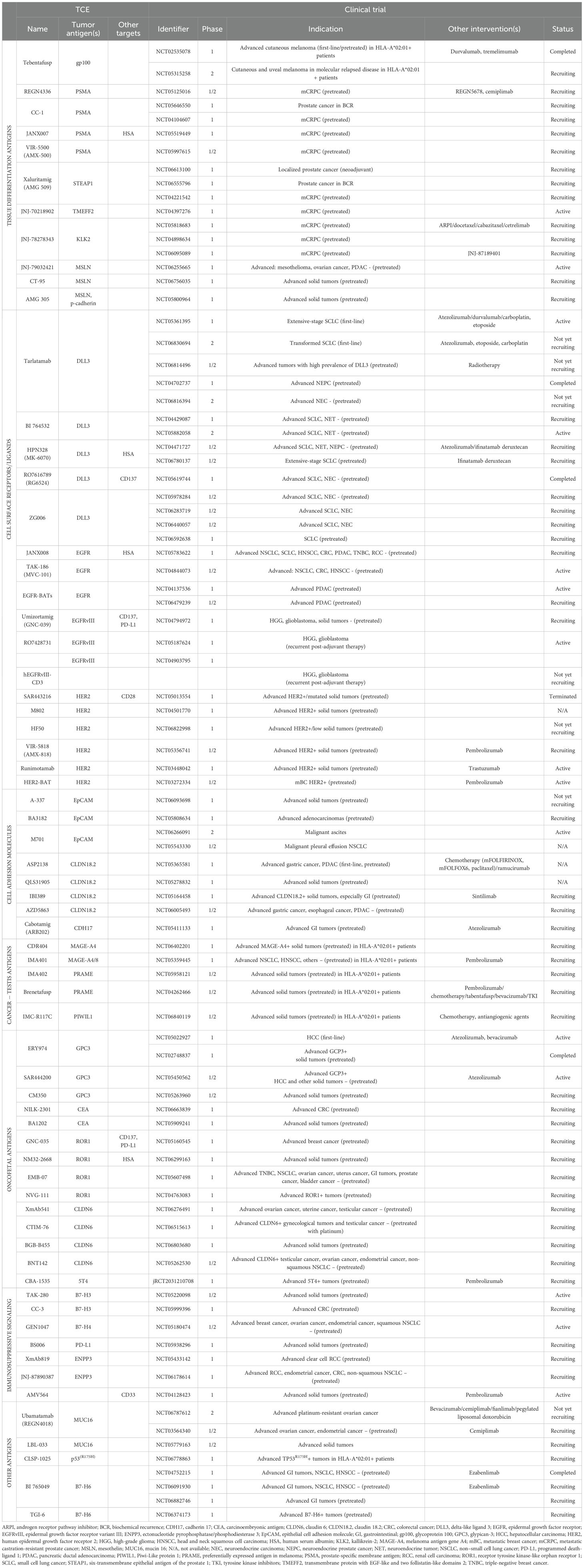
Table 1. T-cell engagers undergoing early clinical development in patients with solid tumors.
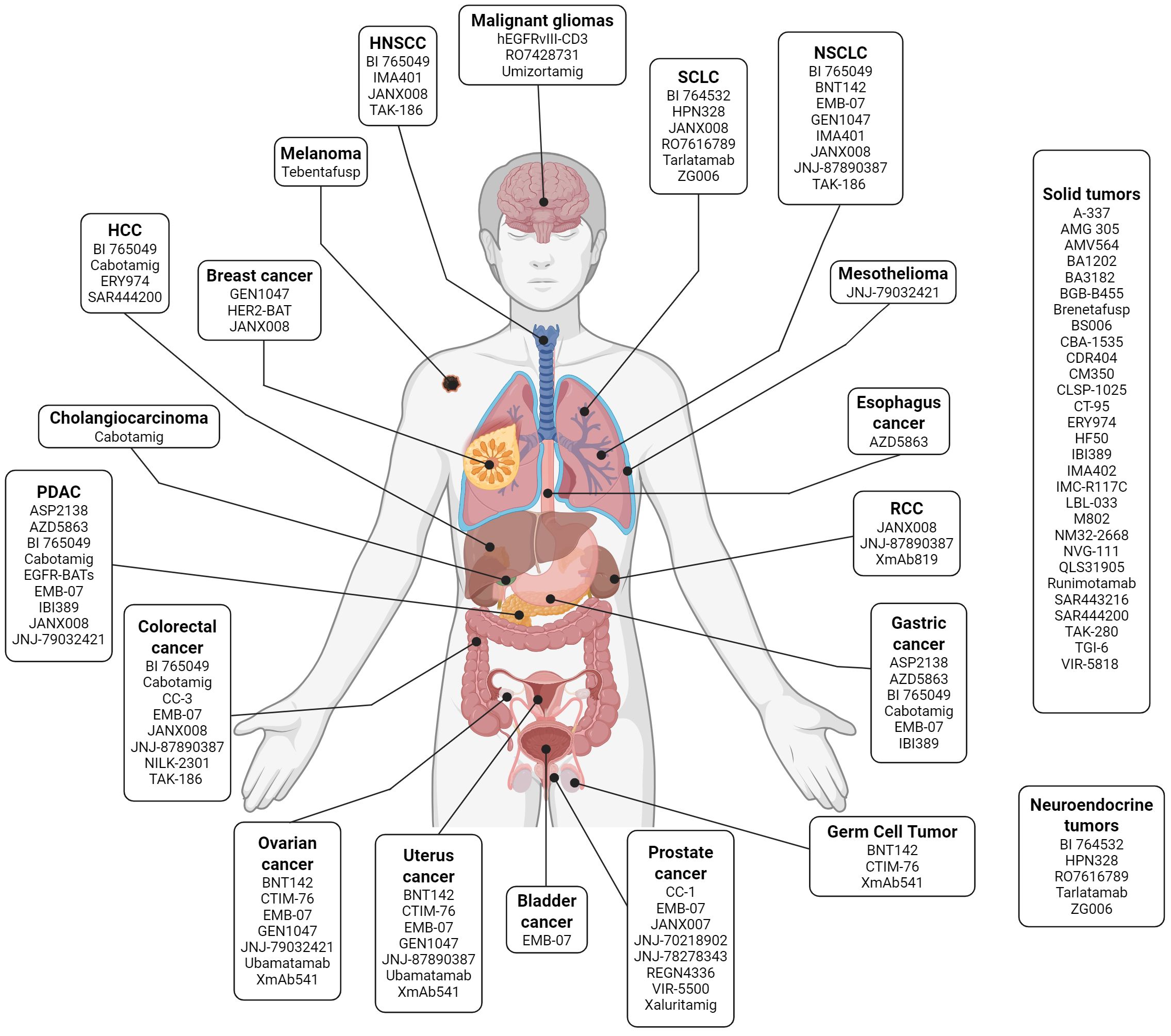
Figure 2. T-cell engagers in clinical development grouped by indication. List of selected T-cell engagers that are currently undergoing early-phase clinical development according to the tumor histology where they have been tested. Image created with Biorender.com. HNSCC, head and neck squamous cell carcinoma; HCC, hepatocellular carcinoma; NSCLC, non-small cell lung cancer; PDAC, pancreatic ductal adenocarcinoma; RCC, renal cell carcinoma; SCLC, small-cell lung cancer.
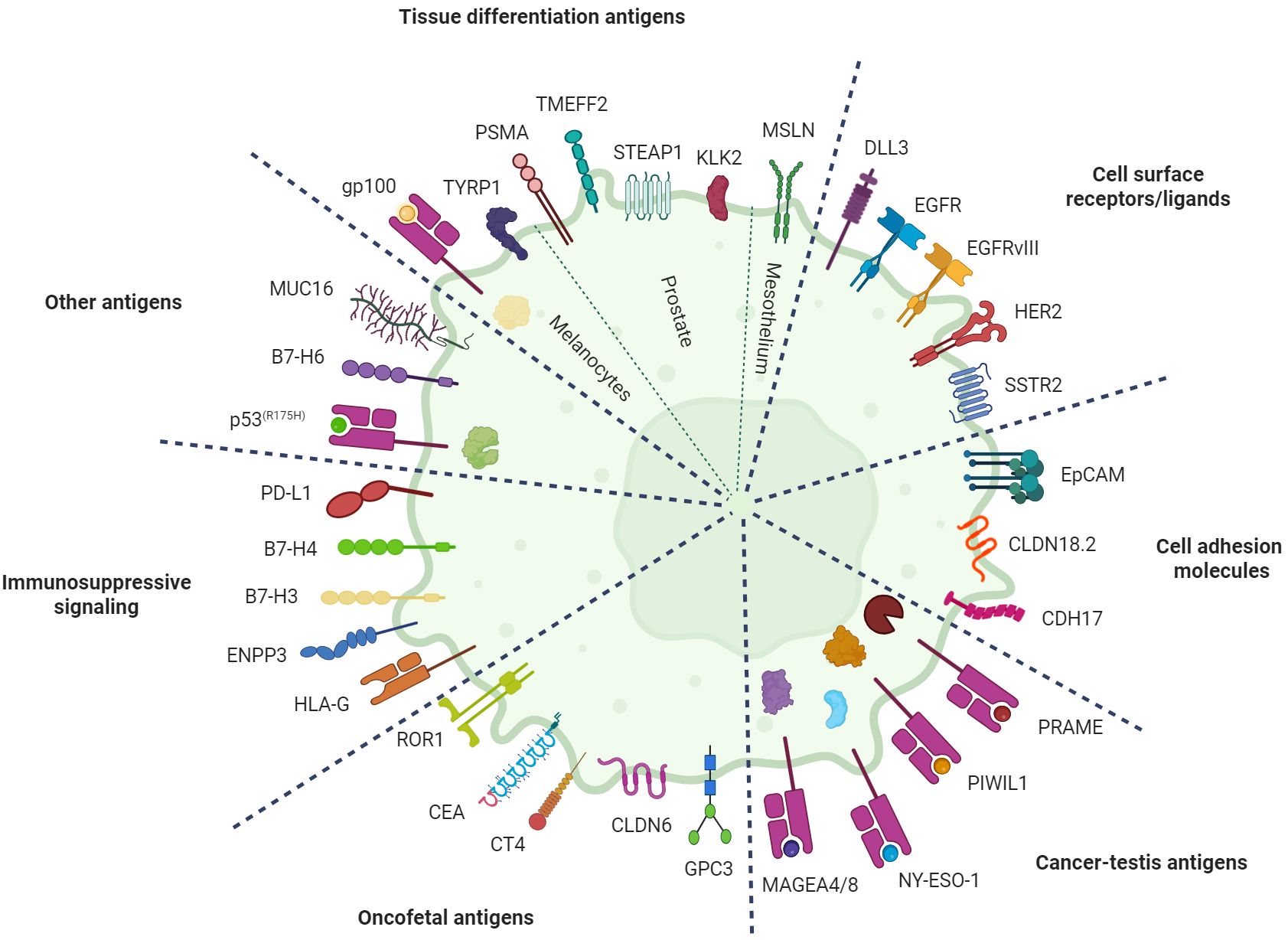
Figure 3. Targets for T-cell engagers in solid tumors. Illustration of the tumor-associated antigens (TAAs) that have been explored as targets for T-cell engagers in patients with solid tumors divided by category. Intracellular antigens are exposed on the plasma membrane as peptide fragments bound to major histocompatibility complex (MHC) class I molecules. Image created with Biorender.com. B7-H3, B7 homolog 3; B7-H4, B7 homolog 4; B7-H6, B7 homolog 6; CEA, carcinoembryonic antigen; CDH17, cadherin 17; CLDN18.2, claudin 18 isoform 2; CLDN6, claudin 6; CT4, cancer/testis antigen 4; DLL3, delta-like ligand 3; EGFR, epidermal growth factor receptor; EGFRvIII, epidermal growth factor receptor variant III; ENPP3, ectonucleotide pyrophosphatase/phosphodiesterase 3; EpCAM, epithelial cell adhesion molecule; GPC3, glypican 3; gp100, glycoprotein 100; HER2, human epidermal growth factor receptor 2; HLA-G, human leukocyte antigen G; KLK2, kallikrein 2; MAGEA4/8, melanoma-associated antigen A4/A8; MUC16, mucin 16; MSLN, mesothelin; NY-ESO-1, New York esophageal squamous cell carcinoma 1; PD-L1, programmed death-ligand 1; PIWIL1, P-element induced wimpy testis-like protein 1; PRAME, preferentially expressed antigen in melanoma; PSMA, prostate-specific membrane antigen; ROR1, receptor tyrosine kinase-like orphan receptor 1; SSTR2, somatostatin receptor 2; STEAP1, six-transmembrane epithelial antigen of the prostate 1; TMEFF2, transmembrane protein with EGF-like and two follistatin-like domains 2; TYRP1, tyrosinase-related protein 1.
2.2 TCEs targeting tissue differentiation antigens
The first milestone in the development of TCEs in solid tumors is approval by the U.S. Food and Drug Administration (FDA) of tebentafusp in January 2022 as first-line therapy for patients with advanced uveal melanoma (20). Tebentafusp is an ImmTAC that recognizes glycoprotein 100 (gp100) fragments mounted on MHC molecules encoded by the allele human leukocyte antigen (HLA)-A*02:01. As an essential component of the melanosome, the melanin-producing organelle in melanocytes, gp100 is also expressed in most cases of malignant tumors of melanocytic lineage (21). Efforts are ongoing to replicate the success obtained by tebentafusp in uveal melanoma in patients with cutaneous melanoma. A phase 1b trial assessing tebentafusp in combination with ICIs in 85 heavily pretreated patients with advanced cutaneous melanoma showed a response rate of 14% and a 1-year overall survival (OS) rate of 76% (22). Tebentafusp is also being evaluated in patients with uveal or cutaneous melanoma with molecular relapsed disease after removal of the primary tumor (NCT05315258).
Another potential melanocyte-specific target of TCEs is tyrosinase-related protein 1 (TYRP1), an additional constituent of the melanosome that is also expressed at low levels at the plasma membrane. Unfortunately, a phase 1 trial of the BsAb RO7293583 in patients with advanced TYRP1-positive cutaneous, uveal, or mucosal melanoma reported no objective response (23). This was attributed, at least in part, to the development of antidrug antibodies (ADAs) that resulted in reduced active drug exposure in a significant proportion of patients.
Prostate cancer is the second most frequent cancer and the fourth cause of cancer-related death in men worldwide (24). Besides the epidemiological relevance, different characteristics make prostate cancer an attractive target for immunotherapies. The prostate is a non-vital organ characterized by the expression of various proteins that are often conserved in prostate neoplasms and are not significantly expressed in other parts of the body. Among these, intense research has focused on prostate-specific membrane antigen (PSMA), a membrane enzyme that is overexpressed in 85%–100% of prostate cancers, especially in patients with metastatic disease (25). Results of various studies assessing TCEs targeting PSMA show limited efficacy of these agents as monotherapy in patients with metastatic castration-resistant prostate cancer (mCRPC). The objective response rate (ORR) ranged from 0% with the BiTe pasotuxizumab (26) or the Fc-silenced BsAb AMG 340 (27) to 7.4% with the half-life extended BiTE acapatamab (28). Only a minority of patients achieved a prostate-specific antigen (PSA) decrease of more than 50% compared with baseline (PSA50), a commonly used efficacy endpoint in clinical trials of mCRPC. Similar results were obtained with HPN424, which contains CD3-, PSMA-, and HSA-binding domains (29). Due to the lack of efficacy and a challenging safety profile, the clinical development of these four TCEs has been discontinued. It is important to highlight that prostate cancer is notoriously a tumor where other forms of immunotherapy have all shown limited efficacy (30, 31). Nonetheless, a new wave of PSMA-targeting TCEs has entered clinical development. Promising preliminary results show that almost all patients with mCRPC treated with the target dose of the BsAb CC-1 achieved some degree of PSA reduction (32). Preliminary data also show a PSA50 decline in 7 of 12 patients treated with the dual-masked TCE VIR-5500, a prodrug that is activated by tumor-associated proteases (33). The safety profile of this agent appears promising, with no patient presenting more than grade 2 cytokine release syndrome (CRS), a common AE associated with the use of TCEs and characterized by a systemic inflammatory response that can potentially lead to death due to respiratory failure, cardiac arrest, or multiorgan failure (34). JANX007 is a TRACTr biologic that contains PSMA- and CD3-binding domains, a peptide mask that prevents CD3 engagement on T cells, an albumin-binding domain attached to the mask, and a cleavable linker. To exert its therapeutic effect, after binding to PSMA-expressing tumor cells, JANX007 undergoes proteolytic cleavage to expose the CD3-engaging domain, while the albumin-binding domain is removed together with the mask to prevent the recirculation of the drug. Encouraging preliminary data from an ongoing phase 1 trial in patients with mCRPC show a PSA50 decline in all patients (n=16) treated with JANX007, with 75% of them maintaining the PSA decline after ≥12 weeks. The ORR was 50% among eight patients with measurable disease, and the drug was well tolerated (35).
Other targets for TCEs under investigation in prostate cancer include six-transmembrane epithelial antigen of prostate 1 (STEAP1), transmembrane protein with EGF-like and two follistatin-like domains 2 (TMEFF2), and kallikrein-2 (KLK2). Xaluritamig is a humanized antibody that contains two identical anti-STEAP1 binding domains, an anti-CD3 domain, and an effectorless Fc region. A phase 1 trial of xaluritamig monotherapy in patients with mCRPC reported that 24% of 67 evaluable patients and 41% of those in the target dose cohorts achieved a partial response (36). The PSA50 decline rates were 49% and 59%, respectively. At a median follow-up of 23.5 months, the median OS was 17.4 months (37). Although treatment-related adverse events (TRAEs) were observed in 97% of patients, they were serious in only 39% of patients. The most frequent were CRS (72% of all grades, 16% grade ≥3), fatigue (45%), and myalgia (34%) (36). Following these results, a phase 3 study (NCT06691984) has been launched to investigate xaluritamig versus cabazitaxel or an antiandrogen therapy in patients with pretreated mCRPC. JNJ-70218902 is a T cell-engaging BsAb targeting TMEFF2 that is being developed in patients with mCRPC. In an ongoing phase 1 study, JNJ-70218902 monotherapy led to a PSA50 decline in only 12.2% of patients, while the ORR among 33 patients with measurable disease was 15.2%. Toxicity was manageable, with 18.3% of patients experiencing TRAEs of grade ≥3, most frequently in the form of fatigue, lymphopenia, and asthenia (38).
2.3 TCEs targeting cell surface receptors/ligands
The second milestone in TCEs’ clinical advancement for solid tumors occurred in May 2024, following the FDA’s accelerated approval of tarlatamab for extensive-stage small-cell lung cancer (SCLC) pretreated with platinum-containing chemotherapy (39). Tarlatamab is a BiTE with an effectorless Fc fragment that targets delta-like ligand 3 (DLL3), a non-canonical inhibitory ligand of the Notch pathway. In normal cells, the expression of DLL3 is low and mainly confined to the Golgi apparatus and cytoplasmic vesicles. However, it is upregulated and relocates at the cell membrane in neuroendocrine neoplasms (40). Studies are now exploring tarlatamab, alone and in association with ICIs and chemotherapy in patients with treatment-naïve SCLC. Tarlatamab is also being studied in patients with extrapulmonary neuroendocrine tumors. For example, in patients with metastatic neuroendocrine prostate cancer (NEPC), preliminary data show that tarlatamab monotherapy resulted in an ORR of 10.5% among all patients and 22.2% in patients with DLL3-positive tumors (41). Additional TCEs targeting DLL3 are being investigated in patients with neuroendocrine tumors. Positive preliminary data from a phase 1/2 study of HPN328, which has a binding domain for HSA, show an ORR of 50% among patients with pretreated SCLC, 44% among patients with neuroendocrine carcinoma, and 36% among patients with NEPC. The drug was well tolerated, and although 59% of patients experienced CRS, it was grade ≥3 in only 3% of them (42). Promising efficacy and tolerability were also recently reported with the BsAb BI 764532 in a similar patient population (43).
The ErbB receptor family comprises four transmembrane receptors with tyrosine kinase activity, named ErbB 1 to 4, that bind to extracellular ligands and play essential roles in the initiation and progression of several types of solid tumors (44). The members of this family that have been more closely implicated in cancer physiopathology are epidermal growth factor receptor (EGFR; ErbB1) and human epidermal growth factor receptor 2 (HER2; ErbB2), with several targeted agents already approved by regulatory agencies. Numerous TCEs targeting EGFR, typically designed as prodrugs, are being tested in patients with advanced, treatment-refractory tumors of epithelial origin. For example, JANX008 is a TRACTr targeting EGFR with a similar structure as JANX007 (see above). Likewise, TAK-186 is a COBRA biologic targeting EGFR that also engages HSA and whose CD3 effector domain is unmasked by tumor proteases (45). CX-904 was a protease-activatable TCE whose clinical development has been discontinued. In a phase 1 study with preliminary data available, CX-904 demonstrated a very favorable safety profile and signs of activity, with an ORR of 33% and a disease control rate of 100% among six patients with pretreated pancreatic cancer (46).
Another strategy to engage T cells against EGFR-expressing tumors involves the use of EGFR-targeting bispecific antibody-armed activated T cells (BATs), consisting of autologous T cells that are expanded and conjugated ex vivo with a BsAb that binds to CD3 and EGFR. Their clinical development is currently focusing on patients with advanced pancreatic cancer. Results from an adaptive trial show clear signs of immune activation following the infusion of EGFR-BATs in patients with treatment-refractory pancreatic carcinoma. However, as monotherapy, the antitumor activity was modest, with no objective responses observed and two patients obtaining stable disease (47).
Mutations in EGFR that lead to a constitutively active receptor are common in certain tumor types. EGFRvIII is a tumor-specific EGFR variant that is detected in up to 19% of patients with glioblastoma (48), an aggressive type of brain tumor. A prematurely terminated phase 1 trial of the BiTE etevritamab showed that among eight patients with recurrent EGFRvIII-positive glioblastoma, only one achieved a partial response and two had a stable disease (49). However, approximately 50% of patients experienced serious AEs, most commonly in the form of headaches and depressed consciousness. Two additional TCEs, the BiTE hEGFRvIII-CD3 and the BsAb RO7428731, are being studied in patients with EGFRvIII-positive high-grade glioma or glioblastoma progressing after adjuvant radiotherapy and temozolomide. Moreover, a tetra-specific antibody construct named umizortamig, with binding domains for CD3, EGFRvIII, the immune checkpoint programmed death-ligand 1 (PD-L1), and the T-cell costimulatory receptor CD137, is being assessed in patients with relapsed or treatment-refractory malignant glioma as well as other solid tumors.
HER2 can act as an oncogene and drive tumor progression through either activating mutations or overexpression. The latter event is frequent in patients with certain types of cancer, such as breast cancer and gastric carcinoma, but can also be found in a minority of patients with other cancer histologies (50). Early attempts to target HER2-expressing tumors with TCEs have shown disappointing results. Two studies assessing the BsAbs ertumaxomab or GBR 1302 as single agents in patients with different types of HER2-positive tumors reported scarce to no objective responses (51, 52). Next-generation TCEs targeting HER2 that have entered clinical testing include the BsAbs M802 and runimotamab, the dual-masked prodrug VIR-5818, and the TRAFsome construct HF50. The latter consists of antibody fragments anchored onto liposomal surfaces with the aim of concentrating the drug in the TME due to enhanced permeability and retention effect compared with healthy tissues. Preliminary data from the cohorts of VIR-5818 monotherapy in a phase 1 study show that 50% of patients experienced some degree of tumor shrinkage, with two of six patients with treatment-refractory colorectal cancer achieving a partial response (33). This agent is well tolerated, with only a minority of patients presenting with grade 1–2 CRS and none with grade 3. SAR443216 is a tri-specific TCE that binds to HER2, CD3, and the T-cell costimulatory receptor CD28. Regrettably, a phase 1 trial showed a disappointing ORR of 0% among 40 patients with heavily pretreated HER2-positive or HER2-mutated tumors treated with SAR443216 (53).
HER2-directed BATs have also been developed. A study published some years ago reported scarce activity of HER2-BAT monotherapy in 23 patients with metastatic HER2-amplified breast cancer, with just one patient obtaining a partial response (54). A phase 1/2 trial is now assessing HER2-BATs in association with the ICI pembrolizumab in a similar patient population.
Finally, tidutamab was a T cell-engaging BsAb with a silent Fc fragment targeting somatostatin receptor 2 (SSTR2) that was tested in patients with advanced, well-differentiated neuroendocrine tumors (NETs). Although tidutamab was well tolerated and induced sustained T-cell activation and cytokine release, no objective response was observed (55). The development of this agent has been discontinued.
2.4 TCEs targeting cell adhesion molecules
Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein mediating cell–cell adhesion in epithelial tissues. High levels of EpCAM expression are found in many carcinomas and have been associated with disease progression (56). Historically, the clinical application of TCEs targeting EpCAM has been hindered by the frequent occurrence of severe toxicities due to target abundance in normal tissues (57, 58). Nevertheless, their clinical development has continued. Preliminary data from a phase 2 randomized study (n=84) show that, after paracentesis, the intraperitoneal infusion of the BsAb M701 was effective in prolonging puncture-free survival compared with paracentesis alone among patients with malignant ascites due to epithelial cancer (59). A non-significant improvement in OS was observed, with 6-month survival rates of 32.3% for M701 and 12.6% for paracentesis alone. This agent was manageable, with 21.7% of patients experiencing serious TRAEs, primarily in the form of anemia, hypokalemia, and hyperglycemia (60). M701 is also being studied in patients with symptomatic malignant pleural effusion due to advanced non-small cell lung cancer (NSCLC), showing promising preliminary signs of activity and good tolerability (61). Another TCE targeting EpCAM is BA3182, a prodrug BsAb that is conditionally activated in the TME by the acidic pH. BA3182 is being tested as a single agent in patients with different types of adenocarcinomas.
Claudins (CLDNs) are a family of transmembrane proteins that are essential components of tight junctions. There are 26 members of this family in humans, with strong tissue- and cell-specific distribution. CLDN expression is dysregulated in various cancers and has been implicated in cancer-cell proliferation, invasion, migration, and metastasis (62). Various TCEs targeting CLDN18.2 are being studied, with a focus on patients with gastrointestinal tumors. Preliminary data show that approximately one-third of patients with treatment-refractory CLDN18.2-positive pancreatic adenocarcinoma or gastric cancer treated with the BsAb IBI389 obtained an objective response. Nevertheless, this was accompanied by significant toxicity, with more than half of patients experiencing grade ≥3 TRAEs (63, 64).
Cadherins (CDHs) are a family of numerous cell–cell adhesion molecules that can have both tumor-suppressive and tumor-promoting roles depending on the context and the specific member (65). Cabotamig, a TCE targeting CDH17, is currently under investigation in association with the ICI atezolizumab in patients with gastrointestinal tumors. The clinical development of PF-06671008, which targets p-cadherin, has been curbed due to a lack of efficacy (66).
2.5 TCEs targeting CTAs
Due to their limited distribution in adult cells and tissues outside the testes and their abundant expression in various tumor types, CTAs have long been considered an attractive target for anticancer therapy, particularly for immune-based interventions. Indeed, since T cells naturally recognize these antigens, efforts have been made to develop CTA-based vaccines. However, despite generating high frequencies of reactive T cells in the blood, no agent has shown consistent activity (67). Various TCEs targeting intracellular CTAs are under investigation in patients who carry the HLA-A*02:01 allele. Brenetafusp is an ImmTAC that engages preferentially expressed antigen in melanoma (PRAME). Preliminary results from an ongoing phase 1/2 study show that, in a cohort of 36 patients with cutaneous melanoma pretreated with ICIs, the ORR to brenetafusp monotherapy was 11%, with no responses observed in patients with PRAME-negative tumors (68). Similarly, in a cohort of 47 patients with platinum-resistant ovarian cancer, brenetafusp showed signs of activity both as monotherapy and in combination with chemotherapy (69). Brenetafusp appeared safe; the most frequently reported AE was grade 1–2 CRS in approximately half of patients in the monotherapy cohorts, predominantly during the first weeks of therapy. The clinical development of another ImmTAC named IMCnyeso which targets New York esophageal squamous cell carcinoma 1 (NY-ESO-1) has been discontinued after a phase 1 trial reported no objective response among 28 patients with solid tumors (70). IMA401 and IMA402 are two TCERs targeting melanoma antigen gene (MAGE)-A4/8-positive or PRAME-positive tumors, respectively. Encouraging early signs of activity accompanied by a manageable safety profile have been recently disclosed, and clinical trials are ongoing (71).
2.6 TCEs targeting oncofetal antigens
Glypican-3 (GPC3) is a membrane-bound heparan sulfate proteoglycan that is overexpressed in most cases of hepatocellular carcinoma (HCC) as well as in up to 45% of patients with squamous NSCLC and a minority of patients with other tumor types (72). A phase 1 trial of ERY974, a BsAb with a silenced Fc region, reported modest efficacy among 29 patients with GPC3-positive solid tumors, with only one patient achieving a partial response. The most frequent TRAEs were CRS, which was the dose-limiting toxicity (DLT), and pyrexia (73). SAR444200 is a GPC3-targeting TCE with two essential characteristics: structurally, it is a tandem of fragments from one domain of a heavy-chain antibody that is based on nanobody technology; functionally, it engages T cells via binding of TCRαβ subunit instead of CD3ϵ. Preliminary data from a phase 1/2 study show a ≥20% decrease in alpha-fetoprotein, a serum marker of HCC, in 2 of 18 patients with advanced HCC treated with SAR444200 (74).
Carcinoembryonic antigen (CEA) is a cell surface glycoprotein that belongs to the immunoglobulin gene superfamily and is often used as a marker in various types of epithelial tumors. Initial attempts to target CEA with TCEs have been negative, with cibisatamab and AMG 211 being discontinued after clinical studies reported scarce efficacy (75–77). New molecules include the BsAbs NILK-2301 and BA1202, which are being tested in patients with colorectal cancer and other solid tumors, respectively.
CLDN6 is a member of the CLDN family (see above) that is expressed especially in patients with gynecological tumors or germ cell tumors. While the development of BiTE AMG 794 has recently been halted, various T cell-engaging BsAbs are being developed. Moreover, BNT142 is a lipid nanoparticle-formulated RNA encoding a T cell-engaging BsAb targeting CLDN6. After intravenous administration, BNT142 accumulates in the liver, where the RNA is translated and the BsAb is self-assembled and then secreted into circulation (78). This formulation is predicted to improve the pharmacokinetic profile by prolonging the systemic availability of the TCE.
2.7 TCEs targeting immunosuppressive signaling
HLA-G is a non-classical MHC molecule with an immunosuppressive function whose physiological expression is restricted to the maternal–fetal interface and immune-privileged organs in the adult but can be aberrantly expressed in cancer (79). The TCE JNJ-78306358 was studied in heavily pretreated patients with tumors with a high prevalence of HLA-G expression. No objective responses were observed, and four patients experienced DLTs (80). The clinical development of JNJ-78306358 has been discontinued.
AMV564 is a tetravalent tandem diabody with two binding sites for CD33, a cell surface glycoprotein expressed by myeloid cells, and two for CD3ϵ (81). It has been tested in both myeloid neoplasms, where it directly induces the elimination of neoplastic cells, and in solid tumors, where it stimulates immune-mediated tumor clearance by depleting immunosuppressive myeloid-derived suppressor cells (MDSCs). Encouraging preliminary results from a phase 1 trial show that, in patients with treatment-refractory solid tumors, AMV564 induced clinical responses when administered both as monotherapy and in combination with pembrolizumab, including a complete response in a monotherapy-treated patient with ovarian cancer (82). Importantly, AMV564 was well tolerated, no DLT was observed, and the maximum tolerated dose (MTD) was not reached.
BS006 is an engineered recombinant type II herpes simplex oncolytic virus that encodes for a BsAb that redirects T cells toward PD-L1-positive tumor cells. Following intratumor injection, BS006-infected tumor cells are instructed to produce and secrete the TCE. Accordingly, BS006 is expected to induce a robust antitumor immune response that combines the inflammatory preconditioning of the TME due to the direct effect of the oncolytic virus plus the T cell-mediated response deriving from the action of the TCE and the inhibition of PD-L1. A phase 1 study is evaluating BS006 as a single agent in patients with advanced treatment-refractory solid tumors. Results are pending.
3 Overcoming the limits
Following the success in patients with B-cell hematologic malignancies and those with uveal melanoma or SCLC, the development of TCEs has generated intense interest and excitement. Nonetheless, most of the currently available TCEs provide only limited benefit in patients with solid tumors (Table 2). Like what has been observed in the field of CAR-T cells (83), TCEs have demonstrated significant success in treating B-cell neoplasms, with seven agents of this class already approved by regulatory agencies (Supplementary Table S1). However, their application in solid tumors presents additional challenges that have limited their efficacy (84) (Figure 4). These include the immunosuppressive TME, which may be devoid of T cells and/or contain immunosuppressive cells such as MDSCs and regulatory T cells (Tregs) that prevent the activation of T cells; the physical barrier posed by the tumor stroma, which contains a dense extracellular matrix and results in poor penetration of drugs; and antigen heterogeneity, which may lead to the emergence of clones of tumor cells that do not express the TAA targeted by the TCE. In addition, tumor-intrinsic resistance mechanisms such as antigen loss, resistance to apoptosis induction, and upregulation of inhibitory immune checkpoints may have contributed to this outcome (84).
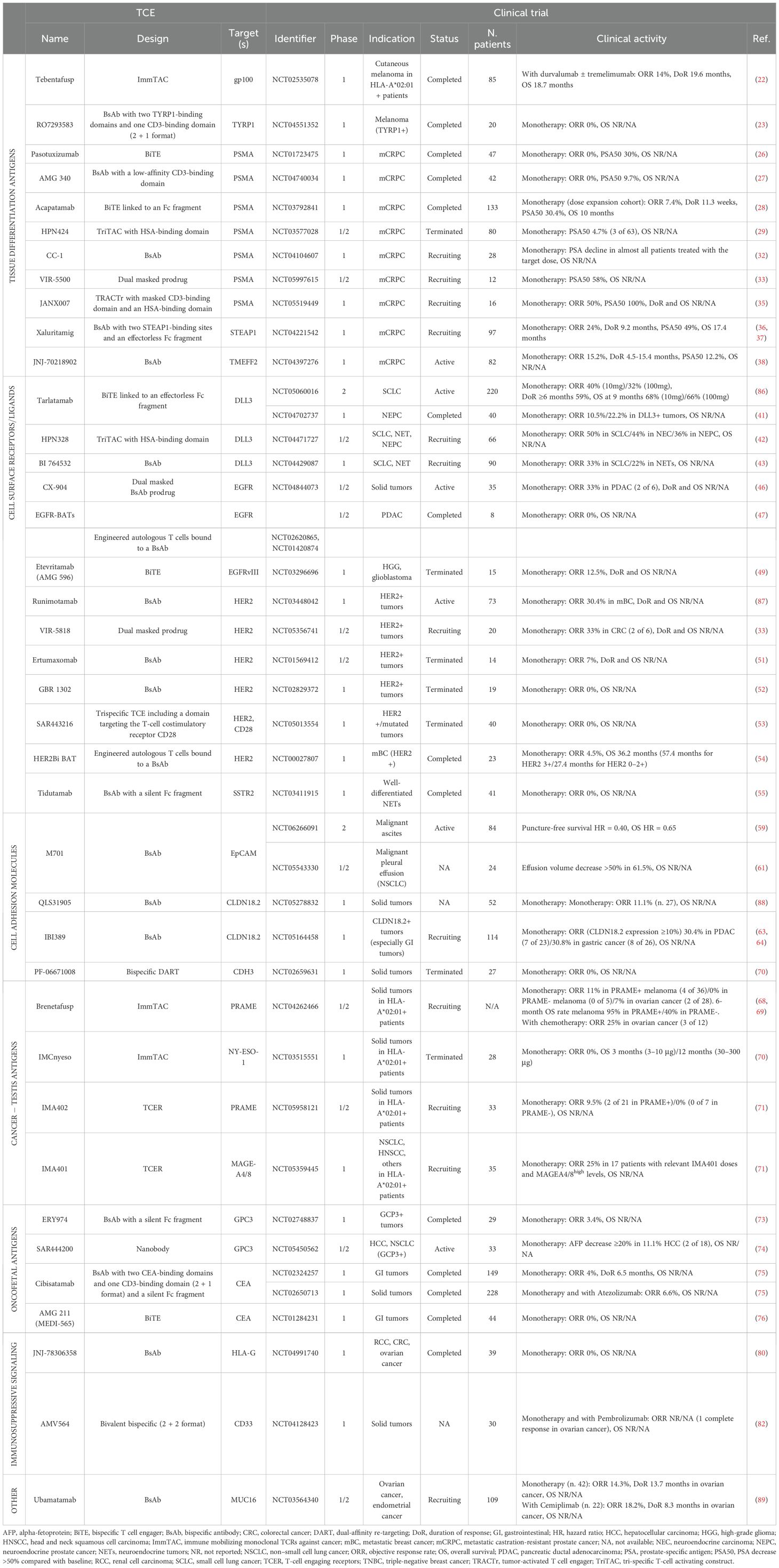
Table 2. Clinical activity of T-cell engagers with final or interim results in advanced solid tumors.
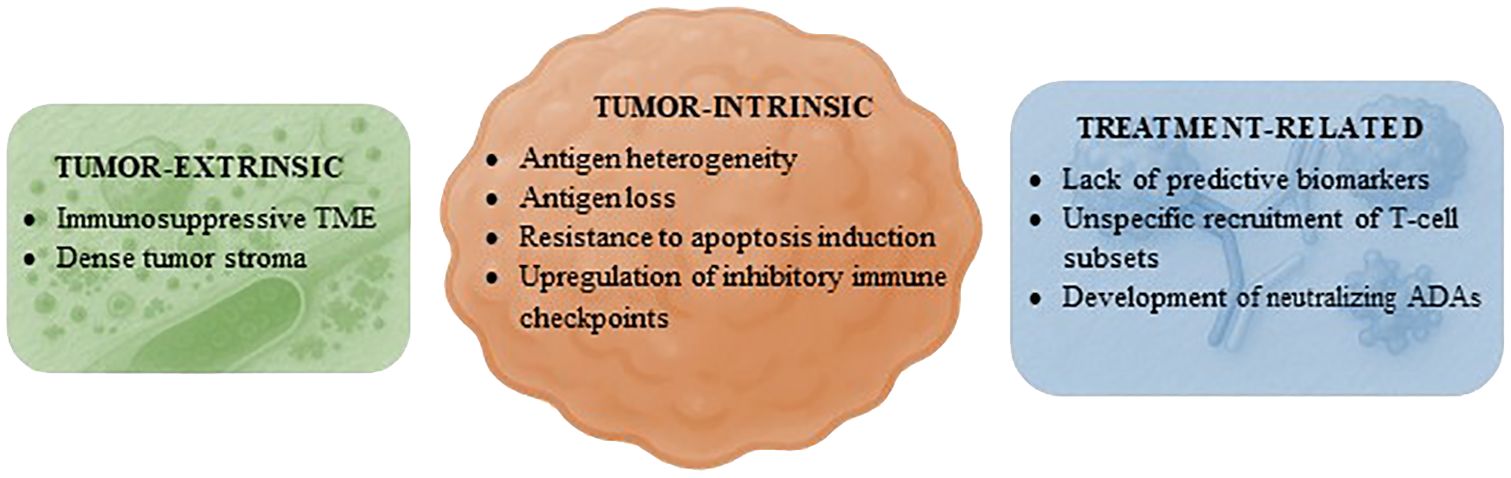
Figure 4. Mechanisms of resistance to T-cell engagers in solid tumors. Proposed factors hindering the efficacy of T-cell engagers in patients with solid tumors grouped by category. ADAs, antidrug antibodies; TME, tumor microenvironment.
Factors inherent to the therapeutic agent or treatment strategy may also have hindered the successful application of TCEs in solid tumors. First, there is a lack of predictive biomarkers for efficacy beyond the expression of the TAA. Second, the administration of a TCE may recruit not only T lymphocytes with effector functions but also various other T-cell subsets. These may have immunosuppressive properties, such as naïve or exhausted T cells as well as Tregs, or they may increase treatment toxicity, including other CD4+ T-cell populations (85). Third, the development of neutralizing ADAs in response to the immunogenicity of TCEs is frequently reported in clinical studies (23, 26, 28, 36, 74, 75, 80). A strategy that has been implemented to attenuate this risk involves the pretreatment of patients with drugs that deplete B cells, such as anti-CD20 antibodies (75). Interestingly, numerous TCEs targeting DLL3 demonstrated consistent clinical activity in tumors of neuroendocrine lineage (39, 41–43, 86). In contrast, a TCE targeting SSTR2 failed to provide benefit in patients with NETs (55). This implies that, at least in some instances, accurately identifying the TAA that T cells should target in each tumor type may be crucial in determining the efficacy of these drugs, regardless of the chemical or pharmacological properties of the TCE.
The dose-finding studies included in this review suggest that the safety profile of current TCEs is not a prominent factor limiting their therapeutic potential. The clinical data made available during the last 5 years from completed or terminated studies, along with those from ongoing clinical trials, reveal that the MTD has been identified in only 19% of these studies, while it was not reached in 61% of them (Supplementary Figure S1). Indeed, various optimization strategies are generally applied to mitigate toxicity and decrease the incidence and severity of class-specific AEs such as CRS and neurotoxicity. Examples include step-up dosing to modulate the intensity of T-cell activation and cytokine release, subcutaneous administration to decrease the maximum concentration and provide a more gradual release of the drug into the bloodstream compared with the intravenous route, and prophylactic treatment with anti-interleukin-6 agents to prevent the cytokine storm (34, 90). An additional strategy involves the design of TCEs with a low-affinity binding site for CD3 to reduce cytokine release (91).
Continued innovation in TCE design, alongside a deeper understanding of its mechanisms of action and interaction with the TME, is essential for realizing its full therapeutic potential. For instance, combining TCEs with other therapeutic approaches such as ICIs, vaccines, or conventional chemotherapy might enhance overall efficacy and potentially prevent resistance. Numerous early-phase clinical trials are already testing TCEs in combination with diverse classes of antitumor medical therapies. The results of these cohorts will be highly informative in refining the therapeutic application of TCEs.
The identification of new TAAs that can be targeted could also facilitate the successful development of TCEs. Apart from TAAs that are common to different tumor histologies or shared by patients with the same tumor type, tailoring TCEs based on individual patient profiles could enhance therapeutic outcomes and minimize adverse effects. For example, after the molecular profiling of a tumor biopsy, a TCE could be designed to target one or more TAAs that are specific to each patient. This procedure could be repeated at the time of tumor progression to redirect treatment toward the emerging tumor-cell clones that drive resistance, thus providing a longitudinal, adaptive approach to cancer immunotherapy.
4 Conclusions
TCEs are a rapidly evolving class of therapeutic agents in oncology with the potential to significantly impact the treatment landscape for solid tumors. They represent a transformative, powerful, and targeted approach to cancer immunotherapy. Although expectations regarding TCEs have not yet been fulfilled in patients with solid tumors, encouraging results from a few contemporary studies bring hope for a change of direction. With a wealth of new TCEs being developed and tested in clinical trials, the next few years will be critical in providing a definitive verdict regarding their utility in solid tumors.
Author contributions
AS: Conceptualization, Methodology, Visualization, Writing – original draft. GI: Visualization, Writing – review & editing. JG: Writing – review & editing. EG: Visualization, Writing – review & editing. MF: Writing – review & editing. SS: Writing – review & editing. FL: Writing – review & editing. CF: Writing – review & editing. GD: Conceptualization, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
We thank Ministero della Salute RC2025 for the support in realizing this work.
Conflict of interest
GD has received consulting fees from Bayer; payment or honoraria from AstraZeneca, Bayer, Gilead, and MSD; and support for travel and accommodation from AstraZeneca and Roche.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1665838/full#supplementary-material
Glossary
ADCC: antibody-dependent cell-mediated cytotoxicity
ADCP: antibody-dependent cell-mediated phagocytosis
AE: adverse event
BATs: bispecific antibody armed activated T cells
BiTE: bispecific T cell engager
BsAb: bispecific antibody
CAR-T: chimeric antigen receptor-T
CDC: complement-dependent cytotoxicity
CDH17: cadherin 17
CEA: carcinoembryonic antigen
CLDN6: claudin 6
CLDN18.2: claudin 18.2
CRS: cytokine release syndrome
DLL3: delta-like ligand 3
COBRA: COnditional bispecific redirected activation
DLT: dose-limiting toxicity
EGFR: epidermal growth factor receptor
EGFRvIII: epidermal growth factor receptor variant III
EpCAM: epithelial cell adhesion molecule
FDA: Food and Drug Administration
gp100: glycoprotein 100
GPC3: glypican-3
HCC: hepatocellular carcinoma
HER2: human epidermal growth factor receptor 2
HLA: human leukocyte antigen
HSA: human serum albumin
ICI: immune checkpoint inhibitor
ImmTAC: immune mobilizing monoclonal TCRs against cancer
KLK2: kallikrein-2
MAGE-A4: melanoma antigen gene A4
mCRPC: metastatic castration-resistant prostate cancer
MDSC: myeloid-derived suppressor cells
MHC: major histocompatibility complex
MTD: maximum tolerated dose
NEPC: neuroendocrine prostate cancer
NET: neuroendocrine tumor
NSCLC: non–small cell lung cancer
NY-ESO-1: New York esophageal squamous cell carcinoma 1
ORR: objective response rate
OS: overall survival
PD-L1: programmed death-ligand 1
PRAME: preferentially expressed antigen in melanoma
PSA: prostate-specific antigen
PSA50: PSA decrease >50% compared with baseline
PSMA: prostate-specific membrane antigen
RCC: renal cell carcinoma
ROR1: receptor tyrosine kinase-like orphan receptor 1
SCLC: small cell lung cancer
SSTR2: somatostatin receptor 2
STEAP1: six-transmembrane epithelial antigen of the prostate 1
TAA: tumor-associated antigen
TCE: T cell engager
TCER: T cell engaging receptors
TCR: T cell receptor
TILs: tumor-infiltrating lymphocytes
TME: tumor microenvironment
TMEFF2: transmembrane protein with EGF-like and two follistatin-like domains 2
TRAE: treatment-related adverse events
TRACTr: tumor activated T cell engager
TRAFsome: T-cell redirecting antibody fragment-anchored liposome
Treg: regulatory T cell
TYRP1: Tyrosinase-related protein 1.
References
1. Lizée G, Overwijk WW, Radvanyi L, Gao J, Sharma P, and Hwu P. Harnessing the power of the immune system to target cancer. Annu Rev Med. (2013) 64:71–90. doi: 10.1146/annurev-med-112311-083918
2. Brudno JN, Maus MV, and Hinrichs CS. CAR T cells and T-cell therapies for cancer: A translational science review. JAMA. (2024) 332:1924–35. doi: 10.1001/jama.2024.19462
3. Qiu J, Cheng Z, Jiang Z, Gan L, Zhang Z, and Xie Z. Immunomodulatory precision: A narrative review exploring the critical role of immune checkpoint inhibitors in cancer treatment. Int J Mol Sci. (2024) 25:5490. doi: 10.3390/ijms25105490
4. Haen SP, Löffler MW, Rammensee HG, and Brossart P. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat Rev Clin Oncol. (2020) 17:595–610. doi: 10.1038/s41571-020-0387-x
5. Tan HL, Yong C, Tan BZ, Fong WJ, Padmanabhan J, Chin A, et al. Conservation of oncofetal antigens on human embryonic stem cells enables discovery of monoclonal antibodies against cancer. Sci Rep. (2018) 8:11608. doi: 10.1038/s41598-018-30070-z
6. Qin X, Ning W, Liu H, Liu X, Luo W, and Xia N. Stepping forward: T-cell redirecting bispecific antibodies in cancer therapy. Acta Pharm Sin B. (2024) 14:2361–77. doi: 10.1016/j.apsb.2024.03.027
7. Vokes NI and Van Allen EM. Tumor evolution: A problem of histocompatibility. Cell. (2017) 171:1252–3. doi: 10.1016/j.cell.2017.11.012
8. Syn NL, Teng MWL, Mok TSK, and Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. (2017) 18:e731–41. doi: 10.1016/S1470-2045(17)30607-1
9. Liu J and Zhu J. Progresses of T-cell-engaging bispecific antibodies in treatment of solid tumors. Int Immunopharmacol. (2024) 138:112609. doi: 10.1016/j.intimp.2024.112609
10. Arvedson T, Bailis JM, Britten CD, Klinger M, Nagorsen D, Coxon A, et al. Targeting solid tumors with bispecific T cell engager immune therapy. Ann Rev Cancer Biol. (2022) 6:17–34. doi: 10.1146/annurev-cancerbio-070620-104325
11. Jin BK, Odongo S, Radwanska M, and Magez S. NANOBODIES®: A review of diagnostic and therapeutic applications. Int J Mol Sci. (2023) 24:5994. doi: 10.3390/ijms24065994
12. Scott AM, Wolchok JD, and Old LJ. Antibody therapy of cancer. Nat Rev Cancer. (2012) 12:278–87. doi: 10.1038/nrc3236
13. Li H, Er Saw P, and Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol. (2020) 17:451–61. doi: 10.1038/s41423-020-0417-8
14. Ai Z, Wang B, Song Y, Cheng P, Liu X, and Sun P. Prodrug-based bispecific antibodies for cancer therapy: advances and future directions. Front Immunol. (2025) 16:1523693. doi: 10.3389/fimmu.2025.1523693
15. Rolin C, Zimmer J, and Seguin-Devaux C. Bridging the gap with multispecific immune cell engagers in cancer and infectious diseases. Cell Mol Immunol. (2024) 21:643–61. doi: 10.1038/s41423-024-01176-4
16. Austin RJ, Lemon BD, Aaron WH, Barath M, Culp PA, DuBridge RB, et al. TriTACs, a novel class of T-cell-engaging protein constructs designed for the treatment of solid tumors. Mol Cancer Ther. (2021) 20:109–20. doi: 10.1158/1535-7163.MCT-20-0061
17. de Souza JE, Galante PA, de Almeida RV, da Cunha JP, Ohara DT, Ohno-MaChado L, et al. SurfaceomeDB: a cancer-orientated database for genes encoding cell surface proteins. Cancer Immun. (2012) 12:15. doi: 10.1158/1424-9634.DCL-15.12.2
18. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med. (2012) 18:980–7. doi: 10.1038/nm.2764
19. Bunk S, Hofmann M, Pszolla G, Hutt M, Schwöbel F, Unverdorben F, et al. IMA402, an off-the-shelf, next-generation TCR bispecific (TCER®) for efficiently targeting an HLA-presented peptide from the pan-cancer antigen PRAME. Blood. (2022) 140:9089–90. doi: 10.1182/blood-2022-165937
20. Available online at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tebentafusp-tebn-unresectable-or-metastatic-uveal-melanoma (Accessed April 30 2025).
21. Wagner SN, Wagner C, Schultewolter T, and Goos M. Analysis of Pmel17/gp100 expression in primary human tissue specimens: implications for melanoma immuno- and gene-therapy. Cancer Immunol Immunother. (1997) 44:239–47. doi: 10.1007/s002620050379
22. Hamid O, Hassel JC, Shoushtari AN, Meier F, Bauer TM, Salama AKS, et al. Tebentafusp in combination with durvalumab and/or tremelimumab in patients with metastatic cutaneous melanoma: a phase 1 study. J Immunother Cancer. (2023) 11:e006747. doi: 10.1136/jitc-2023-006747
23. Spreafico A, Couselo EM, Irmisch A, Bessa J, Au-Yeung G, Bechter O, et al. Phase 1, first-in-human study of TYRP1-TCB (RO7293583), a novel TYRP1-targeting CD3 T-cell engager, in metastatic melanoma: active drug monitoring to assess the impact of immune response on drug exposure. Front Oncol. (2024) 14:1346502. doi: 10.3389/fonc.2024.1346502
24. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
25. Parsi M, Desai MH, Desai D, Singhal S, Khandwala PM, and Potdar RR. PSMA: a game changer in the diagnosis and treatment of advanced prostate cancer. Med Oncol. (2021) 38:89. doi: 10.1007/s12032-021-01537-3
26. Hummel HD, Kufer P, Grüllich C, Seggewiss-Bernhardt R, Deschler-Baier B, Chatterjee M, et al. Pasotuxizumab, a BiTE® immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy. (2021) 13:125–41. doi: 10.2217/imt-2020-0256
27. Falchook GS, McKean M, Kelly WK, Patel MR, Bupathi M, Liaw BCH, et al. Phase 1 clinical trial of AMG 340, a prostate-specific membrane antigen (PSMA)-targeted T-cell engager with a novel low-affinity CD3 binding domain designed to mitigate toxicity for the treatment of metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. (2024) 42:Abstract e14587. doi: 10.1200/JCO.2024.42.16_suppl.e1458. 2024 ASCO Annual Meeting I.
28. Dorff T, Horvath LG, Autio K, Bernard-Tessier A, Rettig MB, Machiels JP, et al. A phase I study of acapatamab, a half-life extended, PSMA-targeting bispecific T-cell engager for metastatic castration-resistant prostate cancer. Clin Cancer Res. (2024) 30:1488–500. doi: 10.1158/1078-0432.CCR-23-2978
29. De Bono JS, Fong L, Beer TM, Gao X, Geynisman DM, Burris HA III, et al. Results of an ongoing phase 1/2a dose escalation study of HPN424, a tri-specific half-life extended PSMA-targeting T-cell engager, in patients with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. (2021) 39. doi: 10.1200/JCO.2021.39.15_suppl.5013. Abstract 5013: 2021 ASCO Annual Meeting.
30. Rizzo A, Mollica V, Cimadamore A, Santoni M, Scarpelli M, Giunchi F, et al. Is there a role for immunotherapy in prostate cancer? Cells. (2020) 9:2051. doi: 10.3390/cells9092051
31. Noori M, Azizi S, Mahjoubfar A, Abbasi Varaki F, Fayyaz F, Mousavian AH, et al. Efficacy and safety of immune checkpoint inhibitors for patients with prostate cancer: a systematic review and meta-analysis. Front Immunol. (2023) 14:1181051. doi: 10.3389/fimmu.2023.1181051
32. Heitmann JS, Hackenbruch C, Walz JS, Jung S, Pflügler M, Schlenk RF, et al. Updated results on the bispecific PSMAxCD3 antibody CC-1 for treatment of prostate cancer. J Clin Oncol. (2024) 42. doi: 10.1200/JCO.2024.42.16_suppl.2536. Abstract 2536: 2024 ASCO Annual Meeting I.
33. Available online at: https://investors.vir.bio/news/news-details/2025/Vir-Biotechnology-Announces-Encouraging-Safety-and-Efficacy-Data-in-Ongoing-Dose-Escalation-Trials-for-Dual-Masked-T-Cell-Engagers-VIR-5818-in-Solid-Tumors-and-VIR-5500-in-mCRPC/default.aspx (Accessed April 30 2025). Press release 08 January 2025.
34. Géraud A, Hueso T, Laparra A, Bige N, Ouali K, Cauquil C, et al. Reactions and adverse events induced by T-cell engagers as anti-cancer immunotherapies, a comprehensive review. Eur J Cancer. (2024) 205:114075. doi: 10.1016/j.ejca.2024.114075
35. Available online at: https://investors.januxrx.com/investor-media/news/news-details/2025/Janux-Therapeutics-Reports-Fourth-Quarter-and-Full-Year-2024-Financial-Results-and-Business-Highlights/default.aspx (Accessed April 30 2025). Press release 27 February 2025.
36. Kelly WK, Danila DC, Lin CC, Lee JL, Matsubara N, Ward PJ, et al. Xaluritamig, a STEAP1 × CD3 xmAb 2 + 1 immune therapy for metastatic castration-resistant prostate cancer: results from dose exploration in a first-in-human study. Cancer Discov. (2024) 14:76–89. doi: 10.1158/2159-8290.CD-23-0964
37. Armstrong AJ, Appleman LJ, Danila DC, Lin C-C, Lee JL, Matsubara N, et al. 1610P - Circulating tumour cell (CTC) enumeration and overall survival (OS) in men with metastatic castration-resistant prostate cancer (mCRPC) treated with xaluritamig. Ann Oncol. (2024) 35:S971–2. doi: 10.1016/j.annonc.2024.08.1691
38. Calvo E, Doger B, Carles J, Peer A, Sarid D, Eigl BJ, et al. A first-in-human study of JNJ-70218902, a bispecific T-cell-redirecting antibody against TMEFF2 in metastatic castration-resistant prostate cancer. Oncologist. (2025) 30:oyae313. doi: 10.1093/oncolo/oyae313
39. Available online at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-tarlatamab-dlle-extensive-stage-small-cell-lung-cancer (Accessed April 30 2025).
40. Peddio A, Pietroluongo E, Lamia MR, Luciano A, Caltavituro A, Buonaiuto R, et al. DLL3 as a potential diagnostic and therapeutic target in neuroendocrine neoplasms: A narrative review. Crit Rev Oncol Hematol. (2024) 204:104524. doi: 10.1016/j.critrevonc.2024.104524
41. Aggarwal RR, Rottey S, Bernard-Tessier A, Mellado-Gonzalez B, Kosaka T, Stadler WM, et al. Phase 1b study of tarlatamab in de novo or treatment-emergent neuroendocrine prostate cancer (NEPC). J Clin Oncol. (2024) 42. doi: 10.1200/JCO.2024.42.16_suppl.5012. Abstract 5012: 2024 ASCO Annual Meeting.
42. Beltran H, Johnson ML, Jain P, Schenk EL, Sanborn RE, Thompson JR, et al. Updated results from a phase 1/2 study of HPN328, a tri-specific, half-life (T1/2) extended DLL3-targeting T-cell engager in patients (pts) with small cell lung cancer (SCLC) and other neuroendocrine cancers (NEC). J Clin Oncol. (2024) 42. doi: 10.1200/JCO.2024.42.16_suppl.8090. Abstract 8090: 2024 ASCO Annual Meeting.
43. Wermke M, Gambardella V, Kuboki Y, Felip E, Sanmamed MF, Alese O, et al. Abstract 670P: Phase I trial of the delta-like ligand-3 (DLL3)/CD3 IgG-Like T cell engager BI 764532 in patients (pts) with DLL3-positive tumors: Updated data. Ann Oncol. (2024) 35:S525–6. doi: 10.1016/j.annonc.2024.08.736
44. Arteaga CL and Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. (2014) 25:282–303. doi: 10.1016/j.ccr.2014.02.025
45. Panchal A, Seto P, Wall R, Hillier BJ, Zhu Y, Krakow J, et al. COBRA™: a highly potent conditionally active T cell engager engineered for the treatment of solid tumors. MAbs. (2020) 12:1792130. doi: 10.1080/19420862.2020.1792130
46. Available online at: https://ir.cytomx.com/news-releases/news-release-details/cytomx-therapeutics-announces-positive-initial-phase-1a-dose (Accessed April 30 2025). Press release 08 May 2024.
47. Lum LG, Thakur A, Choi M, Deol A, Kondadasula V, Schalk D, et al. Clinical and immune responses to anti-CD3 x anti-EGFR bispecific antibody armed activated T cells (EGFR BATs) in pancreatic cancer patients. Oncoimmunology. (2020) 9:1773201. doi: 10.1080/2162402X.2020.1773201
48. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
49. Rosenthal MA, Balana C, van Linde ME, Sayehli C, Fiedler WM, Wermke M, et al. ATIM-49 (LTBK-01). AMG 596, a novel anti-EGFRVIII bispecific T cell engager (BITE®) molecule for the treatment of glioblastoma (GBM): planned interim analysis in recurrent GBM (RGBM). Neuro Oncol. (2019) 21:vi283. doi: 10.1093/neuonc/noz219.1195
50. Cheng X. A comprehensive review of HER2 in cancer biology and therapeutics. Genes (Basel). (2024) 15:903. doi: 10.3390/genes15070903
51. Haense N, Atmaca A, Pauligk C, Steinmetz K, Marmé F, Haag GM, et al. A phase I trial of the trifunctional anti Her2 × anti CD3 antibody ertumaxomab in patients with advanced solid tumors. BMC Cancer. (2016) 16:420. doi: 10.1186/s12885-016-2449-0
52. Wermke M, Alt J, Kauh J, Back J, Salhi Y, Reddy V, et al. Preliminary results from a phase I study of GBR 1302, a bispecific antibody T-cell engager, in HER2 positive cancers. Ann Oncol. (2018) 29:viii408–viii409. doi: 10.1093/annonc/mdy288.020
53. Calvo E, Moreno Garcia V, Oh D-Y, Ryu MH, Garralda E, Chung W-P, et al. Abstract 40O - Phase I/Ib open-label study of an HER2-targeted T cell engager (TCE)–SAR443216 in patients (pts) with advanced solid tumors: Intravenous (IV) dose-escalation results. ESMO Open. (2025) 10:104199. doi: 10.1016/j.esmoop.2025.104199
54. Lum LG, Thakur A, Al-Kadhimi Z, Colvin GA, Cummings FJ, Legare RD, et al. Targeted T-cell therapy in stage IV breast cancer: A phase I clinical trial. Clin Cancer Res. (2015) 21:2305–14. doi: 10.1158/1078-0432.CCR-14-2280
55. El-Rayes B, Hendifar AE, Pant S, Wilky BA, Reilley M, Benson AB, et al. (2021). Preliminary safety, pharmacodynamic, and antitumor activity of tidutamab, an SSTR2 x CD3 bispecific antibody, in subjects with advanced neuroendocrine tumors, in: The North American Neuroendocrine Tumor Society’s (NANETS) Annual Symposium, Abstract 109.
56. Liu Y, Wang Y, Sun S, Chen Z, Xiang S, Ding Z, et al. Understanding the versatile roles and applications of EpCAM in cancers: from bench to bedside. Exp Hematol Oncol. (2022) 11:97. doi: 10.1186/s40164-022-00352-4
57. Kebenko M, Goebeler ME, Wolf M, Hasenburg A, Seggewiss-Bernhardt R, Ritter B, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology. (2018) 7:e1450710. doi: 10.1080/2162402X.2018.1450710
58. Borlak J, Länger F, Spanel R, Schöndorfer G, and Dittrich C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcγ receptors. Oncotarget. (2016) 7:28059–74. doi: 10.18632/oncotarget.8574
59. Liu R, Xu J, Lin R, Li N, Li G, Zhang T, et al. 61O Updated results of a phase II trial evaluating an anti-EpCAM x anti-CD3 bispecific antibody, M701, for the treatment of Malignant ascites. Ann Oncol. (2024) 35:S1427–8. doi: 10.1016/j.annonc.2024.10.082
60. Xu J, Zhoo C, Wang S, Ma S, Wang T, Huang S, et al. 539P Interim results of a phase I study of M701, a recombinant anti-EpCAM and anti-CD3 bispecific antibody in EpCAM-positive cancer patients with Malignant ascites. Ann Oncol. (2021) 32:S603. doi: 10.1016/j.annonc.2021.08.1061
61. Cai J, Zhang F, Song Z, Jin J, Lv D, Pang W, et al. 1371P An anti-EpCAM x CD3 bispecific antibody, M701, for the treatment of Malignant pleural effusion in NSCLC patients: Intermediate results of a prospective multicenter phase Ib trial. Ann Oncol. (2024) 35:S862. doi: 10.1016/j.annonc.2024.08.1426
62. Hana C, Thaw Dar NN, Galo Venegas M, and Vulfovich M. Claudins in cancer: A current and future therapeutic target. Int J Mol Sci. (2024) 25:4634. doi: 10.3390/ijms25094634
63. Hao J, Zheng L, Ruihong D, Jieer Y, Xu Q, Wang L-W, et al. Safety and efficacy of IBI389, an anti-CLDN18.2/CD3 bispecific antibody, in patients with advanced pancreatic ductal adenocarcinoma: Preliminary results from a phase 1 study. J Clin Oncol. (2024) 42. doi: 10.1200/JCO.2024.42.16_suppl.4011. Abstract 4011: 2024 ASCO Annual Meeting.
64. Zheng L, Ruihong D, Jieer Y, Xu Q, Guo Z, Hu C, et al. Safety and preliminary efficacy results of IBI389, an anti-CLDN18.2/CD3 bispecific antibody, in patients with solid tumors and gastric or gastro-esophageal tumors: A phase 1 dose escalation and expansion study. J Clin Oncol. (2024) 42. doi: 10.1200/JCO.2024.42.16_suppl.2519. Abstract 2519: 2024 ASCO Annual Meeting.
65. van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer. (2014) 14:121–34. doi: 10.1038/nrc3647
66. Harding JJ, Garrido-Laguna I, Chen X, Basu C, Dowlati A, Forgie A, et al. A phase 1 dose-escalation study of PF-06671008, a bispecific T-cell-engaging therapy targeting P-cadherin in patients with advanced solid tumors. Front Immunol. (2022) 13:845417. doi: 10.3389/fimmu.2022.845417
67. Ren S, Zhang Z, Li M, Wang D, Guo R, Fang X, et al. Cancer testis antigen subfamilies: Attractive targets for therapeutic vaccine (Review). Int J Oncol. (2023) 62:71. doi: 10.3892/ijo.2023.5519
68. Davar D, Williams A, Lopez J, Olson D, Sato T, Shaw H, et al. Phase I safety and efficacy of brenetafusp, a PRAME × CD3 ImmTAC T bispecific, in post-checkpoint cutaneous melanoma (CM). J Immunother Cancer. (2024) 12:A1–A1683. doi: 10.1136/jitc-2024-sitc2024.0694
69. Friedman C, De Burgh Williams A, Lopez JS, Ouali K, Middleton MR, Thistlethwaite F, et al. 750P - Phase I safety and efficacy of brenetafusp, a PRAME × CD3 ImmTAC T cell engager, in platinum resistant ovarian cancer (PROC). Ann Oncol. (2024) 35:S569–70. doi: 10.1016/j.annonc.2024.08.811
70. Lopez JS, Milhem M, Butler MO, Thistlethwaite F, Van Tine BA, D’Angelo SP, et al. Phase 1 study of IMCnyeso, a T cell receptor bispecific ImmTAC targeting NY-ESO-1-expressing Malignancies. Cell Rep Med. (2025) 6:101994. doi: 10.1016/j.xcrm.2025.101994
71. Available online at: https://investors.immatics.com/news-releases/news-release-details/immatics-announces-full-year-2024-financial-results-and-business (Accessed April 30 2025). Press release 27 March 2025.
72. Moek KL, Fehrmann RSN, van der Vegt B, de Vries EGE, and de Groot DJA. Glypican 3 Overexpression across a Broad Spectrum of Tumor Types Discovered with Functional Genomic mRNA Profiling of a Large Cancer Database. Am J Pathol. (2018) 188:1973–81. doi: 10.1016/j.ajpath.2018.05.014
73. Safran H, Druta M, Morse M, Lynce F, Pintova S, Almhanna K, et al. Abstract CT111: Results of a phase 1 dose escalation study of ERY974, an anti-glypican 3 (GPC3)/CD3 bispecific antibody, in patients with advanced solid tumors. Cancer Res. (2021) 81. doi: 10.1158/1538-7445.AM2021-CT111
74. Almhanna K, Hong JY, Chenard-Poirier M, Ryoo B-Y, Lim DW-T, El-Khoueiry AB, et al. Phase I/II, open-label, first-in-human study of the anti-GPC3 T cell engager SAR444200 in patients with advanced solid tumors: Updated safety and pharmacokinetic analysis. ESMO Open. (2025) 10:104165. doi: 10.1016/j.esmoop.2025.104165
75. Segal NH, Melero I, Moreno V, Steeghs N, Marabelle A, Rohrberg K, et al. CEA-CD3 bispecific antibody cibisatamab with or without atezolizumab in patients with CEA-positive solid tumours: results of two multi-institutional Phase 1 trials. Nat Commun. (2024) 15:4091. doi: 10.1038/s41467-024-48479-8
76. Moek KL, Fiedler WM, von Einem JC, Verheul HM, Seufferlein T, de Groot DJ, et al. 427P - Phase I study of AMG 211/MEDI-565 administered as continuous intravenous infusion (cIV) for relapsed/refractory gastrointestinal (GI) adenocarcinoma. Ann Oncol. (2018) 29:viii139–viii140. doi: 10.1093/annonc/mdy279.414
77. Pishvaian M, Morse MA, McDevitt J, Norton JD, Ren S, Robbie GJ, et al. Phase 1 dose escalation study of MEDI-565, a bispecific T-cell engager that targets human carcinoembryonic antigen, in patients with advanced gastrointestinal adenocarcinomas. Clin Colorectal Cancer. (2016) 15:345–51. doi: 10.1016/j.clcc.2016.07.009
78. Stadler CR, Ellinghaus U, Fischer L, Bähr-Mahmud H, Rao M, Lindemann C, et al. Preclinical efficacy and pharmacokinetics of an RNA-encoded T cell-engaging bispecific antibody targeting human claudin 6. Sci Transl Med. (2024) 16:eadl2720. doi: 10.1126/scitranslmed.adl2720
79. Wang S, Wang J, Xia Y, Zhang L, Jiang Y, Liu M, et al. Harnessing the potential of HLA-G in cancer therapy: advances, challenges, and prospects. J Transl Med. (2024) 22:130. doi: 10.1186/s12967-024-04938-w
80. Geva R, Vieito M, Ramon J, Perets R, Pedregal M, Corral E, et al. Safety and clinical activity of JNJ-78306358, a human leukocyte antigen-G (HLA-G) x CD3 bispecific antibody, for the treatment of advanced stage solid tumors. Cancer Immunol Immunother. (2024) 73:205. doi: 10.1007/s00262-024-03790-7
81. Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res. (2016) 22:5829–38. doi: 10.1158/1078-0432.CCR-16-0350
82. Mettu NB, Starodub A, Piha-Paul SA, Abdul-Karim RM, Tinoco G, Shafique MR, et al. Results of a phase 1 dose-escalation study of AMV564, a novel T-cell engager, alone and in combination with pembrolizumab in patients with relapsed/refractory solid tumors. J Clin Oncol. (2021) 39. doi: 10.1200/JCO.2021.39.15_suppl.2555. Abstract 2555: 2021 ASCO Annual Meeting I.
83. Du B, Qin J, Lin B, Zhang J, Li D, and Liu M. CAR-T therapy in solid tumors. Cancer Cell. (2025) 43:665–79. doi: 10.1016/j.ccell.2025.03.019
84. Cao L, Leclercq-Cohen G, Klein C, Sorrentino A, and Bacac M. Mechanistic insights into resistance mechanisms to T cell engagers. Front Immunol. (2025) 16:1583044. doi: 10.3389/fimmu.2025.1583044
85. Singh A, Dees S, and Grewal IS. Overcoming the challenges associated with CD3+ T-cell redirection in cancer. Br J Cancer. (2021) 124:1037–48. doi: 10.1038/s41416-020-01225-5
86. Ahn MJ, Cho BC, Felip E, Korantzis I, Ohashi K, Majem M, et al. Tarlatamab for patients with previously treated small-cell lung cancer. N Engl J Med. (2023) 389:2063–75. doi: 10.1056/NEJMoa2307980
87. Modi S, Yap TA, Tan TJ, Santoro A, Gambardella V, Cassier P, et al. Abstract CT204: Phase 1a/b study of runimotamab, a HER2 x CD3 T cell-engaging bispecific antibody, administered as a single agent and in combination with trastuzumab in patients with HER2-expressing breast cancer (BC). Cancer Res. (2025) 85:CT204. doi: 10.1158/1538-7445.AM2025-CT204
88. Wang Y, Gong J, Sun Y, Yang S, Zhang M, Cui J, et al. Abstract 132P: A phase I clinical trial of QLS31905 in advanced solid tumors. Immunooncol Technol. (2023) 20. doi: 10.1016/j.iotech.2023.100604
89. Liu J, O’Malley D, Van Nieuwenhuysen E, Moore K, Hamilton E, Yeku O, et al. Abstract PO011LBA/1512: Ubamatamab (MUC16xCD3 bispecific antibody) with or without Cemiplimab (anti-PD-1 antibody) in recurrent ovarian cancer: phase 1 clinical and biomarker results. Int J Gynecol Cancer. (2023) 33:A9–A10. doi: 10.1136/ijgc-2023-IGCS.11
90. Ball K, Dovedi SJ, Vajjah P, and Phipps A. Strategies for clinical dose optimization of T cell-engaging therapies in oncology. MAbs. (2023) 15:2181016. doi: 10.1080/19420862.2023.2181016
Keywords: solid tumors, T-cell engager, immunotherapy, early-phase trials, T lymphocytes
Citation: Spinazzola A, Iannantuono GM, Gulley JL, Giudice E, Filetti M, Sganga S, Bianco FL, Floudas CS and Daniele G (2025) Current landscape of T-cell engagers in early-phase clinical development in solid cancers. Front. Immunol. 16:1665838. doi: 10.3389/fimmu.2025.1665838
Received: 14 July 2025; Accepted: 16 September 2025;
Published: 06 October 2025.
Edited by:
Fernando Torres Andón, Institute of Biomedical Research of A Coruña (INIBIC), SpainReviewed by:
Henry Sutanto, Airlangga University, IndonesiaMaria Turchaninova, Institute of Bioorganic Chemistry (RAS), Russia
Cheorl-Ho Kim, Sungkyunkwan University, Republic of Korea
Copyright © 2025 Spinazzola, Iannantuono, Gulley, Giudice, Filetti, Sganga, Bianco, Floudas and Daniele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gennaro Daniele, Z2VubmFyby5kYW5pZWxlQHBvbGljbGluaWNvZ2VtZWxsaS5pdA==