 Yu Wang
Yu Wang Yue-ping Jia
Yue-ping Jia Hui-min Zeng
Hui-min Zeng- Department of Pediatrics, Peking University People’s Hospital, Peking University, Beijing, China
Background: While CD19-directed chimeric antigen receptor T-cell (CAR-T) therapy demonstrates remarkable efficacy in relapsed/refractory (R/R) ALL, its application in earlier treatment lines requires further investigation. This study aimed to evaluate the efficacy, safety, and cellular kinetics of CD19 CAR-T therapy in pediatric B-cell ALL (B-ALL) patients with minimal residual disease (MRD) positivity or chemotherapy intolerance.
Methods: Between 2017 and 2021, 50 eligible pediatric B-ALL patients (with positive MRD or chemotherapy intolerance) received CD19 CAR-T therapy. Efficacy endpoints included complete remission (CR), MRD-negative CR (MRD-CR), overall survival (OS), and leukemia-free survival (LFS). CAR-T cellular kinetics parameters (Cmax, AUC0-28d, persistence) were quantified via qPCR and correlated with clinical outcomes. Safety assessment covered cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and infections.
Results: At day 28 post-infusion, the CR and MRD-CR rates were 98% and 96%, respectively. With a median follow-up of 68.7 months, the 5-year OS and LFS rates were 74.9% and 67.8%. Multivariate analysis identified prolonged B-cell aplasia (BCA) duration (HR = 0.969, p = 0.021) and female sex (HR = 0.235, p = 0.032) as independent protective factors for LFS. Cellular kinetics analysis showed effective in vivo expansion in 98% of patients, with a median Cmax of 30,860 copies/μg DNA and a median time-to-peak of 10.5 days. The MRD-CR group at day 28 exhibited significantly higher Cmax and AUC0-28d (p = 0.017; p = 0.029) and superior CAR-T persistence (p = 0.030) compared to the non-MRD-CR group. Pre-infusion tumor burden levels did not significantly impact CAR-T expansion or duration. BCA duration positively correlated with CAR-T persistence (r=0.570, p < 0.001), but CAR-T expansion parameters (Cmax and AUC0-28d) did not significantly influence BCA. Regarding safety, grade ≥3 CRS occurred in 16% of patients, and ICANS in 10%. Pre-infusion MRD ≥ 10-3 was an independent predictor of severe CRS.
Conclusion: CD19 CAR-T therapy demonstrates highly effective MRD clearance and provides long-term survival benefits with a manageable safety profile in pediatric B-ALL patients with MRD positivity or chemotherapy intolerance. Effective CAR-T expansion occurs even at low tumor burdens. These findings support the potential for advancing CAR-T therapy into earlier treatment lines, although its value requires further validation in prospective studies.
Introduction
Minimal residual disease (MRD) is a critical prognostic biomarker during the treatment of acute lymphoblastic leukemia (ALL) and is closely associated with risk stratification and long-term outcomes (1, 2). The persistence or re-emergence of MRD during therapy often reflects development of chemotherapy resistance and is associated with a heightened risk of relapse (3). Moreover, a subset of patients develops severe treatment-related complications—such as prolonged cytopenias, life-threatening infections, or organ toxicities—that render them unable to tolerate further conventional chemotherapy, thereby creating an urgent need for alternative therapeutic strategies.
In the era of immunotherapy, CAR-T cell therapy has significantly improved outcomes for patients with relapsed or refractory (R/R) ALL, achieving remarkable complete remission (CR) and MRD negativity rate (4). Building on this success, there is growing interest in exploring CAR-T therapy in earlier treatment phases—including MRD-positive settings or as a consolidation strategy—for patients with high-risk features or chemotherapy intolerance. However, several theoretical and practical concerns remain:
1. The low tumor burden in CR may result in suboptimal CAR-T expansion due to reduced antigen exposure.
2. Clinical experience and robust efficacy data regarding CAR-T use in preemptive or early-line settings are still limited.
Emerging evidence from studies in large B-cell lymphoma (LBCL) supports the feasibility and efficacy of CAR-T even among patients achieving CR prior to infusion (5–7). The ZUMA-12 trial, for example, reported high response rates and durable survival in high-risk LBCL patients receiving axicabtagene ciloleucel (axi-cel) as first-line treatment (8). In ALL, although clinical experience is accumulating, dedicated studies focusing on CAR-T therapy in non-bulky, MRD-positive, or chemotherapy-intolerant patients remain limited. Moreover, the cellular pharmacokinetics and expansion dynamics of CAR-T products in the setting of low disease burden are still poorly characterized.
This study was designed to address these unresolved questions by evaluating the efficacy, safety, and cellular kinetics of CD19 CAR-T therapy in a well-defined, homogeneous cohort of pediatric B-ALL patients with either MRD positivity or chemotherapy intolerance. We provide long-term follow-up data and detailed kinetic analyses that distinguish our study from previous reports, offering new insights into the potential application of CAR-T therapy in earlier lines of treatment.
Materials and methods
Patients
Between June 2017 and March 2021, 50 pediatric B-ALL patients receiving CD19 CAR-T therapy were enrolled in the trial at our center. Inclusion criteria comprised: (1) Persistent or recurrent MRD positivity during conventional chemotherapy, immunotherapy, or hematopoietic stem cell transplantation (HSCT); (2) Inability to continue conventional chemotherapy due to serious treatment-related complications (termed chemotherapy intolerance) was objectively defined by the occurrence of any of the following events attributable to chemotherapy: (i) prolonged severe myelosuppression (neutrophil count < 0.5 × 109/L or platelet count < 20 × 109/L) lasting >4 weeks despite adequate supportive care; (ii) life-threatening infection (e.g., sepsis, fungal pneumonia) during neutropenic phases; (iii) grade ≥3 non-hematologic organ toxicity (e.g., cardiotoxicity, hepatotoxicity, neurotoxicity) according to common terminology criteria for adverse events (CTCAE) v5.0; or (iv) recurrent febrile neutropenia episodes necessitating hospitalization. The study was approved by the Ethics Committee of Peking University People’s Hospital and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each patient’s parents, or their guardians.
Anti-CD19 CAR T-cell manufacture and lymphodepletion regimen
CAR-T cells were derived from peripheral blood mononuclear cells (PBMCs) collected from patient/donor leukapheresis. For patients without HSCT history, PBMCs were autologous. For those with prior HSCT, PBMCs were donor-derived. PBMCs were stimulated with dynabeads coated with anti-CD3 and anti-CD28 mAbs (Thermo Fisher Scientific). Activated T cells were transduced with lentiviral vector encoding the anti-CD19 CAR construct consisting of CD19 recognition domain, transmembrane link domain, 4-1BB intracellular domain, CD3ζ intracellular domain. After lentiviral transduction, the CAR-T-19 cells were cultured in medium supplemented with 500 IU/ml IL-2 at 37 °C/5% CO2 for approximately 5 to 11 days to obtain sufficient cells for infusion. The final product, CAR-T-19, was prepared by diluting CAR-T cells with 2% human albumin-containing saline. The main component of the final product consisted of CAR-positive CAR-T-19 cells, with a small proportion of natural killer cells. The cell preparation process met the product quality and activity requirements outlined in the guidelines for CAR-T cell therapy products. The final product exhibited a cell activity of not less than 70% and remained effective within 12 hours at the temperature ranging from 10°C to 25°C.
From Day -5 to -3 before infusion, the LD regimen was administered: Fludarabine 30 mg/m² and Cyclophosphamide 300 mg/m² daily for 3 days.
Diagnostic definitions, MRD detection, efficacy and adverse reaction assessment
1. Cytogenetic risk stratification: Based on the National Comprehensive Cancer Network (NCCN) 2024 risk stratification criteria (9).
2. MRD re-emergence: Redetection of previously negative MRD on ≥2 consecutive occasions (≥2 weeks apart) with bone marrow blasts <5% and no intervening targeted therapy. MRD positivity defined as: flow cytometry (FCM)-MRD ≥0.01% and/or qPCR detection of previously positive fusion genes.
3. MRD detection: MRD was assessed via FCM (sensitivity 10-4) on day 28 post-infusion, and then subsequently at regular intervals typically every 2 months for the first year, every 3 months from year 2 to 3, and every 6 months from year 3 to 5, with a total monitoring duration of 5 years, or as clinically indicated upon suspicion of relapse. For patients who underwent HSCT after CAR-T therapy, MRD was assessed at 0, 1, 2, 3, 4.5, 6, 9, and 12 months after HSCT, and then every 6 months thereafter until 5 years post-transplantation.
4. B-cell aplasia (BCA): Peripheral blood or bone marrow analysis showing either B cells ≤1% of white blood cells or B cells ≤3% of lymphocytes (10).
5. CRS and ICANS were graded according to the 2018 American Society for Blood and Marrow Transplantation (ASBMT) guidelines (11).
Pharmacokinetics of CAR-T cells
CAR-T cell pharmacokinetics were characterized through quantitative analysis of individual concentration-time profiles in peripheral blood, employing non-compartmental analysis (NCA) methodology. Circulating CAR-T cell levels were quantified via quantitative PCR (qPCR) using transgene-specific primers, with results expressed as copy numbers per microgram (μg) of genomic DNA. PK monitoring was performed pre- and post-infusion at 7 days, 14 days, 28 days, 12 weeks, 26 weeks, 39 weeks, 54 weeks, and 104 weeks post-infusion. From year 3 to year 5 after infusion, monitoring was performed every 6 months until becoming undetectable. Monitoring was terminated when peripheral blood CAR-T levels fell below the lower limit of quantification (LLOQ; 100 transgene copies/μg genomic DNA) on two consecutive assessments. Key PK parameters include:
Cmax: Peak level of genetically modified cells in vivo post-infusion.
Tlast: Duration of detectable CAR-T transgene in peripheral blood.
Tmax: Time to reach peak CAR-T cell expansion.
AUC0–28d: Area under the curve representing total transgene exposure during the expansion phase (Days 0–28).
Statistical analyses
The last follow-up date was June 30, 2025. OS was calculated from the date of CAR-T cell infusion to death from any cause or last follow-up, whichever occurred first. Similarly, leukemia-free survival (LFS) was measured from CAR-T infusion to relapse, death, or last follow-up, whichever came first. Data were not censored when new therapy (including chemotherapy or allo-HSCT) was initiated in the absence of active disease. Continuous variables were expressed as mean ± standard deviation for normally distributed data or median (range) for non-normally distributed data. Categorical variables were presented as counts (percentages). Categorical variables were compared using χ² or Fisher’s exact tests, while continuous variables were analyzed with t-tests (normally distributed data) or Mann-Whitney U tests (non-normally distributed data). Survival analyses employed Kaplan-Meier curves with log-rank tests, and multivariable analysis used Cox proportional hazards models. Multivariable logistic regression identified risk factors for adverse events. Statistical significance was defined as p < 0.05. Cumulative incidence of relapse (CIR) analysis was performed using R software (Bell Labs, New Providence, NJ, USA), other statistical analyses used SPSS 26.0 (SPSS Inc., Chicago, IL, USA), and figures were generated with GraphPad Prism 9.5.1 (GraphPad Software, LLC, USA).
Results
Patient characteristics
A total of 50 consecutive B-ALL patients with MRD positivity or chemotherapy intolerance who underwent CAR T-cell therapy at Peking University People’s Hospital were included in this study. Baseline characteristics are summarized in Supplementary Table S1. Enrolled patients comprised 7 cases (14%) intolerant to chemotherapy due to treatment-related severe complications, 41 cases (82%) with MRD re-emergence during therapy, and 2 cases (4%) persistently MRD-positive after achieving remission with intensive chemotherapy. The median follow-up time post-infusion was 68.7 months (range: 5.9–97.5). Patients received a median CAR-T dose of 3.68 × 106 cells/kg (range: 0.05–6.33 × 106), with a median interval from diagnosis to CAR-T infusion of 15.8 months (range: 1.5–132.0).
Treatment outcomes
Forty-nine patients (98%) achieved CR and/or complete remission with incomplete hematologic recovery (CRi), including 48 (96%) attaining MRD-negative CR/CRi at 28 days after CD19 CAR-T cell infusion. The 5-year OS rate was 74.9% [95% Confidence Interval (CI), 64.8–85.1%], and the 5-year LFS rate was 67.8% (95% CI, 57.5–78.0%). Fifteen patients (30%) experienced relapse post-infusion. The median time from infusion to relapse was 12.0 months (range: 0.5–38.4 months). Among them, 4 patients experienced CD19-negative relapse with a median relapse time of 5.0 months, while the other 11 cases were CD19-positive relapse with a median relapse time of 15.7 months. Based on CAR-T persistence, patient preference, and donor availability, 32 patients bridged to allo-HSCT after CAR-T infusion, with a median bridging interval of 2.6 months (range: 1.3–9.0).
Univariate analysis results are presented in Supplementary Table S2. All variables with p values <0.2 were included in subsequent multivariate Cox regression analyses. In the multivariate analysis, longer BCA duration (HR, 0.969; 95% CI, 0.944–0.995; p = 0.021) and female patients (HR, 0.235; 95% CI, 0.062–0.884; p = 0.032) were associated with longer LFS (Table 1A). While absence of prior transplantation and no ICANS post-infusion showed potential protective effects on OS, these associations were not statistically significant (Table 1B). Notable, no significant association between OS/LFS and the following variables: patient age, genetic risk factors, CAR-T history, pre-infusion MRD status, CAR-T cell dose, severe CRS, bridging to HSCT, or CD19 CAR-T cell expansion.
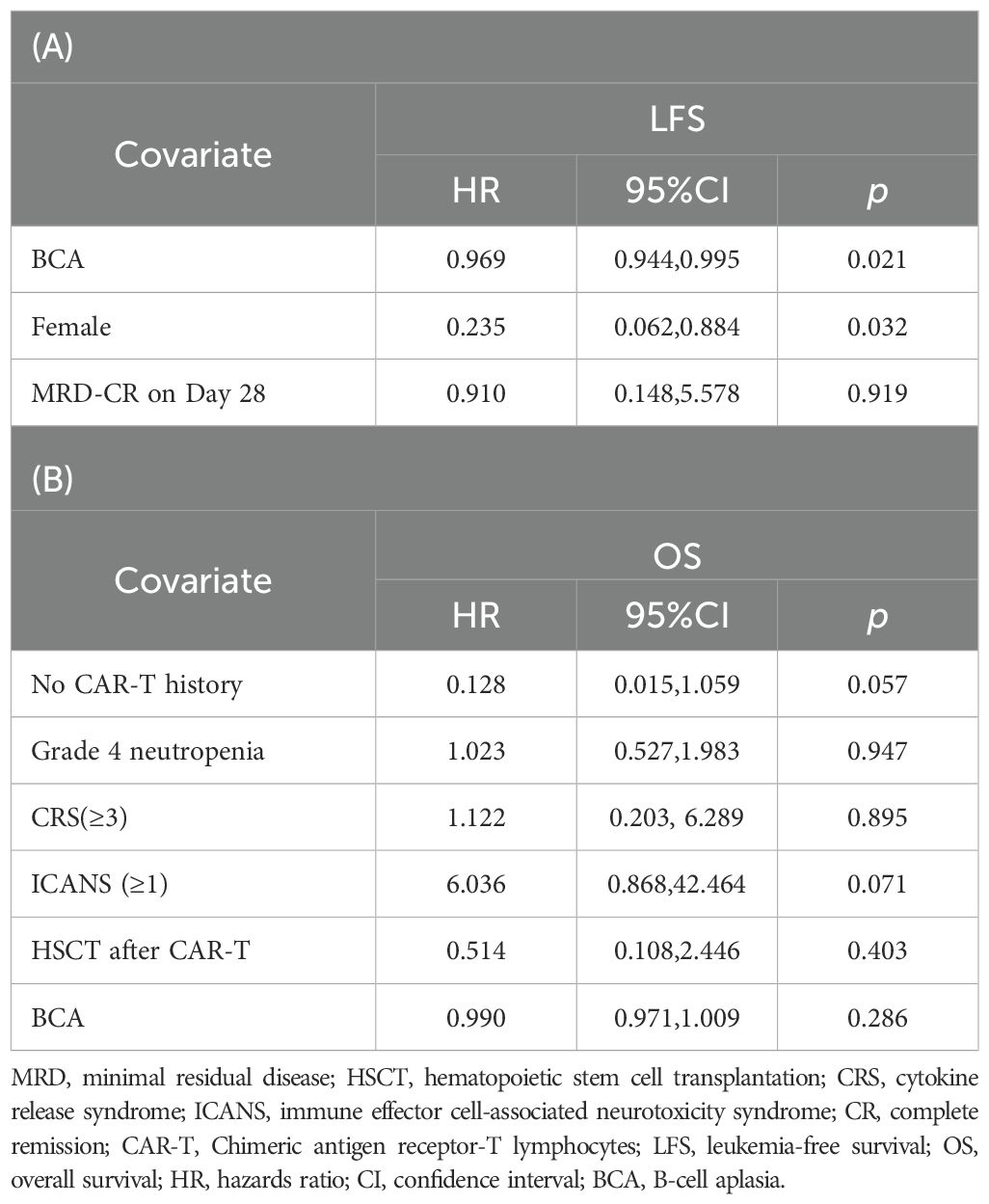
Table 1. Multivariable analysis of LFS (A) and OS (B) for subgroups.
To evaluate the effect of post-infusion bridging transplantation across different pre-CAR-T tumor burdens, bone marrow assessments were conducted at two timepoints: prior to LD (pre-LD) and post-LD. Stratified Kaplan-Meier analysis demonstrated that in patients with low tumor burden (pre-LD MRD < 10-4), bridging transplantation was associated with significantly inferior OS (median OS: 22.2 vs. 67.0 months; p = 0.022) (Figure 1) and higher non-relapse mortality (NRM: 15.4% ± 1.1% vs. 0%). The detrimental impact of transplantation on OS persisted in patients with post-LD MRD <10-4 (median OS: 43.3 vs. 68.3 months; p = 0.031). However, bridging transplantation showed no significant effect on LFS across all tumor burden strata (Figure 1). Subgroup analyses indicated that patients with high tumor burden (MRD ≥ 10-2) in the non-transplant group showed a trend toward reduced OS and LFS, though statistical significance was not reached, likely due to limited sample size (Supplementary Figure S1). Besides, no statistically significant differences were detected between different genetic risk groups (high-risk vs. low/intermediate-risk) (Supplementary Figure S2).
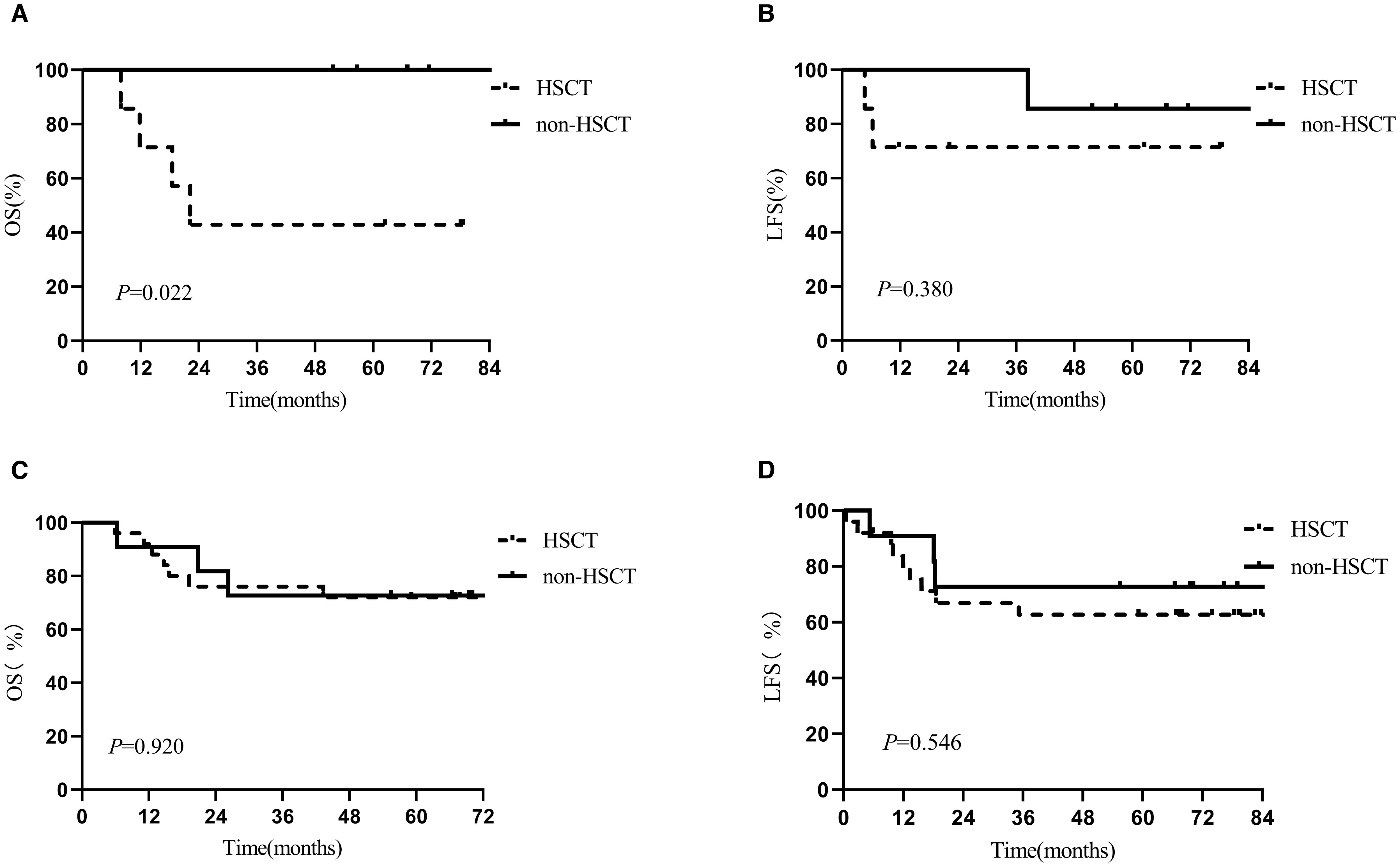
Figure 1. Kaplan-Meier estimates of 5-year outcomes in the HSCT and non-HSCT arms in different MRD group. (A) OS in MRD<10-4, (B) LFS in MRD<10-4, (C) OS in MRD≥10-4, (D) LFS in MRD≥10-4.
Toxicity
Supplementary Table S3 summarizes adverse events within 4 weeks post-CD19 CAR-T infusion. Grade 4 neutropenia was observed in 48% (24/50). The median nadir absolute neutrophil count was 0.53 × 109/L (range: 0.05–1.45 × 109/L). One patient (2%), who had a pre-LD MRD level of 3.22%, developed intermittent high fever, cytopenia, hyperferritinemia, hypofibrinogenemia, and elevated transaminases shortly after infusion, meeting the diagnostic criteria for hemophagocytic lymphohistiocytosis (HLH). Initial treatment with tocilizumab and glucocorticoids was ineffective in controlling the inflammatory response; ultimately, plasma exchange led to symptom resolution. Within the first 4 weeks post-infusion, a total of 9 patients (18.0%) experienced infections of any grade. These included bacterial bloodstream infections (n=4), viral reactivations (CMV n=2, EBV n=1), and invasive fungal infections (n=2). CRS and severe CRS (≥ grade 3) occurred in 68.0% and 16.0% of patients, respectively. ICANS incidence was 10.0%. Fourteen patients (28.0%) received tocilizumab, and 7 (14.0%) required corticosteroid therapy after infusion. None of the 5 patients with prior HSCT developed graft-versus-host disease (GVHD) post-infusion.
Univariate analysis (Table 2, Supplementary Table S4) identified that patients with high pre-infusion tumor burden (defined as MRD ≥10-3 either pre- or post-LD) had significantly increased risks of ICANS and severe CRS. CD19 CAR-T cell dose also influenced CRS severity: patients receiving doses below the median threshold (3.66 × 106 cells/kg) exhibited reduced CRS risk (p = 0.049). Both pre- and post-LD MRD levels retained independent predictive value for severe CRS when incorporated into the multivariate analysis separately. Multivariate analysis for ICANS was precluded by the limited number of events (n=5).
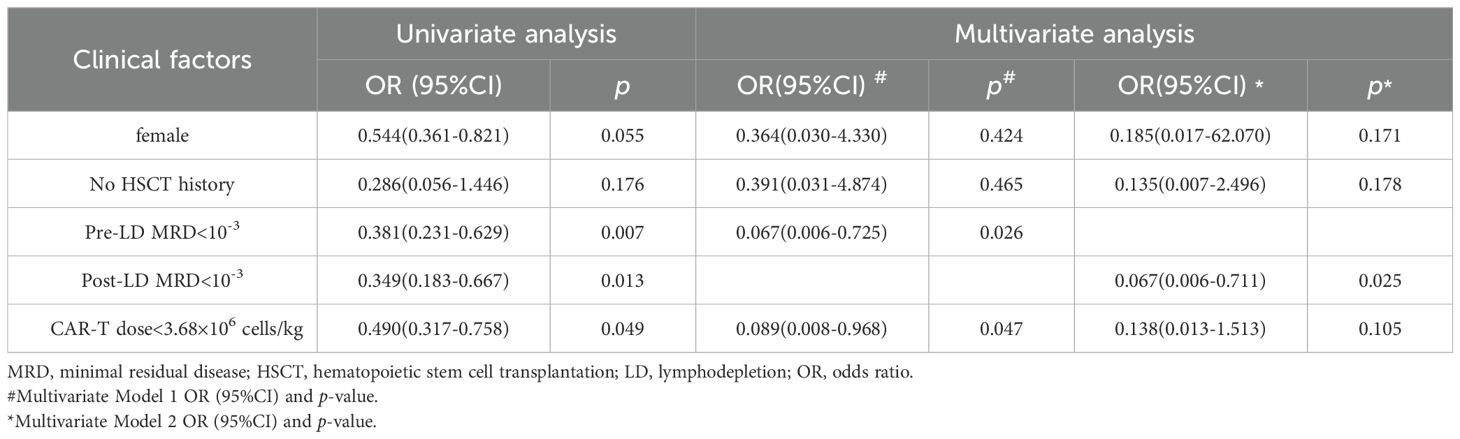
Table 2. Univariate screening (P<0.2) and multivariate analysis of clinical factors associated with grade ≥3 CRS.
Post-infusion, 78% of patients (39/50) developed elevated interleukin-6 (IL-6) levels, with a median peak concentration of 33.3 pg/mL (range 7.6 to >5000) occurring at a median of 8 days (range 1-22). Elevated IL-6 levels showed strong predictive capacity for both severe CRS (OR = 5.250) and ICANS (OR = 9.000) (p < 0.001 for both), suggesting its utility as an early-warning biomarker for CAR-T-associated toxicities.
PK of CAR-T cells
Robust in vivo expansion of CAR-T cells was achieved in 98% of patients (49/50). Pharmacokinetic evaluation demonstrated median time-to-peak expansion (Tmax) at 10.5 days (range 4-21), with median peak CAR-T cell levels (Cmax) reaching 30,860 copies/μg genomic DNA (range 1,602-1,890,000). Significant positive correlations were observed between CAR-T expansion kinetics and in vivo persistence (Tlast) (Cmax: r = 0.349, p = 0.016 and AUC0–28d: r = 0.313, p = 0.032) (Supplementary Figure S3). Responders at day 28 demonstrated significantly enhanced expansion kinetics compared to non-responders, with MRD-negative CR patients (n=48) exhibiting higher geometric mean Cmax (319,016 vs. 863 copies/μg DNA; p = 0.017), greater exposure (AUC0-28d, p = 0.029), and improved CAR-T cell persistence (p = 0.030).
Patients receiving corticosteroids post-infusion showed higher Cmax (p = 0.017) and AUC0–28d (p = 0.012), potentially reflecting more pronounced inflammatory responses. However, corticosteroid use had no significant impact on CAR-T persistence (p = 0.502). Although patients with high tumor burden (Supplementary Figure S4) and those experiencing severe CRS/ICANS (Table 3) showed upward trends in Cmax and AUC0–28d, these differences did not reach statistical significance.
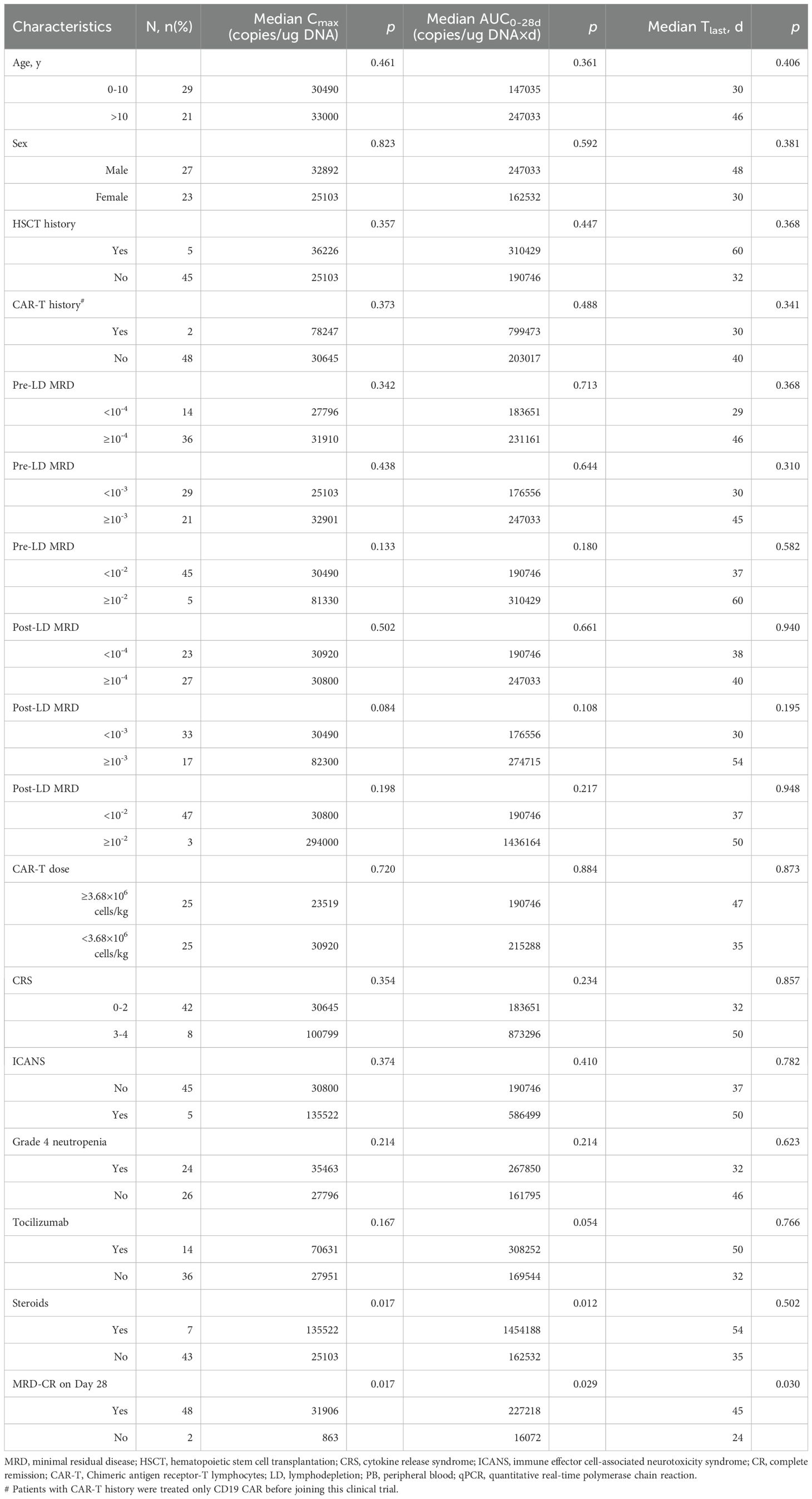
Table 3. Clinical determinants of PB CD19 CAR-T kinetics (qPCR).
BCA developed in 96% of patients (48/50), with a median onset of 1–20 days and a median duration of 59 days. Notably, BCA duration exhibited a strong positive correlation with CAR-T persistence (r = 0.570, p < 0.001), but showed no significant association with CAR-T expansion parameters (Cmax and AUC0–28d; Figure 2).
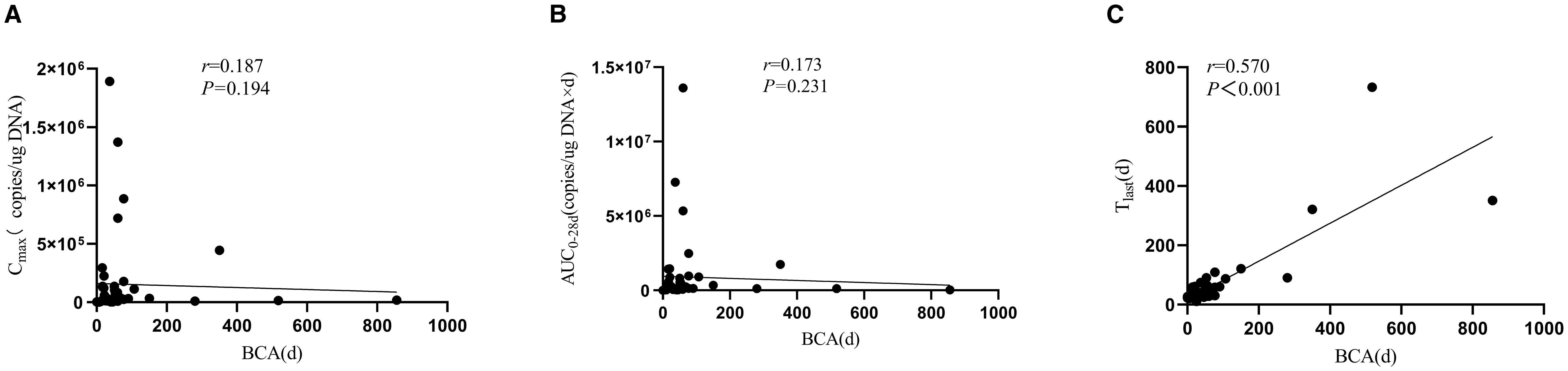
Figure 2. Association between B-cell aplasia duration and CAR-T-19 pharmacokinetic parameters. (A) Cmax, (B) AUC0-28d, (C) Tlast..
Discussion
Accumulating clinical evidence supports the therapeutic efficacy of CAR-T therapy in R/R B-ALL. The global ELIANA trial demonstrated an 81% overall response rate (ORR) with tisagenlecleucel in pediatric/adolescent patients, accompanied by 5-year event-free survival (EFS) and OS rates of 42% and 55%, respectively (12). Importantly, multiple studies consistently identify MRD status as a critical prognostic factor, wherein MRD positivity correlates with significantly elevated risks of relapse and mortality (3, 13). Our institutional data corroborate these findings: patients exhibiting persistent or recurrent MRD during therapy achieved a 5-year OS of 49.8% ± 4.3%, with a CIR of 60.2% ± 4.3% (14).
Building upon this clinical foundation, we conducted a retrospective study of 50 pediatric B-ALL patients with persistent MRD positivity or chemotherapy intolerance who received CD19 CAR-T therapy. Our results demonstrated robust efficacy, with a Day 28 ORR of 98%, MRD-negative CR rate of 96%, and 5-year OS of 74.9%. One important reason about the favorable long-term outcomes compared to historical studies in R/R ALL is that our cohort consisted of patients with MRD positivity or chemotherapy intolerance, not overt, bulky relapsed/refractory disease. This inherently selects for a patient population with a lower disease burden and potentially less aggressive disease biology, including reduced genetic evolution and chemoresistance. And these findings corroborate the ZUMA-3 trial outcomes, wherein earlier CAR-T intervention conferred superior survival benefits: patients receiving CAR-T after one prior therapy line achieved 80% 1-year OS versus 69% in heavily pretreated patients (≥2 prior lines) (15). Growing evidence supports CAR-T integration into earlier-line settings: ① A single-center trial of CAR-T consolidation in elderly B-ALL patients showed durable efficacy; ②Huang et al. reported 2-year OS rates of 92% when combining CAR-T with dasatinib in 29 treatment-naive Ph+ ALL patients (16), and ③ An MD Anderson Cancer Center (MDACC) real-world study using brexucabtagene autoleucel (brexu-cel) as consolidation therapy in 52 adult B-ALL patients demonstrated 1-year relapse-free survival (RFS) and OS rates of 65% and 90%, respectively. Collectively, these data underscore CAR-T’s potential in treatment-naive or minimally pretreated populations. Blinatumomab has also shown efficacy in patients with MRD-positive B-ALL, achieving MRD-negative responses in 97% of cases in the era of immunotherapy (17). In contrast to continuous infusion-based therapies such as blinatumomab, which often require extended hospitalization or the use of portable pumps, CAR-T therapy offers the advantage of a single administration with potential for sustained activity in vivo. The decision between these immunotherapeutic strategies, however, depends on multiple factors, including urgency of response, institutional experience, drug availability, cost, as well as patient and clinician preferences. Further prospective studies are warranted to directly compare the clinical roles of different immunotherapies—such as CAR-T versus blinatumomab—in earlier-line treatment of ALL patients.
Advancing CAR-T therapy to earlier treatment lines offers significant advantages. First, patients in earlier disease stages typically exhibit superior performance status and fewer comorbidities, enhancing tolerance to CAR-T infusion and improving safety outcomes-as evidenced by the 10% ICANS and 16% severe CRS rates in this cohort (18). Second, collecting lymphocytes during earlier disease stages circumvents chemotherapy-induced impairment of T-cell viability and function, thereby increasing the likelihood of generating products enriched with stem cell memory T cells (TSCM) or central memory T cells (TCM) that may enhance therapeutic efficacy (19). Although phenotypic characterization of infused CAR-T cells was not performed in our study, the favorable 5-year OS (74.9%) indirectly supports the clinical benefits of early intervention in B-ALL. Consequently, implementing CAR-T therapy earlier (e.g., at MRD positivity or as earlier-line treatment) reduces exposure to adverse prognostic factors such as high tumor burden and cumulative therapy toxicity (20), thus optimizing both efficacy and safety profiles.
In this cohort, 64.0% of patients underwent HSCT following CD19 CAR-T therapy. The role of post-CAR-T HSCT consolidation remains controversial: while a Phase I trial from Seattle Children’s Hospital reported significantly lower relapse rates with HSCT consolidation versus CAR-T monotherapy (18% vs. 55%) (21), the ELANA trial found no significant impact of HSCT on OS (22). Our data demonstrate that for early-phase B-ALL patients without active relapse, bridging to HSCT post-CAR-T conferred no significant OS or LFS benefit. Subgroup analyses further revealed no significant advantage across pre-infusion MRD strata or genetic risk groups. Notably, HSCT was associated with increased NRM attributable to transplant-related complications-particularly in the MRD < 10-4 group (NRM: 15.4% ± 1.1%)-which may compromise overall survival in transplanted patients, consistent with prior reports (23, 24). Consequently, for patients with low pre-infusion tumor burden (e.g., MRD < 10-4), HSCT should be deferred. Subsequent clinical management can be guided by monitoring peripheral BCA, CAR-T persistence, and MRD assessments (25–27). For high tumor burden patients (MRD ≥10-2), however, HSCT may provide clinical benefit despite the lack of statistical significance in our subgroup analysis, likely due to limited sample size. Furthermore, the high rate of subsequent HSCT bridging (64%) in our cohort, while reflecting real-world clinical practice, represents a key study limitation. The relatively small size of the non-HSCT group reduces the statistical power to evaluate the standalone efficacy of CAR-T therapy and may introduce selection bias, as the decision to proceed to transplant was non-randomized and influenced by factors such as donor availability and physician preference.
Current evidence indicates that sufficient antigenic stimulation is required to activate CAR-T cells and drive clonal expansion (28)—a prerequisite for antitumor efficacy. Consequently, evaluating CD19 CAR-T expansion kinetics in CR patients is imperative. To address this, we investigated key determinants of cellular kinetics governing CAR-T expansion and persistence in CR patients. Our data demonstrate rapid CAR-T cell expansion to peak levels at a median of 10.5 days post-infusion, consistent with established expansion models where maximal activity occurs within two weeks (29, 30). Notably, the in vivo proliferative capacity of CAR-T cells generates PK profiles characteristically distinct from conventional therapeutics. Infusion dose exhibited no significant impact on expansion magnitude or durability, aligning with prior reports (10). Furthermore, varying pre-infusion MRD levels similarly had no significant effect on expansion kinetics or persistence; PK analyses confirmed robust expansion even under low tumor burden conditions. Previous studies in R/R B-ALL established positive correlations between clinical response rates and both peak CAR-T concentrations (Cmax) and area-under-the-curve (AUC0-28d) (15, 29, 30)-a relationship further validated by our findings. These results provide a mechanistic rationale for advancing CAR-T therapy into earlier-line and frontline treatment settings.
CD19 CAR-T cell therapy targets both malignant cells and normal CD19-expressing B cells, inducing BCA. Consequently, BCA serves as a surrogate biomarker for monitoring functional CAR-T persistence in vivo (10). Prior clinical evidence, including the ZUMA-3 trial, demonstrated that loss of peripheral blood BCA coincided with functional CAR-T clearance (15). Our study further establishes that BCA duration significantly correlates with in vivo CAR-T persistence (r=0.570, p < 0.001) and strongly predicts clinical efficacy. Patients with prolonged CAR-T persistence exhibited superior LFS. These results validate BCA as a robust activity biomarker and suggest that enhancing CAR-T persistence promotes durable disease control. While BCA serves as a functional marker of CAR-T functionality, its persistence requires close monitoring and proactive management of hypogammaglobulinemia to reduce infection risk. Although the rate of early infections in our cohort was manageable (18%), this study did not systematically capture late-onset infections, representing another notable limitation. The long-term infection risk associated with sustained BCA underscores the importance of regular assessment of immunoglobulin levels and timely immunoglobulin replacement therapy when indicated.
This study provides the first comprehensive evaluation of CD19 CAR-T cell therapy as earlier-line therapy for pediatric B-ALL patients with MRD positivity or chemotherapy intolerance. We demonstrate robust CAR-T expansion kinetics even in morphologically CR patients, resulting in high MRD clearance rates, durable responses, and manageable severe adverse events. These findings provide compelling evidence supporting earlier-line CAR-T implementation in high-risk pediatric ALL. Multicenter randomized controlled trials (RCTs) are warranted to validate this strategy’s clinical utility.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Institutional Review Board (IRB)/Ethics Committee of our hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
YW: Writing – original draft. Y-PJ: Data curation, Writing – original draft. A-DL: Methodology, Writing – original draft. L-PZ: Data curation, Formal Analysis, Writing – review & editing. Y-JX: Writing – review & editing. H-MZ: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (NSFC, Grant Number 82000151), project (RDJP2023-18) supported by Peking University People’s Hospital Scientific Research Development Funds, and project supported by Peking University People’s Hospital Scientific Research Development Funds (RZ2023-04).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1672509/full#supplementary-material
References
1. Berry DA, Zhou S, Higley H, Mukundan L, Fu S, Reaman GH, et al. Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: A meta-analysis. JAMA Oncol. (2017) 3:e170580. doi: 10.1001/jamaoncol.2017.0580
2. Short NJ, Kantarjian H, Ravandi F, Konopleva M, Jain N, Kanagal-Shamanna R, et al. High-sensitivity next-generation sequencing MRD assessment in ALL identifies patients at very low risk of relapse. Blood Adv. (2022) 6:4006–14. doi: 10.1182/bloodadvances.2022007378
3. Pemmaraju N, Kantarjian H, Jorgensen JL, Jabbour E, Jain N, Thomas D, et al. Significance of recurrence of minimal residual disease detected by multi-parameter flow cytometry in patients with acute lymphoblastic leukemia in morphological remission. Am J Hematol. (2017) 92:279–85. doi: 10.1002/ajh.24629
4. Pasquini MC, Hu ZH, Curran K, Laetsch T, Locke F, Rouce R, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. (2020) 4:5414–24. doi: 10.1182/bloodadvances.2020003092
5. Wang TP, Ahn KW, Shadman M, Kaur M, Ahmed N, Bacher U, et al. Chimeric antigen receptor T-cell infusion for large B-cell lymphoma in complete remission: a center for international blood and marrow transplant research analysis. Leukemia. (2024) 38:1564–9. doi: 10.1038/s41375-024-02242-6
6. Wudhikarn K, Tomas AA, Flynn JR, Devlin SM, Brower J, Bachanova V, et al. Low toxicity and excellent outcomes in patients with DLBCL without residual lymphoma at the time of CD19 CAR T-cell therapy. Blood Adv. (2023) 7:3192–8. doi: 10.1182/bloodadvances.2022008294
7. Jallouk AP, Gouni S, Westin J, Feng L, Mistry H, Steiner RE, et al. Axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma patients in complete metabolic response. Haematologica. (2023) 108:1163–7. doi: 10.3324/haematol.2022.281954
8. Neelapu SS, Dickinson M, Munoz J, Ulrickson ML, Thieblemont C, Oluwole OO, et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: the phase 2 ZUMA-12 trial. Nat Med. (2022) 28:735–42. doi: 10.1038/s41591-022-01731-4
9. Shah B, Mattison RJ, Abboud R, Abdelmessieh P, Aldoss I, Burke PW, et al. Acute lymphoblastic leukemia, version 2.2024, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2024) 22:563–76. doi: 10.6004/jnccn.2024.0051
10. Mueller KT, Waldron E, Grupp SA, Levine JE, Laetsch TW, Pulsipher MA, et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. (2018) 24:6175–84. doi: 10.1158/1078-0432.CCR-18-0758
11. Brown ART and Gutierrez C. Comments regarding “ASBMT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transpl. (2019) 25:e209–10. doi: 10.1016/j.bbmt.2019.02.027
12. Laetsch TW, Maude SL, Rives S, Hiramatsu H, Bittencourt H, Bader P, et al. Three-year update of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia in the ELIANA trial. J Clin Oncol. (2023) 41:1664–9. doi: 10.1200/JCO.22.00642
13. Cheng S, Inghirami G, Cheng S, and Tam W. Simple deep sequencing-based post-remission MRD surveillance predicts clinical relapse in B-ALL. J Hematol Oncol. (2018) 11:105. doi: 10.1186/s13045-018-0652-y
14. Wang Y, Xue YJ, Jia YP, Zuo YX, Lu AD, and Zhang LP. Re-emergence of minimal residual disease detected by flow cytometry predicts an adverse outcome in pediatric acute lymphoblastic leukemia. Front Oncol. (2021) 10:596677. doi: 10.3389/fonc.2020.596677
15. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. (2021) 398:491–502. doi: 10.1016/S0140-6736(21)01222-8
16. Zhang M, Fu S, Feng J, Hong R, Wei G, Zhao H, et al. Dasatinib and CAR T-cell therapy in newly diagnosed philadelphia chromosome-positive acute lymphoblastic leukemia: A nonrandomized clinical trial. JAMA Oncol. (2025) 11:625–9. doi: 10.1001/jamaoncol
17. Hodder A, Mishra AK, Enshaei A, Baird S, Elbeshlawi I, Bonney D, et al. Blinatumomab for first-line treatment of children and young persons with B-ALL. J Clin Oncol. (2024) 42:907–14. doi: 10.1200/JCO.23.01392
18. Corona M, Ip A, Brown S, Luna A, Khatib H, Flynn JR, et al. Treatment failure patterns in early versus late introduction of CAR T-cell therapy in large B-cell lymphoma. Bone Marrow Transpl. (2025) 60:491–8. doi: 10.1038/s41409-025-02519-z
19. Junkuhn C, Schiele P, Walter AL, Hamm F, Obermayer B, Busch D, et al. Prior chemotherapy deteriorates T-cell quality for CAR T-cell therapy in B-cell non-Hodgkin’s lymphoma. J Immunother Cancer. (2025) 13:e010709. doi: 10.1136/jitc-2024-010709
20. Schultz LM, Baggott C, Prabhu S, Pacenta HL, Phillips CL, Rossoff J, et al. Disease burden affects outcomes in pediatric and young adult B-cell lymphoblastic leukemia after commercial tisagenlecleucel: A pediatric real-world chimeric antigen receptor consortium report. J Clin Oncol. (2022) 40:945–55. doi: 10.1200/JCO.20.03585
21. Shalabi H, Yuan CM, Kulshreshtha A, Dulau-Florea A, Salem D, Gupta GK, et al. Disease detection methodologies in relapsed B-cell acute lymphoblastic leukemia: Opportunities for improvement. Pediatr Blood Cancer. (2020) 67:e28149. doi: 10.1002/pbc.28149
22. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
23. Dai HP, Kong DQ, Shen HJ, Cui W, Wang Q, Li Z, et al. CAR-T cell therapy followed by allogenic hematopoietic stem cell transplantation yielded comparable outcome between Ph like ALL and other high-risk ALL. biomark Res. (2023) 11:19. doi: 10.1186/s40364-023-00451-2
24. Shadman M, Gauthier J, Hay KA, Voutsinas JM, Milano F, Li A, et al. Safety of allogeneic hematopoietic cell transplant in adults after CD19-targeted CAR T-cell therapy. Blood Adv. (2019) 3:3062–9. doi: 10.1182/bloodadvances.2019000593
25. Pulsipher MA, Han X, Maude SL, Laetsch TW, Qayed M, Rives S, et al. Next-generation sequencing of minimal residual disease for predicting relapse after tisagenlecleucel in children and young adults with acute lymphoblastic leukemia. Blood Cancer Discov. (2022) 3:66–81. doi: 10.1158/2643-3230.BCD-21-0095
26. Rampotas A and Roddie C. The present and future of CAR T-cell therapy for adult B-cell ALL. Blood. (2025) 145:1485–97. doi: 10.1182/blood.2023022922
27. Gabelli M, Oporto-Espuelas M, Burridge S, Chu J, Farish S, Hedges E, et al. Maintenance therapy for early loss of B-cell aplasia after anti-CD19 CAR T-cell therapy. Blood Adv. (2024) 8:1959–63. doi: 10.1182/bloodadvances.2023011168
28. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
29. Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. (2017) 130:2317–25. doi: 10.1182/blood-2017-06-786129
Keywords: chimeric antigen receptor T-cell, B-cell acute lymphoblastic leukemia, minimal residual disease, earlier-line therapy, pharmacokinetics
Citation: Wang Y, Jia Y-p, Lu A-d, Zhang L-p, Xue Y-j and Zeng H-m (2025) Effective MRD clearance and long-term survival with CD19 CAR-T in pediatric B-ALL patients with MRD positivity or chemotherapy intolerance. Front. Immunol. 16:1672509. doi: 10.3389/fimmu.2025.1672509
Received: 24 July 2025; Accepted: 22 September 2025;
Published: 06 October 2025.
Edited by:
Thomas Böldicke, Helmholtz Association of German Research Centers (HZ), GermanyReviewed by:
Michał Zarobkiewicz, Medical University of Lublin, PolandSongmi Wang, Huazhong University of Science and Technology, China
Copyright © 2025 Wang, Jia, Lu, Zhang, Xue and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-juan Xue, eHVleXVqdWFueWFuemlAMTYzLmNvbQ==; Hui-min Zeng, emVuZ2h1aW1pbnByZXR0eUAxNjMuY29t
†These authors have contributed equally to this work and share last authorship