 Han G. Kim
Han G. Kim Jaimar C. Rincon
Jaimar C. Rincon Philip A. Efron
Philip A. Efron Robert Maile
Robert Maile- Sepsis and Critical Illness Research Center, Department of Surgery, College of Medicine, University of Florida College of Medicine, Gainesville, FL, United States
Innate immune memory, traditionally underappreciated in contrast to adaptive immunity, is now recognized as a critical component of host defense, particularly in the context of sepsis and sterile inflammatory injury. Recent advances have identified a central role for metabolic and epigenetic reprogramming in driving trained immunity (TRIM), where monocytes, macrophages, and other innate cells develop enhanced or tolerized responses to secondary stimuli. This review synthesizes current knowledge of how damage-associated molecular patterns (DAMPs), including oxidized LDL, HMGB1, heme, urate crystals, and mitochondrial DNA, serve as potent inducers of immunometabolic rewiring, often through the mTOR/HIF-1α axis or alternative pathways such as SYK signaling. We highlight distinct epigenetic mechanisms, such as enhancer priming via H3K4me1/H3K27ac, and metabolic shifts like the Warburg effect, succinate accumulation, and fatty acid synthesis, that define the trained or tolerized states. Particular attention is given to the relevance of these mechanisms in the pathophysiology of sepsis, burns, trauma, and other critical illnesses where persistent DAMP exposure may sustain maladaptive inflammation or immunosuppression. We review data linking central (stem cell-level) and peripheral reprogramming to long-term immune dysfunction in various inflammatory disease models, and explore how DAMPs intersect with PAMPs to shape the immune trajectory. Finally, we identify pressing gaps in the field, including the need for standardized TRIM models, validated biomarkers of innate memory, and mechanistic clarity on mitochondrial DAMPs in immune tolerance. These insights provide a foundation for future therapeutic strategies aimed at modulating trained immunity to improve outcomes in critically ill patients.
1 Introduction
Traditionally, immune memory is associated with clonal expansion and antigen specificity that is marked by adaptive immune responses. Meanwhile, the innate immune system has been understood as a nonspecific first line of defense that does not involve immunological memory. Despite a lack of antigen specificity, innate cells exhibit germline-encoded pattern recognition receptors (PRRs) that are able to recognize both Pathogen-Associated Molecular Patterns (PAMPs) as well as Damage-Associated Molecular Patterns (DAMPs). Recent research has demonstrated that these signaling pathways are linked to metabolic/epigenetic rewiring that results in not only short-term peripheral changes in innate immunological function, but also long-term central myeloid-based changes (1) (Figure 1).
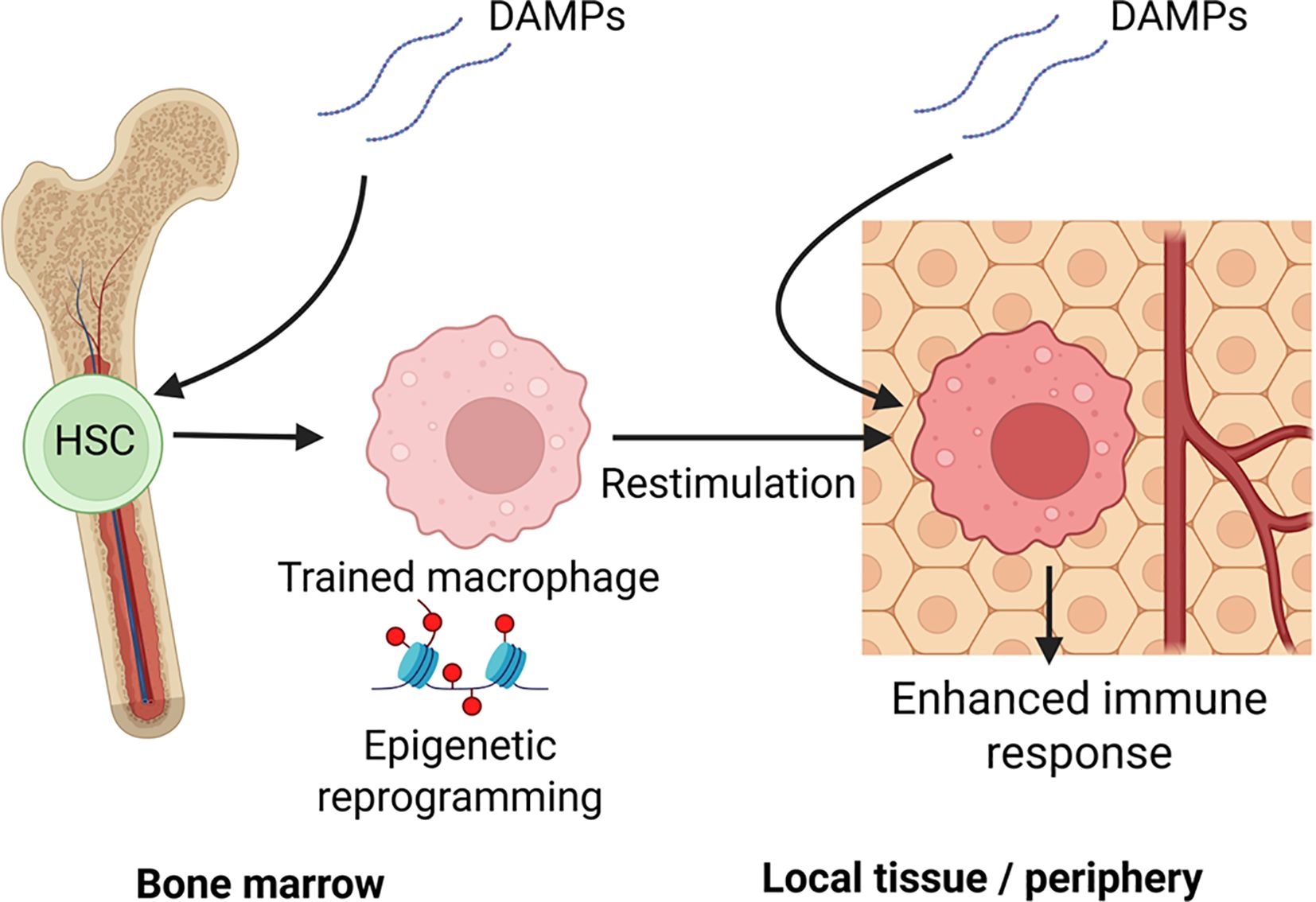
Figure 1. Central and peripheral mechanisms of DAMP-induced innate immune memory: This schematic illustrates how damage-associated molecular patterns (DAMPs), such as those released during trauma, burns, or ischemic injury, can induce trained immunity through both central and peripheral pathways: Central Training (left): DAMPs reaching the bone marrow microenvironment activate pattern recognition receptors (e.g., via IL-1β or systemic oxidized lipids), leading to epigenetic reprogramming of hematopoietic stem and progenitor cells (HSPCs). These changes imprint a trained phenotype in progeny monocytes and macrophages, skewing hematopoiesis toward a myeloid-biased, pro-inflammatory lineage. Peripheral Training (right): In local tissues, resident or recruited monocytes and macrophages encounter DAMPs (e.g., heme, HMGB1, uric acid) and undergo metabolic and epigenetic reprogramming. Upon secondary stimulation (e.g., by infection or reinjury), these cells exhibit altered immune responses; in the figure, trained immunity is enhanced cytokine production, ROS generation, and antimicrobial activity. Together, these establish a durable innate memory state that influences systemic immunity long after the initial DAMP exposure has resolved. Created with BioRender.com.
Three forms of innate immunological memory have now been described: priming, training, and tolerance characterized by distinct function and duration despite common activation using PRRs and epigenetic/metabolic rewiring (Figure 2). Innate priming describes a transient state where restimulation occurs prior to the end of the first stimulation, resulting in an enhanced response. This contrasts with trained immunity (TRIM) where even after the cell returns to metabolic homeostasis, it is able to present an enhanced immune response. Finally, there is innate tolerance which is similar to priming in time-scale, but restimulation results in a decreased ability to mount an immune response. Endotoxin LPS tolerization is a common example for this form of innate memory.
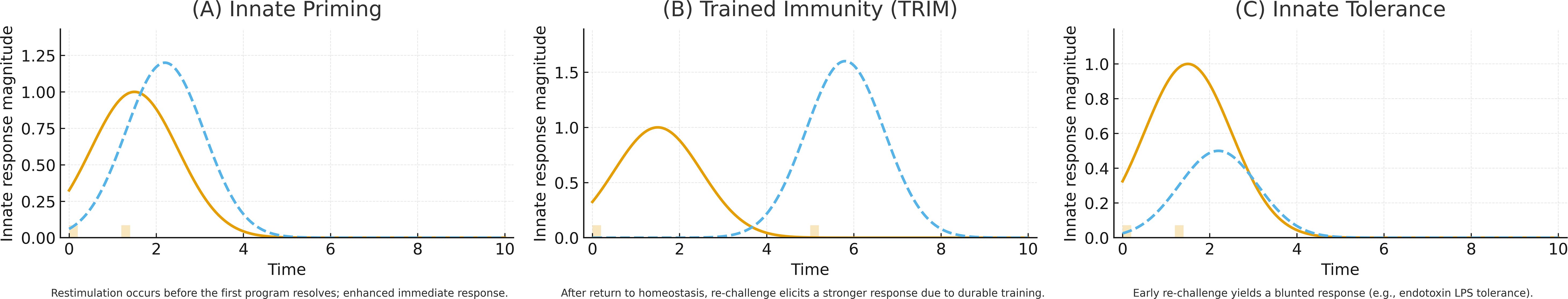
Figure 2. Three forms of innate immunological memory are elicited by common PRR activation and epigenetic/metabolic rewiring but differ in kinetics and functional outcome. (A) Innate priming is transient; re-stimulation before completion of the first transcriptional program yields an enhanced immediate response (orange line, primary response; blue dotted line, re-challenge response). (B) TRIM persists beyond return to metabolic homeostasis, enabling an augmented response to delayed re-challenge. (C) Innate tolerance occurs on a similar timescale to priming, but early re-stimulation produces a blunted response; endotoxin (LPS) tolerization is a canonical example.
Despite differences in presentation, all three types of memory share common underlying mechanisms as evidenced by the ability of one memory type to influence the other. For example, C. albicans cell wall component β-glucan has been shown to reverse epigenetic reprogramming by LPS-tolerization resulting in cells being able to produce robust pro-inflammatory cytokines following LPS re-exposure (2). Studies like these illustrate a need to understand innate memory mechanisms while better defining pathways leading to specific memory responses.
One of the earliest works demonstrating the link between innate memory and epigenetics came from a 2007 study on LPS tolerance. Toll-like Receptor (TLR)-4 mediated LPS signaling of peripheral macrophages induced two gene types with differentiated epigenetic phenotypes; genes that are silenced (tolerizeable) and genes that can be primed (non-tolerizeable) (3). The concept of long-lasting TRIM first began from observations of viral vaccines such as Bacile Calmette-Guérin (BCG) resulting in reduced patient mortality in non-targeted pathogens for months after initial vaccination (4). Further research into BCG and microbial ligands like β-glucan reveal functional reprogramming resulting in nonspecific enhancement of the innate immune system’s ability to respond to subsequent stimuli (5–7). However, this innate functional reprogramming is not exclusive to PAMPs but have also been noted in endogenous DAMPs. For example, oxidized low-density lipoproteins (oxLDL), heme, and vimentin have all been connected to TRIM reprogramming in both autoimmune and sepsis/trauma related disease models (8, 9). Nevertheless, despite evidence of endogenous molecules as inducers of innate training, the difference in mechanism between PAMPs and DAMPs and their coordinated function is still poorly understood.
A fundamental concern in the field is that trained immunity and tolerance lack unified experimental criteria. Different groups use different primary stimuli (from LPS to β-glucan to necrotic cell extracts) and different secondary challenge assays to define “memory.” As a result, what is labeled as trained immunity in one study might not fully match another’s conditions. There are also contradictory findings; for example, some studies report that tolerized macrophages have diminished pathogen killing ability, while others show tolerized cells retaining or even enhancing certain antimicrobial functions. These discrepancies point to unknown modulators (such as cell type, species, or timing differences) that are not accounted for. The lack of standardized models is a problem: without common benchmarks, it is fundamentally difficult to compare results and build a coherent theory. This issue underlies many debates and slows progress in the field.
To understand the role of DAMPs in innate memory, we will outline the key mechanistic underpinnings of innate memory (epigenetics and metabolic changes), and then explore various DAMP-related innate immune training literature to define the current landscape. We will then discuss future directions of study of DAMP-related innate memory.
2 The role of DAMPs in inflammation
DAMPs were first recognized by Polly Matzinger as alarm signals of cellular distress that work alongside pathogen-associated pathways within the immune system (10). These ‘alarmin’ signals can be triggered by various stressors such as radiation, oxidative stress, and chemical agents (11). Both DAMPs and PAMPs act on PRRs which are germ-line proteins that recognize common structures found on bacteria to initiate inflammatory responses (12). There are four main subcategories of PRRs which TLR, Nucleotide-binding oligomerization domain (NOD)- Leucine rich repeat (LRR)-containing receptors (NLRs), Retinoic acid-inducible gene I (RIG-I)-like receptors as well as C-type lectin receptors (CLRs) (13).
In steady-state conditions, these endogenous molecules are thought to not induce inflammatory pathways. However, under infection, stress, or injury, these molecules can be converted into DAMPs via three mechanisms as recently described: displacement from a tolerant position, change of chemical properties, or change in the molecule concentration (14). Generally, DAMPs serve a beneficial role by upregulating immune defense, maintenance of homeostasis, and tissue repair. However, they also facilitate continual upregulation of inflammatory signals that, when pushed to extremes, can result in immune dysfunction. For example, DAMPs like HMGB1 and cell free DNA (cfDNA) from neutrophil NETosis have been correlated with sepsis severity (15–17) Furthermore, DAMPs can play a role in initiating autoimmune conditions with multiple sclerosis studies highlighting the role that high-mobility group box 1 (HMGB1) and heat shock protein 70 (HSP70) play in autoimmune encephalomyelitis (18–20). Moreover, extensive research has been conducted on the DAMP oxLDL for its ability to induce NLRP3-mediated TRIM (21).
However, it is important to note that despite many commonalities in DAMP and PAMP-mediated pathways, there are notable differences in how they interact with the immune system. Although the DAMP HMGB1 has been linked to LPS-mediated TLR4 desensitization (22), studies note DAMPs alone may not be enough to completely inhibit TLR signaling (23). Therefore, DAMPs likely play a synergistic role with PAMPs. For example, the DAMP oxPAPC, the bioactive component of oxLDL, when coactivated with LPS elicits hyperinflammatory metabolic changes compared to isolated treatments (24). Interestingly, although studies involving oxLDL reveal similar training mechanisms to PAMPs like β-glucan, other DAMPs such as heme present divergent pathways resulting in TRIM (8, 9). Furthermore, recent research has proposed alternative methods of endogenous damage signaling. For example, research into extracellular vesicles (EVs) have shown that EVs serve as mediators of inflammatory responses in various traumatic injuries and sepsis (reviewed in (25)). In addition to miRNAs, proteins, mRNA, and lipids, burn EVs possess DAMPs such as HMGB1 (26). Moreover, transfer of EVs isolated from burn patients or mice can induce immunological dysfunction (27–29). These studies illustrate that further research needs to be done to outline key mechanisms behind DAMP’s ability to induce TRIM in disease-relevant models.
3 Central versus peripheral differences in training
Innate memory exists at the level of the local tissue through “peripheral training” while “central training” occurs at the level of Hematopoietic Stem and Progenitor cells (HSPC) such as myeloid progenitor cells in the bone marrow (Figure 1). In BCG-vaccination studies, induction of BCG into bone marrow can induce epigenetically modified macrophages with improved clearance of pathogens (30). However, central reprogramming are not limited to PAMP activity. Duchenne muscular dystrophy (DMD) is a fatal genetic disease linked to trained immunity. In a study using adoptive transfer of bone marrow-derived macrophages from DMD mice to control mice, hyperreactive macrophages could last up to 11 weeks post-transplant (31). DAMPs such as heme can activate hematopoietic stem cells in the bone marrow resulting in both increased percentage and frequency of myeloid lineage differentiation correlating with TRIM-related epigenetic changes and in-vivo resistance to bacterial sepsis (9). Finally, a recent study linked a transient occlusion of the middle cerebral artery to upregulated TRIM responses that persisted in monocytes and macrophages for up to 3 months post-injury (32). This was linked to epigenetic remodeling of bone marrow derived cells, establishing a link between neurological injury to cardiac dysfunction and highlighting a clinical application of DAMP induced training on central immune processes.
4 Epigenetic and metabolic rewiring in the context of innate memory
Divergent innate immune memory states are induced by PAMPs and DAMPs through metabolic and epigenetic reprogramming. In a seminal study, mice lacking T and B cells were protected against reinfection with Candida albicans through monocyte-dependent mechanisms, introducing the term “trained immunity” and linking it to promoter H3K4me3 gains at inflammatory genes (5). Integrative epigenome-transcriptome profiling later showed that monocyte-to-macrophage differentiation and trained immunity involve coordinated promoter and enhancer remodeling, with β-glucan training co-inducing H3K4me3 at promoters and H3K4me1/H3K27ac at distal enhancers (33) Complementing this, β-glucan could partially reverse LPS-induced tolerance in humans and restore cytokine production, underscoring distinct epigenetic routes to training versus tolerance (2).
These observations fit a promoter-enhancer framework in which H3K4me3 marks active promoters, while H3K4me1 and H3K27ac demarcate primed and active enhancers, respectively (34)(Figure 3) The “latent enhancer” model posits that previously unmarked regions acquire transcription factor (TF) binding and enhancer marks upon stimulation and then respond faster upon restimulation (35) Functionally, enhancer acetylation can amplify transcription by modulating burst kinetics, providing a causal link between H3K27ac and increased output (36, 37). Causality is further supported by perturbations of chromatin enzymes. Inhibiting the H3K9 methyltransferase G9a (EHMT2) decreased H3K9me2 at promoters and augmented trained responses to diverse stimuli in human monocytes and in patients receiving BCG (38). Together, these studies support a concise model: β-glucan-induced trained immunity is encoded by coordinated promoter and enhancer rewiring. This is centered on H3K4me3 at promoters and H3K4me1/H3K27ac at enhancers, whose functional consequence is enhanced transcriptional bursting and cytokine output upon rechallenge (reviewed in (39). Non-coding RNAs (ncRNAs) act as upstream “scaffolds” and downstream “switches” that couple metabolism to chromatin in TRIM. A landmark human macrophage study identified UMLILO, a proximal lncRNA at the chemokine locus, which recruits chromatin-modifying machinery to install H3K4me3/H3K27ac and prime CXCL promoters for rapid re-activation. This directly demonstrated an lncRNA requirement for trained responses (1). At the microRNA (miRNA) scale, in β-glucan-trained monocytes, miR-9-5p targets IDH3A, lowers α-KG and favors fumarate accumulation, thereby reinforcing HIF-1α-linked glycolysis and histone methylation that encode training. Genetic loss of miR-9-5p blunted TRIM cytokines in vitro and impairs trained responses in vivo (40). By contrast, miR-146a dampens TLR signaling and is necessary for endotoxin tolerance in human monocytic models (41). These data illustrate how distinct ncRNAs drive trained versus tolerized trajectories.
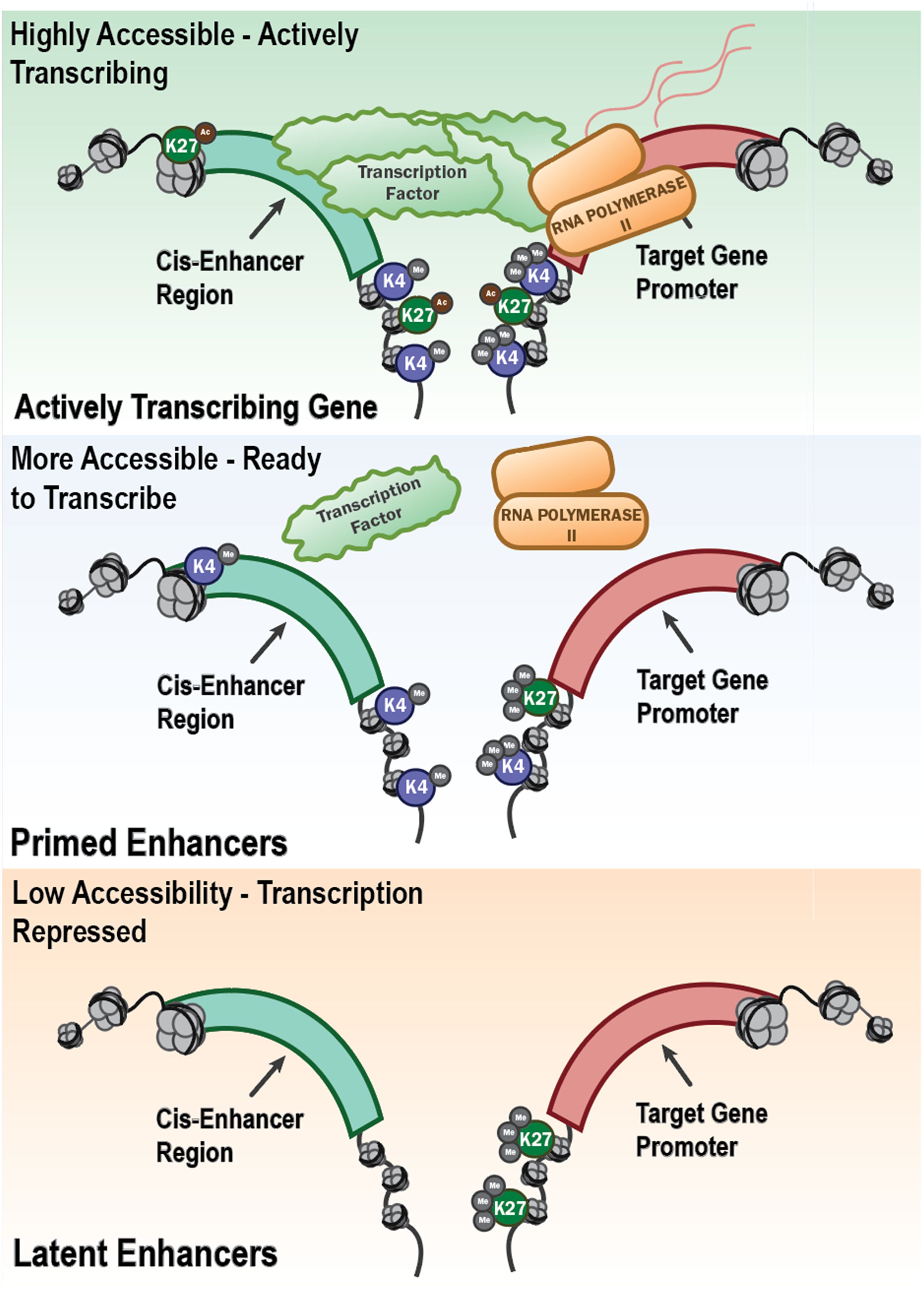
Figure 3. Epigenetic regulation of gene transcription through enhancer accessibility and histone modifications. This schematic illustrates the progressive chromatin states that define the transcriptional potential of innate immune genes. Open chromatin with active histone modifications (e.g., H3K4me3 and H3K27ac) at both the cis-enhancer region and gene promoter allows recruitment of transcription factors and RNA polymerase II, resulting in active transcription. This is shown as “Highly Accessible – Actively Transcribing” (top panel) in the schematic. Other regions, “Primed Enhancers” (middle panel) are regions that are more accessible and marked by H3K4me1 at enhancers and H3K4me3 at promoters, with limited or absent H3K27 acetylation. They are poised for rapid activation but not actively transcribing, representing a key feature of trained innate immunity. In contrast, “Latent Enhancers” (bottom panel) have closed chromatin marked by H3K27me3 or a lack of activating histone marks which are associated with low accessibility and transcriptional repression. These regions may become activated under specific stimuli or remain epigenetically silenced, characteristic of immune tolerance. Together, these chromatin states underlie the long-term transcriptional reprogramming that defines trained immunity or tolerance in innate immune cells.
Upon primary stimulation, monocytes and macrophages also undergo a rapid shift toward aerobic glycolysis with increased glucose uptake and lactate production, which supports ATP generation and biosynthesis required for heightened cytokine output during rechallenge (Figure 4). A seminal mechanistic study established that β-glucan training is driven by the Phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mechanistic target of rapamycin (mTOR) and Hypoxia-Inducible Factor 1α (HIF-1α) axis that installs a glycolytic program; pharmacologic or genetic inhibition of AKT, mTOR, or HIF-1α abrogated both the metabolic shift and trained cytokine responses in vitro and in vivo (42). Consistent human and vaccination data show that BCG training elevates glycolytic flux and glucose uptake, with mTOR as an upstream controller (43). Beyond glycolysis, rewiring of the tricarboxylic-acid network provides signaling metabolites that couple metabolism to chromatin. In β-glucan-trained monocytes, glutaminolysis-driven accumulation of fumarate integrates immunometabolic and epigenetic programs by inhibiting KDM5 histone demethylases. Exogenous fumarate alone can imprint a trained phenotype, whereas blockade of glutaminolysis or cholesterol synthesis impairs training in mice (44). In parallel, the mevalonate branch of cholesterol biosynthesis acts as a pro-training signal: mevalonate activates insulin-like growth factor 1 receptor (IGF1R)-mTOR and promotes histone modifications at inflammatory loci. Statins prevent induction of trained immunity, while mevalonate kinase deficiency is associated with a constitutively “trained” state (45). Diet and sterile DAMPs can similarly program innate memory; notably, short-term Western diet triggers NLRP3-dependent trained immunity and myelopoietic skewing in vivo (21). Tolerance represents a counter-programmed metabolic state characterized by suppression of glycolysis and reliance on oxidative phosphorylation and fatty-acid oxidation, often enforced by anti-inflammatory mediators and stress sensors. Mechanistically, the itaconate pathway limits inflammatory output and contributes to tolerization by directly inhibiting Ten-eleven translocation (TET) DNA dioxygenases, thereby reshaping the epigenome and dampening NF-κB/STAT programs (46). Under glucose restriction or settings favoring catabolism, monocytes compensate with AMPK-driven fatty-acid oxidation and mitochondrial respiration, which sustains core functions while reducing respiratory burst (47) Together, these studies define trained immunity as a coordinated metabolic program centered on mTOR-dependent glycolysis, reinforced by Tricarboxylic Acid Cycle (TCA)-derived signaling metabolites and the mevalonate pathway, that licenses durable functional reprogramming of innate cells. The tolerant trajectory instead engages itaconate-TET2 signaling and AMPK-supported oxidative metabolism to constrain inflammatory outputs.
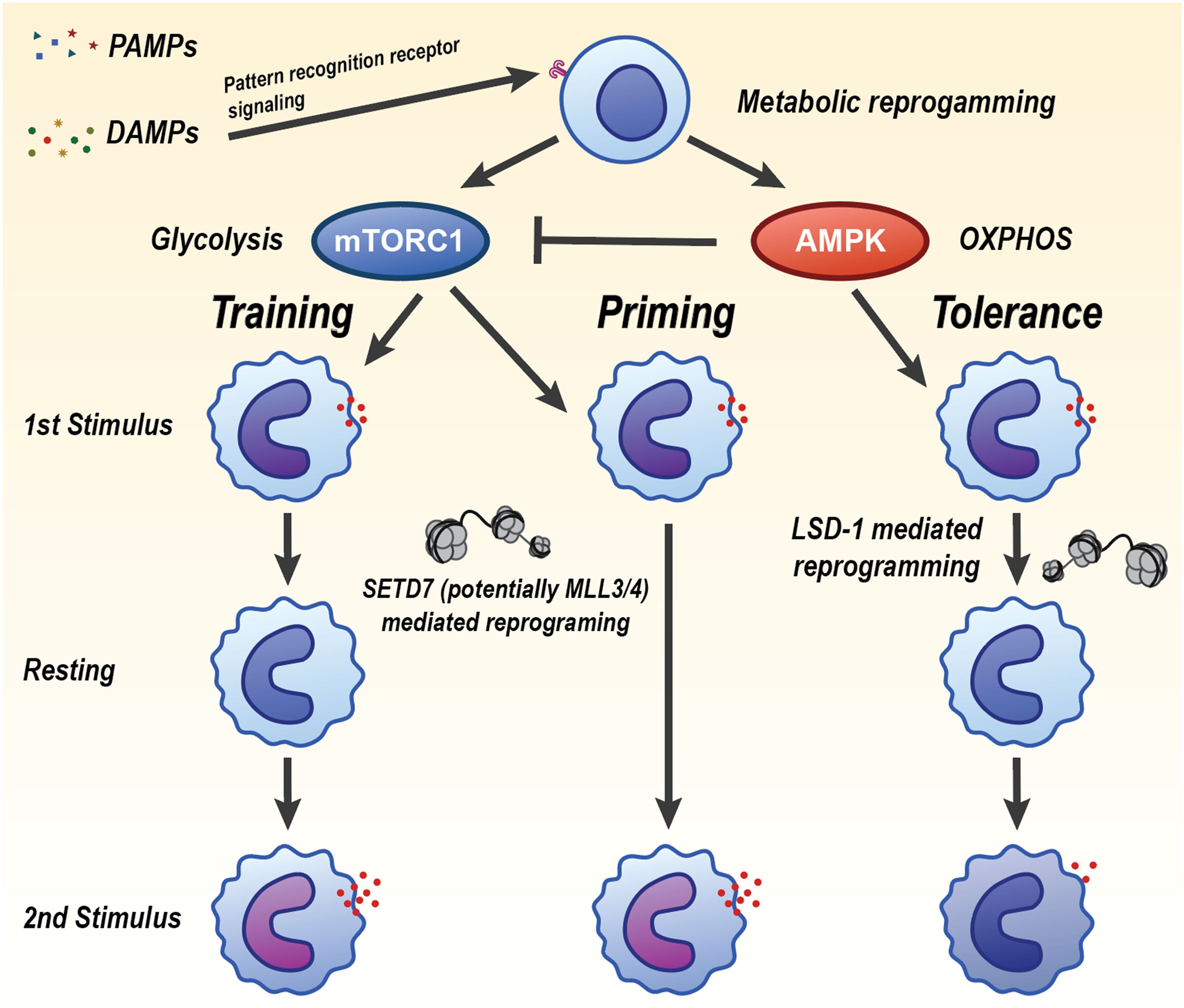
Figure 4. Divergent innate immune memory states induced by PAMPs and DAMPs through metabolic and epigenetic reprogramming. Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) signal through pattern recognition receptors to initiate metabolic reprogramming in innate immune cells. The balance of mTORC1 and AMPK activity may determine whether the cell undergoes trained immunity, priming, or tolerance. Trained immunity is driven by mTORC1 activation and increased glycolysis, leading to heightened inflammatory responses upon secondary challenge. This is maintained by SETD7- and possibly MLL3/4-mediated epigenetic reprogramming. Tolerance is characterized by AMPK-driven oxidative phosphorylation (OXPHOS), reduced inflammatory cytokine production, and is reinforced by LSD1-mediated chromatin remodeling. Priming represents an intermediate or poised state that can resolve into training or tolerance depending on context and secondary cues. Each state is associated with a distinct transcriptional and functional response upon restimulation, regulated by metabolic pathways and histone-modifying enzymes that mediate long-term gene expression changes.
5 Specific DAMPs as modulators of innate memory
DAMPs-mediated innate memory compared to PAMPs, is poorly understood with limited research in its role on both short-term priming and long-term immunometabolic reprogramming. Many studies have since explored DAMPs’ role in TRIM while outlining metabolic and epigenetic reprogramming of both priming and TRIM (48) (Table 1). Recent studies into PAMPs also demonstrate a synergy in PAMP-driven TRIM responses enhanced by the release of DAMPs (49).
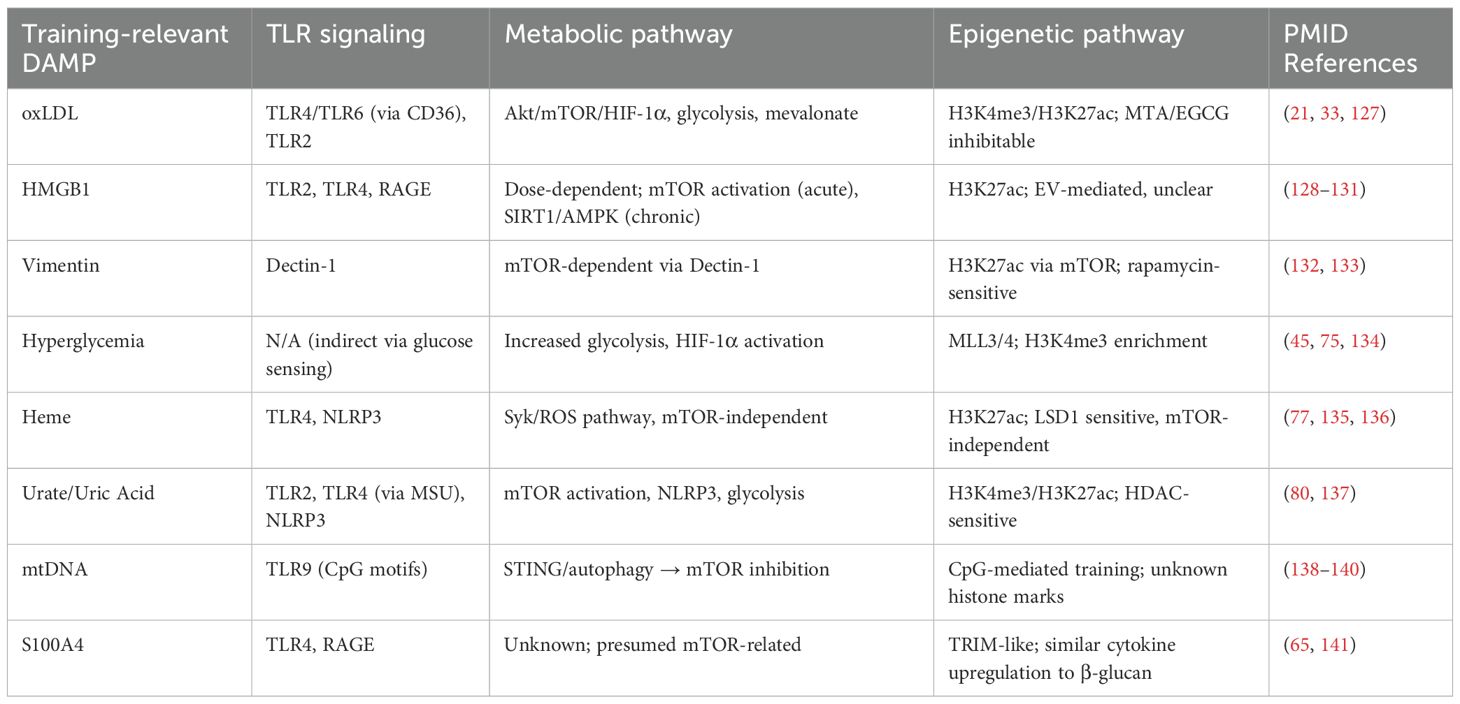
Table 1. Key pattern recognition receptors, metabolic programs, and epigenetic mechanisms involved in DAMP-induced trained immunity.
An important consideration when discussing PAMPs/DAMPs driven innate memory is dosage (Figure 5). Low-level stressors such as LPS engage the dectin-1-mTOR-HIF-1α axis promoting resistance while high-level dosages can activate AMPK and induce tolerance (50). However, DAMPs by their characterization as disequilibrium endogenous molecules involve a dose-dependent consideration in their mechanism. Therefore, although DAMPs-dosing is important, the following section will focus primarily on the roles of different stressors in driving innate memory. Finally, although traditional understanding of DAMP-induced training excludes cytoplasmatic PRR activation (9), research into EVs provides an alternative mechanism for DAMP-induced TRIM involving pathways such as NLR and RIG-1. These EVs may provide an alternative training mechanism as PAMP-based models have shown that gut-derived EVs are linked to MyD88-mediated training of bone marrow-derived neutrophils (51). In the following sections we will explore the role of DAMPs in modulating epigenetic and metabolic reprogramming, and the pathways they may activate in establishing innate memory (Figures 6, 7).
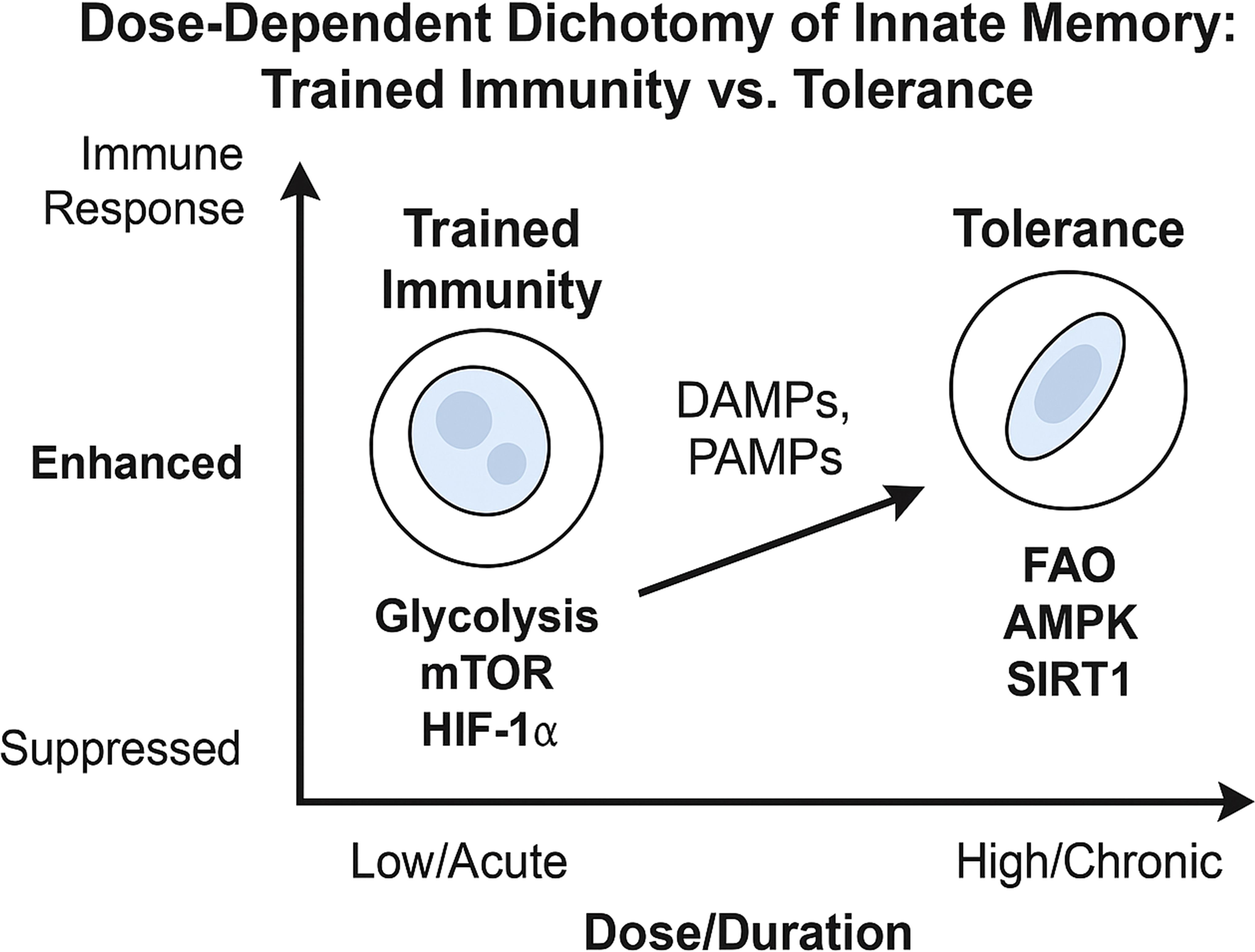
Figure 5. Dose-Dependent dichotomy of trained immunity vs. tolerance. This schematic depicts how low/acute vs. high/chronic doses of DAMPs (or PAMPs) drive innate cells toward either a trained or tolerant phenotype. Low-dose or acute exposure to PAMPs or DAMPs typically activates mTORC1 and HIF-1α–driven glycolysis, leading to trained immunity, characterized by enhanced cytokine production, increased metabolic activity, and epigenetic remodeling (e.g., H3K4me3, H3K27ac) that primes the cell for heightened responses upon secondary stimulation. High-dose or repeated stimulation instead promotes activation of AMPK, SIRT1, and autophagy pathways, resulting in immune tolerance. This state features suppressed inflammatory responses, increased fatty acid oxidation (FAO), and repressive epigenetic modifications (e.g., H3K9me3), which collectively dampen reactivity to subsequent challenges.
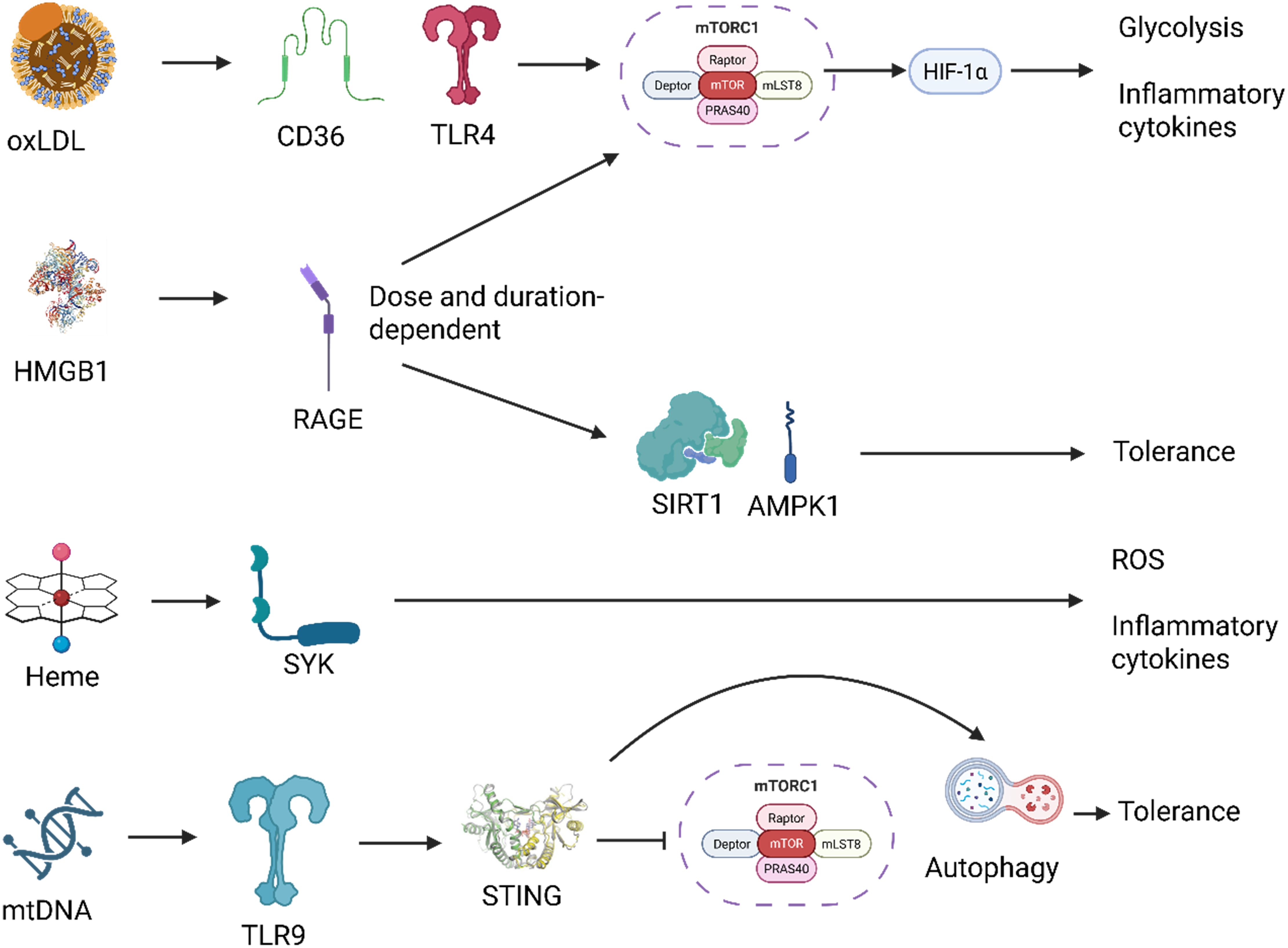
Figure 6. Distinct signaling pathways downstream of DAMPs mediate context-specific innate immune reprogramming. Damage-associated molecular patterns (DAMPs) activate unique receptor-mediated pathways that converge on distinct metabolic and transcriptional responses. For example, oxidized LDL (oxLDL) signals through CD36 and TLR4, activating the mTOR-HIF-1α axis and promoting trained immunity characterized by glycolysis and pro-inflammatory cytokine production. HMGB1, depending on dose and context, activates RAGE, leading to either AMPK/SIRT1-mediated tolerance or mTOR-driven training. Heme triggers SYK kinase signaling, driving reactive oxygen species (ROS) generation and a trained phenotype via a mTOR-independent route. Mitochondrial DNA (mtDNA) is sensed via TLR9, engaging the STING pathway, which intersects with autophagy and inhibits mTOR, promoting an autophagy-high, tolerant state. These divergent signaling routes illustrate how specific DAMPs imprint tailored innate immune memory programs depending on receptor usage, signal integration, and cellular context. Created with BioRender.com.
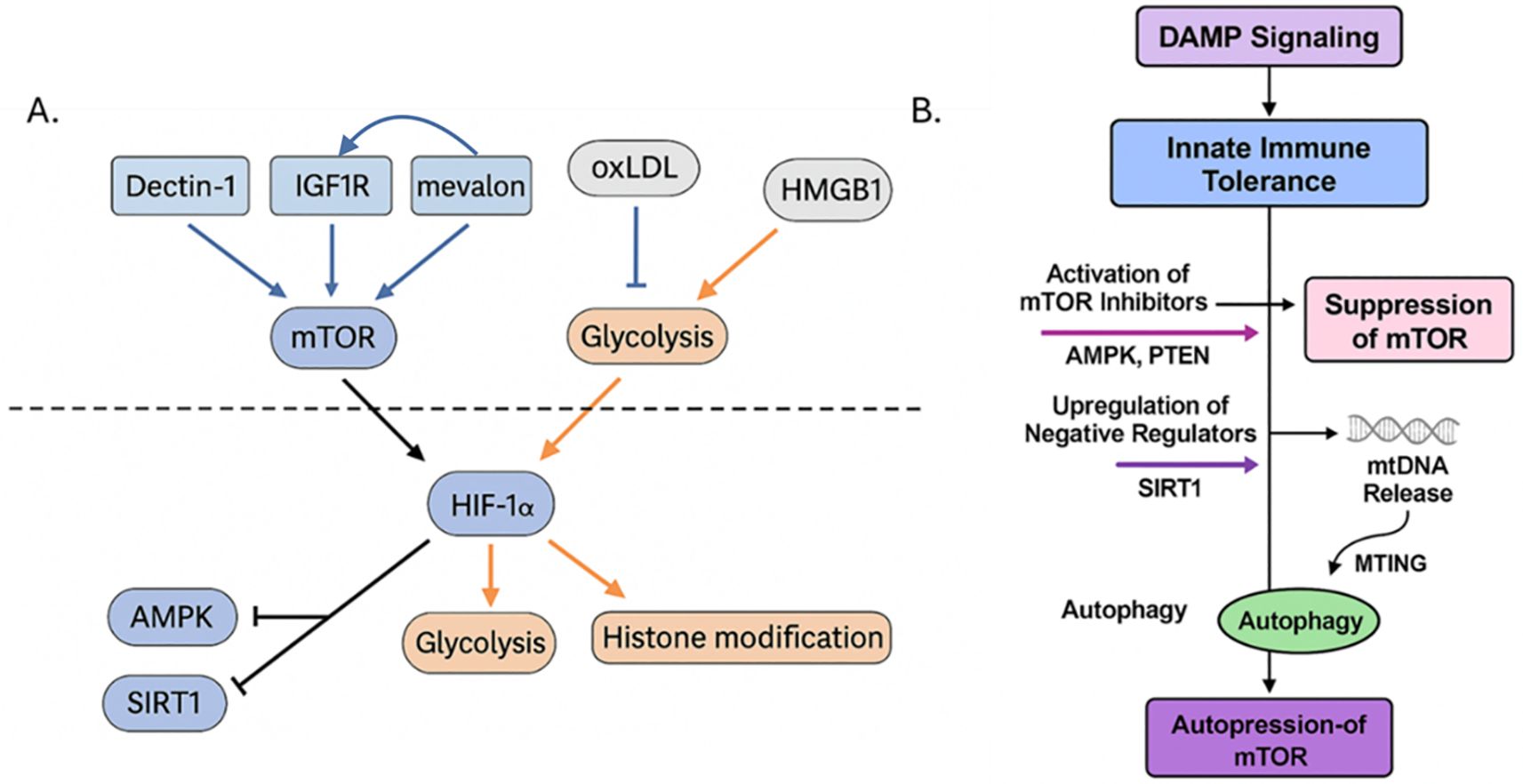
Figure 7. (A) The mTOR signaling axis integrates metabolic and epigenetic cues to regulate trained versus tolerized innate immune states. This diagram illustrates the central role of the mechanistic target of rapamycin (mTOR) in determining whether innate immune cells adopt a trained or tolerized phenotype following DAMP stimulation. Upstream activators such as oxLDL engages CD36/TLR4 and enhances IGF1R signaling via mevalonate (“mevalon”) accumulation, leading to PI3K–AKT–mTOR activation. Dectin-1, often engaged by fungal β-glucan and possibly some DAMPs, also activates mTOR via Syk and downstream AKT. Downstream of mTOR, activation of HIF-1α, glycolysis, and histone acetylation/methylation (e.g., H3K4me3, H3K27ac) leads to enhanced inflammatory gene transcription and the trained immunity phenotype. Various negative feedback and inhibitory loops exist; for example, in tolerized states, AMPK, SIRT1, and STING/autophagy pathways are activated by cellular stress, energy depletion, or chronic DAMP signaling (e.g., high-dose HMGB1 or mtDNA), which suppress mTOR activity. (B) Key inhibitory nodes are activated, such as AMPK and PTEN, which directly suppress mTOR activity, and SIRT1, are activated that are negative regulators that promote a tolerogenic program. In parallel, mitochondrial DNA (mtDNA) release during cellular stress activates STING and related pathways, which further inhibit mTOR through mitochondrial training-induced genes (MTING) and enhance autophagy. Together, these processes reinforce a feedback loop of autophagy-driven mTOR repression, leading to sustained immune tolerance and reduced inflammatory responsiveness. This promotes oxidative metabolism, histone deacetylation or repression, and reduced cytokine expression, resulting in immune tolerance. Created with BioRender.com.
5.1 Oxidized LDL
Atherosclerosis involves the accumulation of oxidized lipids which have been shown to recruit and differentiate monocyte into macrophages within the intimal layer of the artery. It was first demonstrated that oxLDL could induce TRIM in monocytes with TLR4/TLR2 restimulation resulting in an upregulation of TNF-α and IL-6 as well as promoting CD36 and SR-A scavenger receptors resulting in the increase in foam-cell formation (8). Epigenetic analysis demonstrates H3K4me3 enrichment on promoters akin to β-glucan induced training that could be inhibited by methyltransferase inhibitors. Finally, the inhibition of PI3K and ERK could decrease TRIM. Other studies link innate memory of oxLDL with mTOR and NF-kB (52, 53). Considering that oxLDL-induced TRIM can be inhibited by using Torin1 and Raptor/Rictor siRNA gene silencing, this points to oxLDL-TRIM being activated by the Akt/mTOR/HIF-1α axis, a key pathway for β-glucan TRIM (54). However, subsequent studies highlighted an alternative mechanism where Liver X Receptor (LXR) activation can also induce TRIM phenotype (55). LXR agonists could augment oxLDL-induced TRIM while inhibitors reduced IL-6 and TNF-α cytokine response and H3K4me3/H3K27ac enrichment on promoters (56). These activators of HIF-1α result in a mevalonate/IGF1-R feedback loop known to drive TRIM epigenetic remodeling (45).
The “Warburg effect” glycolytic shift, increased cytokine production, and H3K4me3 and H3K27me3 enrichment on the TNF-α promoter is consistent in monocytes isolated from clinical patients with symptomatic atherosclerosis (57). Another focus of oxLDL has been in the Western Diet model with induction of NLRP3-mediated TRIM linked to the systemic inflammation characteristic of a Western Diet (21). Interestingly, a recent study demonstrates that a transient Western Diet followed by a healthier diet can induce TRIM which downregulates subsequent inflammatory response to DSS colitis and Citrobacter rodentium-induced colitis (58). OxLDL TRIM can impact intracellular steroid hormone regulation in monocytes with progesterone inhibiting oxLDL’s TNF-α and IL-6 TRIM response (59). Finally, studies note that epigenetic & metabolic rewiring of TRIM is not exclusive to innate immune cells with studies indicating that oxLDL can induce a mTOR/TLR2/TLR4-mediated TRIM phenotype in both aortic endothelial and coronary smooth muscle cells (60).
5.2 HMGB1 and vimentin
HMGB1 is a nonhistone protein from the nucleus that serves as a chaperone in the nucleus while promoting autophagy in the cytoplasm (61). HMGB1 can be released both actively as well as passively as a DAMP through various cell-death mechanisms such as pyroptosis, necrosis, and apoptosis. When interacting with PRRs TLR2 and TLR4, HMGB1 induces NF-kB dependent transcription (62). HMGB1 meanwhile binds with Mac-1 and RAGE to activate NF-kB and drive neutrophil recruitment in tissue injury (63). Incubation of splenocytes from healthy mice for 8 days with HMGB1, results in splenocytes that can elicit enhanced response to subsequent LPS stimulation (64). In a study comparing TRIM capability of multiple DAMPs to β-glucan, HMGB1 was found to exhibit TRIM properties albeit to a lower extent (65). While the mechanisms of HMGB1 and its different oxidation forms in inducing innate memory are uncertain, recent reports demonstrate how macrophages stimulated with dsHMGB1 present different transcriptomic profile to LPS and LPS/IFN-γ stimulated cells (66).
Vimentin meanwhile is an intermediate filament protein that acts on the dectin-1 receptor (67). Both endogenous vimentin and HMGB1 are released during transplantation and are a focus of post-organ transplantation ischemia-reperfusion injury TRIM. HMGB1 is upregulated during allogenic transplants in mice a week post-surgery (68). A 2018 paper from Braza et al. demonstrate that initial stimulation by vimentin followed by restimulation using HMGB1 could create a TRIM response that was similar to TRIM from β-glucan (69). In the same paper, the team developed rapamycin-based nanobiologics (mTORi-HDL), which could prevent innate training in an in vitro β-glucan monocyte model. Afterwards, they confirmed decreased restimulation by LPS in graft-infiltrating macrophages in mice from the nanobiologics following an allogeneic heart transplant. In studies, HMGB1 has been found to be enriched in EVs from both human and mice burn and express a biphasic immune response characteristic of burn immune dysfunction (26, 70). While no studies have yet linked the role that DAMPs-carrying EVs play in innate memory, more research is needed to understand the mechanisms in which these processes occur.
5.3 Hyperglycemia
In addition to causing complications such as atherosclerotic diseases and increased infection risk, evidence of the link between diabetes and inflammation has continued to grow over the years (71). In 1993, it was discovered that TNF plays a role in insulin resistance in mice, while epidemiological markers such as fibrinogen, IL-6, sialic acid, and C-reactive protein increase in humans with Type 2 diabetes (72). One driver of diabetes complications is hyperglycemia as transient levels of increased glycated hemoglobin levels correlate with long-term complications (73). This has resulted in the development of a concept known as “hyperglycemic memory”.
In a 2021 study by Thiem et al, STZ-treated hyperglycemic mice and in-vitro glucose stimulated/β-glucan co-stimulated monocytes demonstrate how glucose can exacerbate immune training and induce low levels of TRIM on its own with increased transcription of glycolysis-related genes (74). Furthemore, inhibition of the KMT2A epigenetic histone writers reduce TNF TRIM response (74). Hyperglycemia results in central innate memory training that can be adoptively transferred to healthy mice inducing a pro-inflammatory response mediated by the transcription factor Runx1 (75). Interestingly, glucose may also play a role in neurological innate training as in vivo murine Streptozotocin (STZ)-treatment can result in a TRIM-like inflammatory profile following LPS restimulation in brain microglia and astrocytes (76).
5.4 Heme
Heme is a tetrapyrrole that gets released by broken red blood cells following hemolysis or tissue damage. Heme can accumulate as “labile” heme in the plasma by containing a central iron atom that can change from a ferric (Fe3+) to ferrous (Fe2+) oxidation state. In severe cases when heme is unable to be managed, it results in free radical production, serves as a pro-oxidant, and can become a DAMP by activating the PRRs TLR4 and NLRP3. The classic TRIM monocyte-macrophage in vitro model was used to determine that specific PRR-recognized pattern of heme, not its oxidative properties, induces a characteristic TRIM immune response (9). The team notes H3K27ac epigenetic changes caused by heme through an alternative pathway to β-glucan. Unlike HMGB1 or oxLDL, TRIM induced by heme does not rely on mTOR with rapamycin treatment having no effect on training. Instead, SYK inhibition using R406 blocks heme-training corroborating other heme-related studies (77).
5.6 Monosodium urate crystals and uric acid
Urate serves as the final oxidation product in purine catabolism with high levels leading to the autoimmune gout pathologies. Moderate levels of urates are important for neuroprotective properties in Parkison’s, Alzheimer’s, multiple sclerosis, and amyotrophic lateral sclerosis (78). However, high levels of urate can result in nucleation of monosodium urate (MSU) crystals that can act as immunologically active DAMPs (14). The inflammation mechanism of urate involves priming by either MSU crystals or soluble urate to activate NF-kB through a MyD88-dependent pathway and initiate transcription of pro-IL-1β (79). Afterwards, MSU crystals induce the NLRP3 inflammasome pathway creating pro-caspase-1 which cleaves pro-IL-1β that in turn drives IL-1 mediated inflammation (80).
The effects of MSU and soluble urate results in downstream epigenetic/metabolic changes linked to innate priming which suggests the existence of TRIM-related training (81). Consistent with other mTOR training models, MSU crystals induce mTOR activation in monocytes as rapamycin or metformin treatment can inhibit inflammasome activity and subsequent inflammation (82). Epigenetically, Histone Deacetylase (HDAC) inhibition of MSU induced cells can downregulate MSU-induced proinflammatory cytokines (83). Meanwhile ChIP-seq analysis reveal that soluble urate can induce H3K27ac and H3K4me3 reprogramming in monocytes (81). Stimulation of PBMCs by uric acid followed by LPS restimulation results in improved IL-1β production via innate priming that is reversable through the use of methyltransferase inhibitors (84).
5.6 mtDNA/TLR9-mediated training
In shock-related disease models such as sepsis and trauma, mitochondrial DNA (mtDNA) has been linked to immunodysregulation (85, 86). In one model of mitochondrial herniation, release of mtDNA into the cytosol is mediated by BAK/BAX pores independent of apoptotic caspases (87). Similar to bacterial DNA, mtDNA contains unmethylated CpG’s that can target TLR9 and inflammasomes such as NLRP3 and AIM2 (86). Although no studies have yet linked mtDNA to innate memory, MyD88-mediated TLR9 has become a target of TRIM with findings that CpG-ODN can prime innate cells for improved clearance of P. aeruginosa infection (88). In a recent study it was found that TRIM from TLR9/MyD88-mediated CpG-ODN trained bone marrow derived monocytes can be adoptively transferred to wild type mice resulting in improved bacterial clearance (89). Like other TRIM models, mTOR was a key mediator in CpG-ODN TRIM with treatment by rapamycin inhibiting the trained phenotype. Although it is unclear whether CpG-rich mtDNA can result in similar training phenotypes, continued research can help elucidate the role of TLR9-mediated TRIM and mtDNA in shock-related disease models.
5.7 S100A4
S100A4 is a calcium binding protein that is involved with the process of tissue fibrosis. It acts via TLR4 and RAGE to upregulate proinflammatory cytokines such as IL-1 and IL-6 via a MyD88-mediated response (90). In a TRIM screening involving multiple DAMPs such as HMGB1, fibrinogen, and HSP90, only S100A4 was able to induce TRIM-like response at the levels of β-glucan (65). Further research needs to be done to understand whether DAMPs like S100A4 exhibit classical TRIM or if their enhanced immune response is a product of an elongated priming state.
6 Mechanisms of DAMP-specific metabolic reprogramming
DAMP exposure fundamentally alters the metabolic programming of innate immune cells, including how mitochondria handle the TCA cycle and OXPHOS. For example, oxLDL, a prototypical sterile DAMP in atherosclerosis, drives a strong mitochondrial response in trained cells. Monocytes briefly exposed to oxLDL show broad upregulation of mitochondrial genes and accumulation of TCA cycle metabolites during the training period with oxLDL-trained macrophages develop enlarged, hyperfunctional mitochondria, indicative of mitochondrial reprogramming (91). In contrast, other DAMPs may rely less on mitochondrial respiration. Aldosterone, an endogenous danger signal in cardiovascular stress, can induce trained immunity without significant changes in glycolytic flux or OXPHOS activity although persistent mTOR-dependent ROS production was found. This indicates that certain DAMP- stimuli achieve a trained state through alternative metabolic routes, such as fatty acid synthesis, discussed below. Many DAMPs that drive acute inflammation end up promoting glycolytic suppression if they persist. For example, prolonged exposure to HMGB1 or S100 alarmins can trigger autocrine IL-10 and metabolic brake mechanisms that shut down glycolysis over time (though acute HMGB1 is pro-glycolytic, chronic HMGB1 may lead to feedback inhibition) (92). In septic patients (who experience both PAMPs and DAMPs), monocytes often display a “glycolytic paralysis” consistent with endotoxin tolerance, which is linked to worse outcomes (93).
Many DAMPs also directly perturb cholesterol or fatty acid pathways to sustain the trained state. For example, oxLDL-conditioned monocytes adopt a long-lasting pro-atherogenic phenotype characterized by foam cell formation and amplified cytokine release (8). Mechanistically, oxLDL engages PRR that initiate these changes, such as minimally oxidized LDL binds to CD14 and TLR2/4 on monocytes, while heavily oxidized LDL binds to scavenger receptors like CD36 and LOX-1. These inputs drive cholesterol accumulation in macrophages (foam cell formation) and provoke inflammatory gene expression, creating a “primed” state. Trained immunity triggered by oxLDL is associated with increased cholesterol synthesis and storage as well as pro-inflammatory output. In fact, acute oxLDL exposure induces canonical trained-immunity traits: activation of mTOR-HIF1α signaling, elevated glycolysis, and histone H3K4 methylation at inflammatory gene promoters.
Cholesterol crystals also activate NLRP3, triggering IL-1β release that can act on hematopoietic stem cells to expand myelopoiesis (21). Indeed, as discussed above, Western diet failed to induce trained immunity in Nlrp3-deficient mice.
Aldosterone, a steroid hormone, was recently shown to train human monocytes in vitro by engaging the mineralocorticoid receptor and upregulating fatty acid synthesis enzymes. Transcriptomic and metabolic analyses revealed that fatty acid synthesis (FAS) is crucial for this trained phenotype: aldosterone-treated monocytes strongly induced expression of lipid anabolic enzymes. Inhibition of the fatty acid synthesis pathway (e.g. with an acetyl-CoA carboxylase inhibitor) completely abrogated aldosterone-induced trained immunity (94). At the chromatin level, aldosterone priming led to enrichment of the activating histone mark H3K4me3 at promoters of fatty acid metabolism genes highlighting how metabolic and epigenetic effects converge in this model. Notably, aldosterone-trained cells did not exhibit the typical glycolytic switch seen with many PAMPs; instead, glycolysis and OXPHOS remained unchanged while anabolic lipid pathways were elevated (95). This dependence on FAS is unique; while the PAMP β-glucan also causes some increase in fatty acid synthesis, inhibition of FAS does not block β-glucan-induced training, whereas it abolishes aldosterone-induced training. Aldosterone may also promote intracellular cholesterol loading in monocytes, compounding its pro-atherogenic effect.
A recent study by Wang et al. suggests that DAMPs can modulate lipid metabolism in innate cells, thereby influencing their inflammatory setpoint (96). In ulcerative colitis (a condition with chronic HMGB1 release), the DAMP HMGB1 was found to negatively regulate CPT1a (the rate-limiting enzyme for mitochondrial fatty acid oxidation, FAO) in macrophages. HMGB1 overexpression drove macrophages to reduce FAO and instead favor glycolysis and pro-inflammatory polarization. Knockdown of HMGB1 had the opposite effect, enhancing FAO and promoting an anti-inflammatory profile. These data suggest that high levels of certain DAMPs can actively suppress the tolerant metabolic program. In summary, tolerant innate cells exhibit increased fatty acid oxidation and mitochondrial support, which reinforces their low-inflammatory state.
Host-derived signals such as DAMPs also commonly utilize mTOR to rewire cellular metabolism toward a trained phenotype. Certain DAMPs, by engaging specific receptors, could feed into PI3K-Akt pathways that transiently activate mTOR (as seen with HMGB1 or IL-33 in acute inflammation), but the chronic presence of DAMPs often leads to feedback inhibition of mTOR. For instance, prolonged HMGB1-RAGE signaling can increase SIRT1 and AMPK activity in macrophages which tips the balance towards mTOR suppression. Mitochondrial DAMPs like mtDNA activate cytosolic stress responses (e.g. STING) that intersect with autophagy pathways, indirectly inhibiting mTOR and promoting a tolerant, autophagy-high state to dispose of damaged organelles (97, 98). The oxLDL-induced training of human monocytes as described above depends on persistent mTOR activation, as evidenced by mTOR phosphorylation and corresponding accumulation of HIF-1α and its target genes (54). Inhibition of mTORC1 by rapamycin or Raptor knockdown, oxLDL-trained monocytes no longer produce elevated TNFα/IL-6 upon a second stimulus. mTOR activity in this context was shown to drive ROS generation and a positive feedback loop, as mTOR-dependent cytosolic ROS further stabilized HIF-1α. In contrast, and discussed below, recent work showed that heme, another DAMP, induces trained immunity in macrophages independently of mTOR, relying instead on Syk kinase signaling (77) through an unknown mechanism. Endogenous metabolic signals themselves can feed into mTOR pathway. For example, that mevalonate/cholesterol pathway discussed above generates mevalonate which can act as a danger signal, activating IGF1 receptor and downstream PI3K-mTOR in monocytes (45). This implies an interesting autocrine loop, with a cell’s own metabolic output (mevalonate) reinforcing mTOR signaling and thus the trained state (Figure 7).
The role of mTOR in modulating immune cell functions during both the acute and chronic phases post-injury has been examined (99–101). Burn injuries induce a chronic hyper-responsive training state in the innate immune system, characterized by increased recruitment of neutrophils to the lungs and enhanced reactive oxygen and nitrogen species (RONS) production. Administration of rapamycin, reversed this hyper-responsiveness at time of injury, leading to decreased neutrophil oxidative burst and impaired bacterial clearance, thereby increasing mortality in burn-injured mice. This study underscores the critical role of mTOR in promoting long lasting neutrophil antimicrobial functions post-injury.
7 DAMPs and innate memory as possible drivers of immune dysfunction
As illustrated thus far, the mechanisms underlying innate memory are complex which is further complicated when translating in vitro research to clinical applications. In addition to vaccine-like promise in PAMPs-based TRIM through agents like β-glucan or BCG, in disease models such as sepsis there is an interplay between PAMPs and DAMPs that may modulate severity of disease progression. Evolutionarily adaptive DAMP-mediated inflammatory feedback loops that help to clear infections can become maladaptive when taken to extremes as suggested in Systemic Inflammatory Response Syndrome (SIRS) and Persistent Inflammation, Immunosuppression, and Catabolism Syndrome (PICS) models (102, 103). As demonstrated in oxLDL atherosclerosis and vimentin transplantation models, DAMPs are capable of inducing TRIM without a pathogen stimulus. The western diet has been a common target of DAMP-mediated TRIM.
“Dirty mice,” or mice exposed to diverse microbial environments, have emerged as valuable models for studying immune responses that closely resemble those in humans (reviewed in (104)). Unlike specific pathogen-free (SPF) mice, dirty mice possess an immune system that has matured through natural microbial exposures, leading to a more experienced and responsive immune profile. These recently developed platforms underscore the importance of considering environmental microbial exposures when studying trained immunity and its implications for disease models.
Translating DAMP-driven TRIM from mice to patients requires caution. Baseline myeloid populations differ with mice having lower circulating neutrophil fractions whereas humans are neutrophil-dominant with CD14/CD16 subsets (105). PRR thresholds are species-specific (e.g., TLR4/LPS sensitivity, Dectin-1 isoforms, inflammasome activation), so the same DAMP can bias toward training versus tolerance across species (106, 107). HSPC training shows species and age effects (G-CSF responsiveness, niche cytokines, lifespan of trained progeny (30, 108)), and human clinical modifiers such as sex, comorbidities, transfusions, sedatives, and drugs (statins, steroids, metformin) can either amplify or decrease TRIM signatures (21, 45, 109).
In addition to autoimmune frameworks, other DAMP-relevant TRIM have been proposed in sterile critical care models such as burns and trauma where sepsis-like paradigms are relevant. After burn injury, despite the return to more normal transcriptional activity following acute inflammatory states, macrophages and neutrophils exhibit a hyperinflammatory response when rechallenged by Pseudomonas aeruginosa (99). Other studies have demonstrated epigenetic reprogramming in circulating macrophages following trauma and burn while a recent study linked a central TRIM-mediated response to brain trauma that was shown to exacerbate inflammatory cardiac dysfunction (32, 110). Continued research into DAMP-driven TRIM in context of both autoimmune and critical care frameworks is crucial to be able to deliver future TRIM-targeting therapies. While mTOR activation enhances antimicrobial functions of neutrophils, contributing to improved pathogen clearance, it also predisposes to immune dysregulation and increased susceptibility to secondary infections during the chronic phase. These findings suggest that therapeutic modulation of mTOR signaling requires a delicate balance to harness its protective effects without exacerbating immune dysfunction. Further research is warranted to develop targeted interventions that can fine-tune mTOR activity, optimizing immune responses for better clinical outcomes in burn patients.
As research continues, integrating DAMP-related signals into the framework of innate immune memory will be crucial for devising interventions that can fine-tune immune responses in a range of sterile and infectious inflammatory conditions.
8 Discussion
8.1 Conceptual gaps-in-knowledge
As research continues, integrating DAMP-related signals into the framework of innate immune memory will be crucial for devising interventions that can fine-tune immune responses in a range of sterile and infectious inflammatory conditions. It is yet to be determined whether the outcome (training versus tolerance) is intrinsically dictated by the stimulus type; for example, exogenous signals, eg PAMPS (β-glucan, BCG vaccine) or DAMPs (oxLDL, heme) predominantly evoke a trained immunity phenotype, whereas others such as high-dose LPS invariably drive tolerance (39, 111, 112). An opposing view is that any PAMP or DAMP can lead to training or tolerance depending on exposure conditions; low/acute doses trigger a heightened responsive state, whereas high/repetitive doses induce a refractory state. This “dose-dependent” model is supported by evidence that even classical tolerogens like endotoxin can produce a trained response if given in subclinical amounts (50, 113). It remains unclear why certain DAMPs induce robust innate memory whereas others do not. Many sterile injury-associated DAMPs (e.g. mtDNA, ATP, nuclear histones) have not been shown to cause a lasting trained immunity response – some instead drive transient activation or tolerance. The field needs to determine what features of a stimulus (molecular structure, receptor engagement pattern, etc.) dictate the epigenetic outcome. One particularly understudied area is the role of mitochondrial danger signals in shaping innate immune memory. Mitochondria-derived DAMPs such as mtDNA, N-formyl peptides, and cardiolipin can mimic infection signals but recent data suggest they may push innate immunity toward a suppressed state; patients with acute myocardial infarction show elevated mitochondrial DNA in circulation associated with monocyte endotoxin tolerance (114). Do mtDAMPs actively reprogram monocytes via pattern recognition receptors (e.g. TLR9 for DNA) to become tolerant, and if so, through what epigenetic marks? Is this effect transient or long-lasting? It is also unknown whether mitochondrial signals could ever lead to a trained phenotype under different contexts; they have been mostly linked to immunoparalysis in studies to date. This represents a gap at the intersection of metabolism, cell death, and innate memory: deciphering how mitochondrial stress signals modulate myeloid training vs. tolerance is an important research priority. Another gap in our knowledge is how innate immune training interacts with the adaptive immune system. Innate training can influence the environment in which adaptive responses develop (for example, trained monocytes secrete more IL-1β, which could enhance T-cell activation). Conversely, tolerized innate cells produce more IL-10 and less IL-12, potentially skewing T-cell polarization towards regulatory or Th2 responses. Do trained macrophages or NK cells translate into better vaccine efficacy or improved heterologous immunity in humans (beyond correlative evidence)? Conversely, does a state of innate tolerance contribute to failures in vaccination or reactivation of latent infections? Bridging this gap will require integrated studies of innate and adaptive responses in models of infection, vaccination, and disease.
An important nuance is the dose- and context-dependence (“hormesis”) of innate memory and how this plays out in tissue-specific macrophage lineages. As outlined by Jentho and colleagues (115), low-level/short-lived danger signals can enhance cellular fitness and responsiveness, whereas high-level/chronic exposure skews toward adaptive suppression. Notably, functional readouts of ‘training’ are not uniform across lineages: while monocytes and neutrophils often display increased cytokine output and phagocytosis after training, microglia provide a counterexample in which phagocytic capacity is preferentially augmented in tolerized states. Work has demonstrated that repeated or chronic stimulation in the CNS can imprint an immune-depressed but debris-clearing microglial phenotype (116–118). These observations argue that outcome definitions (training vs tolerance) should be interpreted alongside stimulus dose and cell identity.
There is also debate about whether inducing innate immune memory is desirable or harmful. Trained immunity provides enhanced protection against infections and has been linked to improved outcomes (e.g. survival benefits of BCG or fungal exposure beyond specific pathogens). On the other hand, others point out that inappropriate or chronic activation of trained immunity by endogenous DAMPs may contribute to pathology, such as atherosclerosis (inflammatory foam cell formation from oxLDL-trained macrophages) and neurodegeneration (such as exacerbation of Alzheimer’s pathology by trained microglia (117)) Conversely, endotoxin tolerance can be protective by preventing hyperinflammation (as in sepsis), but if excessive it leaves the host immunosuppressed and susceptible to secondary infections. Another unresolved question is how the innate immune system balances repair mechanisms versus defense activation. Trained immunity skews cells toward inflammation (high IL-1β, TNFα, etc.), whereas tolerance reduces inflammation to facilitate recovery. For instance, after severe trauma or ischemia, DAMP-triggered tolerance might prevent further tissue damage by inflammatory overreaction – but at the cost of leaving the host open to infection (as seen in trauma and stroke patients with subsequent immune suppression) (119). Conversely, if DAMPs provoke trained immunity, they might clear debris and pathogens faster, but potentially at the expense of collateral damage and fibrosis. We do not know under which conditions DAMP-induced training is beneficial and when it becomes detrimental. Understanding this balance is critical for therapy, to know whether inducing a trained response in a sterile injury (e.g. heart attack) helps healing or worsens it through excess inflammation.
Current evidence points to reprogramming of bone marrow HSPCs to reconcile the fact that many innate cells have a short lifespan (days/weeks) yet innate memory can last for months or years. Such “central training” then continuously generate pre-trained myeloid cells (120). However, the mechanisms by which signals in peripheral tissues (or circulation) feed back to HSPCs are not fully elucidated. Cytokine mediators (like IL-1β, GM-CSF) and DAMP-induced flux of growth factors are implicated in conveying the memory imprint to the bone marrow niche, but many questions remain. How do tolerogenic marks (e.g. suppressed histone acetylation at inflammatory gene promoters) remain stable over time, and are they actively maintained by feedback loops or just slowly decay? A related issue is the potential fitness cost or trade-off for the stem cell pool; does carrying an epigenetic memory of past stress alter HSPC function or exhaustion over the long term? Key gaps include how long trained immunity lasts in people (some evidence suggests BCG effects persist for years, but how reliably)? and whether repeated or chronic exposures produce cumulative effects. For example, does an initial trained immunity from one insult (say, a mild infection or injury) alter the course of a completely different disease months later? Epidemiological hints (e.g. certain vaccines reducing all-cause mortality via innate mechanisms) are intriguing but mechanistically vague. Conversely, does a period of endotoxin tolerance (such as after severe sepsis) increase cancer risk or allow latent infections to flare? These are difficult questions to study, and thus gaps in knowledge. We also do not fully understand the reversibility/plasticity of innate memory states; can a tolerized immune system be “retrained” back to normal, and can a trained system be reset to avoid chronic inflammation? Addressing these gaps will likely require longitudinal human studies and novel experimental models that more faithfully replicate the human immune system (121).
8.2 Lack of biomarkers of training:
We currently lack readily measurable biomarkers of trained immunity or tolerance in clinical settings and developing a panel of innate memory biomarkers is an identified gap in our current knowledge (122). In the research lab, genome-wide histone modification patterns (like H3K27ac or H3K4me3 peaks at promoters) and transcriptomic profiles are used to identify trained versus tolerized cells. Metabolically, trained cells show increased glycolytic flux or certain metabolite accumulation (e.g. fumarate, succinate), whereas tolerant cells have high FAO and itaconate production. However, these signatures are difficult to assess in patients. There is a pressing need for verified surrogate markers that could indicate an individual’s innate immune state, such as specific cytokine patterns, surface markers, or metabolic indicators in blood tests. The lack of such biomarkers is a knowledge gap that hampers clinical translation. It also limits our ability to survey populations for trained immunity (e.g. after vaccination or diet changes) or to diagnose maladaptive tolerance (e.g. in sepsis). Research is ongoing to find candidate markers (such as circulating IL-1 receptor antagonist levels, or monocyte expression of certain cell-surface PRRs and checkpoint molecules in tolerance), but no consensus has been reached.
8.3 Therapeutic modulation of innate memory:
A future direction is the manipulation of trained immunity or tolerance as a therapeutic in various diseases. In cancer, for instance, activating trained immunity in tumor-associated macrophages or NK cells has potential to boost anti-tumor activity. There is interest in using compounds like beta-glucans or new synthetic agonists to reprogram innate cells in cancer patients towards a more inflammatory, tumoricidal state (123). On the other hand, chronic inflammatory and autoimmune diseases might benefit from inducing innate tolerance. For example, in conditions like gout or atherosclerosis where DAMP-driven trained immunity sustains pathology, drugs that promote a tolerogenic reprogramming of myeloid cells could alleviate disease (15). Early preclinical work shows that nanomedicines targeting monocytes can encourage tolerance and improve outcomes in organ transplantation models. In sepsis and critical illness, a two-pronged therapeutic strategy might emerge, with transient “training block” signals early on to avoid cytokine storm, then later promote immune training when patients become immunosuppressed. Immunomodulators like mTOR inhibitors (rapamycin) or AMPK activators (metformin) could be repurposed to tip the balance towards tolerance or training as needed as we have shown in burn injury (99). Given that metabolic reprogramming and epigenetic remodeling are at the heart of innate immune memory, future interventions may precisely target these processes. We anticipate further development of epigenetic drugs (inhibitors or enhancers of specific histone modifiers) to modulate trained immunity. For instance, blockers of the histone methyltransferases or demethylases that control key inflammatory gene loci might be used to prevent excessive training in inflammatory diseases (124). Conversely, HDAC inhibitors or bromodomain inhibitors could potentially be used in carefully titrated ways to sustain beneficial trained immunity in immunocompromised individuals. On the metabolic side, regulating the mTOR pathway is a clear avenue; transient mTOR inhibition might enforce a tolerant program (useful in cytokine storm or autoimmunity), whereas mTOR activation (for example via IGF1 or amino acid supplementation) might enhance training when needed for protection (though direct mTOR agonism is complex). Another target is the cellular metabolism of succinate, fumarate, and itaconate. These metabolites are known to influence immune memory. Drugs that mimic the effect of fumarate accumulation (which promotes training via HIF-1α stabilization) or that elevate endogenous antioxidative pathways (to modulate ROS and avoid excessive training) are under consideration (42). We expect to see trials of metabolic adjuvants or epigenetic therapies aimed at recalibrating innate immune memory in diseases ranging from sepsis to neurodegenerative disorders.
Another translational application of DAMP research will be strategies to manage the immune aftermath of sterile tissue injury (trauma, surgery, myocardial infarction, stroke). As discussed, increases of DAMPs in these settings can drive a profound innate immune tolerance, contributing to high infection rates in patients after injury (125). Future developments may include therapies to neutralize or clear DAMPs immediately after major tissue damage; for example, using extracellular DNA scavengers (DNAse enzymes to digest released mitochondrial DNA) or blocking DAMP receptors (such as antagonists to HMGB1 or IL-1 receptors), preventing an overshoot into tolerance. Monitoring patients innate immune status via biomarkers might enable personalized interventions; if a trauma patient shows signs of dangerous tolerance, one could administer trained immunity boosters (like recombinant GM-CSF or TLR agonists at low dose) to restore functionality. Conversely, if an individual is trending towards a hyper-trained, inflammatory state that could cause complications, targeted anti-inflammatory treatments could be applied. In essence, dynamic control of innate memory after sterile injuries could improve recovery and reduce complications. This precision medicine approach, treating innate immunity as a adjustable parameter, represents a forward-looking development as our understanding of DAMPs and innate memory deepens.
Lastly, future research is likely to adopt a more integrative view of innate immune memory across the lifespan and across systems. There is growing interest in how factors like aging, microbiome composition, stress, trauma and chronic metabolic status (e.g. obesity, diabetes) influence trained immunity versus tolerance. For example, aging is associated with “inflammaging,” possibly due to cumulative trained immunity and chronic low-grade DAMP release, and interventions like caloric restriction might mitigate this by promoting a more tolerant anti-inflammatory baseline (reviewed in (126)). We may see development of lifestyle or nutritional interventions that modulate innate memory (for instance, specific diets or microbiome-derived products that favor beneficial training without tipping into pathology). Additionally, cross-talk between different organ systems’ innate immunity (brain microglia, liver Kupffer cells, etc.) will be explored, which could open up new preventive strategies for complex diseases. The duality of trained immunity and tolerance provides a conceptual framework that future work will apply to diverse conditions from Alzheimer’s disease to cancer metastasis to vaccine responses in infants. By filling current knowledge gaps, the DAMP-driven innate memory can be translated into medical advances.
Author contributions
HK: Writing – original draft, Writing – review & editing, Conceptualization. JR: Writing – review & editing. PE: Writing – review & editing. RM: Conceptualization, Supervision, Funding acquisition, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. Supported by NIH NIGMS R01-GM146134 (RM) and NIGMS R35-GM140806 (PAE).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fanucchi S, Fok ET, Dalla E, Shibayama Y, Borner K, Chang EY, et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet. (2019) 51:138–50. doi: 10.1038/s41588-018-0298-2
2. Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, et al. beta-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. (2016) 167:1354–68 e14. doi: 10.1016/j.cell.2016.09.034
3. Foster SL, Hargreaves DC, and Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. (2007) 447:972–8. doi: 10.1038/nature05836
4. Segura E and Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol. (2013) 34:440–5. doi: 10.1016/j.it.2013.06.001
5. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
6. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A. (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
7. Riksen NP and Netea MG. Immunometabolic control of trained immunity. Mol Aspects Med. (2021) 77:100897. doi: 10.1016/j.mam.2020.100897
8. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, and Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
9. Jentho E, Ruiz-Moreno C, Novakovic B, Kourtzelis I, Megchelenbrink WL, Martins R, et al. Trained innate immunity, long-lasting epigenetic modulation, and skewed myelopoiesis by heme. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2102698118
10. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
11. Fulda S, Gorman AM, Hori O, and Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. (2010) 2010:214074. doi: 10.1155/2010/214074
12. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. (1989) 54 Pt 1:1–13. doi: 10.1101/sqb.1989.054.01.003
13. Walsh D, McCarthy J, O’Driscoll C, and Melgar S. Pattern recognition receptors–molecular orchestrators of inflammation in inflammatory bowel disease. Cytokine Growth Factor Rev. (2013) 24:91–104. doi: 10.1016/j.cytogfr.2012.09.003
14. Ma M, Jiang W, and Zhou R. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity. (2024) 57:752–71. doi: 10.1016/j.immuni.2024.03.002
15. Li L and Lu YQ. The regulatory role of high-mobility group protein 1 in sepsis-related immunity. Front Immunol. (2020) 11:601815. doi: 10.3389/fimmu.2020.601815
16. Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD, and Diener KR. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep. (2017) 7:5850. doi: 10.1038/s41598-017-06205-z
17. Margraf S, Logters T, Reipen J, Altrichter J, Scholz M, and Windolf J. Neutrophil-derived circulating free DNA (cf-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock. (2008) 30:352–8. doi: 10.1097/SHK.0b013e31816a6bb1
18. Land WG. Role of DAMPs and cell death in autoimmune diseases: the example of multiple sclerosis. Genes Immun. (2023) 24:57–70. doi: 10.1038/s41435-023-00198-8
19. Shi J, Xiao Y, Zhang N, Jiao M, Tang X, Dai C, et al. HMGB1 from astrocytes promotes EAE by influencing the immune cell infiltration-associated functions of BMECs in mice. Neurosci Bull. (2022) 38:1303–14. doi: 10.1007/s12264-022-00890-1
20. Mansilla MJ, Comabella M, Rio J, Castillo J, Castillo M, Martin R, et al. Up-regulation of inducible heat shock protein-70 expression in multiple sclerosis patients. Autoimmunity. (2014) 47:127–33. doi: 10.3109/08916934.2013.866104
21. Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–75 e14. doi: 10.1016/j.cell.2017.12.013
22. Aneja RK, Tsung A, Sjodin H, Gefter JV, Delude RL, Billiar TR, et al. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol. (2008) 84:1326–34. doi: 10.1189/jlb.0108030
23. Eppensteiner J, Kwun J, Scheuermann U, Barbas A, Limkakeng AT, Kuchibhatla M, et al. Damage- and pathogen-associated molecular patterns play differential roles in late mortality after critical illness. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.127925
24. Di Gioia M, Spreafico R, Springstead JR, Mendelson MM, Joehanes R, Levy D, et al. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat Immunol. (2020) 21:42–53. doi: 10.1038/s41590-019-0539-2
25. Seim RF, Herring LE, Mordant AL, Willis ML, Wallet SM, Coleman LG Jr., et al. Involvement of extracellular vesicles in the progression, diagnosis, treatment, and prevention of whole-body ionizing radiation-induced immune dysfunction. Front Immunol. (2023) 14:1188830. doi: 10.3389/fimmu.2023.1188830
26. Coleman LG Jr., Maile R, Jones SW, Cairns BA, and Crews FT. HMGB1/IL-1beta complexes in plasma microvesicles modulate immune responses to burn injury. PloS One. (2018) 13:e0195335. doi: 10.1371/journal.pone.0195335
27. Willis ML, Mahung C, Wallet SM, Barnett A, Cairns BA, Coleman LG Jr., et al. Plasma extracellular vesicles released after severe burn injury modulate macrophage phenotype and function. J Leukoc Biol. (2022) 111:33–49. doi: 10.1002/JLB.3MIA0321-150RR
28. Maile R, Willis ML, Herring LE, Prevatte A, Mahung C, C BA, et al. Burn injury induces proinflammatory plasma extracellular vesicles that associate with length of hospital stay in women: CRP and SAA1 as potential prognostic indicators. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms221810083
29. Saccu G, Menchise V, Gai C, Bertolin M, Ferrari S, Giordano C, et al. Bone marrow mesenchymal stromal/stem cell-derived extracellular vesicles promote corneal wound repair by regulating inflammation and angiogenesis. Cells. (2022) 11. doi: 10.3390/cells11233892
30. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. (2018) 172:176–90 e19. doi: 10.1016/j.cell.2017.12.031
31. Bhattarai S, Li Q, Ding J, Liang F, Gusev E, Lapohos O, et al. TLR4 is a regulator of trained immunity in a murine model of Duchenne muscular dystrophy. Nat Commun. (2022) 13:879. doi: 10.1038/s41467-022-28531-1
32. Simats A, Zhang S, Messerer D, Chong F, Beskardes S, Chivukula AS, et al. Innate immune memory after brain injury drives inflammatory cardiac dysfunction. Cell. (2024) 187:4637–55 e26. doi: 10.1016/j.cell.2024.06.028
33. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
34. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. (2012) 489:57–74. doi: 10.1038/nature11247
35. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. (2013) 152:157–71. doi: 10.1016/j.cell.2012.12.018
36. Chen LF, Lin YT, Gallegos DA, Hazlett MF, Gomez-Schiavon M, Yang MG, et al. Enhancer histone acetylation modulates transcriptional bursting dynamics of neuronal activity-inducible genes. Cell Rep. (2019) 26:1174–88 e5. doi: 10.1016/j.celrep.2019.01.032
37. Raj A, Peskin CS, TranChina D, Vargas DY, and Tyagi S. Stochastic mRNA synthesis in mammalian cells. PloS Biol. (2006) 4:e309. doi: 10.1371/journal.pbio.0040309
38. Mourits VP, van Puffelen JH, Novakovic B, Bruno M, Ferreira AV, Arts RJ, et al. Lysine methyltransferase G9a is an important modulator of trained immunity. Clin Transl Immunol. (2021) 10:e1253. doi: 10.1002/cti2.1253
39. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
40. Su H, Liang Z, Weng S, Sun C, Huang J, Zhang T, et al. miR-9-5p regulates immunometabolic and epigenetic pathways in beta-glucan-trained immunity via IDH3alpha. JCI Insight. (2021) 6. doi: 10.1172/jci.insight.144260
41. Nahid MA, Pauley KM, Satoh M, and Chan EK. miR-146a is critical for endotoxin-induced tolerance: IMPLICATION IN INNATE IMMUNITY. J Biol Chem. (2009) 284:34590–9. doi: 10.1074/jbc.M109.056317
42. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
43. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. (2016) 17:2562–71. doi: 10.1016/j.celrep.2016.11.011
44. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
45. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–46 e9. doi: 10.1016/j.cell.2017.11.025
46. Chen LL, Morcelle C, Cheng ZL, Chen X, Xu Y, Gao Y, et al. Itaconate inhibits TET DNA dioxygenases to dampen inflammatory responses. Nat Cell Biol. (2022) 24:353–63. doi: 10.1038/s41556-022-00853-8
47. Raulien N, Friedrich K, Strobel S, Rubner S, Baumann S, von Bergen M, et al. Fatty acid oxidation compensates for lipopolysaccharide-induced warburg effect in glucose-deprived monocytes. Front Immunol. (2017) 8:609. doi: 10.3389/fimmu.2017.00609
48. Crisan TO, Netea MG, and Joosten LA. Innate immune memory: Implications for host responses to damage-associated molecular patterns. Eur J Immunol. (2016) 46:817–28. doi: 10.1002/eji.201545497
49. Stephens M, Liao S, and von der Weid PY. Mesenteric lymphatic alterations observed during DSS induced intestinal inflammation are driven in a TLR4-PAMP/DAMP discriminative manner. Front Immunol. (2019) 10:557. doi: 10.3389/fimmu.2019.00557
50. Bauer M, Weis S, Netea MG, and Wetzker R. Remembering pathogen dose: long-term adaptation in innate immunity. Trends Immunol. (2018) 39:438–45. doi: 10.1016/j.it.2018.04.001
51. Lajqi T, Kostlin-Gille N, Hillmer S, Braun M, Kranig SA, Dietz S, et al. Gut microbiota-derived small extracellular vesicles endorse memory-like inflammatory responses in murine neutrophils. Biomedicines. (2022) 10. doi: 10.3390/biomedicines10020442
52. Gao S, Liu W, Zhuo X, Wang L, Wang G, Sun T, et al. The activation of mTOR is required for monocyte pro-inflammatory response in patients with coronary artery disease. Clin Sci (Lond). (2015) 128:517–26. doi: 10.1042/CS20140427
53. Wang YC, Hu YW, Sha YH, Gao JJ, Ma X, Li SF, et al. Ox-LDL upregulates IL-6 expression by enhancing NF-kappaB in an IGF2-dependent manner in THP-1 macrophages. Inflammation. (2015) 38:2116–23. doi: 10.1007/s10753-015-0194-1
54. Sohrabi Y, Lagache SMM, Schnack L, Godfrey R, Kahles F, Bruemmer D, et al. mTOR-dependent oxidative stress regulates oxLDL-induced trained innate immunity in human monocytes. Front Immunol. (2018) 9:3155. doi: 10.3389/fimmu.2018.03155
55. Sohrabi Y, Sonntag GVH, Braun LC, Lagache SMM, Liebmann M, Klotz L, et al. LXR activation induces a proinflammatory trained innate immunity-phenotype in human monocytes. Front Immunol. (2020) 11:353. doi: 10.3389/fimmu.2020.00353
56. Findeisen HM, Voges VC, Braun LC, Sonnenberg J, Schwarz D, Korner H, et al. LXRalpha regulates oxLDL-induced trained immunity in macrophages. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23116166
57. Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. (2016) 254:228–36. doi: 10.1016/j.atherosclerosis.2016.10.019
58. Wu D, Shi Y, Zhang H, and Miao C. Epigenetic mechanisms of Immune remodeling in sepsis: targeting histone modification. Cell Death Dis. (2023) 14:112. doi: 10.1038/s41419-023-05656-9
59. Groh LA, Verel DE, van der Heijden C, Matzaraki V, Moorlag S, de Bree LC, et al. Immune modulatory effects of progesterone on oxLDL-induced trained immunity in monocytes. J Leukoc Biol. (2022) 112:279–88. doi: 10.1002/JLB.3AB1220-846R
60. Schnack L, Sohrabi Y, Lagache SMM, Kahles F, Bruemmer D, Waltenberger J, et al. Mechanisms of trained innate immunity in oxLDL primed human coronary smooth muscle cells. Front Immunol. (2019) 10:13. doi: 10.3389/fimmu.2019.00013
61. Chen R, Kang R, and Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. (2022) 54:91–102. doi: 10.1038/s12276-022-00736-w
62. Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. (2006) 290:C917–24. doi: 10.1152/ajpcell.00401.2005
63. Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. (2007) 26:1129–39. doi: 10.1038/sj.emboj.7601552
64. Valdes-Ferrer SI, Rosas-Ballina M, Olofsson PS, Lu B, Dancho ME, Li J, et al. High-mobility group box 1 mediates persistent splenocyte priming in sepsis survivors: evidence from a murine model. Shock. (2013) 40:492–5. doi: 10.1097/SHK.0000000000000050
65. Neidhart M, Pajak A, Laskari K, Riksen NP, Joosten LAB, Netea MG, et al. Oligomeric S100A4 is associated with monocyte innate immune memory and bypass of tolerance to subsequent stimulation with lipopolysaccharides. Front Immunol. (2019) 10:791. doi: 10.3389/fimmu.2019.00791
66. Qu H, Heinback R, Salo H, Ewing E, Espinosa A, Aulin C, et al. Transcriptomic profiling reveals that HMGB1 induces macrophage polarization different from classical M1. Biomolecules. (2022) 12. doi: 10.3390/biom12060779
67. Thiagarajan PS, Yakubenko VP, Elsori DH, Yadav SP, Willard B, Tan CD, et al. Vimentin is an endogenous ligand for the pattern recognition receptor Dectin-1. Cardiovasc Res. (2013) 99:494–504. doi: 10.1093/cvr/cvt117
68. Huang Y, Yin H, Han J, Huang B, Xu J, Zheng F, et al. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. (2007) 7:799–808. doi: 10.1111/j.1600-6143.2007.01734.x
69. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Perez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity. (2018) 49:819–28 e6. doi: 10.1016/j.immuni.2018.09.008
70. Willis ML, Mahung C, Wallet SM, Barnett A, Cairns BA, Coleman LG Jr., et al. Plasma extracellular vesicles released after severe burn injury modulate macrophage phenotype and function. J Leukoc Biol. (2021) 111(1):33–49. doi: 10.1002/JLB.3MIA0321-150RR
71. Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis GA, Vogiatzi G, Papaioannou S, et al. The role of inflammation in diabetes: current concepts and future perspectives. Eur Cardiol. (2019) 14:50–9. doi: 10.15420/ecr.2018.33.1
72. Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes. (2003) 52:1799–805. doi: 10.2337/diabetes.52.7.1799
73. Holman RR, Paul SK, Bethel MA, Matthews DR, and Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. (2008) 359:1577–89. doi: 10.1056/NEJMoa0806470
74. Thiem K, Keating ST, Netea MG, Riksen NP, Tack CJ, van Diepen J, et al. Hyperglycemic memory of innate immune cells promotes in vitro proinflammatory responses of human monocytes and murine macrophages. J Immunol. (2021) 206:807–13. doi: 10.4049/jimmunol.1901348
75. Edgar L, Akbar N, Braithwaite AT, Krausgruber T, Gallart-Ayala H, Bailey J, et al. Hyperglycemia induces trained immunity in macrophages and their precursors and promotes atherosclerosis. Circulation. (2021) 144:961–82. doi: 10.1161/CIRCULATIONAHA.120.046464
76. Lee KS, Yoon SH, Hwang I, Ma JH, Yang E, Kim RH, et al. Hyperglycemia enhances brain susceptibility to lipopolysaccharide-induced neuroinflammation via astrocyte reprogramming. J Neuroinflamm. (2024) 21:137. doi: 10.1186/s12974-024-03136-1
77. Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourao-Sa D, Fortes GB, et al. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J Biol Chem. (2010) 285:32844–51. doi: 10.1074/jbc.M110.146076
78. Kutzing MK and Firestein BL. Altered uric acid levels and disease states. J Pharmacol Exp Ther. (2008) 324:1–7. doi: 10.1124/jpet.107.129031
79. Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. (2006) 116:2262–71. doi: 10.1172/JCI28075
80. Martinon F, Petrilli V, Mayor A, Tardivel A, and Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. doi: 10.1038/nature04516
81. Badii M, Gaal OI, Cleophas MC, Kluck V, Davar R, Habibi E, et al. Urate-induced epigenetic modifications in myeloid cells. Arthritis Res Ther. (2021) 23:202. doi: 10.1186/s13075-021-02580-1
82. Chung YH, Kim DH, and Lee WW. Monosodium urate crystal-induced pro-interleukin-1beta production is post-transcriptionally regulated via the p38 signaling pathway in human monocytes. Sci Rep. (2016) 6:34533. doi: 10.1038/srep34533
83. Cleophas MCP, Crisan TO, Kluck V, Hoogerbrugge N, Netea-Maier RT, Dinarello CA, et al. Romidepsin suppresses monosodium urate crystal-induced cytokine production through upregulation of suppressor of cytokine signaling 1 expression. Arthritis Res Ther. (2019) 21:50. doi: 10.1186/s13075-019-1834-x
84. Crisan TO, Cleophas MC, Oosting M, Lemmers H, Toenhake-Dijkstra H, Netea MG, et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann Rheum Dis. (2016) 75:755–62. doi: 10.1136/annrheumdis-2014-206564
85. Grazioli S and Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol. (2018) 9:832. doi: 10.3389/fimmu.2018.00832
86. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
87. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. (2018) 359. doi: 10.1126/science.aao6047
88. Fensterheim BA, Guo Y, Sherwood ER, and Bohannon JK. The cytokine response to lipopolysaccharide does not predict the host response to infection. J Immunol. (2017) 198:3264–73. doi: 10.4049/jimmunol.1602106
89. Owen AM, Luan L, Burelbach KR, McBride MA, Stothers CL, Boykin OA, et al. MyD88-dependent signaling drives toll-like receptor-induced trained immunity in macrophages. Front Immunol. (2022) 13:1044662. doi: 10.3389/fimmu.2022.1044662
90. O’Reilly S. S100A4 a classical DAMP as a therapeutic target in fibrosis. Matrix Biol. (2024) 127:1–7. doi: 10.1016/j.matbio.2024.01.002
91. Groh LA, Ferreira AV, Helder L, van der Heijden C, Novakovic B, van de Westerlo E, et al. oxLDL-induced trained immunity is dependent on mitochondrial metabolic reprogramming. Immunometabolism. (2021) 3:e210025. doi: 10.20900/immunometab20210025
92. Watanabe H and Son M. The immune tolerance role of the HMGB1-RAGE axis. Cells. (2021) 10. doi: 10.3390/cells10030564
93. Liu J, Zhou G, Wang X, and Liu D. Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell Mol Life Sci. (2022) 79:456. doi: 10.1007/s00018-022-04490-0
94. Gyssens IC and Netea MG. Heterologous effects of vaccination and trained immunity. Clin Microbiol Infect. (2019) 25:1457–8. doi: 10.1016/j.cmi.2019.05.024
95. Zhong C, Yang X, Feng Y, and Yu J. Trained immunity: an underlying driver of inflammatory atherosclerosis. Front Immunol. (2020) 11:284. doi: 10.3389/fimmu.2020.00284
96. Wang F, Luo L, Wu Z, Wan L, Li F, and Wen Z. HMGB1 modulates macrophage metabolism and polarization in ulcerative colitis by inhibiting cpt1a expression. Front Biosci (Landmark Ed). (2024) 29:387. doi: 10.31083/j.fbl2911387
97. Decout A, Katz JD, Venkatraman S, and Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
98. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
99. Dunn JLM, Kartchner LB, Gast K, Sessions M, Hunter RA, Thurlow L, et al. Mammalian target of rapamycin regulates a hyperresponsive state in pulmonary neutrophils late after burn injury. J Leukoc Biol. (2018) 103:909–18. doi: 10.1002/JLB.3AB0616-251RRR
100. Hall HR, Mahung C, Dunn JLM, Kartchner LM, Seim RF, Cairns BA, et al. Characterization of the basal and mTOR-dependent acute pulmonary and systemic immune response in a murine model of combined burn and inhalation injury. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23158779
101. Mahung C, Wallet SM, Jacobs JE, Zhou LY, Zhou H, Cairns BA, et al. Multiplexed human gene expression analysis reveals a central role of the TLR/mTOR/PPARgamma and NFkB axes in burn and inhalation injury-induced changes in systemic immunometabolism and long-term patient outcomes. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23169418
102. Denning NL, Aziz M, Gurien SD, and Wang P. DAMPs and NETs in sepsis. Front Immunol. (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
103. Chen GY and Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
104. Hamilton SE, Badovinac VP, Beura LK, Pierson M, Jameson SC, Masopust D, et al. New insights into the immune system using dirty mice. J Immunol. (2020) 205:3–11. doi: 10.4049/jimmunol.2000171
105. Mestas J and Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
106. Vaure C and Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. (2014) 5:316. doi: 10.3389/fimmu.2014.00316
107. Walsh C, Gangloff M, Monie T, Smyth T, Wei B, McKinley TJ, et al. Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J Immunol. (2008) 181:1245–54. doi: 10.4049/jimmunol.181.2.1245
108. Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, and Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PloS Biol. (2007) 5:e201. doi: 10.1371/journal.pbio.0050201
109. Sun SN, Zhou Y, Fu X, Zheng YZ, Xie C, Qin GY, et al. A pilot study of the differentiated landscape of peripheral blood mononuclear cells from children with incomplete versus complete Kawasaki disease. World J Pediatr. (2024) 20:189–200. doi: 10.1007/s12519-023-00752-4
110. Chen T, Conroy J, Wang X, Situ M, Namas RA, Vodovotz Y, et al. The independent prognostic value of global epigenetic alterations: An analysis of single-cell ATAC-seq of circulating leukocytes from trauma patients followed by validation in whole blood leukocyte transcriptomes across three etiologies of critical illness. EBioMedicine. (2022) 76:103860. doi: 10.1016/j.ebiom.2022.103860
111. Netea MG, Quintin J, and van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
112. Ochando J, Fayad ZA, Madsen JC, Netea MG, and Mulder WJM. Trained immunity in organ transplantation. Am J Transplant. (2020) 20:10–8. doi: 10.1111/ajt.15620
113. Lajqi T, Poschl J, Frommhold D, and Hudalla H. The role of microbiota in neutrophil regulation and adaptation in newborns. Front Immunol. (2020) 11:568685. doi: 10.3389/fimmu.2020.568685
114. Fernandez-Ruiz I, Arnalich F, Cubillos-Zapata C, Hernandez-Jimenez E, Moreno-Gonzalez R, Toledano V, et al. Mitochondrial DAMPs induce endotoxin tolerance in human monocytes: an observation in patients with myocardial infarction. PloS One. (2014) 9:e95073. doi: 10.1371/journal.pone.0095073
115. Jentho E, Lajqi T, Yang K, Winkler R, Stojiljkovic M, Wetzker R, et al. Pathogen-induced hormetic responses. In: Rattan SIS and Kyriazis M, editors. The science of hormesis in health and longevity. Elsevier Academic Press, Amsterdam, Netherlands (2019). p. 161–70.
116. Lajqi T, Lang GP, Haas F, Williams DL, Hudalla H, Bauer M, et al. Memory-like inflammatory responses of microglia to rising doses of LPS: key role of PI3Kgamma. Front Immunol. (2019) 10:2492. doi: 10.3389/fimmu.2019.02492
117. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
118. Schaafsma W, Zhang X, van Zomeren KC, Jacobs S, Georgieva PB, Wolf SA, et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav Immun. (2015) 48:205–21. doi: 10.1016/j.bbi.2015.03.013
119. Hernandez-Jimenez E, Gutierrez-Fernandez M, Cubillos-Zapata C, Otero-Ortega L, Rodriguez-Frutos B, Toledano V, et al. Circulating monocytes exhibit an endotoxin tolerance status after acute ischemic stroke: mitochondrial DNA as a putative explanation for poststroke infections. J Immunol. (2017) 198:2038–46. doi: 10.4049/jimmunol.1601594
120. Zmonarski SC, Banasik M, Madziarska K, Mazanowska O, and Krajewska M. The role of toll-like receptors in multifactorial mechanisms of early and late renal allotransplant injury, with a focus on the TLR4 receptor and mononuclear cells. Adv Clin Exp Med. (2019) 28:981–7. doi: 10.17219/acem/94139
121. Bartsch R, Singer CF, Pfeiler G, Hubalek M, Stoeger H, Pichler A, et al. Conventional versus reverse sequence of neoadjuvant epirubicin/cyclophosphamide and docetaxel: sequencing results from ABCSG-34. Br J Cancer. (2021) 124:1795–802. doi: 10.1038/s41416-021-01284-2
122. Vail M, Beaufrere H, Gallini S, Paluch H, Brandao J, and DiGeronimo PM. Hematologic and plasma biochemical prognostic indicators for stranded free-ranging phocids presented for rehabilitation. Sci Rep. (2022) 12:10546. doi: 10.1038/s41598-022-14923-2
123. Jan JT, Cheng TR, Juang YP, Ma HH, Wu YT, Yang WB, et al. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS-CoV-2 infection. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2021579118
124. Yano K, Morinaka Y, Wang F, Huang P, Takehara S, Hirai T, et al. GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture. Proc Natl Acad Sci U S A. (2019) 116:21262–7. doi: 10.1073/pnas.1904964116
125. Timmermans K, Kox M, Vaneker M, van den Berg M, John A, van Laarhoven A, et al. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med. (2016) 42:551–61. doi: 10.1007/s00134-015-4205-3
126. Lajqi T, Kostlin-Gille N, Bauer R, Zarogiannis SG, Lajqi E, Ajeti V, et al. Training vs. Tolerance: the yin/yang of the innate immune system. Biomedicines. (2023) 11. doi: 10.3390/biomedicines11030766
127. van der Heijden C, Keating ST, Groh L, Joosten LAB, Netea MG, and Riksen NP. Aldosterone induces trained immunity: the role of fatty acid synthesis. Cardiovasc Res. (2020) 116:317–28. doi: 10.1093/cvr/cvz137
128. Yang WS, Kim JJ, Lee MJ, Lee EK, and Park SK. Ectodomain shedding of RAGE and TLR4 as a negative feedback regulation in high-mobility group box 1-activated aortic endothelial cells. Cell Physiol Biochem. (2018) 51:1632–44. doi: 10.1159/000495651
129. Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. (2004) 101:296–301. doi: 10.1073/pnas.2434651100
130. Andersson U and Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. (2011) 29:139–62. doi: 10.1146/annurev-immunol-030409-101323
131. Lotze MT and Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. (2005) 5:331–42. doi: 10.1038/nri1594
132. dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun. (2015) 6:6574. doi: 10.1038/ncomms7574
133. Kang YY, Kim DY, Lee SY, Kim HJ, Kim T, Cho JA, et al. Innate immune training initiates efferocytosis to protect against lung injury. Adv Sci (Weinh). (2024) 11:e2308978. doi: 10.1002/advs.202308978
134. Mossel DM, Moganti K, Riabov V, Weiss C, Kopf S, Cordero J, et al. Epigenetic regulation of S100A9 and S100A12 expression in monocyte-macrophage system in hyperglycemic conditions. Front Immunol. (2020) 11:1071. doi: 10.3389/fimmu.2020.01071
135. Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. (2012) 119:2368–75. doi: 10.1182/blood-2011-08-375303
136. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A. (2014) 111:E4110–8. doi: 10.1073/pnas.1405023111
137. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
138. Fang C, Mo F, Liu L, Du J, Luo M, Men K, et al. Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell Mol Immunol. (2021) 18:2211–23. doi: 10.1038/s41423-020-0456-1
139. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. (2016) 164:896–910. doi: 10.1016/j.cell.2015.12.057
140. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
Keywords: innate training, DAMPs, trauma, epigenetics, innate immunity
Citation: Kim HG, Rincon JC, Efron PA and Maile R (2025) DAMP-driven trained immunity: metabolic and epigenetic reprogramming in critical illness and chronic inflammation. Front. Immunol. 16:1669054. doi: 10.3389/fimmu.2025.1669054
Received: 18 July 2025; Accepted: 10 November 2025; Revised: 30 October 2025;
Published: 24 November 2025.
Edited by:
Yihui Wang, Shanghai Jiao Tong University, ChinaReviewed by:
Trim Lajqi, Heidelberg University Hospital, GermanyTingting Pan, Shanghai Jiao Tong University, China
Renske Johanna De Jong, University Hospital RWTH Aachen, Germany
Copyright © 2025 Kim, Rincon, Efron and Maile. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Maile, cm9iZXJ0Lm1haWxlQHN1cmdlcnkudWZsLmVkdQ==