 Jessica Kümmel
Jessica Kümmel Nicolas Schlegel
Nicolas Schlegel Johanna C. Wagner
Johanna C. Wagner- Department of General, Visceral, Transplantation, Vascular and Pediatric Surgery, University Hospital Würzburg, Würzburg, Germany
Inflammatory bowel diseases (IBD), including Crohn´s disease (CD) and ulcerative colitis (UC) remain highly prevalent and are associated with a reduced quality of life in affected patients. The pathophysiology of IBD is multifactorial since genetic predisposition, altered immune function, changes in intestinal microbiota, environmental factors, and loss of intestinal barrier function together induce disease manifestation. A critical key factor is the dysregulation of the immune system which explains that all medical therapeutic approaches target the immune response. However, the success of these therapies is limited and associated with severe side effects which demonstrates the need for novel therapeutic approaches. Previous research demonstrated that CD4+ regulatory T (Treg) cells are important regulators of intestinal homeostasis but are reduced in number and function relative to effector T cells in IBD. This led to the concept that genetically engineered, antigen-specific Tregs may represent a promising strategy to address immune dysregulation in IBD. Due to their antigen specificity, chimeric antigen receptors (CARs) enable additional target-dependent activation and migration of Tregs at disease sites. While CARs are increasingly established for the generation of antigen-specificity for T cell therapies in cancer, the implementation of CARs for IBD is in a preliminary state. Nonetheless, CAR constructs specific to circulating carcinoembryonic antigen (CEA), flagellin, or IL23R have been developed recently for potential application in IBD. Based on these novel developments, this review will discuss the role of Tregs in IBD disorders and present the current state of CAR Treg models.
1 Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are the major types of inflammatory bowel disease (IBD). The prevalence and incidence of these inflammatory disorders have risen over the past decade (1, 2). Currently, a stable, yet high incidence level was observed in countries such as North America and Europe, while the incidence increased significantly in newly industrialized countries, including Africa, Asia, and South American countries, such as Brazil (1).
CD is an inflammatory disease that involves all layers of the intestinal wall and can affect any part of the gastrointestinal tract. It is characterized by episodes of remission and exacerbation (3). CD can have extra-intestinal manifestations, such as oral, musculoskeletal, dermatological, or ocular manifestations (4, 5). Both sexes are equally affected and the median age of onset is 30 years, with two peaks of onset between 15 and 30 years and around 50 years. Symptoms include diarrhea, abdominal pain, weight loss, and nausea. Oral manifestation symptoms contain diffuse lip or mouth swelling, esophageal lesions redness and edema (4, 6). UC is often diagnosed in late adolescence and early adulthood affecting both sexes equally (7). Inflammation is typically restricted to the mucosa, begins in the rectum and may extend continuously through the colon with variable extent or involve the entire mucosal surface (7, 8). Over time, there is often an alternation between periods of progression and regression of the inflammation (7). Symptoms include rectal bleeding, bloody diarrhea, rectal urgency, anemia, weight loss, fever, tachycardia, tenesmus, or nausea (7, 9).
The pathogenesis of IBD is multifactorial as genetic factors, such as heredity or mutations and non-coding variations, as well as environmental factors, including diet, microbiota, and medication influence the development of disease (10, 11). Another key factor is the dysregulation of the immune system, including the initiation of inflammation by triggering a pro- and anti-inflammatory cascade. Immune cells, such as B cells, T cells, macrophages, dendritic cells, and neutrophil granulocytes infiltrate the effector site where the immune response is dysregulated. The infiltrating cells secrete cytokines, such as TNF, interferon-gamma (IFN-γ), interleukins, and other cytokines that activate pro-inflammatory pathways (11, 12). The loss of Treg number and function in the gut wall significantly contributes to the onset of inflammation in IBD (13–15). Current medical treatments for IBD are limited in their efficacy (16), so novel therapeutic approaches are necessary.
Consequently, Tregs have emerged as a promising target for therapeutic interventions in IBD. Thus, in order to achieve disease specificity and re-direct Tregs to the site of inflammation, Tregs with a genetically engineered chimeric antigen receptor (CAR) have been developed (17). Up to now, a few promising pre-clinical studies in IBD models have produced encouraging results, paving the way for subsequent clinical studies. Those already developed CAR Treg models include CEA-specific, Flagellin-specific, and IL23R-CAR Tregs. Prior to clinical translation, it is imperative to address safety concerns, limitations in availability, prevention of CAR Treg exhaustion, and enhancements in phenotypic stability and suppressive function (17, 18).
The following review will give an overview of Tregs and the current state of research on Tregs in IBD as well as the various engineered CAR Treg models that have been developed for the treatment of IBD. With this we highlight the emerging potential of CAR-engineered Tregs as a novel therapeutic option.
2 Regulatory T cells in health and IBD disorders
2.1 Regulatory T cells as immune regulators
In the context of IBD, a dysregulation of pro-inflammatory and anti-inflammatory immune cells has been observed (19, 20). The transition to a pro-inflammatory milieu leads to the increased recruitment of pro-inflammatory immune cells, including neutrophils, macrophages, B cells, and T effector cells (21, 22). This results in a cytokine-rich environment, replete with pro-inflammatory cytokines, such as TNF-α, IL-1ß, and IL-6 (23). Therefore, in order to counteract this pro-inflammatory milieu, it is imperative to enhance the anti-inflammatory response. Therefore, immune cells with anti-inflammatory properties are pivotal actors in this process, such as regulatory T cells (24).
CD4+ regulatory T cells play a pivotal role in the mechanism of immune tolerance and comprise approximately 10% of the total CD4+ T cell population (25, 26). The defining characteristics of Tregs include the expression of cell surface receptors CD25 (IL-2 receptor-α chain) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), along with low expression of CD127 (27–29). Another molecular marker is the Treg master lineage factor forkhead box protein P3 (FOXP3). This transcription factor is encoded by the X chromosome and is crucial for the development and function of Tregs (27, 28). The differentiation process of Treg cells is associated with the induction of FOXP3, for instance, through T cell receptor (TCR) signaling strength or by cytokines such as IL-2, IL-7, or IL-15, which act through the gamma chain of the cytokine receptor. This facilitates the induction of FOXP3 and, consequently, the differentiation process (30, 31). Co-stimulatory signals through CD28, particularly the Lck-binding domain of the CD28 cytoplasmic tail, appear to be involved in the induction of the transcription factor FOXP3 expression and the activation of Tregs (32).
In general, the population of CD4+ regulatory T cells comprises two distinct subsets: tTregs and pTregs, as illustrated in Figure 1. Thymus-derived Tregs (tTregs) are derived from CD4+ T cells within the thymus (Figure 1A) (33). In contrast, peripheral Tregs (pTregs) develop as naïve CD4+ T cells and transform into FOXP3-expressing regulatory T cells in peripheral tissues upon TGF-ß and IL-2 stimulation (Figure 1B) (19, 34, 35). Furthermore, differences in the expression of neuropilin 1 (NRP1) and the member of the Ikaros transcription factor HELIOS have been observed among different Treg populations, with tTregs expressing these proteins constitutively (36, 37). Helios+ Tregs have been shown to exhibit heightened suppressive activity (38). The FOXP3 expression is enhanced by this transcription factor through binding on the FOXP3-promotor (39). In addition, Helios may be a marker for thymic-derived Tregs (40).
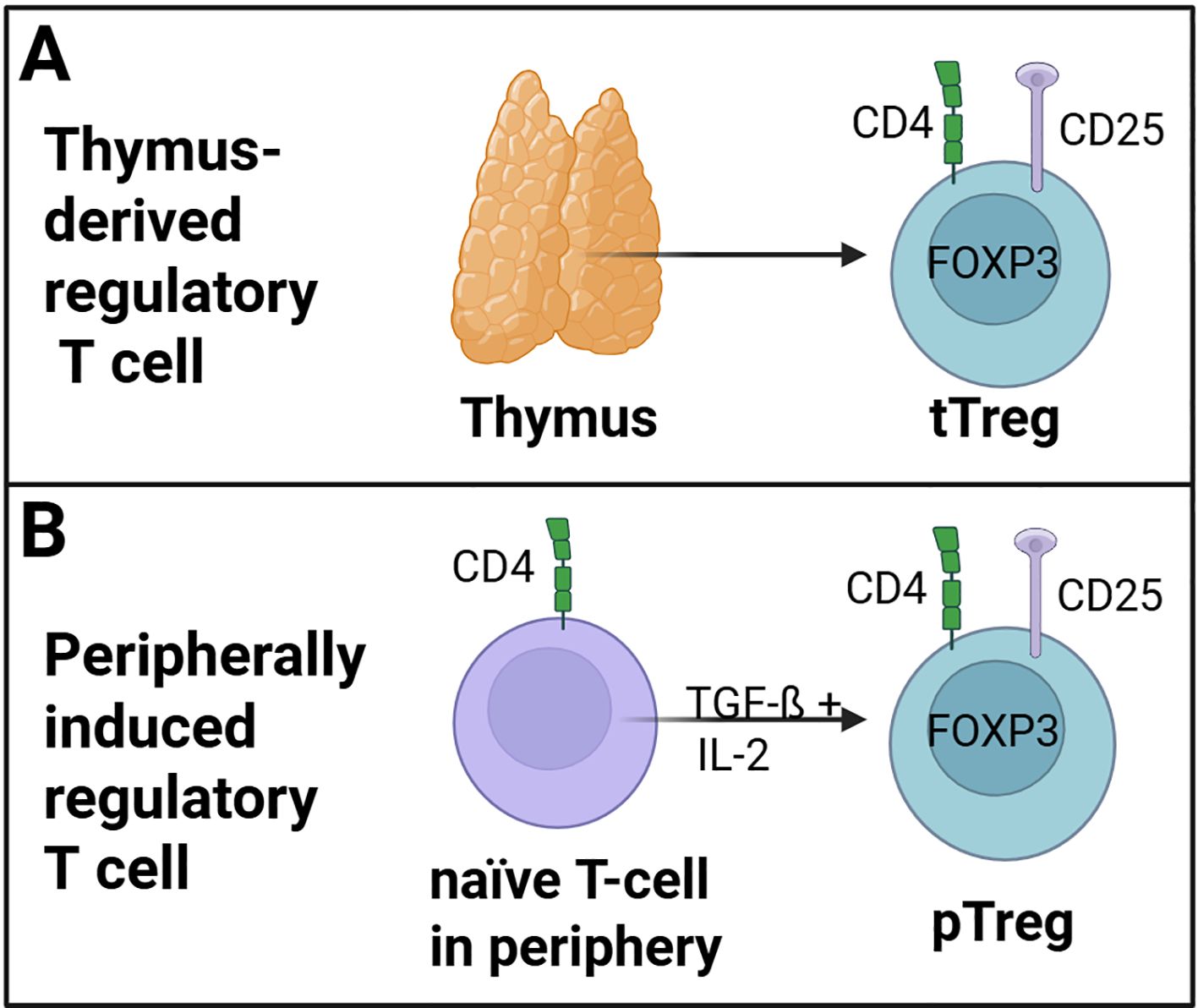
Figure 1. Regulatory T cell (Treg) subsets. (A) tTregs are derived from naïve CD4+ T cells within the thymus. (B) pTregs developed as naïve CD4+ T cells and transformed into FOXP3-expressing pTregs cells in the peripheral tissue trough TGF-ß and IL-2 stimulation. (Created in https://BioRender.com).
The anti-inflammatory potential of Tregs is mediated through a variety of mechanisms, that are summarized in Figure 2 (19, 41–43). One of these mechanisms involves the secretion of factors, such as inducible T cell co-stimulator (ICOS) and inhibitory cytokines, such as IL-10, IL-35, and TGF-ß (Figure 2A) (44–46). Furthermore, Tregs have been demonstrated to suppress the function of CD4+ effector T cells (Teffs) by blocking Teff protein synthesis through the secretion of IL-10 and TGF-ß (47).
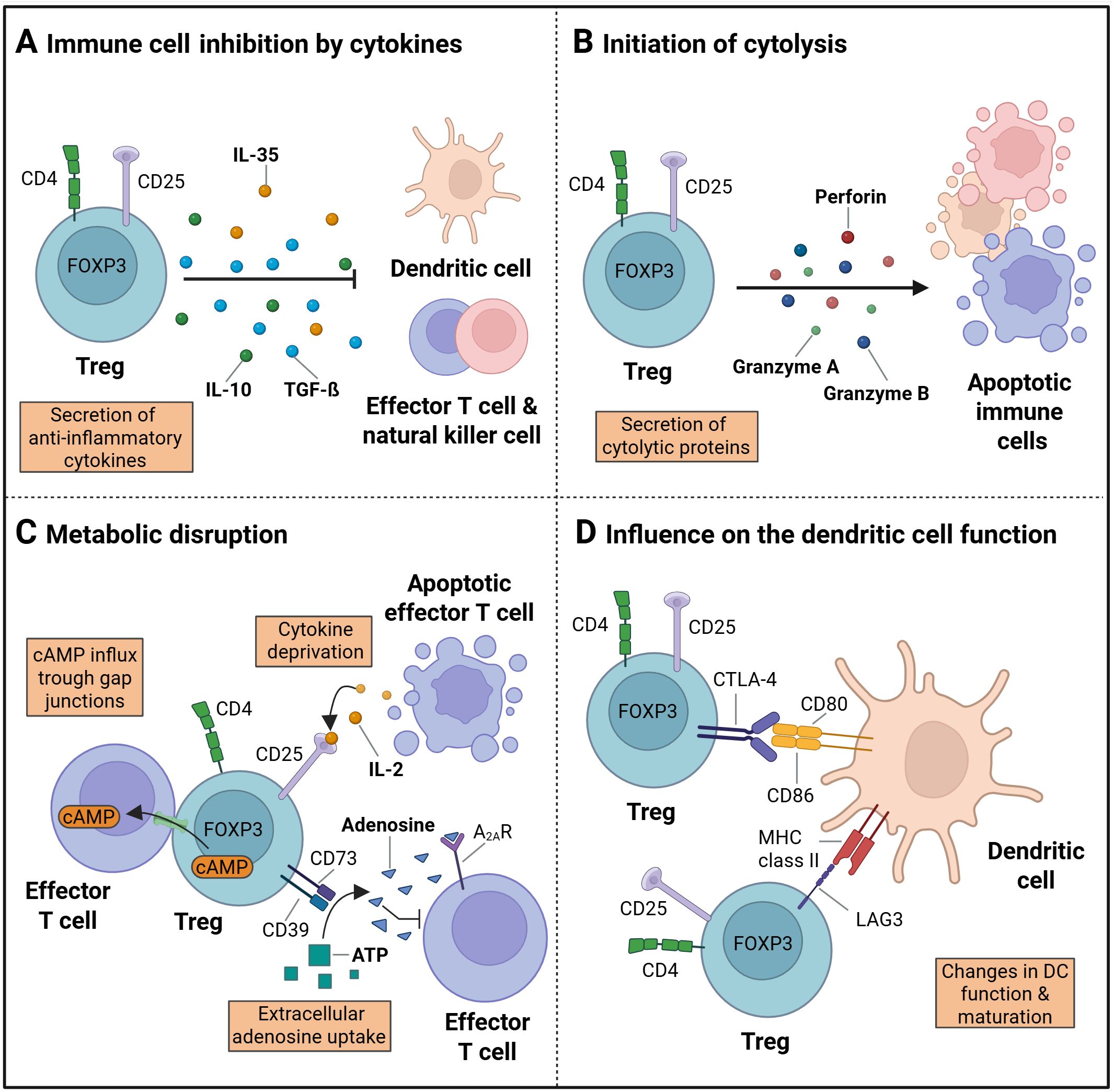
Figure 2. Treg immune regulation. (A) The process of immune suppression by regulatory T cells (Tregs) can be achieved through the secretion of cytokines, such as IL-10, IL-35 and TGF-ß. (B) The initiation of apoptosis following cytolysis is accompanied by the secretion of granzyme A and B, as well as perforins. (C) The disruption of metabolism is facilitated by the influx of cAMP into target cells via gap junctions, the deprivation of cytokines, and the production of extracellular adenosines. (D) The maturation and function of dendritic cells (DC) can be influenced by cell surface receptor proteins, including CTLA-4 and LAG3, which are expressed on Treg cells. (Created in https://BioRender.com). Adapted from “How regulatory T cells work” by Vignali, (D) et al, 2008, Nat Rev Immunol 8, 523-532.
Another mode of action is the suppression by cytolysis via secretion of granzyme A or B and perforin (Figure 2B). Granzymes A and B have been identified as critical mediators of cytolysis in effector T cells, NK cells, and CD8+ cytotoxic T cells (41, 48). Additional regulatory mechanisms encompass metabolic disruption (Figure 2C) (44). Metabolic disruption in the target cells is initiated through a variety of mechanisms, including cyclic AMP (cAMP)-mediated inhibition (49) or apoptosis caused by cytokine deprivation, specifically IL2 deprivation as Tregs exert a high IL-2 consumption (19). The presence of extracellular adenosine nucleosides has been observed to result in the suppression of effector T cell function, NK cell function, and DC maturation. Consequently, the adenosine A2A receptor (A2AR) is activated, and the process of extracellular adenosine formation from ATP by CD39 and CD73 is initiated (50, 51).
A further mechanism by which Treg cells modify the action of immune system cells involves the modulation of DC maturation and function (44). The suppression of DCs can be achieved via secreted cytokines (Figure 2A), initiation of cytolysis (Figure 2B), an uptake of extracellular adenosine nucleosides (Figure 2C), or the removal of co-stimulatory molecules (Figure 2D). The later has been shown to reduce the ability of DCs to stimulate effector T cells by binding of the CTLA-4 Treg surface receptor to CD80 and CD86 on the DC surface (52, 53). In addition, the maturation of DCs can be influenced and their function can be suppressed by binding of lymphocyte-activation gene 3 (LAG3) to MHC class II molecules on the DC surface (54–56).
2.2 Treg dysfunction in inflammatory bowel diseases
Tregs play a key role in the regulation of immune tolerance and in the control of inflammatory lesions in patients with IBD (Figure 3A). A defective function of Tregs in IBD can be a result of inherent defects and extrinsic and intrinsic alterations (57).
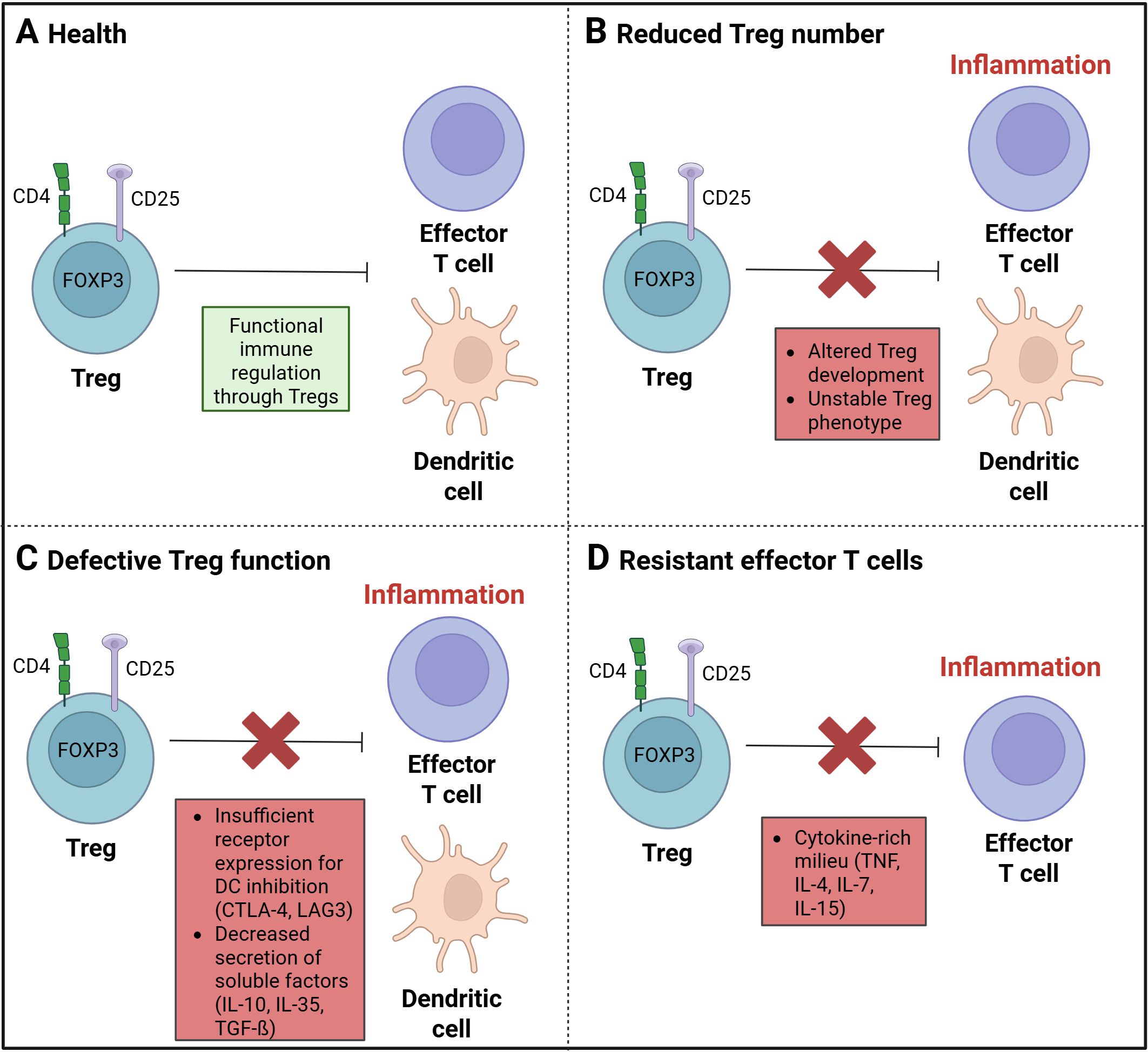
Figure 3. Treg dysregulation in inflammatory diseases. (A) A balanced and functional immune regulation by regulatory T cells (Tregs) is found in a state of health. (B) A disruption in immune regulation, resulting from a decrease in Treg cells, has been linked to a state of imbalance and dysfunction. Such a disturbance may be attributed to either an aberrant Treg development process or an unstable Treg phenotype. (C) The function of Treg cells can be affected by insufficient receptor expression for the DC inhibition through CTLA-4 or LAG3. A decrease in the secretion of soluble factors, such as IL-10, IL-35, or TGF-ß, may also result in dysfunction rather than significant anti-inflammatory effects. (D) A cytokine-rich milieu, comprising TNF, IL-4, IL-7, and IL-15, may improve the development of effector T cells (Teffs) and suppress the Treg function against Teffs. (Created in https://BioRender.com).
In some IBD patients, a reduced number of peripheral Tregs (20) (Figure 3B) as well as an increased level of soluble IL2Rα (CD25) in serum has been observed (58–60). Soluble IL2Rα has been identified as an inflammation marker, resulting from the proteolytic cleavage of the membrane-bound chain, a process known as shedding. This process contributes to the activation of T cells, as observed in the context of inflammation. Conventional CD4+ T cells have been found to produce a higher amount of soluble IL2Rα compared to Tregs (61, 62). The reduced number of regulatory T cells in active IBD (20) could be influenced by an altered thymic development and peripheral homeostasis through factors like CD28, IL-2, or TGF-ß (63, 64) as Tregs are IL-2 and CD28-dependent for their development, activation and function (64–66) and TGF-ß is necessary for maintenance in the periphery (67, 68). A stable Treg phenotype is also important for the immunosuppressive function. The loss of characteristics including the constant expression of the Treg marker FOXP3, leads to a reduction of the anti-inflammatory and immune regulatory function (69).
In patients with active CD, the level of Tregs in the lamina propria was found to be increased, indicating a resistance to Treg-mediated suppression or defective function (70, 71). A defective Treg function (Figure 3C) could result from insufficient expression of cell surface receptors, such as CTLA-4 or LAG3, which are important for the inhibition of dendritic cells. Another dysfunction in regulating immune responses could occur due to a failure of the production and secretion of granzyme A or B, as well as soluble factors, such as IL-10, IL-35, or TGF-ß (63, 72). The deficiency of IL-10 has been demonstrated to induce intestinal inflammation, and a Treg lacking IL-10 receptors has been observed to result in immune-mediated colitis (73).
Through the high level of pro-inflammatory immune cells at the site of intestinal inflammation, altered levels of cytokines such as, IL-1ß, IL-6, IL-12, and TNF-α are observed in the inflamed mucosa or lamina propria (74–77). The higher levels of such pro-inflammatory molecules have been shown to promote the proliferation of pro-inflammatory immune cells, such as effector T cells and, consequently, enhance resistance to Treg suppression (78, 79). It is hypothesized that the presence of a cytokine-rich milieu on the other side could potentially enable a limited function of Tregs (72) and could decrease a stable FOXP3 expression (69). Therefore, the efficacy of Tregs to suppress Teffs is reduced (Figure 3D).
In the context of therapeutic development, it is essential to expand Tregs and enhance the stability of their phenotype. Furthermore, it is necessary to support their suppressive function. Moreover, the specific guidance of improved Tregs to the inflammatory site may be necessary. This can be achieved by engineering of CARs that respond to proteins overexpressed at the site of inflammation. The engineered Treg specificity has the potential to result in more specific migration of anti-inflammatory cells to the effector site and thus increasing the local anti-inflammatory milieu. This could potentially increase their therapeutic success.
3 Chimeric antigen receptors for antigen-specificity
One novel tool to generate antigen-specificity is the use of chimeric antigen receptors (CARs). CARs are synthetically designed to bind to specific target antigens. The advent of CAR T cells can be traced back to 1989, when Zelig Eshar and Gideon Gross first developed engineered T cells (80, 81). Since then, there has been significant progress in the development of CAR constructs, with the emergence of more than four generations of these engineered receptors (Figure 4). The utilization of CARs confers numerous benefits, including the capacity to overcome HLA restrictions and enhance antigen specificity (18, 82, 83).
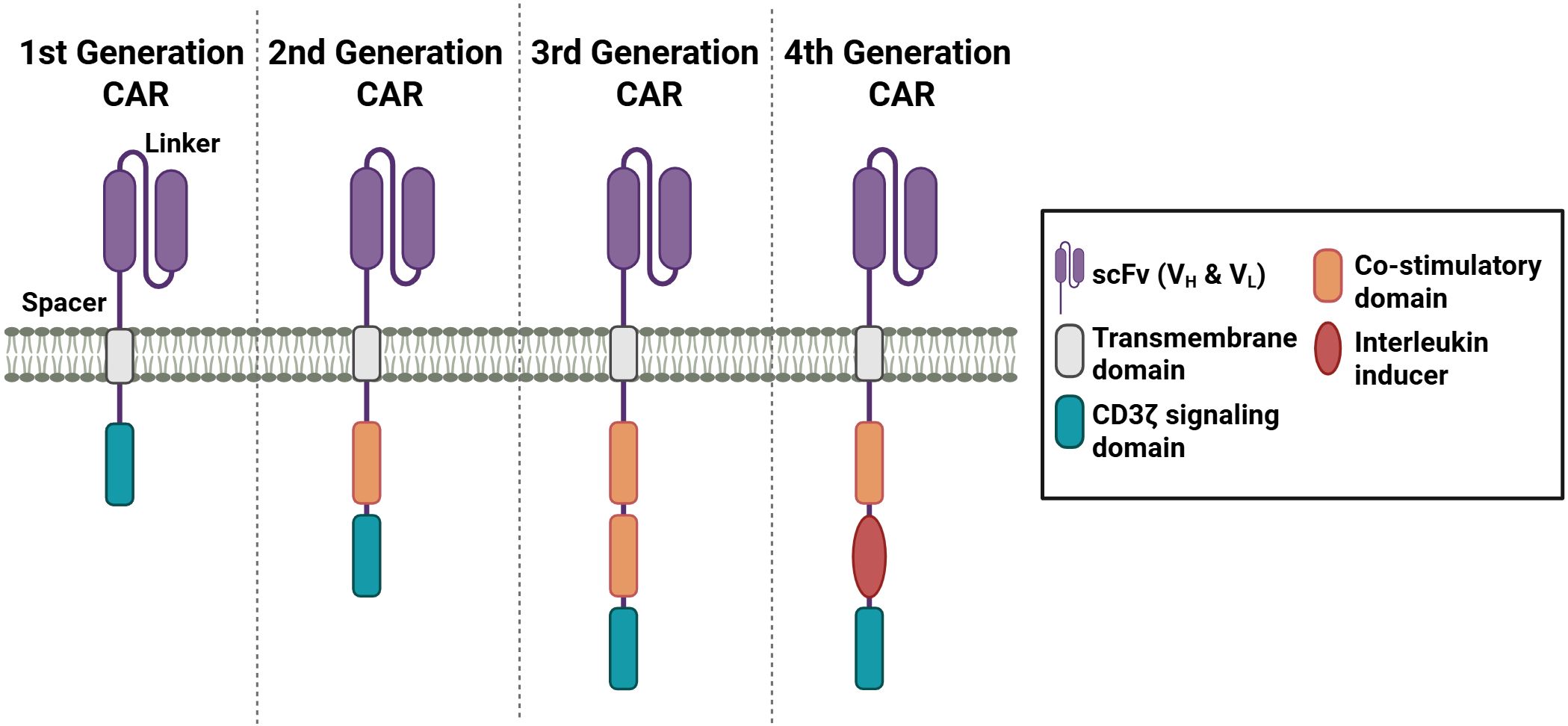
Figure 4. Different generations of CAR receptors. CAR receptors contain a scFv domain, which is connected through a spacer sequence to the transmembrane domain. Depending on the generation, one or more co-stimulatory domains and a CD3 signaling domain are incorporated. The fourth generation of CAR receptors contains an interleukin inducer domain. (Created in https://BioRender.com).
The target antigen is bound through a single-chain variable fragment (scFv) recognition domain originating from an antibody, which allows the specificity of the CAR construct. The scFv domain contains a light (VL) and heavy (VH) variable fragment, which are connected through a linker sequence. The order of the variable fragments, as well as the length of the linker sequences, has been demonstrated to influence the affinity and the stability of the receptor (84–86). The spacer domain contains sequences from CD28 or CD8α or from CH2 or CH3 domains of IgG1, 2, or 4 (87, 88). The length of the spacer domain has been shown to influence the distance between the effector and target cell (84). To anchor the CAR construct, a hydrophobic alpha helix transmembrane (TM) domain is connected to the spacer domain. The TM sequence of the first generation of CARs is derived from the CD3 TM domain (89). In the subsequent generations of CAR constructs, transmembrane sequences from CD4, CD8α, or CD28 were employed (87).
The first generation of CARs only consists of the T cell receptor (TCR) signaling chain CD3ζ, which functions to mediate T cell activation (90). However, the stimulation proved insufficient to achieve T cell expansion and elicit substantial therapeutic responses. In the subsequent second generation, an intracellular co-stimulatory domain, such as CD28 or 4-1BB, was incorporated. Studies have demonstrated that engineered CAR constructs with 4-1BB exhibited advantages in the persistence, while CARs with CD28 co-stimulatory domain revealed more rapid T cell activation (91–93). Third generation of CAR constructs were designed to enhance the T cell activating signal. Thus, two co-stimulatory domains from CD28, 4-1BB, OX40, CD27, or ICOS were combined (94). The incorporation of CD28 and an intracellular domain from the TNFR family into third-generation CAR constructs has been shown to facilitate the simultaneous activation of disparate signaling pathways (95, 96). The fourth CAR generation is based on the second-generation CAR and includes one co-stimulatory domain and an interleukin inducer domain, which enables the inducible release of IL-12 and IL-18 upon CAR activation (97, 98). This release of IL-12 or IL-18 creates a pro-inflammatory environment by recruiting and activating other immune cells, which in turn generate an immune response through further cytokine secretion and the promotion of exocytosis processes via granzyme or perforin. The fourth generation of CAR T cells are also referred to in the literature as TRUCKS, an acronym for “T cells redirected for antigen-unrestricted cytokine-initiated killing” (97–100). Subsequent exploration of additional CAR constructs was conducted in the context of next-generation CARs. One notable example involves the incorporation of a truncated IL-2 receptor β-chain domain, accompanied by a STAT3 binding site, within a next-generation CAR design (99).
The initial application of engineered CAR T cells was in the context of cancer therapies. Consequently, the initial documented success in these therapeutic interventions pertains to the administration of anti-CD19-CAR T cells in patients with advanced leukemia (101). Since then, the possibilities of CAR-T cell therapy have also been transferred to other diseases including allergic asthma (102), diabetes (103, 104), and IBD (105–107). Pre-clinical studies encompass an expanding array of CAR Treg models. A notable example is the development of CAR Tregs specific to β-amyloid for the treatment of Alzheimer’s disease. The efficacy of these cells has been assessed using murine brain lysates in preliminary studies (108). CAR Treg models for diabetes, including insulin B chain (InsB)-specific CAR Tregs, were developed and demonstrated suppressive effects in an adoptive transfer model of T cell-induced autoimmune diabetes in immunodeficient NOD mice, as reported by Spanier et al. (104). A number of HLA-A2-specific CAR Treg models have been engineered for utilization in transplant research, with an objective to avert graft-versus-host disease (GvHD) (109, 110). A number of Phase 1 studies of CAR Treg models are currently underway, including a study of CD6-CAR Tregs for the treatment of patients with chronic GvHD after an allogeneic hematopoietic cell transplantation (HCT) (111). The objective of this Phase 1 study is to ascertain the safety and tolerability of the intervention, as well as to assess its feasibility. In addition, the CAR Treg activity and persistence, as well as the alterations in immune biomarkers, will be monitored (111). A further clinical Phase 1/2 study includes HLA-A2 CAR Tregs (TX200-TR101) for a treatment of living doner kidney transplant recipients (112). The study’s objective is to assess the safety and tolerability of the treatment regimen, in addition to the range of doses that are administered. Additionally, immunosuppression and CAR Treg localization through renal transplant biopsies will be observed (112). A study of a similar nature utilizes HLA-A2 CAR Tregs (QEL-001) in order to prevent liver transplant rejection, as well in a clinical Phase 1/2 study (113). A further example is a clinical Phase 1 study with CAR Tregs specific for citrullinated proteins (Cit-P) for the treatment of refractory rheumatoid arthritis (114). As demonstrated in a series of pre-clinical and early clinical trials, CAR Tregs exhibit therapeutic potential for further applications in immune-related diseases.
4 Antigen-specific CAR Tregs in IBD therapies
As previously stated, Tregs possess the capacity to modulate the activities of immune cells, thereby regulating the inflammatory process in an anti-inflammatory manner. To this end, various models of genetically engineered CAR Tregs have been developed for the treatment of IBD using three different antigens upregulated in patients with IBD, including CEA-specific, Flagellin-specific CAR Tregs and IL23R-CAR Tregs.
4.1 CEA-specific CAR Tregs
The circulating carcinoembryonic antigen (CEA) has been studied as a tumor marker for colorectal cancer (115). However, in patients with inflammatory bowel disease, elevated CEA levels have also been observed (105). The levels were associated with the type, duration, and severity of the disease, as well as with the age and surgical status of the patients (115). Thus, Blat et al. generated a CAR targeting CEA (105).
In the study, the analysis of CEA-specific CAR Tregs was undertaken to ascertain their therapeutic potential in patients suffering from colitis (105). A stable phenotype of these cells, characterized by the expression of FOXP3 in the engineered CAR Tregs, was observed. They demonstrated the characteristic enhancement in IL-2 and IFN-γ secretion by anti-CEA CAR T cell activation and an enhancement of the IL-10 level following anti-CEA CAR Treg activation upon antigen exposure. Subsequent suppression assays showed an improved suppression of CD4+ effector T cells through anti-CEA CAR Tregs when stimulated by CEA antigen. In this study, a decrease in IL-2 levels and a suppression of Teff proliferation were observed. Furthermore, the study noted that i.p. injected CEA-specific CAR Tregs accumulated in the colon of diseased CEABAC-10 mice (105). CEABAC is a human “bacterial artificial chromosome” that contains part of the CEA family gene cluster, which consists of the CEACAM5, CEACAM3, CEACAM6, and CEACAM7 genes (116). The name CEABAC-10 refers to the number of copies of the CEABAC transgene in the mouse model’s genome (116). The expression of CEA is predominantly observed within the gastrointestinal tract (117, 118). Membrane anchorage of CEA is attributed to two distinct groups: glycophosphatidylinositol (GPI)-anchored members, including CEACAM5, CEACAM6, and CEACAM7, and transmembrane members, such as CEACAM3 (116). In a transfer colitis mouse model, which is achieved by injection of anti-CEA CAR CD4+ Teffs, suppressive effects and inhibitory effects on abdominal accumulation of the anti-CAR CD4+ Teffs were achieved by treatment with CEA-specific CAR Tregs. The CAR Tregs showed also in the AOM-DSS model colitis-suppressive effects (105).
4.2 Flagellin-specific CAR Tregs
An aberrant composition of gut microbiota has been associated with the onset of inflammatory bowel diseases. IBD lesions are frequently observed in the terminal ileum and colon, which are known to harbor the highest concentrations of bacteria (119). The identification of dominant bacterial antigens in IBD has highlighted flagellin as a pivotal antigen, particularly in Crohn’s disease. The major subunit of the bacterial flagella is flagellin (119, 120). The flagellum enables the bacteria to move into the intestinal mucus layer (121). The antigen flagellin has been identified as a ligand for Toll-like receptor (TLR) 5 on the surface of innate and adaptive immune cells, including Tregs, and epithelial cells (106). Through interaction of flagellin with the TLR5 receptor, the production and secretion of cytokines and co-stimulatory molecules were initiated in immune cells, such as T cells, monocytes or NK cells (122). This results in an inflammation reaction. It is notable that the colon mucosa is remarkably susceptible to colon flagellin, while an intact colonic mucosa remains unresponsive to luminal flagellin (123). As previously mentioned, TLR5 is also expressed in Tregs, which are known for their immune regulatory and anti-inflammatory functions (124). This characteristic renders flagellin a potential target for Treg therapy.
Boardman et al. (106) engineered a second-generation CAR construct targeting flagellins, which derived from Escherichia coli H18 (FliC). Following transduction with the CAR receptor, the engineered Tregs exhibited a stable phenotype, characterized by the persistent expression of FOXP3 and Helios. The activation of the developed FliC-CARs was achieved in the presence of the target antigen, flagellin. This was observed by an upregulation of CD69, PD1, and GARP, as well as by increased secretion of IL-10. The immunosuppressive reactions were documented by the inhibition of the proliferation of CD4+ and CD8+ responder T cells. In order to investigate the potential of the FliC-specific CARs to guide the engineered cells to damaged intestinal tissues, FliC-CAR T cells were injected intravenously in immunodeficient NSG mice. An accumulation of the FliC-CAR T cells in the colon was observed (106). The study by Boardman et al. (106) demonstrates the potential of specific genetically engineered CAR constructs on regulatory T cells for IBD therapies, based on identified overexpressed or highly available target antigens at the inflammation site.
4.3 IL23R-CAR Tregs
Genome-wide studies have revealed an association of Crohn’s disease with the IL-23 signaling pathway through the IL-23 receptor (IL23R) (125), thereby emphasizing an essential role of IL-23 in the pathogenesis and manifestation of IBD (126). The IL23R expression has been found to be elevated in the intestinal tracts of patients with Crohn’s disease (107). Furthermore, the presence of specific T cell subsets at the intestinal inflammatory site is influenced by secreted IL-23 cytokines. In patients with ulcerative colitis (UC) and Crohn’s disease (CD), an increased production of IL-23 by myeloid cells or neutrophils has been observed (126, 127). Additionally, IL23R signaling in T cells has been shown to inhibit FOXP3 expression, thereby resulting in the inhibition of differentiation of peripheral naive T cells to pTregs (128).
Cui et al. (107) generated specific CAR Tregs targeting IL23R for the treatment of CD disorders. It was observed that the IL23R-CAR expression did not modify FOXP3 expression in the Tregs. The study further demonstrated the capacity of these CAR Tregs to induce antigen-specific immunosuppression through the inhibition of CD4+ T cell proliferation. Subsequent analyses further confirmed the ability of engineered CAR Tregs to suppress DC function and maturation. The study emphasized that the selection of CAR constructs with the lowest tonic signal in the absence of stimulation was crucial to prevent Treg exhaustion and the loss of suppression function. This finding underscores the crucial role of CAR design in modulating the function of CAR Tregs. In this regard, IL23R-CAR Tregs have emerged as a promising therapeutic candidate for the treatment of CD, exhibiting characteristics such as antigen-specific immunosuppression and in vivo migration into IL23R-expressing tissue (107).
These three CAR Treg cell products show the immense potential of CAR Treg therapy for patients in IBD (Table 1). Although all three antigen-targets show a disease-specific increase in expression level, the risk of off-site effects remains a problem. For example, CEA is also highly expressed in colon cancer (115), limiting the therapeutic window for this CAR-construct. Similarly, off-target effects can be caused by the CAR Treg with flagellin specificity, the major subunit of bacterial flagella, due to the high bacterial concentration in the gut (119). The IL-23 receptor is expressed on the surface of T cells (128), which could influence immune homeostasis at sites without inflammation.
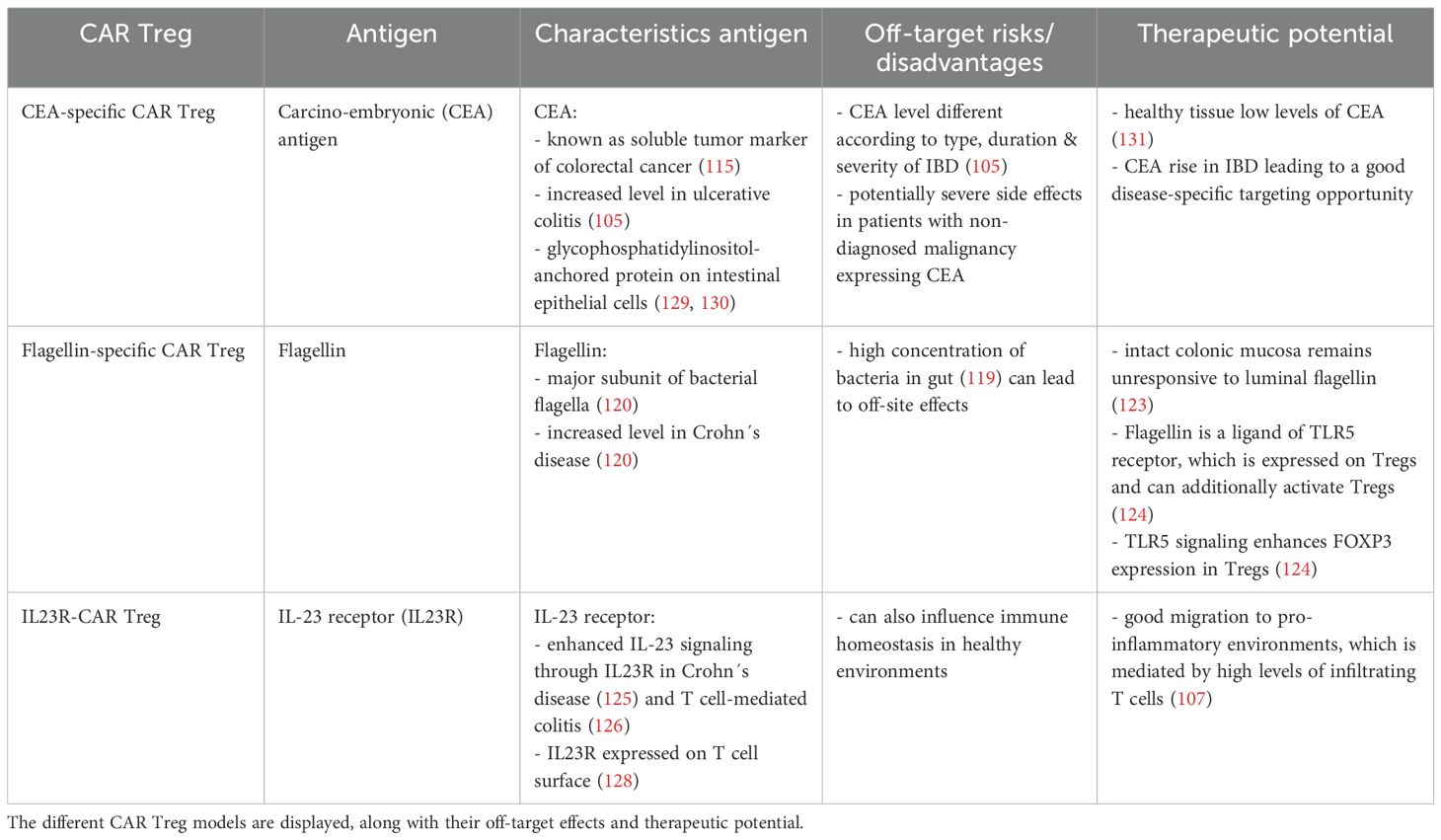
Table 1. CAR Treg model summary.
5 Current challenges and future possibilities
Current therapeutic interventions for IBD, encompassing anti-cytokine treatments and immune cell trafficking/signaling blockers, exhibit limited efficacy (16). Consequently, the use of Tregs has emerged as a promising therapeutic strategy. As antigen-specific Tregs are more potent, genetically engineering CARs for antigen-specificity is a promising therapeutic approach. CARs can be tailored to suit specific requirements. The incorporation of a 4-1BB co-stimulatory domain has been shown to enhance persistence, while a CD28 co-stimulatory domain facilitates more expeditious signaling activation. The modular system can be utilized to develop new and optimized CAR T cells with longer persistence, improved stability, and faster signaling activation or coupling of other mechanisms. Genome-wide studies of IBD have enabled the identification of antigens associated with the disorder, which could be used for additional antigen-specific CAR constructs (125, 132, 133). Therefore, the potential use of IBD-dysregulated epithelial barrier proteins as antigens is possible. Examples include mucus layer protein Muc1 (Mucin-1) and desmosomal proteins such as Dsg3 (Desmoglein-3) (133).
For the regulatory function of Treg cells, the stable expression of FOXP3 as a master transcription factor is essential (134). It has been shown that repetitive in vitro stimulation of human Tregs with anti-CD3 and anti CD28 coated beads leads to loss of FOXP3 expression (135–137). Thus, the application of a Treg therapy in a disease of chronic inflammation could lead to a loss of FOXP3. The stabilization of the expression of this transcription factor could be addressed through the targeting of epigenetic modifiers, such as DNA methyltransferases, histone deacetylases, histone demethylases or methyltransferases, including EZH2 (138, 139). The expression patterns subsequent to Treg activation are influenced by EZH2 methyltransferase (139). Additionally, it has been demonstrated to directly regulate FOXP3 expression (139, 140). A further study observed enhanced expansion and an improvement in FOXP3 expression in response to a combination of IL-2, rapamycin, histone deacetylase, and DNA methyltransferase inhibitors (141). In other studies, an improved and stabilized FOXP3 expression was reported through the addition of vitamin C (142). Treg destabilization upon repeated TCR-stimulation might be rare (143–145), but the effect of repeated CAR stimulation on Tregs remains to be fully examined. Thus, it is worth further investigation considering the supraphysiological signaling through a CAR (146, 147).
In order for CAR Treg treatment to be applied in the clinic, there are a number of further key issues that need to be addressed before its clinical translation. Aside from showing the efficacy of CAR Treg treatment, the safety of this cell therapy will become paramount. The implementation of safety mechanisms, such as suicide programs, which have been developed for CAR effector T cell therapies, can be transferred to CAR Treg therapies. The incorporation of suicide cassettes has been demonstrated to serve the purpose of preventing dysfunctions or depleting the transferred engineered cells (138). Suicide genes that are currently under development include truncated epidermal growth factor receptor (EGFRt) and RQR8 (148). EGFRt is co-expressed with the CAR receptor, and the transferred cells can be eliminated with the application of an EGFRt-directed antibody, cetuximab (149). RQR8 combines parts from CD34 and CD20 antigens. The administration of CD20-directed antibodies, such as rituximab, has been demonstrated to enhance the susceptibility of target cells to lysis (148). Additionally, cells expressing RQR8 could be sorted by CD34 (148). The engineered CaspaCIDe, a safety switch technology, possesses the capability to, through the addition of rimiducid as the trigger, induce the dimerization of the CID domains in cases of toxicity. The subsequent initiation of the apoptosis pathway is catalyzed by iCasp9 (Caspase9) (150).
In other studies, the administration of dasatinib, a tyrosine kinase inhibitor, has been demonstrated to exert effects that block T cell function, with these effects being reversible upon removal (151). The efficacy of this effect may also extend to CAR Tregs.
In addition to efficacy and safety, ensuring the universal accessibility of CAR Treg therapy is imperative. A notable constraint pertains to the financial burden associated with CAR Treg therapy, in addition to the necessity for specialized equipment (17, 152). The provision of CAR Tregs as personalized products is a process that necessitates a substantial duration of production out of patient blood, accompanied by challenges related to the quality of the expanded cell product. Furthermore, IBD patients exhibit reduced Treg cell numbers, which consequently leads to challenges in isolating a sufficient number of cells and formulating a personalized therapy (152, 153). An enhancement in accessibility might be achievable through the standardization of the product and the process as well as the implementation of cryopreserved batches (154, 155). Off-the-shelf products are consistently available. However, they also reveal issues related to cell number, quality, and the time required to generate them. Additionally, HLA (human leukocyte antigen) compatibility is a critical factor in this process. In order to prevent alloreactivity, the generation of banks with different common HLA haplotypes is a potential solution (155). Alternatively, gene editing methods, such as the CRISPR-Cas9 system, can be utilized to eliminate T cell receptors (TCRs). One method involves the direct encoding of CAR DNA in the TRAC locus (156). Other methods involve the rendering of cells HLA-deficient (154, 157).
Many preclinical and clinical trials will have to be conducted before the ideal CAR Treg therapy for patients with IBD can become standard-of-care. This ideal CAR Treg product not only should be efficient, persistent, and safe but lead to a significant reduction in symptom and disease burden while remaining accessible and affordable (Figure 5). Whether an off-the-shelf product or a personalized product with autologous Treg will be a more feasible approach needs to be addressed in future research.
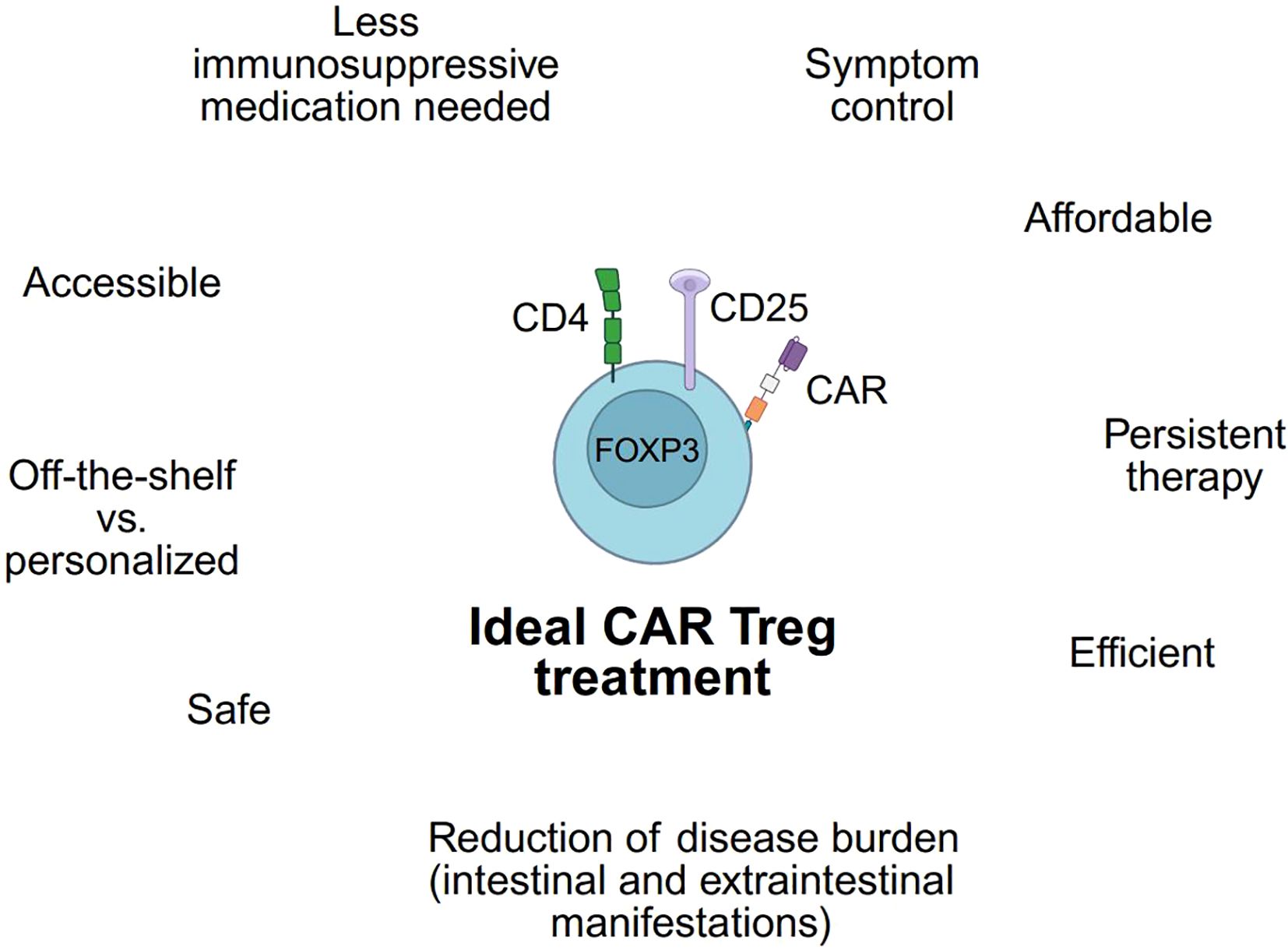
Figure 5. Requirements of an ideal CAR Treg. Utilizing a CAR Treg product to treat IBD patients depends on fulfilling numerous criteria. These requirements are presented for an ideal CAR Treg. (Created in https://BioRender.com).
6 Conclusion
Current IBD therapies are associated with limited success and severe side effects. As Tregs exert anti-inflammatory effects and the ability to suppress pro-inflammatory immune cells, the use of Tregs for the treatment of IBD is a promising approach. Using genetically engineered CAR constructs to guide Tregs to the site of inflammation can significantly enhance their function and therapeutic potential. These CARs have been developed and improved in function and efficacy in several CAR generations in recent years. The design of an appropriate CAR construct with respect to the antigen-specific target and the use in IBD opens new therapeutic opportunities. In preclinical studies, the anti-CEA, anti-flagellin, and anti-IL23R-CAR Tregs have been shown to be promising first developments for the treatment of IBD. Nevertheless, safety concerns must be addressed for clinical translation. The development of suicide programs, such as the incorporation of suicide genes (e.g., EGFRt and RQR8) or the utilization of CaspaCIDe technology in engineered Tregs to enhance safety, has emerged as a pivotal area of focus in contemporary studies, with the objective of facilitating clinical translation. Moreover, the issue of accessibility must be addressed by considering universal useable CAR Tregs that are devoid of allorejection reactions. In addition, the development of strategies to prevent CAR Treg exhaustion and to enhance a stable phenotype and suppressive function could be addressed through the addition of factors against epigenetic modifiers. Such modifiers include DNA methyltransferases, histone deacetylases, histone demethylases, and methyltransferases.
After considering the current limitations, the use of targeted, engineered Tregs to treat inflammation bowel diseases appears to be a promising therapeutic approach to overcome the limitation of current therapies.
Author contributions
JK: Visualization, Writing – original draft. NS: Conceptualization, Writing – review & editing. JW: Writing – review & editing, Funding acquisition, Supervision, Conceptualization.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This publication was funded by the University of Wuerzburg in the funding program Open Access Publishing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. (2017) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
2. GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. (2020) 5(1):17–30. doi: 10.1016/S2468-1253(19)30333-4
3. Ranasinghe IR, Tian C, and Hsu R. Crohn Disease. In: StatPearls. StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC, Treasure Island (FL (2025).
4. Pecci-Lloret MP, Ramirez-Santisteban E, Hergueta-Castillo A, Guerrero-Gironés J, and Oñate-Sánchez RE. Oral manifestations of crohn’s disease: A systematic review. J Clin Med. (2023) 12(20):6450. doi: 10.3390/jcm12206450
5. Sange AH, Srinivas N, Sarnaik MK, Modi S, Pisipati Y, Vaidya S, et al. Extra-intestinal manifestations of inflammatory bowel disease. Cureus. (2021) 13:e17187. doi: 10.7759/cureus.17187
6. Feuerstein JD and Cheifetz AS. Crohn disease: epidemiology, diagnosis, and management. Mayo Clinic Proc. (2017) 92:1088–103. doi: 10.1016/j.mayocp.2017.04.010
7. Magro F, Gionchetti P, Eliakim R, Ardizzone S, Armuzzi A, Barreiro-de Acosta M, et al. Third european evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohn’s Colitis. (2017) 11:649–70. doi: 10.1093/ecco-jcc/jjx008
8. Feuerstein JD, Moss AC, and Farraye FA. Ulcerative colitis. Mayo Clin Proc. (2019) 94:1357–73. doi: 10.1016/j.mayocp.2019.01.018
9. Gajendran M, Loganathan P, Jimenez G, Catinella AP, Ng N, Umapathy C, et al. A comprehensive review and update on ulcerative colitis. Disease-a-Month. (2019) 65:100851. doi: 10.1016/j.disamonth.2019.02.004
10. Shouval DS and Rufo PA. The role of environmental factors in the pathogenesis of inflammatory bowel diseases: A review. JAMA Pediatr. (2017) 171:999–1005. doi: 10.1001/jamapediatrics.2017.2571
11. Ramos GP and Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. (2019) 94:155–65. doi: 10.1016/j.mayocp.2018.09.013
12. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. (2019) 2019:7247238. doi: 10.1155/2019/7247238
13. Mortezaee K. Microbiota interaction with Tregs: a target for colitis. Clin Trans Oncol. (2025). doi: 10.1007/s12094-025-03974-2
14. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. Individual intestinal symbionts induce a distinct population of RORy+ regulatory T cells. Science. (2015) 349:993–7. doi: 10.1126/science.aaa9420
15. Makita S, Kanai T, Nemoto Y, Totsuka T, Okamoto R, Tsuchiya K, et al. Intestinal lamina propria retaining CD4+CD25+ Regulatory T cells is A suppressive site of intestinal inflammation1. J Immunol. (2007) 178:4937–46. doi: 10.4049/jimmunol.178.8.4937
16. Neurath MF, Sands BE, and Rieder F. Cellular immunotherapies and immune cell depleting therapies in inflammatory bowel diseases: the next magic bullet? Gut. (2024) 74(1):9–14. doi: 10.1136/gutjnl-2024-332919
17. Arjomandnejad M, Kopec AL, and Keeler AM. CAR-T regulatory (CAR-Treg) cells: engineering and applications. Biomedicines. (2022) 10(2):287. doi: 10.3390/biomedicines10020287
18. Mohseni YR, Tung SL, Dudreuilh C, Lechler RI, Fruhwirth GO, and Lombardi G. The future of regulatory T cell therapy: promises and challenges of implementing CAR technology. Front Immunol. (2020) 11:1608. doi: 10.3389/fimmu.2020.01608
19. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. (2016) 17:1322–33. doi: 10.1038/ni.3540
20. Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A, et al. Peripheral and intestinal regulatory CD4+CD25high T cells in inflammatory bowel disease. Gastroenterology. (2005) 128:1868–78. doi: 10.1053/j.gastro.2005.03.043
21. Salem M, El Azreq M-A, Pelletier J, Robaye B, Aoudjit F, and Sévigny J. Exacerbated intestinal inflammation in P2Y6 deficient mice is associated with Th17 activation. Biochim Biophys Acta, Mol Basis Dis. (2019) 1865(10):2595–605. doi: 10.1016/j.bbadis.2019.06.019
22. Noronha AM, Liang Y, Hetzel JT, Hasturk H, Kantarci A, Stucchi A, et al. Hyperactivated B cells in human inflammatory bowel disease. J Leukoc Biol. (2009) 86:1007–16. doi: 10.1189/jlb.0309203
23. Raddatz D, Bockemühl M, and Ramadori G. Quantitative measurement of cytokine mRNA in inflammatory bowel disease: relation to clinical and endoscopic activity and outcome. Eur J Gastroenterol Hepatol. (2005) 17(5):547–57. doi: 10.1097/00042737-200505000-00012
24. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. (2011) 117:3921–8. doi: 10.1182/blood-2010-10-311894
25. Tanoue T, Atarashi K, and Honda K. Development and maintenance of intestinal regulatory T cells. Nat Rev Immunol. (2016) 16:295–309. doi: 10.1038/nri.2016.36
26. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, and Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
27. Kosinsky RL, Gonzalez MM, Saul D, Barros LL, Sagstetter MR, Fedyshyn Y, et al. The FOXP3(+) pro-inflammatory T cell: A potential therapeutic target in crohn’s disease. Gastroenterology. (2024) 166:631–44 e17. doi: 10.1053/j.gastro.2024.01.007
28. Fontenot JD, Gavin MA, and Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
29. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. (2006) 203:1701–11. doi: 10.1084/jem.20060772
30. Li X and Zheng Y. Regulatory T cell identity: formation and maintenance. Trends Immunol. (2015) 36:344–53. doi: 10.1016/j.it.2015.04.006
31. Josefowicz SZ, Lu LF, and Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
32. Tai X, Cowan M, Feigenbaum L, and Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. (2005) 6:152–62. doi: 10.1038/ni1160
33. Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol. (1999) 162:5317–26. doi: 10.4049/jimmunol.162.9.5317
34. Selvaraj RK and Geiger TL. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-β. J Immunol. (2007) 179(2):1390. doi: 10.4049/jimmunol.179.2.1390-b
35. Freudenberg K, Lindner N, Dohnke S, Garbe AI, Schallenberg S, and Kretschmer K. Critical role of TGF-β and IL-2 receptor signaling in foxp3 induction by an inhibitor of DNA methylation. Front Immunol. (2018) 9:125. doi: 10.3389/fimmu.2018.00125
36. Ohkura N, Kitagawa Y, and Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. (2013) 38:414–23. doi: 10.1016/j.immuni.2013.03.002
37. Santosh Nirmala S, Kayani K, Gliwinski M, Hu Y, Iwaszkiewicz-Grzes D, Piotrowska-Mieczkowska M, et al. Beyond FOXP3: a 20-year journey unravelling human regulatory T-cell heterogeneity. Front Immunol. (2023) 14:1321228. doi: 10.3389/fimmu.2023.1321228
38. Elkord E, Abd Al Samid M, and Chaudhary B. Helios, and not FoxP3, is the marker of activated Tregs expressing GARP/LAP. Oncotarget. (2015) 6:20026–36. doi: 10.18632/oncotarget.4771
39. Getnet D, Grosso JF, Goldberg MV, Harris TJ, Yen HR, Bruno TC, et al. A role for the transcription factor Helios in human CD4(+)CD25(+) regulatory T cells. Mol Immunol. (2010) 47:1595–600. doi: 10.1016/j.molimm.2010.02.001
40. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. (2010) 184:3433–41. doi: 10.4049/jimmunol.0904028
41. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, and Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
42. Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, et al. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. (2006) 25:129–41. doi: 10.1016/j.immuni.2006.04.015
43. Caridade M, Graca L, and Ribeiro RM. Mechanisms underlying CD4+ Treg immune regulation in the adult: from experiments to models. Front Immunol. (2013) 4:378. doi: 10.3389/fimmu.2013.00378
44. Vignali DAA, Collison LW, and Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
45. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
46. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, and Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res. (2007) 13:4345–54. doi: 10.1158/1078-0432.CCR-07-0472
47. So L, Obata-Ninomiya K, Hu A, Muir VS, Takamori A, Song J, et al. Regulatory T cells suppress CD4+ effector T cell activation by controlling protein synthesis. J Exp Med. (2023) 220(3):e20221676. doi: 10.1084/jem.20221676
48. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, and Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. (2005) 174:1783–6. doi: 10.4049/jimmunol.174.4.1783
49. Rueda CM, Jackson CM, and Chougnet CA. Regulatory T-cell-mediated suppression of conventional T-cells and dendritic cells by different cAMP intracellular pathways. Front Immunol. (2016) 7:216. doi: 10.3389/fimmu.2016.00216
50. Ben Addi A, Lefort A, Hua X, Libert F, Communi D, Ledent C, et al. Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A(2B) receptor. Eur J Immunol. (2008) 38(6):1610–20. doi: 10.1002/eji.200737781
51. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
52. Huang Q, Lam AJ, Boardman DA, Dawson NAJ, and Levings MK. Suppression of human dendritic cells by regulatory T cells. Bio Protoc. (2021) 11:e4217. doi: 10.21769/BioProtoc.4217
53. Walker LSK and Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. (2011) 11:852–63. doi: 10.1038/nri3108
54. Wardell CM, MacDonald KN, Levings MK, and Cook L. Cross talk between human regulatory T cells and antigen-presenting cells: Lessons for clinical applications. Eur J Immunol. (2021) 51:27–38. doi: 10.1002/eji.202048746
55. Bayry J, Triebel F, Kaveri SV, and Tough DF. Human dendritic cells acquire a semimature phenotype and lymph node homing potential through interaction with CD4+CD25+ regulatory T cells. J Immunol. (2007) 178:4184–93. doi: 10.4049/jimmunol.178.7.4184
56. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. (2008) 180:5916–26. doi: 10.4049/jimmunol.180.9.5916
57. Lin W, Truong N, Grossman WJ, Haribhai D, Williams CB, Wang J, et al. Allergic dysregulation and hyperimmunoglobulinemia E in Foxp3 mutant mice. J Allergy Clin Immunol. (2005) 116:1106–15. doi: 10.1016/j.jaci.2005.08.046
58. Yamada A, Arakaki R, Saito M, Tsunematsu T, Kudo Y, and Ishimaru N. Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J Gastroenterol. (2016) 22:2195–205. doi: 10.3748/wjg.v22.i7.2195
59. Louis E, Belaiche J, Van Kemseke C, Schaaf N, Mahieu P, and Mary JY. Soluble interleukin-2 receptor in Crohn’s disease. Digestive Dis Sci. (1995) 40:1750–6. doi: 10.1007/BF02212697
60. Jalalvand M, Enayati S, Akhtari M, Madreseh E, Jamshidi A, Farhadi E, et al. Blood regulatory T cells in inflammatory bowel disease, a systematic review, and meta-analysis. Int Immunopharmacol. (2023) 117:109824. doi: 10.1016/j.intimp.2023.109824
61. Damoiseaux J. The IL-2 – IL-2 receptor pathway in health and disease: The role of the soluble IL-2 receptor. Clin Immunol. (2020) 218:108515. doi: 10.1016/j.clim.2020.108515
62. Kirman I, Nielsen OH, Kjaersgaard E, and Brynskov J. Interleukin-2 receptor α and β chain expression by circulating αβ and γδ T cells in inflammatory bowel disease. Digestive Dis Sci. (1995) 40:291–5. doi: 10.1007/BF02065412
63. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. (2010) 10:849–59. doi: 10.1038/nri2889
64. Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. (2009) 206:1853–62. doi: 10.1084/jem.20090746
65. Setoguchi R, Hori S, Takahashi T, and Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. (2005) 201:723–35. doi: 10.1084/jem.20041982
66. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ Immunoregulatory T cells that control autoimmune diabetes. Immunity. (2000) 12:431–40. doi: 10.1016/S1074-7613(00)80195-8
67. Liston A and Rudensky AY. Thymic development and peripheral homeostasis of regulatory T cells. Curr Opin Immunol. (2007) 19:176–85. doi: 10.1016/j.coi.2007.02.005
68. Boden EK and Snapper SB. Regulatory T cells in inflammatory bowel disease. Curr Opin Gastroenterol. (2008) 24(6):733–741. doi: 10.1097/MOG.0b013e328311f26e
69. Li Z, Arijs I, De Hertogh G, Vermeire S, Noman M, Bullens D, et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm Bowel Dis. (2010) 16(8):1299–310. doi: 10.1002/ibd.21229
70. Reikvam DH, Perminow G, Lyckander LG, Gran JM, Brandtzaeg P, Vatn M, et al. Increase of regulatory T cells in ileal mucosa of untreated pediatric Crohn’s disease patients. Scand J Gastroenterol. (2011) 46:550–60. doi: 10.3109/00365521.2011.551887
71. Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology. (2009) 136(4):1308–e3. doi: 10.1053/j.gastro.2008.12.053
72. Mayne CG and Williams CB. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflammation Bowel Dis. (2013) 19:1772–88. doi: 10.1097/MIB.0b013e318281f5a3
73. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. (2011) 34:566–78. doi: 10.1016/j.immuni.2011.03.018
74. Rao A, Strauss O, Kokkinou E, Bruchard M, Tripathi KP, Schlums H, et al. Cytokines regulate the antigen-presenting characteristics of human circulating and tissue-resident intestinal ILCs. Nat Commun. (2020) 11:2049. doi: 10.1038/s41467-020-15695-x
75. Ng SC, Benjamin JL, McCarthy NE, Hedin CRH, Koutsoumpas A, Plamondon S, et al. Relationship between human intestinal dendritic cells, gut microbiota, and disease activity in Crohn’s disease. Inflamm Bowel Dis. (2010) 17(10):2027–37. doi: 10.1002/ibd.21590
76. Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, and Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol. (1995) 154:2434–40. doi: 10.4049/jimmunol.154.5.2434
77. Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med. (2004) 351:2069–79. doi: 10.1056/NEJMoa033402
78. Walker LS. Regulatory T cells overturned: the effectors fight back. Immunology. (2009) 126:466–74. doi: 10.1111/j.1365-2567.2009.03053.x
79. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
80. Gross G, Gorochov G, Waks T, and Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. (1989) 21:127–30.
81. Gross G, Waks T, and Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. (1989) 86(24):10024–8. doi: 10.1073/pnas.86.24.10024
82. Feins S, Kong W, Williams EF, Milone MC, and Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. (2019) 94:S3–9. doi: 10.1002/ajh.25418
83. Tomasik J, Jasinski M, and Basak GW. Next generations of CAR-T cells - new therapeutic opportunities in hematology? Front Immunol. (2022) 13:1034707. doi: 10.3389/fimmu.2022.1034707
84. Yoo HJ and Harapan BN. Chimeric antigen receptor (CAR) immunotherapy: basic principles, current advances, and future prospects in neuro-oncology. Immunol Res. (2021) 69:471–86. doi: 10.1007/s12026-021-09236-x
85. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, and Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. (2019) 20(6):1283. doi: 10.3390/ijms20061283
86. Whitlow M, Bell BA, Feng S-L, Filpula D, Hardman KD, Hubert SL, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. (1993) 6(8):989–95. doi: 10.1093/protein/6.8.989
87. Guedan S, Calderon H, Posey AD Jr., and Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. (2019) 12:145–56. doi: 10.1016/j.omtm.2018.12.009
88. Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. (2005) 28:203–11. doi: 10.1097/01.cji.0000161397.96582.59
89. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, and Gilham DE. The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3ζ Transmembrane Domain Is Dependent upon Incorporation of the Receptor into the Endogenous TCR/CD3 Complex. J Immunol. (2010) 184:6938–49. doi: 10.4049/jimmunol.0901766
90. Eshhar Z, Waks T, Gross G, and Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
91. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. (2009) 106(9):3360–5. doi: 10.1073/pnas.0813101106
92. Sadelain M, Brentjens R, and Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
93. van der Stegen SJ, Hamieh M, and Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. (2015) 14:499–509. doi: 10.1038/nrd4597
94. Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. (2018) 3(1):e96976. doi: 10.1172/jci.insight.96976
95. Enblad G, Karlsson H, Gammelgard G, Wenthe J, Lovgren T, Amini RM, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. (2018) 24:6185–94. doi: 10.1158/1078-0432.CCR-18-0426
96. Zhong XS, Matsushita M, Plotkin J, Riviere I, and Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. (2010) 18:413–20. doi: 10.1038/mt.2009.210
97. Chmielewski M and Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
98. Glienke W, Dragon AC, Zimmermann K, Martyniszyn-Eiben A, Mertens M, Abken H, et al. GMP-compliant manufacturing of TRUCKs: CAR T cells targeting GD(2) and releasing inducible IL-18. Front Immunol. (2022) 13:839783. doi: 10.3389/fimmu.2022.839783
99. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, and Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
100. Chmielewski M, Kopecky C, Hombach AA, and Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. (2011) 71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103
101. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
102. Skuljec J, Chmielewski M, Happle C, Habener A, Busse M, Abken H, et al. Chimeric antigen receptor-redirected regulatory T cells suppress experimental allergic airway inflammation, a model of asthma. Front Immunol. (2017) 8:1125. doi: 10.3389/fimmu.2017.01125
103. Obarorakpor N, Patel D, Boyarov R, Amarsaikhan N, Cepeda JR, Eastes D, et al. Regulatory T cells targeting a pathogenic MHC class II: Insulin peptide epitope postpone spontaneous autoimmune diabetes. Front Immunol. (2023) 14:1207108. doi: 10.3389/fimmu.2023.1207108
104. Spanier JA, Fung V, Wardell CM, Alkhatib MH, Chen Y, Swanson LA, et al. Tregs with an MHC class II peptide-specific chimeric antigen receptor prevent autoimmune diabetes in mice. J Clin Invest. (2023) 133(18):e168601. doi: 10.1172/JCI168601
105. Blat D, Zigmond E, Alteber Z, Waks T, and Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. (2014) 22:1018–28. doi: 10.1038/mt.2014.41
106. Boardman DA, Wong MQ, Rees WD, Wu D, Himmel ME, Orban PC, et al. Flagellin-specific human CAR Tregs for immune regulation in IBD. J Autoimmun. (2023) 134:102961. doi: 10.1016/j.jaut.2022.102961
107. Cui Y, David M, Bouchareychas L, Rouquier S, Sajuthi S, Ayrault M, et al. IL23R-specific CAR Tregs for the treatment of Crohn’s disease. J Crohns Colitis. (2024) 19(3):jjae135. doi: 10.1093/ecco-jcc/jjae135
108. Saetzler V, Riet T, Schienke A, Henschel P, Freitag K, Haake A, et al. Development of beta-amyloid-specific CAR-tregs for the treatment of alzheimer’s disease. Cells. (2023) 12(16):2115. doi: 10.3390/cells12162115
109. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126(4):1413–24. doi: 10.1172/JCI82771
110. Henschel P, Landwehr-Kenzel S, Engels N, Schienke A, Kremer J, Riet T, et al. Supraphysiological FOXP3 expression in human CAR-Tregs results in improved stability, efficacy, and safety of CAR-Treg products for clinical application. J Autoimmun. (2023) 138:103057. doi: 10.1016/j.jaut.2023.103057
111. Allogeneic CD6 Chimeric Antigen Receptor T Regulatory Cells (CD6-CAR Tregs) for the Treatment of Patients With Chronic Graft Versus Host Disease After Allogeneic Hematopoietic Cell Transplantation (2024). ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/study/NCT05993611 (Accessed October 06, 2025)
112. Safety & Tolerability Study of Chimeric Antigen Receptor T-Reg Cell Therapy in Living Donor Renal Transplant Recipients (STEADFAST) (2021). ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/study/NCT04817774 (Accessed October 06, 2025).
113. Safety and Clinical Activity of QEL-001 in A2-mismatch Liver Transplant Patients (LIBERATE) (2022). ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/study/NCT05234190 (Accessed October 06, 2025).
114. Baxter SK, Moreland L, Griffith M, Massarotti E, Sparks JA, Katsumoto TR, et al. POS0034 REGULATE-RA: A phase 1 study of CAR-T regulatory cells targeting citrullinated proteins in refractory rheumatoid arthritis: interim safety and tolerability. Ann Rheumatic Diseases. (2025) 84:348. doi: 10.1016/j.ard.2025.05.431
115. Rule AH, Goleski-Reilly C, Sachar DB, Vandevoorde J, and Janowitz HD. Circulating carcinoembryonic antigen (CEA): relationship to clinical status of patients with inflammatory bowel disease. Gut. (1973) 14:880–4. doi: 10.1136/gut.14.11.880
116. Chan CHF and Stanners CP. Novel mouse model for carcinoembryonic antigen-based therapy. Mol Ther. (2004) 9:775–85. doi: 10.1016/j.ymthe.2004.03.009
117. Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and Malignant tissues. Semin Cancer Biol. (1999) 9:67–81. doi: 10.1006/scbi.1998.0119
118. Stanners CP. Cell Adhesion and Communication Mediated by the CEA Family (1st ed.). CRC Press: London (1998). p 324. doi: 10.1201/9781482283402
119. Himmel ME, Hardenberg G, Piccirillo CA, Steiner TS, and Levings MK. The role of T-regulatory cells and Toll-like receptors in the pathogenesis of human inflammatory bowel disease. Immunology. (2008) 125:145–53. doi: 10.1111/j.1365-2567.2008.02939.x
120. Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. (2004) 113(9):1296–306. doi: 10.1172/JCI20295
121. Alexander KL, Zhao Q, Reif M, Rosenberg AF, Mannon PJ, Duck LW, et al. Human microbiota flagellins drive adaptive immune responses in crohn’s disease. Gastroenterology. (2021) 161:522–35 e6. doi: 10.1053/j.gastro.2021.03.064
122. Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. (2005) 175:1551–7. doi: 10.4049/jimmunol.175.3.1551
123. Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, and Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci U S A. (2005) 102(38):13610–5. doi: 10.1073/pnas.0502174102
124. Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, and Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. (2005) 175:8051–9. doi: 10.4049/jimmunol.175.12.8051
125. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. (2006) 314:1461–3. doi: 10.1126/science.1135245
126. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. (2006) 116(6):1310–6. doi: 10.1172/JCI21404
127. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. (2019) 20:970–9. doi: 10.1038/s41590-019-0415-0
128. Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. (2010) 33:279–88. doi: 10.1016/j.immuni.2010.08.010
129. Bramswig KH, Poettler M, Unseld M, Wrba F, Uhrin P, Zimmermann W, et al. Soluble carcinoembryonic antigen activates endothelial cells and tumor angiogenesis. Cancer Res. (2013) 73:6584–96. doi: 10.1158/0008-5472.CAN-13-0123
130. Saiz-Gonzalo G, Hanrahan N, Rossini V, Singh R, Ahern M, Kelleher M, et al. Regulation of CEACAM family members by IBD-associated triggers in intestinal epithelial cells, their correlation to inflammation and relevance to IBD pathogenesis. Front Immunol. (2021) 12:655960. doi: 10.3389/fimmu.2021.655960
131. Aldilaijan AF, Kim YI, Kim CW, Yoon YS, Park IJ, Lim S-B, et al. Clinical implication of tissue carcinoembryonic antigen expression in association with serum carcinoembryonic antigen in colorectal cancer. Sci Rep. (2023) 13:7616. doi: 10.1038/s41598-023-34855-9
132. Bai Z, Hao J, Chen M, Yao K, Zheng L, Liu L, et al. Integrating plasma proteomics with genome-wide association data to identify novel drug targets for inflammatory bowel disease. Sci Rep. (2024) 14:16251. doi: 10.1038/s41598-024-66780-w
133. Vancamelbeke M, Vanuytsel T, Farré R, Verstockt S, Ferrante M, Van Assche G, et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflammation Bowel Dis. (2017) 23:1718–29. doi: 10.1097/MIB.0000000000001246
134. Barbi J, Pardoll D, and Pan F. Treg functional stability and its responsiveness to the microenvironment. Immunol Rev. (2014) 259:115–39. doi: 10.1111/imr.12172
135. Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol. (2009) 39:1088–97. doi: 10.1002/eji.200838904
136. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (Tregs) with minimal loss of in vivo functional activity. Sci Trans Med. (2011) 3(83):83ra41. doi: 10.1126/scitranslmed.3001809
137. Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplantation. (2016) 16(1):58–71. doi: 10.1111/ajt.13456
138. Raffin C, Vo LT, and Bluestone JA. T(reg) cell-based therapies: challenges and perspectives. Nat Rev Immunol. (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
139. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity. (2015) 42:227–38. doi: 10.1016/j.immuni.2015.01.007
140. Xiong Y, Khanna S, Grzenda AL, Sarmento OF, Svingen PA, Lomberk GA, et al. Polycomb antagonizes p300/CREB-binding protein-associated factor to silence FOXP3 in a Kruppel-like factor-dependent manner. J Biol Chem. (2012) 287:34372–85. doi: 10.1074/jbc.M111.325332
141. Miyara M, Chader D, Burlion A, Goldstein J, Sterlin D, Norol F, et al. Combination of IL-2, rapamycin, DNA methyltransferase and histone deacetylase inhibitors for the expansion of human regulatory T cells. Oncotarget. (2017) 8:104733–44. doi: 10.18632/oncotarget.10914
142. Kasahara H, Kondo T, Nakatsukasa H, Chikuma S, Ito M, Ando M, et al. Generation of allo-antigen-specific induced Treg stabilized by vitamin C treatment and its application for prevention of acute graft versus host disease model. Int Immunol. (2017) 29:457–69. doi: 10.1093/intimm/dxx060
143. Zhou X, Bailey-Bucktrout S, Jeker LT, and Bluestone JA. Plasticity of CD4+ FoxP3+ T cells. Curr Opin Immunol. (2009) 21:281–5. doi: 10.1016/j.coi.2009.05.007
144. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science. (2010) 329:1667–71. doi: 10.1126/science.1191996
145. Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, and Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A. (2009) 106(6):1903–8. doi: 10.1073/pnas.0811556106
146. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
147. Lamarche C, Ward-Hartstonge K, Mi T, Lin DTS, Huang Q, Brown A, et al. Tonic-signaling chimeric antigen receptors drive human regulatory T cell exhaustion. Proc Natl Acad Sci U S A. (2023) 120:e2219086120. doi: 10.1073/pnas.2219086120
148. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
149. Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. (2011) 118:1255–63. doi: 10.1182/blood-2011-02-337360
150. Gargett T and Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. (2014) 5:235. doi: 10.3389/fphar.2014.00235
151. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. (2019) 11(499):eaau5907. doi: 10.1126/scitranslmed.aau5907
152. Balcerek J, Shy BR, Putnam AL, Masiello LM, Lares A, Dekovic F, et al. Polyclonal regulatory T cell manufacturing under cGMP: A decade of experience. Front Immunol. (2021) 12:744763. doi: 10.3389/fimmu.2021.744763
153. Voskens CJ, Fischer A, Roessner S, Lorenz C, Hirschmann S, Atreya R, et al. Characterization and expansion of autologous GMP-ready regulatory T cells for TREG-based cell therapy in patients with ulcerative colitis. Inflammatory Bowel Diseases. (2017) 23:1348–59. doi: 10.1097/MIB.0000000000001192
154. Depil S, Duchateau P, Grupp SA, Mufti G, and Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. (2020) 19(3):185–99. doi: 10.1038/s41573-019-0051-2
155. Taylor CJ, Peacock S, Chaudhry AN, Bradley JA, and Bolton EM. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell. (2012) 11:147–52. doi: 10.1016/j.stem.2012.07.014
156. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
Keywords: regulatory T cell, CAR receptor, inflammatory bowel diseases, flagellin- specific CAR, IL23R-CAR, cell therapy, immunotherapy
Citation: Kümmel J, Schlegel N and Wagner JC (2025) Opportunities and challenges harnessing antigen-specific CD4+ regulatory T cells in inflammatory bowel disease. Front. Immunol. 16:1667053. doi: 10.3389/fimmu.2025.1667053
Received: 16 July 2025; Accepted: 16 October 2025;
Published: 29 October 2025.
Edited by:
Apostolos Zaravinos, European University Cyprus, CyprusReviewed by:
Vini John, Children’s Hospital of Los Angeles, United StatesXin Xia, Jiangsu University, China
Copyright © 2025 Kümmel, Schlegel and Wagner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johanna C. Wagner, V2FnbmVyX0o2QHVrdy5kZQ==