 Qiongrong Xia
Qiongrong Xia Xiaohua Liu
Xiaohua Liu Huacui Huang†
Huacui Huang†- Department of Medical Laboratory, Xindu District People’s Hospital of Chengdu, Chengdu, Sichuan, China
Viral infections pose a major threat to global health, causing severe pneumonia, meningitis, hepatitis, and fatal complications. Viruses are highly dependent on host cellular factors to complete their life cycle, and host proteases, as one of the core regulatory hubs, profoundly influence the progression of infection and pathogenicity. Viruses rely on specific host proteases (e.g., transmembrane serine proteases [TMPRSS family], furin, cathepsins, and others such as caspases and metalloproteases) to precisely cleave and activate viral surface glycoproteins and internal precursor proteins, thereby facilitating efficient invasion, replication, release, and immune evasion. Meanwhile, host proteases participate bidirectionally in immune regulation. They can be exploited by viruses to exacerbate pathological damage (e.g., triggering cytokine storms), yet also act as key defense components by directly cleaving viral proteins to inhibit infection. Different viruses have evolved sophisticated strategies to hijack host proteases, whose activity, specificity, and tissue distribution directly determine the viral tissue tropism and pathogenic potential. Compared to highly mutable viral targets, host proteases serve as ideal targets for developing host-directed antiviral drugs (HADs) due to their genetic stability and conserved mechanisms, but their toxicity requires careful evaluation because of their physiological roles. Inhibitor strategies targeting host proteases have demonstrated promising breakthrough potential in circumventing drug resistance and exerting broad-spectrum inhibitory activity against diverse viruses. Elucidating the multidimensional roles of host proteases in infection is crucial for designing the next-generation of broad-spectrum, anti-drug resistance antiviral strategies. This review systematically summarizes the regulatory mechanisms of host proteases at various stages of viral infection and advances in targeted intervention strategies, providing theoretical support for the development of resistance-resistant and broad-spectrum antiviral therapeutics.
1 Introduction
Viral infectious diseases persistently threaten global public health, with over 200 viruses known to cause human diseases, yet currently approved antiviral drugs effectively target only approximately 10 viral pathogens (1). Over the past decade, the frequency of emerging disease outbreaks has increased. From the Ebola virus (EBOV) epidemic to the coronavirus disease 2019 (COVID-19) pandemic, these events reveal the severe impact of viral infections. Chronic infections such as human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV) have cumulatively affected over 350 million people and claimed more than 40 million lives to date (2, 3). Meanwhile, acute pathogens such as influenza virus (FluV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infect over 1 billion people annually, leading to more than 5 million severe cases and over 10 million related deaths (4, 5). Current viral protease-targeting antiviral therapies face two major challenges. First, the issue of drug resistance targeting viral proteases is becoming increasingly prominent. The treatment failure rate of HIV-1 protease inhibitors (e.g., lopinavir) has reached 20% (6, 7), oseltamivir resistance rates in circulating hemagglutinin 1 neuraminidase 1 (H1N1) strains reached 3.76% (8), and the Glu166Val (E166V) mutation in SARS-CoV-2 main protease (Mpro, also known as 3CLpro) reduces nirmatrelvir activity by 100-fold, often causing treatment failure (9–11). Second, narrow-spectrum activity limits efficacy against highly variable viruses with multiple serotypes, such as dengue virus (DENV) (1). These critical challenges urgently require innovative antiviral strategies and targets.
Viruses depend entirely on host cells to accomplish critical life cycle steps, including invasion, replication, maturation, and release. During this process, host proteases, as key enzymes responsible for protein degradation and modification, serve as pivotal regulators at every infection stage, positioning them as high-potential breakthrough targets. Their core value is first evidenced by conserved cleavage mechanisms across viral families. Furin mediates the cleavage of HIV envelope glycoprotein gp160 into gp120/gp41 subunits to initiate membrane fusion (12). It also hydrolyzes human papillomavirus (HPV) late protein 2 (L2) at Arg470, enabling viral genome translocation across the nuclear membrane (13). Such protease-mediated conformational rearrangements are essential for cross-family viral infections. This compartmentalization ensures precise activation of viral proteins at critical subcellular sites. EBOV requires endosomal cathepsins B/L (CTSB/L) under low-pH conditions to trim glycoprotein 1 (GP1) subunits and expose the receptor-binding domain (14–17), while coronaviruses (CoVs) undergo furin-mediated pre-cleavage of spike (S) proteins in the Golgi compartments to enhance infectivity (18, 19). Compartment-specific proteases (e.g., transmembrane protease serine 2 (TMPRSS2)) spatiotemporally optimize cleavage (20, 21). In addition to acting as viral cofactors, host proteases further influence viral proliferation and transmission by modulating host cell signaling (22, 23), immune responses (24, 25), and apoptosis (26, 27). Consequently, targeting host proteases not only blocks the viral life cycle but also pioneers innovative therapeutic strategies by regulating host responses.
Despite the substantial potential of host proteases as antiviral targets, their intrinsic biological properties pose significant challenges for targeted interventions. The same protease often exhibits functional pleiotropy in distinct infection contexts. For example, furin enhances viral particle maturation in HIV infection (28, 29), yet generates incompletely cleaved immature particles in DENV infection, facilitating host cell invasion via non-canonical pathways and exacerbating severe disease (30). Similarly, TMPRSS2 drives respiratory viral infections through its high expression in the bronchial epithelium (31), yet in prostate tissue, it is androgen-regulated and participates in epithelial differentiation and tissue remodeling. Aberrant TMPRSS2 expression may promote prostatic hyperplasia or carcinoma (31). These complexities necessitate intervention strategies with precisely regulated spatiotemporal specificities. A deeper layer of challenge stems from the essential physiological functions performed by these proteases themselves. Taking furin as an example, this critical precursor protein convertase participates in multiple core physiological processes. It regulates neuronal activity by processing β-nerve growth factor (β-NGF) (32); within the skeletal system, it is responsible for the maturation of the pro-hormone pro-osteocalcin (pro-OCN), modulating its activation and endocrine function, while also influencing the secretion of osteoblast-derived metabolic hormones (33). Similarly, TMPRSS2 plays a pivotal role across multiple tissues. In the kidneys, it participates in processing the epithelial sodium channel (ENaC) to regulate sodium reabsorption (34, 35). In the prostate, it is highly expressed and regulates prostatic fluid secretion and sperm function (36). And in the intestine, it maintains intestinal barrier integrity by cleaving the tight junction protein occluding (34). Collectively, these complex biological characteristics underscore that antiviral strategies targeting host proteases must establish a delicate balance between antiviral efficacy and physiological safety. Any intervention crucially requires precise avoidance of interference with their normal physiological functions.
This review focuses on the multifaceted roles of host proteases in viral infections, systematically outlining their core functions across four key stages. These stages include viral entry activation, replication and assembly, release and dissemination, and immune evasion. It also provides an in-depth assessment of innovative intervention strategies targeting these proteases. By analyzing the multiple functions of these proteases, this review aims to pioneer novel therapeutic approaches to suppress viral transmission and pathogenicity and provide a new perspective and theoretical basis for future viral therapeutic research.
2 Classification and function of host proteases
Host proteases serve as key effector molecules in virus-host interactions, exerting multifaceted regulatory roles via the viral life cycle through specific peptide bond hydrolysis. Their primary functions include (1) cleavage and activation of viral precursor proteins (e.g., glycoproteins, polyproteins) to confer infectivity or replication capacity; (2) modulation of host signaling pathways to alter cellular states for viral replication or immune evasion; and (3) remodeling of the extracellular matrix or membrane structures to create a favorable microenvironment for viral entry, assembly, or dissemination. Specifically, viruses have evolved a three-pronged strategy to manipulate this system: (i) targeted exploitation of tissue- or cell-specifically expressed protease isoforms [e.g., TMPRSS2 is highly expressed in the respiratory epithelium (37)]; (ii) induction of aberrant protease expression or activation in the infection microenvironment[(e.g., furin cleaves influenza virus HA0 (38, 39)]; (iii) evolution of specific cleavage motifs in viral proteins that are recognized by host proteases [e.g., HIV gp120 contains an REKR cleavage motif (40)]. Based on catalytic mechanisms, host proteases are classified into three major classes (serine proteases, cysteine proteases, and metalloproteases), in addition to aspartic proteases and threonine proteases. Their functional diversity originates from evolutionary divergence in active-site residues and substrate-binding pocket architectures.
2.1 Serine proteases
Serine proteases are characterized by a highly conserved Ser-His-Asp catalytic triad (41), as exemplified by TMPRSS2 (Figure 1A). The structural basis for their functional divergence lies in the diverse stereoconformations of substrate-binding pockets. For instance, the catalytic groove of TMPRSS2 precisely accommodates the receptor-binding domain (RBD) of coronavirus spike proteins (e.g., human coronavirus HKU1 (HCoV-HKU1)), inducing conformational changes that trigger membrane fusion (42). Conversely, furin recognizes multibasic cleavage motifs (e.g., Arg-X-X-Arg↓) within the polyproteins of diverse pathogens through its distinctive substrate-binding cleft, mediating their maturation and infectivity (40). During viral infections, serine proteases are extensively exploited to activate both surface glycoproteins (e.g., influenza HA, CoVs S, and HIV gp160) and internal precursor polyproteins—serving as critical rate-limiting steps in viral entry and maturation.
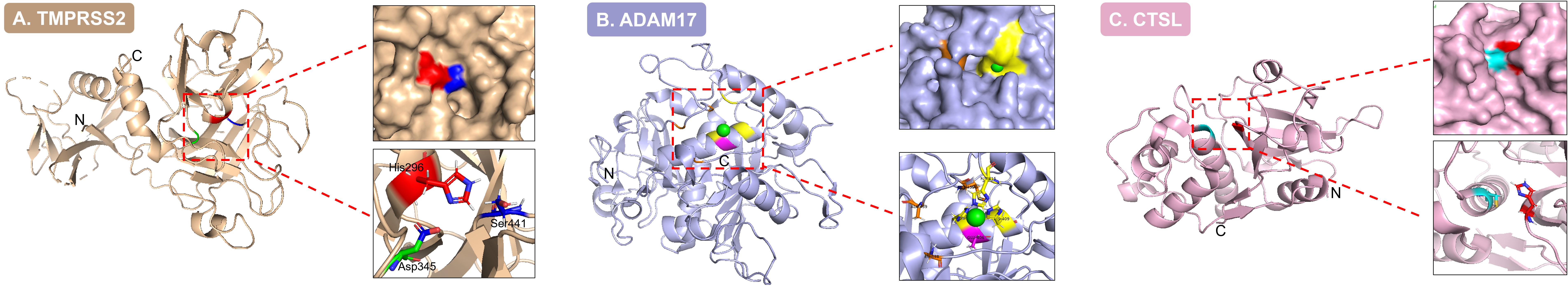
Figure 1. Three-dimensional structures and catalytic center characteristics of three types of core host proteases. (A). In the TMPRSS2 structure (serine protease; PDB: 7MEQ), key residues of the catalytic triad—His296 (red), Asp345 (green), and Ser441 (blue)—are positioned at the active site, demonstrating the conserved catalytic architecture characteristic of serine proteases. (B). The ADAM17 structure (metalloprotease; PDB: 9O54) reveals the catalytic Zn²+ ion (green sphere) coordinated by residues Glu406 (magenta), Met435, Ala439, and Leu348 (orange), illustrating the metal ion-dependent catalytic mechanism. (C). Within the CTSL structure (cysteine protease; PDB: 7W34), the catalytic dyad residues Cys25 (blue) and His163 (red) localize to the active pocket, underpinning the catalytic functionality of cysteine proteases.
2.2 Metalloproteases
Metalloproteases are characterized by an active center coordinated through specific metal ions (e.g., Zn²+) coordinated by surrounding amino acid residues (e.g., histidine, glutamate, or aspartate) (43), as exemplified by a disintegrin and metalloproteinase 17 (ADAM17) (Figure 1B). Their functional diversity arises from the structural plasticity of the metal-binding and catalytic domains. For example, matrix metalloprotease-2 (MMP-2) and matrix metalloprotease-9 (MMP-9) exemplify this diversity, utilizing catalytic domains to degrade collagen networks with high efficiency (44), while ADAM17 precisely cleaves transmembrane signaling molecules using its zinc-finger motif (45). Under physiological conditions, metalloproteases maintain homeostasis by regulating extracellular matrix remodeling, growth factor release, and the migration of inflammatory cells. Viruses disrupt this balance through dual mechanisms: (1) inducing aberrant overexpression of metalloproteases in the inflammatory microenvironment, such as HBV infection upregulating MMP-9 to promote liver fibrosis (46), or (2) hijacking proteolytic functions to facilitate dissemination routes, as evidenced by HIV exploiting metalloprotease-mediated degradation of extracellular matrix components to enhance cell-to-cell spread (47).
2.3 Cysteine proteases
Cysteine proteases feature a cysteine residue at their active site, functioning through a Cys-His catalytic dyad (48), exemplified by cathepsin L (CTSL) (Figure 1C). Their activity is tightly regulated by subcellular localization (e.g., pH, redox status). For instance, cathepsin B (CTSB) and CTSL activate viral fusion proteins in the acidic environment of endosomes (15), whereas caspase-3 executes apoptotic cascades in the cytoplasm (49). These enzymes dominate critical host processes—including lysosomal antigen processing, irreversible initiation of programmed cell death, and dynamic cytoskeletal remodeling—all precisely driven by the redox-sensitive thiol activity of their cysteine residues. Viruses hijack these mechanisms through spatiotemporally precise strategies. In the acidic endosomal microenvironment, activated CTSB/CTSL promotes viral entry [e.g., EBOV (14–17)]; in the cytoplasm, they regulate apoptosis-related proteases such as caspases to inhibit or promote cell death, creating a metabolic environment conducive to viral replication [e.g., adenovirus delays apoptosis by inhibiting caspase-3 activity (27)]. The mechanisms by which these microenvironmental molecular interactions determine infection outcomes will be dissected in subsequent mechanistic investigations.
In addition to the above, the host protease network also includes aspartic proteases and threonine proteases. Aspartic proteases utilize an active center formed by two conserved aspartic acid residues (Asp) (50), which enables the specific recognition and cleavage of viral polyprotein substrates in acidic microenvironments. For example, cathepsin D (CTSD) mediates the conformational rearrangement of HIV gp120, thereby facilitating direct interaction with coreceptors and enhancing the efficiency of viral membrane fusion (51). Threonine proteases employ threonine residues as nucleophilic attack centers to drive proteolytic cascades (52). The threonine hydrolase activity of the proteasome plays dual regulatory roles in the rotavirus life cycle, mediating viral capsid uncoating for genome release during invasion, and optimizing viral particle maturation through degradation of host restriction factors during assembly (53, 54). Through substrate-specific cleavage, these two protease classes cooperatively regulate key nodes of the viral replication cycle alongside previously described proteases.
From a molecular evolutionary perspective, the functional diversity of host proteases reflects a dynamic equilibrium forged through protracted virus-host coevolution. For example, the acquisition of furin cleavage sites (e.g., PRRA insertion in SARS-CoV-2) by coronavirus spike proteins likely reflects the adaptive evolution of viruses to exploit furin, which is highly expressed in the respiratory mucosa. In response, hosts have evolved defense mechanisms, such as the serine protease inhibitor (serpin) family, to continuously counteract the viral hijacking of proteases. This persistent evolutionary pressure drives the structural plasticity of protease substrate-binding domains, enabling specific viral families to utilize distinct protease subtypes to complete their life cycles, while also shaping the complexity of host defense networks (elaborated in Section 3) (Table 1).
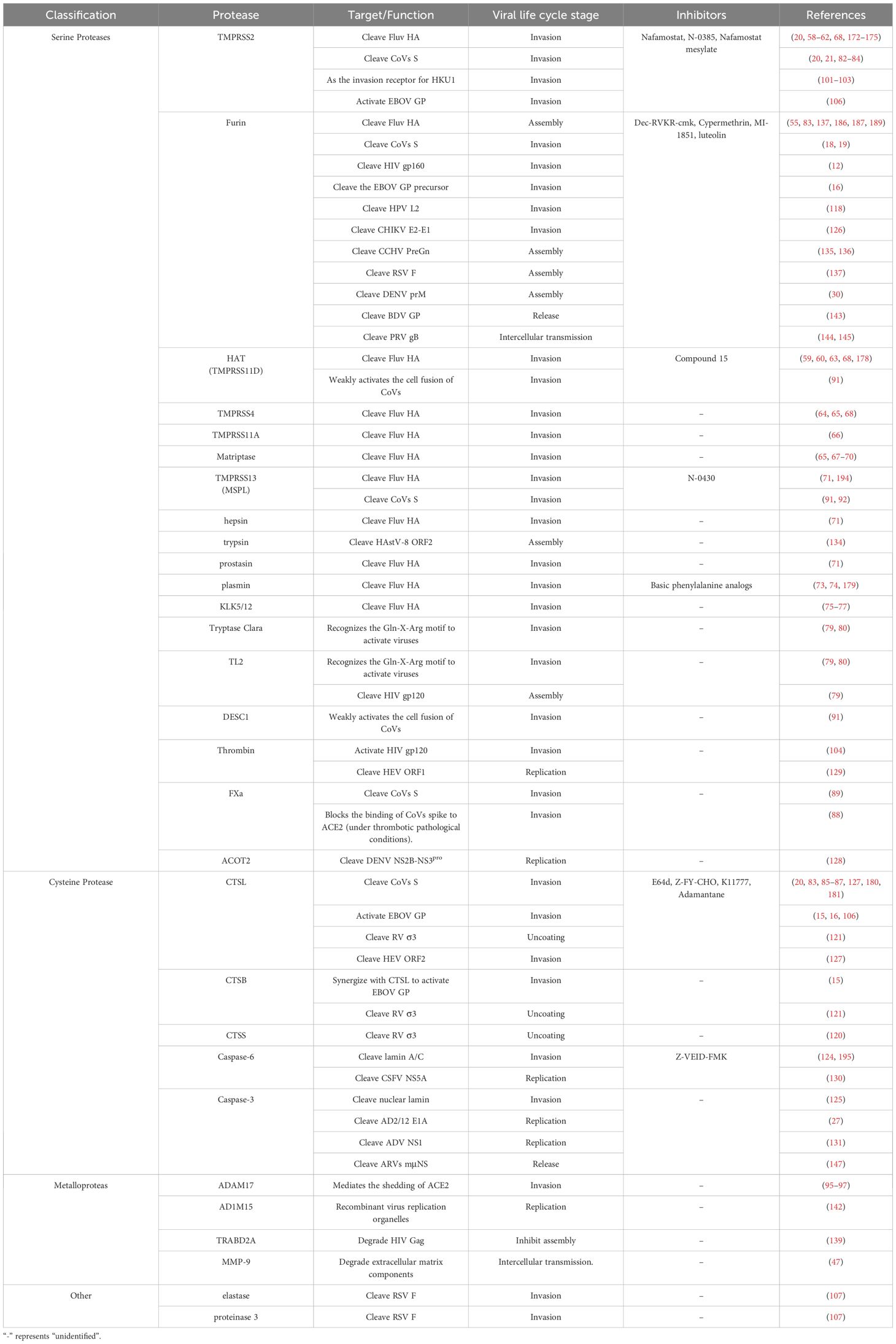
Table 1. Roles of host proteases in the viral life cycle.
3 Core mechanistic roles of host proteases in viral infection
3.1 Host protease regulatory networks in viral invasion
The process of viral invasion into host cells constitutes a dynamic interplay between pathogens and the host protease system. Conformational activation of viral surface glycoproteins, driven by host protease-mediated site-specific proteolytic cleavage, constitutes the essential initial step in the infection cascade. This regulatory strategy exhibits remarkable viral specificity and evolutionary adaptability.
3.1.1 Host protease-dependent activation mechanisms of influenza viruses
The influenza virus hemagglutinin (HA) glycoprotein functions as the principal mediator of viral entry, and its proteolytic processing determines infection efficiency, exhibiting significant subcellular localization specificity and viral subtype dependency. The HA precursor (HA0) must be cleaved by host proteases to generate HA1/HA2 subunits, thereby exposing the N-terminal fusion peptide (GLFGAIAGFIE) and enabling the virus to acquire membrane fusion capability—it is particularly emphasized that although uncleaved HA0 can bind to sialic acid receptors on host cells, it completely loses the ability to drive membrane fusion (38, 39, 55, 56). This critical cleavage event occurs at two stages of the viral life cycle. Furin primarily cleaves HA0 (especially in highly pathogenic H5/H7 subtypes) in the host cell’s Golgi apparatus during viral assembly and release, allowing newly formed virions to acquire fusion potential before release (55, 57); in contrast, TMPRSS2 and others complete the cleavage on the surface of host cell membranes during the viral entry stage (58–62). This activation process is orchestrated through cooperative actions within the host protease network, including human airway trypsin-like protease (HAT, also referred to as TMPRSS11D) (59, 60, 63), TMPRSS2 (58–62), TMPRSS4 (64, 65), TMPRSS11A (66), and matriptase (ST14 gene) (65, 67–70) (Figure 2A), which catalyze HA cleavage on the cell membrane surface to induce fusogenic conformational changes, thereby facilitating infection across diverse influenza subtypes. Notably, influenza subtypes exhibit distinct protease dependencies. TMPRSS2 is indispensable for HA activation of H7N9 and H1N1pdm in primary human bronchial epithelial cells, consistent with its high expression in respiratory mucosal epithelia. In contrast, TMPRSS4 dominates HA processing of H3N2 and influenza B viruses in murine alveolar type II epithelial cells due to its specific distribution in lung parenchymal cells (37, 71). In the absence of TMPRSS2, TMPRSS13 (alias MSPL, matriptase-like protease), hepsin, and prostasin maintain viral infectivity through compensatory cleavage (71). This segregation of protease function likely reflects the divergent expression profiles of type II transmembrane serine protease (TTSP) family members.
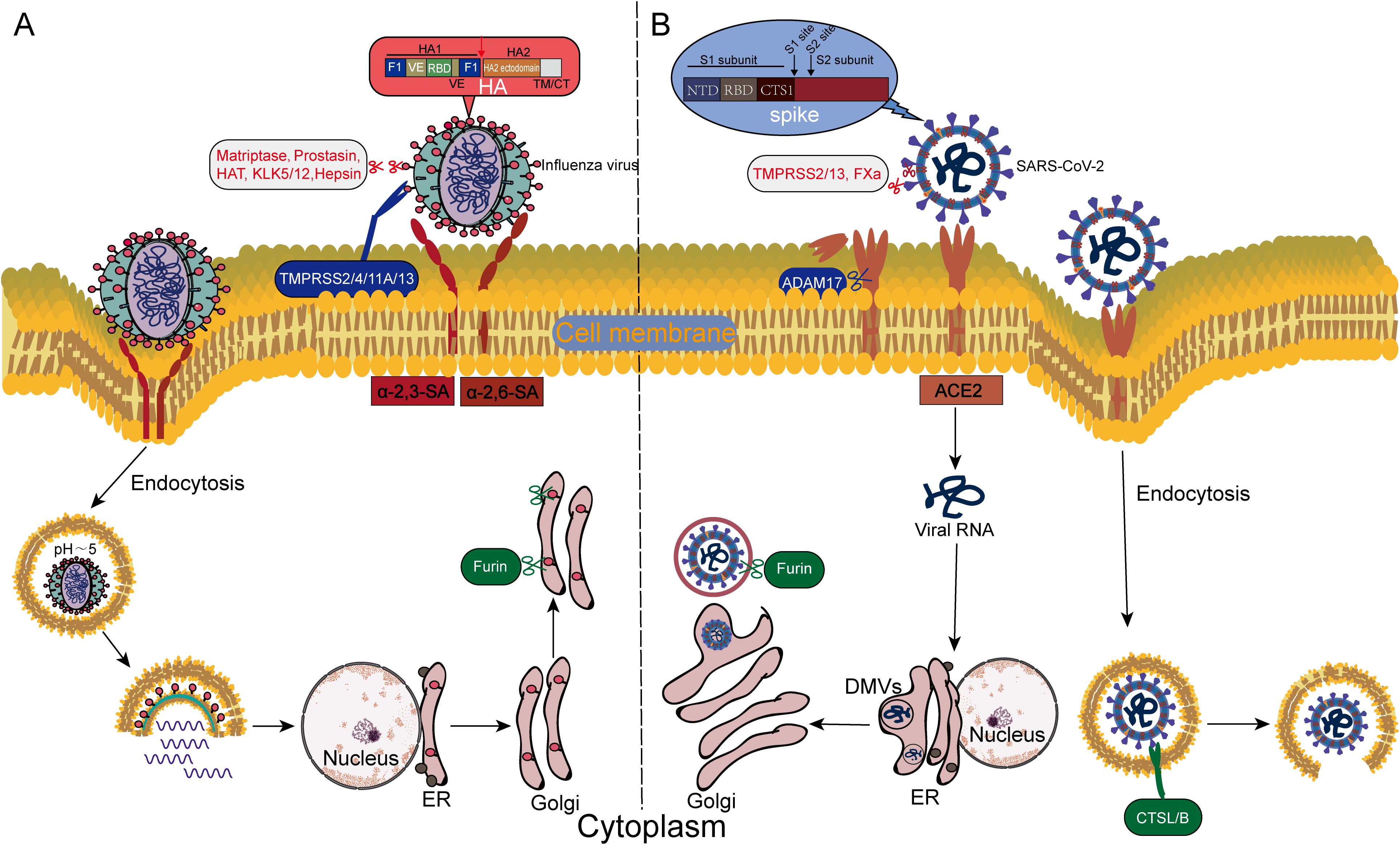
Figure 2. Differences in the host protease-dependent invasion mechanisms of influenza virus and SARS-CoV-2. (A) Influenza virus invades via the endosome-dependent pathway: Host proteases (e.g., TMPRSS2, cathepsins) cleave the HA, exposing the fusion peptide, which mediates the fusion of the viral envelope with the endosomal membrane in the acidic endosomal environment. (B) SARS-CoV-2 employs a dual-pathway strategy: On the plasma membrane surface, furin, TMPRSS2/13, and FXa cleave the S1/S2 or S2’ sites of the spike protein (S), directly triggering immediate fusion between the viral envelope and the host cell membrane; In the endosomal pathway, CTSL/B cleave the S protein, driving fusion between the viral envelope and the endosomal membrane.
Influenza C virus (ICV) employs a distinct mechanism, wherein its hemagglutinin-esterase (HE) fusion protein, which has dual functions in receptor binding and destruction, strictly relies on TMPRSS2 for activation on the cell membrane surface (72), exemplifying viral adaptive evolution to host systems. Moreover, secretory protease networks [e.g., plasmin (73, 74), kallikrein (KLK) (75, 76), and KLK12 (77)] can promote the spread of avian influenza viruses by specifically cleaving HA subtypes in the extracellular environment (such as respiratory secretions). Notably, TMPRSS13 plays a unique role in activating highly pathogenic avian influenza virus (HPAIV, e.g., H5N1/H7N9 subtypes) through its broad-spectrum cleavage capability (processing both monobasic and polybasic sites) and calcium-independent catalytic activity (78). Tryptase Clara and tryptase TL2 specifically recognize the consensus cleavage motif Gln (Glu)-X-Arg in influenza A and Sendai viruses to activate the viruses in the extracellular microenvironment of respiratory epithelial cells (79, 80). Conversely, some viruses (e.g., H1N1 subtypes) utilize endosomal proteases to accomplish HA cleavage after endocytosis (81).
3.1.2 Multi-layered host protease regulatory networks orchestrate coronavirus invasion
Coronavirus entry is governed by the spatiotemporal coordinated activation of the spike (S) glycoprotein through host protease interplay. In SARS-CoV-2, priming cleavage at the S1/S2 junction (multibasic PRRAR motif) by furin and TMPRSS2 within the Golgi apparatus enhances virion maturation and infectivity (18, 19, 82, 83). Subsequent activation diverges into dual pathways. At the plasma membrane surface, TMPRSS2-mediated cleavage of the S2’ site exposes the fusion peptide to drive immediate virus-host membrane fusion (20, 21, 84). Endocytosed virions rely on endosomal CTSL for S protein processing (85–87) (Figure 2B). Notably, host protease-mediated viral entry exhibits significant bidirectionality. Coagulation factor Xa (FXa) can inhibit viral entry and infection by cleaving specific domains of the S protein, thereby blocking its binding to the ACE2 receptor (88); however, it paradoxically enhances membrane fusion efficiency through S1/S2 or S2’ cleavage in thrombotic microenvironments alongside thrombin (89). Additionally, porcine epidemic diarrhea virus (PEDV) enters cells through clathrin-mediated endocytosis in synergy with serine proteases (90), suggesting evolutionarily conserved strategies among different coronaviruses in utilizing host factors.
Host proteases exhibit functional divergence in viral invasion. TMPRSS2 and TMPRSS13 play central roles in both virus-cell fusion and subsequent cell-cell fusion stages. In contrast, HAT and differentially expressed in squamous cell carcinoma gene 1 (DESC1) show significantly weaker activation efficiency in these two fusion processes (91). TMPRSS13 has been shown to specifically promote the membrane fusion of swine acute diarrhea syndrome coronavirus (SADS-CoV) (92), suggesting the potential regulatory properties of TTSP members in determining viral host range. Metalloproteases enhance viral attachment by cleaving coronavirus spikes and ACE2 receptors, while ADAM17 facilitates viral endocytosis and is associated with inflammatory damage by mediating ACE2 shedding (93–97). Evolutionary analyses reveal that the E484 mutation enables SARS-CoV-2 to acquire cross-binding capacity with the MERS-CoV receptor dipeptidyl peptidase 4 (DPP4, also known as CD26) (98, 99), a receptor plasticity potentially attributable to furin-mediated optimization of spike protein conformation. Studies indicate that the binding of DPP4 receptors to MERS-CoV and the infection process are species-dependent. Differences in glycosylation patterns of mouse DPP4 restrict viral infection, whereas DPP4 receptors from bats, camels, and humans can support efficient viral infection (100). Notably, TMPRSS2 can also act as a receptor to bind the RBD of the human coronavirus HKU1 spike protein, inducing its conformational changes to trigger fusion (101–103), highlighting the critical role of TTSPs in cross-species transmission.
3.1.3 Host protease utilization strategies in other viral families
The utilization of host proteases represents a universal strategy for both enveloped and non-enveloped viruses during invasion (Figure 3). Among enveloped viruses, furin, as the core enzyme mediating the cleavage of HIV gp160, recognizes the conserved R-X-K/R-R motif in its sequence to cleave gp160 into gp120 and gp41, thereby activating viral invasion capability (12); meanwhile, thrombin enhances virus-induced cell fusion by activating HIV gp120 (104). During the initiation stage of cell fusion in placental development, the human endogenous retroviruses (HERVs) envelope protein Syncytin-1 similarly relies on furin cleavage to activate its fusion function, while Syncytin-2 maintains fusion activity via processing by the proprotein convertase subtilisin/kexin type 7 (PCSK7) (105). Filoviruses, such as EBOV, employ a proteolytic cascade activation strategy for their glycoprotein (GP). Furin mediates initial cleavage of the GP precursor in the secretory pathway, and endosomal CTSB/CTSL further trim the GP1 subunit to expose the human endosomal receptor Niemann-Pick C1 (NPC1) receptor-binding domain (14–17). Studies have confirmed that TMPRSS2 and CTSL can form a redundant mechanism to compensate for furin functional defects (106), highlighting the complexity of host protease networks. The activation of the fusion (F) protein of paramyxoviruses universally depends on host proteases. Respiratory syncytial virus (RSV) requires elastase and proteinase 3 for F protein cleavage (107), whereas human parainfluenza virus 3 (HPIV3) and mumps virus (MuV) utilize trypsin-like proteases or furin (108, 109). In-depth research reveals that TMPRSS2 and TMPRSS13 in the lung epithelium can directly cleave the HPIV3 F protein, regulating the release efficiency of infectious virions (108). Notably, furin cleavage sites exhibit cross-species conservation within the Paramyxoviridae family. The F proteins of HPIV3, HPIV5, virulent Newcastle disease virus (NDV) strains, measles virus (MV), and RSV all contain such sites (110–115), implying their universal value as key molecular switches. Structural conservation extends to fusion mechanisms. The post-fusion core conformations of enveloped viral fusion proteins—including SARS S, murine hepatitis virus (MHV) S, EBOV GP2, influenza virus HA2, HIV gp41, and paramyxovirus F2—exhibit striking homology (116), revealing deep evolutionary convergence across viral families. This conservation reflects a shared evolutionary strategy, where host protease activation (e.g., furin cleavage, cathepsin trimming) serves as a molecular switch that triggers conformational rearrangements from metastable pre-fusion states to stable post-fusion cores, ensuring spatiotemporally regulated membrane fusion across viral families.
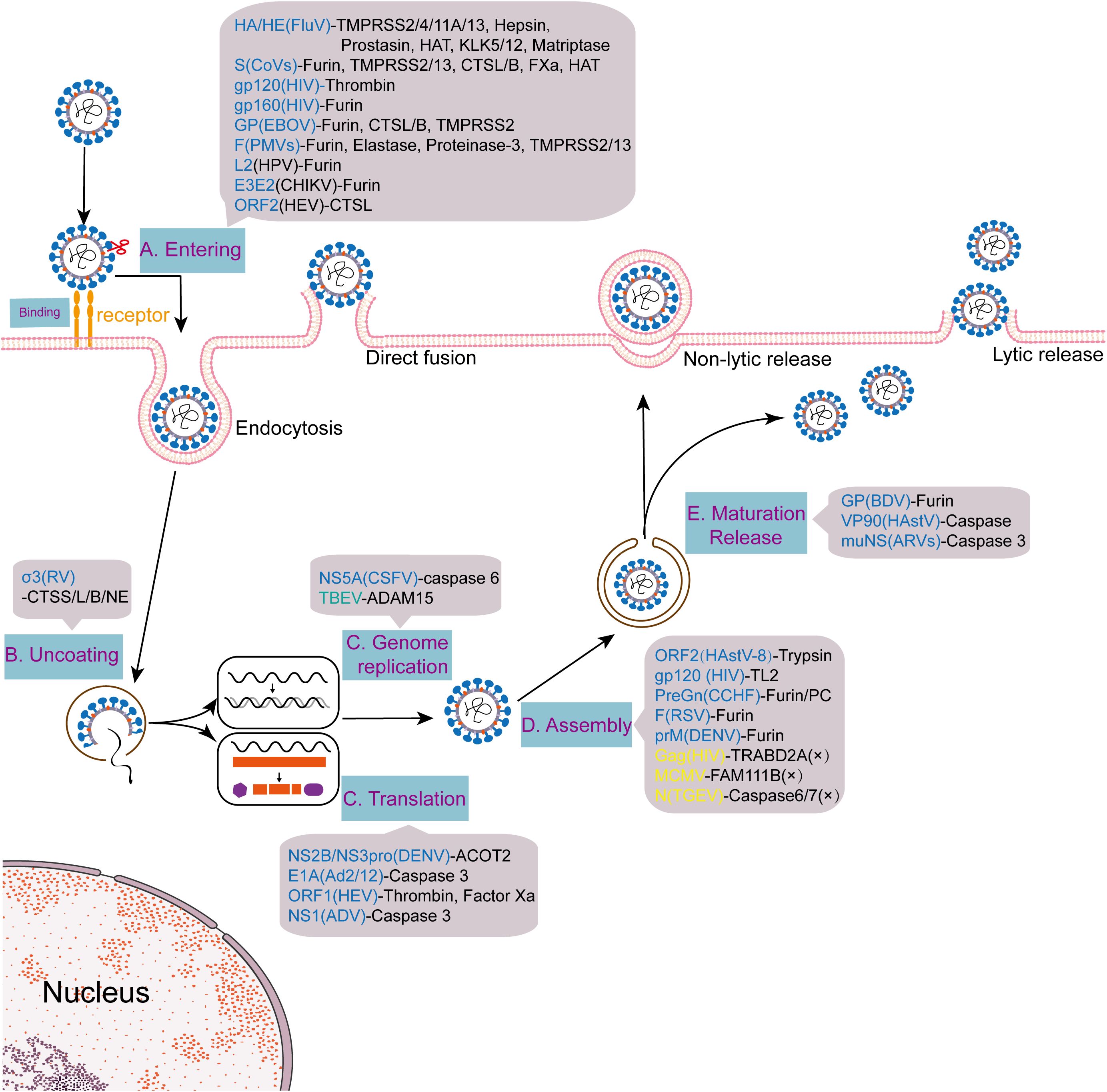
Figure 3. Multinodal regulation of viral life cycles by host proteases. Viruses such as influenza and SARS-CoV-2 hijack host proteases (e.g., TMPRSS2, furin) during the stages of entry, replication, assembly, and release, achieving infection through a multi-node regulatory pattern.
Among non-enveloped viruses, HPV relies on furin-mediated cleavage of the minor capsid protein L2. The released C-terminal peptide (L2CT) not only mediates viral genome escape from endosomes but also recruits the nuclear transport factor karyopherin alpha2 (KPNA2) to guide DNA across the nuclear membrane barrier (13, 117). While L2 cleavage-deficient mutants do not affect viral attachment and endocytosis, they result in complete loss of infectivity due to failed nuclear import, indicating that furin-mediated cleavage of L2 is a critical rate-limiting step in the HPV infection cycle (118). Within the Reoviridae family, cleavage of the rotavirus VP4 spike protein by trypsin significantly enhances its membrane fusion capacity (119). For reoviruses, the σ3 capsid protein requires cleavage by cathepsin S (CTSS) or CTSL/B to facilitate viral uncoating and genome release (120, 121). Additionally, neutrophil elastase can promote rotavirus uncoating and infection in U937 promonocytes, substituting for CTSL to mediate a non-canonical infection pathway (122). Enteroviruses may be inactivated through conformational changes induced by serine proteases, such as subtilisin A, via capsid binding or direct cleavage, causing viral disintegration (123). Notably, polyomavirus SV40 employs a distinct strategy in quiescent cells. It activates host caspase-6 to cleave nuclear lamin A/C, inducing transient nuclear membrane deformation and dephosphorylation. This process establishes a locally softened “nuclear membrane window” to facilitate direct transport of the viral genome from the endoplasmic reticulum into the nucleus (124). This finding reveals a novel pathway by which viruses utilize host proteases to remodel the nuclear physical barriers. Similarly, the parvovirus minute virus of mice (MVM) induces caspase-3-mediated cleavage of the nuclear lamina to form physical pores, promoting capsid nuclear entry (125). Together, these findings highlight the innovative evolutionary adaptations of non-enveloped viruses in the mechanisms of nuclear membrane traversal.
Alphaviruses, such as chikungunya virus (CHIKV), rely on furin-mediated processing of their envelope protein precursor E3E2 to form functional E2-E1 heterodimers. The receptor-binding activity of these heterodimers, together with low pH-induced conformational instability, collectively drives the viral membrane fusion process. Geographic isolation has driven the evolutionary divergence of protease utilization. African strains specifically depend on membrane-bound cilia proteases and PC5B for cleavage of E3E2 at the HRQRR64/ST site, whereas Asian strains achieve cleavage at the RRQRR64/SI site via membrane-bound/soluble cilia proteases, PC5A, PC5B, and PACE4. Notably, PC7 and SKI-1 lack cleavage activity against both strain types (126), reflecting the adaptive evolution of viruses to regional host microenvironments. Additionally, hepatitis E virus (HEV) entry into hepatocytes depends on CTSL-mediated processing of viral particles and cleavage of the glycosylated ORF2 protein (127), confirming the universal role of cysteine proteases in the invasion of enveloped viruses.
3.2 Dynamic regulatory mechanisms of host proteases in viral replication and assembly
Host proteases precisely regulate viral replication and assembly through specific cleavage events, exhibiting multidimensional coordination and dynamic evolutionary characteristics. During the viral replication phase, host proteases play a central role in regulating key processes, such as the activation of viral precursor proteins and the formation of replication complexes (Figure 3). For instance, in RNA viruses, the dengue virus NS3 serine protease requires cooperation with the host serine protease acyl-CoA thioesterase 2 (ACOT2) to cleave polyproteins for functional replication complex formation (128). HEV initiates genome replication through thrombin-mediated cleavage at conserved sites of the ORF1 polyprotein (129). The caspase-6 cleavage motif (DTTD/272) in the non-structural protein NS5A of classical swine fever virus (CSFV) further confirms its regulatory role in viral replication (130). In DNA virus systems, caspase-mediated cleavage events exhibit bidirectional regulation of viral replication. Cleavage of adenovirus 2/12 (Ad2/12) early region 1A (E1A) protein by caspase-3 results in the loss of transcriptional activation function, impairing the transcriptional program necessary for efficient replication (27). In contrast, removal of the nuclear localization sequence from the NS1 protein of Aleutian mink disease parvovirus (AMDV) by caspase-3 promotes the cytoplasmic transport of ribonucleoprotein complexes, facilitating replication-related processes (131). These findings reveal the multi-target regulatory characteristics of host proteases in the viral replication process.
During virus assembly, host proteases primarily exert precise regulation by mediating the modification and maturation of viral structural proteins. The nucleocapsid protein (N) of transmissible gastroenteritis virus (TGEV) and IAV loses its genome-binding capacity after caspase-6/7 cleavage, resulting in a dramatic decrease in infectious virus particle yield (132, 133); the human astrovirus type 8 (HAstV-8) ORF2 polyprotein is specifically cleaved by trypsin to generate functional fragments that participate in capsid assembly and replication, respectively (134). Notably, protease processing strategies are viral species-specific. HIV-1 promotes the exposure of the gp41 fusion domain and the correct assembly of Env proteins through TL2 serine protease-mediated cleavage of the gp120 V3 loop (79). While furin/PC protease processing of the Crimean-Congo hemorrhagic fever (CCHF) glycoprotein precursor produces the GP38 glycoprotein, which may optimize the viral assembly environment through membrane remodeling mechanisms (135, 136). Furin cleavage of the RSV F protein is not essential for its transport but can significantly enhance viral particle assembly efficiency (137), highlighting their precise regulation of viral morphogenesis. In addition, caspase-3 mediates the nucleocytoplasmic transport of the influenza virus ribonucleoprotein complex (RNP), and its inhibition leads to RNP retention in the nucleus and triggers assembly defects. The enzyme cleaves the nuclear lamina protein Lamin A/C via a non-apoptotic pathway, remodels the nuclear membrane structure to facilitate RNP transport to the cytoplasm, and provides key components for viral particle assembly (138), further confirming the multifaceted regulatory mechanism of host proteases in viral morphogenesis.
The host-virus interaction network at the protease level is characterized by dynamic interplay and coevolution. Host factors, such as TRAB domain-containing protein 2A (TRABD2A), can inhibit viral assembly by degrading the Gag protein of HIV-1 (139), while the primate-specific restriction factor FAM111B inhibits the replication of mouse cytomegalovirus (MCMV) in human cells by enriching in viral replication regions—a restriction stemming from the fact that MCMV has not evolved strategies against family with sequence similarity 111 member B (FAM111B) in its natural hosts (rodents) (140). Studies on cross-species transmission reveal that viruses can actively utilize host proteases to break through barriers. The conserved cleavage of HEV pORF1 by thrombin and FXa is a key basis for its ability to cross host boundaries (141). Tick-borne encephalitis virus (TBEV) reorganizes the membrane system by relocating host ADAM15 protease to its replication region, thereby optimizing its own replication environment (142). Such adaptive strategies frequently drive systematic mutations in the cleavage sites of viral proteases.
3.3 Diverse regulatory mechanisms of host proteases in viral release and transmission
Host proteases profoundly enhance viral particle release and transmission efficiency through precise regulation of viral maturation and microenvironment remodeling (Figure 3). The core function of furin is evolutionarily conserved across viral families during terminal maturation. For example, furin-mediated cleavage of HIV gp160 enhances viral particle infectivity and induces conformational rearrangements to evade neutralizing antibodies (28, 29); flaviviruses (e.g., DENV and ZIKV) require furin cleavage of prM to M protein for mature particle formation, though incompletely cleaved immature particles retain infectivity via non-canonical entry pathways that exacerbate disease severity (30). While Borna disease virus (BDV) glycoprotein (GP, encoded by ORF-IV) strictly depends on site-specific furin cleavage at Arg249 to maintain bioactivity (143). These collective mechanisms underscore how proteolytic processing fine-tunes viral dissemination strategies across diverse species.
Viral cell-to-cell transmission involves a more extensive proteolytic regulatory network. Although furin cleavage of pseudorabies virus glycoprotein B (gB) is dispensable for in vitro viral replication, it remains critical for mediating membrane fusion and syncytium formation (144, 145). MMP-9 significantly enhances HIV cell-to-cell transmission by degrading extracellular matrix components (47). Additionally, caspase family members play pivotal roles in facilitating viral release. They cleave the human astrovirus capsid precursor VP90 to form mature capsids, thereby promoting viral release (146); while they dissolve cytoplasmic inclusion bodies maintained by Avian reoviruses (ARVs) mμNS protein to expel mature particles (147). Collectively, these diverse mechanisms highlight the intricate deep coevolutionary relationship between viruses and the host protease system.
3.4 Host protease regulatory networks in immune response and evasion
Host proteases construct multidimensional regulatory networks spanning molecular cleavage to systemic immunity during viral immune responses and evasion strategies (Table 2). Viruses achieve immune evasion by hijacking the protease activity. For instance, influenza viruses employ TMPRSS2 not only to enhance viral membrane fusion efficiency, but also to promote vascular permeability by activating the “influenza virus-cytokine-trypsin” cycle. The upregulated trypsin and pro-inflammatory cytokines exacerbate tissue destruction and immune suppression, enabling the virus to evade immune clearance and continue to replicate (148). Similarly, cathepsin G (CTSG) recruits monocytes/macrophages to inflammatory sites during HIV-1 infection and heightens their viral susceptibility, establishing a positive feedback loop (25). Conversely, the host has evolved protease-based antiviral defenses—neutrophil serine proteases (NE/PR3/CTSG) directly cleave the SARS-CoV-2 spike protein to block viral entry (149), while myeloid-specific serine proteases interfere with NF-κB activation by processing its p65 subunit, thereby inhibiting critical HIV replication processes (24). This bidirectional protease warfare underscores the evolutionary arms race at the host-pathogen interface.
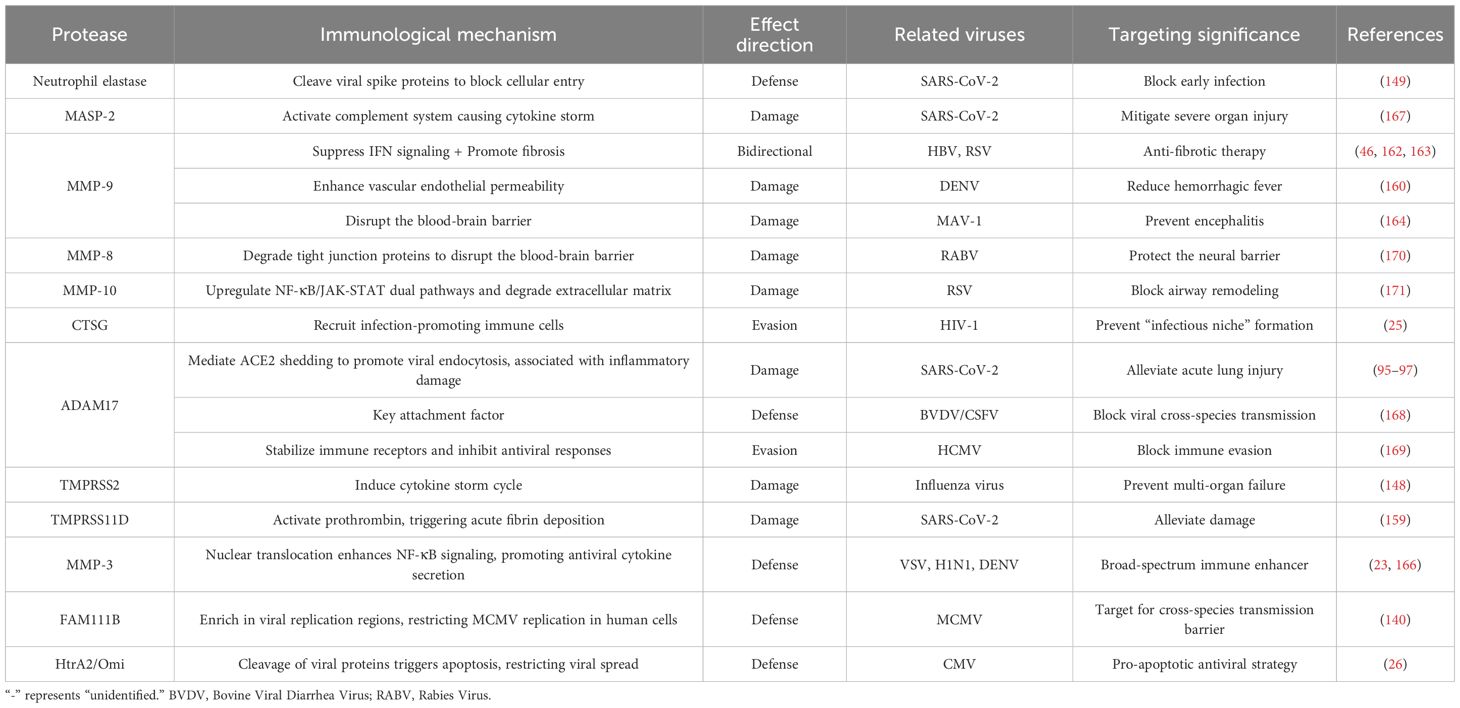
Table 2. Immune regulatory functions of host proteases in viral infections.
The precise regulation of host protease activity by host factors constitutes a critical mechanism in antiviral defense. Interferon-induced guanylate binding proteins (GBPs, such as GBP2 and GBP5) inhibit furin and PCSK family activity, impeding the maturation of viral envelope glycoprotein precursors, including HIV-1 gp160, and significantly reducing the infectivity of various viruses, including HIV-1, ZIKV, MV, and IAV (150). Alpha-soluble NSF attachment protein (α-SNAP) binds to the P-domain of furin, inhibiting cleavage of the SARS-CoV-2 spike protein and other furin-dependent viral glycoproteins (151). Members of the serine protease inhibitor superfamily (SERPIN) also play essential roles, alpha (1)-antitrypsin (A1AT) inhibits TMPRSS2 and ADAM17, blocking SARS-CoV-2 spike protein activation and ACE2 shedding (152–154), while plasminogen activator inhibitor-1 (PAI-1) regulates fibrinolysis and additionally inhibits the enzymatic activity of proteases including FXIIa and TMPRSS2 (155). Furthermore, cystatin C (CST3) competitively inhibits the activity of CTSB/CTSL through its N-terminal region, while its dimer form enhances CTSB activity by binding to a structure-specific allosteric pocket of CTSB (156). Interleukin-1β (IL-1β) activates ADAM17 through phosphorylation (157). Collectively, these regulatory networks of host factors over protease activities profoundly influence viral infection processes.
The functions of host proteases frequently exhibit tissue- and cell-compartment specificity, and profoundly influence systemic immune responses. In renal cells, SARS-CoV-2 evades the inhibitory effect of the host restriction factor nuclear coactivator 7 (NCOA7) via a TMPRSS2-mediated non-endosomal pathway (158). Conversely, when SARS-CoV-2 infects human bronchial epithelial cells, it induces ST14/TMPRSS11D to activate prothrombin, triggering acute fibrin deposition (159). At the systemic level, SARS-CoV-2 activates the NETs-PAD-4 pathway to induce lung epithelial cell death (22), whereas dengue virus is directly linked to imbalances in the coagulation-fibrinolytic system through a metalloproteinase-mediated vascular leakage mechanism (160). Moreover, the degree of coagulation and fibrinolytic activation induced by it is positively correlated with disease severity (161).
The matrix metalloproteinase (MMP) family exhibits complex and differentiated functions in viral immunomodulation. In HBV infection, MMP-9 promotes viral replication and hepatic fibrosis by inhibiting interferon signaling (46). RSV infection efficiently stimulates MMP-9 expression in vivo and in vitro (162), while disruption of the MMP-9/TIMP-1 balance drives airway remodeling—a key pathogenic feature of chronic pulmonary fibrosis (163). In a neuroinvasive model, mouse adenovirus (MAV-1) activates microglial MMP-2/MMP-9 to disrupt the blood-brain barrier, representing a critical pathological mechanism underlying encephalitis development (164). Studies on MHV infection further revealed that increased viral replication during lethal infection is closely associated with significantly elevated expression levels of MMPs, TIMPs, and chemokine genes (165). Conversely, MMP-3 exerts broad-spectrum antiviral activity against vesicular stomatitis virus (VSV), H1N1, and HSV-1 through NF-κB signaling potentiation via nuclear translocation, while simultaneously enhancing anti-dengue immune responses (23, 166).
Complement system regulation represents another critical battleground for viral immune evasion. Aberrant interactions between the mannose-binding lectin-associated serine protease 2 (MASP-2) and the SARS-CoV-2 N protein drive complement hyperactivation, fueling cytokine storms and multiorgan damage—a mechanism particularly prominent in severe COVID-19 (167). Conversely, the high-temperature requirement protein A2 (HtrA2/Omi) effectively limits cytomegalovirus (CMV) spread by triggering apoptotic pathways through cleavage of key viral or host proteins (26). Long-term host-virus coevolution has forged dynamic equilibria in proteolytic cleavage sites. ADAM17, a pivotal immunoregulatory node, plays essential roles in host defense against pestiviruses (168), while being targeted by cytomegaloviruses to remodel the cell surface proteome (169). Neuroinvasive viruses exploit MMP-8 to degrade tight junctions in the blood-brain barrier (170), whereas RSV infection induces MMP-10 expression in nasal epithelial cells and modulates the immune microenvironment through NF-κB/JAK-STAT crosstalk (171). These mechanisms collectively demonstrate how viruses achieve immune microenvironment remodeling via multidimensional regulation of proteolytic networks.
4 Antiviral intervention strategies targeting host proteases: from single-target inhibition to multidimensional synergistic regulation
Compared with the drug resistance challenges posed by high-frequency mutations in viral genomes, host proteases, due to the high genetic stability of their encoding genes, emerge as highly attractive targets for developing host-directed antiviral drugs (HADs). This approach significantly mitigates risks of viral escape mutations. Furthermore, inhibitors targeting host proteases generally exhibit broad-spectrum antiviral potential, providing a feasible approach to combat multiple viral infections. With the deepening understanding of the viral infection complexity and immune evasion mechanisms, intervention strategies targeting these critical regulatory nodes in the viral life cycle are dynamically evolving from traditional single-target inhibition to multidimensional synergistic regulation.
4.1 Continuous advancement in single-target inhibition research
Currently, research on single-target inhibitors targeting key host proteases continues to deepen. Small-molecule inhibitors remain the primary focus due to their favorable drugability and high maturity in development. In the field of targeting transmembrane serine proteases, the TMPRSS2 inhibitor nafamostat (Figure 4A) blocks 93% of SARS-CoV-2 plasma membrane invasion but exhibits limited inhibitory effects on TMPRSS4-dependent MERS-CoV (20, 172). The new-generation inhibitor N-0385, with low nanomolar potency, effectively inhibits the invasion of variants, including Omicron (173, 174). While nafamostat mesylate can reduce viral load in murine lungs (175), its clinical efficacy is limited by rapid cleavage and inactivation by TMPRSS11D (176). Other broad-spectrum serine protease inhibitors, such as 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF) and N-alpha-tosyl-L-phenylalanyl chloromethyl ketone (TPCK), can inhibit RSV infection, among which AEBSF acts primarily at the early stage of viral entry (177). Structurally optimized HAT serine protease inhibitors (e.g., compound 15, Ki = 15 nM) enhance selectivity through novaricine modification, thereby effectively inhibiting influenza virus replication (178). Additionally, basic phenylalanine analogs reduce the titers of West Nile virus (WNV) and DENV by 10,000-fold via plasmin inhibition (179).
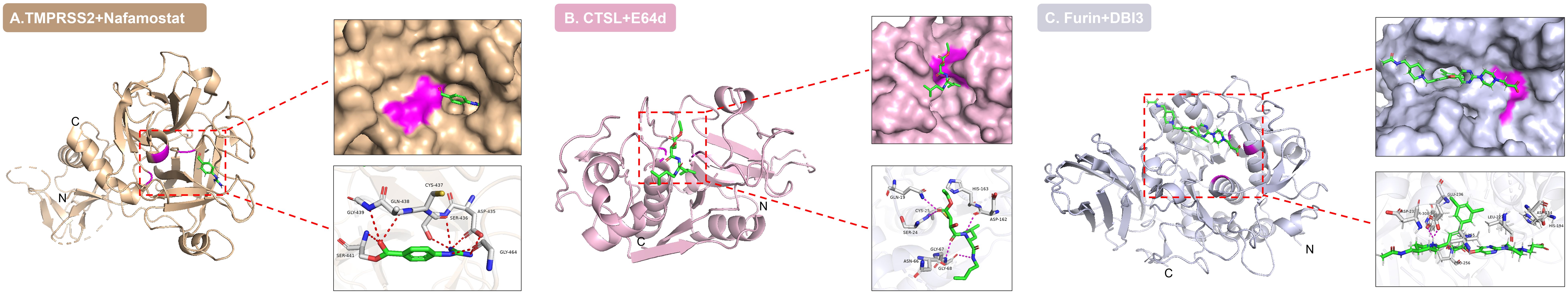
Figure 4. Active site binding modes of host protease-inhibitor complexes. Three-dimensional structures of (A) TMPRSS2-nafamostat (PDB: 7MEQ), (B) CTSL-E64d (PDB: 8HET), and (C) furin in complex with dichlorophenylpyridine-based inhibitor 3 (DBI3, PDB: 7QXY) are shown. Inhibitors (green) targetedly insert into the active sites of proteases, forming key interactions with catalytic residues and surrounding amino acids (e.g., Ser441 in TMPRSS2, Cys25 in CTSL, Asp154 in furin) via hydrogen bonds and hydrophobic interactions. The catalytic centers are highlighted in magenta.
Classical cysteine protease inhibitors targeting CTSL demonstrate broad-spectrum antiviral efficacy. In vitro models, inhibitors E64d (Figure 4B) and Z-FY-CHO effectively suppress SARS-CoV-2 pseudovirus infection (20, 83, 180, 181), while the broad-spectrum inhibitor K11777 exhibits potent inhibitory activity against HEV, with an EC50 of approximately 0.02 nM (127). In vivo, rotavirus capsid disassembly strictly depends on CTSL, and treatment with the inhibitor Z-Phe-Tyr(t-Bu)-diazomethyl ketone causes drastic viral yield reduction (121). Additionally, amantadine, an anti-influenza drug, blocks SARS-CoV-2 infection by inhibiting CTSL activity (87), while the natural product gallinamide A and its analogs also exhibit potent antiviral activity (182).
Furin inhibitor development has achieved significant advances across multiple fronts. The competitive inhibitor dec-RVKR-cmk can block F protein cleavage and viral budding of RSV (137), while exerting anti-SARS-CoV-2 activity by inhibiting spike protein cleavage and syncytium formation (183), though it is ineffective against filoviruses (184). This compound additionally suppresses flavivirus release without affecting RNA replication (185). In terms of allosteric inhibitors, cypermethrin exhibits broad-spectrum activity against drug-resistant SARS-CoV-2 by binding to a novel allosteric pocket (186), while the reversible inhibitor MI-1851 reduces viral load by 190-fold (83, 187). It is worth noting that protease inhibitors designed based on the (3,5-dichlorophenyl) pyridine skeleton (Figure 4C) exhibit high potency and antiviral activity against SARS-CoV-2 at the cellular level (188), providing new insights for broad-spectrum antiviral treatment. The natural product luteolin inhibits furin in a non-competitive manner and significantly reduces DENV viremia in vivo (189). Emerging approaches explore novel paradigms. Targeting the off-state of furin opens new avenues for the design of selective inhibitors (190). Decanoyl-RVKR-CMK effectively blocks E3E2 precursor cleavage by CHIKV (30). And furin conformation provides new opportunities for structure-based drug discovery (186), demonstrating the feasibility of developing customized inhibitors for specific viruses.
In addition to small-molecule inhibitors, peptide-based compounds, endogenous regulatory factors, and biologics serve as vital complements to single-target inhibition strategies due to their targeting specificity and biocompatibility advantages. In peptide inhibitor research, polyarginine repeat sequences function as competitive inhibitors of furin substrate cleavage, effectively inhibiting HIV infection by blockade of gp160 protein processing (191). Peptides P9 and P9R significantly reduce SARS-CoV-2 viral load in hamster models by impairing CTSL activity (192, 193). The TMPRSS13 peptidomimetic inhibitor N-0430 blocks SARS-CoV-2 pseudovirus entry (194), while the caspase-6 inhibitor Z-VEID-FMK alleviates pathological damage in SARS-CoV-2 and MERS-CoV animal models (195). Recombinant applications of endogenous inhibitors demonstrate substantial progress. Interferon-induced GBP5 protein inhibits furin activity, markedly reducing infectivity of multiple pathogens, including HIV-1, Zika virus (ZIKV), MV, and IAV (150). Serpin family B member 8 (Serpin B8, also known as PI8 and CAP2) binds to and inhibits the proprotein convertase furin (28). Furthermore, α-SNAP suppresses furin-dependent viral glycoprotein cleavage through binding to the furin P-domain (149). In the field of biologics, nanobodies exhibit exceptional advantages due to their high specificity. The anti-TMPRSS2 nanobodies inhibit the enzymatic activity of TMPRSS2 and hinder HKU1 pseudovirus entry using S441A TMPRSS2 (101). Dromedary heavy-chain-derived nanobodies specifically inhibit the catalytic activity of furin, blocking its cleavage of two critical substrates, transforming growth factor beta (TGFβ) and glypican-3 (GPC3) (196).
4.2 Translational breakthroughs in inhibitor synergy strategies
Multi-target synergistic strategies are overcoming the limitations of single-inhibitor therapies. Clinical studies have shown that the TMPRSS2 inhibitor N-0385, when combined with the antiviral drugs remdesivir or nirmatrelvir, exhibits broad-spectrum synergistic activity against Omicron subvariants (174). In chronic hepatitis B treatment, entecavir coupled with furin inhibitors concurrently suppresses viral replication and hepatitis B e antigen (HBeAg) secretion (197). Spironolactone enhances antiviral effects by antagonizing TMPRSS2/ADAM17 to reduce soluble ACE2, synergizing with DPP-4 inhibitors to improve clinical outcomes in COVID-19 patients (198). Additionally, xanthan gum combined with camostat significantly enhances anti-influenza virus potency (199). Mechanistic studies further confirmed that non-toxic furin inhibitors combined with TMPRSS2 inhibitors block 95% of lung cell infections (200), demonstrating the translational potential of inhibitor synergy strategies in multistep blockade of viral invasion.
4.3 Innovative waves in multi-target drug development
Dual- and multi-target therapeutics are spearheading novel antiviral strategies. Compound BAPA exhibits an EC50 of 0.3 μM against H1N1 by inhibiting HAT/TMPRSS2 (201). The tri-targeting peptidomimetic MM3122 simultaneously inhibits TMPRSS2, matriptase, and hepsin, maintains sub-nanomolar potency against the SARS-CoV-2 EG.5.1 variant, and significantly attenuates pulmonary edema in mice (202). Delivery system innovations propel the development of bispecific compound 212-148, which simultaneously inhibits TMPRSS2 and CTSL/CTSB (203), with nanoerythrocyte carriers substantially enhancing delivery efficiency (204). Diazoxide inhibits TMPRSS2/furin (IC50=1.35/13.2 μM), while compound MI-1148 blocks transmission of highly pathogenic avian influenza (HPAI) and canine distemper virus by targeting PC1/3 (205). Notably, the mechanisms of action of protease inhibitors nafamostat and camostat may extend beyond TMPRSS2 inhibition itself, involving coagulation cascade-induced cleavage of spike proteins. Given the centrality of anticoagulation management in COVID-19 therapy, early intervention may provide synergistic benefits by blocking viral entry (89).
Structure-guided design has achieved pivotal breakthroughs. The α-ketoamide inhibitors 14a/14b exhibit potent broad-spectrum anti-coronaviral activity through covalent binding to CTSL and calpain-1 (CAPN1), achieving exceptional potency against SARS-CoV-2 variants (EC50 as low as 0.80 nM) (206). The natural product omicsynin B4 demonstrates pan-coronaviral activity against human coronavirus 229E (HCoV-229E), human coronavirus OC43 (HCoV-OC43), and SARS-CoV-2 prototype/variants by dual blockade of CTSL/TMPRSS2 (207). At the level of respiratory protease regulation, influenza HA activation mediated by human eosinophils and DESC1 (but not TMPRSS11A) is specifically inhibited by hepatocyte growth factor activator inhibitor 1 (HAI-1) (66). The endogenous regulator serine protease inhibitor Kazal‐type 6 (SPINK6) inhibits HAT/KLK5 to restrict influenza virion maturation (208), while dichlorobiphenyl-containing matriptase inhibitors achieve ultrahigh potency (Ki < 3 nM) through chemical optimization, demonstrating exceptional thrombin selectivity and concentration-dependent inhibition of H9N2 viral replication in MDCK(II) cells (209). Among matriptase/TMPRSS2 inhibitors evaluated by Gamba, D. et al., MI-463 and MI-1900 exhibit antiviral effects against H1N1/H9N2 at concentrations of 20-50 µM, suggesting that they block viral entry by inhibiting host protease-mediated cleavage (210). The oral dual-target drug olgotrelvir, which simultaneously inhibits SARS-CoV-2 Mpro and CTSL, has emerged as a new paradigm for clinical translation (211).
4.4 Translational challenges and cutting-edge strategies
Despite extensive development of host protease inhibitors demonstrating antiviral potential in preclinical models, few have successfully transitioned to clinical application. The current translational bottlenecks primarily stem from three key challenges. First, host compensatory escape—viruses can not only bypass inhibition by activating functionally redundant host proteases, such as SARS-CoV-2 switching from TMPRSS2-dependent entry to CTSL-mediated invasion pathways (85–87); they can also utilize their own encoded proteases to compensate for critical functions. For instance, HCV relies solely on its NS3/4A serine protease with NS4A as a cofactor to independently cleave viral polyproteins (212). Similarly, DENV requires its NS3 protease—an essential component for nonstructural protein hydrolysis—which functions with its own NS2B cofactor (213). Meanwhile, SARS-CoV-2 processes polyproteins pp1a and pp1ab through its Mpro to generate 16 mature nonstructural proteins (nsp1-nsp16), which collectively form the replication/transcription complex that provides core support for viral replication (214). Second, off-target toxicity—broad-spectrum inhibitors (e.g., camostat) inhibit TMPRSS2 while interfering with proteases involved in coagulation, inflammation, and blood pressure regulation, significantly increasing the risk of serious adverse events in clinical treatment groups (215); third, tissue delivery obstacles—small-molecule inhibitors struggle to penetrate specific compartments (e.g., inactivation in the acidic lysosomal environment, blockage by the blood-brain barrier).
It is noteworthy that multi-target synergistic strategies aimed at enhancing antiviral efficacy, such as dual-target proteolysis-targeting chimera (PROTAC) degraders, may increase off-target risks due to the expanded range of target molecules. Current research seeks breakthroughs through two precision-optimized design approaches. One leverages spatial precision by confining activity release ranges using tissue-microenvironment-responsive carriers (216), while another employs conformational precision through allosteric site engineering to selectively engage inactive states of host proteases, thereby enhancing specificity (217). These approaches pave a critical pathway for balancing synergistic potency and safety. Thus, developing novel inhibitors with high selectivity, resistance barriers, and microenvironmental adaptability has become an urgent need to address the challenges of viral evolution. Cutting-edge strategies focus on three breakthroughs. One is the exploration of allosteric inhibitory sites (e.g., targeting furin exosite-III). Another is the design of dual-target PROTAC degraders (e.g., simultaneous degradation of TMPRSS2/CTSB). The third is the development of smart responsive nanocarriers (e.g., pH-sensitive liposomes loaded with cystatin C targeting endosomes).
5 Conclusion
Viral infections can trigger various severe diseases, such as pneumonia, meningitis, hepatitis, and cardiovascular diseases, posing a significant threat to human health. The regulatory role of host proteases in viral infections has transcended traditional understanding. As one of the core dynamic hubs in the virus-host interaction network, these enzymes not only directly drive critical processes, including viral entry, replication, and immune evasion, but also profoundly reshape infection progression through spatiotemporal activity regulation. Viruses typically hijack host protease activity to facilitate infection, with their specificity and activity directly determining viral pathogenicity. Research has elucidated three core viral evolutionary strategies enabling cross-species transmission—hijacking tissue-enriched proteases (e.g., TMPRSS2 in respiratory epithelial cells); inducing abnormal activation of microenvironment proteases (e.g., inflammation-driven MMP-9 overexpression); and optimizing adaptability of cleavage sites (e.g., the PRRA motif in the SARS-CoV-2 S protein). These mechanisms provide molecular foundations for understanding viral pathogenicity variations. In therapeutic development, host protease targeting is transitioning from single-inhibition approaches toward multidimensional synergistic paradigms. Innovative designs, including dual-target PROTAC degraders, allosteric inhibitors, and intelligent delivery systems, mark a significant turning point in the field. However, clinical translation encounters three persistent challenges—host compensatory escape mechanisms, off-target toxicity, and delivery barriers. Future breakthroughs require a focus on space-conformation precision technologies, such as employing microenvironment-responsive carriers to restrict active compound distribution or designing selective binding to inactive states of proteases based on allosteric sites, thereby balancing efficacy with physiological safety.
Looking forward, host protease-targeted therapy progress will focus on three interconnected dimensions. At the mechanism-elucidation level, cryo-EM and molecular dynamics simulations reveal dynamic conformational changes of protease-substrate complexes (e.g., the transient intermediates formed during furin cleaves the S protein), providing atomic-resolution blueprints for allosteric inhibitor design. At the technological development level, an AI-driven multi-target degradation agent screening for multi-target degraders integrates, host proteomics and viral evolution data to predict optimal target combinations (e.g., the combined intervention of TMPRSS2 and CTSL). At the clinical translation level, it requires establishing tiered organoid-animal model evaluation systems to assess tissue-specific toxicity in human-mimetic microenvironments (e.g., the long-term consequences of prostate TMPRSS2 suppression), alongside exploring sequential therapies against viral escape. The paramount value of these advances lies not only in significantly reducing the risk of target mutation-driven drug resistance—the genetic stability of host proteases makes them an “anchor” for controlling highly variable viruses (such as HIV and DENV)—but also in providing broad-spectrum countermeasures for emerging viral outbreaks. From respiratory to neuroinvasive viruses, host protease-targeting strategies are transforming antiviral development paradigms. Realizing this vision demands deep integration of virology, structural biology, and nanomedicine. Consequently, developing novel intervention strategies targeting host proteases holds broad application prospects and significant research value in the field of antiviral therapy.
Author contributions
QX: Funding acquisition, Project administration, Writing – original draft, Writing – review & editing. XL: Funding acquisition, Investigation, Project administration, Writing – review & editing. HH: Investigation, Supervision, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The study was supported by grants from the Health Commission of Chengdu (Grant No.2025682), Chengdu Medical Research Program (Grant No.2024323)and Sichuan Hospital Association County Hospital Research Special Project (Grant No.2024LC001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
α-SNAP: alpha-soluble NSF attachment protein
A1AT: αlpha (1)-antitrypsin
ACOT2: acyl-CoA thioesterase 2
ADAM17: a disintegrin and metalloproteinase 17
Ad2/12: adenovirus 2/12
AEBSF: 4-(2-aminoethyl) benzenesulfonyl fluoride
AMDV: Aleutian mink disease parvovirus
ARVs: Avian reoviruses
Asp: aspartic
β-NGF: β-nerve growth factor
BDV: Borna disease virus
CAPN1: calpain-1
CCHF: Crimean-Congo hemorrhagic fever
CHIKV: chikungunya virus
CMV: cytomegalovirus
COVID-19: coronavirus disease 2019
cryo-EM: cryo-electron microscopy
CSFV: classical swine fever virus
CST3: cystatin C
CTSB: cathepsin B
CTSD: cathepsin D
CTSG: cathepsin G
CTSL: cathepsin L
CTSS: cathepsin S
DENV: dengue virus
DESC1: differentially expressed in squamous cell carcinoma gene 1
DPP4: dipeptidyl peptidase 4
E166V: Glu166Val
E1A: region 1A
EBOV: Ebola virus
ENaC: epithelial sodium channel
FAM111B: family with sequence similarity 111 member B
FluV: influenza virus
Fxa: coagulation factor Xa
gB: glycoprotein B
GBPs: guanylate binding proteins
GP: glycoprotein
GP1: glycoprotein 1
GPC3: glypican-3
H1N1: hemagglutinin 1 neuraminidase 1
HA: hemagglutinin
HADs: host-directed antiviral drugs
HAI-1: hepatocyte growth factor activator inhibitor 1
HAstV-8: human astrovirus type 8
HAT: human airway trypsin-like protease
HBeAg: hepatitis B e antigen
HBV: hepatitis B virus
HCV: hepatitis C virus
HCoV-229E: human coronavirus 229E
HCoV-HKU1: human coronavirus HKU1
HCoV-OC43: human coronavirus OC43
HE: hemagglutinin-esterase
HERVs: human endogenous retroviruses
HEV: hepatitis E virus
HIV: human immunodeficiency virus
HPIV3: human parainfluenza virus 3
HPV: human papillomavirus
ICV: Influenza C virus
IL-1β: interleukin-1β
KPNA2: karyopherin alpha2
L2: late protein 2
MASP-2: Mannose-binding lectin-associated serine protease 2
MAV-1: mouse adenovirus
MCMV: mouse cytomegalovirus
MHV: murine hepatitis virus
MMP: matrix metalloproteinase
MMP-2: matrix metalloprotease-2
MMP-9: matrix metalloprotease-9
MuV: mumps virus
MV: measles virus
MVM: minute virus of mice
NCOA7: Nuclear receptor coactivator 7
NDV: Newcastle disease virus
NPC1: the human endosomal receptor Niemann-Pick C1
PAI-1: plasminogen activator inhibitor-1
PCSK7: proprotein convertase subtilisin/kexin type 7
PEDV: porcine epidemic diarrhea virus
pro-OCN: pro-osteocalcin
PROTAC: proteolysis-targeting chimera
RBD: receptor-binding domain
RNP: ribonucleoprotein
RSV: Respiratory syncytial virus
SADS-CoV: swine acute diarrhea syndrome coronavirus
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
SERPIN: serine protease inhibitor superfamily
SPINK6: Serine protease inhibitor Kazal‐type 6
TBEV: tick-borne encephalitis virus
TGFβ: transforming growth factor beta
TGEV: transmissible gastroenteritis virus
TMPRSS: transmembrane serine protease
TPCK: N-alpha-tosyl-L-phenylalanyl chloromethyl ketone
TRABD2A: TRAB domain-containing protein 2A
TTSP: type II transmembrane serine protease
VSV: Vesicular Stomatitis Virus
WNV: West Nile virus
ZIKV: Zika virus
References
1. Karim M, Lo CW, and Einav S. Preparing for the next viral threat with broad-spectrum antivirals. J Clin Invest. (2023) 133:e170236. doi: 10.1172/JCI170236
2. Available online at: https://www.who.int/teams/global-hiv-hepatitis-and-stis-programmes/hiv/strategic-information/hiv-data-and-statistics (Accessed June 19, 2025).
3. Available online at: https://www.who.int/data/gho/data/themes/chronic-viral-hepatitis (Accessed June 19, 2025).
4. Available online at: https://www.who.int/zh/news-room/fact-sheets/detail/influenza-(seasonal) (Accessed June 19, 2025).
5. Available online at: https://data.who.int/dashboards/covid19/deaths?n=c (Accessed June 19, 2025).
6. Carr A, Mackie NE, Paredes R, and Ruxrungtham K. HIV drug resistance in the era of contemporary antiretroviral therapy: A clinical perspective. Antivir Ther. (2023) 28:13596535231201162. doi: 10.1177/13596535231201162
7. Datir R, Kemp S, El Bouzidi K, Mlchocova P, Goldstein R, Breuer J, et al. In vivo emergence of a novel protease inhibitor resistance signature in HIV-1 matrix. mBio. (2020) 11:e02036–20. doi: 10.1128/mBio.02036-20
8. Zhang W, Xu H, Guan S, Wang C, and Dong G. Frequency and distribution of H1N1 influenza A viruses with oseltamivir-resistant mutations worldwide before and after the 2009 pandemic. J Med Virol. (2022) 94:4406–16. doi: 10.1002/jmv.27870
9. Iketani S, Mohri H, Culbertson B, Hong SJ, Duan Y, Luck MI, et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature. (2023) 613:558–64. doi: 10.1038/s41586-022-05514-2
10. Huang C, Shuai H, Qiao J, Hou Y, Zeng R, Xia A, et al. A new generation M(pro) inhibitor with potent activity against SARS-CoV-2 Omicron variants. Signal Transduct Target Ther. (2023) 8:128. doi: 10.1038/s41392-023-01392-w
11. Duan Y, Zhou H, Liu X, Iketani S, Lin M, Zhang X, et al. Molecular mechanisms of SARS-CoV-2 resistance to nirmatrelvir. Nature. (2023) 622:376–82. doi: 10.1038/s41586-023-06609-0
12. Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, and Garten W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. (1992) 360:358–61. doi: 10.1038/360358a0
13. Cruz L, Biryukov J, Conway MJ, and Meyers C. Cleavage of the HPV16 Minor Capsid Protein L2 during Virion Morphogenesis Ablates the Requirement for Cellular Furin during De Novo Infection. Viruses. (2015) 7:5813–30. doi: 10.3390/v7112910
14. Lee JE and Saphire EO. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. (2009) 4:621–35. doi: 10.2217/fvl.09.56
15. Chandran K, Sullivan NJ, Felbor U, Whelan SP, and Cunningham JM. Endosomal proteolysis of the ebola virus glycoprotein is necessary for infection. Science. (2005) 308:1643–5. doi: 10.1126/science.1110656
16. Hofmann-Winkler H, Kaup F, and Pöhlmann S. Host cell factors in filovirus entry: novel players, new insights. Viruses. (2012) 4:3336–62. doi: 10.3390/v4123336
17. Hunt CL, Lennemann NJ, and Maury W. Filovirus entry: A novelty in the viral fusion world. Viruses. (2012) 4:258–75. doi: 10.3390/v4020258
18. Papa G, Mallery DL, Albecka A, Welch LG, Cattin-Ortolá J, Luptak J, et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PloS Pathog. (2021) 17:e1009246. doi: 10.1371/journal.ppat.1009246
19. Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. (2021) 591:293–9. doi: 10.1038/s41586-021-03237-4
20. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052
21. Beumer J, Geurts MH, Lamers MM, Puschhof J, Zhang J, van der Vaart J, et al. A CRISPR/Cas9 genetically engineered organoid biobank reveals essential host factors for coronaviruses. Nat Commun. (2021) 12:5498. doi: 10.1038/s41467-021-25729-7
22. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217:e20201129. doi: 10.1084/jem.20201129
23. Feng T, Tong H, Ming Z, Deng L, Liu J, Wu J, et al. Matrix metalloproteinase 3 restricts viral infection by enhancing host antiviral immunity. Antiviral Res. (2022) 206:105388. doi: 10.1016/j.antiviral.2022.105388
24. Franzoso G, Biswas P, Poli G, Carlson LM, Brown KD, Tomita-Yamaguchi M, et al. A family of serine proteases expressed exclusively in myelo-monocytic cells specifically processes the nuclear factor-kappa B subunit p65 in vitro and may impair human immunodeficiency virus replication in these cells. J Exp Med. (1994) 180:1445–56. doi: 10.1084/jem.180.4.1445
25. Moriuchi H, Moriuchi M, and Fauci AS. Cathepsin G, a neutrophil-derived serine protease, increases susceptibility of macrophages to acute human immunodeficiency virus type 1 infection. J Virol. (2000) 74:6849–55. doi: 10.1128/JVI.74.15.6849-6855.2000
26. McCormick AL, Roback L, and Mocarski ES. HtrA2/Omi terminates cytomegalovirus infection and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA). PloS Pathog. (2008) 4:e1000063. doi: 10.1371/journal.ppat.1000063
27. Grand RJ, Schmeiser K, Gordon EM, Zhang X, Gallimore PH, and Turnell AS. Caspase-mediated cleavage of adenovirus early region 1A proteins. Virology. (2002) 301:255–71. doi: 10.1006/viro.2002.1586
28. Petersen M, Lotke R, Hopfensperger K, Victoria S, Haußmann I, Burster T, et al. Inhibition of infectious HIV-1 production by rerouting the cellular furin inhibitor serpin B8. J Virol. (2023) 97:e0029423. doi: 10.1128/jvi.00294-23
29. Moulard M, Hallenberger S, Garten W, and Klenk HD. Processing and routage of HIV glycoproteins by furin to the cell surface. Virus Res. (1999) 60:55–65. doi: 10.1016/S0168-1702(99)00002-7
30. Rodenhuis-Zybert IA, Moesker B, da Silva Voorham JM, van der Ende-Metselaar H, Diamond MS, Wilschut J, et al. A fusion-loop antibody enhances the infectious properties of immature flavivirus particles. J Virol. (2011) 85:11800–8. doi: 10.1128/JVI.05237-11
31. Clinckemalie L, Spans L, Dubois V, Laurent M, Helsen C, Joniau S, et al. Androgen regulation of the TMPRSS2 gene and the effect of a SNP in an androgen response element. Mol Endocrinol. (2013) 27:2028–40. doi: 10.1210/me.2013-1098
32. Bresnahan PA, Leduc R, Thomas L, Thorner J, Gibson HL, Brake AJ, et al. Human fur gene encodes a yeast KEX2-like endoprotease that cleaves pro-beta-NGF in vivo. J Cell Biol. (1990) 111:2851–9. doi: 10.1083/jcb.111.6.2851
33. Al Rifai O, Chow J, Lacombe J, Julien C, Faubert D, Susan-Resiga D, et al. Proprotein convertase furin regulates osteocalcin and bone endocrine function. J Clin Invest. (2017) 127:4104–17. doi: 10.1172/JCI93437
34. Rickman OJ, Guignard E, Chabanon T, Bertoldi G, Auberson M, and Hummler E. Tmprss2 maintains epithelial barrier integrity and transepithelial sodium transport. Life Sci Alliance. (2024) 7:e202302304. doi: 10.26508/lsa.202302304
35. Sure F, Afonso S, Essigke D, Schmidt P, Kalo MZ, Nesterov V, et al. Transmembrane serine protease 2 and proteolytic activation of the epithelial sodium channel in mouse kidney. J Am Soc Nephrol. (2025) 36:420–34. doi: 10.1681/ASN.0000000521
36. Chen YW, Lee MS, Lucht A, Chou FP, Huang W, Havighurst TC, et al. TMPRSS2, a serine protease expressed in the prostate on the apical surface of luminal epithelial cells and released into semen in prostasomes, is misregulated in prostate cancer cells. Am J Pathol. (2010) 176:2986–96. doi: 10.2353/ajpath.2010.090665
37. Limburg H, Harbig A, Bestle D, Stein DA, Moulton HM, Jaeger J, et al. TMPRSS2 is the major activating protease of influenza A virus in primary human airway cells and influenza B virus in human type II pneumocytes. J Virol. (2019) 93:e00649–19. doi: 10.1128/JVI.00649-19
38. Le TQ, Kawachi M, Yamada H, Shiota M, Okumura Y, and Kido H. Identification of trypsin I as a candidate for influenza A virus and Sendai virus envelope glycoprotein processing protease in rat brain. Biol Chem. (2006) 387:467–75. doi: 10.1515/BC.2006.062
39. Kido H, Okumura Y, Yamada H, Le TQ, and Yano M. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr Pharm Des. (2007) 13:405–14. doi: 10.2174/138161207780162971
40. Schneck NA, Ivleva VB, Cai CX, Cooper JW, and Lei QP. Characterization of the furin cleavage motif for HIV-1 trimeric envelope glycoprotein by intact LC-MS analysis. Analyst. (2020) 145:1636–40. doi: 10.1039/C9AN02098E
41. Blow DM, Birktoft JJ, and Hartley BS. Role of a buried acid group in the mechanism of action of chymotrypsin. Nature. (1969) 221:337–40. doi: 10.1038/221337a0
42. Fernández I, Saunders N, Duquerroy S, Bolland WH, Arbabian A, Baquero E, et al. Structural basis of TMPRSS2 zymogen activation and recognition by the HKU1 seasonal coronavirus. Cell. (2024) 187:4246–4260.e16. doi: 10.1016/j.cell.2024.06.007
43. Bode W, Gomis-Rüth FX, and Stöckler W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. (1993) 331:134–40. doi: 10.1016/0014-5793(93)80312-I
44. Allan JA, Docherty AJ, Barker PJ, Huskisson NS, Reynolds JJ, and Murphy G. Binding of gelatinases A and B to type-I collagen and other matrix components. Biochem J. (1995) 309:299–306. doi: 10.1042/bj3090299
45. Saad MI and Jenkins BJ. The protease ADAM17 at the crossroads of disease: revisiting its significance in inflammation, cancer, and beyond. FEBS J. (2024) 291:10–24. doi: 10.1111/febs.16923
46. Chen J, Xu W, Chen Y, Xie X, Zhang Y, Ma C, et al. Matrix metalloproteinase 9 facilitates hepatitis B virus replication through binding with type I interferon (IFN) receptor 1 to repress IFN/JAK/STAT signaling. J Virol. (2017) 91:e01824–16. doi: 10.1128/JVI.01824-16
47. Muratori C, Sistigu A, Ruggiero E, Falchi M, Bacigalupo I, Palladino C, et al. Macrophages transmit human immunodeficiency virus type 1 products to CD4-negative cells: involvement of matrix metalloproteinase 9. J Virol. (2007) 81:9078–87. doi: 10.1128/JVI.00675-07
48. Verma S, Dixit R, and Pandey KC. Cysteine proteases: modes of activation and future prospects as pharmacological targets. Front Pharmacol. (2016) 7:107. doi: 10.3389/fphar.2016.00107
49. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
50. Dunn BM. Structure and mechanism of the pepsin-like family of aspartic peptidases. Chem Rev. (2002) 102:4431–58. doi: 10.1021/cr010167q
51. El Messaoudi K, Sistigu A, Ruggiero E, Falchi M, Bacigalupo I, and Palladino C. A human milk factor susceptible to cathepsin D inhibitors enhances human immunodeficiency virus type 1 infectivity and allows virus entry into a mammary epithelial cell line. J Virol. (2000) 74:1004–7. doi: 10.1128/JVI.74.2.1004-1007.2000
52. Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. (1997) 386:463–71. doi: 10.1038/386463a0
53. Contin R, Arnoldi F, Mano M, and Burrone OR. Rotavirus replication requires a functional proteasome for effective assembly of viroplasms. J Virol. (2011) 85:2781–92. doi: 10.1128/JVI.01631-10
54. Abad AT, McNamara AJ, and Danthi P. Proteasome activity is required for reovirus entry into cells. J Virol. (2023) 97:e0134823. doi: 10.1128/jvi.01348-23
55. Stieneke-Gröber A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, et al. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. (1992) 11:2407–14. doi: 10.1002/j.1460-2075.1992.tb05305.x
56. Skehel JJ and Waterfield MD. Studies on the primary structure of the influenza virus hemagglutinin. Proc Natl Acad Sci U.S.A. (1975) 72:93–7. doi: 10.1073/pnas.72.1.93
57. Horimoto T, Nakayama K, Smeekens SP, and Kawaoka Y. Proprotein-processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. J Virol. (1994) 68:6074–8. doi: 10.1128/jvi.68.9.6074-6078.1994
58. Meyer D, Sielaff F, Hammami M, Böttcher-Friebertshäuser E, Garten W, and Steinmetzer T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem J. (2013) 452:331–43. doi: 10.1042/BJ20130101
59. Böttcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, and Matrosovich M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol. (2006) 80:9896–8. doi: 10.1128/JVI.01118-06
60. Böttcher-Friebertshäuser E, Freuer C, Sielaff F, Schmidt S, Eickmann M, Uhlendorff J, et al. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J Virol. (2010) 84:5605–14. doi: 10.1128/JVI.00140-10
61. Bestle D, Limburg H, Kruhl D, Harbig A, Stein DA, Moulton H, et al. Hemagglutinins of avian influenza viruses are proteolytically activated by TMPRSS2 in human and murine airway cells. J Virol. (2021) 95:e0090621. doi: 10.1128/JVI.00906-21
62. Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pöhlmann S, et al. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PloS Pathog. (2013) 9:e1003774. doi: 10.1371/journal.ppat.1003774
63. Kido H, Beppu Y, Sakai K, and Towatari T. Molecular basis of proteolytic activation of Sendai virus infection and the defensive compounds for infection. Biol Chem. (1997) 378:255–63. doi: 10.1515/bchm.1997.378.3-4.255
64. Kühn N, Bergmann S, Kösterke N, Lambertz RLO, Keppner A, van den Brand JMA, et al. The proteolytic activation of (H3N2) influenza A virus hemagglutinin is facilitated by different type II transmembrane serine proteases. J Virol. (2016) 90:4298–307. doi: 10.1128/JVI.02693-15
65. Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol. (2009) 83:3200–11. doi: 10.1128/JVI.02205-08
66. Zmora P, Hoffmann M, Kollmus H, Moldenhauer AS, Danov O, Braun A, et al. TMPRSS11A activates the influenza A virus hemagglutinin and the MERS coronavirus spike protein and is insensitive against blockade by HAI-1. J Biol Chem. (2018) 293:13863–73. doi: 10.1074/jbc.RA118.001273
67. Beaulieu A, Gravel É, Cloutier A, Marois I, Colombo É, Désilets A, et al. Matriptase proteolytically activates influenza virus and promotes multicycle replication in the human airway epithelium. J Virol. (2013) 87:4237–51. doi: 10.1128/JVI.03005-12
68. Baron J, Tarnow C, Mayoli-Nüssle D, Schilling E, Meyer D, Hammami M, et al. Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A viruses. J Virol. (2013) 87:1811–20. doi: 10.1128/JVI.02320-12
69. Hamilton BS, Gludish DW, and Whittaker GR. Cleavage activation of the human-adapted influenza virus subtypes by matriptase reveals both subtype and strain specificities. J Virol. (2012) 86:10579–86. doi: 10.1128/JVI.00306-12
70. Whittaker GR and Straus MR. Human matriptase/ST 14 proteolytically cleaves H7N9 hemagglutinin and facilitates the activation of influenza A/Shanghai/2/2013 virus in cell culture. Influenza Other Respir Viruses. (2020) 14:189–95. doi: 10.1111/irv.12707
71. Harbig A, Mernberger M, Bittel L, Pleschka S, Schughart K, Steinmetzer T, et al. Transcriptome profiling and protease inhibition experiments identify proteases that activate H3N2 influenza A and influenza B viruses in murine airways. J Biol Chem. (2020) 295:11388–407. doi: 10.1074/jbc.RA120.012635
72. Sato K, Hayashi H, Shimotai Y, Yamaya M, Hongo S, Kawakami K, et al. TMPRSS2 activates hemagglutinin-esterase glycoprotein of influenza C virus. J Virol. (2021) 95:e0129621. doi: 10.1128/JVI.01296-21
73. Murakami M, Towatari T, Ohuchi M, Shiota M, Akao M, Okumura Y, et al. Mini-plasmin found in the epithelial cells of bronchioles triggers infection by broad-spectrum influenza A viruses and Sendai virus. Eur J Biochem. (2001) 268:2847–55. doi: 10.1046/j.1432-1327.2001.02166.x
74. Li S, Towatari T, Ohuchi M, Shiota M, Akao M, and Okumura Y. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J Virol. (1993) 67:6667–73. doi: 10.1128/jvi.67.11.6667-6673.1993
75. Magnen M, Gueugnon F, Guillon A, Baranek T, Thibault VC, Petit-Courty A, et al. Kallikrein-related peptidase 5 contributes to H3N2 influenza virus infection in human lungs. J Virol. (2017) 91:e00421–17. doi: 10.1128/JVI.00421-17
76. Magnen M, Elsässer BM, Zbodakova O, Kasparek P, Gueugnon F, Petit-Courty A, et al. Kallikrein-related peptidase 5 and seasonal influenza viruses, limitations of the experimental models for activating proteases. Biol Chem. (2018) 399:1053–64. doi: 10.1515/hsz-2017-0340
77. Hamilton BS and Whittaker GR. Cleavage activation of human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J Biol Chem. (2013) 288:17399–407. doi: 10.1074/jbc.M112.440362
78. Okumura Y, Takahashi E, Yano M, Ohuchi M, Daidoji T, Nakaya T, et al. Novel type II transmembrane serine proteases, MSPL and TMPRSS13, Proteolytically activate membrane fusion activity of the hemagglutinin of highly pathogenic avian influenza viruses and induce their multicycle replication. J Virol. (2010) 84:5089–96. doi: 10.1128/JVI.02605-09
79. Kido H, Niwa Y, Beppu Y, and Towatari T. Cellular proteases involved in the pathogenicity of enveloped animal viruses, human immunodeficiency virus, influenza virus A and Sendai virus. Adv Enzyme Regul. (1996) 36:325–47. doi: 10.1016/0065-2571(95)00016-X
80. Kido H, Murakami M, Oba K, Chen Y, and Towatari T. Cellular proteinases trigger the infectivity of the influenza A and Sendai viruses. Mol Cells. (1999) 9:235–44. doi: 10.1016/S1016-8478(23)13535-7
81. Boycott R, Klenk HD, and Ohuchi M. Cell tropism of influenza virus mediated by hemagglutinin activation at the stage of virus entry. Virology. (1994) 203:313–9. doi: 10.1006/viro.1994.1489
82. Mykytyn AZ, Breugem TI, Riesebosch S, Schipper D, van den Doel PB, Rottier RJ, et al. SARS-CoV-2 entry into human airway organoids is serine protease-mediated and facilitated by the multibasic cleavage site. Elife. (2021) 10:e64508. doi: 10.7554/eLife.64508
83. Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulton H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. (2020) 3:e202000786. doi: 10.26508/lsa.202000786
84. Zang R, Gomez Castro MF, McCune BT, Zeng Q, Rothlauf PW, Sonnek NM, et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci Immunol. (2020) 5:eabc3582. doi: 10.1126/sciimmunol.abc3582
85. Hoffmann M, Mösbauer K, Hofmann-Winkler H, Kaul A, Kleine-Weber H, Krüger N, et al. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature. (2020) 585:588–90. doi: 10.1038/s41586-020-2575-3
86. Jackson CB, Farzan M, Chen B, and Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
87. Zhao MM, Yang WL, Yang FY, Zhang L, Huang WJ, and Hou W. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther. (2021) 6:134. doi: 10.1038/s41392-021-00558-8
88. Dong W, Wang J, Tian L, Zhang J, Settles EW, Qin C, et al. Factor Xa cleaves SARS-CoV-2 spike protein to block viral entry and infection. spike Protein to block Viral entry infection. Nat Commun. (2023) 14:1936. doi: 10.1038/s41467-023-37336-9
89. Kastenhuber ER, Mercadante M, Nilsson-Payant B, Johnson JL, Jaimes JA, Muecksch F, et al. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. Elife. (2022) 11:e77444. doi: 10.7554/eLife.77444
90. Park JE, Cruz DJ, and Shin HJ. Clathrin- and serine proteases-dependent uptake of porcine epidemic diarrhea virus into Vero cells. Virus Res. (2014) 191:21–9. doi: 10.1016/j.virusres.2014.07.022
91. Shi W, Fan W, Bai J, Tang Y, Wang L, Jiang Y, et al. TMPRSS2 and MSPL facilitate trypsin-independent porcine epidemic diarrhea virus replication in vero cells. Viruses. (2017) 9:114. doi: 10.3390/v9050114
92. Han Y, Ma Y, Wang Z, Feng F, Zhou L, Feng H, et al. TMPRSS13 promotes the cell entry of swine acute diarrhea syndrome coronavirus. J Med Virol. (2024) 96:e29712. doi: 10.1002/jmv.29712
93. Harte JV, Wakerlin SL, Lindsay AJ, McCarthy JV, and Coleman-Vaughan C. Metalloprotease-dependent S2’-activation promotes cell-cell fusion and syncytiation of SARS-CoV-2. Viruses. (2022) 14:2094. doi: 10.3390/v14102094
94. Chan JF, Huang X, Hu B, Chai Y, Shi H, Zhu T, et al. Altered host protease determinants for SARS-CoV-2 Omicron. Sci Adv. (2023) 9:eadd3867. doi: 10.1126/sciadv.add3867
95. Healy EF and Lilic M. A model for COVID-19-induced dysregulation of ACE2 shedding by ADAM17. Biochem Biophys Res Commun. (2021) 573:158–63. doi: 10.1016/j.bbrc.2021.08.040
96. Jocher G, Grass V, Tschirner SK, Riepler L, Breimann S, Kaya T, et al. ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion. EMBO Rep. (2022) 23:e54305. doi: 10.15252/embr.202154305
97. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, and Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. (2014) 88:1293–307. doi: 10.1128/JVI.02202-13
98. Peck KM, Cockrell AS, Yount BL, Scobey T, Baric RS, and Heise MT. Glycosylation of mouse DPP4 plays a role in inhibiting Middle East respiratory syndrome coronavirus infection. J Virol. (2015) 89:4696–9. doi: 10.1128/JVI.03445-14
99. Li Y, Zhang Z, Yang L, Lian X, Xie Y, Li S, et al. The MERS-CoV receptor DPP4 as a candidate binding target of the SARS-CoV-2 spike. iScience. (2020) 23:101160. doi: 10.1016/j.isci.2020.101160
100. Peck KM, Scobey T, Swanstrom J, Jensen KL, Burch CL, Baric RS, et al. Permissivity of dipeptidyl peptidase 4 orthologs to Middle East respiratory syndrome coronavirus is governed by glycosylation and other complex determinants. J Virol. (2017) 91:e00534–17. doi: 10.1128/JVI.00534-17
101. Saunders N, Fernandez I, Planchais C, Michel V, Rajah MM, Baquero Salazar E, et al. TMPRSS2 is a functional receptor for human coronavirus HKU1. Nature. (2023) 624:207–14. doi: 10.1038/s41586-023-06761-7
102. McCallum M, Park YJ, Stewart C, Sprouse KR, Addetia A, Brown J, et al. Human coronavirus HKU1 recognition of the TMPRSS2 host receptor. Cell. (2024) 187:4231–4245.e13. doi: 10.1016/j.cell.2024.06.006
103. Wang H, Liu X, Zhang X, Zhao Z, Lu Y, Pu D, et al. TMPRSS2 and glycan receptors synergistically facilitate coronavirus entry. Cell. (2024) 187:4261–4271.e17. doi: 10.1016/j.cell.2024.06.016
104. Ling H, Xiao P, Usami O, and Hattori T. Thrombin activates envelope glycoproteins of HIV type 1 and enhances fusion. Microbes Infect. (2004) 6:414–20. doi: 10.1016/j.micinf.2004.01.010
105. Krchlikova V, Braun E, Weiss J, Stafl K, Jech L, Badarinarayan SS, et al. Inhibition of placental trophoblast fusion by guanylate-binding protein 5. Sci Adv. (2025) 11:eadt5388. doi: 10.1126/sciadv.adt5388
106. Bestle D, Bittel L, Werner AD, Kämper L, Dolnik O, Krähling V, et al. Novel proteolytic activation of Ebolavirus glycoprotein GP by TMPRSS2 and cathepsin L at an uncharted position can compensate for furin cleavage. Virus Res. (2024) 347:199430. doi: 10.1016/j.virusres.2024.199430
107. Lopes BRP, da Silva GS, de Lima Menezes G, de Oliveira J, Watanabe ASA, Porto BN, et al. Serine proteases in neutrophil extracellular traps exhibit anti-Respiratory Syncytial Virus activity. Int Immunopharmacol. (2022) 106:108573. doi: 10.1016/j.intimp.2022.108573
108. Stearns K, Lampe G, Hanan R, Marcink T, Niewiesk S, Sternberg SH, et al. Human parainfluenza virus 3 field strains undergo extracellular fusion protein cleavage to activate entry. mBio. (2024) 15:e0232724. doi: 10.1128/mbio.02327-24
109. Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. (2002) 3:753–66. doi: 10.1038/nrm934
110. Lamb RA and Kolakofsky D. Paramyxoviridae: the viruses and their replication. Fields Virol. (1996) 1177–1204.
111. Spriggs MK and Collins PL. Human parainfluenza virus type 3: messenger RNAs, polypeptide coding assignments, intergenic sequences, and genetic map. J Virol. (1986) 59:646–54. doi: 10.1128/jvi.59.3.646-654.1986
112. Paterson RG, Harris TJ, and Lamb RA. Fusion protein of the paramyxovirus simian virus 5: nucleotide sequence of mRNA predicts a highly hydrophobic glycoprotein. Proc Natl Acad Sci U.S.A. (1984) 81:6706–10. doi: 10.1073/pnas.81
113. Toyoda T, Sakaguchi T, Imai K, Inocencio NM, Gotoh B, Hamaguchi M, et al. Structural comparison of the cleavage-activation site of the fusion glycoprotein between virulent and avirulent strains of Newcastle disease virus. Virology. (1987) 158:242–7. doi: 10.1016/0042-6822(87)90261-3
114. Richardson C, Hull D, Greer P, Hasel K, Berkovich A, Englund G, et al. The nucleotide sequence of the mRNA encoding the fusion protein of measles virus (Edmonston strain): a comparison of fusion proteins from several different paramyxoviruses. Virology. (1986) 155:508–23. doi: 10.1016/0042-6822(86)90212-6
115. Collins PL, Huang YT, and Wertz GW. Nucleotide sequence of the gene encoding the fusion (F) glycoprotein of human respiratory syncytial virus. Proc Natl Acad Sci U.S.A. (1984) 81:7683–7. doi: 10.1073/pnas.81.24.7683
116. Lamb RA and Jardetzky TS. Structural basis of viral invasion: lessons from paramyxovirus F. Curr Opin Struct Biol. (2007) 17:427–36. doi: 10.1016/j.sbi.2007.08.016
117. Richards RM, Lowy DR, Schiller JT, and Day PM. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U.S.A. (2006) 103:1522–7. doi: 10.1073/pnas.0508815103
118. Bronnimann MP, Calton CM, Chiquette SF, Li S, Lu M, Chapman JA, et al. Furin cleavage of L2 during papillomavirus infection: minimal dependence on cyclophilins. J Virol. (2016) 90:6224–34. doi: 10.1128/JVI.00038-16
119. Crawford SE, Mukherjee SK, Estes MK, Lawton JA, Shaw AL, Ramig RF, et al. Trypsin cleavage stabilizes the rotavirus VP4 spike. J Virol. (2001) 75:6052–61. doi: 10.1128/JVI.75.13.6052-6061.2001
120. Golden JW, Bahe JA, Lucas WT, Nibert ML, and Schiff LA. Cathepsin S supports acid-independent infection by some reoviruses. J Biol Chem. (2004) 279:8547–57. doi: 10.1074/jbc.M309758200
121. Ebert DH, Deussing J, Peters C, and Dermody TS. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. (2002) 277:24609–17. doi: 10.1074/jbc.M201107200
122. Golden JW and Schiff LA. Neutrophil elastase, an acid-independent serine protease, facilitates reovirus uncoating and infection in U937 promonocyte cells. Virol J. (2005) 2:48. doi: 10.1186/1743-422X-2-48
123. Corre MH, Rey B, David SC, Torii S, Chiappe D, and Kohn T. The early communication stages between serine proteases and enterovirus capsids in the race for viral disintegration. Commun Biol. (2024) 7:969. doi: 10.1038/s42003-024-06627-2
124. Butin-Israeli V, Ben-nun-Shaul O, Kopatz I, Adam SA, Shimi T, Goldman RD, et al. Simian virus 40 induces lamin A/C fluctuations and nuclear envelope deformation during cell entry. Nucleus. (2011) 2:320–30. doi: 10.4161/nucl.2.4.16371
125. Cohen S, Marr AK, Garcin P, and Panté N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J Virol. (2011) 85:4863–74. doi: 10.1128/JVI.01999-10
126. Ozden S, Lucas-Hourani M, Ceccaldi PE, Basak A, Valentine M, Benjannet S, et al. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J Biol Chem. (2008) 283:21899–908. doi: 10.1074/jbc.M802444200
127. Klöhn M, Burkard T, Janzen J, Haase JA, Gömer A, Fu R, et al. Targeting cellular cathepsins inhibits hepatitis E virus entry. Hepatology. (2024) 80:1239–51. doi: 10.1097/HEP.0000000000000912
128. Ma S, Shi S, Xu B, Liu M, Xie L, Su Y, et al. Host serine protease ACOT2 assists DENV proliferation by hydrolyzing viral polyproteins. mSystems. (2024) 9:e0097323. doi: 10.1128/msystems.00973-23
129. Pierce DM, Buchanan FJT, Macrae FL, Mills JT, Cox A, Abualsaoud KM, et al. Thrombin cleavage of the hepatitis E virus polyprotein at multiple conserved locations is required for genome replication. PloS Pathog. (2023) 19:e1011529. doi: 10.1371/journal.ppat.1011529
130. Xie J, Guo H, Gong W, Jiang D, Zhang L, Jia J, et al. Identification of cleavage of NS5A of C-strain classical swine fever virus. Arch Virol. (2017) 162:391–400. doi: 10.1007/s00705-016-3117-z
131. Best SM, Shelton JF, Pompey JM, Wolfinbarger JB, and Bloom ME. Caspase cleavage of the nonstructural protein NS1 mediates replication of Aleutian mink disease parvovirus. J Virol. (2003) 77:5305–12. doi: 10.1128/JVI.77.9.5305-5312.2003
132. Zhirnov OP, Konakova TE, Garten W, and Klenk H. Caspase-dependent N-terminal cleavage of influenza virus nucleocapsid protein in infected cells. J Virol. (1999) 73:10158–63. doi: 10.1128/JVI.73.12.10158-10163.1999
133. Eléouët JF, Slee EA, Saurini F, Castagné N, Poncet D, Garrido C, et al. The viral nucleocapsid protein of transmissible gastroenteritis coronavirus (TGEV) is cleaved by caspase-6 and -7 during TGEV-induced apoptosis. J Virol. (2000) 74:3975–83. doi: 10.1128/JVI.74.9.3975-3983.2000
134. Méndez E, Fernández-Luna T, López S, Méndez-Toss M, and Arias CF. Proteolytic processing of a serotype 8 human astrovirus ORF2 polyprotein. J Virol. (2002) 76:7996–8002. doi: 10.1128/JVI.76.16.7996-8002.2002
135. Sanchez AJ, Vincent MJ, Erickson BR, and Nichol ST. Crimean-congo hemorrhagic fever virus glycoprotein precursor is cleaved by Furin-like and SKI-1 proteases to generate a novel 38-kilodalton glycoprotein. J Virol. (2006) 80:514–25. doi: 10.1128/JVI.80.1.514-525.2006
136. Bergeron É, Zivcec M, Chakrabarti AK, Nichol ST, Albariño CG, and Spiropoulou CF. Recovery of recombinant Crimean Congo hemorrhagic fever virus reveals a function for non-structural glycoproteins cleavage by furin. PloS Pathog. (2015) 11:e1004879. doi: 10.1371/journal.ppat.1004879
137. Sugrue RJ, Brown C, Brown G, Aitken J, and Mc LRHW. Furin cleavage of the respiratory syncytial virus fusion protein is not a requirement for its transport to the surface of virus-infected cells. J Gen Virol. (2001) 82:1375–86. doi: 10.1099/0022-1317-82-6-1375
138. Wurzer WJ, Planz O, Ehrhardt C, Giner M, Silberzahn T, Pleschka S, et al. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J. (2003) 22:2717–28. doi: 10.1093/emboj/cdg279
139. Liang G, Zhao L, Qiao Y, Geng W, Zhang X, Liu M, et al. Membrane metalloprotease TRABD2A restricts HIV-1 progeny production in resting CD4(+) T cells by degrading viral Gag polyprotein. Nat Immunol. (2019) 20:711–23. doi: 10.1038/s41590-019-0385-2
140. Ostermann E, Luoto LM, Clausen M, Virdi S, and Brune W. E2F3-dependent activation of FAM111B restricts mouse cytomegalovirus replication in primate cells. J Virol. (2024) 98:e0134924. doi: 10.1128/jvi.01349-24
141. Kanade GD, Pingale KD, and Karpe YA. Activities of thrombin and factor xa are essential for replication of hepatitis E virus and are possibly implicated in ORF1 polyprotein processing. J Virol. (2018) 92:e01853–17. doi: 10.1128/JVI.01853-17
142. Yang Q, Pei R, Wang Y, Zhou Y, Yang M, Chen X, et al. ADAM15 participates in tick-borne encephalitis virus replication. J Virol. (2021) 95:e01926–20. doi: 10.1128/JVI.01926-20
143. Richt JA, Fürbringer T, Koch A, Pfeuffer I, Herden C, Bause-Niedrig I, et al. Processing of the Borna disease virus glycoprotein gp94 by the subtilisin-like endoprotease furin. J Virol. (1998) 72:4528–33. doi: 10.1128/JVI.72.5.4528-4533.1998
144. Okazaki K. Proteolytic cleavage of glycoprotein B is dispensable for in vitro replication, but required for syncytium formation of pseudorabies virus. J Gen Virol. (2007) 88:1859–65. doi: 10.1099/vir.0.82610-0
145. Chen M, Wang MH, Shen XG, Liu H, Zhang YY, Peng JM, et al. Neuropilin-1 facilitates pseudorabies virus replication and viral glycoprotein B promotes its degradation in a furin-dependent manner. J Virol. (2022) 96:e0131822. doi: 10.1128/jvi.01318-22
146. Méndez E, Salas-Ocampo E, and Arias CF. Caspases mediate processing of the capsid precursor and cell release of human astroviruses. J Virol. (2004) 78:8601–8. doi: 10.1128/JVI.78.16.8601-8608.2004
147. Rodríguez-Grille J, Busch LK, Martínez-Costas J, and Benavente J. Avian reovirus-triggered apoptosis enhances both virus spread and the processing of the viral nonstructural muNS protein. Virology. (2014) 462-463:49–59. doi: 10.1016/j.virol.2014.04.039
148. Wang S, Le TQ, Kurihara N, Chida J, Cisse Y, Yano M, et al. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J Infect Dis. (2010) 202:991–1001. doi: 10.1086/656044
149. Leborgne NG, Devisme C, Kozarac N, Berenguer Veiga I, Ebert N, Godel A, et al. Neutrophil proteases are protective against SARS-CoV-2 by degrading the spike protein and dampening virus-mediated inflammation. JCI Insight. (2024) 9:e174133. doi: 10.1172/jci.insight.174133
150. Braun E, Hotter D, Koepke L, Zech F, Groß R, Sparrer KMJ, et al. Guanylate-binding proteins 2 and 5 exert broad antiviral activity by inhibiting furin-mediated processing of viral envelope proteins. Cell Rep. (2019) 27:2092–104. doi: 10.1016/j.celrep.2019.04.063
151. Wang J, Luo J, Wen Z, Wang X, Shuai L, Zhong G, et al. Alpha-soluble NSF attachment protein prevents the cleavage of the SARS-CoV-2 spike protein by functioning as an interferon-upregulated furin inhibitor. mBio. (2022) 13:e0244321. doi: 10.1128/mbio.02443-21
152. Azouz NP, Klingler AM, Callahan V, Akhrymuk IV, Elez K, Raich L, et al. Alpha 1 antitrypsin is an inhibitor of the SARS-CoV-2-priming protease TMPRSS2. Pathog Immun. (2021) 6:55–74. doi: 10.20411/pai.v6i1.408
153. Jedicke N, Struever N, Aggrawal N, Welte T, Manns MP, Malek NP, et al. α-1-antitrypsin inhibits acute liver failure in mice. Hepatology. (2014) 59:2299–308. doi: 10.1002/hep.27024
154. Wettstein L, Weil T, Conzelmann CA, Müller JA, Groß R, Hirschenberger M, et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat Commun. (2021) 12:1726. doi: 10.1038/s41467-021-21972-0
155. Haynes LM, Holding ML, DiGiovanni HL, Siemieniak D, and Ginsburg D. High-throughput amino acid-level characterization of the interactions of plasminogen activator inhibitor-1 with variably divergent proteases. Protein Sci. (2025) 34:e70088. doi: 10.1002/pro.70088
156. Modenbach JM, Möller C, Asgarbeik S, Geist N, Rimkus N, Dörr M, et al. Biochemical analyses of cystatin-C dimers and cathepsin-B reveals a trypsin-driven feedback mechanism in acute pancreatitis. Nat Commun. (2025) 16:1702. doi: 10.1038/s41467-025-56875-x
157. Hall KC and Blobel CP. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PloS One. (2012) 7:e31600. doi: 10.1371/journal.pone.0031600
158. Khan H, Winstone H, Jimenez-Guardeño JM, Graham C, Doores KJ, Goujon C, et al. TMPRSS2 promotes SARS-CoV-2 evasion from NCOA7-mediated restriction. PloS Pathog. (2021) 17:e1009820. doi: 10.1371/journal.ppat.1009820
159. Erickson R, Huang C, Allen C, Ireland J, Roth G, Zou Z, et al. SARS-CoV-2 infection of human lung epithelial cells induces TMPRSS-mediated acute fibrin deposition. Nat Commun. (2023) 14:6380. doi: 10.1038/s41467-023-42140-6
160. Luplertlop N, Missé D, Bray D, Deleuze V, Gonzalez JP, Leardkamolkarn V, et al. Dengue-virus-infected dendritic cells trigger vascular leakage through metalloproteinase overproduction. EMBO Rep. (2006) 7:1176–81. doi: 10.1038/sj.embor.7400814
161. Huang YH, Liu CC, Wang ST, Lei HY, Liu HL, Lin YS, et al. Activation of coagulation and fibrinolysis during dengue virus infection. J Med Virol. (2001) 63:247–51. doi: 10.1002/1096-9071(200103)63:3<247::AID-JMV1008>3.0.CO;2-F
162. Kong MY, Whitley RJ, Peng N, Oster R, Schoeb TR, Sullender W, et al. Matrix metalloproteinase-9 mediates RSV infection in vitro and in vivo. Viruses. (2015) 7:4230–53. doi: 10.3390/v7082817
163. Li W and Shen HH. Effect of respiratory syncytial virus on the activity of matrix metalloproteinase in mice. Chin Med J (Engl). (2007) 120:5–11. doi: 10.1097/00029330-200701010-00002
164. Diemer C, Schneider M, Seebach J, Quaas J, Frösner G, Schätzl HM, et al. Cell type-specific cleavage of nucleocapsid protein by effector caspases during SARS coronavirus infection. J Mol Biol. (2008) 376:23–34. doi: 10.1016/j.jmb.2007.11.081
165. Zhou J, Stohlman SA, Atkinson R, Hinton DR, and Marten NW. Matrix metalloproteinase expression correlates with virulence following neurotropic mouse hepatitis virus infection. J Virol. (2002) 76:7374–84. doi: 10.1128/JVI.76.15.7374-7384.2002
166. Zuo X, Pan W, Feng T, Shi X, and Dai J. Matrix metalloproteinase 3 promotes cellular anti-dengue virus response via interaction with transcription factor NFκB in cell nucleus. PloS One. (2014) 9:e84748. doi: 10.1371/journal.pone.0084748
167. Bally I, Drumont G, Rossi V, Guseva S, Botova M, Reiser JB, et al. Revisiting the interaction between complement lectin pathway protease MASP-2 and SARS-CoV-2 nucleoprotein. Front Immunol. (2024) 15:1419165. doi: 10.3389/fimmu.2024.1419165
168. Zaruba M, Chen HW, Pietsch OF, Szakmary-Braendle K, Auer A, Mötz M, et al. ADAM17 is an essential factor for the infection of bovine cells with pestiviruses. Viruses. (2022) 14:381. doi: 10.3390/v14020381
169. Rubina A, Patel M, Nightingale K, Potts M, Fielding CA, Kollnberger S, et al. ADAM17 targeting by human cytomegalovirus remodels the cell surface proteome to simultaneously regulate multiple immune pathways. Proc Natl Acad Sci U.S.A. (2023) 120:e2303155120. doi: 10.1073/pnas.2303155120
170. Fang A, Yuan Y, Huang F, Wang C, Tian D, Zhou R, et al. Lab-attenuated rabies virus facilitates opening of the blood-brain barrier by inducing matrix metallopeptidase 8. J Virol. (2022) 96:e0105022. doi: 10.1128/jvi.01050-22
171. Hirakawa S, Kojima T, Obata K, Okabayashi T, Yokota S, Nomura K, et al. Marked induction of matrix metalloproteinase-10 by respiratory syncytial virus infection in human nasal epithelial cells. J Med Virol. (2013) 85:2141–50. doi: 10.1002/jmv.23718
172. Shirato K, Kawase M, and Matsuyama S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol. (2013) 87:12552–61. doi: 10.1128/JVI.01890-13
173. Shapira T, Monreal IA, Dion SP, Buchholz DW, Imbiakha B, Olmstead AD, et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature. (2022) 605:340–8. doi: 10.1038/s41586-022-04661-w
174. Pérez-Vargas J, Lemieux G, Thompson CAH, Désilets A, Ennis S, Gao G, et al. Nanomolar anti-SARS-CoV-2 Omicron activity of the host-directed TMPRSS2 inhibitor N-0385 and synergistic action with direct-acting antivirals. Antiviral Res. (2024) 225:105869. doi: 10.1016/j.antiviral.2024.105869
175. Li K, Meyerholz DK, Bartlett JA, and McCray PB, Jr. The TMPRSS2 inhibitor nafamostat reduces SARS-CoV-2 pulmonary infection in mouse models of COVID-19. mBio. (2021) 12:e0097021. doi: 10.1128/mBio.00970-21
176. Fraser BJ, Wilson RP, Ferková S, Ilyassov O, Lac J, Dong A, et al. Structural basis of TMPRSS11D specificity and autocleavage activation. Nat Commun. (2025) 16:4351. doi: 10.1038/s41467-025-59677-3
177. Van der Gucht W, Leemans A, De Schryver M, Heykers A, Caljon G, Maes L, et al. Respiratory syncytial virus (RSV) entry is inhibited by serine protease inhibitor AEBSF when present during an early stage of infection. Virol J. (2017) 14:157. doi: 10.1186/s12985-017-0824-3
178. Sielaff F, Böttcher-Friebertshäuser E, Meyer D, Saupe SM, Volk IM, Garten W, et al. Development of substrate analogue inhibitors for the human airway trypsin-like protease HAT. Bioorg Med Chem Lett. (2011) 21:4860–4. doi: 10.1016/j.bmcl.2011.06.033
179. Kouretova J, Hammamy MZ, Epp A, Hardes K, Kallis S, Zhang L, et al. Effects of NS2B-NS3 protease and furin inhibition on West Nile and Dengue virus replication. J Enzyme Inhib Med Chem. (2017) 32:712–21. doi: 10.1080/14756366.2017.1306521
180. Hoffmann M, Kleine-Weber H, and Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. (2020) 78:779–784.e5. doi: 10.1016/j.molcel.2020.04.022
181. Zhao MM, Zhu Y, Zhang L, Zhong G, Tai L, Liu S, et al. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies. Cell Discov. (2022) 8:53. doi: 10.1038/s41421-022-00419-w
182. Ashhurst AS, Tang AH, Fajtová P, Yoon MC, Aggarwal A, Bedding MJ, et al. Potent anti-SARS-coV-2 activity by the natural product gallinamide A and analogues via inhibition of cathepsin L. J Med Chem. (2022) 65:2956–70. doi: 10.1021/acs.jmedchem.1c01494
183. Cheng YW, Chao TL, Li CL, Chiu MF, Kao HC, Wang SH, et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. (2020) 33:108254. doi: 10.1016/j.celrep.2020.108254
184. Ströher U, Willihnganz L, Jean F, and Feldmann H. Blockage of filoviral glycoprotein processing by use of a protein-based inhibitor. J Infect Dis. (2007) 196 Suppl 2:S271–5. doi: 10.1086/520592
185. Imran M, Saleemi MK, Chen Z, Wang X, Zhou D, Li Y, et al. Decanoyl-Arg-Val-Lys-Arg-Chloromethylketone: An Antiviral Compound That Acts against Flaviviruses through the Inhibition of Furin-Mediated prM Cleavage. Viruses. (2019) 11:1011. doi: 10.3390/v11111011
186. Feng D, Ren L, Wu J, Guo L, Han Z, Yang J, et al. Permethrin as a potential furin inhibitor through a novel non-competitive allosteric inhibition. Molecules. (2023) 28:1883. doi: 10.3390/molecules28041883
187. Lam van TV, Heindl MR, Schlutt C, Böttcher-Friebertshäuser E, Bartenschlager R, Klebe G, et al. The basicity makes the difference: improved canavanine-derived inhibitors of the proprotein convertase furin. ACS Med Chem Lett. (2021) 12:426–32. doi: 10.1021/acsmedchemlett.0c00651
188. Dahms SO, Schnapp G, Winter M, Büttner FH, Schlepütz M, Gnamm C, et al. Dichlorophenylpyridine-based molecules inhibit furin through an induced-fit mechanism. ACS Chem Biol. (2022) 17:816–21. doi: 10.1021/acschembio.2c00103
189. Peng M, Watanabe S, Chan KWK, He Q, Zhao Y, Zhang Z, et al. Luteolin restricts dengue virus replication through inhibition of the proprotein convertase furin. Antiviral Res. (2017) 143:176–85. doi: 10.1016/j.antiviral.2017.03.026
190. Dahms SO, Haider T, Klebe G, Steinmetzer T, and Brandstetter H. OFF-state-specific inhibition of the proprotein convertase furin. ACS Chem Biol. (2021) 16:1692–700. doi: 10.1021/acschembio.1c00411
191. Kibler KV, Miyazato A, Yedavalli VS, Dayton AI, Jacobs BL, Dapolito G, et al. Polyarginine inhibits gp160 processing by furin and suppresses productive human immunodeficiency virus type 1 infection. J Biol Chem. (2004) 279:49055–63. doi: 10.1074/jbc.M403394200
192. Zhao H, To KKW, Sze KH, Yung TT, Bian M, Lam H, et al. A broad-spectrum virus- and host-targeting peptide against respiratory viruses including influenza virus and SARS-CoV-2. Nat Commun. (2020) 11:4252. doi: 10.1038/s41467-020-17986-9
193. Zhao H, To KKW, Lam H, Zhou X, Chan JF, Peng Z, et al. Cross-linking peptide and repurposed drugs inhibit both entry pathways of SARS-CoV-2. Nat Commun. (2021) 12:1517. doi: 10.1038/s41467-021-21825-w
194. Joushomme A, Désilets A, Champagne W, Hassanzadeh M, Lemieux G, Gravel-Trudeau A, et al. Development of ketobenzothiazole-based peptidomimetic TMPRSS13 inhibitors with low nanomolar potency. J Enzyme Inhib Med Chem. (2025) 40:2466841. doi: 10.1080/14756366.2025.2466841
195. Chu H, Hou Y, Yang D, Wen L, Shuai H, Yoon C, et al. Coronaviruses exploit a host cysteine-aspartic protease for replication. Nature. (2022) 609:785–92. doi: 10.1038/s41586-022-05148-4
196. Zhu J, Declercq J, Roucourt B, Ghassabeh GH, Meulemans S, Kinne J, et al. Generation and characterization of non-competitive furin-inhibiting nanobodies. Biochem J. (2012) 448:73–82. doi: 10.1042/BJ20120537
197. Yang HY, Zheng NQ, Li DM, Gu L, and Peng XM. Entecavir combined with furin inhibitor simultaneously reduces hepatitis B virus replication and e antigen secretion. Virol J. (2014) 11:165. doi: 10.1186/1743-422X-11-165
198. Asadipooya K, Asadipooya A, and Adatorwovor R. Combination of spironolactone and DPP-4 inhibitors for treatment of SARS-CoV-2 infection: a literature review. Arch Virol. (2024) 169:122. doi: 10.1007/s00705-024-06043-1
199. Chun K, Na Y, Kim B, Lee D, Choi J, Kim G, et al. Synergistic antiviral activity of xanthan gum and camostat against influenza virus infection. Viruses. (2025) 17:301. doi: 10.3390/v17030301
200. Essalmani R, Jain J, Susan-Resiga D, Andréo U, Evagelidis A, Derbali RM, et al. Distinctive roles of furin and TMPRSS2 in SARS-CoV-2 infectivity. J Virol. (2022) 96:e0012822. doi: 10.1128/jvi.00128-22
201. Böttcher-Friebertshäuser E, Lu Y, Meyer D, Sielaff F, Steinmetzer T, Klenk HD, et al. Hemagglutinin activating host cell proteases provide promising drug targets for the treatment of influenza A and B virus infections. Vaccine. (2012) 30:7374–80. doi: 10.1016/j.vaccine.2012.10.001
202. Boon ACM, Bricker TL, Fritch EJ, Leist SR, Gully K, Baric RS, et al. Efficacy of host cell serine protease inhibitor MM3122 against SARS-CoV-2 for treatment and prevention of COVID-19. J Virol. (2024) 98:e0190323. doi: 10.1128/jvi.01903-23
203. Wang H, Yang Q, Liu X, Xu Z, Shao M, Li D, et al. Structure-based discovery of dual pathway inhibitors for SARS-CoV-2 entry. Nat Commun. (2023) 14:7574. doi: 10.1038/s41467-023-42527-5
204. Yang H, Zhou JN, Zhang XM, Ling DD, Sun YB, Li CY, et al. Nanoengineered red blood cells loaded with TMPRSS2 and cathepsin L inhibitors block SARS-CoV-2 pseudovirus entry into lung ACE2(+) cells. Adv Mater. (2024) 36:e2310306. doi: 10.1002/adma.202310306
205. Hardes K, Becker GL, Lu Y, Dahms SO, Köhler S, Beyer W, et al. Novel furin inhibitors with potent anti-infectious activity. ChemMedChem. (2015) 10:1218–31. doi: 10.1002/cmdc.201500103
206. Xie X, Lan Q, Zhao J, Zhang S, Liu L, Zhang Y, et al. Structure-based design of pan-coronavirus inhibitors targeting host cathepsin L and calpain-1. Signal Transduct Target Ther. (2024) 9:54. doi: 10.1038/s41392-024-01758-8
207. Li Y, Wang K, Sun H, Wu S, Wang H, Shi Y, et al. Omicsynin B4 potently blocks coronavirus infection by inhibiting host proteases cathepsin L and TMPRSS2. Antiviral Res. (2023) 214:105606. doi: 10.1016/j.antiviral.2023.105606
208. Wang D, Li C, Chiu MC, Yu Y, Liu X, Zhao X, et al. SPINK6 inhibits human airway serine proteases and restricts influenza virus activation. EMBO Mol Med. (2022) 14:e14485. doi: 10.15252/emmm.202114485
209. Pilgram O, Keils A, Benary GE, Müller J, Merkl S, Ngaha S, et al. Improving the selectivity of 3-amidinophenylalanine-derived matriptase inhibitors. Eur J Med Chem. (2022) 238:114437. doi: 10.1016/j.ejmech.2022.114437
210. Gamba D, van Eijk N, Lányi K, Monostory K, Steinmetzer T, Marosi A, et al. PK/PD investigation of antiviral host matriptase/TMPRSS2 inhibitors in cell models. Sci Rep. (2024) 14:16621. doi: 10.1038/s41598-024-67633-2
211. Mao L, Shaabani N, Zhang X, Jin C, Xu W, Argent C, et al. Olgotrelvir, a dual inhibitor of SARS-CoV-2 M(pro) and cathepsin L, as a standalone antiviral oral intervention candidate for COVID-19. Med. (2024) 5:42–61.e23. doi: 10.1016/j.medj.2024.01.013
212. Kolykhalov AA, Agapov EV, and Rice CM. Specificity of the hepatitis C virus NS3 serine protease: effects of substitutions at the 3/4A, 4A/4B, 4B/5A, and 5A/5B cleavage sites on polyprotein processing. J Virol. (1994) 68:7525–33. doi: 10.1128/jvi.68.11.7525-7533.1994
213. Falgout B, Pethel M, Zhang YM, and Lai CJ. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol. (1991) 65:2467–75. doi: 10.1128/jvi.65.5.2467-2475.1991
214. Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. (2020) 368:1331–5. doi: 10.1126/science.abb4489
215. Khan U, Mubariz M, Khlidj Y, Nasir MM, Ramadan S, Saeed F, et al. Safety and Efficacy of Camostat Mesylate for Covid-19: a systematic review and Meta-analysis of Randomized controlled trials. BMC Infect Dis. (2024) 24:709. doi: 10.1186/s12879-024-09468-w
216. Chen J, Song Y, Yang W, Guo J, Zhang S, Wan D, et al. Enzyme and reduction dual-responsive peptide micelles as nanocarriers for smart drug delivery. ACS Appl Nano Mater. (2023) 6:16179–88. doi: 10.1021/acsanm.3c02059
217. Li L, Li Y, Ma X, Li P, Zeng S, Jiang F, et al. A pomalidomide-based gefitinib PROTAC degrader effectively inhibits lung cancer progression in EGFR-TKIs-acquired resistant models by targeting EGFR degradation and ETFA-mediated ATP generation. Bioorg Chem. (2025) 164:108864. doi: 10.1016/j.bioorg.2025.108864
Keywords: host proteases, viral infection, immune response, regulatory mechanism, broad-spectrum antiviral agents
Citation: Xia Q, Liu X and Huang H (2025) Host proteases: key regulators in viral infection and therapeutic targeting. Front. Immunol. 16:1671173. doi: 10.3389/fimmu.2025.1671173
Received: 22 July 2025; Accepted: 25 August 2025;
Published: 18 September 2025.
Edited by:
Ralph A Tripp, University System of Georgia, United StatesReviewed by:
Narva Deshwar Kushwaha, Wayne State University, United StatesRishikesh Lotke, Fraunhofer Institute for Cell Therapy and Immunology (IZI), Germany
Jacco Boon, WashU, United States
Copyright © 2025 Xia, Liu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiongrong Xia, eGlhcXJAc3R1LmNkdXRjbS5lZHUuY24=; Xiaohua Liu, eGlhb2h1YWxpdTIwMjNAMTYzLmNvbQ==
†These authors have contributed equally to this work