 Chao Lei
Chao Lei Dongjie Wu
Dongjie Wu Houyan Zhang
Houyan Zhang Zhiya Yang2
Zhiya Yang2 Bohao Huang
Bohao Huang Wenliang Lv
Wenliang Lv- 1Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Department of Oncology, Wangjing Hospital, Chinese Academy of Traditional Chinese Medicine, Beijing, China
- 3School of Traditional Chinese Medicine, Beijing University of Traditional Chinese Medicine, Beijing, China
Organ fibrosis represents a final common pathway of chronic tissue injury, characterized by persistent extracellular matrix (ECM) accumulation and progressive loss of organ function. While canonical inflammatory and profibrotic cascades have been extensively studied, emerging evidence highlights the pivotal role of mechanotransduction-the process by which cells sense and transduce biomechanical cues-in orchestrating immune responses and driving fibrotic remodeling. This review conceptualizes the mechanotransduction-immune axis as a dual regulatory network wherein mechanical forces not only activate profibrotic signaling in resident stromal cells but also dynamically reprogram immune cell phenotypes and functions. We systematically delineate the molecular and cellular mechanisms by which matrix stiffness, shear stress, and mechanical stretch engage integrins, focal adhesion kinase, Piezo1, and TRPV4 to coordinate inflammatory signaling and ECM remodeling. Additionally, we discuss how immune cells, including macrophages, T cells, and neutrophils, sense and respond to mechanical inputs to amplify profibrotic responses. Finally, we summarize emerging translational therapeutic perspectives targeting this mechanotransduction-immune interplay, encompassing small-molecule inhibitors, nanomedicine approaches, gene editing technologies, and cell therapies. By integrating mechanistic insights and translational strategies, this review aims to provide a comprehensive framework for understanding and therapeutically targeting the mechanotransduction-immune axis in organ fibrosis.
Highlights
● Matrix stiffness, shear stress, and tensile forces activate key profibrotic mechanosensors.
● The mechanotransduction-immune axis forms an integrated regulatory network in fibrosis.
● Immune-mechanical crosstalk amplifies profibrotic signaling in organ fibrosis.
● Emerging therapeutic strategies and smart nanocarriers targeting this axis offer promising precision interventions for fibrosis across organs.
1 Introduction
Organ fibrosis represents a core pathological process in end-stage organ failure across multiple chronic diseases, commonly affecting vital organs such as the heart, liver, lungs, and kidneys. It is primarily characterized by excessive deposition and aberrant remodeling of the extracellular matrix (ECM), ultimately leading to loss of tissue elasticity and progressive functional decline (1, 2). In recent years, with in-depth investigation into fibrogenesis, the mechanical properties of tissues have emerged as a critical focus in fibrosis research. Mechanotransduction refers to the biological process wherein cells convert external mechanical stimuli-including matrix stiffness, fluid shear stress, and tensile forces-into biochemical signals. This conversion activates specific signaling pathways, thereby modulating cellular proliferation, migration, polarization, and gene expression (3). The pivotal role of mechanical stimuli in fibrosis progression is widely recognized, and their function in regulating inflammation and immune responses is increasingly emphasized (4). Under pathological conditions, significant alterations in tissue biomechanics occur. Concurrently, the structure and function of mechanotransduction molecules at the cellular level-such as adhesion molecules, ion channels, and cytoskeletal proteins-are modified, thereby influencing the activity of multiple associated signaling pathways. Such biomechanical dysregulation manifests across fibrotic processes in various organs. Myofibroblasts serve as the primary ECM producers, and their activation constitutes a critical step in fibrosis development. However, this process does not occur in isolation. The immune system, particularly innate and adaptive immune cells, plays an indispensable role in regulating myofibroblast activation and fibrotic responses (5).
In recent years, with the advancement of the emerging interdisciplinary field of mechanoimmunology, it has become increasingly recognized that mechanical signals not only play crucial roles in embryonic development, cardiovascular homeostasis, and bone metabolism but also profoundly participate in inflammation and immune regulation (6). During circulation, immune cells are continually exposed to diverse mechanical environments, modulated by forces such as stretch, shear, and compression. Concurrently, tissue-resident immune cells persistently experience mechanical stimuli from their local microenvironment. Furthermore, the structure and spatial architecture of the ECM constitute complex mechanical inputs that further influence immune cell functional states. These mechanical signals are transmitted through mechanosensors on the cell membrane and intracellularly, integrated into mechanotransduction networks, and subsequently activate multiple signaling pathways. Aberrant mechanical environments are considered key triggers for dysregulated interplay between structural cells and immune cells. This dual-dimensional regulatory mechanism-simultaneously involving both mechanical and immunological aspects-represents the core focus of mechanoimmunology research.
This review will systematically delineate the driving mechanisms of mechanical signals in fibrosis progression and their regulatory effects on immune cell behavior. We will specifically emphasize the response patterns of different immune cells to mechanical stimuli and their functional roles within the fibrotic microenvironment. By integrating current research advances, we aim to provide novel perspectives for understanding the impact of the mechanical microenvironment on immune regulation and fibrotic pathology, thereby establishing a theoretical framework for developing relevant therapeutic strategies (Figure 1).
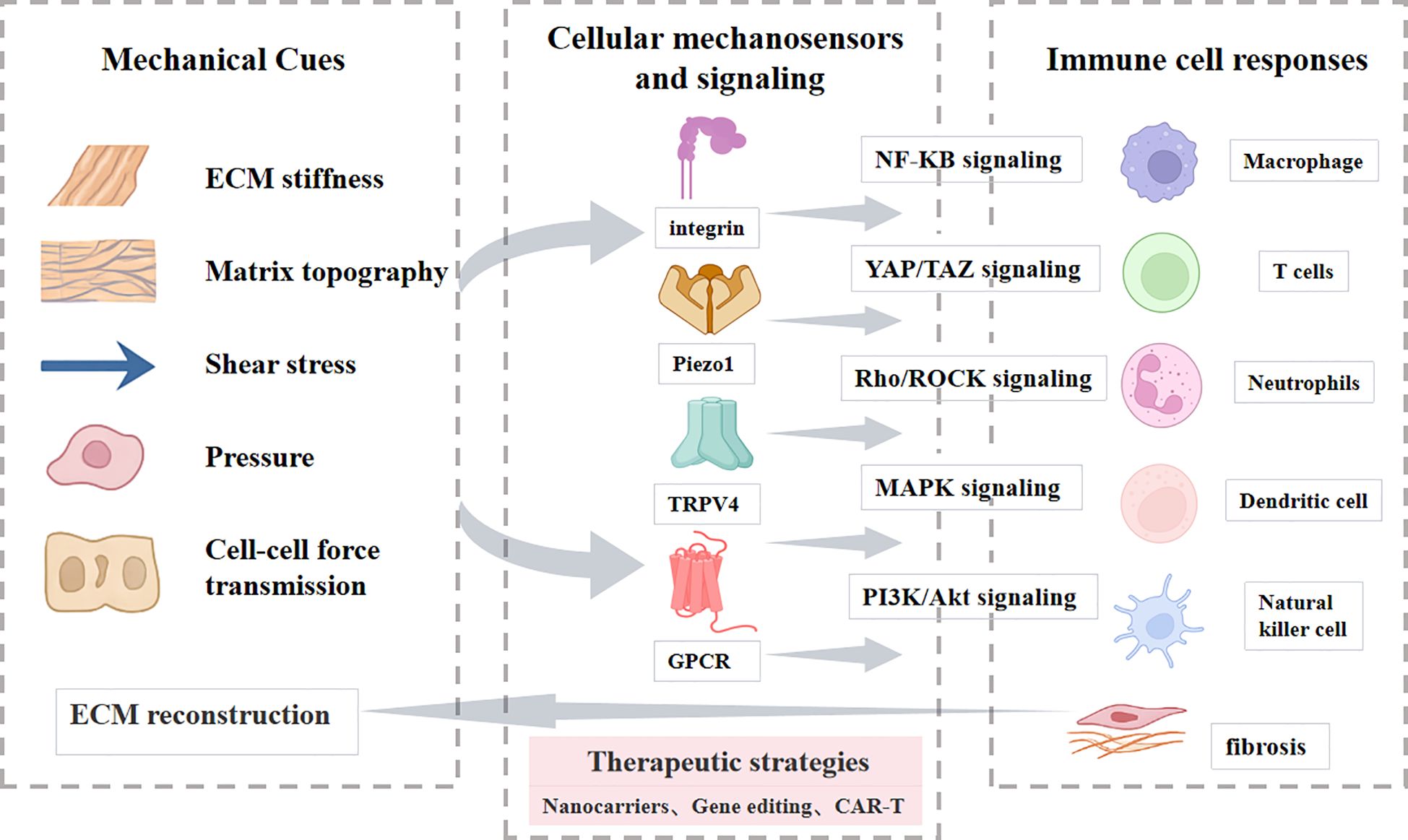
Figure 1. Overview of the review structure. A schematic illustration guiding readers through the main sections and topics covered in this review.
2 Mechanosensation and fibrosis-driving mechanisms
Mechanotransduction converts external physical stimuli into intracellular signaling events, ultimately inducing alterations in cellular behaviors such as proliferation, migration, gene expression regulation, and differentiation (7, 8). Cellular responses to mechanical stimuli rely on mechanosensors distributed across the extracellular space, cytoplasm, and nucleus to perceive and integrate mechanical cues, thereby activating specific biological reactions. Key mechanosensors identified to date include Integrins, FAK, Mechanosensitive ion channels, Proteoglycan complexes, Cytoskeletal components, and Nuclear structural proteins (9–12). These sensors detect extracellular mechanical changes and transduce them into biochemical signals through multiple downstream pathways, including ROCK, YAP/TAZ, MAPK, PI3K/Akt pathway, and NF-κB (13–15).
2.1 ECM stiffness and topography sensing
2.1.1 Matrix stiffness
In healthy tissues, the ECM maintains moderate elasticity and organized fiber alignment. Fibrotic remodeling leads to excessive deposition of collagen and other matrix components, markedly increasing stiffness and altering microarchitecture (9, 16). Cells sense increased rigidity mainly via integrins, which cluster upon ligand engagement and recruit focal adhesion kinase FAK and Src family kinases (17–20). This activation drives RhoA/ROCK signaling, promoting stress fiber assembly and reinforcing cytoskeletal tension. Elevated intracellular force facilitates the nuclear translocation of mechanosensitive transcription factors, which upregulate profibrotic genes including ACTA2 and COL1A1. Concurrently, matrix stiffening promotes the release and activation of latent TGF-β, which synergizes with integrin signaling to amplify Smad-dependent transcriptional programs (21–23). For example, in hepatic stellate cells, stiff substrates enhance FAK–YAP cooperation, sustaining ECM production and myofibroblast activation (24, 25). These events illustrate how matrix stiffening not only drives intracellular mechanotransduction but also establishes a vicious cycle in which integrin–FAK–ROCK and YAP/TAZ–Smad pathways reinforce each other. This positive feedback perpetuates fibroblast activation and excessive ECM deposition, thereby locking tissues into a progressively fibrotic state.
2.1.2 Matrix topography
Matrix topography refers to nanoscale and microscale features such as fiber alignment, roughness, and porosity. In fibrosis, highly oriented collagen bundles guide fibroblast migration along fiber axes, reinforce directional traction forces, and further remodel ECM architecture. Nanoscale surface roughness alters membrane curvature and activates mechanosensitive ion channels, including Piezo1 and TRPV4, triggering calcium influx and downstream calcineurin-NFAT signaling (26–29). These pathways cooperate with YAP/TAZ to drive contractile differentiation. Surface roughness also modulates integrin clustering and focal adhesion distribution (30), enhancing fibroblast contractility and α-SMA expression. Additionally, reduced matrix porosity constrains cell spreading, increases cytoskeletal tension, and promotes YAP nuclear localization, while simultaneously limiting immune cell infiltration and facilitating local accumulation of profibrotic mediators (31, 32). These micro- and nanoscale topographical changes couple structural remodeling of the ECM to intracellular mechanotransduction. By synchronously regulating fibroblast activation and immune cell accessibility, altered topography reinforces ECM deposition and sustains a microenvironment conducive to progressive fibrosis.
Taken together, cell–matrix interactions transform extracellular stiffness or architecture into intracellular signaling events that activate fibroblasts and immune cells. Through integrin, FAK/Src, and downstream RhoA–ROCK and MAPK cascades, these signals converge on enhanced cytoskeletal contractility and YAP/TAZ-dependent transcription. The outcome is excessive ECM production, which in turn stiffens the matrix and perpetuates a profibrotic feedback loop.
2.2 Shear stress sensing
Shear stress is one of the primary fluid mechanical forces that shape cellular responses and tissue remodeling (33). Cells perceive shear forces through various membrane-associated structures, including primary cilia, microvilli, the glycocalyx, intercellular junctions, integrins, GPCRs, and mechanosensitive ion channels (34, 35). These structures transduce mechanical cues by physical deformation, tension shifts, or conformational activation, initiating mechanochemical signaling cascades (36, 37).
The primary cilium, a microtubule-based organelle, is a critical shear sensor. Under flow, its basal body and associated ion channels trigger calcium influx and downstream signaling (3, 34, 38). Microvilli and the glycocalyx increase the effective surface area for shear force capture and maintain barrier function, particularly in endothelial cells (35, 39). Integrins sense shear via focal adhesion complexes, activating FAK-Src and RhoA pathways to regulate cytoskeletal tension (40–42). GPCRs respond to membrane deformation through conformational shifts that initiate G protein–mediated signaling cascades. Recent studies have identified OXGR1, a GPCR responsive to oxoglutarate, as a critical regulator of fibroblast function. Upon activation, OXGR1 engages the PI3K/Akt pathway, thereby promoting fibroblast proliferation and contributing to extracellular matrix remodeling. This mechanistic link highlights the role of GPCR-mediated PI3K/Akt activation in the pathogenesis of fibrotic tissue dynamics (43, 44). Piezo1 detects shear stress to mediate calcium entry and regulate vascular tone, while TRPV4 contributes to volume regulation and pro-inflammatory amplification, aberrant activation of these channels is implicated in fibrosis across organs (45). Collectively, these diverse shear-sensing mechanisms converge on PI3K/Akt, RhoA–ROCK, and NF-κB pathways, orchestrating fibroblast activation, immune modulation, and ECM remodeling. By integrating mechanical flow cues into cellular signaling, shear stress acts as a central driver of the self-reinforcing cycle that underlies chronic fibrotic progression.
2.3 Pressure sensing
In contrast, pressure exerts perpendicular forces on cells, including hydrostatic pressure, trans-epithelial or trans-endothelial gradients, and interstitial fluid pressure (IFP). Pressure alters cell morphology and tension states, activating mechanosensors and downstream pathways. Abnormal pressure contributes to perfusion deficits, apoptosis, autophagy, and inflammatory mediator release in hepatic (46), pulmonary (47), renal (48), and scleral fibrosis (49).
Hydrostatic pressure, common in perfused tissues, increases with fluid accumulation and transmits load through sinusoids or interstitial spaces. For example, portal hypertension in liver fibrosis impairs liver sinusoidal endothelial cells (LSEC) barrier function and activates hepatic stellate cells (HSCs) mechanically (46, 50). Hydrostatic pressure enhances cytoskeletal remodeling and fibrotic gene expression via RhoA/ROCK and YAP/TAZ pathways, exacerbating tissue stiffening and fibrogenesis (51).
Trans-epithelial and trans-endothelial gradients are prevalent in renal tubules, alveoli, and bile ducts. Elevated gradients increase mechanical load, trigger stress responses, and disrupt barriers. In kidney injury, intraluminal pressure activates Piezo1, leading to calcium influx, EMT induction, and collagen deposition (52).
IFP is persistently elevated in fibrotic tissues due to matrix accumulation, crosslinking, and lymphatic dysfunction, forming an “interstitial pressure trap.” This environment upregulates hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF), and matrix metalloproteinases (MMPs), altering metabolism and oxidative stress (53–55). Sustained compression reorganizes F-actin and promotes YAP/TAZ and MRTF-A nuclear translocation, activating TGF-β autocrine signaling and Smad-dependent transcription to establish profibrotic feedback loops (49, 56). These distinct forms of pressure sensing converge on cytoskeletal remodeling, mechanosensitive ion channel activation, and YAP/TAZ–Smad signaling. By coordinating fibroblast activation, barrier dysfunction, and metabolic reprogramming, pressure not only reflects tissue injury but also acts as a potent driver of the self-reinforcing cycle that sustains fibrosis progression.
In summary, shear stress and interstitial flow are sensed by mechanosensitive channels such as Piezo1 and TRPV4, which activate calcium influx, NF-κB, and PI3K/Akt signaling. These events shape macrophage polarization and endothelial activation, thereby amplifying local inflammation and fibroblast activation. Ultimately, fluid mechanical cues reinforce ECM deposition and accelerate fibrotic progression.
2.4 Cell-cell mechanical coupling
2.4.1 Cadherin-mediated mechanotransduction
Beyond cell-matrix interactions, direct cell-cell mechanical communication, primarily mediated by cadherins, plays a critical role in the pathophysiology of organ fibrosis. Cadherins are calcium-dependent transmembrane glycoproteins that function as dynamic mechanosensing and signal transduction hubs, converting physical forces into biochemical signals (57, 58). The core mechanism involves force-dependent conformational changes that enhance trans-cellular binding and allosterically propagate tension intracellularly, stabilizing β-catenin and recruiting vinculin to bridge the cadherin-catenin complex to the actin cytoskeleton (59–62). This reinforced adhesion platform recruits and activates FAK and Src kinases, initiating downstream RhoA/ROCK signaling to amplify actomyosin contractility and regulate the nucleocytoplasmic shuttling of YAP/TAZ (13).
Emerging evidence positions cadherin-mediated coupling, particularly via Cadherin-11, as a pivotal mechano-immune bridge in fibrosis. A seminal study demonstrated that Cadherin-11 facilitates robust adhesion between M2-like macrophages and myofibroblasts, creating a niche enriched with active TGF-β (3, 57, 63, 64). This heterotypic adhesion not only enables macrophages to mechanically activate fibroblasts but also potentially reinforces the pro-fibrotic polarization of macrophages themselves through sustained mechanical feedback. Further illuminating this paradigm, a recent study by Astrab et al. reveals that Cadherin-11 directly interacts with the mechanosensor Piezo1 and cooperates with IL-6 signaling to drive fibroblast activation (65). This emerging model posits Cadherin-11 as a signaling node that integrates mechanical stress perception (via Piezo1) with inflammatory cues to amplify pro-fibrotic responses.
In summary, cadherin-mediated mechanotransduction provides a fundamental pathway for bi-directional mechanical and chemical crosstalk between stromal and immune cells. Its role as a mechano-immune bridge offers a compelling molecular framework for understanding how physical cues sculpt the immune landscape in fibrosis, presenting a promising target for dual-mechanism therapeutic interventions.
2.4.2 Paratensile signaling
Paratensile signaling represents a sophisticated mode of long-range mechanical communication in fibrosis, operating through three sequential phases: force generation, propagation, and cellular response (66, 67). The process initiates with force generation by activated myofibroblasts, which exert substantial contractile forces on the ECM via integrin-mediated focal adhesions. These forces are propagated through stiffened and aligned collagen bundles, which serve as conduits for tensile stress over distances exceeding several cell lengths. The efficiency of this transmission depends critically on collagen cross-linking by enzymes such as LOX (1, 68). Distal cells then sense these mechanical cues via mechanosensors like Piezo1 or discoidin domain receptor 2 (DDR2), activating pro-fibrotic transcriptional programs.
Experimental models have provided direct evidence for paratensile signaling. For instance, in vitro studies using atomic force microscopy (AFM) to apply localized force via collagen fibers have shown that remotely located fibroblasts upregulate pro-fibrotic genes—a response abolished upon inhibition of Piezo1 or DDR2 (66). Ex vivo models of the fibrotic–nonfibrotic interface further demonstrate that mechanical force propagation through collagen networks is necessary for the expansion of fibrotic lesions. Laser ablation of intervening collagen fibers between activated myofibroblasts and quiescent fibroblasts abrogates this activation, confirming the ECM’s role as a mechanical conduit (67, 69).
For example, in liver fibrosis, portal hypertension and inflammation remodel liver sinusoidal endothelial cells and enhance their attachment to perisinusoidal collagen. Tensile forces transmitted along collagen bundles mechanically activate hepatic stellate cells, increasing profibrotic gene expression via integrin-FAK-YAP/TAZ pathways (50, 70). Thus, paratensile signaling establishes the remodeled ECM as a long-range communication network that drives fibrosis progression by enabling localized mechanical activation to spread radially beyond injury sites (71). This concept expands our understanding of stromal–mechanical crosstalk and suggests new therapeutic strategies aimed at disrupting pathological force transmission, such as targeting LOX or Piezo1.
2.4.3 Cell-matrix-cell interactions
A further level of coupling arises from cell–matrix–cell interactions. Here, cells remodel their local ECM while transmitting mechanical signals to other cells anchored within the same matrix. In pulmonary fibrosis, fibroblast-generated traction reorganizes and stiffens collagen networks, enhancing integrin clustering and focal adhesion maturation in adjacent fibroblasts. This mechanical relay synchronizes myofibroblast activation and contractile differentiation across tissues (32, 72). Similarly, in cardiac fibrosis, fibroblast tension increases regional matrix stiffness and promotes sustained YAP/TAZ activation, establishing a feedforward loop of fibrosis progression (73).
Overall, force transmission between neighboring cells through adherens junctions, gap junctions, and cytoskeletal connections creates a multicellular amplification system. Mechanical forces propagate from fibroblasts to immune cells and back, sustaining ROCK, YAP/TAZ, and NF-κB activation across cell populations. This cross-talk expands fibrotic foci and drives the transition from local injury to widespread fibrosis. While the previous sections focused on how mechanical cues are sensed and transmitted at the cellular level, the next sections will dissect the regulation of immune cells in the mechano-fibrotic axis. Figure 2 and Figure 3 provide an integrated overview of the major mechanical cues and mechanotransduction pathways involved in fibrosis progression.
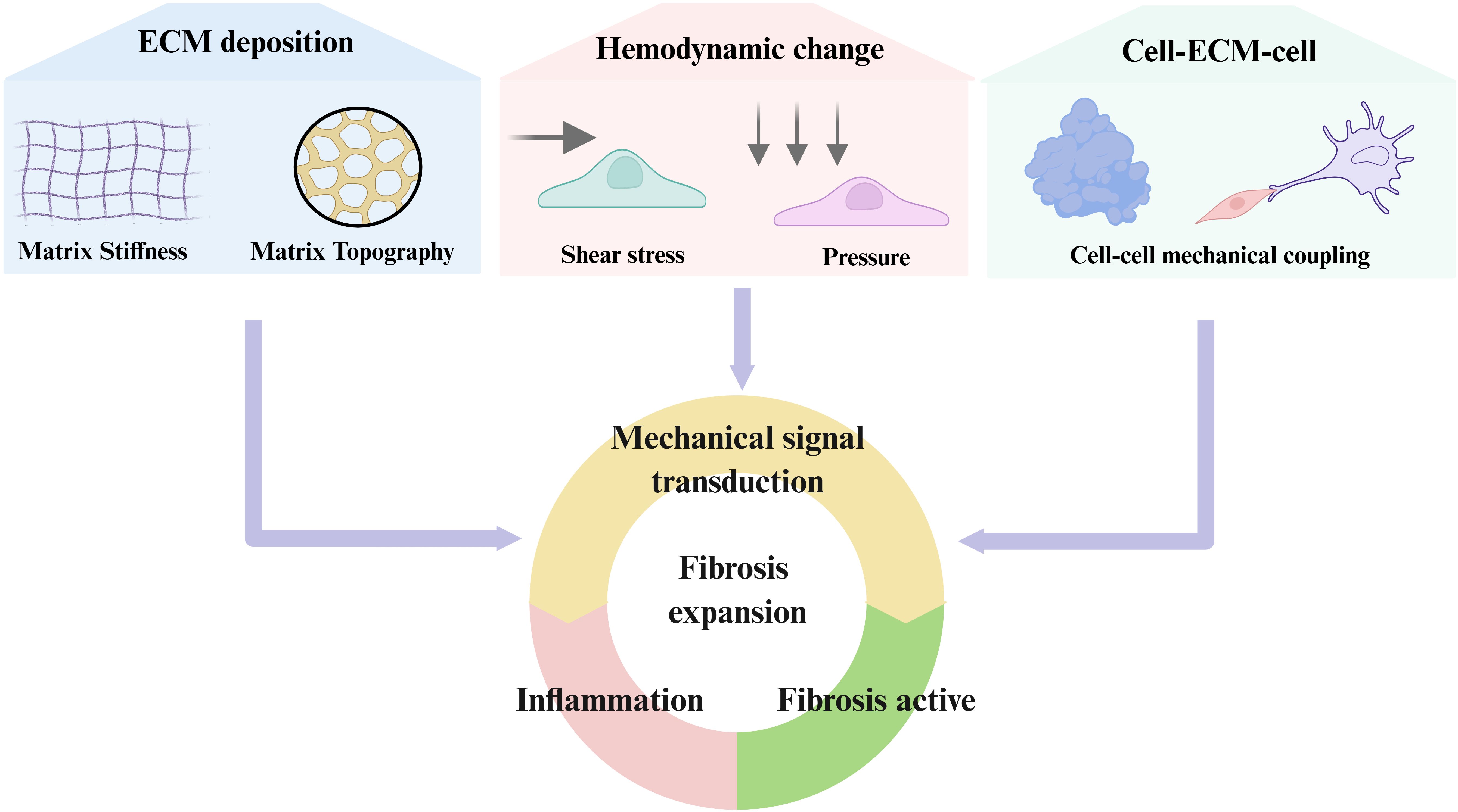
Figure 2. Principal biomechanical cues driving fibrotic activation. Fibrosis is orchestrated by dynamic changes in extracellular matrix stiffness, matrix topography, aberrant hemodynamics (shear and pressure), and mechanical crosstalk between cells and the ECM. These biomechanical cues are sensed through integrins, cadherins, GPCRs, mechanosensitive ion channels, and cytoskeletal linkers, which activate downstream signaling cascades including RhoA/ROCK, YAP/TAZ, MAPK, PI3K/Akt, and NF-κB. Collectively, these pathways converge on fibroblast activation, immune modulation, and excessive ECM deposition, thereby establishing self-reinforcing feedback loops that drive and sustain progressive fibrosis.
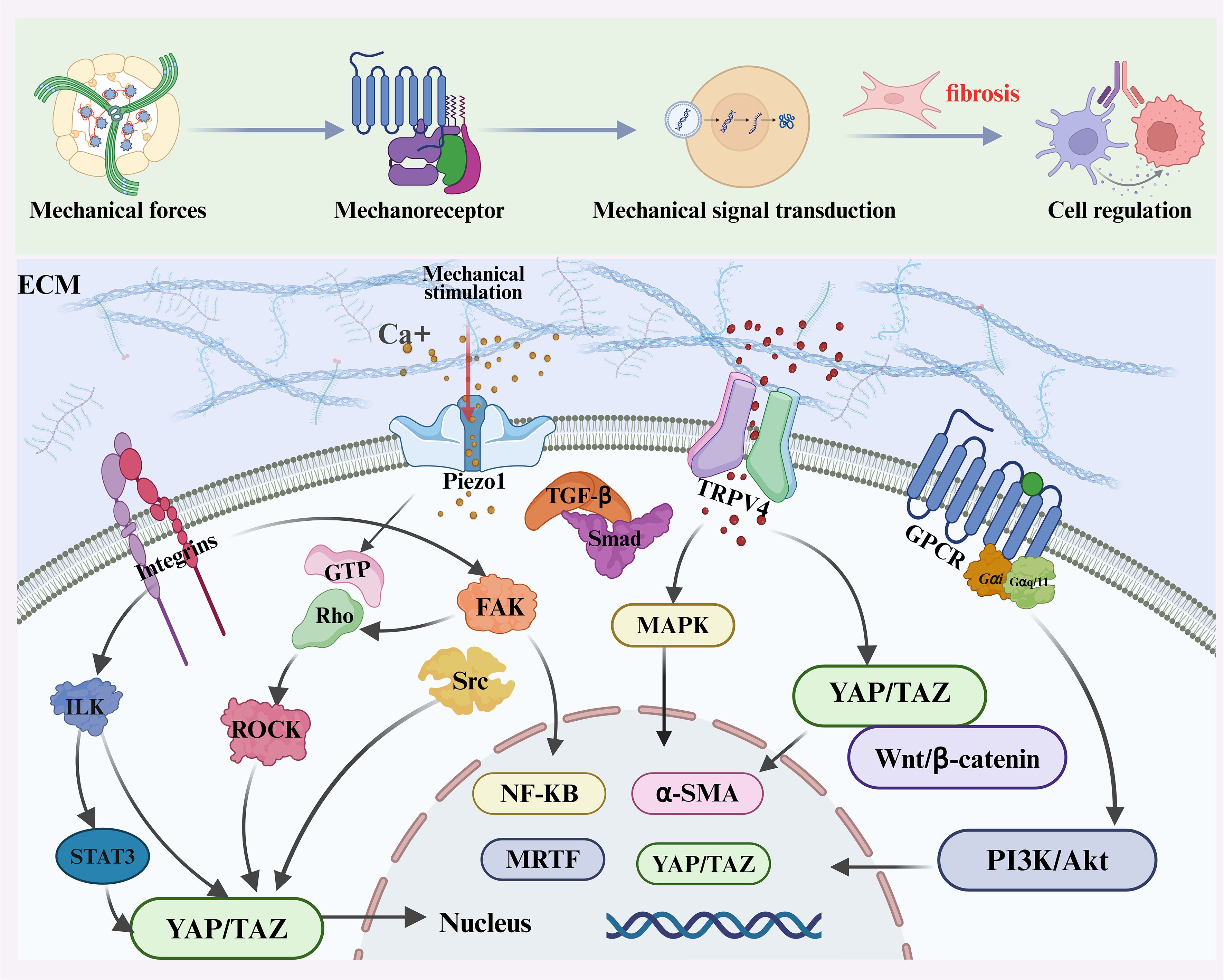
Figure 3. Overview of mechanotransduction and immune regulation in fibrosis. Mechanical forces—including stiffness, shear stress, pressure, stretch, and topography—are sensed by mechanoreceptors such as Integrins, Piezo1, TRPV4, and G protein–coupled receptors (GPCRs). These receptors activate downstream signaling cascades including focal adhesion kinase (FAK)/Src, Rho/ROCK, MAPK, PI3K/Akt, TGF-β/Smad, and YAP/TAZ pathways, promoting cytoskeletal remodeling, profibrotic gene expression, and ECM deposition.
3 Regulation of immune cells in the mechano-fibrotic axis
Notably, mechanically driven remodeling of the extracellular matrix also creates a mechanically adaptive microenvironment for immune cells. Immune cells are not only key regulators of inflammation and tissue repair but also highly sensitive to changes in the mechanical microenvironment. This mechanosensitivity enables them to sense variations in matrix stiffness, fluid shear stress, and intercellular tension, thereby modulating their migration, activation, secretory profile, and differentiation. In this context, immune cells act as both responders and amplifiers of mechanical signals.
3.1 Macrophage Mechanosensing and Polarization
Macrophage functional plasticity, encompassing their capacity to polarize into pro-inflammatory (M1) or pro-repair/pro-fibrotic (M2) states, is critically implicated in fibrosis pathogenesis. Beyond biochemical cues, the biomechanical properties of the fibrotic microenvironment—notably increased matrix stiffness, fluid shear stress, and intercellular tension—are now recognized as active instructors of macrophage polarization (6, 74, 75). This mechanical instruction is decoded via a sophisticated mechanotransduction network that converts physical forces into biochemical signals, ultimately reprogramming macrophage gene expression and function (76). The core mechanosensing apparatus in macrophages comprises integrin-based focal adhesions and mechanosensitive ion channels. Upon adhesion to pathologically stiff matrices, integrin clustering activates FAK, initiating downstream RhoA/ROCK signaling to enhance actomyosin contractility and cytoskeletal tension (17, 77). Concurrently, mechanical stimuli directly activate channels such as Piezo1 and TRPV4, inducing rapid Ca2+ influx that serves as a pivotal second messenger (78, 79).
These proximal sensing events converge on key signaling hubs that cooperatively dictate polarization fate. The transcriptional co-activators YAP/TAZ are central mediators. Under high cytoskeletal tension, inhibition of the LATS1/2 kinases in the Hippo pathway permits YAP/TAZ nuclear translocation. There, they partner with transcription factors like TEAD to drive pro-fibrotic (e.g., CTGF, CYR61) and pro-inflammatory gene expression (13, 80). Stiffness-driven M1 polarization is facilitated by the Piezo1-YAP axis, while YAP/TAZ inhibition attenuates this inflammatory response (81). Complementarily, NF-κB signaling is potently activated by mechanical cues; Piezo1-mediated Ca2+ influx can engage the calcineurin-NFAT pathway, synergizing with canonical IKK-NF-κB signaling to upregulate classic M1 markers like TNF-α and IL-1β (81, 82).
As fibrosis progresses, sustained mechanical stimulation can promote a shift toward an M2 phenotype. Persistent RhoA/ROCK signaling on stiff substrates not only maintains YAP/TAZ activation but also cooperates with cytokine-activated STAT6 to induce expression of M2 markers (e.g., Arg1, CD206) and enhance secretion of TGF-β1 (78, 82, 83). This M2-polarized state, in turn, activates fibroblasts via paracrine signaling, exacerbating ECM deposition and matrix stiffness—thereby establishing a self-perpetuating “mechano-immune positive feedback loop”. Notably, these mechanically induced phenotypic shifts can be stabilized through epigenetic modifications, conferring a “mechanical memory” that persists even after the initial mechanical insult is removed (84).
In summary, the M1/M2 transition in macrophages is precisely orchestrated by an integrated mechanotransduction network. This network employs integrins/FAK and Piezo1/TRPV4 as primary sensors, with YAP/TAZ, NF-κB, and ROCK serving as core signaling axes to translate biomechanical cues into transcriptional programs that guide immune responses. A deeper dissection of this mechanism is crucial for developing novel therapies targeting the mechano-immune axis in fibrosis.
3.2 T cells mechanotransduction
T cell mechanosensing primarily occurs via T cell receptor (TCR) engagement, integrin-mediated adhesion, and the cytoskeletal machinery. Stiff matrices activate ILK–STAT3 and YAP/TAZ signaling, enhancing RORγt expression and promoting Th17 differentiation. Th17 cells secrete IL-17A, IL-22, and GM-CSF, which induce EMT, fibroblast activation, and inflammatory loops (85–87). Matrix viscoelasticity modulates AP-1 protein expression, generating distinct T cell subsets (88). In addition, shear stress induces sustained T cell activation and reprogramming through Piezo1-mediated calcium influx, which in turn enhances downstream signaling pathways such as NF-κB and NFAT (4). Moderate shear also increases major histocompatibility complex (MHC)-I and CD86 expression and cytokine secretion, enhancing T cell activation capacity (89, 90).
In contrast, Tregs are central to maintaining homeostasis and suppressing inflammation, but their stability is also mechanically regulated. Soft, low-tension environments support Foxp3 expression and suppressive function. In stiffened matrices, YAP/TAZ activation downregulates Foxp3, driving Treg conversion toward Th17/Th1 phenotypes and loss of immunoregulatory capacity (91, 92). In liver fibrosis models, Treg instability often coexists with Th17 expansion, suggesting that mechanical disturbance underlies (93, 94).
3.3 Neutrophils mechanotransduction
Neutrophils are highly responsive to mechanical cues, particularly matrix stiffness and viscoelasticity. Stiff substrates promote increased neutrophil spreading, ROS production, and the release of neutrophil extracellular traps (NETs), a process termed NETosis (95, 96). In stiff or stretched microenvironments, neutrophils are prone to NETosis, releasing DNA webs enriched in tissue factor (TF), histones, myeloperoxidase (MPO), and neutrophil elastase (97–99). In fibrotic tissues, excessive NET formation has been implicated in sustaining chronic inflammation and stimulating profibrotic. For example, in murine models, NET inhibition reduces fibroblast activation and collagen accumulation, validating their pathological role (100).
Mechanosensitive ion channels such as Piezo1 and TRPV4 also contribute to T cell and neutrophil responses by regulating intracellular calcium dynamics and cytoskeletal remodeling (98, 101–103). Although most studies have focused on acute inflammatory contexts, these pathways are likely relevant to chronic fibrotic settings, where persistent mechanical remodeling creates proinflammatory microenvironments.
3.4 Dendritic cells mechanotransduction
Dendritic cells (DCs) are highly sensitive to mechanical cues in their microenvironment, including matrix stiffness, viscoelasticity, and interstitial flow (104). Increased substrate stiffness promotes DC spreading, actin cytoskeletal reorganization, and enhanced maturation, characterized by upregulated expression of MHC-II, CD80, and CD86, which facilitates T cell priming (105, 106). Mechanical forces also influence DC cytokine secretion: stiffer matrices enhance the production of IL-6, TNF-α, and TGF-β, potentially contributing to fibroblast activation and extracellular matrix deposition (105). Mechanosensitive pathways, such as Piezo1-mediated Ca2+ influx, regulate DC motility and antigen-presenting functions, allowing DCs to act as both sensors and modulators of mechanically altered fibrotic microenvironments (107, 108). DCs translate mechanical perturbations into immunomodulatory signals that can either exacerbate or resolve fibrosis, positioning them as critical mechano-immune hubs in fibrotic progression and potential therapeutic targets.
3.5 Natural killer cells mechanotransduction
Natural killer (NK) cells, crucial effectors of innate immunity, are increasingly recognized as responsive mechanosensors whose cytotoxic and migratory functions are tuned by mechanical cues within the tissue microenvironment (6). In fibrotic contexts, pathologically stiffened ECM enhances NK cell cytotoxicity, a process mediated significantly by the mechanosensitive ion channel Piezo1. Piezo1 activation facilitates calcium influx, promoting polarization of the microtubule-organizing center and directed release of cytotoxic granules toward target cells (109). Beyond static stiffness, fluid shear stress dynamically regulates NK cell function. Notably, the activating receptor NKG2D has been implicated in mechanosensation. Under physiological shear stress, ligand engagement induces conformational changes in NKG2D, triggering downstream signaling involving VAV1 and PI3K phosphorylation, which specifically enhances granzyme B delivery to target cells without broadly upregulating cytokine secretion (110).
Within the fibrotic niche, characterized by progressive matrix stiffening and altered interstitial flow, the functional impact of these mechanical pathways on NK cells remains an open question. While direct evidence for a mechanically-driven pro-fibrotic NK phenotype is scarce, it is plausible that chronic mechanical stimulation could modulate their activity, potentially shifting the balance between cytotoxic and immunomodulatory functions (6, 111). In summary, NK cells are active participants in mechano-immunological crosstalk (112). Understanding how Piezo1 and NKG2D integrate mechanical signals in the context of fibrotic architecture is essential to fully delineate their role in the mechano-immune axis of organ fibrosis. Figure 4 demonstrates that immune cell subsets contribute to fibrosis by releasing pro-inflammatory cytokines and engaging in direct or paracrine interactions with fibroblasts.
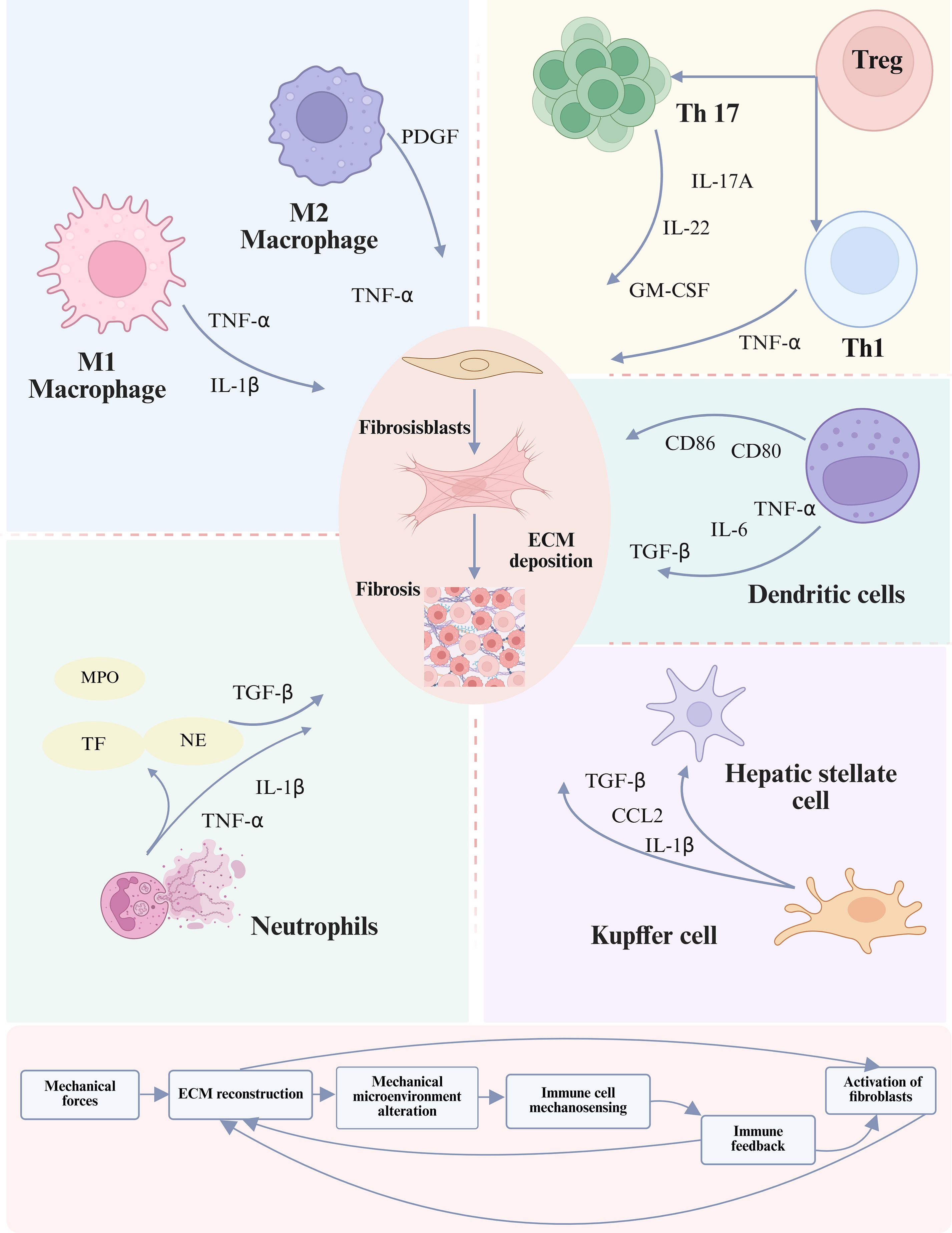
Figure 4. Role of the immune microenvironment in fibrosis progression. Immune cells both contribute to and regulate fibrotic remodeling by modulating the activation of fibroblasts and HSCs. This regulation occurs directly through cell-cell contact or indirectly via the release of cytokines, chemokines, and ligand-receptor signaling, thereby shaping the trajectory of extracellular matrix deposition and tissue scarring (113). The lower panel illustrates a positive feedback loop in which activated fibroblasts not only produce excessive ECM but also stimulate the mechanotransduction–immune axis, thereby perpetuating fibrosis progression through iterative interactions between mechanical cues and immune responses (105). To date no definitive evidence shows that mechanical cues such as ECM stiffness directly switch NK cells to a pro-fibrotic mode, representing a gap in the mechanotransduction-immune axis understanding.
3.6 Immune cell feedback loops reinforcing fibrotic mechanotransduction
3.6.1 Immune cell-extracellular matrix feedback amplifies mechanical signal transduction
Immune cells not only passively respond to changes in the mechanical microenvironment but also actively remodel the mechanical properties of the ECM, forming an “immune–matrix–mechanical” closed feedback loop that drives fibrosis progression. As local ECM stiffness increases, immune cells undergo adaptive remodeling at multiple levels-including morphology, adhesion molecule expression, and activation of mechanosensitive pathways-to enhance their mechanical responsiveness. For example, in high-stiffness microenvironments, classically activated M1 macrophages significantly upregulate integrin αMβ2 (CD11b/CD18), strengthening their adhesion to ECM components such as type I collagen and fibronectin. This high-affinity binding activates the integrin-FAK-ROCK signaling axis, promoting actomyosin contractility. The resulting active traction forces are transmitted into the surrounding matrix, stimulating adjacent fibroblasts through YAP/TAZ and RhoA signaling to adopt a myofibroblastic phenotype (114, 115). Activated macrophages also modulate ECM mechanics through secreted factors. M2 macrophages secrete TGF-β1, which can activate fibroblasts to produce LOX, thereby promoting collagen crosslinking and matrix stiffening (68). This chemical-mechanical coupling drives early matrix soft-to-stiff transitions and maintains high matrix rigidity during advanced fibrosis. Importantly, the mechanical signals generated by immune cells reinforce their own profibrotic polarization. In stiff matrices, macrophages sustain M2 phenotypes and secretion of TGF-β1 and PDGF via persistent ROCK2 activation, creating a positive feedback loop that amplifies mechanoinflammatory signaling (116). In animal models of pulmonary fibrosis, macrophage-specific ROCK2 inhibition reduces contractile force generation, suppresses profibrotic signaling, attenuates fibroblast activation (83).
3.6.2 Immune cells establish force-transmitting networks via cell-cell adhesion
Immune cells propagate mechanical signals through direct physical contacts, forming structural networks that coordinate mechanotransduction across tissues. Adhesion molecules form the basis of these “force transmission networks,” simultaneously mediating biochemical signaling and coordinating intercellular mechanical stress. A classical example is LFA-1 (CD11a/CD18) binding to ICAM-1. LFA-1–ICAM-1 interactions stabilize immune synapses under shear stress (117, 118), supporting persistent immune activation in fibrotic lungs (119, 120). Beyond classical synapses, heterotypic mechanical contacts between immune and stromal cells are critical in fibrotic disease. In lung, liver, and kidney fibrosis, M2 macrophages form stable adhesions with activated fibroblasts via N-cadherin–β-catenin complexes, enabling bidirectional mechanical signaling that promotes myofibroblast differentiation and matrix remodeling (59, 60, 65).
3.7 Epigenetic regulation in the mechano-immune axis of fibrosis
Emerging evidence underscores that epigenetic regulation serves as a critical molecular interface translating transient mechanical cues into sustained pro-fibrotic phenotypes, thereby establishing a “mechanical memory” that perpetuates fibrosis even after the initial insult is resolved (121). This mechano-epigenetic axis operates as a self-reinforcing loop across both stromal and immune cells, constituting the core of the mechano-immune axis.
The process is initiated by mechanical force sensing through integrins and ion channels (e.g., Piezo1), leading to the activation of downstream effectors such as YAP/TAZ and MRTF in stromal cells. These transcriptional co-activators recruit chromatin-modifying complexes to profibrotic gene loci (122). A key mechanistic insight comes from the recently identified EZH2–YAP feedback loop. In fibrotic kidneys, upregulated EZH2—a histone methyltransferase—deposits the repressive mark H3K27me3 at the promoter of LATS1, a core inhibitor of the Hippo pathway. This epigenetic silencing promotes YAP nuclear translocation, which in turn reinforces EZH2 expression, forming a vicious cycle that amplifies fibrotic signaling (122). Concurrently, mechanical stress induces nuclear deformation, directly increasing chromatin accessibility at mechanosensitive enhancers near genes such as ACTA2 and COL1A1 (123–125). These changes are further stabilized by stiffness-induced metabolic shifts, which alter the availability of metabolites such as α-ketoglutarate (α-KG) and S-adenosylmethionine (SAM), thereby influencing the activity of DNA methylation enzymes including DNMTs and TETs (126).
This mechanical reprogramming extends to immune cells within the fibrotic niche. In macrophages, substrate stiffness triggers a cytoskeleton–Src–p300 axis that drives histone H3 acetylation (H3Ac) at promoters of pro-inflammatory genes (e.g., IL1B, TNF), reinforcing M1-like polarization (77). The ensuing secretion of IL-1β and TNF-α not only amplifies inflammation but also directly activates surrounding fibroblasts, creating a feedforward loop. Evidence also suggests that mechanical cues, in concert with epigenetic regulators such as specific non-coding RNAs, may promote monocyte transdifferentiation toward a myofibroblast-like phenotype, directly expanding the pool of matrix-producing cells (127). In T cells, mechanical forces during T-cell receptor engagement can induce epigenetic modifications at cytokine loci, skewing the balance toward a pro-fibrotic Th17 response over a regulatory Treg phenotype, thereby sustaining chronic inflammation (128).
In summary, epigenetic regulation stabilizes the persistent cellular activation that characterizes fibrosis. The mechano-immune axis, driven by these epigenetic programs, ensures that mechanical insults evolve into a sustained pathological dialogue between stromal and immune cells. Future studies should focus on dissecting cell-type-specific epigenetic codes and developing strategies to disrupt this dialogue and reverse the pathological “mechanical memory” for true disease regression. A schematic summarizing these epigenetic mechanisms within the mechano-immune axis is proposed in Figure 5.
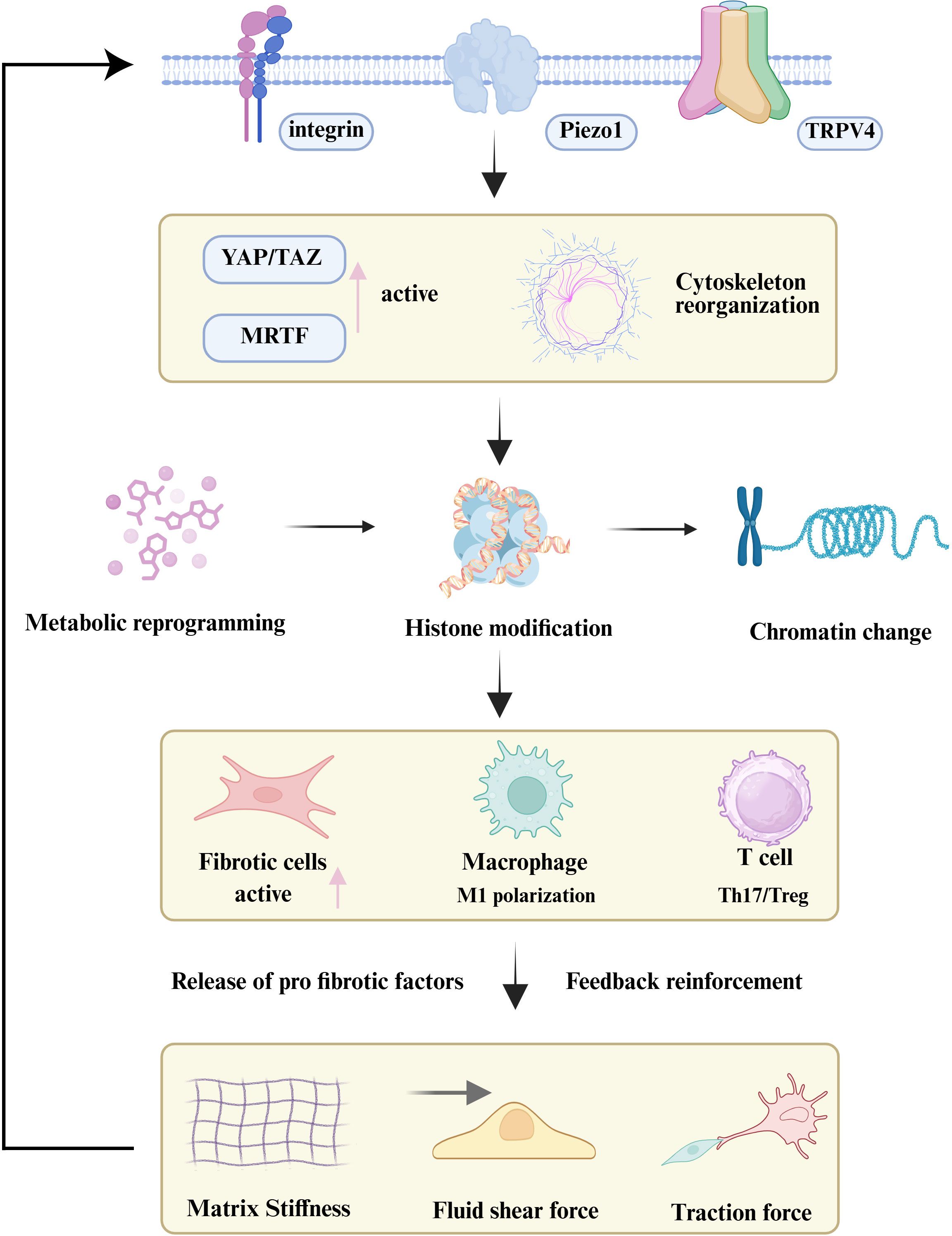
Figure 5. The mechano-epigenetic-immune axis in organ fibrosis. Persistent mechanical stimuli are sensed by cellular mechanoreceptors (e.g., Integrins, Piezo1), activating downstream signaling (YAP/TAZ) and inducing cytoskeletal reorganization. This force transmission leads to nuclear remodeling and widespread epigenetic reprogramming, which locks both stromal and immune cells into profibrotic activated states. The resulting secretion of factors creates a positive feedback loop that reinforces the fibrotic microenvironment, perpetuating disease progression.
4 Emerging therapeutic strategies targeting mechanotransduction-immune interactions in fibrosis
Traditional antifibrotic therapies have primarily focused on controlling inflammation, inhibiting fibroblast activation, and blocking collagen synthesis. However, their clinical efficacy has often been limited. In recent years, as understanding of fibrotic pathogenesis has deepened-particularly with the elucidation of the intersecting pathways of mechanotransduction and immune regulation-targeting the “mechanotransduction–immune axis” has emerged as a promising novel intervention strategy. This axis encompasses the entire cascade from mechanical stimulus sensing and intracellular signal transduction to immune cell activation and functional reprogramming, thereby providing a theoretical foundation and practical potential for multi-target synergistic therapies.
4.1 Signal pathway interventions targeting the mechanotransduction-immune axis
4.1.1 Integrin signaling pathway
In fibrotic tissues, integrin expression is markedly upregulated, especially among the αv integrin family such as αvβ1, αvβ3, αvβ6, which can sense matrix stiffness and activate TGF-β, thereby inducing immune cell chemotaxis and fibroblast activation. By disrupting mechanical force transmission between cells and the ECM, integrin inhibitors not only attenuate TGF-β activation but also modulate immune cell adhesion and migration, effectively dampening pro-fibrotic inflammation at its source (129). For example, Cilengitide, an αvβ3/β5 antagonist, blocks immune cell adhesion and migration,inhibiting TGF-β–dependent myofibroblast differentiation, significantly reducing inflammatory infiltration and collagen deposition in liver and lung fibrosis models (130, 131). The αvβ6 antagonist Bexotegrast (PLN-74809) has advanced into Phase IIb/III clinical trials for idiopathic pulmonary fibrosis (IPF). Its mechanism involves specifically interrupting epithelial cell-derived TGF-β activation by the αvβ6 integrin, thereby disrupting the αvβ6/TGF-β positive feedback loop driving fibrosis progression (132). As primary mechanosensors at the “mechanotransduction-immune-fibrosis” axis, integrins represent promising targets for early intervention strategies.
4.1.2 Mechanosensitive ion channels
Mechanosensitive ion channels are highly responsive to mechanical cues and constitute critical nodes linking biomechanical stimuli to immune regulation. Piezo1 inhibitors such as GsMTx4 suppress shear-induced macrophage M1 polarization and fibroblast activation in animal models (133). TRPV4 channel activation leads to calcium influx, which subsequently activates MAPK and NF-κB pathways to drive myofibroblast differentiation and ECM accumulation (134). TRPV4 antagonists, such as GSK2193874, have demonstrated potent antifibrotic effects in preclinical bleomycin-induced lung fibrosis models, significantly reducing IL-6 and collagen expression and lowering inflammation scores (135). Furthermore, TRPV4 activation contributes to ROCK pathway activation. This promotes cytoskeletal remodeling, increases cellular tension, and enhances fibroblast migration, thereby establishing the biomechanical foundation for fibroblast recruitment and activation (134). As key molecular integrators of mechanotransduction and immune/inflammatory signaling, TRPV4 channels represent compelling therapeutic targets for fibrosis.
4.1.3 Rho/ROCK signaling pathway
Y-27632, a classical ROCK inhibitor, effectively suppresses TGF-β-driven fibroblast activation, attenuates myofibroblast differentiation and collagen deposition. For example, Y-27632 has been shown to prevent dimethylnitrosamine-induced hepatic fibrosis in rats (136). Additionally, several studies suggest that Y-27632 may influence macrophage polarization and dampen inflammatory responses (137, 138). Next-generation, highly selective ROCK2 inhibitors such as KD025 have demonstrated the ability to modulate STAT3 signaling,correct immune dysregulation, and suppress immune-mediated fibrotic progression (139–141).
4.1.4 YAP/TAZ signaling
Targeting the YAP/TAZ signaling axis has emerged as a promising antifibrotic strategy. In a rat bleomycin-induced pulmonary fibrosis model, Zeyada et al. reported that trigonelline significantly inhibited YAP activity, evidenced by reduced nuclear translocation and downregulation of multiple pro-fibrotic genes (142). Haak et al. demonstrated that dihydrexidine (DHX), by activating dopamine D1 receptors, selectively suppresses YAP/TAZ activity, attenuating the profibrotic activation of pulmonary fibroblasts and hepatic stellate cells. In mouse models of pulmonary and cholestatic liver fibrosis, DHX treatment substantially attenuated collagen deposition and inflammation while preserving epithelial regenerative capacity (143). Statins, including simvastatin, have also shown antifibrotic potential in preclinical studies by promoting YAP cytoplasmic retention and inactivation, offering opportunities for drug repurposing (144, 145). Verteporfin, a direct inhibitor of YAP-TEAD binding and transcriptional activity, significantly reduced collagen I and fibronectin expression and ameliorated fibrosis severity in a unilateral ureteral obstruction (UUO) kidney fibrosis model, while improving epithelial structural integrity (146, 147). Emerging evidence also suggests that YAP/TAZ signaling modulates macrophage polarization and cytokine production, thereby linking mechanical cues to immune-driven fibrosis (148).
4.2 Smart delivery systems and responsive biomaterials targeting the mechanotransduction-immune axis
Achieving precise interventions targeting the mechanotransduction–immune axis requires overcoming the limitations of conventional pharmacotherapy, such as widespread biodistribution, limited specificity, and dose-limiting toxicity. In recent years, intelligent delivery platforms and engineered biomaterials have demonstrated significant advantages in controlling tissue specificity, cellular selectivity, and microenvironmental responsiveness (149–152). They are emerging as critical bridges linking mechanical signal modulation, immune regulation, and antifibrotic therapy. Particularly in fibrotic tissues characterized by pronounced aberrant mechanical cues, the development of nanocarriers with mechano-responsive properties has become a frontier in precision drug delivery. For example, shear stress–sensitive liposomes and polymeric nanoparticles can release anti-inflammatory or antifibrotic agents specifically at sites with elevated hemodynamic shear stress, enabling localized drug release triggered by mechanical forces (153, 154). In parallel, stiffness-sensitive nanocarriers selectively recognize regions of increased matrix rigidity and achieve spatial targeting through microenvironmental mechanical signals. This “mechanically responsive release” mechanism significantly enhances drug accumulation and therapeutic efficacy within fibrotic lesions (155).
The drug-loading capability of such materials further reinforces their role in targeted interventions. Nanocarriers can leverage the enhanced permeability and retention (EPR) effect in highly vascularized fibrotic foci, while active targeting ligands (e.g., peptides, antibodies) further enhance cellular specificity (156). For instance, mannose-modified albumin nanoparticles delivering TGF-β1 siRNA have been shown to effectively attenuate pulmonary fibrosis severity in murine models (157). In regenerative applications, bioengineered scaffolds can be combined with healthy cells or organoids to form implantable constructs that provide adhesive substrates and support functional tissue replacement. Proof-of-concept studies demonstrate that decellularized splenic matrix (DSM) can be repurposed to engineer 3D hepatic constructs with metabolic activity (158).
4.3 Advances in gene and cell engineering for mechanotransduction-immune precision therapy in liver fibrosis
Synthetic biology and gene engineering technologies are pioneering cell- and gene-level interventions to precisely target the mechanotransduction–immune axis in liver fibrosis. This disease exhibits high spatial heterogeneity and a dynamically evolving microenvironment where conventional small-molecule therapies often lack cellular specificity and sustained efficacy. Gene editing tools such as CRISPR/Cas9 now enable cell type-specific modulation of mechanosensitive pathways. For example, macrophage-specific knockout of Piezo1 or TRPV4 significantly reduces proinflammatory cytokine production and M1 polarization, thereby alleviating the persistent inflammatory stimulation of HSCs (159–161). Similarly, silencing of downstream mechanotransduction effectors like YAP/TAZ effectively suppresses ECM synthesis and abnormal immune activation (76). Animal studies employing lipid nanoparticles or viral vectors to deliver such gene-editing tools have demonstrated localized hepatic expression, low immunogenicity, and promising antifibrotic effects.
In terms of cell engineering, immune cells such as macrophages and T cells have been bioengineered to integrate both mechanosensory and immunoregulatory functions, broadening the potential of cell-based therapies in fibrosis. For instance, chimeric antigen receptor (CAR)-T cell technology, originally developed for cancer immunotherapy, is now being explored in fibrotic diseases. CAR-T cells designed to recognize fibroblast activation protein (FAP) on pathogenic fibroblasts and engineered to modulate mechanical signal transduction pathways may achieve selective clearance of fibrogenic cells while adapting to the altered mechanical microenvironment (162, 163).
4.4 Clinical challenges in mechanotransduction-targeted antifibrotic therapies
Despite the promising antifibrotic potential of targeting the mechanotransduction-mmune axis in animal models and early-phase clinical studies, the translation of these findings into clinical applications faces multiple challenges. For example, Bexotegrast has demonstrated good safety and favorable phase II outcomes in IPF, including attenuation of lung function decline, reduction of collagen metabolism biomarkers, and improvement in radiographic fibrosis scores. However, its ability to improve clinically meaningful endpoints such as overall survival remains to be validated in phase III trials. Furthermore, although receptor occupancy studies have confirmed Bexotegrast’s dose-dependent binding activity to αvβ6 integrin, clinical efficacy data are still lacking (164). The repositioning of integrin antagonists such as Cilengitide for fibrotic diseases has also encountered setbacks. In liver fibrosis models (e.g., TAA- or BDL-induced), Cilengitide unexpectedly exacerbated fibrotic progression, as evidenced by increased collagen septa thickness, upregulation of profibrotic genes TGF-β1 and TIMP-1/2 (165). These results underscore critical pharmacological limitations of Cilengitide—particularly suboptimal dosing regimens, lack of organ-specific targeting, and potential off-target profibrotic effects—which collectively hinder its repurposing for antifibrotic therapy. ROCK inhibitors such as KD025 offer a promising example of mechanotransduction-targeted therapies achieving regulatory approval for chronic graft versus host disease (GVHD), yet their long-term safety and efficacy in fibrotic diseases remain uncertain (166).
In addition, organ-specific pathological and physiological differences pose major barriers to cross-organ translation. In liver fibrosis, key challenges include low drug delivery efficiency, widespread intrahepatic distribution, and potential target-related hepatotoxicity. In kidney fibrosis, high perfusion rates and steep pressure gradients complicate drug pharmacokinetics and targeting specificity. Other organs such as the lung, heart, and skin present additional barriers, including structural heterogeneity and distinct cellular targets. A common challenge across organs is the lack of strategies for precise delivery to fibrotic lesions. The multifocal, multi-organ distribution and heterogeneity of fibrotic foci remain major obstacles to efficient and specific targeting. Although advanced delivery platforms such as nanocarriers and gene therapies have emerged, most remain at the preclinical stage and face translational and safety concerns. Moreover, the field lacks standardized, clinically relevant endpoints. Current studies rely heavily on surrogate biomarkers, with insufficient emphasis on long-term outcomes such as survival and organ function. The translational gap between animal models and human disease is also substantial, particularly in terms of tissue stiffness, chronic inflammation, and microenvironmental complexity, all of which limit the predictive value of preclinical results. Figure 6 and Table 1 present emerging antifibrotic therapies that target mechanotransductive and immune pathways, alongside key translational barriers and representative agents under investigation across fibrotic organ systems.

Figure 6. Emerging therapeutic approaches targeting fibrosis-related mechanotransduction and immune responses. Key strategies include inhibition of mechanoreceptors (Cilengitide [integrin inhibitor], Bexotegrast [integrin modulator], GSK2193874 [TRPV4 antagonist], GsMTx4 [Piezo1 inhibitor]), blockade of mechanical signaling (Y-27632, KD025 [ROCK inhibitor], verteporfin, dihydrexidine, trigonelline [YAP/TAZ inhibitor]), and modulation of immune activation via liposomes, nanoparticles, CRISPR/Cas9 editing, and CAR-T cell therapy to attenuate ECM deposition and myofibroblast activation.
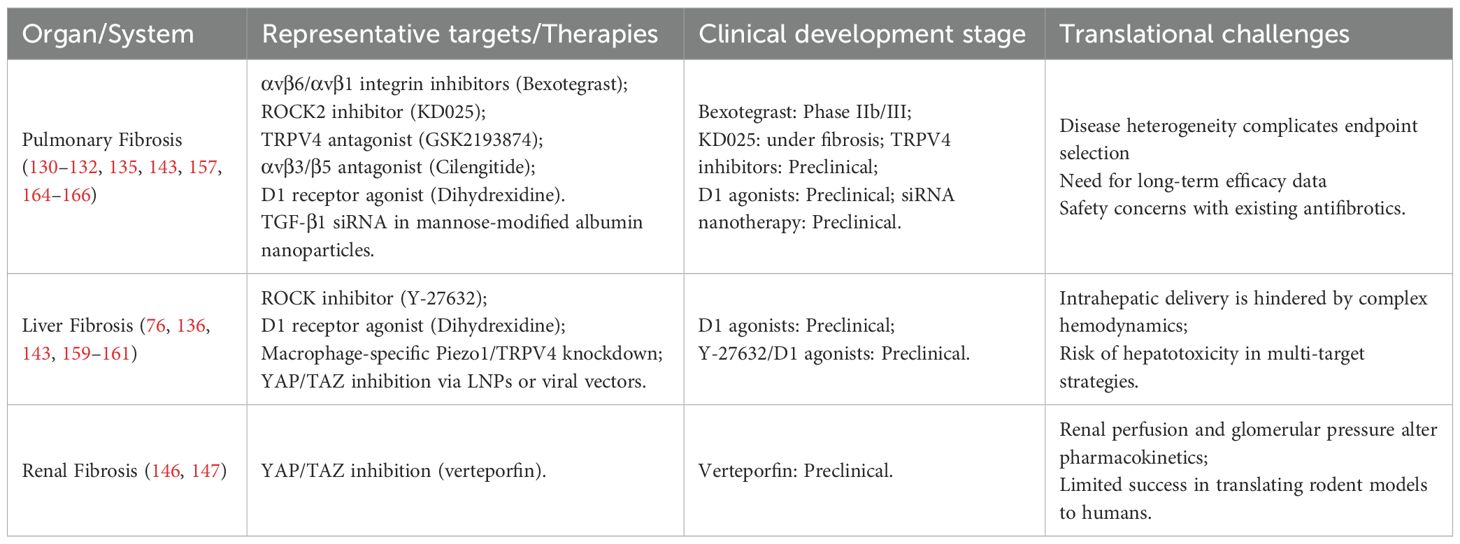
Table 1. Translational challenges and representative therapies targeting the mechanotransduction-immune axis in organ fibrosis.
5 Conclusion and perspectives
Organ fibrosis is increasingly recognized not merely as the end result of immune overactivation or chronic inflammation, but as a dynamic pathophysiological process governed by biomechanical regulation and immunological reprogramming. The evolving fibrotic microenvironment generates sustained mechanical cues—including matrix stiffening, interstitial shear stress, and architectural distortion—that not only activate fibroblasts but also reshape immune cell behavior, differentiation, and metabolism. In this context, the mechanotransduction–immune axis has emerged as a unifying framework integrating physical and immunological inputs into a self-reinforcing fibrotic circuit. Based on this understanding, a conceptual stage-specific model can be envisioned, aligning mechanical and immune processes with fibrosis evolution. In the early stage, tissue injury triggers damage associated molecular patterns (DAMP)-mediated immune priming and initial fibroblast recruitment. Concurrent subtle ECM remodeling begins to engage mechanosensors like Piezo1, TRPV4, and integrins. Interventions during this phase may benefit from combined targeting of immune polarization and mechanosensors, as suggested by recent findings (159, 161). During the amplification phase, mechanotransductive signals converge with inflammatory pathways, YAP/TAZ activation synergizes with TGF-β signaling to promote myofibroblast transition and immune polarization (167–169). This stage is characterized by a pathological “mechanical–immune–fibroblast” positive feedback loop. In the late stage, extensive ECM crosslinking induces irreversible stiffening and “mechanical memory,” sustaining α-SMA myofibroblast activation and impeding immunomodulation and tissue repair (168). Effective treatments must reverse stiff ECM and eliminate fibrogenic cells, while minimizing collateral tissue damage. Such a stage-target-mechanism paradigm may offer a framework for precision interventions across fibrotic diseases. Viewing fibrosis as an adaptive, rhythm-dependent, mechanobiological network underscores the necessity of time-adaptive mechanism-guided interventions for true reversal. Table 2 provides a comparative summary of current and emerging anti-fibrotic therapies, organized by molecular targets, mechanisms of action, applicable fibrosis stages, and translational readiness, offering a concise reference for researchers and clinicians.
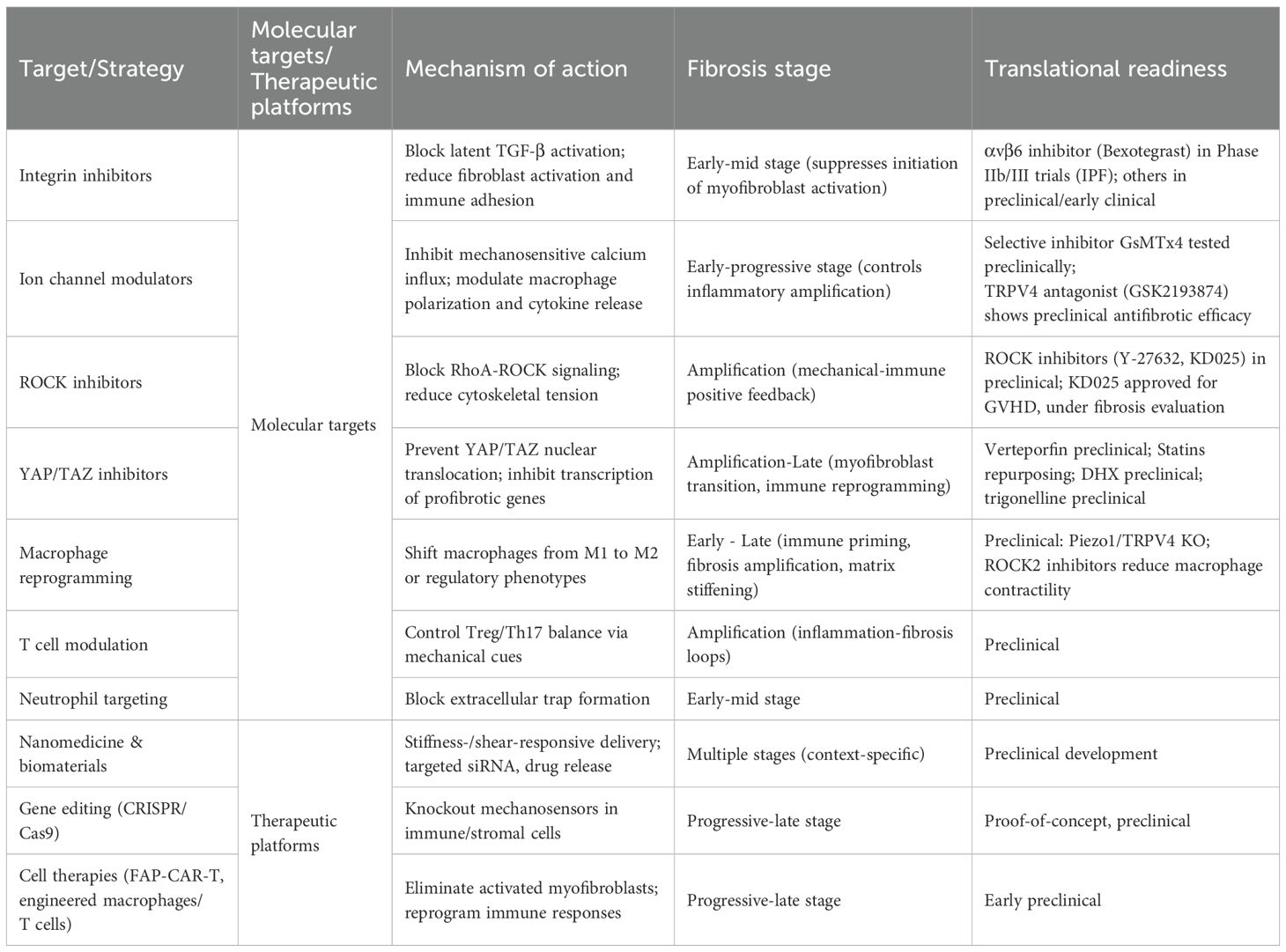
Table 2. Comparative overview of fibrosis-targeted therapeutic strategies: molecular targets, mechanisms of action, fibrosis stage applicability, and translational readiness.
Future work should focus on defining immune-subtype-specific mechanosensing mechanisms, engineering adaptive therapeutics responsive to mechano-immune signals, and developing mechanobiological biomarkers that integrate tissue stiffness imaging, interstitial flow dynamics, and immune cell mechanosensitivity thresholds. In this emerging paradigm, fibrosis is not a static scarring process, but a dynamic mechano-immune dialogue with encoded structural and informational memory, which is ultimately interceptable through biomechanically-tuned interventions. Such an approach promises to transform fibrotic disease management into one that is precise, plastic, and cross-organally applicable.
Author contributions
CL: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. DW: Writing – original draft, Writing – review & editing. HZ: Writing – original draft, Writing – review & editing. ZY: Writing – original draft, Writing – review & editing. BH: Writing – original draft, Writing – review & editing. JW: Writing – original draft, Writing – review & editing. YL: Writing – original draft, Writing – review & editing. WL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by National Natural Science Foundation of China (No. 82374332); the Key Collaborative Research Project of the Scientific and Technological Innovation Project of the China Academy of Chinese Medical Sciences (No. CI2023C026YL); the Clinical Research and Translational Capacity Improvement Project for High-Level Traditional Chinese Medicine Hospitals (Nos. HLCMHPP2023086 and HLCMHPP2023013); and the Sanming Project of Medicine in Shenzhen (No. SZZYSM202311014).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CAR-T, Chimeric Antigen Receptor T Cell; DSM, Decellularized Splenic Matrix; ECM, Extracellular Matrix; EMT, Epithelial–Mesenchymal Transition; GPCR, G Protein-Coupled Receptor; FAK, Focal Adhesion Kinase; HSCs, Hepatic Stellate Cells; IFP, Interstitial Fluid Pressure; LOX, Lysyl Oxidase; LSECs, Liver Sinusoidal Endothelial Cells; MMPs, Matrix Metalloproteinases; MRTF-A, Myocardin-Related Transcription Factor A; NET, Neutrophil Extracellular Trap; PDGF, Platelet-Derived Growth Factor; ROS, Reactive Oxygen Species; TF, Tissue Factor; MPO, Myeloperoxidase.
References
1. Theocharis AD, Manou D, and Karamanos NK. The extracellular matrix as a multitasking player in disease. FEBS J. (2019) 286:2830–69. doi: 10.1111/febs.14818
2. Hao M, Han X, Yao Z, Zhang H, Zhao M, Peng M, et al. The pathogenesis of organ fibrosis: Focus on necroptosis. Br J Pharmacol. (2023) 180:2862–79. doi: 10.1111/bph.15952
3. Mascharak S, Guo JL, Griffin M, Berry CE, Wan DC, and Longaker MT. Modelling and targeting mechanical forces in organ fibrosis. Nat Rev bioengineering. (2024) 2:305–23. doi: 10.1038/s44222-023-00144-3
4. Lei M and Chen G. Integration of mechanics and immunology: Perspective for understanding fibrotic disease mechanisms and innovating therapeutic strategies. Acta Biomater. (2025) 199:35–49. doi: 10.1016/j.actbio.2025.05.011
5. Kendall RT and Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. (2014) 5:123. doi: 10.3389/fphar.2014.00123
6. Mittelheisser V, Gensbittel V, Bonati L, Li W, Tang L, and Goetz JG. Evidence and therapeutic implications of biomechanically regulated immunosurveillance in cancer and other diseases. Nat nanotechnol. (2024) 19:281–97. doi: 10.1038/s41565-023-01535-8
7. Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. (2006) 20:811–27. doi: 10.1096/fj.05-5424rev
8. Jaalouk DE and Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. (2009) 10:63–73. doi: 10.1038/nrm2597
9. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. (2002) 110:673–87. doi: 10.1016/S0092-8674(02)00971-6
10. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. (2010) 330:55–60. doi: 10.1126/science.1193270
11. White JP, Cibelli M, Urban L, Nilius B, McGeown JG, and Nagy I. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev. (2016) 96:911–73. doi: 10.1152/physrev.00016.2015
12. Dahl KN, Ribeiro AJ, and Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ Res. (2008) 102:1307–18. doi: 10.1161/CIRCRESAHA.108.173989
13. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. (2011) 474:179–83. doi: 10.1038/nature10137
14. Chen CS. Mechanotransduction - a field pulling together. J Cell Sci. (2008) 121:3285–92. doi: 10.1242/jcs.023507
15. Ohashi K, Fujiwara S, and Mizuno K. Roles of the cytoskeleton, cell adhesion and rho signalling in mechanosensing and mechanotransduction. J Biochem. (2017) 161:245–54. doi: 10.1093/jb/mvw082
16. De Boer AA, Monk JM, Liddle DM, Hutchinson AL, Power KA, Ma DW, et al. Fish-oil-derived n-3 polyunsaturated fatty acids reduce NLRP3 inflammasome activity and obesity-related inflammatory cross-talk between adipocytes and CD11b(+) macrophages. J Nutr Biochem. (2016) 34:61–72. doi: 10.1016/j.jnutbio.2016.04.004
17. Sun Z, Guo SS, and Fässler R. Integrin-mediated mechanotransduction. J Cell Biol. (2016) 215:445–56. doi: 10.1083/jcb.201609037
18. Humphries JD, Byron A, and Humphries MJ. Integrin ligands at a glance. J Cell Sci. (2006) 119:3901–3. doi: 10.1242/jcs.03098
19. Calderwood DA, Campbell ID, and Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol. (2013) 14:503–17. doi: 10.1038/nrm3624
20. Geiger B, Spatz JP, and Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. (2009) 10:21–33. doi: 10.1038/nrm2593
21. Wu H, Zhang Y, Xu Z, Zhang J, Zhang X, and Zhao Z. The expression of integrin alpha5beta1 and transforming growth factor-beta in pulmonary fibrosis of rat. Zhonghua Bing Li Xue Za Zhi. (1999) 28:427–31.
22. Zhao XK, Cheng Y, Liang Cheng M, Yu L, Mu M, Li H, et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Sci Rep. (2016) 6:19276. doi: 10.1038/srep19276
23. Shochet GE, Brook E, Bardenstein-Wald B, Grobe H, Edelstein E, Israeli-Shani L, et al. Integrin alpha-5 silencing leads to myofibroblastic differentiation in IPF-derived human lung fibroblasts. Ther Adv Chronic Dis. (2020) 11:2040622320936023. doi: 10.1177/2040622320936023
24. Tschumperlin DJ, Ligresti G, Hilscher MB, and Shah VH. Mechanosensing and fibrosis. J Clin Invest. (2018) 128:74–84. doi: 10.1172/JCI93561
25. Zhao XK, Yu L, Cheng ML, Che P, Lu YY, Zhang Q, et al. Focal adhesion kinase regulates hepatic stellate cell activation and liver fibrosis. Sci Rep. (2017) 7:4032. doi: 10.1038/s41598-017-04317-0
26. Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DT, et al. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc Natl Acad Sci U S A. (2014) 111:16148–53. doi: 10.1073/pnas.1409802111
27. Cox CD, Bae C, Ziegler L, Hartley S, Nikolova-Krstevski V, Rohde PR, et al. Removal of the mechanoprotective influence of the cytoskeleton reveals PIEZO1 is gated by bilayer tension. Nat Commun. (2016) 7:10366. doi: 10.1038/ncomms10366
28. Beech DJ and Xiao B. Piezo channel mechanisms in health and disease. J Physiol. (2018) 596:965–7. doi: 10.1113/JP274395
29. Yim EK, Darling EM, Kulangara K, Guilak F, and Leong KW. Nanotopography-induced changes in focal adhesions, cytoskeletal organization, and mechanical properties of human mesenchymal stem cells. Biomaterials. (2010) 31:1299–306. doi: 10.1016/j.biomaterials.2009.10.037
30. Reichelt J. Mechanotransduction of keratinocytes in culture and in the epidermis. Eur J Cell Biol. (2007) 86:807–16. doi: 10.1016/j.ejcb.2007.06.004
31. Hui E, Moretti L, Barker TH, and Caliari SR. The combined influence of viscoelastic and adhesive cues on fibroblast spreading and focal adhesion organization. Cell Mol Bioeng. (2021) 14:427–40. doi: 10.1007/s12195-021-00672-1
32. Wipff PJ, Rifkin DB, Meister JJ, and Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. (2007) 179:1311–23. doi: 10.1083/jcb.200704042
33. Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. (2007) 292:H1209–24. doi: 10.1152/ajpheart.01047.2006
34. Nauli SM and Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. (2004) 26:844–56. doi: 10.1002/bies.20069
35. Weinbaum S, Tarbell JM, and Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev BioMed Eng. (2007) 9:121–67. doi: 10.1146/annurev.bioeng.9.060906.151959
36. Li YS, Haga JH, and Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. (2005) 38:1949–71. doi: 10.1016/j.jbiomech.2004.09.030
37. Hahn C and Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. (2009) 10:53–62. doi: 10.1038/nrm2596
38. Delling M, DeCaen PG, Doerner JF, Febvay S, and Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. (2013) 504:311–4. doi: 10.1038/nature12833
39. Villalba N, Baby S, and Yuan SY. The endothelial glycocalyx as a double-edged sword in microvascular homeostasis and pathogenesis. Front Cell Dev Biol. (2021) 9:711003. doi: 10.3389/fcell.2021.711003
40. Katoh K. Integrin and its associated proteins as a mediator for mechano-signal transduction. Biomolecules. (2025) 15:166. doi: 10.3390/biom15020166
41. Xu J, Mathur J, Vessières E, Hammack S, Nonomura K, Favre J, et al. GPR68 senses flow and is essential for vascular physiology. Cell. (2018) 173:762–75.e16. doi: 10.1016/j.cell.2018.03.076
42. Makino A, Prossnitz ER, Bünemann M, Wang JM, Yao W, and Schmid-Schönbein GW. G protein-coupled receptors serve as mechanosensors for fluid shear stress in neutrophils. Am J Physiol Cell Physiol. (2006) 290:C1633–9. doi: 10.1152/ajpcell.00576.2005
43. Wang Q, Zhang Q, Zhang Y, and Zhao X. Yak OXGR1 promotes fibroblast proliferation via the PI3K/AKT pathways. J Cell Biochem. (2019) 120:6729–40. doi: 10.1002/jcb.27970
44. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. (2004) 6:499–506. doi: 10.1038/ncb1137
45. Adapala RK, Katari V, Teegala LR, Thodeti S, Paruchuri S, and Thodeti CK. TRPV4 mechanotransduction in fibrosis. Cells. (2021) 10:3053. doi: 10.3390/cells10113053
46. Huang Z, Khalifa MO, Gu W, and Li TS. Hydrostatic pressure induces profibrotic properties in hepatic stellate cells via the RhoA/ROCK signaling pathway. FEBS Open Bio. (2022) 12:1230–40. doi: 10.1002/2211-5463.13405
47. Rahman Z, Bordoloi AD, Rouhana H, Tavasso M, van der Zon G, Garbin V, et al. Interstitial flow potentiates TGF-β/Smad-signaling activity in lung cancer spheroids in a 3D-microfluidic chip. Lab Chip. (2024) 24:422–33. doi: 10.1039/D3LC00886J
48. Li FY, Xie XS, Fan JM, Li Z, Wu J, and Zheng R. Hydraulic pressure inducing renal tubular epithelial-myofibroblast transdifferentiation in vitro. J Zhejiang Univ Sci B. (2009) 10:659–67. doi: 10.1631/jzus.B0920110
49. Wiedenmann CJ, Gottwald C, Zeqiri K, Frömmichen J, Bungert E, Gläser M, et al. Slow interstitial fluid flow activates TGF-β Signaling and drives fibrotic responses in human tenon fibroblasts. Cells. (2023) 12:2205. doi: 10.3390/cells12172205
50. Zhao YQ, Deng XW, Xu GQ, Lin J, Lu HZ, and Chen J. Mechanical homeostasis imbalance in hepatic stellate cells activation and hepatic fibrosis. Front Mol Biosci. (2023) 10:1183808. doi: 10.3389/fmolb.2023.1183808
51. Liu Z, Li S, Qian X, Li L, Zhang H, and Liu Z. RhoA/ROCK-YAP/TAZ axis regulates the fibrotic activity in dexamethasone-treated human trabecular meshwork cells. Front Mol Biosci. (2021) 8:728932. doi: 10.3389/fmolb.2021.728932
52. Zhao X, Kong Y, Liang B, Xu J, Lin Y, Zhou N, et al. Mechanosensitive Piezo1 channels mediate renal fibrosis. JCI Insight. (2022) 7:e152330. doi: 10.1172/jci.insight.152330
53. Zhu Y, Chen J, Chen C, Tang R, Xu J, Shi S, et al. Deciphering mechanical cues in the microenvironment: from non-malignant settings to tumor progression. biomark Res. (2025) 13:11. doi: 10.1186/s40364-025-00727-9
54. Xin Y, Li K, Huang M, Liang C, Siemann D, Wu L, et al. Biophysics in tumor growth and progression: from single mechano-sensitive molecules to mechanomedicine. Oncogene. (2023) 42:3457–90. doi: 10.1038/s41388-023-02844-x
55. Mao J, Liu J, Zhou M, Wang G, Xiong X, and Deng Y. Hypoxia-induced interstitial transformation of microvascular endothelial cells by mediating HIF-1α/VEGF signaling in systemic sclerosis. PLoS One. (2022) 17:e0263369. doi: 10.1371/journal.pone.0263369
56. Vallée A and Lecarpentier Y. TGF-β in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. (2019) 9:98. doi: 10.1186/s13578-019-0362-3
57. Hu Q, Saleem K, Pandey J, Charania AN, Zhou Y, and He C. Cell adhesion molecules in fibrotic diseases. Biomedicines. (2023) 11:1995. doi: 10.3390/biomedicines11071995
58. Beaven E, Kumar R, Bhatt HN, Esquivel SV, and Nurunnabi M. Myofibroblast specific targeting approaches to improve fibrosis treatment. Chem Commun (Cambridge England). (2022) 58:13556–71. doi: 10.1039/D2CC04825F
59. Schroer AK and Merryman WD. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J Cell Sci. (2015) 128:1865–75. doi: 10.1242/jcs.162891
60. Ko KS, Arora PD, and McCulloch CA. Cadherins mediate intercellular mechanical signaling in fibroblasts by activation of stretch-sensitive calcium-permeable channels. J Biol Chem. (2001) 276:35967–77. doi: 10.1074/jbc.M104106200
61. Kehrberg RJ and DeMali KA. E-Cadherin: A conductor of cellular signaling. Curr Opin Cell Biol. (2025) 95:102559. doi: 10.1016/j.ceb.2025.102559
62. Wang A, Dunn AR, and Weis WI. Mechanism of the cadherin-catenin F-actin catch bond interaction. eLife. (2022) 11:e80130. doi: 10.7554/eLife.80130
63. Campàs O, Noordstra I, and Yap AS. Adherens junctions as molecular regulators of emergent tissue mechanics. Nat Rev Mol Cell Biol. (2024) 25:252–69. doi: 10.1038/s41580-023-00688-7
64. Lodyga M, Cambridge E, Karvonen HM, Pakshir P, Wu B, Boo S, et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci Signaling. (2019) 12:eaao3469. doi: 10.1126/scisignal.aao3469
65. Astrab LR, Skelton ML, and Caliari SR. Direct M2 macrophage co-culture overrides viscoelastic hydrogel mechanics to promote fibroblast activation. bioRxiv. (2024). doi: 10.1101/2024.10.13.618034
66. Liu L, Yu H, Zhao H, Wu Z, Long Y, Zhang J, et al. Matrix-transmitted paratensile signaling enables myofibroblast-fibroblast cross talk in fibrosis expansion. Proc Natl Acad Sci U S A. (2020) 117:10832–8. doi: 10.1073/pnas.1910650117
67. Long Y, Niu Y, Liang K, and Du Y. Mechanical communication in fibrosis progression. Trends Cell Biol. (2022) 32:70–90. doi: 10.1016/j.tcb.2021.10.002
68. Xiong J, Xiao R, Zhao J, Zhao Q, Luo M, Li F, et al. Matrix stiffness affects tumor-associated macrophage functional polarization and its potential in tumor therapy. J Trans Med. (2024) 22:85. doi: 10.1186/s12967-023-04810-3
69. Fan Q, Zheng Y, Wang X, Xie R, Ding Y, Wang B, et al. Dynamically re-organized collagen fiber bundles transmit mechanical signals and induce strongly correlated cell migration and self-organization. Angewandte Chemie (International Ed English). (2021) 60:11858–67. doi: 10.1002/anie.202016084
70. Natarajan V, Harris EN, and Kidambi S. SECs (Sinusoidal endothelial cells), liver microenvironment, and fibrosis. BioMed Res Int. (2017) 2017:4097205. doi: 10.1155/2017/4097205
71. Wang H, Abhilash AS, Chen CS, Wells RG, and Shenoy VB. Long-range force transmission in fibrous matrices enabled by tension-driven alignment of fibers. Biophys J. (2014) 107:2592–603. doi: 10.1016/j.bpj.2014.09.044
72. Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. (2010) 190:693–706. doi: 10.1083/jcb.201004082
73. Kural MH and Billiar KL. Regulating tension in three-dimensional culture environments. Exp Cell Res. (2013) 319:2447–59. doi: 10.1016/j.yexcr.2013.06.019
74. Wynn TA and Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
75. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
76. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L344–57. doi: 10.1152/ajplung.00300.2014
77. Veerasubramanian PK, Shao H, Meli VS, Phan T, Luu TU, Liu WF, et al. A Src-H3 acetylation signaling axis integrates macrophage mechanosensation with inflammatory response. Biomaterials. (2021) 279:121236. doi: 10.1016/j.biomaterials.2021.121236
78. Tang Y, Zhao C, Zhuang Y, Zhong A, Wang M, Zhang W, et al. Mechanosensitive Piezo1 protein as a novel regulator in macrophages and macrophage-mediated inflammatory diseases. Front Immunol. (2023) 14:1149336. doi: 10.3389/fimmu.2023.1149336
79. Du S and Liu K. Mechanosensitive ion channels and inflammation: key links in cellular signal transduction. Inflammation Res. (2025) 74:104. doi: 10.1007/s00011-025-02057-w
80. Zhang Z, He P, Yang L, Gong J, Qin R, and Wang M. Posttranslational modifications of YAP/TAZ: molecular mechanisms and therapeutic opportunities. Cell Mol Biol Lett. (2025) 30:83. doi: 10.1186/s11658-025-00760-4
81. Mei F, Guo Y, Wang Y, Zhou Y, Heng BC, Xie M, et al. Matrix stiffness regulates macrophage polarisation via the Piezo1-YAP signalling axis. Cell proliferation. (2024) 57:e13640. doi: 10.1111/cpr.13640
82. Fish A, Forster J 3rd, Malik V, and Kulkarni A. Shear-stress initiates signal two of NLRP3 inflammasome activation in LPS-primed macrophages through piezo1. ACS Appl materials interfaces. (2025) 17:7363–76. doi: 10.1021/acsami.4c18845
83. Li Q, Cheng Y, Zhang Z, Bi Z, Ma X, Wei Y, et al. Inhibition of ROCK ameliorates pulmonary fibrosis by suppressing M2 macrophage polarisation through phosphorylation of STAT3. Clin Trans Med. (2022) 12:e1036. doi: 10.1002/ctm2.1036
84. Walker CJ, Crocini C, Ramirez D, Killaars AR, Grim JC, Aguado BA, et al. Nuclear mechanosensing drives chromatin remodelling in persistently activated fibroblasts. Nat Biomed Eng. (2021) 5:1485–99. doi: 10.1038/s41551-021-00709-w
85. O'Shea JJ, Steward-Tharp SM, Laurence A, Watford WT, Wei L, Adamson AS, et al. Signal transduction and Th17 cell differentiation. Microbes Infect. (2009) 11:599–611. doi: 10.1016/j.micinf.2009.04.007
86. Sisto M and Lisi S. Targeting interleukin-17 as a novel treatment option for fibrotic diseases. J Clin Med. (2023) 13:164. doi: 10.3390/jcm13010164
87. Barron A MS, Fabre T, and De S. Distinct fibroblast functions associated with fibrotic and immune-mediated inflammatory diseases and their implications for therapeutic development. F1000Res. (2024) 13:54. doi: 10.12688/f1000research.143472.1
88. Adu-Berchie K, Liu Y, Zhang DKY, Freedman BR, Brockman JM, Vining KH, et al. Generation of functionally distinct T-cell populations by altering the viscoelasticity of their extracellular matrix. Nat BioMed Eng. (2023) 7:1374–91. doi: 10.1038/s41551-023-01052-y
89. Puri M and Sonawane S. Liver sinusoidal endothelial cells in the regulation of immune responses and fibrosis in metabolic dysfunction-associated fatty liver disease. Int J Mol Sci. (2025) 26:3988. doi: 10.3390/ijms26093988
90. Dombroski JA, Rowland SJ, Fabiano AR, Knoblauch SV, Hope JM, and King MR. Fluid shear stress enhances dendritic cell activation. Immunobiology. (2023) 228:152744. doi: 10.1016/j.imbio.2023.152744
91. Shi L, Lim JY, and Kam LC. Substrate stiffness enhances human regulatory T cell induction and metabolism. Biomaterials. (2023) 292:121928. doi: 10.1016/j.biomaterials.2022.121928
92. Liu L, Yang J, Zu B, Wang J, Sheng K, Zhao L, et al. Acacetin regulated the reciprocal differentiation of Th17 cells and Treg cells and mitigated the symptoms of collagen-induced arthritis in mice. Scand J Immunol. (2018) 88:e12712. doi: 10.1111/sji.12712
93. He B, Wu L, Xie W, Shao Y, Jiang J, Zhao Z, et al. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. (2017) 18:33. doi: 10.1186/s12865-017-0215-y
94. Sun XF, Gu L, Deng WS, and Xu Q. Impaired balance of T helper 17/T regulatory cells in carbon tetrachloride-induced liver fibrosis in mice. World J Gastroenterol. (2014) 20:2062–70. doi: 10.3748/wjg.v20.i8.2062
95. Pang X, He X, Qiu Z, Zhang H, Xie R, Liu Z, et al. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct Target Ther. (2023) 8:1. doi: 10.1038/s41392-022-01259-6
96. Niethammer P. Neutrophil mechanotransduction: A GEF to sense fluid shear stress. J Cell Biol. (2016) 215:13–4. doi: 10.1083/jcb.201609101
97. Khanmohammadi M, Danish H, Sekar NC, Suarez SA, Chheang C, Peter K, et al. Cyclic stretch enhances neutrophil extracellular trap formation. BMC Biol. (2024) 22:209. doi: 10.1186/s12915-024-02009-6
98. Baratchi S, Danish H, Chheang C, Zhou Y, Huang A, Lai A, et al. Piezo1 expression in neutrophils regulates shear-induced NETosis. Nat Commun. (2024) 15:7023. doi: 10.1038/s41467-024-51211-1
99. Yan S, Li M, Liu B, Ma Z, and Yang Q. Neutrophil extracellular traps and pulmonary fibrosis: an update. J Inflammation (Lond). (2023) 20:2. doi: 10.1186/s12950-023-00329-y
100. Suzuki M, Ikari J, Anazawa R, Tanaka N, Katsumata Y, Shimada A, et al. PAD4 deficiency improves bleomycin-induced neutrophil extracellular traps and fibrosis in mouse lung. Am J Respir Cell Mol Biol. (2020) 63:806–18. doi: 10.1165/rcmb.2019-0433OC
101. Wu Y, Lu K, Lu Y, Liao J, Zhang S, Yang S, et al. Transient receptor potential vanilloid 4 (TRPV4) in neutrophils enhances myocardial ischemia/reperfusion injury. J Leukoc Biol. (2023) 114:266–79. doi: 10.1093/jleuko/qiad063
102. Zhu Y, Wang T, Yang Y, Wang Z, Chen X, Wang L, et al. Low shear stress exacerbates atherosclerosis by inducing the generation of neutrophil extracellular traps via Piezo1-mediated mechanosensation. Atherosclerosis. (2024) 391:117473. doi: 10.1016/j.atherosclerosis.2024.117473
103. Mihara H, Boudaka A, Tominaga M, and Sugiyama T. Transient receptor potential vanilloid 4 regulation of adenosine triphosphate release by the adenosine triphosphate transporter vesicular nucleotide transporter, a novel therapeutic target for gastrointestinal baroreception and chronic inflammation. Digestion. (2020) 101:6–11. doi: 10.1159/000504021
104. Lee M, Du H, Winer DA, Clemente-Casares X, and Tsai S. Mechanosensing in macrophages and dendritic cells in steady-state and disease. Front Cell Dev Biol. (2022) 10:1044729. doi: 10.3389/fcell.2022.1044729
105. Chakraborty M, Chu K, Shrestha A, Revelo XS, Zhang X, Gold MJ, et al. Mechanical stiffness controls dendritic cell metabolism and function. Cell Rep. (2021) 34:108609. doi: 10.1016/j.celrep.2020.108609
106. Alraies Z, Rivera CA, Delgado MG, Sanséau D, Maurin M, Amadio R, et al. Cell shape sensing licenses dendritic cells for homeostatic migration to lymph nodes. Nat Immunol. (2024) 25:1193–206. doi: 10.1038/s41590-024-01856-3
107. Janssen E, van den Dries K, Ventre M, and Cambi A. Mechanobiology of myeloid cells. Curr Opin Cell Biol. (2024) 86:102311. doi: 10.1016/j.ceb.2023.102311
108. Wang Y, Yang H, Jia A, Wang Y, Yang Q, Dong Y, et al. Dendritic cell Piezo1 directs the differentiation of TH1 and Treg cells in cancer. eLife. (2022) 11:1–23. doi: 10.7554/eLife.79957
109. Yanamandra AK, Zhang J, Montalvo G, Zhou X, Biedenweg D, Zhao R, et al. PIEZO1-mediated mechanosensing governs NK-cell killing efficiency and infiltration in three-dimensional matrices. Eur J Immunol. (2024) 54:e2350693. doi: 10.1002/eji.202350693
110. Hu B, Xin Y, Hu G, Li K, and Tan Y. Fluid shear stress enhances natural killer cell’s cytotoxicity toward circulating tumor cells through NKG2D-mediated mechanosensing. APL bioengineering. (2023) 7:036108. doi: 10.1063/5.0156628
111. Glässner A, Eisenhardt M, Krämer B, Körner C, Coenen M, Sauerbruch T, et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest. (2012) 92:967–77. doi: 10.1038/labinvest.2012.54
112. Vogel V and Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. (2006) 7:265–75. doi: 10.1038/nrm1890
113. Henderson NC, Rieder F, and Wynn TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
114. Sun M, Spill F, and Zaman MH. A computational model of YAP/TAZ mechanosensing. Biophys J. (2016) 110:2540–50. doi: 10.1016/j.bpj.2016.04.040
115. Sándor N, Lukácsi S, Ungai-Salánki R, Orgován N, Szabó B, Horváth R, et al. CD11c/CD18 dominates adhesion of human monocytes, macrophages and dendritic cells over CD11b/CD18. PLoS One. (2016) 11:e0163120. doi: 10.1371/journal.pone.0163120
116. Xu Y, Ying L, Lang JK, Hinz B, and Zhao R. Modeling mechanical activation of macrophages during pulmonary fibrogenesis for targeted anti-fibrosis therapy. Sci Adv. (2024) 10:eadj9559. doi: 10.1126/sciadv.adj9559
117. Piechocka IK, Keary S, Sosa-Costa A, Lau L, Mohan N, Stanisavljevic J, et al. Shear forces induce ICAM-1 nanoclustering on endothelial cells that impact on T-cell migration. Biophys J. (2021) 120:2644–56. doi: 10.1016/j.bpj.2021.05.016
118. Celli L, Ryckewaert JJ, Delachanal E, and Duperray A. Evidence of a functional role for interaction between ICAM-1 and nonmuscle alpha-actinins in leukocyte diapedesis. J Immunol. (2006) 177:4113–21. doi: 10.4049/jimmunol.177.6.4113
119. Nakao A, Hasegawa Y, Tsuchiya Y, and Shimokata K. Expression of cell adhesion molecules in the lungs of patients with idiopathic pulmonary fibrosis. Chest. (1995) 108:233–9. doi: 10.1378/chest.108.1.233
120. Steiner O, Coisne C, Cecchelli R, Boscacci R, Deutsch U, Engelhardt B, et al. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood-brain barrier endothelium. J Immunol. (2010) 185:4846–55. doi: 10.4049/jimmunol.0903732
121. Xiong J and Zhu B. Epigenetic preparation of future gene induction kinetics. Annu Rev Genet. (2025) 2025.591.1-1.25. doi: 10.1146/annurev-genet-012825-093148
122. Xia Q, Xu F, Zhang L, Ding W, Liu J, Liu J, et al. The EZH2 selective inhibitor ZLD1039 attenuates UUO-induced renal fibrosis by suppressing YAP activation. Mol biomed. (2025) 6:36. doi: 10.1186/s43556-025-00276-5
123. Kalukula Y, Stephens AD, Lammerding J, and Gabriele S. Mechanics and functional consequences of nuclear deformations. Nat Rev Mol Cell Biol. (2022) 23:583–602. doi: 10.1038/s41580-022-00480-z
124. Sen B, Xie Z, Thomas MD, Pattenden SG, Howard S, McGrath C, et al. Nuclear actin structure regulates chromatin accessibility. Nat Commun. (2024) 15:4095. doi: 10.1038/s41467-024-48580-y
125. Randall LJ, Bajan S, Tran TD, Harvey RJ, and Russell FD. Propolis compound inhibits profibrotic TGF-β1/SMAD signalling in human fibroblasts. Sci Rep. (2025) 15:27260. doi: 10.1038/s41598-025-07918-2
126. Suo X, Ge Q, Peng L, Zhu Q, Zhang M, Cheng X, et al. Emerging epigenetic modifications in renal fibrosis: From mechanisms to treatments. Acta Pharm Sin B. (2025). doi: 10.1016/j.apsb.2025.09.012
127. Balan OV, Malysheva IE, and Fedorenko OM. Monocytes/macrophages as one of the sources of myofibroblasts in the development of tissue fibrosis: the role of noncoding RNAs. Russian J Genet. (2025) 61:263–70. doi: 10.1134/S1022795424701643
128. Ghazanfari D, Ren L, Cantú MS, and King MR. Cellular mechanoactivation of antigen-presenting cells and T cells for cancer immunotherapy. Curr Opin Biomed Eng. (2025) 36:100619. doi: 10.1016/j.cobme.2025.100619
129. Margadant C and Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. (2010) 11:97–105. doi: 10.1038/embor.2009.276
130. Bagnato GL, Irrera N, Pizzino G, Santoro D, Roberts WN, Bagnato G, et al. Dual αvβ3 and αvβ5 blockade attenuates fibrotic and vascular alterations in a murine model of systemic sclerosis. Clin Sci (Lond). (2018) 132:231–42. doi: 10.1042/CS20171426
131. Yi M, Yuan Y, Ma L, Li L, Qin W, Wu B, et al. Inhibition of TGFβ1 activation prevents radiation-induced lung fibrosis. Clin Transl Med. (2024) 14:e1546. doi: 10.1002/ctm2.1546
132. Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. (2008) 177:56–65. doi: 10.1164/rccm.200706-805OC
133. Wang Y, Chu T, Meng C, Bian Y, and Li J. Piezo1-specific deletion in macrophage protects the progression of chronic inflammatory bowel disease in mice. Cell Mol Gastroenterol Hepatol. (2025) 19:101495. doi: 10.1016/j.jcmgh.2025.101495
134. Rahaman SO, Grove LM, Paruchuri S, Southern BD, Abraham S, Niese KA, et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest. (2014) 124:5225–38. doi: 10.1172/JCI75331
135. Pairet N, Mang S, Fois G, Keck M, Kühnbach M, Gindele J, et al. TRPV4 inhibition attenuates stretch-induced inflammatory cellular responses and lung barrier dysfunction during mechanical ventilation. PLoS One. (2018) 13:e0196055. doi: 10.1371/journal.pone.0196055
136. Tada S, Iwamoto H, Nakamuta M, Sugimoto R, Enjoji M, Nakashima Y, et al. A selective ROCK inhibitor, Y27632, prevents dimethylnitrosamine-induced hepatic fibrosis in rats. J Hepatol. (2001) 34:529–36. doi: 10.1016/S0168-8278(00)00059-3
137. Tu PC, Pan YL, Liang ZQ, Yang GL, Wu CJ, Zeng L, et al. Mechanical stretch promotes macrophage polarization and inflammation via the rhoA-ROCK/NF-κB pathway. BioMed Res Int. (2022) 2022:6871269. doi: 10.1155/2022/6871269
138. Liu Y, Tejpal N, You J, Li XC, Ghobrial RM, and Kloc M. ROCK inhibition impedes macrophage polarity and functions. Cell Immunol. (2016) 300:54–62. doi: 10.1016/j.cellimm.2015.12.005
139. Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, Chen W, Scher JU, Mo R, et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A. (2014) 111:16814–9. doi: 10.1073/pnas.1414189111
140. Zanin-Zhorov A and Blazar BR. ROCK2, a critical regulator of immune modulation and fibrosis has emerged as a therapeutic target in chronic graft-versus-host disease. Clin Immunol. (2021) 230:108823. doi: 10.1016/j.clim.2021.108823
141. Nalkurthi C, Schroder WA, Melino M, Irvine KM, Nyuydzefe M, Chen W, et al. ROCK2 inhibition attenuates profibrogenic immune cell function to reverse thioacetamide-induced liver fibrosis. JHEP Rep. (2022) 4:100386. doi: 10.1016/j.jhepr.2021.100386
142. Zeyada MS, Eraky SM, and El-Shishtawy MM. Trigonelline mitigates bleomycin-induced pulmonary inflammation and fibrosis: Insight into NLRP3 inflammasome and SPHK1/S1P/Hippo signaling modulation. Life Sci. (2024) 336:122272. doi: 10.1016/j.lfs.2023.122272
143. Haak AJ, Kostallari E, Sicard D, Ligresti G, Choi KM, Caporarello N, et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med. (2019) 11:eaau6296. doi: 10.1183/13993003.congress-2019.OA2113
144. Hao F, Xu Q, Wang J, Yu S, Chang HH, Sinnett-Smith J, et al. Lipophilic statins inhibit YAP nuclear localization, co-activator activity and colony formation in pancreatic cancer cells and prevent the initial stages of pancreatic ductal adenocarcinoma in KrasG12D mice. PLoS One. (2019) 14:e0216603. doi: 10.1371/journal.pone.0216603
145. Santos DM, Pantano L, Pronzati G, Grasberger P, Probst CK, Black KE, et al. Screening for YAP inhibitors identifies statins as modulators of fibrosis. Am J Respir Cell Mol Biol. (2020) 62:479–92. doi: 10.1165/rcmb.2019-0296OC
146. Liang M, Yu M, Xia R, Song K, Wang J, Luo J, et al. Yap/Taz deletion in Gli(+) cell-derived myofibroblasts attenuates fibrosis. J Am Soc Nephrol. (2017) 28:3278–90. doi: 10.1681/ASN.2015121354
147. Jin J, Wang T, Park W, Li W, Kim W, Park SK, et al. Inhibition of yes-associated protein by verteporfin ameliorates unilateral ureteral obstruction-induced renal tubulointerstitial inflammation and fibrosis. Int J Mol Sci. (2020) 21:8184. doi: 10.3390/ijms21218184
148. Meli VS, Atcha H, Veerasubramanian PK, Nagalla RR, Luu TU, Chen EY, et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci Adv. (2020) 6:eabb8471. doi: 10.1126/sciadv.abb8471
149. Guan X, Shen Y, Zhao C, Li X, Li X, Lu D, et al. Cascade-responsive nanoprodrug disrupts immune-fibroblast communications for potentiated cancer mechanoimmunotherapy. Advanced healthcare materials. (2025) 14:e2500176. doi: 10.1002/adhm.202500176
150. Vlasova KY, Kerr A, Pennock ND, Jozic A, Sahel DK, Gautam M, et al. Synthesis of ionizable lipopolymers using split-Ugi reaction for pulmonary delivery of various size RNAs and gene editing. Nat Commun. (2025) 16:4021. doi: 10.1038/s41467-025-59136-z
151. Lv X, Min J, Huang J, Wang H, Wei S, Huang C, et al. Simultaneously controlling inflammation and infection by smart nanomedicine responding to the inflammatory microenvironment. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2024) 11:e2403934. doi: 10.1002/advs.202403934
152. Liu J, Liu J, Mu W, Ma Q, Zhai X, Jin B, et al. Delivery strategy to enhance the therapeutic efficacy of liver fibrosis via nanoparticle drug delivery systems. ACS nano. (2024) 18:20861–85. doi: 10.1021/acsnano.4c02380
153. Wang J, Kaplan JA, Colson YL, and Grinstaff MW. Mechanoresponsive materials for drug delivery: Harnessing forces for controlled release. Adv Drug Delivery Rev. (2017) 108:68–82. doi: 10.1016/j.addr.2016.11.001
154. Wang Y, Pisapati AV, Zhang XF, and Cheng X. Recent developments in nanomaterial-based shear-sensitive drug delivery systems. Adv Healthc Mater. (2021) 10:e2002196. doi: 10.1002/adhm.202002196
155. Athirasala A, Patel S, Menezes PP, Kim J, Tahayeri A, et al. Matrix stiffness regulates lipid nanoparticle-mRNA delivery in cell-laden hydrogels. Nanomedicine. (2022) 42:102550. doi: 10.1016/j.nano.2022.102550
156. Nehoff H, Parayath NN, Domanovitch L, Taurin S, and Greish K. Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int J Nanomed. (2014) 9:2539–55. doi: 10.2147/IJN.S47129
157. Singh A, Chakraborty S, Wong SW, Hefner NA, Stuart A, Qadir AS, et al. Nanoparticle targeting of de novo profibrotic macrophages mitigates lung fibrosis. Proc Natl Acad Sci U S A. (2022) 119:e2121098119. doi: 10.1073/pnas.2121098119
158. Liu P, Xiang JX, Zheng XL, Su JB, Dong DH, Yang LF, et al. Implantation strategy of tissue-engineered liver based on decellularized spleen matrix in rats. Nan Fang Yi Ke Da Xue Xue Bao. (2018) 38:698–703. doi: 10.3969/j.issn.1673-4254.2018.06.09
159. Wang Y, Wang J, Zhang J, Wang Y, Wang Y, Kang H, et al. Stiffness sensing via Piezo1 enhances macrophage efferocytosis and promotes the resolution of liver fibrosis. Sci Adv. (2024) 10:eadj3289. doi: 10.1126/sciadv.adj3289
160. Luo S, Zhao X, Jiang J, Deng B, Liu S, Xu H, et al. Piezo1 specific deletion in macrophage protects the progression of liver fibrosis in mice. Theranostics. (2023) 13:5418–34. doi: 10.7150/thno.86103
161. Dutta B, Goswami R, and Rahaman SO. TRPV4 plays a role in matrix stiffness-induced macrophage polarization. Front Immunol. (2020) 11:570195. doi: 10.3389/fimmu.2020.570195
162. Yan J, Wang SY, Su Q, Zou MW, Zhou ZY, Shou J, et al. Targeted immunotherapy rescues pulmonary fibrosis by reducing activated fibroblasts and regulating alveolar cell profile. Nat Commun. (2025) 16:3748. doi: 10.1038/s41467-025-59093-7
163. Yashaswini CN, Cogliati B, Qin T, To T, Williamson T, Papp TE, et al. In vivo anti-FAP CAR T therapy reduces fibrosis and restores liver homeostasis in metabolic dysfunction-associated steatohepatitis. bioRxiv. (2025). doi: 10.1101/2025.02.25.640143
164. Mooney JJ, Jacobs S, Lefebvre ÉA, Cosgrove GP, Clark A, Turner SM, et al. Bexotegrast shows dose-dependent integrin αvβ6 receptor occupancy in lungs of participants with idiopathic pulmonary fibrosis: A phase 2, open-label clinical trial. Ann Am Thorac Soc. (2025) 22:350–8. doi: 10.1513/AnnalsATS.202409-969OC
165. Patsenker E, Popov Y, Stickel F, Schneider V, Ledermann M, Sägesser H, et al. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. (2009) 50:1501–11. doi: 10.1002/hep.23144
166. Jagasia M, Lazaryan A, Bachier CR, Salhotra A, Weisdorf DJ, Zoghi B, et al. ROCK2 inhibition with belumosudil (KD025) for the treatment of chronic graft-versus-host disease. J Clin Oncol. (2021) 39:1888–98. doi: 10.1200/JCO.20.02754
167. He X, Tolosa MF, Zhang T, Goru SK, Ulloa Severino L, Misra PS, et al. Myofibroblast YAP/TAZ activation is a key step in organ fibrogenesis. JCI Insight. (2022) 7:e146243. doi: 10.1172/jci.insight.146243
168. Noom A, Sawitzki B, Knaus P, and Duda GN. A two-way street - cellular metabolism and myofibroblast contraction. NPJ Regener Med. (2024) 9:15. doi: 10.1038/s41536-024-00359-x
Keywords: organ fibrosis, mechanotransduction, immune, extracellular matrix, therapeutic target
Citation: Lei C, Wu D, Zhang H, Yang Z, Huang B, Wang J, Li Y and Lv W (2025) The mechanotransduction-immune axis in organ fibrosis: dual regulatory mechanisms and translational therapeutic perspectives. Front. Immunol. 16:1670213. doi: 10.3389/fimmu.2025.1670213
Received: 21 July 2025; Accepted: 10 October 2025;
Published: 24 October 2025.
Edited by:
Michael V. Volin, Midwestern University, United StatesReviewed by:
Priyanka Verma, University of Michigan, United StatesKusum Devi, The University of Iowa, United States
Copyright © 2025 Lei, Wu, Zhang, Yang, Huang, Wang, Li and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenliang Lv, bHZ3ZW5saWFuZ0Bzb2h1LmNvbQ==; Yanbo Li, bHliODUyMEAxMjYuY29t; Jie Wang, amlld2FuZzkxM0AxMjYuY29t