 Feng Gao
Feng Gao Xujie Liu
Xujie Liu Fan Sun
Fan Sun Yadong Xiao1,2
Yadong Xiao1,2 Zhaoxia Qu
Zhaoxia Qu- 1Department of Molecular Microbiology and Immunology, Hastings Center for Pulmonary Research, Norris Comprehensive Cancer Center, University of Southern California Keck School of Medicine, Los Angeles, CA, United States
- 2Department of Microbiology and Molecular Genetics, UPMC Hillman Cancer Center, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Introduction: The PDZ-LIM domain-containing protein PDLIM2 serves as a unique tumor suppressor and immune modulator. Its repression in either lung epithelial or myeloid cells has been shown to promote lung cancer and therapy resistance. However, whether PDLIM2 plays a broader role in other lung diseases remains unclear.
Methods: Gene expression data on human samples were exploited to investigate if PDLIM2 is repressed in the lung of patients with chronic obstructive pulmonary disease (COPD) or interstitial lung disease (ILD/idiopathic pulmonary fibrosis (IPF). PDLIM2 conditional knockout (KO) mice and wild type (WT) control mice were intratracheally instilled with the bacterial endotoxin lipopolysaccharide (LPS) to induce acute lung injury (ALI), a murine model of human acute respiratory distress syndrome (ARDS) that can also provide mechanistic insights into COPD, pulmonary fibrosis (PF) and infectious disease. Kaplan-Meier estimator was used to determine animal survival rate, and histological analysis and single-cell RNA sequencing (scRNA-seq) of mouse lung tissues were performed to systematically define the roles of PDLIM2 at the population and single-cell level. Ex vivo phagocytosis and neutrophil extracellular trap (NET) formation assays were also performed to validate the scRNA-seq analysis.
Results: PDLIM2 was repressed in the lungs of COPD and ILD/IPF patients, and this repression was associated with disease severity. Selective deletion of PDLIM2 in myeloid cells rendered mice more vulnerable to lung injury and mortality by LPS intratracheal instillation. The increased susceptibility was linked to exacerbated pro-inflammation signaling and diminished anti-inflammation signaling in the lung, and particularly, in lung macrophages and neutrophils.
Conclusions: PDLIM2 plays an indispensable role in preventing ALI/ARDS and death, and its repression is associated with COPD and ILD progression. These data suggest that PDLIM2 repression, especially in lung myeloid cells, is a common mechanism driving COPD, ILD/IPF, and lung cancer and increasing patients’ susceptibility to infection.
Introduction
COPD and lung cancer are the leading causes of respiratory mortality and cancer death, respectively (1, 2). On the other hand, ILD is a term for a large group of lung diseases characterized by fibrosis and scarring in the lung, with IPF as the most common and severe form (3–5). While those fatal diseases are fundamentally different, they are often associated with aberrant lung inflammation (6–9). Moreover, they all make patients much more susceptible to lung infection (10–13). Interestingly, both COPD and ILD/IPF are significant independent risk factors and have long been assumed to be pre-stage diseases and causative drivers for lung cancer (6, 14–18). However, the common mechanisms and molecular links for these malignancies remain largely unknown. As a matter of fact, our knowledge of either COPD or ILD/IPF is poor, and accordingly, the treatment options and outcomes for those incurable diseases are very limited.
In this regard, the immune modulator and tumor suppressor PDLIM2, which is expressed highest in the lung under normal conditions, is repressed in more than 90% of all human lung cancer cases when 50% of the expression level of lung tissues adjacent to tumors is used as the cut-off (19–30). Human and mouse studies demonstrate PDLIM2 repression as a causative driver of lung cancer and therapy resistance (20, 21). For example, lung epithelial- or myeloid-specific PDLIM2 deletion or global PDLIM2 deletion in mice promotes lung cancer development, chemoresistance, and/or causes complete resistance to immune checkpoint inhibitors (ICIs) (20–22). Remarkably, clinically feasible nano-delivery of PDLIM2 (nanoPDLIM2) shows a promising efficacy as a monotherapy, and in combination with ICIs and chemo drugs, completely eradicates all tumors in most animals without adding toxicity, in the preclinical model of refractory lung cancer (21, 31). It is thus both scientifically and clinically important to investigate the roles of PDLIM2 in COPD, ILD/IPF, and lung infectious diseases.
Materials and methods
Animals and LPS intratracheal instillation
PDLIM2flox/flox/Lysozyme M-Cre+/- (PDLIM2 mKO), PDLIM2flox/flox, Lysozyme M-Cre+/-, and WT mice have been described before (20–22, 32–34). All mice were under a pure FVB/NJ background. Mice of 6-8-week-old were intratracheally instilled with LPS (4 mg/g body weight, Sigma-Aldrich) or phosphate-buffered saline (PBS), followed by daily body weight monitoring. When the weight loss of an infected mouse was more than 20% of the initial body weight, the mouse was humanely euthanized and counted as dead. Some mice were euthanized for bronchioalveolar lavage (BAL) collection, lung histological analysis, and scRNA-seq assay at 48 hours after LPS treatment. All animals were maintained under a specific pathogen-free condition and used according to protocols approved by the IACUC of the University of Pittsburgh and the University of Southern California.
Lung histological assays
Lung tissues were excised, fixed in formalin, embedded in paraffin, and cut into 4-5μm thick sections. Sections were stained with H&E or Trichrome, and images were analyzed using ImageJ software (NIH). The histological lung injury was scored according to the standard scoring system (35, 36), which includes four parameters: (A) alveolar space neutrophils; (B) interstitial neutrophils; (C) proteinaceous debris in alveolar space; and (D) alveolar septal thickening. The total score was calculated as: ((20 x A) + (14 x B) + (7 x C) + (2 x D))/86. A total of 20 random high-power fields (400x total magnification) of each mouse were scored by the blinded researcher. The final score per mouse was calculated by averaging the scores of the 20 fields, resulting in an overall score between zero (no lung injury) and one (severe lung injury). The collagen score was calculated as previously described to grade lung fibrosis (37, 38). In brief, a paraffin section of lung, stained by a trichrome method, was systematically scanned in a microscope using a 10x objective. Each successive field was individually assessed for severity of interstitial fibrosis and allotted a score between 0 and 8 using a predetermined scale of severity. After examining the whole section, the mean score of all the fields was taken as the collagen score for that section and expressed correct to two decimal places.
BAL
Upon euthanasia, the mice lungs were lavaged with 1 ml PBS four times. The recovered BAL fluids (about 3.4 ml) were centrifuged, and pelleted cells were visualized and counted on Hema 3-stained cytocentrifuge slides (39–42).
scRNA-seq analysis
A lab protocol was developed to combine the 10x Genomics single-cell 5’ kit with customized sample multiplexing. Lungs freshly dissected from euthanized mice were dissociated into a single-cell suspension (43, 44). The cell samples of four mice from each group were pooled together and stained with TotalSeq CD45 antibody conjugated with unique oligo barcodes purchased from BioLegend. All hashtagged samples were pooled together for Gel Bead-in-Emulsion (GEM) partitioning and library construction using 10x genomics single cell 5’ kit (CG000330 Rev A). The resulting gene expression and cell hashing libraries were sequenced at a respective depth of 250 and 50 million reads with paired-ends on an Illumina NovaSeq S4 instrument. The 10x Genomics’ Cell Ranger pipeline was used to demultiplex hashtags into separate samples, perform alignment and filtering, decode cellular barcodes, count unique molecular identifiers (UMIs), normalize counts, and generate feature-barcode matrices. The derived matrices were further processed in Seurat. Quality control was firstly pursued to remove cells with low reads (UMIs<=500, Features<=200) and excess mitochondrial expression (>=12%) and batch effects, followed by dimensionality reduction, cell clustering, cell type classification, cell counting, and gene expression comparative analysis among cell clusters where differentiated gene expression was assessed at the population level and the cellular level. Differentially expressed genes (DEGs) derived above were imported into Ingenuity Pathway Analysis (IPA) (QIAGEN) for pathway and functional analysis.
scRNA-seq data in Lung Cancer Atlas (LuCA) were also analyzed to examine PDLIM2 expression in the lung macrophages of COPD patients versus normal individuals using the scANVI pipeline (45). The RDS file from the core atlas was downloaded from LuCA and imported into Seurat. Cells of interest were extracted by assay (10x 3’ v2), cell type (Macrophage), disease (Normal Control/COPD), and feature (PDLIM2). PDLIM2 expression in macrophages from each patient was calculated as the average of its cellular expression. Patients with fewer macrophage cells were skipped. Analysis of PDLIM2 expression in neutrophils was aborted as not enough cells were characterized in the original atlas data.
Gene microarray and bulk RNA-seq analysis
Lung Genomics Research Consortium (LGRC) gene expression microarray data were analyzed to examine PDLIM2 expression in the lungs of patients with COPD or ILD. The data had been normalized using a pairwise cyclic loess approach, and the probes were collapsed to one probe per gene by selecting the probe with the highest average signal (46–49).
Ex vivo phagocytosis assays
As described before (50), fresh mouse lung tissues were minced into small pieces, gently pressed with the syringe plunger top, and filtered with 40μm cell strainers. Cells passed through the strainer were seeded in a 24-well ultra-low attachment plate (Corning Inc., Corning, NY, USA) with 400μl culture medium containing the indicated antibody for 20 minutes. The cells were spun down at 1800 rpm for 5 min, and the supernatant was replaced with 400μl pHrodo Green S. aureus Bioparticles Conjugate (Thermo Fisher Scientific, Waltham, MA, USA. 1mg/ml) in Live Cell Imaging Solution (Thermo Fisher Scientific, Waltham, MA, USA). Two hours later, the phagocytic abilities of lung alveolar macrophages (AMs) were determined by flow cytometry (50, 51).
Ex vivo NET formation assays
Bone marrow cells were flushed from the femurs of the indicated mice and pelleted via centrifugation at 427 x g for 7 minutes at 4 °C. After red blood cell lysis, neutrophils were isolated at the interface of the Histopaque 1119 and Histopaque 1077 layers via density gradient centrifugation (52). Isolated neutrophils were cultured in duplicates in a 6-well plate at a concentration of 10 million cells per well. One duplicate set was stimulated with 50 ug/mL LPS overnight, while the other served as untreated control. To quantify NET formation, 100ul of culture supernatant was aspirated post-centrifugation and transferred to a 96-well plate. The samples were stained with PicoGreen (1:10,000 dilution), and fluorescence was measured on SpectraMAX ID3. The final NET quantification was determined by subtracting the reads of the untreated controls from the reads of the LPS-treated wells.
Statistics
Student’s t test (2-tailed, unpaired) was used to assess the significance of differences between 2 groups. Ordinary 1-way ANOVA was used to assess the significance of differences among groups of more than 2. All bars in the figures represent mean ± SEM. P values less than 0.05 and 0.01 were considered statistically significant and highly statistically significant, respectively.
Results
PDLIM2 repression in human lung diseases
Analysis of LGRC database revealed that PDLIM2 expression was decreased in the lungs of patients with COPD or ILD/IPF (Figure 1A, Supplementary Table S1). Of note, PDLIM2 repression was associated with the severity of COPD and ILD/IPF (Figures 1B, C). Analysis of the scRNA-seq data in LuCA further revealed that PDLIM2 was repressed in the lung macrophages of COPD patients (Figure 1D). Macrophages are the most abundant immune cells within the lung and serve as the key sentinels in the lung. They are also the main culprits of various pathogenic conditions, including COPD, ILD/IPF, lung cancer, and pulmonary infectious disease (7–9, 22, 32–34, 53, 54). Currently, it remains largely unknown how the important immune cells are transformed from lung guards into perpetrators. These data identified PDLIM2 repression as a common phenotype of COPD and ILD/IPF that may cause the pathogenic transformation of lung macrophages for disease progression.
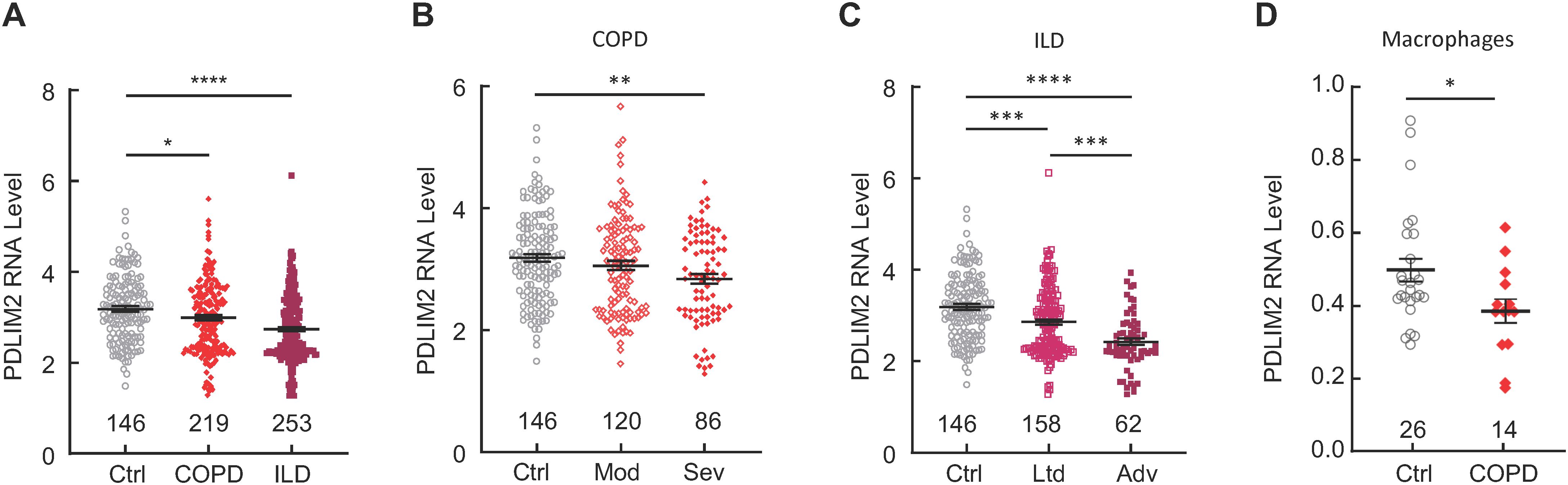
Figure 1. PDLIM2 repression in human COPD and ILD/IPF. (A) Analysis of LGRC database showing PDLIM2 repression in human COPD and ILD/IPF. (B) LGRC data showing association between PDLIM2 repression and COPD severity. (C) LGRC data showing association between PDLIM2 repression and ILD/IPF severity. (D) Analysis of the LuCA scRNA-seq data showing PDLIM2 repression in the lung macrophages of COPD patients. Ctrl stands for normal lung tissues. Sample numbers are listed above the x-axis. Each data point represents one patient. Data represent means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ANOVA or Student’s t test.
Increased susceptibility to LPS-induced lung injury and death by myeloid PDLIM2 deletion
To examine the role of lung macrophage intrinsic PDLIM2 in lung diseases, PDLIM2 mKO mice, in which PDLIM2 is selectively deleted in lung macrophages and other myeloid cells, were subjected to LPS model of ALI/ARDS (55). This model can also provide mechanistic insights into COPD, PF and lung infectious disease (54–57). In consistent with previous studies (58), LysM-Cre+/- mice showed no difference in LPS-induced ALI as FVB/N mice and PDLIM2flox/flox mice, all of which have normal PDLIM2 expression and are included in the WT control group of the studies (Figure 2A). PDLIM2 mKO mice showed markedly decreased survival after LPS treatment at the dose of 4 mg/kg body weight, compared to the WT control mice. Consistently, the lung tissues of PDLIM2 mKO mice showed exacerbated lung injury and much more severe inflammatory reactions, including inflammatory cell infiltration, alveolar congestion, alveolar wall thickening, hemorrhage, and collagen accumulation (Figures 2B–D). These data suggested that PDLIM2 repression, particularly in lung macrophages and neutrophils, is a key mechanism driving pathogenic inflammation and rendering patients much more vulnerable to infection, lung damage, and mortality.

Figure 2. Indispensable role of myeloid PDLIM2 in preventing lung injury and death induced by LPS. (A) Kaplan-Meier curve showing increased mortality of PDLIM2 mKO mice treated with LPS. (B) H&E staining showing worsen lung damage and augmented immune cell lung infiltration in PDLIM2 mKO mice treated with LPS (n = 3). (C) Trichrome staining showing higher collagen accumulation in the lung of PDLIM2 mKO mice treated with LPS (n = 6). (D) Hema 3 staining of BAL cells on cytocentrifuge slides showing more immune cells in the lung of PDLIM2 mKO mice treated with LPS (WT: n = 5; mKO: n = 4). Scale bars: 25 µm. Data represent means ± SEM. *P < 0.05; **P < 0.01; Student’s t test.
Imbalanced activation of pro- versus anti- inflammatory signaling pathways in the total lung immune cell population of myeloid PDLIM2 deletion mice by LPS
To systematically define the molecular mechanisms underlying the super-sensitivity of PDLIM2 mKO mice to LPS, immune cells enriched from the lung tissues of PDLIM2 mKO and WT mice treated with LPS or PBS were subjected to scRNA-seq. In line with no abnormalities of PDLIM2 mKO mice under pathogen-free conditions, scRNA-seq data showed no significant difference in the numbers and composition of immune cells, including macrophages and neutrophils in the lung of PDLIM2 mKO and WT mice treated with PBS (Supplementary Figure S1). As expected, LPS treatment induced dramatic but overall similar changes in both PDLIM2 mKO and WT mice. The top upstream regulators activated by LPS are also the same in both mice (Supplementary Figure S2).
Consistent with the histological studies above, notably, the scRNA-seq data indicated that LPS treatment led to more macrophages, neutrophils, T and B cells in the lung of PDLIM2 mKO mice in comparison to WT mice (Supplementary Figure S1D). In further support of this, the pathways and functions for leukocyte recruitment were activated by LPS at a higher level in all pulmonary immune cells of PDLIM2 mKO mice as a whole population (Figures 3A, B). Furthermore, the inflammatory signaling pathways were more activated at the total population level of lung immune cells in the PDLIM2 mKO mice, as evidenced by the higher activated pathogen-induced cytokine storm signaling, IL-17 signaling, IL-33 signaling, and HMGB1 signaling (Figures 3A, C). In contrast, activation of the acute phase response (APR) signaling pathway, which plays a central role in restoring tissue homeostasis, was activated at lower levels (Figures 3A, D). On the other hand, the anti-inflammatory LXR/RXR signaling pathway was repressed to a greater extent (Figure 3A). Similar patterns were seen for these signaling pathways in the lung of patients with COPD, ILD, or ALI/ARDS (Supplementary Figure S3). These data suggested that PDLIM2 repression, particularly in lung myeloid cells, disrupts the immune balance toward excessive inflammation in the lung in response to lung infection.

Figure 3. Over-activated pro-inflammatory signaling pathways in total lung immune cell population of LPS-treated myeloid PDLIM2 deletion mice. (A) Pathway and function analysis showing higher activated pro-inflammatory signaling pathways but less activated or more repressed anti-inflammatory signaling pathways in whole immune cells in the lung of LPS-treated PDLIM2 mKO mice. (B) Gene expression heatmap of component genes in lung immune cells showing increased leukocyte recruitment activation in the lung of LPS-treated PDLIM2 mKO mice. (C) Gene expression heatmap of component genes in lung immune cells showing increased pathogen-induced cytokine storm signaling in the lung of LPS-treated PDLIM2 mKO mice. (D) Gene expression heatmap of component genes in lung immune cells showing decreased acute phase response signaling in the lung of LPS-treated PDLIM2 mKO mice.
Overactive NF-κB and pathogenic activation of lung macrophages in myeloid PDLIM2 deletion mice by LPS
Logically, the phenotypes in LPS-treated PDLIM2 mKO mice were originally initiated and mostly attributed to PDLIM2 deficiency in lung macrophages, because they are the first immune cells that encounter LPS. Thus, the molecular difference of lung macrophages in LPS-treated PDLIM2 mKO and WT mice was characterized. In line with the role of PDLIM2 in repressing the master pro-inflammation transcription factor NF-κB (19–25, 59–66), scRNA-seq data showed much higher activation of the NF-κB signaling pathway in the lung macrophages of PDLIM2 mKO mice treated with LPS (Figure 4A). The proinflammatory macrophage classical activation signaling pathway was also more activated in the lung macrophages of LPS-treated PDLIM2 mKO mice (Figures 4A, B). However, the phagosome formation and inducible nitric oxide synthetase (iNOS) signaling pathways, which are critical for the most important innate immune function of macrophages to directly kill and clear pathogens, were less activated (Figures 4A, C). Consistently, AMs from LPS-treated PDLIM2 mKO mice showed a reduced phagocytotic ability ex vivo (Figure 4D).
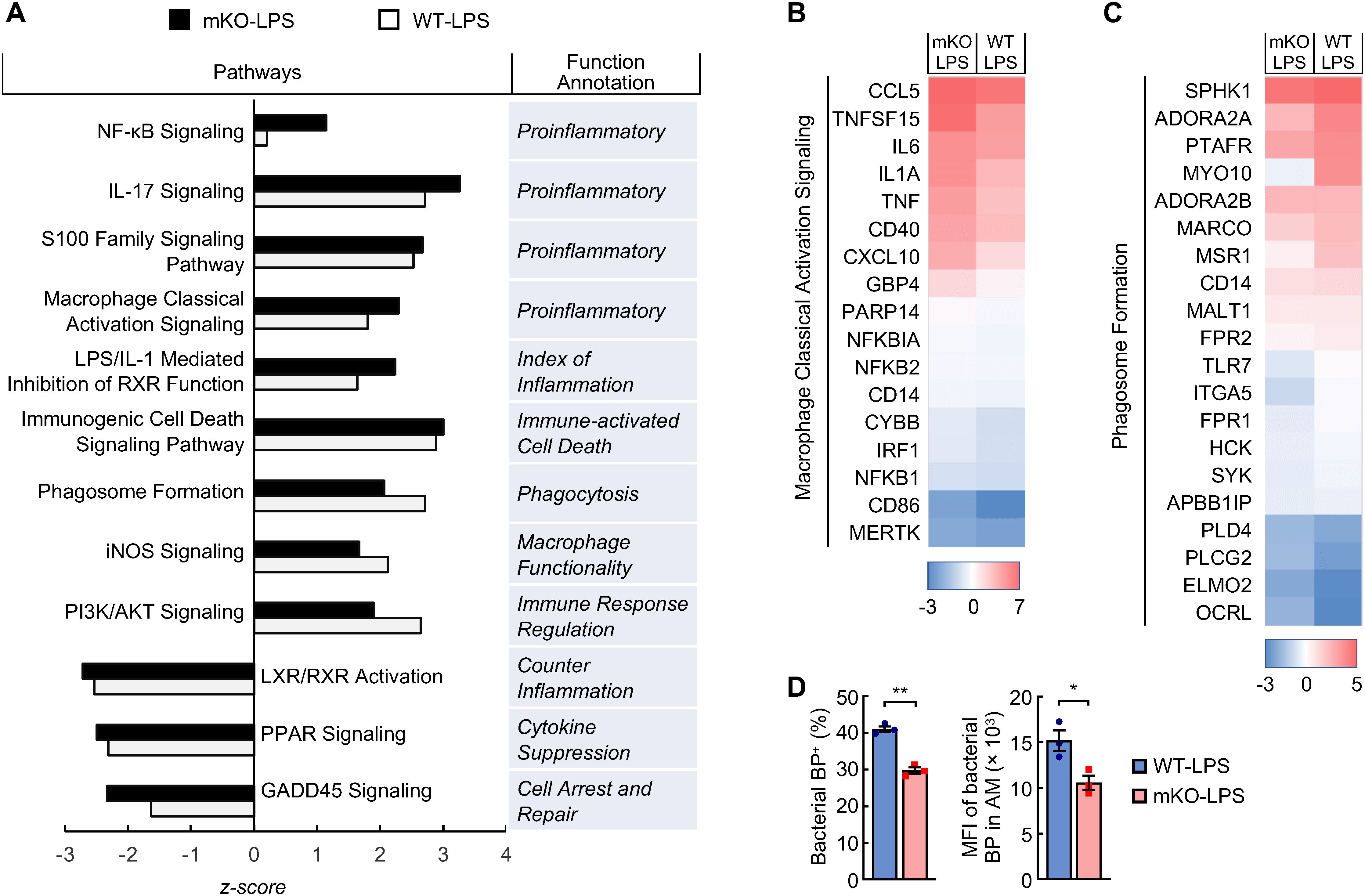
Figure 4. Pathogenic activation of signaling pathways in the lung macrophages of LPS-treated myeloid PDLIM2 deletion mice. (A) Pathway and function analysis of lung macrophages showing hyper-activated pro-inflammatory signaling pathways but less activated or more repressed signaling pathways involved in anti-inflammation or macrophage innate immunity function in LPS-treated PDLIM2 mKO mice. (B) Gene expression heatmap of component genes showing increased macrophage classical activation signaling pathway in the lung macrophages of LPS-treated PDLIM2 mKO mice. (C) Gene expression heatmap of component genes showing lower phagosome formation ability of the lung macrophages in LPS-treated PDLIM2 mKO mice. (D) Ex vivo phagocytosis assays showing defective ability of AMs from LPS-treated PDLIM2mKO mice in phagocytizing pHrodo green S. aureus bioparticles (BP) (n = 3). Data represent means ± SEM. *P < 0.05; **P < 0.01; Student’s t test.
Many other inflammation pathways were also found to be more activated in the lung macrophages of the LPS-treated PDLIM2 mKO mice, such as IL-17 signaling, LPS/IL-1 mediated inhibition of RXR function, S100 family signaling, and immunogenic cell death signaling (Figure 4A). In sharp contrast, several signaling pathways known to counter and repress inflammation and/or facilitate the innate immunity and wound repairing of macrophages, i.e., PI3K/AKT, LXR/RXR, PPAR, and GADD45 signaling, were either less activated or more repressed. These data suggested that PDLIM2 repression leads to NF-κB over-activation induced by LPS, eventually resulting in aberrant activation of lung macrophages for pathogenic inflammation and fatal lung injury.
Exaggerated NF-κB activity and divergent activation of lung neutrophils in myeloid PDLIM2 deletion mice by LPS
Following lung macrophage activation by pulmonary infection, neutrophils are the first to be recruited into the lung and become predominant. Like macrophages, neutrophils are major lung sentinels that could also be pathogenic drivers. Consistent with their PDLIM2 deficiency, neutrophils in the lung of LPS-treated PDLIM2 mKO mice, like lung macrophages, had much higher activation of the NF-κB signaling pathway in comparison to WT mice (Figure 5A). Several common pro-inflammatory signaling pathways, including toll-like receptor, LPS/IL-1 mediated inhibition of RXR function, and pyroptosis signaling pathways, were more activated in the neutrophils of the PDLIM2 mKO mice (Figures 5A, B). Activation of the triggering receptor expressed on myeloid cells 1 (TREM1) signaling pathway was also much higher (Figures 5A, C). TREM1 is predominantly expressed on neutrophils, and its activation leads to proinflammatory immune responses (67). However, the neutrophil extracellular trap (NET) signaling pathway was dramatically lower (Figures 5A, D). NET is an important mechanism that neutrophils use to immobilize and kill pathogens (68). In support of this, PDLIM2-deficient neutrophils treated with LPS exhibited a reduced NET formation ability ex vivo in comparison with WT neutrophils with the same LPS treatment (Figure 5E). These data suggested that PDLIM2 repression in neutrophils dampens their host-protective functions against lung infection and unleashes their pro-inflammatory activity to cause excessive inflammation and lethal lung injury.

Figure 5. Aberrant activation of signaling pathways in the lung neutrophils of LPS-treated myeloid PDLIM2 deletion mice. (A) Pathway and function analysis of lung neutrophils showing superior activated pro-inflammatory signaling pathways but less activated signaling pathways involved in neutrophil innate immunity function in LPS-treated PDLIM2 mKO mice. (B) Gene expression heatmap of component genes showing elevated activation of the pyroptosis signaling pathway in the lung neutrophils of LPS-treated PDLIM2 mKO mice. (C) Gene expression heatmap of component genes showing elevated TREM1 signaling activation in the lung neutrophils of LPS-treated PDLIM2 mKO mice. (D) Gene expression heatmap of component genes showing less activated NET signaling in the lung neutrophils in LPS-treated PDLIM2 mKO mice. (E) Ex vivo NET assays showing defective NET ability of LPS-treated neutrophils deficient in PDLIM2 (n = 3). Data represent means ± SEM. *P < 0.05; Student’s t test.
Discussion
The studies above provide the first evidence linking PDLIM2 to ALI/ARDS, COPD, ILD/IPF, and lung infection and infectious disease. As a matter of fact, they are the first investigation on the role of PDLIM2 in the lung diseases other than lung cancer. They have identified PDLIM2 repression as a common phenomenon of human COPD, ILD/IPF, and lung cancer. Using the LPS mouse model, they have further shown that repression of PDLIM2 in myeloid cells, especially in lung macrophages, is a causal mechanism underlying infection-induced ALI/ARDS and death.
PDLIM2 protects against lung infection by limiting inflammation from damage while simultaneously promoting pathogen killing (Supplementary Figure S4). PDLIM2 repression in lung macrophages and neutrophils results in uncontrolled activation of NF-κB in these crucial immune cells, thereby unleashing their pro-inflammatory activity and dampening their host-protective functions against lung infection. The pathogenic activation of lung macrophages and neutrophils causes excessive inflammation in the lung and subsequently lung damage and animal death.
It seems that myeloid PDLIM2 repression is dispensable for or only plays an insufficient role in the initiation or even the early stages of COPD and ILD/IPF, since PDLIM2 mKO mice are healthy and show no phenotypes under pathogen-free conditions, including no COPD and ILD/IPF development. Nevertheless, PDLIM2 expression is largely intact in the lung of COPD and ILD/IPF patients at the early disease stages. Except for rendering patients vulnerable to lung infections, however, the aberrant inflammation caused by PDLIM2 repression may also contribute to COPD, ILD/IPF lung disease progression, as suggested by the association of PDLIM2 repression with the severity of COPD, ILD/IPF in humans and the excessive lung inflammation and damage caused by myeloid PDLIM2 deficiency in mice treated with LPS, which in COPD, ILD/IPF may also cause disease progression.
Recent studies indicate that PDLIM2 repression in the lung macrophages of mice with lung cancer is attributed to the transcription repressor BACH1 activated by reactive oxygen species (ROS) (22). Given the high ROS levels in the lung of patients with COPD and ILD/IPF (69), it is highly plausible that PDLIM2 repression in COPD and ILD/IPF is also mediated by ROS-activated BACH1. Interestingly, ROS inhibitors can restore PDLIM2 expression in lung macrophages and prevents lung cancer in vivo (22). Furthermore, delivery of exogenous PDLIM2 by clinically feasible nanoparticles shows high therapeutic efficacy in the mouse model of refractory lung cancer (21). Thus, therapeutic strategies for targeting PDLIM2 should be further explored for the prevention and treatment of COPD, ILD/ILD, ALI/ARDS, and infectious disease.
Data availability statement
From https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000624.v1.p1, we obtained LGRC data on human COPD and ILD/IPF. The scRNA-seq data on human COPD were obtained from LuCA at https://cellxgene.cziscience.com/collections/edb893ee-4066-4128-9aec2785eb2b03f8287. All data generated or analyzed during this study are included in this article and its additional files. The mouse scRNA-seq data produced in this paper are deposited in the online NCBI Gene Expression Omnibus (GSE249800) for free access. The Extra data that support the findings of this study are available from the corresponding authors upon reasonable request.
Ethics statement
The studies involving humans were approved by the Institutional Animal Care and Use Committee (IACAUC) of the University of Southern California. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The animal study was approved by the Institutional Animal Care and Use Committee (IACAUC) of the University of Southern California. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
FG: Methodology, Writing – review & editing, Formal Analysis, Investigation, Software, Resources, Validation, Data curation, Visualization. XL: Methodology, Investigation, Data curation, Validation, Writing – review & editing. FS: Writing – review & editing, Investigation, Data curation, Validation, Methodology. YX: Methodology, Validation, Data curation, Investigation, Writing – review & editing. GX: Methodology, Writing – review & editing, Supervision, Validation, Investigation, Resources, Writing – original draft, Formal Analysis, Visualization, Project administration, Conceptualization, Funding acquisition. ZQ: Visualization, Resources, Formal Analysis, Writing – original draft, Project administration, Funding acquisition, Methodology, Conceptualization, Data curation, Investigation, Validation, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study is supported in part by the NIH National Institute of General Medical Sciences (NIGMS) grant R01 GM144890, National Cancer Institute (NCI) grant R01 CA258614, National Heart, Lung, and Blood Institute R01 HL177140, American Cancer Society (ACS) Research Scholar grant RSG-19-166-01-TBG, American Lung Association (ALA) Lung Cancer Discovery Award 821321, and Tobacco Related-Disease Research Program (TRDRP) Research Award T33IR6461.
Acknowledgments
We thank S.D.S. for his advice and constructive feedback.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1669117/full#supplementary-material
Supplementary Figure 1 | Experimental design and outline on multiplexed scRNA-seq data analysis. (A) Overall experimental design. (B) Demultiplexing of hashtagged samples. (C) Cell population profiling of immune cells in the lung of PDLIM2 mKO and WT mice treated with LPS or PBS. (D) Cell population profiling showing increased immune cells in the lung of LPS-treated PDLIM2 mKO mice.
Supplementary Figure 2 | Top upstream regulators identified for LPS-induced gene expression changes in lung immune cells of PDLIM2 mKO and WT mice.
Supplementary Figure 3 | Top pathways altered in the lung of patients with ILD, COPD or ARDS.
Supplementary Figure 4 | Graphic summary of PDLIM2’s dual roles in the case of lung infection.
Supplementary Table 1 | The clinical attributes data, downloaded from the Lung Genomics Research Consortium (LGRC) website, for all patients included in the analyses shown in Figures 1A–C.
References
1. Syamlal G, Kurth LM, Dodd KE, Blackley DJ, Hall NB, and Mazurek JM. Chronic obstructive pulmonary disease mortality by industry and occupation — United States, 2020. MMWR Morb Mortal Wkly Rep. (2022) 71:1550–4. doi: 10.15585/mmwr.mm7149a3
2. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, and Jemal A. Cancer statistics, 2025. CA Cancer J Clin. (2025) 75:10–45. doi: 10.3322/caac.21871
3. Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. (2018) 27:180076. doi: 10.1183/16000617.0076-2018
4. Shaw J, Marshall T, Morris H, and Chaudhuri N. Idiopathic pulmonary fibrosis: a clinical update. Br J Gen Pract. (2018) 68:249–50. doi: 10.3399/bjgp18X696149
5. Kalchiem-Dekel O, Galvin JR, Burke AP, Atamas SP, and Todd NW. Interstitial lung disease and pulmonary fibrosis: a practical approach for general medicine physicians with focus on the medical history. J Clin Med. (2018) 7:476. doi: 10.3390/jcm7120476
6. Parris BA, O'Farrell HE, Fong KM, and Yang IA. Chronic obstructive pulmonary disease (COPD) and lung cancer: common pathways for pathogenesis. J Thorac Dis. (2019) 11:S2155–72. doi: 10.21037/jtd.2019.10.54
7. Bagnato G and Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. (2015) 24:102–14. doi: 10.1183/09059180.00003214
8. Heukels P, Moor CC, von der Thüsen JH, Wijsenbeek MS, and Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med. (2019) 147:79–91. doi: 10.1016/j.rmed.2018.12.015
9. Shenderov K, Collins SL, Powell JD, and Horton MR. Immune dysregulation as a driver of idiopathic pulmonary fibrosis. J Clin Invest. (2021) 131:e143226. doi: 10.1172/JCI143226
10. Perlin E, Bang KM, Shah A, Hursey PD, Whittingham WL, Hashmi K, et al. The impact of pulmonary infections on the survival of lung cancer patients. Cancer. (1990) 66:593–6. doi: 10.1002/1097-0142(19900801)66:3<593::AID-CNCR2820660331>3.0.CO;2-R
11. Akinosoglou KS, Karkoulias K, and Marangos M. Infectious complications in patients with lung cancer. Eur Rev Med Pharmacol Sci. (2013) 17:8–18.
12. Lange P. Chronic obstructive pulmonary disease and risk of infection. Pneumonol Alergol Pol. (2009) 77:284–8. doi: 10.5603/ARM.27817
13. Margaritopoulos GA, Antoniou KM, and Wells AU. Comorbidities in interstitial lung diseases. Eur Respir Rev. (2017) 26:160027. doi: 10.1183/16000617.0027-2016
14. Sekine Y, Katsura H, Koh E, Hiroshima K, and Fujisawa T. Early detection of COPD is important for lung cancer surveillance. Eur Respir J. (2012) 39:1230–40. doi: 10.1183/09031936.00126011
15. Durham AL and Adcock IM. The relationship between COPD and lung cancer. Lung Cancer. (2015) 90:121–7. doi: 10.1016/j.lungcan.2015.08.017
16. Naccache JM, Gibiot Q, Monnet I, Antoine M, Wislez M, Chouaid C, et al. Lung cancer and interstitial lung disease: a literature review. J Thorac Dis. (2018) 10:3829–44. doi: 10.21037/jtd.2018.05.75
17. Ballester B, Milara J, and Cortijo J. Idiopathic pulmonary fibrosis and lung cancer: mechanisms and molecular targets. Int J Mol Sci. (2019) 20:593. doi: 10.3390/ijms20030593
18. Abu Qubo A, Numan J, Snijder J, Padilla M, Austin JHM, Capaccione KM, et al. Idiopathic pulmonary fibrosis and lung cancer: future directions and challenges. Breathe (Sheff). (2022) 18:220147. doi: 10.1183/20734735.0147-2022
19. Guo ZS and Qu Z. PDLIM2: Signaling pathways and functions in cancer suppression and host immunity. Biochim Biophys Acta Rev Cancer. (2021) 1876:188630. doi: 10.1016/j.bbcan.2021.188630
20. Sun F, Li L, Yan P, Zhou J, Shapiro SD, Xiao G, et al. Causative role of PDLIM2 epigenetic repression in lung cancer and therapeutic resistance. Nat Commun. (2019) 10:5324. doi: 10.1038/s41467-019-13331-x
21. Sun F, Yan P, Xiao Y, Zhang H, Shapiro SD, Xiao G, et al. Improving PD-1 blockade plus chemotherapy for complete remission of lung cancer by nanoPDLIM2. eLife. (2024) 9:RP89638R1. doi: 10.7554/eLife.89638.2
22. Li L, Sun F, Han L, Liu X, Xiao Y, Gregory AD, et al. PDLIM2 repression by ROS in alveolar macrophages promotes lung tumorigenesis. JCI Insight. (2021) 6:e144394. doi: 10.1172/jci.insight.144394
23. Qu Z, Yan P, Fu J, Jiang J, Grusby MJ, Smithgall TE, et al. DNA methylation-dependent repression of PDZ-LIM domain-containing protein 2 in colon cancer and its role as a potential therapeutic target. Cancer Res. (2010) 70:1766–72. doi: 10.1158/0008-5472.CAN-09-3263
24. Qu Z, Fu J, Yan P, Hu J, Cheng SY, and Xiao G. Epigenetic repression of PDZ-LIM domain-containing protein 2: implications for the biology and treatment of breast cancer. J Biol Chem. (2010) 285:11786–92. doi: 10.1074/jbc.M109.086561
25. Sun F, Xiao Y, and Qu Z. Oncovirus Kaposi sarcoma herpesvirus (KSHV) represses tumor suppressor PDLIM2 to persistently activate nuclear factor κB (NF-κB) and STAT3 transcription factors for tumorigenesis and tumor maintenance. J Biol Chem. (2015) 290:7362–8. doi: 10.1074/jbc.C115.637918
26. Yan P, Qu Z, Ishikawa C, Mori N, and Xiao G. Human T-cell leukemia virus type I-mediated repression of PDZ-LIM domain-containing protein 2 involves DNA methylation but independent of the viral oncoprotein tax. Neoplasia. (2009) 11:1036–41. doi: 10.1593/neo.09752
27. Tanaka T, Soriano MA, and Grusby MJ. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity. (2005) 22:729–36. doi: 10.1016/j.immuni.2005.04.008
28. Torrado M, Senatorov VV, Trivedi R, Fariss RN, and Tomarev SI. Pdlim2, a novel PDZ-LIM domain protein, interacts with alpha-actinins and filamin A. Invest Ophthalmol Vis Sci. (2004) 45:3955–63. doi: 10.1167/iovs.04-0721
29. Loughran G, Healy NC, Kiely PA, Huigsloot M, Kedersha NL, and O'Connor R. Mystique is a new insulin-like growth factor-I-regulated PDZ-LIM domain protein that promotes cell attachment and migration and suppresses anchorage-independent growth. Mol Biol Cell. (2005) 16:1811–22. doi: 10.1091/mbc.e04-12-1052
30. Ashouri K, Krause H, Eilliott A, Liu SV, Ma PC, Halmos B, et al. Characterization of PDLIM2 in non-small cell lung cancer. Cancer Res. (2024) 84:5201. doi: 10.1158/1538-7445.AM2024-5201
31. Le TH, Sun F, Xiao G, and Qu Z. NanoPDLIM2-based combination therapy for lung cancer treatment in mouse preclinical studies. Bio Protoc. (2025) 15:e5437. doi: 10.21769/BioProtoc.5437
32. Zhou J, Qu Z, Sun F, Han L, Li L, Yan S, et al. Myeloid STAT3 promotes lung tumorigenesis by transforming tumor immunosurveillance into tumor-promoting inflammation. Cancer Immunol Res. (2017) 5:257–68. doi: 10.1158/2326-6066.CIR-16-0073
33. Li L, Han L, Sun F, Zhou J, Ohaegbulam KC, Tang X, et al. NF-κB RelA renders tumor-associated macrophages resistant to and capable of directly suppressing CD8+ T cells for tumor promotion. Oncoimmunology. (2018) 7:e1435250. doi: 10.1080/2162402X.2018.1435250
34. Sun F, Guo ZS, Gregory AD, Shapiro SD, Xiao G, and Qu Z. Dual but not single PD-1 or TIM-3 blockade enhances oncolytic virotherapy in refractory lung cancer. J Immunother Cancer. (2020) 8:e000294. doi: 10.1136/jitc-2019-000294
35. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, et al. Acute lung injury in animals study group. an official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. (2011) 44:725–38. doi: 10.1165/rcmb.2009-0210ST
36. Zhang H, Xiao Y, Gao F, Le TH, Shapiro SD, Xiao G, et al. Alveolar epithelial NF-κB/RelA guards the lung against bacterial infection. J Immunol. (In press).
37. Ashcroft T, Simpson JM, and Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. (1988) 41:467–70. doi: 10.1136/jcp.41.4.467
38. Stritt M, Stalder AK, and Vezzali E. Orbit image analysis: an open-source whole slide image analysis tool. PloS Comput Biol. (2020) 16:e1007313. doi: 10.1371/journal.pcbi.1007313
39. Sun F, Xiao G, and Qu Z. Murine bronchoalveolar lavage. Bio Protoc. (2017) 7:e2287. doi: 10.21769/BioProtoc.2287
40. Zhou J, Qu Z, Yan S, Sun F, Whitsett JA, Shapiro SD, et al. Differential roles of STAT3 in the initiation and growth of lung cancer. Oncogene. (2015) 34:3804–14. doi: 10.1038/onc.2014.318
41. Qu Z, Sun F, Zhou J, Li L, Shapiro SD, and Xiao G. Interleukin-6 prevents the initiation but enhances the progression of lung cancer. Cancer Res. (2015) 75:3209–15. doi: 10.1158/0008-5472.CAN-14-3042
42. Sun F, Xiao Y, Shapiro SD, Qu Z, and Xiao G. Critical and distinct roles of cell type-specific NF-κB2 in lung cancer. JCI Insight. (2024) 9:e164188. doi: 10.1172/jci.insight.164188
43. Sun F, Qu Z, and Xiao G. Methods to Detect NF-κB Activity in tumor-associated macrophage (TAM) populations. Methods Mol Biol. (2021) 2366:213–41. doi: 10.1007/978-1-0716-1669-7_13
44. Gao F, Li C, Smith SM, Peinado N, Kohbodi G, Tran E, et al. Decoding the IGF1 signaling gene regulatory network behind alveologenesis from a mouse model of bronchopulmonary dysplasia. eLife. (2022) 11:e77522. doi: 10.7554/eLife.77522.sa2
45. Salcher S, Sturm G, Horvath L, Untergasser G, Kuempers C, Fotakis G, et al. High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. Cancer Cell. (2022) 40:1503–20. doi: 10.1016/j.ccell.2022.10.008
46. Bauer Y, Tedrow J, de Bernard S, Birker-Robaczewska M, Gibson KF, Guardela BJ, et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. (2015) 52:217–31. doi: 10.1165/rcmb.2013-0310OC
47. Qing G, Qu Z, and Xiao G. Endoproteolytic processing of C-terminally truncated NF-κB2 precursors at kappaB-containing promoters. Proc Natl Acad Sci U S A. (2007) 104:5324–9. doi: 10.1073/pnas.0609914104
48. Qu Z, Sun D, and Young W. Lithium promotes neural precursor cell proliferation: evidence for the involvement of the non-conical GSK3β-NF-AT signaling. Cell Biosci. (2011) 1:18. doi: 10.1186/2045-3701-1-18
49. Sun F, Qu Z, Xiao Y, Zhou J, Burns TF, Stabile LP, et al. NF-κB1 p105 suppresses lung tumorigenesis through the Tpl2 kinase but independently of its NF-κB function. Oncogene. (2016) 35:2299–310. doi: 10.1038/onc.2015.299
50. Sun F, Li L, Xiao Y, Gregory AD, Shapiro SD, Xiao G, et al. Alveolar macrophages inherently express programmed death-1 ligand 1 for optimal protective immunity and tolerance. J Immunol. (2021) 207:110–4. doi: 10.4049/jimmunol.2100046
51. Qu Z, Fu J, Ma H, Zhou J, Jin M, Mapara MY, et al. PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease. Cell Biosci. (2012) 2:23. doi: 10.1186/2045-3701-2-23
52. Swamydas M, Luo Y, Dorf ME, and Lionakis MS. Isolation of mouse neutrophils. Curr Protoc Immunol. (2015) 110:3.20.1–3.20.15. doi: 10.1002/0471142735.im0320s110
53. Malainou C, Abdin SM, Lachmann N, Matt U, and Herold S. Alveolar macrophages in tissue homeostasis, inflammation, and infection: evolving concepts of therapeutic targeting. J Clin Invest. (2023) 133:e170501. doi: 10.1172/JCI170501
54. Vlahos R and Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. (2014) 5:435. doi: 10.3389/fimmu.2014.00435
55. Matute-Bello G, Frevert CW, and Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L379–99. doi: 10.1152/ajplung.00010.2008
56. Wright JL, Cosio M, and Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L1–15. doi: 10.1152/ajplung.90200.2008
57. Marshall RP, Bellingan G, Webb S, Puddicombe A, Goldsack N, McAnulty RJ, et al. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med. (2000) 162:1783–8. doi: 10.1164/ajrccm.162.5.2001061
58. Gong KQ, Frevert C, and Manicone AM. Deletion of LysM in LysMCre recombinase homozygous mice is non-contributory in LPS-induced acute lung injury. Lung. (2019) 197:819–23. doi: 10.1007/s00408-019-00286-5
59. Xiao G, Rabson AB, Young W, Qing G, and Qu Z. Alternative pathways of NF-κB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. (2006) 17:281–93. doi: 10.1016/j.cytogfr.2006.04.005
61. Tanaka T, Grusby MJ, and Kaisho T. PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. (2007) 8:584–91. doi: 10.1038/ni1464
62. Gao F, Chen YW, Shapiro SD, Xiao G, and Qu Z. PDLIM2 repression: a common mechanism in viral lung infection. BioRxiv. (2025). doi: 10.1101/2025.09.12.675949
63. Sun F, Xiao G, and Qu Z. PDLIM2 is a novel E5 ubiquitin ligase enhancer that stabilizes ROC1 and recruits the ROC1-SCF ubiquitin ligase to ubiquitinate and degrade NF-κB RelA. Cell Biosci. (2024) 14:99. doi: 10.1186/s13578-024-01281-x
64. Li L, Han L, and Qu Z. NF-κB RelA is a cell-intrinsic metabolic checkpoint restricting glycolysis. Cell Biosci. (2024) 14:11. doi: 10.1186/s13578-024-01196-7
65. Fu J, Qu Z, Yan P, Ishikawa C, Aqeilan RI, Rabson AB, et al. The tumor suppressor gene WWOX links the canonical and noncanonical NF-κB pathways in HTLV-I Tax-mediated tumorigenesis. Blood. (2011) 117:1652–61. doi: 10.1182/blood-2010-08-303073
66. Song X and Qu Z. NF-κB1 deficiency promotes macrophage-derived adrenal tumors but decreases neurofibromas in HTLV-I LTR-Tax transgenic mice. PloS One. (2024) 19:e0303138. doi: 10.1371/journal.pone.0303138
67. Siskind S, Brenner M, and Wang P. TREM-1 modulation strategies for sepsis. Front Immunol. (2022) 13:907387. doi: 10.3389/fimmu.2022.907387
68. Branzk N and Papayannopoulos V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol. (2013) 35:513–30. doi: 10.1007/s00281-013-0384-6
Keywords: PDLIM2, NF-κB, RelA/p65, lung infection, COPD, ILD/IPF, lung cancer, LPS
Citation: Gao F, Liu X, Sun F, Xiao Y, Xiao G and Qu Z (2025) Myeloid PDLIM2 repression as a common mechanism of infection susceptibility in lung diseases. Front. Immunol. 16:1669117. doi: 10.3389/fimmu.2025.1669117
Received: 18 July 2025; Accepted: 03 November 2025;
Published: 19 November 2025.
Edited by:
Kunal Bhattacharya, Patanjali Research Institute, IndiaReviewed by:
Xin Zhu, First Affiliated Hospital of Chongqing Medical University, ChinaJulie Gunnells Ledford, University of Arizona, United States
Copyright © 2025 Gao, Liu, Sun, Xiao, Xiao and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaoxia Qu, Wmhhb3hpYS5RdUBtZWQudXNjLmVkdQ==
†These authors have contributed equally to this work and share first authorship