 Tingyan He
Tingyan He Junbin Ou2†
Junbin Ou2† Linlin Wang
Linlin Wang Jun Yang
Jun Yang- 1Department of Rheumatology and Immunology, Shenzhen Children’s Hospital, Shenzhen, China
- 2Department of Pediatric Rheumatology and Immunology, Boai Hospital of Zhongshan, Zhongshan, China
Background: UNC93B1 is a transmembrane protein essential for regulating toll-like receptors (TLRs). Pathogenic variants in human UNC93B1 have recently been described in a limited number of patients with childhood systemic lupus erythematosus and chilblain lupus.
Methods: Demographic data, medical history, and physical examination findings were obtained. Whole-exome sequencing and Sanger sequencing were performed. The interferon-stimulated gene (ISG) score was analyzed.
Results: We report four patients with a novel UNC93B1 c.1007G>A p.R336H variant, including three presenting with juvenile arthritis or rheumatoid arthritis, and one with a predominant phenotype of ITP. In addition to arthritis, these patients presented with interstitial pneumonia as the dominant feature. ISG expression analysis during active disease revealed overexpression of IFN-stimulated cytokine genes and an elevated ISG score in P4. To date, 25 cases with UNC93B1 pathogenic mutations have been reported, including 13 with childhood-onset systemic lupus erythematosus (SLE) and 12 with cutaneous lupus. Management of these patients has varied based on clinical manifestations.
Conclusion: UNC93B1-mutation-associated disease should be considered in the context of early-onset autoimmune disease, especially childhood-onset SLE, juvenile arthritis, and rheumatoid arthritis. Pulmonary involvement should also be monitored in these patients.
Introduction
Toll-like receptors (TLRs) recognize pathogen-derived nucleic acids and initiate signals critical for immune responses to infection. A subset of TLRs, such as TLR7 and TLR9, also recognizes self-RNA and self-DNA, respectively. Enhanced TLR7 signaling contributes to autoimmune diseases such as systemic lupus erythematosus (SLE) (1). UNC93B1 is a transmembrane protein essential for regulating TLRs, particularly TLR7, TLR8, and TLR9. UNC93B1 regulates the trafficking of TLRs from the endoplasmic reticulum to endosomes and is contributes to the lysosomal degradation of TLR7 and TLR8 (1–3). UNC93B1 pathogenic mutations may impair TLR degradation, leading to receptor accumulation, hyper-responsiveness to self-nucleic acids, and hyperactivation of type I interferon and NF-κB signaling pathways (4). Gain-of-function (GOF) human UNC93B1 variants have recently been described in a limited number of patients with childhood systemic lupus erythematosus and chilblain lupus (1, 4–8). Juvenile arthritis was observed in only one patient (5). To date, as with other newly identified inborn errors of immunity (IEIs), the phenotype of UNC93B1-mutation-associated disease is not fully understood. Herein, we describe the clinical manifestations of four patients with a novel UNC93B1 variant to expand the clinical spectrum of UNC93B1-mutation-associated disease.
Methods
Subjects
Demographic data, medical history, and physical examination findings of the patients were obtained through direct interviews and review of patients’ clinical records in Shenzhen Children’s Hospital and Zhongshan Boai Hospital. Written informed consent was obtained based on the principles of the ethics committee of Shenzhen Children’s Hospital.
Whole exome sequencing
Genomic DNA was extracted from isolated peripheral blood cells. Whole-exome sequencing and Sanger sequencing were performed by MyGenostics (Beijing, China). The FASTQ files were mapped to the human reference genome (hg19). The functional effects of variants were predicted using PolyPhen-2 and MutationTaster, and amino acid conservation across species was analyzed. The structural effects of variants were predicted using the AlphaFold Protein Structure Database and PyMOL.
Interferon-stimulated gene expression analysis
ISG expression and ISG score were analyzed according to a previously described protocol (9). Total RNA was extracted from PBMCs using the RNAqueous Kit (Thermo Fisher, US) and cDNA was synthesized using Transcriptor First Strand cDNA Synthesis Kit (Roche, Germany) according to the manufacturer’s instructions. The expression of six ISGs and one housekeeping gene was measured by RT-PCR using TaqMan Fast Advanced Master Mix and Gene Expression Assay (FAM) (Thermo Fisher, US) with the following primers: IFI27 (Hs01086370_m1), IFI44L (Hs00199115_m1), IFIT1 (Hs00356631_g1), ISG15 (Hs00192713_m1), RSAD2 (Hs01057264_m1), SIGLEC1 (Hs00988063_m1), and GAPDH (Hs03929097_g1). The relative expression of each gene was normalized to GAPDH and compared with a control sample (a pooled sample from 10 healthy controls), using the 2−ΔΔCt method. The median relative expression of all six ISGs was used to calculate the IFN score for each sample.
Results
Clinical manifestations of four patients
The proband, P1, a six-year-old girl, was born at 34 weeks’ gestational age to non-consanguineous parents. At 14 months of age, developed bilateral knee arthritis, accompanied by touch intolerance and claudication, and gradually developed pain and limited mobility in multiple interphalangeal joints (Figure 1A). She was admitted to the Women’s and Children’s Medical Center. Laboratory findings included positive ANA, rheumatoid factor (RF), and anti-CCP antibodies, along with elevated levels of C-reactive protein (CRP; maximum of 48.21 mg/L) and erythrocyte sedimentation rate (ESR; maximum of 43 mm/h). MRI showed effusions and marked synovitis in multiple joints, including the knees, interphalangeal, wrist, ankle, and sacroiliac joints (Figure 1C). She was diagnosed with juvenile idiopathic arthritis and treatment with methylprednisolone, methotrexate (MTX), and Enacept, achieving clinical remission for more than 6 months. However, after discontinuing Enacept on her own for three months, she experienced a disease flare and switched to adalimumab, which rapidly resolved the arthralgia. After 13 months of maintenance therapy with adalimumab, she discontinued treatment for financial reasons and redeveloped arthralgia and polyarticular mobility limitation accompanied by interstitial pneumonia. Since then, she has been treated with Enacept and tofacitinib. She had no joint symptoms or signs other than transient wrist and ankle joint pain following respiratory infections.
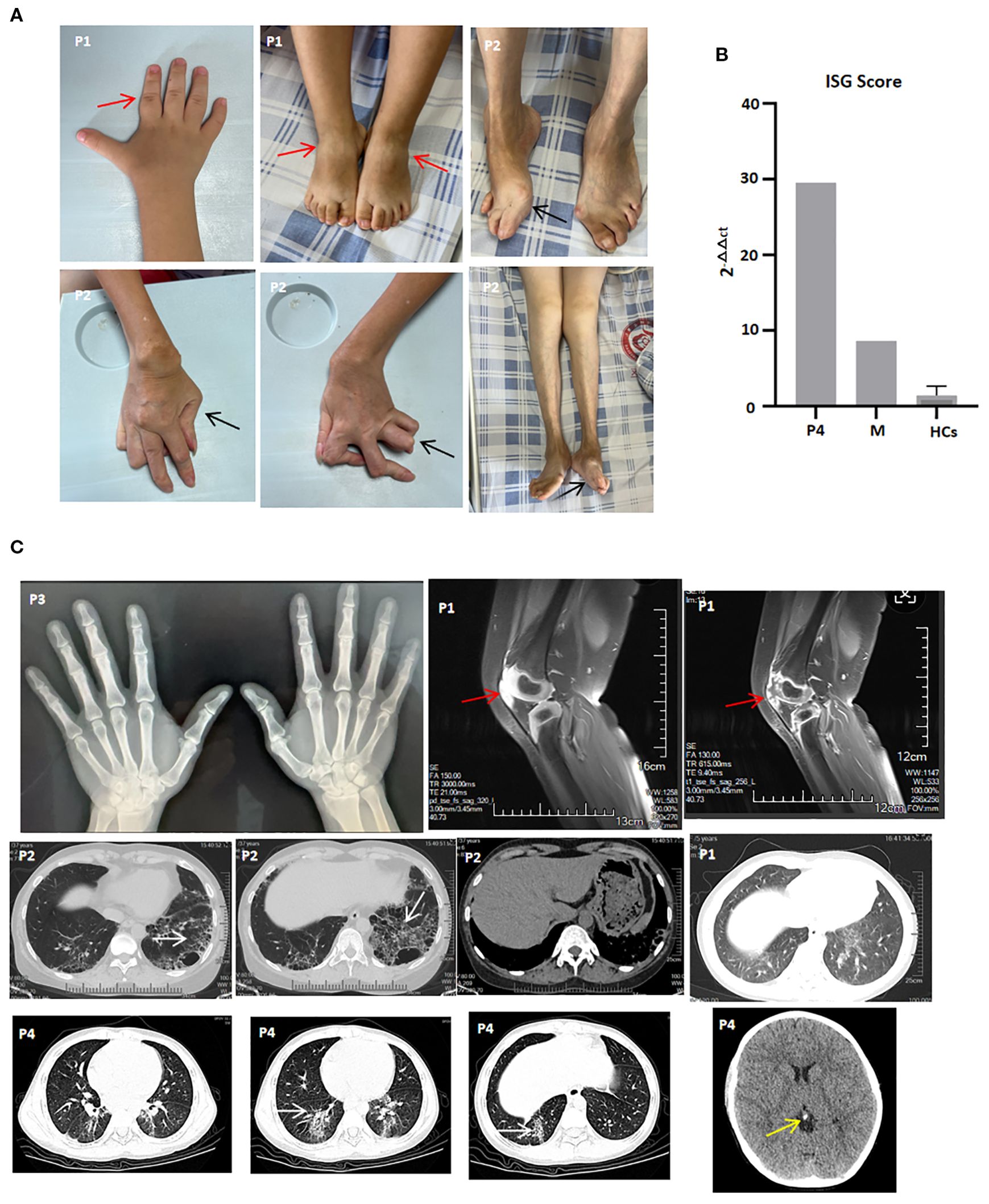
Figure 1. Clinical features and imaging manifestations of patients with the UNC93B1 c.1007G>A p.R336H heterozygous variation. (A) Active arthritis (P1) and joint sequelae (P2). Red arrow indicates joint swelling, and black arrow indicates joint deformity; (B) ISG score of P4, his mother (M) and healthy controls (HCs), shown as one representative result of two repeated experiments; (C) X-ray of both hands (P3), knee MRI (P1), lung CT (P1, P2, and P4), and brain CT (P4). White arrow indicates interstitial lung diseases, red arrow indicates joint effusions and synovitis, and yellow arrow indicates calcification.
P2, the proband, was P1’s mother. At two years of age, she began to experience polyarthralgia and subsequently developed polyarticular deformities She had previously received irregular treatment with prednisone, MTX, and nonsteroidal anti-inflammatory drugs (NSAIDs). At 32 years of age, she was referred to Zhongshan Bo Ai Hospital, presenting with ulnar deviation of the hands and feet; swan neck and button deformity of both hands; generalized joint tenderness; and swelling of both knees (Figure 1A). Laboratory findings included positive ANA, rheumatoid factor (RF), and anti-CCP antibodies, along with elevated ESR (maximum of 65 mm/h). High-resolution CT of the lungs showed central emphysema in both lobes, multiple bullae, and interstitial fibrosis in the upper and lower lobes of both lungs (Figure 1C). During the last two years of follow-up, she did not achieve ACR 30 remission and responded poorly to prednisone, MTX, Enacept, and JAK inhibitors, including tofacitinib, baricitinib, and upadacitinib.
P3, a 40-year-old man, was the proband’s uncle and P2’s older brother. During adolescence, he began to experience arthralgia in the index finger and knee joints. He had previously received prednisone and MTX as initial treatment for three years before self-discontinuing MTX. He was treated with low-dose prednisone and hydroxychloroquine as maintenance therapy. At the last follow-up, although X-rays of both hands showed narrowing of the proximal interphalangeal joint space of the 2nd to 5th fingers, he denied symptoms such as arthralgia, cough, or articular mobility limitation (Figure 1C).
P4, a nine-year-old boy, was P3’s son. At the age of seven, he presented with skin purpura and was diagnosed with immune thrombocytopenia (ITP). Laboratory findings included positive ANA and decreased serum C4 levels. CRP and ESR levels were normal. He received intravenous immunoglobulin (IVIG) treatment, after which his thrombocyte levels returned to normal. However, because thrombocytopenia recurred frequently, prednisone and cyclosporin were added as maintenance therapy. Interferon-stimulated gene (ISG) expression analysis during the active disease state revealed overexpression of IFN-stimulated cytokine genes and an elevated IFN score (Figure 1B). He switched to mycophenolate mofetil (MMF), tofacitinib, and finally tacrolimus due to dose-dependent prednisone use, poor response to cyclosporin, MMF, and tofacitinib, and the development of interstitial pneumonia (Figure 1C). At the last follow-up, he denied symptoms such as arthralgia, cough, or shortness of breath; his thrombocyte levels had returned to normal, and interstitial pneumonia was partially improved.
UNC93B1 variation
Whole-exome sequencing revealed a heterozygous UNC93B1 c.1007G>A p.R336H variant in the four patients, but no variants in healthy family members (Figures 2A–C). The predicted values from PolyPhen_2, Mutation Taster, and GERP++ were 0.997, 1, and 5.09, respectively, suggesting probably damaging effects, disease causation, and high conservation (Figure 2D). This heterozygous variant was absent from ESP6500si, ExAC_ALL, ExAC_EAS, or Inhouse databases and was further confirmed in all patients by Sanger sequencing. No pathogenic variants were identified in other genes related to autoinflammation or autoimmunity.
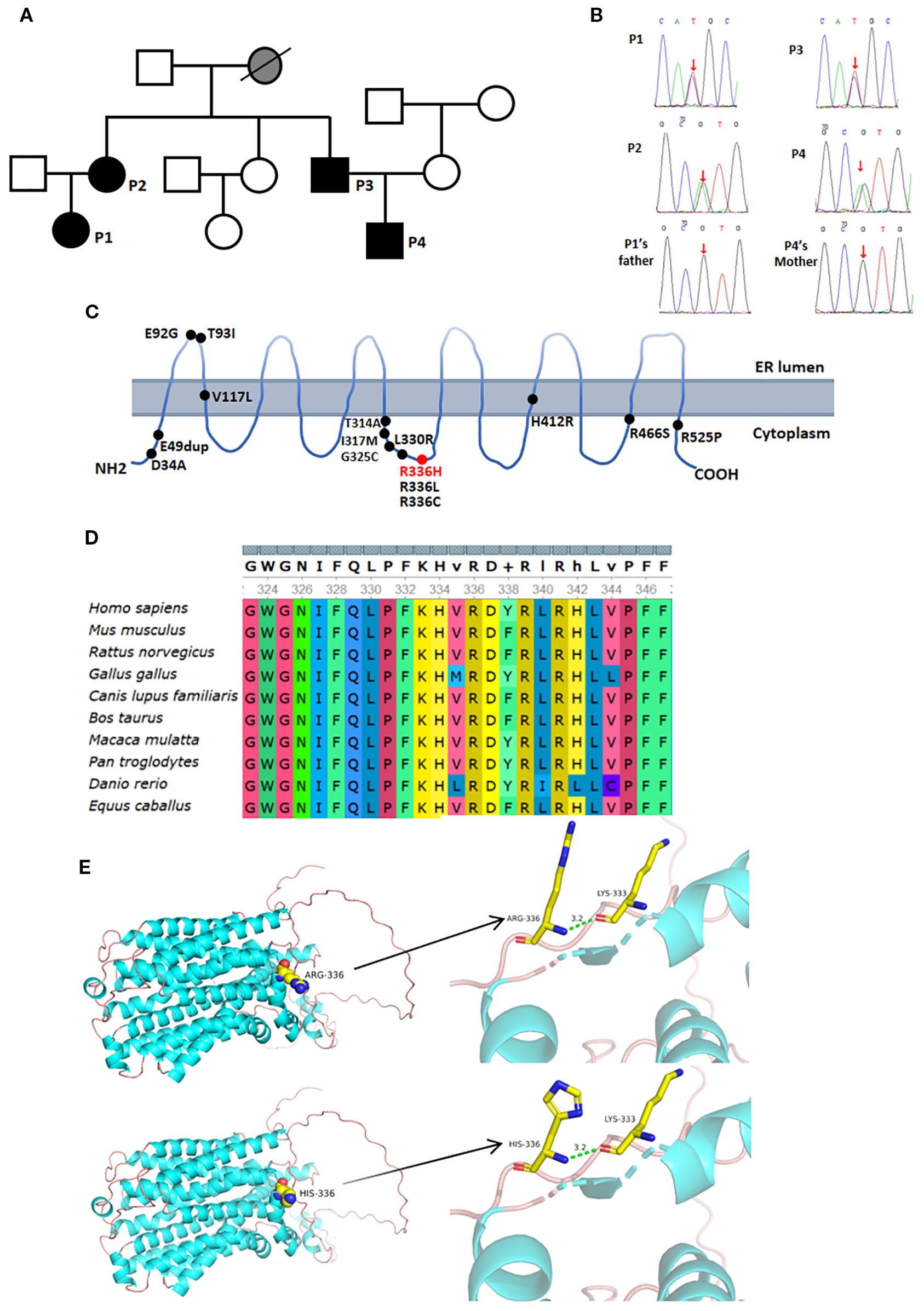
Figure 2. Genetic analysis and imaging manifestations of the patient. (A) Family pedigree, black shows patients with the UNC93B1 c.1007G>A p.R336H heterozygous variation; (B) Red arrow indicates the UNC93B1 c.1007G>A p.R336H heterozygous variation identified by Sanger sequencing; (C) Red font denotes the variation in our patients, while black font indicates previously reported variations; (D) High conservation of p.R336 across species; (E) Protein models of UNC93B1 with and without the c.1007G>A p.R336H variation.
The UNC93B1 c.1007G>A heterozygous variant resulted in an Arg-to-His substitution at amino acid 336, which was predicted by PyMOL not to affect hydrogen bonding (Figure 2E).
Review of literature
To date, 25 cases of UNC93B1 pathogenic mutation have been reported across five studies, including eight males and 17 females (Table 1) (1, 4–7). All patients with available data developed autoimmune disease in childhood. Nearly half of the patients (12 of 25) presented with childhood-onset systemic lupus erythematosus (SLE). Another 12 patients had cutaneous lupus, including one with extended oligoarticular juvenile idiopathic arthritis and a neuroinflammatory movement disorder. One patient presented with autoimmune hemolytic anemia alone. The major immunological abnormalities included positive autoantibodies, decreased C3 and C4 levels, and a positive Coombs’ test. Five patients with cutaneous lupus or familial chilblain lupus were mainly treated with JAK1/2 inhibition. None of the patients received HSCT. Further details are available in Table 1.
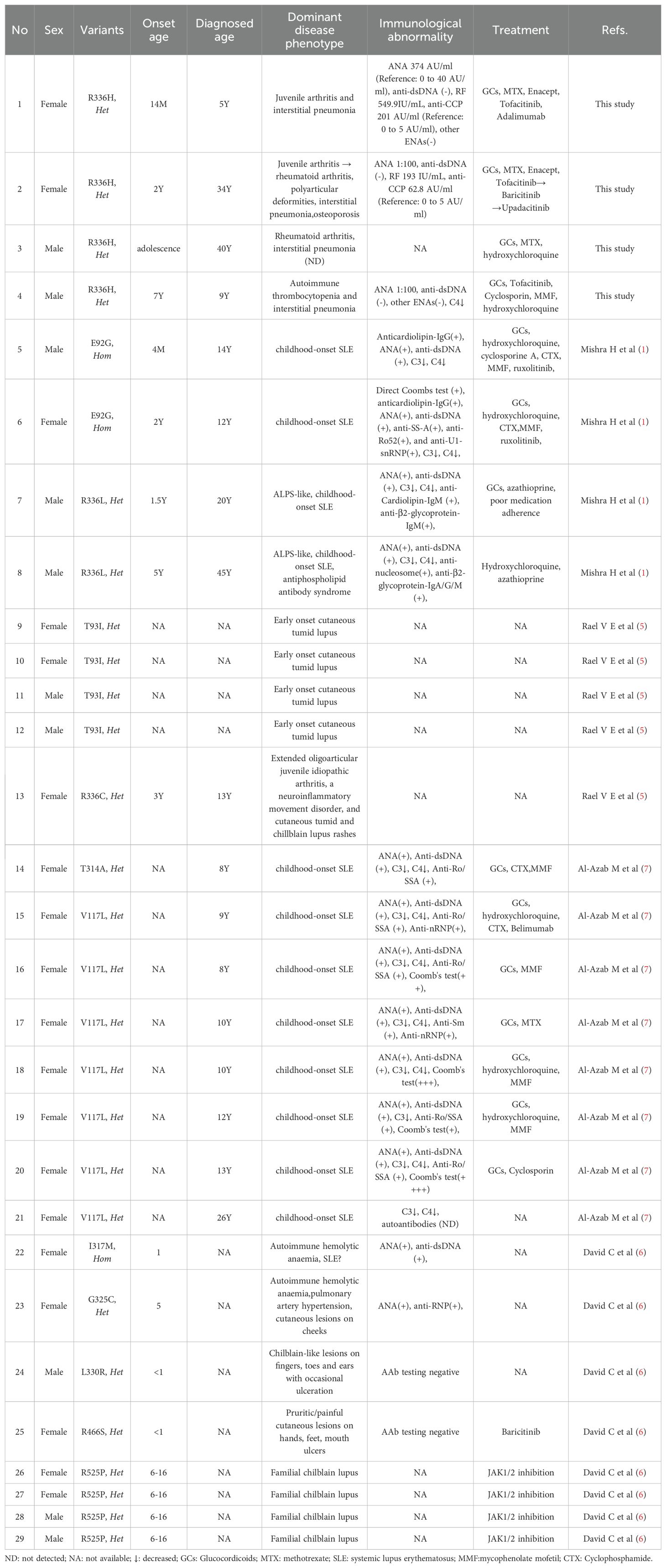
Table 1. An overview of the clinical and immunological findings of patients with UNC93B1 pathogenic mutation.
Discussion
Herein, we report four patients harboring a novel heterozygous c.1007G>A p.R336H variant in UNC93B1, suggesting autosomal dominant inheritance. This variation was absent in healthy family members, is highly conserved, and not found in population databases. At the same amino location, both p.R336L and p.R336C variants in UNC93B1 have been reported to increase TLR7 and TLR8 responses without affecting TLR9 responses (1, 5, 10). According to the American College of Medical Genetics and Genomics, pathogenicity classification was defined as pathogenic (1 PS + 3 PM + 4 PP) (11). Therefore, the heterozygous p.R336H variant may account for this familial clustering of autoimmune diseases and represents a novel pathogenic variant in UNC93B1.
In this study, three patients had Juvenile arthritis or rheumatoid arthritis, except for P4, whose predominant disease phenotype was ITP. JIA has also been reported in a patient with a different p.R336C variant at the same locus. This observation suggests a possible association between pathogenic variants at the p.R336 locus and juvenile arthritis or rheumatoid arthritis. By contrast, pathogenic variants distributed across other domains have been associated with SLE and cutaneous lupus. In summary, identical pathogenic variants may result in varied clinical phenotypes, and different variants may cause the same disease phenotype. Inclusion of additional patients will help clarify potential genotype–phenotype correlations in UNC93B1-variant-associated disease.
In addition to arthritis, our patients presented with dominant interstitial pneumonia, a feature common in STING-associated vasculopathy with onset in infancy (SAVI) and COPA syndrome (12, 13). Previous studies have shown that UNC93B1 interacts with STING and suppresses STING-activated downstream signaling by delivering STING to the lysosomes for degradation (14, 15). Pathogenic variants at the p.R336 locus may impair STING degradation, leading to overactivation of the cGAS–STING signaling pathway, which could partly contribute to the development of interstitial pneumonia.
UNC93B1 variant-associated effects include Toll-like receptor-related responses, hyperactivation of the type I interferon response, and overactivation of the NF-κB and cGAS-STING signaling pathways (4). Thus, in addition to autoimmune disease phenotypes such as rheumatoid arthritis and SLE, UNC93B1 pathogenic variations may theoretically cause autoinflammatory diseases. Inclusion of additional patients in the future will help confirm this possibility.
Proband P1 concurrently received etanercept and tofacitinib for systemic severe inflammation and arthritis. P3 was maintained on low-dose prednisone and hydroxychloroquine. Therefore, management of patients with UNC93B1-mutation-associated diseases should be individualized according to clinical manifestations. Patients with an SLE phenotype can be treated in line with the ACR SLE guidelines. Patients with chilblain lupus phenotype may receive JAK inhibitors as first-line therapy, which can also be used in cases of interstitial pneumonia, refractory SLE phenotype, or refractory arthritis phenotype. Anifrolimumab, a type I IFN receptor antagonist, may serve as an alternative effective treatment for patients with these phenotypes. Hematopoietic stem cell transplantation has not yet been reported, and gene therapy is not currently available. Further clinical trials with larger patient cohorts will help optimize disease management.
The limitation of this study is the lack of experimental validation for this variation, and the functional link to pathogenicity was defined only by in silico predictions and analogy with other variants in UNC93B1.
Conclusion
We have described four patients with a novel pathogenic variant in UNC93B1. Their disease phenotypes, including interstitial pneumonia, juvenile arthritis, and rheumatoid arthritis, expand the clinical phenotype spectrum of UNC93B1-mutation-associated diseases. Management should be individualized.
Data availability statement
The names of the repository/repositories and accession number(s) can be found in the article/supplementary material. Further inquiries about the datasets can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the ethics committee of Shenzhen Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
TH: Writing – original draft, Funding acquisition, Formal analysis. JO: Data curation, Writing – original draft. LH: Writing – original draft, Data curation. XZ: Writing – original draft, Data curation. LW: Investigation, Methodology, Writing – original draft. XL: Writing – review & editing. JY: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. TH received a grant from the Shenzhen Clinical Research Center (20220819113341005), a grant from the Shenzhen Clinical Research Center for Child Health and Disease (szcrc2024_004), and a grant from the National Natural Science Foundation of China (82302056). JY received a grant from the Sanming Project of Medicine in Shenzhen (SZSM202411012).
Acknowledgments
We thank the patients and their family members for their participation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mishra H, Schlack-Leigers C, Lim EL, Thieck O, Magg T, Raedler J, et al. Disrupted degradative sorting of TLR7 is associated with human lupus. Sci Immunol. (2024) 9:eadi9575. doi: 10.1126/sciimmunol.adi9575
2. Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, et al. The chaperone UNC93B1 regulates toll-like receptor stability independently of endosomal TLR transport. Immunity. (2018) 48:911–22. doi: 10.1016/j.immuni.2018.04.011
3. Majer O, Liu B, Woo BJ, Kreuk L, Van Dis E, Barton GM, et al. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature. (2019) 575:371–4. doi: 10.1038/s41586-019-1611-7
4. Wolf C, Lim EL, Mokhtari M, Kind B, Odainic A, Lara-Villacanas E, et al. UNC93B1 variants underlie TLR7-dependent autoimmunity. Sci Immunol. (2024) 9:eadi9769. doi: 10.1126/sciimmunol.adi9769
5. Rael VE, Yano JA, Huizar JP, Slayden LC, Weiss MA, Turcotte EA, et al. Large-scale mutational analysis identifies UNC93B1 variants that drive TLR-mediated autoimmunity in mice and humans. J Exp Med. (2024) 221(8):e20232005. doi: 10.1084/jem.20232005
6. David C, Arango-Franco CA, Badonyi M, Fouchet J, Rice GI, Didry-Barca B, et al. Gain-of-function human UNC93B1 variants cause systemic lupus erythematosus and chilblain lupus. J Exp Med. (2024) 221(8):e20232066. doi: 10.1084/jem.20232066
7. Al-Azab M, Idiiatullina E, Liu Z, Lin M, Hrovat-Schaale K, Xian H, et al. Genetic variants in UNC93B1 predispose to childhood-onset systemic lupus erythematosus. Nat Immunol. (2024) 25:969–80. doi: 10.1038/s41590-024-01846-5
8. Ellyard JI and Gantier MP. Uncovering new causes of monogenic systemic lupus erythematosus. Immunol Cell Biol. (2024) 102:651–4. doi: 10.1111/imcb.12807
9. He T, Wang L, Huang X, Weng R, and Yang J. LACC1 deficiency leading to juvenile arthritis and anemia. Clin Immunol. (2024) 265:110290. doi: 10.1016/j.clim.2024.110290
10. Tsokos GC. The emergence of SLE-causing UNC93B1 variants in 2024. Nat Rev Rheumatol. (2025) 21:67–8. doi: 10.1038/s41584-024-01192-8
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Gul A, Aksentijevich I, Brogan P, Gattorno M, Grayson PC, Ozen S, et al. The pathogenesis, clinical presentations and treatment of monogenic systemic vasculitis. Nat Rev Rheumatol. (2025) 21:414–25. doi: 10.1038/s41584-025-01250-9
13. Melki I, Rose Y, Uggenti C, Van Eyck L, Fremond ML, Kitabayashi N, et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J Allergy Clin Immunol. (2017) 140:543–52. doi: 10.1016/j.jaci.2016.10.031
14. Zhu H, Zhang R, Yi L, Tang YD, and Zheng C. UNC93B1 attenuates the cGAS-STING signaling pathway by targeting STING for autophagy-lysosome degradation. J Med Virol. (2022) 94:4490–501. doi: 10.1002/jmv.27860
Keywords: autoimmune disease, systemic lupus erythematosus, monogenic lupus, UNC93B1, arthritis
Citation: He T, Ou J, Huang L, Zhou X, Wang L, Li X and Yang J (2025) Case Report: Novel UNC93B1 variant causes rheumatoid arthritis and interstitial pneumonia. Front. Immunol. 16:1671984. doi: 10.3389/fimmu.2025.1671984
Received: 23 July 2025; Accepted: 30 August 2025;
Published: 01 October 2025.
Edited by:
Eleonora Gambineri, University of Florence, ItalyReviewed by:
Shirkhan Amikishiyev, Biruni University, TürkiyeLeon Chang, University of Leeds, United Kingdom
Copyright © 2025 He, Ou, Huang, Zhou, Wang, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Yang, cm9nYXNhbnN6QDE2My5jb20=; Xiaolin Li, bW9vbnJpdmVyNjY2NjY2QDE2My5jb20=; Tingyan He, aGV0aW5neWFuMjAxN0BvdXRsb29rLmNvbQ==
†These authors share first authorship