 Pingping Wang†
Pingping Wang† Nan Su
Nan Su Xiaojing Yan
Xiaojing Yan Yan Zhang
Yan Zhang- Department of Hematology, The First Hospital of China Medical University, Shenyang, China
Circulating plasma cells (CPCs) represent an accessible subset of antibody-secreting cells that provide valuable insights into immune activation and regulation. This review presents the first comprehensive synthesis of CPC biology, with a particular focus on the mechanisms governing their generation under physiological conditions and the distinct pathways that drive their formation within tumor microenvironments. We further summarize the broader clinical relevance of CPCs as potential biomarkers across infections, autoimmune diseases, and plasma cell disorders. With the rapid advancement of liquid biopsy technologies, CPC detection has garnered increasing attention in clinical practice. Here, we evaluate current and emerging methods for CPC detection, highlighting their respective advantages and limitations. Finally, we discuss the translational potential of CPCs and outline future research directions to support more precise diagnosis and treatment strategies in CPC-associated conditions.
1 Introduction
Plasma cells, the terminal effectors of the B-cell lineage, are key effector cells in humoral immunity through their production of antibodies and maintenance of immune memory. While most plasma cells reside in specialized niches such as bone marrow, spleen, and mucosa-associated lymphoid tissues, a small population of circulating plasma cells (CPCs) persists in peripheral blood (1). Traditionally considered short-lived and rare, CPCs were thought to reflect routine immune turnover under steady-state conditions, often representing a transitory state between plasmablasts, long-lived plasma cells (LLPCs), or apoptotic cells, particularly in non-disease states. However, CPC populations fluctuate significantly in response to infections, autoimmune diseases, and plasma cell disorders. Recent evidence demonstrates that CPCs expand following vaccination and infection, implicating their involvement in systemic immune surveillance and adaptive memory formation (2, 3). In autoimmune diseases such as systemic lupus erythematosus (SLE), elevated CPC levels correlate with autoantibody production and chronic inflammation, highlighting their potential as dynamic biomarkers for disease activity and therapeutic response (4). In plasma cell disorders, particularly multiple myeloma (MM), CPCs are associated with tumor burden and have shown promise as minimally invasive indicators for early screening and diagnosis, prognostic evaluation, response assessment and therapeutic guidance (5, 6). In MM, CPCs typically reflect clonal plasma cells that have exited bone marrow due to niche remodeling, rather than newly formed CPCs from B-cell activation. Although plasma cell biology has been extensively studied, most existing reviews have concentrated on LLPCs or precursor states rather than CPCs. For instance, Nguyen et al. summarized intrinsic programs such as apoptosis resistance, autophagy, and metabolic regulation, together with extrinsic factors like bone marrow stromal support and cytokine signaling, as key determinants of LLPC survival (7). Manakkat Vijay & Singh synthesized recent insights into the temporal dynamics of generation of plasma cell precursors during germinal center responses (8). While these contributions have advanced our understanding of plasma cell maturation, the biology of CPCs remains insufficiently defined. In particular, the mechanisms that regulate CPC generation under physiological conditions, as opposed to those driving their aberrant expansion within tumor-associated microenvironments, are still unclear. This gap in knowledge has hindered the translation of CPC biology into clinical practice, despite their considerable potential to serve as minimally invasive biomarkers across diverse disease settings.
In this review, we present the first comprehensive synthesis of CPC biology, systematically detailing the mechanisms underlying CPC generation under both physiological conditions and tumor-associated microenvironments, offering an integrated perspective that extends beyond the scope of previous plasma cell-focused reviews. We also highlight recent advances in CPCs research and broaden their clinical relevance beyond MM to include autoimmune diseases, emphasizing their emerging value as biomarkers at the interface of immunology, hematology, and oncology. Finally, we examine current and next-generation CPC detection technologies, and discuss their translational potential in precision medicine, along with future directions for basic and clinical research.
2 Biological characteristics of normal plasma cells
2.1 Origin and development
B cells differentiate into plasmablasts, which are precursors of both short-lived and LLPCs; the latter represent the terminal stage of B cell differentiation. This transition is accompanied by extensive transcriptional reprogramming and has been viewed as a shift from a proliferative plasmablast stage to a non-dividing, antibody-secreting plasma cell state. Notably, recent findings indicate that only a small fraction (~10%) of circulating human plasmablasts are proliferative, while most have already exited the cell cycle (9). Following their generation in the lymph nodes, plasmablasts transiently circulate and subsequently home to specific microenvironments like the bone marrow, spleen, mucosa-associated lymphoid tissue (MALT), or lymph nodes. These microenvironments constitute niches that provide essential survival signals to early-stage plasma cells in peripheral blood.
In general, plasma cell differentiation initiates with the activation of B cells in secondary lymphoid organs, typically characterized by a reprogramming of transcriptional networks: the down-regulation of B cell-promoting factors like Pax5, Bach2, and Bcl6, and the up-regulation of plasma cell-promoting factors including Blimp-1, Xbp-1, and IRF-4 (10–13). Blimp-1, Xbp-1 and IRF-4 are considered the pivotal drivers of plasma cell development (Figure 1). Throughout the activation and directed differentiation of B cells into plasma cells, the suppression of Pax5 expression relieves its inhibitory effect on the plasma cell transcriptional program, thereby accelerating the expression of Blimp-1 and Xbp-1 (10). Blimp-1 predominantly drives plasma cell differentiation by suppressing c-Myc, class II transactivator (CIITA), Pax5, and B-cell receptor signaling components (e.g., Spi-B, Id3), inhibiting immunoglobulin class-switch recombination by down-regulating activation-induced cytidine deaminase (AID), Ku70, Ku86, DNA-dependent protein kinase catalytic subunit (DNA-PKcs), and signal transducer and activator of transcription 6 (STAT6), strengthening the expression of pro-immunoglobulin secretion genes such as ELL2, while permitting the expression of crucial plasma cell genes like Xbp-1 (14–17). Meanwhile, Blimp1 bound to and activated 20 genes coding for proteins implicated in ER function and ER stress control in plasmablasts, directly contributing to the regulation of immunoglobulin secretion (17). IRF-4 promotes plasma cell development by enhancing Prdm1 transcription (encoding for Blimp-1), leading to elevated Blimp-1 expression (18). As a downstream effector of Blimp-1, Xbp-1 orchestrates plasma cell function and immunoglobulin secretion through the unfolded protein response (UPR) (19). Genes like ERdj3 and OBF-1 have been identified as direct Xbp-1 targets involved in both plasma cell differentiation and classical UPR pathways (19). Xbp-1 also regulates transcription of the immunoglobulin heavy chain by controlling heavy chain–specific transcription factors (19). In Blimp-1-deficient B cells, Xbp-1 fails to be up-regulated, resulting in impaired plasma cell function and defective antibody secretion (20). Newly generated plasma cells exit secondary lymphoid organs via S1P1, up-regulated by Klf2, and enter circulation (21, 22). Afterwards, they migrate to bone marrow under the guidance of chemokines such as C-X-C motif chemokine ligand 12 (CXCL12) (23).
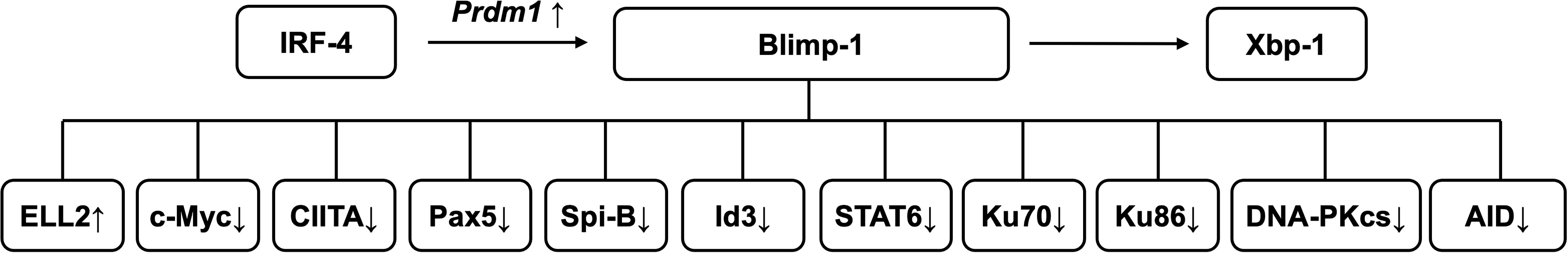
Figure 1. The critical drivers of plasma cell generation. The transcription factors Blimp-1, Xbp-1, and IRF-4 are critical regulators of B cell differentiation into plasma cells. IRF-4 indirectly promotes plasma cell development by up-regulating Prdm1 transcription, leading to increased Blimp-1 expression. In turn, Blimp-1 drives plasma cell differentiation by suppressing the expression of genes involved in B cell receptor signaling (Pax5, Spi-B, Id3, STAT6), germinal center B cell function (Ku70, Ku86, DNA-PKcs, AID), proliferation (c-Myc), and MHC-II presentation (CIITA). Concurrently, Blimp-1 promotes the expression of genes essential for plasma cell identity, such as Xbp-1 and ELL2. Xbp-1 further modulates plasma cell differentiation through the unfolded protein response (UPR).
While most plasma cells are short-lived, residing primarily in the medullary cords of lymph nodes and the red pulp of the spleen, a small subset migrates to the bone marrow, where they further differentiate into long-lived mature plasma cells under the influence of bone marrow-derived survival signals. Recent mechanistic advances have further elucidated the developmental continuum from germinal center B cells to LLPCs (24). Manakkat Vijay et al. identified a TIGIT+ transitional plasma cell precursor population generated during the late phase of the germinal center response, which preferentially gives rise to long-lived bone marrow plasma cells (24). Liu et al. demonstrated that LLPCs exhibit isotype-specific phenotypes, with IgA LLPCs being Ly6AhiTIGIT⁻ and IgG/IgM LLPCs EpCAMhiCXCR3⁻, underscoring that TIGIT expression and survival pathways are differentially regulated across isotypes (25). Collectively, these studies provide mechanistic insights into plasma cell differentiation.
2.2 Functions
A hallmark characteristic of plasma cells is their remarkable capacity for antibody synthesis and secretion. Based on their lifespan, plasma cells are generally classified into two main types: short-lived plasma cells, which are proliferative and survive for days to months, and LLPCs, which are non-proliferative and can persist for decades (26). LLPCs, predominantly generated throughout germinal center reactions, reside long-term in the bone marrow, where they secrete high-affinity, class-switched antibodies such as immunoglobulin G (IgG), immunoglobulin A (IgA) and immunoglobulin E (IgE) and establish stable immunological memory, enabling rapid and high antibody responses upon re-exposure to the same antigen. In contrast, short-lived plasma cells are typically formed in the extrafollicular regions of secondary lymphoid organs and predominantly generate low-affinity IgM antibodies, ultimately facilitating rapid primary immune responses (27). Other than their well-known function of antibody secretion, plasma cells are key contributors to the intricate process of immune response modulation (28). Recent studies suggest that plasma cells can inhibit the development of follicular helper T cells (28). Moreover, certain subsets of plasma cells produce anti-inflammatory cytokines such as interleukin-10 (IL-10) and IL-35, contributing to immune regulation throughout infection (29). In patients with inflammatory bowel disease, some mucosal plasma cells secrete granzyme B, exerting cytotoxic effects (30). Furthermore, IgA+ mucosal plasma cells have been demonstrated to facilitate tumor necrosis factor-alpha (TNF-α) and inducible nitric oxide synthase (iNOS), contributing to local inflammatory responses (31).
2.3 Survival and proliferation
The long-term survival of plasma cells is dependent on a specialized microenvironment, often referred to as the “niche”, which is composed of stromal and hematopoietic-derived cells (32, 33). Nevertheless, the precise cellular composition of this niche remains controversial. Multiple studies have validated that CXCL12-producing stromal cells, megakaryocytes, eosinophils, basophils, and T cells support plasma cell survival through the secretion of soluble factors (34–38). A proliferation-inducing ligand (APRIL) and IL-6 are critical pro-survival cytokines for plasma cells. Both megakaryocytes and eosinophils in the bone marrow can produce APRIL and IL-6 (35, 36), while basophils contribute to plasma cell survival in vivo and in vitro via IL-4 and IL-6 secretion (37). In addition, recent findings indicate that antibody-secreting cells (ASCs) in patients with SLE can also produce APRIL in an autocrine manner, which may further support their survival (39). Regulatory T cells (Tregs) in the bone marrow also play a critical role by expressing high levels of effector molecules. Nonetheless, deletion of CTLA-4 brings about abnormal plasma cell expansion (38). As demonstrated by Cassese et al., a combination of signaling molecules, comprising IL-5, IL-6, stromal cell-derived factor-1α (SDF-1α), TNF-α, and CD44 ligands, is essential for maintaining plasma cell longevity (40). Intrinsically, cytokines such as IL-7 and stem cell factors (SCFs) are critical in the early stages of B cell development, but fail to play a role in sustaining the survival of plasma cells. This observation suggests that plasma cells have a unique set of survival prerequisites, distinct from those of early B cells. In comparison with resting B cells, plasma cells demonstrate high expression of B cell maturation antigen (BCMA), while the expression of transmembrane activator and cyclophilin ligand interactor (TACI) and B cell-activating factor receptor (BAFF-R) is reduced, suggesting that BCMA plays a critical role in plasma cell survival (41). Although BCMA has been proposed to play a critical role in plasma cell survival by mediating APRIL- and BAFF-dependent signals (42), recent evidence suggests that BCMA is dispensable for the survival of LLPCs in mice (43). Menzel et al. demonstrated that BCMA-deficient mice maintain comparable numbers of antigen-specific LLPCs, indicating that BCMA may function primarily as a soluble decoy receptor regulating plasma cell population size rather than serving as an essential survival factor (43). Notably, while BCMA is dispensable for the survival of LLPCs, it nonetheless represents a critical therapeutic target in MM.
In addition to cytokine signaling, nutrient uptake is pivotal for sustaining the high metabolic demands of antibody production. LLPCs exhibit more enhanced glucose uptake than their short-lived plasma cells, underlining the critical role of metabolic support in maintaining their long-term survival (44). Notably, plasma cells can survive for decades in the hypoxic environment of the bone marrow. In vitro studies further reveal that hypoxic conditions enhance plasma cell viability, implicating hypoxia as a potential pivotal factor in promoting long-term survival of LLPCs (45).
In line with the model of replicative self-renewal, LLPCs reside in a quiescent state within the bone marrow niche. While they express a diverse array of cell cycle regulators, they can occasionally be triggered by cellular or immune signals to undergo rare and transient divisions (occurrence <1%) (46). For any given antigen-specific plasma cell, this replicative self-renewal occurs very rarely, with minimal impact on overall antibody titers (46). Nevertheless, the threshold for cell cycle re-entry is substantially reduced in cases where plasma cell precursors harbor oncogenic mutations (46). The finely regulated self-renewal mechanism becomes disrupted, with malignantly transformed plasma cells acquiring uncontrolled proliferative capacity in plasma cell disorders such as MM. This phenomenon leads to clonal plasma cell expansion in the bone marrow microenvironment, followed by further immune evasion through accumulated acquired mutations and microenvironment remodeling, ultimately entering peripheral blood.
2.4 Maturation and maintenance
Plasma cell maturation begins with the differentiation of activated B cells into ASCs, which transiently circulate in the blood as plasmablasts before either undergoing apoptosis or migrating to specialized tissue niches, most notably the bone marrow, where they mature into LLPCs (47). This maturation process involves morphological, transcriptional, and epigenetic changes within the bone marrow microniche (48). Joyner et al. demonstrated that early-minted blood ASCs acquire LLPC-like features through expansion of the endoplasmic reticulum, increased mitochondrial content, and upregulation of pro-survival genes (BCL2, MCL1, BCL-XL), conferring resistance to apoptosis (48). Duan et al. further revealed distinct LLPC maturation trajectories through single-cell transcriptomic analyses, highlighting metabolic reprogramming and the involvement of TNF–NF-κB and mammalian target of rapamycin (mTOR) signaling pathways in LLPC maintenance (47). Functionally, antibody secretion capacity increases as ASCs mature, with Nguyen et al. showing that bone marrow LLPCs produce significantly more IgG per cell than circulating early-minted ASCs (49). Importantly, Schulz et al. identified BCMA as a critical determinant of terminal plasma cell maturation (50). Using a BCMA reporter mouse, they demonstrated that BCMA expression increases with plasma cell maturity and varies by immunoglobulin isotype, emphasizing the essential role of the in vivo microenvironment and APRIL-mediated BCMA signaling in sustaining LLPC survival (50). Collectively, these studies underscore that plasma cell maturation and maintenance are tightly orchestrated processes integrating transcriptional, metabolic, and microenvironmental programs, ultimately ensuring the persistence of protective humoral immunity.
2.5 Roles in immune responses
2.5.1 Roles throughout infection
Throughout the early stages of infection, plasma cells transiently emerge in peripheral blood, bolstering initial immune defense through rapid antibody secretion. Afterwards, they migrate to the bone marrow or spleen, where they differentiate into LLPCs that sustain antibody levels and confer long-term immune protection. Early studies demonstrated that infection-induced inflammatory signals cause the mobilization of plasma cells from the marrow, and this efflux reduces the size of the existing LLPC populations, with concomitant reduction in circulating antibodies derived from these plasma cell populations. This was associated with a dramatic drop in CXCL12 levels and loss of eosinophils in the bone marrow of infected mice (51). Plasma cells and their secreted antibodies play a central role in the long-term protection against chronic viral infection. During chronic or persistent infections, plasma cells undergo clonal expansion and somatic hypermutation, resulting in a diverse antibody repertoire with varying affinities and specificities, including cross-reactivity to multiple antigens (52, 53). Chronic infection is characterized by a longer-lasting germinal center reaction and a continuous differentiation of plasma cells, resulting in the emergence of higher-affinity plasma cells exhibiting increased antibody secretion rates (54). A recent report in Science highlighted the pivotal role of plasma cells in antibody affinity maturation, whereby clonal expansion enhances antibody affinity. This finding offers new insights for vaccine development, suggesting that the design of antigens capable of efficiently driving plasma cell expansion could represent an important strategy to strengthen immune responses (55).
As already suggested by recent investigation, the functions of plasma cells extend beyond antibody production. Specifically, certain subsets can secrete anti-inflammatory cytokines such as IL-10 and IL-35, thereby modulating immune responses throughout infection. For instance, Shen et al. reported that, a subset of B cells suppresses antimicrobial immune responses via IL-10 and IL-35 production during Salmonella infection (29). Phenotypic analysis identified these cells as IgM+CD138hiTACI+CXCR4+CD1dintTim1int plasma cells. Aside from their protective roles, plasma cells may also act as viral targets that expedite infection. Alomari et al. reported that during chronic viral infection, the differentiation of new plasma cells is involved in the early stages of viral infection in B cells, mediated by IL-21 signaling and promoting viral dissemination at early stages (56).
2.5.2 Roles in vaccination and maintaining immune memory
The role of plasma cells in the immune response to vaccination is to serve as the primary source of antigen-specific antibody production, thereby mediating both the immediate and long-term humoral protection elicited by vaccines. The primary goal of vaccination is to induce the generation of plasma cells, particularly LLPCs, to provide durable humoral immunity. During natural infections, such as influenza, measles, or mumps, LLPCs can be established in the bone marrow, sustaining antibody production for decades (57). However, recent studies on SARS-CoV-2 mRNA vaccination suggest that antigen-specific plasma cells may not consistently form long-lived phenotypes in the bone marrow (57). Nguyen et al. reported that, within 2.5 to 33 months after SARS-CoV-2 mRNA vaccination, influenza- and tetanus-specific ASCs were widely detected in the LLPC compartment, whereas SARS-CoV-2-specific ASCs were primarily found in non-LLPC subsets (57). This suggests that SARS-CoV-2-specific plasma cells may not efficiently establish durable bone marrow residency. These findings indicate that the generation and maintenance of plasma cells following vaccination may differ from those induced by natural infection. Understanding the dynamics of plasma cell development and maintenance after vaccination is therefore crucial for optimizing vaccine design and improving the durability of protective immunity.
Long-lasting humoral immunity rests significantly with tightly regulated mechanisms that govern the generation, survival, and homeostasis of plasma cells. Because of their extended lifespan, LLPCs resident in the bone marrow sustain humoral immunity autonomously, independent of memory B cells, T cell help, or persistent antigen presentation. LLPCs are generated following robust germinal center reactions, which provide high-affinity, class-switched antibodies crucial for durable humoral immunity. The mechanisms by which plasma cells maintain long-term immune memory rely on specialized survival niches and extrinsic factors, as detailed in Section 2.3.
3 CPC formation and detection
3.1 Distinct immunophenotypic features of CPCs
Under physiological conditions, recently released CPCs progressively acquire typical plasma cell characteristics (Figure 2). In comparison with bone marrow plasma cells, CPCs display lower surface expression levels of adhesion molecules (CD11a, CD31, CD49d, CD49e, CD49f, and CD56) and activation markers (CD38 and CD27), while expressing higher levels of CD362 (58). In addition, CPCs differ from bone marrow plasma cells by expressing CD62L and intermediate levels of CD138, revealing lower expression of CXC chemokine receptor 4 (CXCR4) and CC chemokine receptor 2 (CCR2), and lacking CD9 expression (59). Compared with plasmablasts, CPCs display distinct phenotypic alterations, including changes in activation markers (decreased CD53, CD45, and CD9, with increased CD27), adhesion molecules (reduced CD47, CD11a, and CD50, with increased CD31, CD49d and CD329), B-cell receptor signaling molecules (decreased CD22, CD19, and HLA-DR), complement receptors (reduced CD58), co-stimulatory molecules (decreased CD40 and CD130, but increased CD86, CD272, CD126, CD32, and CD85j), and plasma cell survival–associated molecules (decreased CD268, with elevated CD270 and CD95) (58). CPCs with this immunophenotype are newly generated plasma cells migrating from lymphoid organs to the bone marrow or tissues (59).
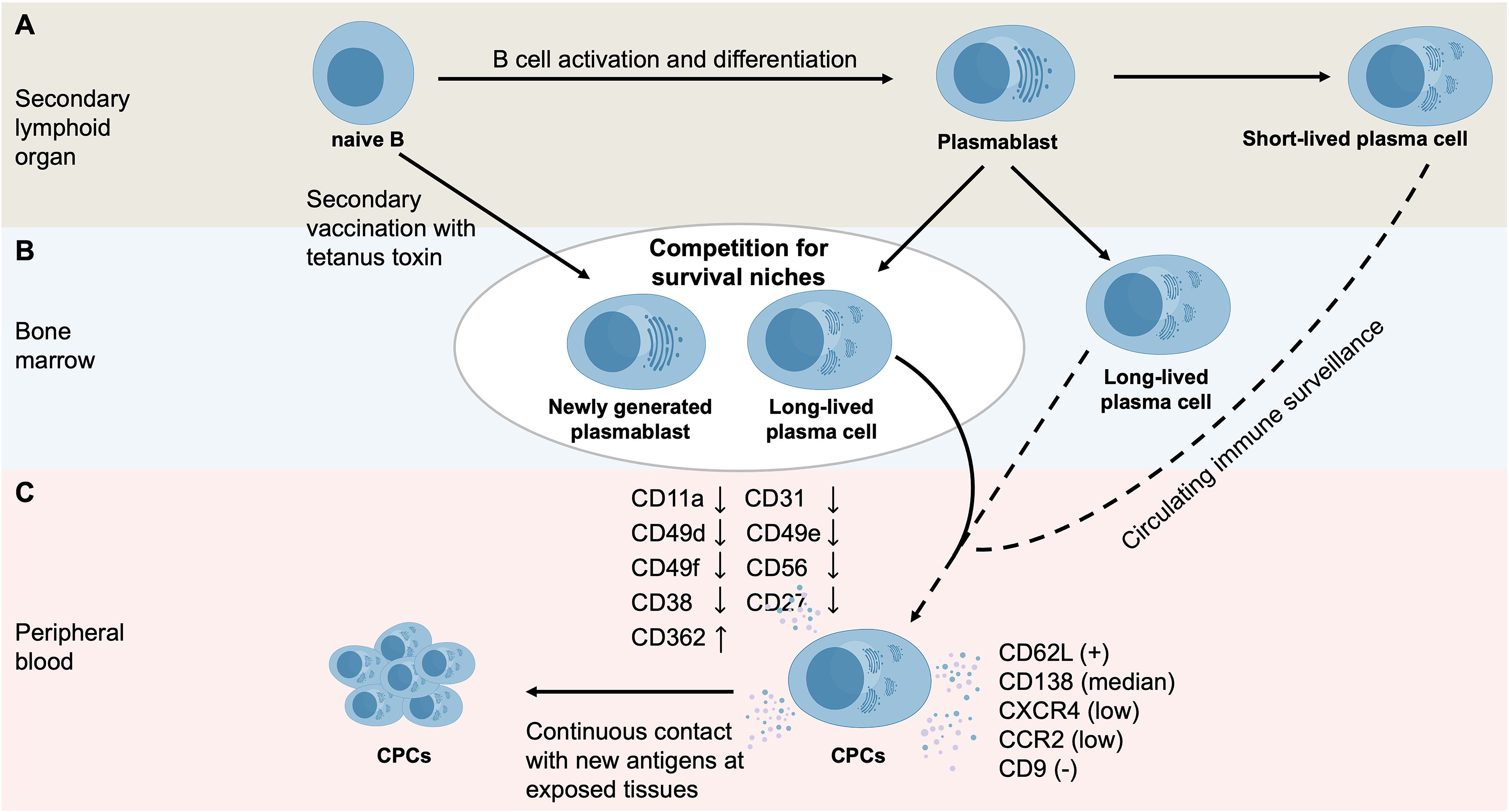
Figure 2. The mechanisms underlying normal CPCs formation. (A) In secondary lymphoid organs, antigen exposure results in B cell activation and differentiation, resulting in the generation of plasmablasts and plasma cells. Short-lived plasma cells mainly exist in peripheral lymphoid tissues, while LLPCs migrate to the bone marrow for long-term survival. Plasma cells may be released into the peripheral blood to participate in circulating immune surveillance. (B) Following immunization (e.g., secondary vaccination with tetanus toxin), recently generated plasmablasts must compete with resident plasma cells for survival niches in the bone marrow, driving the re-entry of resident plasma cells into the peripheral blood. (C) In comparison with bone marrow plasma cells, circulating normal plasma cells display a distinct immunophenotype, including: (i) reduced molecules markers, with e.g., less substantial CD11a, CD31, CD49d, CD49e, CD49f and CD56 expression; (ii) activation markers, with e.g., lower CD38 and CD27 expression; (iii) higher CD362 expression; and (iv) positive CD62L expression, intermediate CD138 levels, low CXCR4 and CCR2 levels, and absence of CD9.
3.2 Exposure to novel antigens accelerates CPC generation
Normal CPCs are undetectable in fetal umbilical cord blood (58). Nonetheless, after birth, as neonates are exposed to novel antigens in mucosal barrier tissues, such as the respiratory and gastrointestinal tracts, the number of CPCs in peripheral blood increases rapidly throughout the first weeks to months of life, reaching a peak between one and two years of age; thereafter, their numbers gradually decline throughout adulthood (58). As already demonstrated by associated studies, CPCs generally present an exceedingly activated phenotype, with 66.8~76.2% of these expressing the proliferation marker Ki-67, illustrating that local antigenic stimulation may trigger CPCs re-entry into the peripheral blood (1).
3.3 Competition for bone marrow niches promotes CPC re-entry into the peripheral blood
Odendahl et al. investigated the dynamics of plasmablasts and plasma cells following a secondary vaccination with tetanus toxin (2). On days 6 and 7 post-immunization, a large number of tetanus toxin-specific plasmablasts (CD19+/CD27high/intracellular IgGhigh/HLA-DRhigh/CD38high/CD20–/CD95+) were released into the peripheral blood from secondary lymphoid organs (2). These cells responded to chemotactic signals mediated by CXCR3 and CXCR4 ligands, probably guiding them to the bone marrow or inflamed tissue (2). Simultaneously, a population of plasma cells appeared in peripheral blood, marked by a long-lived phenotype (CD19+/CD27high/intracellular IgGhigh/HLA-DRlow/CD38+/CD20–/CD95+), secreting unknown, non-tetanus toxin-specific antibodies (2). The detection of these cells in peripheral blood demonstrates that nascent plasmablasts compete with resident plasma cells for survival niches in the bone marrow (2). This competition may facilitate the re-emergence of resident plasma cells into the peripheral circulation, representing a potential mechanism contributing to CPC formation (Figure 2).
3.4 Factors influencing CPC numbers
CPC numbers are influenced by the combined effects of intrinsic cellular determinants, microenvironmental cues, and the host’s physiological or pathological state. In healthy adults, CPCs are very rare, typically exhibiting a concentration of approximately 2 CPCs per microliter of blood (1). Nonetheless, the number of CPCs can increase markedly under certain pathophysiological circumstances. Such elevations may involve either reactive plasma cells, as seen in acute responses following vaccination (2), reactive plasmacytosis triggered by viral infections (3), and autoimmune diseases such as SLE (4), or clonal plasma cells, as observed in malignant plasma cell disorders, including MM and plasma cell leukemia (5, 6). Under such circumstances, abnormal fluctuations in CPC numbers may have significant diagnostic and prognostic value for related disorders. In the context of reactive plasma cells, antigenic stimulation stands as a pivotal driver of plasma cell production, initiating and sustaining immune responses (58). The intensity and duration of these immune responses exert a direct influence on the levels of reactive CPC.
3.5 A possible hematopoietic stem cell-like recirculation mechanism
Hematopoietic stem cells (HSCs) are validated to circulate in the bloodstream under steady-state conditions. Circulating HSCs and their progenitors fluctuate in antiphase with the expression of the chemokine CXCL12 in the bone marrow microenvironment and follow circadian rhythms (60). These circulating HSCs have important implications for immunosurveillance (61). The bone marrow harbors specialized microenvironments for HSCs and their progenitor cells, and plasma cells, with plasma cells and HSCs sharing a similar stromal microenvironment (62). In particular, the concentration of CD34+ cells in the peripheral blood of healthy adults is comparable to that of CPCs under steady-state conditions (1, 63), which suggests that bone marrow plasma cells and HSCs may be regulated by similar recirculation mechanisms.
3.6 Detection of CPCs
3.6.1 Current CPC detection methods
CPC detection techniques vary widely in sensitivity, specificity, applicability, and feasibility. Traditional cytology, immunocytochemistry (IMC), multiparameter flow cytometry (MFC), next-generation flow cytometry (NGF), allele-specific oligonucleotide quantitative PCR (ASO-qPCR), next-generation sequencing (NGS), surface-enhanced Raman spectroscopy (SERS), and inductively coupled plasma mass spectrometry (ICP-MS) each have unique strengths and limitations (Table 1).
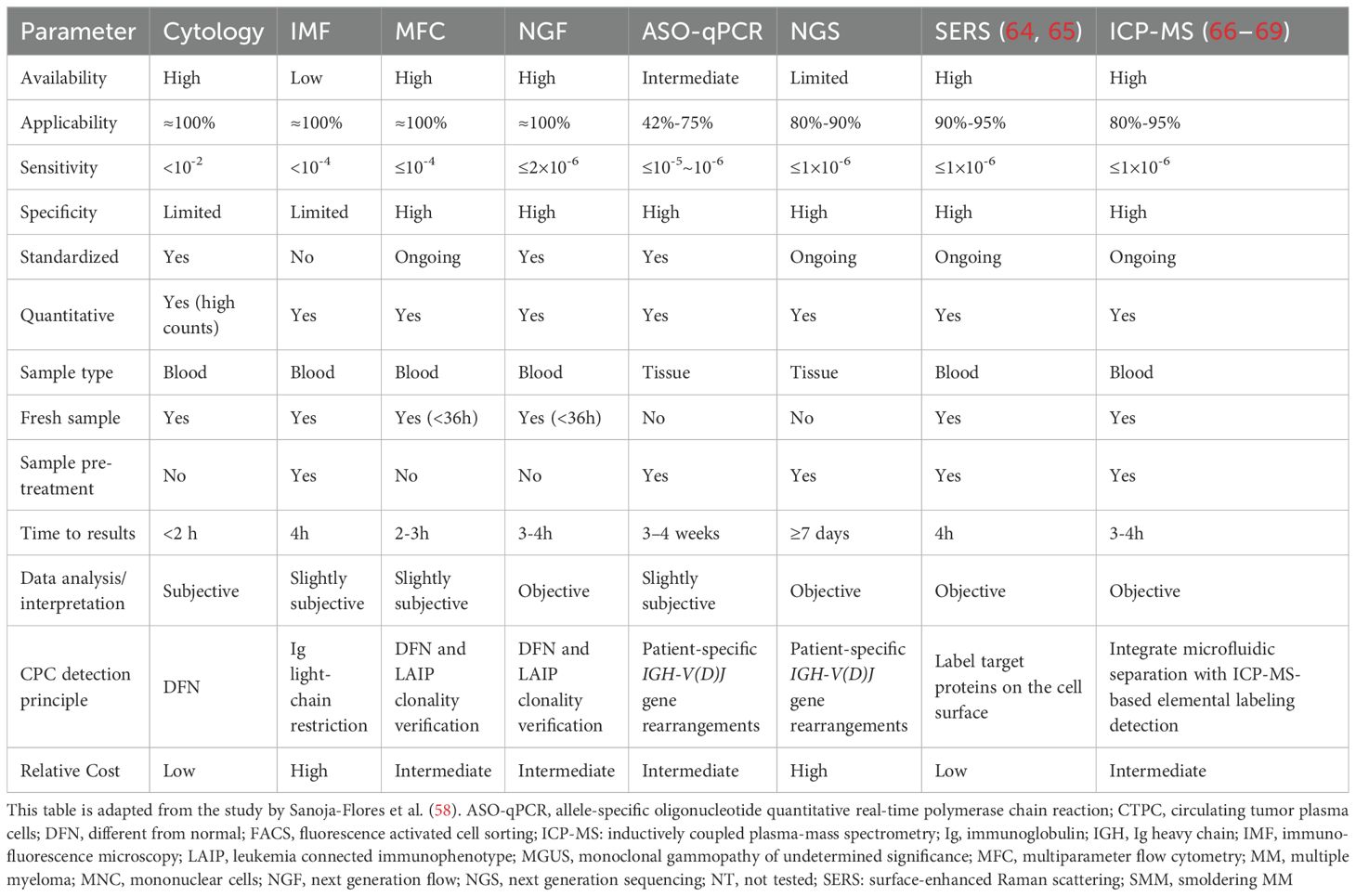
Table 1. Advantages and disadvantages of the most frequently used methods for detection of CPCs.
Cytology remains the most accessible and inexpensive approach, but its low sensitivity (<10⁻2, particularly at low CPC levels), limited specificity, and inter-observer variability and the difficulty of identifying atypical CPC morphologies further restrict its reliability (70, 71). Immunocytochemical methodologies allow for more detailed characterization of monoclonal plasma cells in the blood, particularly through assessment of restricted light chain expression (72). However, it is labor-intensive and lacks standardized protocols. Flow cytometry has emerged as the primary technique for CPC detection due to its high throughput, sensitivity, reliability, and accuracy. CPCs are commonly assessed using flow cytometry in numerous MM treatment guidelines and clinical studies (73–75).
3.6.2 Emerging CPC detection technologies
A wide spectrum of advanced techniques has been developed to improve both the sensitivity and specificity of CPC detection, including MFC, NGF, qPCR and NGS (58). In contrast to bone marrow aspiration cytology and core biopsy, liquid biopsy approaches, such as the analysis of CTCs and circulating tumor DNA (ctDNA), are increasingly used in hematologic malignancies due to their minimally invasive nature and compatibility with multiple detection platforms. Of these methods, MFC plays a central role in differential diagnosis by providing relatively rapid and reliable results, and is used to distinguish malignant from reactive conditions and the classification of multiple diseases such as MM and MGUS (76–78). The standardization of flow cytometry for CPC detection has improved significantly in recent years. Collaborative initiatives, such as the EuroFlow consortium, have established standardized antibody panels, sample preparation protocols, and quality control guidelines, greatly reducing inter-laboratory variability. However, it requires fresh samples within 36 hours and specialized instrumentation. NGF further enhances sensitivity (≤2×10-6) and has benefited from international standardization efforts such as EuroFlow. In contrast to conventional flow cytometry and immunocytochemistry, NGF approximately doubles the detection rate of CPCs in peripheral blood (MGUS: 19%-37% vs. 59%; SMM: 15%-50% vs. 100%; MM: 50%-73% vs. 100%) (79). However, it should be noted that the 100% detection rates reported for SMM and MM are based on specific studies and may not be universally applicable. Other studies have reported lower detection rates for CPCs in these conditions, highlighting variability across NGF-based assessments. In terms of prognosis, CPC quantification by NGF has been useful for effectively distinguishing high-risk MGUS cases that are likely to progress to MM from low-risk cases, and in anticipating survival outcomes in NDMM patients. However, in a subset of MM patients, morphology assessment alone is insufficient to detect CPCs among 2,000 analyzed cells per smear, especially when the level of peripheral blood infiltration is below 0.1%. At the same time, the implementation of NGF demands substantial technical expertise and costly infrastructure. Molecular approaches, including ASO-qPCR and NGS, provide high specificity and detailed molecular resolution, yet are limited by complex sample preparation, restricted availability, longer turnaround times, and higher costs.
As artificial intelligence (AI)-assisted digital pathology continues to evolve, AI-driven automated cellular analysis has emerged as a rapidly advancing field. Chinese researchers have developed the AI-based Morphogo system for digitizing peripheral blood smear samples (70). This system identifies and classifies nucleated cells (approximately 500 to 2000 cells per smear), demonstrating superior sensitivity (89.03%), specificity (99.68%), and accuracy (99.64%) in CPC detection, and facilitates efficient CPC screening in MM patients. Integration of AI with surface-reinforced Raman spectroscopy (SERS) further enhances CPC detection sensitivity. SERS employs gold-coated magnetic nanoparticles functionalized with anti-CD138 and anti-CD38 antibodies to detect CPCs in peripheral blood. Machine learning algorithms applied to SERS signals have proven effective in identifying MM patients with high accuracy (64). Furthermore, the inductively coupled plasma mass spectrometry (ICP-MS), as a high-throughput analytical technique that integrates inductively coupled plasma with mass spectrometry, can overcome major limitations in the detection of CPCs, such as the rarity, heterogeneity, and interference from complex blood matrices (e.g., leukocytes) (66), when combined with microfluidic chip technology. This integrated approach enables the high-purity isolation of CPCs and significantly improves the detection rate of CPCs in clinical samples (100%) (67–69). It offers rapid analysis (5 minutes per 1 mL of blood) and cost efficiency (66) (Table 1).
Beyond AI-assisted cytology, sequencing-based approaches provide complementary insights into CPC biology and tumor burden. Sequencing workflow to interrogate few tumor cells (SWIFT-seq), a single-cell sequencing workflow, applying single-cell RNA and B cell receptor sequencing to paired bone marrow and CTC samples from MM patients, this approach allows detection of cytogenetic abnormalities, assessment of proliferative indices, and tracking of clonal dynamics (80). A circulatory dynamics model incorporating tumor burden, proliferation, cytogenetics, and circulatory capacity can further explain CTC levels in blood. Table 1 clearly summarizes the relative advantages and limitations of these emerging methods, highlighting key factors such as cost, time to results, sample type, and standardization status.
4 CPCs and plasma cell disorders
4.1 Pathogenesis of plasma cell disorders
Plasma cell disorders are a group of clonal plasma cell proliferative diseases, comprising monoclonal gammopathy of undetermined significance (MGUS), MM, plasmacytomas (solitary bone plasmacytoma and extramedullary plasmacytoma), immunoglobulin deposition diseases (primary light chain amyloidosis, light and heavy chain deposition disease), and POEMS syndrome (73). The central feature of plasma cell disorders is the clonal expansion of premalignant or malignant plasma cells, characterized by monoclonal immunoglobulin secretion (81, 82). The tumor microenvironment (TME) plays a critical role in the initiation and progression of plasma cell disorders. The TME is a highly dynamic and complex microenvironment that facilitates tumor growth, increases drug resistance, and compromises immune surveillance (83). Immune remodeling within the bone marrow microenvironment significantly accelerates the progression of plasma cell disorders. Various cellular components function redundantly and compensatory to support the survival of malignant plasma cells. Bone marrow mesenchymal stem cells (BM-MSCs) are critical participants, MSCs secrete CXCL12, the ligand for CXCR4, facilitating plasma cell homing to the bone marrow, providing contact-dependent support through integrins, and secreting pro-survival, anti-apoptotic, and pro-angiogenic cytokines such as IL-6, vascular endothelial growth factor (VEGF), and insulin-like growth factor 1 (IGF-1). In addition to MSCs, other factors promoting disease progression comprise the progressive impairment of tumor-suppressive immune cells (e.g., anti-MM T cells) and the accumulation of pro-tumorigenic immune cells, such as Tregs, Th17 cells, tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and immunosuppressive dendritic cells (DCs) (84). Plasma cell disorders are also characterized by extensive chromosomal abnormalities, which are commonly present from the early stages of the disease. For instance, structural centrosome aberrations may drive early aneuploidy and contribute to malignant transformation (81). Regarding clonal evolution, the transformation of post-germinal center B cells or plasma cells into MGUS and subsequently into MM involves an initiating event followed by multiple secondary genetic alterations. Initiating events typically comprise immunoglobulin heavy chain (IgH) translocations or hyperdiploidy, while secondary events include copy number variations, somatic mutations, and epigenetic alterations, all of which are advantageous for disease progression. Therefore, the clonal evolution from MGUS to MM is driven by a combination of secondary translocations, copy number alterations, oncogenic mutations, epigenetic changes, and tumor microenvironmental remodeling in premalignant plasma cell clones (85).
4.2 The generation of CPCs in plasma cell disorders
Throughout the progression of plasma cell disorders, malignant plasma cells migrate from the bone marrow into the peripheral blood, forming CPCs, which play a key role in tumor dissemination (73). Multiple studies have investigated the mechanisms of CPC generation in MM. The generation of CPCs in MM patients may involve three primary mechanisms (Figure 3).
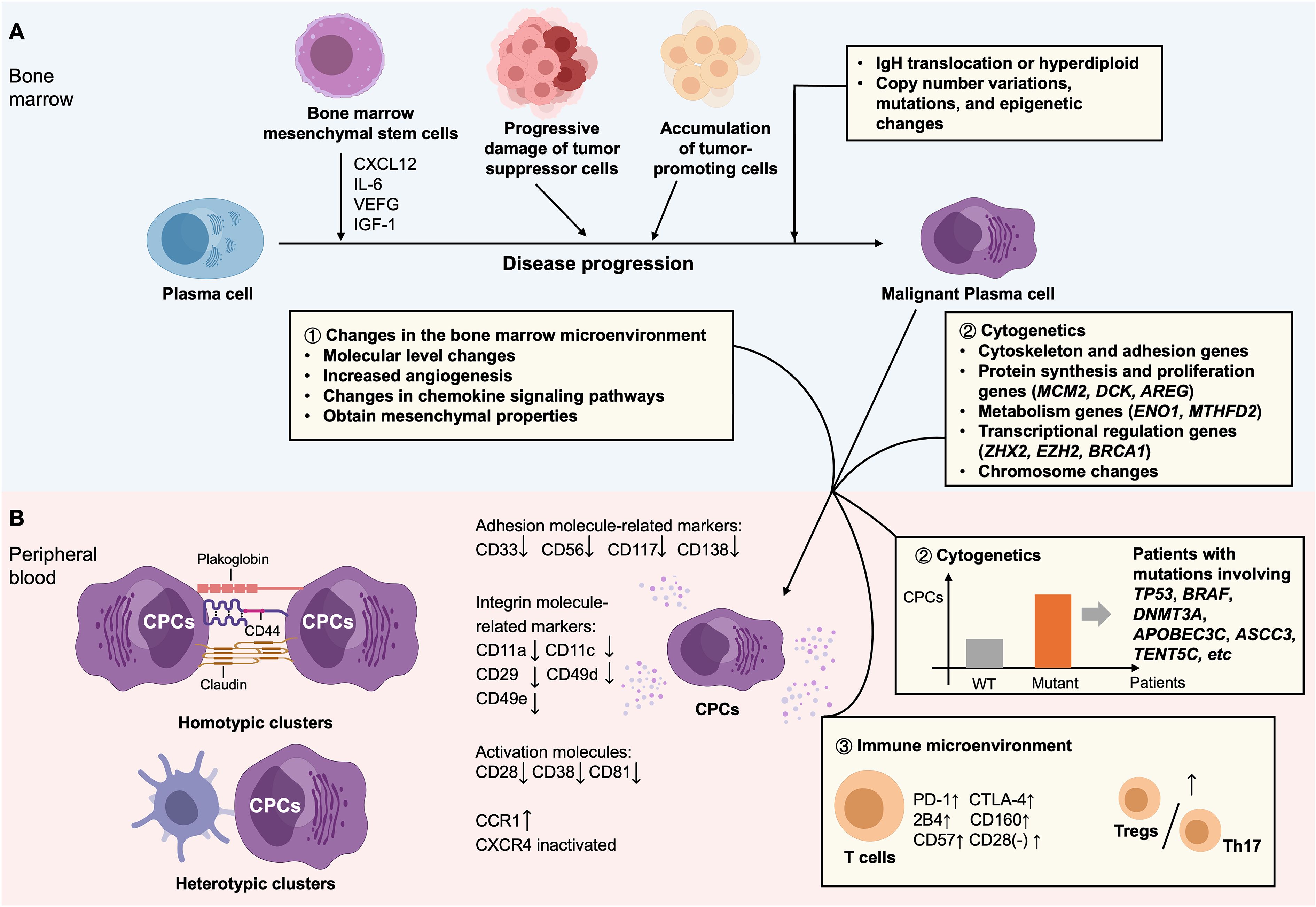
Figure 3. Proposed mechanisms driving the formation of CPCs in tumor. (A) Multiple factors within the bone marrow microenvironment that facilitate the malignant transformation of plasma cells: (i) a complex cellular milieu supports the expansion and survival of malignant plasma cells, with bone marrow-derived mesenchymal stem cells playing a key role; (ii) progressive impairment of tumor-suppressive cell populations, accompanied by the enrichment of tumor-promoting cells subsets; and (iii) multiple genetic events, initiating with primary events such as immunoglobulin heavy chain (IgH) translocations or hyperdiploidy, and followed by secondary events comprising copy number variations, somatic mutations, and epigenetic modifications. (B) Mechanisms facilitating the dissemination of malignant plasma cells from the bone marrow into the peripheral circulation: (i) remodeling of the tumor microenvironment, including alterations at the molecular level, increased angiogenesis, dysregulation of chemokine signaling pathways, and acquisition of mesenchymal-like properties; (ii) cytogenetic aberrations, involving the involvement of hub genes that may enable malignant plasma cells to survive and proliferate outside of the bone marrow microenvironment, as well as specific gene mutations and chromosomal alterations; (iii) immune microenvironmental dysregulation, particularly impaired T-cell function, characterized by T-cell exhaustion or senescence, and an imbalance between Tregs and Th17 cells; and (iv) dissemination mechanisms resembling those of CTC clusters, promoting metastasis formation, which are implicated in the initiation and promotion of distant metastases.
4.2.1 Tumor microenvironment
Alterations in the bone marrow microenvironment promote the egress of malignant plasma cells into the peripheral circulation. Current models of MM dissemination suggest that regions of severe hypoxia and pro-inflammatory conditions within, the bone marrow drive malignant plasma cells into a quiescent state, prompting their migration into the circulation to access alternative supportive niches (86). In comparison with intramedullary myeloma cells, CPCs exhibit decreased expression of adhesion molecules, integrins and activation molecules related markers. These alterations, along with elevated angiogenesis, disrupt adhesion between malignant plasma cells and the bone marrow endothelium, facilitating their intravasation into peripheral blood (87, 88). Chemokine signaling alterations are also critical in extramedullary dissemination. More importantly, hypoxia-inducible factor-2α (HIF-2α) is activated during chronic hypoxia, inducing expression of chemokine receptor CCR1 in MM cells. The interaction between up-regulated CCR1 and inactivated CXCR4 signaling (the CCR1/CXCR4 axis) promotes the egress of MM cells from the marrow, thereby facilitating to dissemination (89). Additionally, epithelial-derived CPCs undergo epithelial to mesenchymal transition-mesenchymal-to-epithelial transition (EMT-MET), characterized by changes in cell adhesion, motility, invasiveness, and loss of epithelial markers, transforming into mesenchymal-like cells that acquire mesenchymal traits enabling vascular invasion (90).
4.2.2 Cytogenetic alterations
CPCs share similar genetic profiles with bone marrow plasma cells. MinimuMM-seq analysis of enriched CPCs demonstrated that CPCs are malignant, and exhibit copy number abnormalities consistent with bone marrow samples (91). Another study employing second-generation flow cytometry analyzed 116 matched samples (55 bone marrow, 53 peripheral blood, and 8 extramedullary plasmacytomas), and demonstrated that approximately 22% of CPCs may originate from distant marrow sites (92). Moreover, 86% and 87% of mutations detected in bone marrow and plasmacytoma cells, separately, were also present in CPCs. 82% of bone marrow mutations were also detected in CPCs, as evidenced by Gene expression analysis of paired CPCs and bone marrow plasma cells in individual patients. Cytogenetic abnormalities may confer plasma cells with immune evasion capabilities and enhanced proliferative potential, thereby facilitating their entry into circulation. Recent studies have identified 41 hub genes involved in CPC biology (93). In addition to genes associated with cytoskeleton and adhesion, these comprise genes involved in protein synthesis and proliferation (e.g., MCM2, DCK, AREG), metabolic processes such as glycolysis and lactate production (e.g., ENO1, MTHFD2), and transcriptional regulation (e.g., ZHX2, EZH2, BRCA1), many of which may promote plasma cells to survive independently of the bone marrow microenvironment (93). A study employing next-generation sequencing (NGS) in a Chinese cohort showed that patients harboring mutations in genes such as TP53, BRAF, DNMT3A, APOBEC3C, ASCC3, and TENT5C exhibited significantly higher CPC levels (94). Down-regulation of TP53 may lower expression of E-cadherin and simultaneously facilitate EMT regulators, eventually lessening cell adhesion to the extracellular matrix. TP53 loss may also up-regulate microRNA-19a/CXCR5 signaling, enhancing invasiveness of myeloma cells (95). Additionally, CPCs have been linked to several chromosomal abnormalities, including t(4;14), del(13q), del(17p), t(11;14), and t(14;16) (58). Among these, high-risk cytogenetics—particularly del(17p13)—are strongly associated with increased CPC counts and play a key role in the development of secondary plasma cell leukemia (PCL), when it emerges as a novel cytogenetic finding during clonal evolution (96).
4.2.3 Immune microenvironment
Immune dysfunction, particularly impaired T cell activity, may facilitate their escape into peripheral blood because malignant plasma cells can be recognized and eliminated by cytotoxic T cells (84). As shown in (97), in MGUS, T cell clusters are enriched in stem-like memory (TCF1hi) and tissue-resident memory phenotypes. The persistence of these populations may be crucial for maintaining tumor immune surveillance throughout the MGUS stage. In contrast, MM is marked by a loss of TCF1+ memory T cells and elevated expression of cytolytic and senescence markers in T cells, indicating a breakdown of protective immunity and immune surveillance. Enhanced T-cell senescence and exhaustion in MM have been confirmed in other studies as well. Markers correlated with T-cell exhaustion (PD-1, CTLA-4, 2B4, CD160) and senescence (CD57, and loss of CD28 expression) are significantly up-regulated in both the peripheral blood and bone marrow of MM patients, where a more pronounced elevation was observed in bone marrow T cells (98). Therefore, the immunosuppressive tumor microenvironment contributes to immune evasion in MM (98). Additionally, T-helper 17 (Th17) cells can impair tumor immune surveillance through cytokine secretion (84). The Treg/Th17 cell ratio, a key indicator of immune regulation, is significantly increased in MM compared with MGUS, supporting the presence of a more immunosuppressive milieu in MM (99).
In addition, CPCs may contribute to metastasis formation through mechanisms like those observed in circulating tumor cell (CTC) cluster formation. These clusters can be homotypic, which consist solely of tumor cells, or heterotypic, which involve tumor cells in association with other cell types. Homotypic CTC clusters form characteristic oligoclonal aggregates, in which cell adhesion molecules such as plakoglobin, claudins, and CD44 are collectively critical for maintaining intercellular connections (100). Hypoxic conditions have been demonstrated to upregulate the expression of these molecules and accelerate cluster formation (100). These homotypic clusters can lead to epigenetic alterations, such as hypomethylation at binding sites for OCT4, NANOG, and SOX2, thereby conferring stem cell-like properties that facilitate metastasis (100). Heterotypic CTC clusters, formed between tumor cells and other cell types such as cancer-correlated fibroblasts, or platelets, display enhanced proliferative, invasive, and homing capabilities at metastatic sites (100). Furthermore, these clusters are also more resistant to immune surveillance (100). Similarly, CTCs interact extensively with a vast spectrum of immune cells. They form CTC-neutrophil and CTC-MDSCs clusters that promote extravasation, differentiation and proliferation. CTC clusters can modulate dendritic cell function and exhibit exceptional resistance to natural killer (NK) cell cytotoxicity or evade NK cell attack through specific molecular pathways. CTCs can directly interact with CD4+ helper T cells and CD8+ cytotoxic T cells, initiating immunosuppressive responses that support tumor cell survival (101). CPCs may adopt similar mechanisms, interacting with diverse immune cells to escape immune surveillance and facilitate dissemination and metastasis. Therefore, elevated CPC levels may reflect impaired immune function and an increased risk of tumor progression and metastasis.
In summary, malignant plasma cells in MM can enter the peripheral circulation by acquiring microenvironmental remodeling capabilities, genetic abnormalities, and immune escape mechanisms. Conversely, MGUS represents an earlier disease stage in which these mechanisms are not fully developed. For instance, while effector immune function is progressively impaired in both MGUS and MM (84), the immune abnormalities in MGUS are milder and may be insufficient to enable CPCs escape into peripheral blood.
4.3 CPCs in diagnosis, prognosis and treatment of plasma cell disorders
4.3.1 Early detection
CPCs can be detected in the peripheral blood from the earliest stages of premalignant transformation, with levels varying across various stages of plasma cell disorders, including MGUS, Smoldering Multiple Myeloma (SMM), MM, and PCL. CPCs analysis offers a minimally invasive, low-risk, and reproducible approach for assessing early disease progression risk.
In MGUS, about 1% of patients progress annually to malignant disease (58), highlighting the need for early monitoring. A European study showed that a CPC level of ≥0.058 cells/μL in peripheral blood could differentiate MGUS from MM with 88% accuracy and was associated with significantly higher 30-month progression risk (79). Similarly, a prospective study of 254 asymptomatic patients in Athens demonstrated that CPCs positivity was significantly associated with increased risk of progression to symptomatic MM (HR: 2.99, P = 0.024) (102).
In SMM, elevated CPC levels in peripheral blood are both indicative of malignant transformation and predictive of disease progression in SMM (73). The multicenter iMMunocell study reported that patients with CPCs >0.015% displayed a significantly higher progression rate in contrast to those with CPCs ≤0.015% (37.5% vs. 4%, P < 0.001) (103). Retrospective data further confirmed that patients fulfilling high CPC levels criterion displayed a significantly elevated risk of progression to active MM within 2 years (71% vs. 24%, P = 0.001) and 3 years (86% vs. 34%, P < 0.001), in contrast to those with lower CPC levels (104).
In more advanced disease, primary plasma cell leukemia (pPCL) is characterized by markedly increased plasma cells in peripheral blood, and its diagnostic criteria have been revised in recent years. In 2013, the International Myeloma Working Group (IMWG) defined pPCL as CPCs ≥20% and/or ≥2×109/L (105). Subsequent evidence showed that patients with ≥5% CPCs had similarly poor outcomes and higher pPCL detection rates, leading the IMWG in 2021 to lower the threshold to ≥5% (6, 106). More recently, Czech researchers used multiparameter flow cytometry and proposed a lower threshold of CPCs ≥2% to identify a subset of ultra-high-risk newly diagnosed MM (NDMM) patients with pPCL-like characteristics (107). These findings suggest that future refinements may adopt CPCs ≥2% as a diagnostic threshold to facilitate earlier detection and intervention.
4.3.2 Prognosis
4.3.2.1 Risk stratification
Accurate risk stratification is essential for prognosis and treatment planning. In SMM, the iMMunocell multicenter study identified 0.015% CPCs as a critical threshold for risk stratification and proposed the “20/2/0.015” SMM model (defined by serum free light chain (sFLC) ratio >20, M-protein >2 g/dL, CPCs >0.015%), which outperformed the traditional 20/2/20 system (103). In addition, CPCs should be taken into consideration alongside conventional markers such as sFLC ratio and M-protein throughout SMM diagnosis to better identify high-risk patients (73).
Similarly, in MM, CPCs have been shown to enhance existing systems. An Italian cohort showed that R-ISS II patients with ≥1 CPCs had worse outcomes than CPC-negative patients, but better than R-ISS III patients. Likewise, in a single-center study of 336 NDMM patients, adding CPCs ≥0.05% improved the discriminatory power of R2-ISS (108). A meta-analysis in China confirmed that elevated CPC levels correlate with advanced ISS/R-ISS stages and high-risk cytogenetic, indicating more aggressive disease features (5). Furthermore, a novel prognostic algorithm integrating CPCs, the Phenotypic Classification System (PCS) and R-ISS achieved superior accuracy compared with any individual system or pairwise combination in stratifying NDMM risk (109).
4.3.2.2 Prognostic prediction
Beyond risk stratification, CPCs themselves are recognized as an independent adverse prognostic factor in NDMM (110). Several studies have proposed effective thresholds: CPCs ≥0.165% predicted inferior OS, 71% vs. 87% in CPCs <0.165% at 3 years (111); CPCs ≥0.105% was linked with markedly shorter PFS and OS (110); CPCs ≥0.038% effectively distinguished high tumor burden and low remission rate populations, and served as an independent predictor of PFS and OS (112). Over the past two years, numerous in-depth studies further validated the prognostic utility of CPCs in MM. As reported by a comprehensive meta-analysis, increased CPC levels were significantly correlated with shorter OS and PFS across all subgroups, irrespective of geographic region, sample size, cutoff values, detection timing, initial treatment regimens, or data types (5). A prospective multicenter study also revealed that CPC-negative NDMM patients at diagnosis had a 5-year OS of 42%, in contrast to 25% in CPC-positive patients (P < 0.05), with prognosis determined more by the mere detectability of CPCs than by their absolute levels (113). In a study of Chinese patients, those with ≥2% CTCs showed significantly worse PFS (P < 0.001; 49 months vs. 25 months) and OS (P < 0.001; NR vs. 38 months) compared to those with <2% CTCs, indicating that a 2% CTC threshold might serve as an indicator of ultra-high-risk MM (114). CPC levels in MM patients are also linked to minimal residual disease (MRD). Patients with CPCs >0.02% had a 2-year MRD-negative rate of 45%, in contrast to 67% and 74% for patients with <0.02% CPCs or undetectable CPCs, respectively (115). Moreover, the median time to achieve MRD negativity was significantly longer in the high CPC group than in the low CPC and CPC-negative groups (34 vs. 17 vs. 13 months, P < 0.001) (79). In PCL, CPCs likewise demonstrate significant prognostic value. In a study involving 33 patients suffering from primary and secondary PCL, complete loss of CD20 expression on CPCs was associated with increased mortality (116). Patients with ≤5% CD20+CPCs had significantly shorter OS than those with >5% CD20+ CPCs (3.4 vs. 47.4 months, P = 0.044). Further studies are required to validate this finding (116).
4.3.3 Correlation with treatment outcome
A prospective study of 141 NDMM patients assessed CPC levels at diagnosis and after 3 (Peripheral blood MRD, PBMRD1) and 6 cycles (PBMRD2) of chemotherapy (117). PBMRD positivity, defined as CPCs ≥0.0001%, was significantly associated with inferior event-free survival and OS, whereas PBMRD negativity independently predicted favorable event-free survival at any time. These findings support routine CPCs monitoring after MM chemotherapy to identify patients at risk of poor response.
Autologous hematopoietic stem cell transplantation (auto-HSCT) remains a cornerstone in the treatment of MM. Multiple studies have demonstrated that CPC status before and after auto-HSCT serves as an independent prognostic factor for PFS, OS, and TTP (118–120). Accordingly, the Chinese expert consensus recommends CPC testing in peripheral blood before auto-HSCT and at day 100 post-transplant (73). In line with this, Chakraborty et al. stratified patients into four groups according to CPC status at diagnosis and pre-transplant: CPC−/−, CPC−/+, CPC+/−, and CPC+/+ (120). In multivariate analysis for overall survival, the CPC-positive groups exhibited substantially higher risk of death compared with the CPC−/− group. Importantly, the prognostic impact of CPCs extends to both transplant-eligible (TE) and transplant-ineligible (TI) NDMM patients. A Greek study including both TE and TI NDMM patients identified an optimal cutoff of 0.02% (115). Patients with CPCs ≥0.02% showed significantly shorter median PFS compared with CPCs <0.02%. Multivariate analysis further confirmed that elevated CPCs above this threshold had an independent prognostic impact on PFS, conferring even greater risk of progression than ISS stage III or high-risk cytogenetics.
4.3.4 Recent therapeutic advance
Recent advancements in therapeutic approaches have significantly improved outcomes for MM patients, especially those with relapsed or refractory disease. Novel therapeutic strategies include monoclonal antibodies, small molecules, and autologous cell-based immunotherapies such as chimeric antigen receptor T-cell (CAR-T) therapy and bispecific antibodies (121). To date, BCMA-targeted CAR-T therapy has shown remarkable efficacy for MM treatment (122), with two FDA-approved products: Idecabtagene vicleucel (ide-cel) and Ciltacabtagene autoloeucel (cilta-cel) for relapsed/refractory MM (121). Additional therapeutic agents include CD38-targeting antibodies (daratumumab, isatuximab), BCMA-targeted agents (belantamab mafodotin, teclistamab), immunomodulatory drugs (lenalidomide, pomalidomide), proteasome inhibitors (bortezomib, carfilzomib), monoclonal antibody elotuzumab, exportin 1 (XPO1) inhibitor selinexor, and various vaccine-based therapies (121, 123, 124). Approved both domestically and internationally, these treatments now provide clinicians with more than ten therapeutic options. The clinical utility of CPCs within the context of these innovative therapies warrants further investigation. At the 2024 ASH Annual Meeting, researchers from the Netherlands presented findings from the Perseus study, identifying CPCs>0.175% as a biomarker for poor prognosis in transplant-eligible, high-risk NDMM patients, who received bortezomib, lenalidomide and dexamethasone with or without daratumumab throughout induction/consolidation, and lenalidomide with or without daratumumab during maintenance (125). In addition, PBMRD monitoring, alongside bone marrow MRD assessment, has been increasingly recognized as an important tool for evaluating novel therapies in MM, with the potential to serve as an independent indicator in the future. Nonetheless, several pivotal questions remain unaddressed despite these promising findings. For instance, while CPCs are thought to arise from malignant bone marrow cells migrating into peripheral circulation, few studies have directly investigated how bone marrow tumor burden correlates with CPC levels. Moreover, recent research has underscored the potential of circulating microRNAs (miRNAs) and cell-free DNA (cfDNA) as biomarkers for MM diagnosis and prognosis (126–128). Given that CPCs, miRNAs, and cfDNA all serve as indicators in liquid biopsy, future studies should investigate their potential interplay, which could provide new insights into MM pathophysiology and risk stratification.
4.3.5 CPCs as clinical biomarkers
In MM, malignant plasma cells exhibit patchy infiltration within the bone marrow. Studies have confirmed that cytogenetic alterations and mutations vary across different tumor sites, reflecting the spatial genetic heterogeneity (129). Although bone marrow plasma cell assessment remains the gold standard for evaluating tumor burden in MM patients, biopsy from a single bone marrow site cannot fully capture the spatial genetic heterogeneity of the disease and may fail to reflect the full disease heterogeneity. Malignant plasma cells in MM disseminate from diverse bone marrow regions or extramedullary sites into the peripheral blood. As a result, peripheral blood sampling may provide a more comprehensive reflection of the overall tumor burden when compared to samples acquired from a single-site bone marrow aspiration. Genetic analyses support this view, revealing that CPCs in peripheral blood offer genetic insights comparable to those of bone marrow plasma cells and may even be undetectable in bone marrow specimens (130). Although approximately 15% of MM patients demonstrate phenotypic discordance between bone marrow clonal plasma cells and matched CPCs, studies have confirmed and extended this finding from a phenotypic standpoint. This discrepancy is more prevalent in patients with elevated CPC levels, suggesting that the increase in CPCs may result from the dissemination of myeloma cells with distinct phenotypic (and possibly genetic) features from diverse bone marrow compartments into the peripheral circulation (115). Overall, for tumor burden assessment, CPC detection in peripheral blood provides a more comprehensive assessment compared to single-site bone marrow biopsy, offering advantages such as simpler sample acquisition, minimal invasiveness, and high reproducibility.
4.4 International consensus and advances in MM-related CPC assessment
As clinical research advances and new evidence accumulates, recommendations regarding CPCs in peripheral blood have undergone multiple updates in both national and international clinical guidelines. In China, the 2015 edition of the MM diagnosis and treatment guidelines was the first to include the percentage of peripheral blood CPCs as a mandatory assessment parameter. This requirement has been retained in all subsequent versions (2017, 2020, 2022, and 2024), with the 2024 edition further incorporating CPCs percentage into the MM prognostic stratification system (73). Specifically, patients with CPCs ≥2% are classified as ultra-high risk, while those with CPCs ≥0.07% are categorized as high-risk (73).
Internationally, the 2023.v2 edition of the National Comprehensive Cancer Network (NCCN) guidelines recognized CPCs as a high-risk clinical factor in MM. In the updated 2025.v1 version, CPCs have also been recognized as a risk factor for MM relapse (131). IMWG included elevated peripheral blood CPCs as a potential diagnostic marker in their 2014 diagnostic criteria (132). In 2021, the IMWG published a consensus on pPCL, revising its diagnostic threshold to CPCs ≥5% (6). Furthermore, the 2022 and 2024 guidelines on MM management both identified elevated peripheral blood CPC levels as a high-risk feature for SMM (133, 134). Furthermore, a 2021 consensus statement by the European Myeloma Network also acknowledged the prognostic relevance of CPCs in peripheral blood (135).
5 CPCs and autoimmune diseases
5.1 Pathogenesis of autoimmune diseases
Autoimmune diseases are characterized by aberrant immune responses in which the immune system mistakenly targets self-tissues, leading to organ damage or dysfunction. Representative examples include SLE, rheumatoid arthritis (RA), and multiple sclerosis. Their pathogenesis involves a complex interplay of genetic predisposition, environmental triggers, and dysregulated immune mechanisms, among which B cells play a pivotal role. Disease activity in autoimmune disorders is closely associated with autoantibodies secreted by plasmablasts and plasma cells. During disease flares, waves of newly generated autoreactive plasma cells might contribute to the occupation of plasma cell niches by autoreactive LLPCs, replacing old, protective plasma cells. CPCs play a central role in the pathogenesis of autoimmune diseases by continuously producing autoantibodies, which are directly implicated in the initiation and perpetuation of chronic inflammation and tissue damage. Plasma cells, particularly the long-lived subset, can escape normal tolerance checkpoints and persist in the circulation and inflamed tissues, secreting high-affinity autoantibodies that target self-antigens. The sustained presence of these autoantibodies leads to immune complex formation, complement activation, and recruitment of inflammatory cells, thereby driving chronic inflammation and organ-specific or systemic tissue injury (136, 137). In comparison with short-lived plasma cells, LLPCs exhibit marked resistance to conventional therapies and persistently secrete pathogenic antibodies, which drive the chronicity and relapse of autoimmune diseases (138).
Clinical observations further support the pathogenic role of plasma cells in autoimmunity, as approximately 30–35% of patients with lupus and RA show elevated levels of plasma cells (139). Consequently, effective depletion of autoreactive plasma cells may represent a key strategy for curative treatment of autoimmune diseases. However, although B cell depletion therapy can eliminate most circulating B cells in peripheral blood (including CPCs), clinical outcomes vary considerably among individuals, likely due to differential activation or survival signals for B cells provided by tissue microenvironment.
5.2 CPCs in SLE prognosis and treatment monitoring
SLE is a chronic inflammatory autoimmune disease characterized by hyperactivation of B cells and an increased frequency of CPCs (4). Simultaneously, terminally differentiated B cells, namely plasmablasts and LLPCs, experience clonal expansion and secrete large amounts of autoantibodies. These antibodies not only mediate the formation of immune complexes but also trigger the production of pro-inflammatory cytokines through type I interferon signaling pathways. This cascade exacerbates tissue injury, sustains a protracted inflammatory milieu, and leads to multi-organ involvement in affected patients (140). Previous studies have suggested that circulating plasmablasts serve as biomarkers for assessing SLE disease activity, anticipating disease flares, and guiding therapeutic decisions (141–143). Numerous research supports the potential role of CPCs in both disease monitoring and treatment evaluation in SLE (144–146). Deng-Ho et al. reported that the percentage of peripheral CD27high plasma cells significantly correlates with SLE Disease Activity Index (SLEDAI) scores, anti-dsDNA antibody titers, and complement (C3/C4) levels in non-infected SLE patients (144). Nevertheless, this association was no longer observed in those SLE patients with concurrent infections, suggesting that the frequency of peripheral CD27high plasma cells may serve as a differentiation marker to distinguish between lupus flares and infections. Moreover, data from several phase III clinical trials of belimumab analyzed by Parodis et al. suggested that treatment responders exhibited a greater decrease in peripheral CD19+CD20⁻CD138+ LLPCs in comparison with non-responders (−48.2% vs. −37.1%; P = 0.024) (145). In a comparative analysis of patients receiving belimumab versus placebo, only the treatment group exhibited a rapid decline in LLPCs, indicating a greater protective effect against disease flares (146). Notably, this trend was absent in the placebo cohort (146). Taken together, these findings suggest that monitoring CPC levels may function as a useful tool for evaluating treatment response and anticipating disease relapse in SLE. Nonetheless, further studies are imperative to validate these observations.
6 Conclusions
In summary, the literature indicates that CPCs may enter the peripheral blood from the bone marrow stroma due to multiple factors, among which tumor microenvironment and cytogenetic abnormalities play crucial roles in this process. The presence of CPCs represents a valuable approach for assessing disease status and treatment efficacy. In plasma cell neoplasms, particularly MM and PCL, CPCs have been firmly established as important prognostic and diagnostic biomarkers. In MM, quantification of CPCs is now recognized as a key risk stratification tool, with thresholds as low as 0.038% predicting adverse outcomes, whereas ≥0.02% correlated with lower MRD negativity, and ≥2% CPCs as a marker of ultra-high-risk MM with pPCL-like features. Although the current diagnostic criterion for pPCL is ≥5% CPCs in peripheral blood, accumulating evidence suggests that even lower levels are clinically relevant and should be considered in the design of future clinical trials. In autoimmune diseases, the role of CPCs remains poorly defined. Although plasma cells are pivotal for autoantibody production and disease pathogenesis in conditions such as SLE, current research predominantly emphasizes tissue-resident and LLPCs rather than circulating subsets. Investigations into plasma cell heterogeneity and targeted depletion strategies are ongoing; however, the clinical utility of CPC quantification outside hematologic malignancies is still limited. Technological advances have improved sensitivity and standardization of CPC detection, enabling minimally invasive monitoring and investigation of CPC biology. Future research should prioritize the optimization and standardization of CPC detection methodologies. Furthermore, integrating these findings into precision medicine and targeted therapeutic strategies could facilitate more individualized and effective clinical management for patients.
Author contributions
PW: Methodology, Conceptualization, Writing – original draft, Investigation. NS: Methodology, Writing – original draft, Investigation. XY: Writing – review & editing, Supervision. FX: Writing – review & editing, Supervision. YZ: Visualization, Writing – review & editing, Conceptualization, Supervision.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Caraux A, Klein B, Paiva B, Bret C, Schmitz A, Fuhler GM, et al. Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica. (2010) 95:1016–20. doi: 10.3324/haematol.2009.018689
2. Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. (2005) 105:1614–21. doi: 10.1182/blood-2004-07-2507
3. Zhu L, Yang P, Zhao Y, Zhuang Z, Wang Z, Song R, et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and influenza patients. Immunity. (2020) 53:685–696.e3. doi: 10.1016/j.immuni.2020.07.009
4. Lugar PL, Love C, Grammer AC, Dave SS, and Lipsky PE. Molecular characterization of circulating plasma cells in patients with active systemic lupus erythematosus. PLoS One. (2012) 7:e44362. doi: 10.1371/journal.pone.0044362
5. Li Q, Ai L, Zuo L, Li J, Zhao F, Xu A, et al. Circulating plasma cells as a predictive biomarker in Multiple myeloma: an updated systematic review and meta-analysis. Ann Med. (2024) 56:2338604. doi: 10.1080/07853890.2024.2338604
6. Fernández de Larrea C, Kyle R, Rosiñol L, Paiva B, Engelhardt M, Usmani S, et al. Primary plasma cell leukemia: consensus definition by the International Myeloma Working Group according to peripheral blood plasma cell percentage. Blood Cancer J. (2021) 11:192. doi: 10.1038/s41408-021-00587-0
7. Nguyen DC, Joyner CJ, Sanz I, and Lee FE. Factors affecting early antibody secreting cell maturation into long-lived plasma cells. Front Immunol. (2019) 10:2138. doi: 10.3389/fimmu.2019.02138
8. Manakkat Vijay GK and Singh H. Cell fate dynamics and genomic programming of plasma cell precursors. Immunol Rev. (2021) 303:62–71. doi: 10.1111/imr.13010
9. Nguyen DC, Saney C, Hentenaar IT, Cabrera-Mora M, Capric V, Woodruff MC, et al. Majority of human circulating IgG plasmablasts stop blasting in a cell-free pro-survival culture. Sci Rep. (2024) 14:3616. doi: 10.1038/s41598-024-53977-2
10. Nera KP, Kohonen P, Narvi E, Peippo A, Mustonen L, Terho P, et al. Loss of Pax5 promotes plasma cell differentiation. Immunity. (2006) 24:283–93. doi: 10.1016/j.immuni.2006.02.003
11. Muto A, Tashiro S, Nakajima O, Hoshino H, Takahashi S, Sakoda E, et al. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature. (2004) 429:566–71. doi: 10.1038/nature02596
12. Huang C, Geng H, Boss I, Wang L, and Melnick A. Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood. (2014) 123:1012–20. doi: 10.1182/blood-2013-07-518605
13. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. (2006) 7:773–82. doi: 10.1038/ni1357
14. Lin Y, Wong K, and Calame K. Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science. (1997) 276:596–9. doi: 10.1126/science.276.5312.596
15. Lin KI, Angelin-Duclos C, Kuo TC, and Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. (2002) 22:4771–80. doi: 10.1128/MCB.22.13.4771-4780.2002
16. Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. (2002) 17:51–62. doi: 10.1016/s1074-7613(02)00335-7
17. Minnich M, Tagoh H, Bönelt P, Axelsson E, Fischer M, Cebolla B, et al. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol. (2016) 17:331–43. doi: 10.1038/ni.3349
18. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, and Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
19. Shen Y and Hendershot LM. Identification of ERdj3 and OBF-1/BOB-1/OCA-B as direct targets of XBP-1 during plasma cell differentiation. J Immunol. (2007) 179:2969–78. doi: 10.4049/jimmunol.179.5.2969
20. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. (2004) 21:81–93. doi: 10.1016/j.immuni.2004.06.010
21. Winkelmann R, Sandrock L, Porstner M, Roth E, Mathews M, Hobeika E, et al. B cell homeostasis and plasma cell homing controlled by Krüppel-like factor 2. Proc Natl Acad Sci U S A. (2011) 108:710–5. doi: 10.1073/pnas.1012858108
22. Kabashima K, Haynes NM, Xu Y, Nutt SL, Allende ML, Proia RL, et al. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J Exp Med. (2006) 203:2683–90. doi: 10.1084/jem.20061289
23. Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, et al. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol. (2002) 169:1277–82. doi: 10.4049/jimmunol.169.3.1277
24. Manakkat Vijay GK, Zhou M, Thakkar K, Rothrauff A, Chawla AS, Chen D, et al. Temporal dynamics and genomic programming of plasma cell fates. Nat Immunol. (2024) 25:1097–109. doi: 10.1038/s41590-024-01831-y
25. Liu X, Yao J, Zhao Y, Wang J, and Qi H. Heterogeneous plasma cells and long-lived subsets in response to immunization, autoantigen and microbiota. Nat Immunol. (2022) 23:1564–76. doi: 10.1038/s41590-022-01345-5
26. Simons BD and Karin O. Tuning of plasma cell lifespan by competition explains the longevity and heterogeneity of antibody persistence. Immunity. (2024) 57:600–611.e6. doi: 10.1016/j.immuni.2024.02.005
27. Bortnick A and Allman D. What is and what should always have been: long-lived plasma cells induced by T cell-independent antigens. J Immunol. (2013) 190:5913–8. doi: 10.4049/jimmunol.1300161
28. Pelletier N, McHeyzer-Williams LJ, Wong KA, Urich E, Fazilleau N, and McHeyzer-Williams MG. Plasma cells negatively regulate the follicular helper T cell program. Nat Immunol. (2010) 11:1110–8. doi: 10.1038/ni.1954
29. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. (2014) 507:366–70. doi: 10.1038/nature12979
30. Cupi ML, Sarra M, Marafini I, Monteleone I, Franzè E, Ortenzi A, et al. Plasma cells in the mucosa of patients with inflammatory bowel disease produce granzyme B and possess cytotoxic activities. J Immunol. (2014) 192:6083–91. doi: 10.4049/jimmunol.1302238
31. Fritz JH, Rojas OL, Simard N, McCarthy DD, Hapfelmeier S, Rubino S, et al. Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature. (2011) 481:199–203. doi: 10.1038/nature10698
32. Minges Wols HA, Underhill GH, Kansas GS, and Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. (2002) 169:4213–21. doi: 10.4049/jimmunol.169.8.4213
33. Khodadadi L, Cheng Q, Radbruch A, and Hiepe F. The maintenance of memory plasma cells. Front Immunol. (2019) 10:721. doi: 10.3389/fimmu.2019.00721
34. Belnoue E, Tougne C, Rochat AF, Lambert PH, Pinschewer DD, and Siegrist CA. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. J Immunol. (2012) 188:1283–91. doi: 10.4049/jimmunol.1103169
35. Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood. (2010) 116:1867–75. doi: 10.1182/blood-2009-12-259457
36. Chu VT, Beller A, Rausch S, Strandmark J, Zänker M, Arbach O, et al. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity. (2014) 40:582–93. doi: 10.1016/j.immuni.2014.02.014
37. Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, and Mack M. Basophils support the survival of plasma cells in mice. J Immunol. (2010) 185:7180–5. doi: 10.4049/jimmunol.1002319
38. Glatman Zaretsky A, Konradt C, Dépis F, Wing JB, Goenka R, Atria DG, et al. T regulatory cells support plasma cell populations in the bone marrow. Cell Rep. (2017) 18:1906–16. doi: 10.1016/j.celrep.2017.01.067
39. Chen W, Hong SH, Jenks SA, Anam FA, Tipton CM, Woodruff MC, et al. Distinct transcriptomes and autocrine cytokines underpin maturation and survival of antibody-secreting cells in systemic lupus erythematosus. Nat Commun. (2024) 15:1899. doi: 10.1038/s41467-024-46053-w
40. Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. (2003) 171:1684–90. doi: 10.4049/jimmunol.171.4.1684
41. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. (2004) 199:91–8. doi: 10.1084/jem.20031330
42. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. (2008) 180:3655–9. doi: 10.4049/jimmunol.180.6.3655
43. Menzel SR, Roth E, Wittner J, Brey S, Weckwerth L, Thomas J, et al. B cell maturation antigen (BCMA) is dispensable for the survival of long-lived plasma cells. Nat Commun. (2025) 16:7106. doi: 10.1038/s41467-025-62530-2
44. Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, et al. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. (2016) 45:60–73. doi: 10.1016/j.immuni.2016.06.011
45. Nguyen DC, Garimalla S, Xiao H, Kyu S, Albizua I, Galipeau J, et al. Factors of the bone marrow microniche that support human plasma cell survival and immunoglobulin secretion. Nat Commun. (2018) 9:3698. doi: 10.1038/s41467-018-05853-7
46. Tooze RM. A replicative self-renewal model for long-lived plasma cells: questioning irreversible cell cycle exit. Front Immunol. (2013) 4:460. doi: 10.3389/fimmu.2013.00460
47. Duan M, Nguyen DC, Joyner CJ, Saney CL, Tipton CM, Andrews J, et al. Understanding heterogeneity of human bone marrow plasma cell maturation and survival pathways by single-cell analyses. Cell Rep. (2023) 42:112682. doi: 10.1016/j.celrep.2023.112682
48. Joyner CJ, Ley AM, Nguyen DC, Ali M, Corrado A, Tipton C, et al. Generation of human long-lived plasma cells by developmentally regulated epigenetic imprinting. Life Sci Alliance. (2021) 5:e202101285. doi: 10.26508/lsa.202101285
49. Nguyen DC, Hentenaar IT, Cabrera-Mora M, Kyu S, Morrison-Porter A, Haddad NS, et al. Maturation of human early-minted blood antibody-secreting cells is coupled with increased IgG secretion rates. Front Immunol. (2025) 16:1644102. doi: 10.3389/fimmu.2025.1644102
50. Schulz SR, Menzel SR, Wittner J, Ulbricht C, Grofe AT, Roth E, et al. Decoding plasma cell maturation dynamics with BCMA. Front Immunol. (2025) 16:1539773. doi: 10.3389/fimmu.2025.1539773
51. Slocombe T, Brown S, Miles K, Gray M, Barr TA, and Gray D. Plasma cell homeostasis: the effects of chronic antigen stimulation and inflammation. J Immunol. (2013) 191:3128–38. doi: 10.4049/jimmunol.1301163
52. Sprumont A, Rodrigues A, McGowan SJ, Bannard C, and Bannard O. Germinal centers output clonally diverse plasma cell populations expressing high- and low-affinity antibodies. Cell. (2023) 186:5486–5499.e13. doi: 10.1016/j.cell.2023.10.022
53. Neumeier D, Pedrioli A, Genovese A, Sandu I, Ehling R, Hong KL, et al. Profiling the specificity of clonally expanded plasma cells during chronic viral infection by single-cell analysis. Eur J Immunol. (2022) 52:297–311. doi: 10.1002/eji.202149331
54. Kräutler NJ, Yermanos A, Pedrioli A, Welten SPM, Lorgé D, Greczmiel U, et al. Quantitative and qualitative analysis of humoral immunity reveals continued and personalized evolution in chronic viral infection. Cell Rep. (2020) 30:997–1012.e6. doi: 10.1016/j.celrep.2019.12.088
55. MacLean AJ, Deimel LP, Zhou P, ElTanbouly MA, Merkenschlager J, Ramos V, et al. Affinity maturation of antibody responses is mediated by differential plasma cell proliferation. Science. (2025) 387:413–20. doi: 10.1126/science.adr6896
56. Alomari N and Totonchy J. IL-21 signaling promotes the establishment of KSHV infection in human tonsil lymphocytes by increasing differentiation and targeting of plasma cells. Front Immunol. (2022) 13:1010274. doi: 10.3389/fimmu.2022.1010274
57. Nguyen DC, Hentenaar IT, Morrison-Porter A, Solano D, Haddad NS, Castrillon C, et al. SARS-CoV-2-specific plasma cells are not durably established in the bone marrow long-lived compartment after mRNA vaccination. Nat Med. (2025) 31:235–44. doi: 10.1038/s41591-024-03278-y
58. Sanoja-Flores L, Flores-Montero J, Pérez-Andrés M, Puig N, and Orfao A. Detection of circulating tumor plasma cells in monoclonal gammopathies: methods, pathogenic role, and clinical implications. Cancers (Basel). (2020) 12:1499. doi: 10.3390/cancers12061499
59. Jourdan M, Caraux A, De Vos J, Fiol G, Larroque M, Cognot C, et al. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood. (2009) 114:5173–81. doi: 10.1182/blood-2009-07-235960
60. Méndez-, Lucas D, Battista M, and Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. (2008) 452:442–7. doi: 10.1038/nature06685
61. Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. (2007) 131:994–1008. doi: 10.1016/j.cell.2007.09.047
62. de Jong MME, Chen L, Raaijmakers MHGP, and Cupedo T. Bone marrow inflammation in haematological Malignancies. Nat Rev Immunol. (2024) 24:543–58. doi: 10.1038/s41577-024-01003-x
63. Mandraffino G, Aragona CO, Basile G, Cairo V, Mamone F, Morace C, et al. CD34+ cell count predicts long lasting life in the oldest old. Mech Ageing Dev. (2017) 164:139–45. doi: 10.1016/j.mad.2017.03.003
64. Zhang D, Chen X, Lin J, Jiang S, Fan M, Liu N, et al. Ultrasensitive detection of circulating plasma cells using surface-enhanced Raman spectroscopy and machine learning for multiple myeloma monitoring. Anal Chem. (2025) 97:4101–10. doi: 10.1021/acs.analchem.4c06244
65. Lin J, Zheng J, and Wu A. An efficient strategy for circulating tumor cell detection: surface-enhanced Raman spectroscopy. J Mater Chem B. (2020) 8:3316–26. doi: 10.1039/c9tb02327e
66. Cai J, Chen B, He M, Yuan G, and Hu B. An integrated inertial-magnetophoresis microfluidic chip online-coupled with ICP-MS for rapid separation and precise detection of circulating tumor cells. Anal Chem. (2024) 96:14222–9. doi: 10.1021/acs.analchem.4c02876
67. Fu X, Chen B, He M, Yuan G, and Hu B. Facile cascaded negative magnetophoresis chip combined with ICP-MS for efficient sorting and online detection of circulating tumor cells. Anal Chem. (2025) 97:6702–10. doi: 10.1021/acs.analchem.4c06889
68. Xu Y, Chen B, He M, Cui Z, and Hu B. All-in-one microfluidic chip for online labeling, separating, and focusing rare circulating tumor cells from blood samples followed by inductively coupled plasma mass spectrometry detection. Anal Chem. (2023) 95:14061–7. doi: 10.1021/acs.analchem.3c02680
69. Nian M, Chen B, He M, and Hu B. A cascaded phase-transfer microfluidic chip with magnetic probe for high-activity sorting, purification, release, and detection of circulating tumor cells. Anal Chem. (2024) 96:766–74. doi: 10.1021/acs.analchem.3c03971
70. Chen P, Zhang L, Cao X, Jin X, Chen N, Zhang L, et al. Detection of circulating plasma cells in peripheral blood using deep learning-based morphological analysis. Cancer. (2024) 130:1884–93. doi: 10.1002/cncr.35202
71. van de Donk NWCJ. How we manage newly diagnosed multiple myeloma with circulating tumor cells. J Clin Oncol. (2023) 41:1342–9. doi: 10.1200/JCO.22.02114
72. Kumar S, Rajkumar SV, Kyle RA, Lacy MQ, Dispenzieri A, Fonseca R, et al. Prognostic value of circulating plasma cells in monoclonal gammopathy of undetermined significance. J Clin Oncol. (2005) 23:5668–74. doi: 10.1200/JCO.2005.03.159
73. Plasma Cell Disease Group, Chinese Society of Hematology, Chinese Medical Association; Laboratory Diagnosis Group, Chinese Society of Hematology, Chinese Medical Association. Chinese expert consensus on the application of flow cytometry for the detectin of circulating plasma cells in plasma cell disorders. Zhonghua Xue Ye Xue Za Zhi. (2024) 45:313–21. doi: 10.3760/cma.j.cn121090-20240117-00026
74. Liu X, Wu F, Ye W, Deng J, Zhang M, Zhang C, et al. Prognostic value of circulating plasma cells detected by flow cytometry in newly diagnosed multiple myeloma patients: a systematic review and meta-analysis. BMJ Open. (2024) 14:e071548. doi: 10.1136/bmjopen-2022-071548
75. Sathya P, Kayal S, Srinivas BH, Hamide A, and Kar R. Quantification of circulating clonal plasma cells by multiparametric flow cytometry as a prognostic marker in patients with newly diagnosed multiple myeloma. Int J Lab Hematol. (2023) 45:917–26. doi: 10.1111/ijlh.14156
76. Jelinek T, Bezdekova R, Zatopkova M, Burgos L, Simicek M, Sevcikova T, et al. Current applications of multiparameter flow cytometry in plasma cell disorders. Blood Cancer J. (2017) 7:e617. doi: 10.1038/bcj.2017.90
77. Gonsalves WI, Jevremovic D, Nandakumar B, Dispenzieri A, Buadi FK, Dingli D, et al. Enhancing the R-ISS classification of newly diagnosed multiple myeloma by quantifying circulating clonal plasma cells. Am J Hematol. (2020) 95:310–5. doi: 10.1002/ajh.25709
78. Periago A, Campillo JA, Mrowiec A, Gimeno L, Montes NR, Martínez-Sánchez MV, et al. Circulating aberrant plasma cells allow risk stratification of patients with myeloma. Am J Hematol. (2016) 91:E353–5. doi: 10.1002/ajh.24431
79. Sanoja-Flores L, Flores-Montero J, Garcés JJ, Paiva B, Puig N, García-Mateo A, et al. Next generation flow for minimally-invasive blood characterization of MGUS and multiple myeloma at diagnosis based on circulating tumor plasma cells (CTPC). Blood Cancer J. (2018) 8:117. doi: 10.1038/s41408-018-0153-9
80. Lightbody ED, Sklavenitis-Pistofidis R, Wu T, Tsuji J, Firer DT, Agius MP, et al. SWIFT-seq enables comprehensive single-cell transcriptomic profiling of circulating tumor cells in multiple myeloma and its precursors. Nat Cancer. (2025) 6:1595–611. doi: 10.1038/s43018-025-01006-0
81. Köhrer S, Dittrich T, Schorb M, Weinhold N, Haberbosch I, Börmel M, et al. High-throughput electron tomography identifies centriole over-elongation as an early event in plasma cell disorders. Leukemia. (2023) 37:2468–78. doi: 10.1038/s41375-023-02056-y
82. Fend F, Dogan A, and Cook JR. Plasma cell neoplasms and related entities-evolution in diagnosis and classification. Virchows Arch. (2023) 482:163–77. doi: 10.1007/s00428-022-03431-3
83. Radhakrishnan V, Golla U, and Kudva AK. Role of immune cells and immunotherapy in multiple myeloma. Life (Basel). (2024) 14:461. doi: 10.3390/life14040461
84. Lagreca I, Riva G, Nasillo V, Barozzi P, Castelli I, Basso S, et al. The role of T cell immunity in monoclonal gammopathy and multiple myeloma: from immunopathogenesis to novel therapeutic approaches. Int J Mol Sci. (2022) 23:5242. doi: 10.3390/ijms23095242
85. van Nieuwenhuijzen N, Spaan I, Raymakers R, and Peperzak V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. (2018) 78:2449–56. doi: 10.1158/0008-5472.CAN-17-3115
86. Rodriguez-Otero P, Paiva B, and San-Miguel JF. Roadmap to cure multiple myeloma. Cancer Treat Rev. (2021) 100:102284. doi: 10.1016/j.ctrv.2021.102284
87. Paiva B, Paino T, Sayagues JM, Garayoa M, San-Segundo L, Martín M, et al. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood. (2013) 122:3591–8. doi: 10.1182/blood-2013-06-510453
88. Testa U and Leone G. Plasma cell neoplasms with spreading in the blood and tissues: extramedullary myeloma disease, a rare aggressive form of multiple myeloma (first of two parts). Mediterr J Hematol Infect Dis. (2025) 17:e2025005. doi: 10.4084/MJHID.2025.005
89. Vandyke K, Zeissig MN, Hewett DR, Martin SK, Mrozik KM, Cheong CM, et al. HIF-2α promotes dissemination of plasma cells in multiple myeloma by regulating CXCL12/CXCR4 and CCR1. Cancer Res. (2017) 77:5452–63. doi: 10.1158/0008-5472.CAN-17-0115
90. Kölbl AC, Jeschke U, and Andergassen U. The significance of epithelial-to-mesenchymal transition for circulating tumor cells. Int J Mol Sci. (2016) 17:1308. doi: 10.3390/ijms17081308
91. Dutta AK, Alberge JB, Lightbody ED, Boehner CJ, Dunford A, Sklavenitis-Pistofidis R, et al. MinimuMM-seq: genome sequencing of circulating tumor cells for minimally invasive molecular characterization of multiple myeloma pathology. Cancer Discov. (2023) 13:348–63. doi: 10.1158/2159-8290.CD-22-0482
92. Garcés JJ, Bretones G, Burgos L, Valdes-Mas R, Puig N, Cedena MT, et al. Circulating tumor cells for comprehensive and multiregional non-invasive genetic characterization of multiple myeloma. Leukemia. (2020) 34:3007–18. doi: 10.1038/s41375-020-0883-0
93. Jelinek T, Nenarokov S, Sevcikova T, Radova E, Venglar O, Kapustova V, et al. Insights into biological mechanisms responsible for egression of circulating tumor plasma cells (CTCs) in multiple myeloma. Blood. (2024) 144:3270. doi: 10.1182/blood-2024-206825
94. Xia Y, Shen N, Zhang R, Wu Y, Shi Q, Li J, et al. High-risk multiple myeloma predicted by circulating plasma cells and its genetic characteristics. Front Oncol. (2023) 13:1083053. doi: 10.3389/fonc.2023.1083053
95. Yue Z, Zhou Y, Zhao P, Chen Y, Yuan Y, Jing Y, et al. p53 Deletion promotes myeloma cells invasion by up-regulating miR19a/CXCR5. Leuk Res. (2017) 60:115–22. doi: 10.1016/j.leukres.2017.07.003
96. Papadhimitriou SI, Terpos E, Liapis K, Pavlidis D, Marinakis T, Kastritis E, et al. The cytogenetic profile of primary and secondary plasma cell leukemia: etiopathogenetic perspectives, prognostic impact and clinical relevance to newly diagnosed multiple myeloma with differential circulating clonal plasma cells. Biomedicines. (2022) 10:209. doi: 10.3390/biomedicines10020209
97. Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight. (2019) 5:e127807. doi: 10.1172/jci.insight.127807
98. Zelle-Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. (2016) 9:116. doi: 10.1186/s13045-016-0345-3
99. Favaloro J, Brown R, Aklilu E, Yang S, Suen H, Hart D, et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk Lymphoma. (2014) 55:1090–8. doi: 10.3109/10428194.2013.825905
100. Ring A, Nguyen-Sträuli BD, Wicki A, and Aceto N. Biology, vulnerabilities and clinical applications of circulating tumour cells. Nat Rev Cancer. (2023) 23:95–111. doi: 10.1038/s41568-022-00536-4
101. Pereira-Veiga T, Schneegans S, Pantel K, and Wikman H. Circulating tumor cell-blood cell crosstalk: Biology and clinical relevance. Cell Rep. (2022) 40:111298. doi: 10.1016/j.celrep.2022.111298
102. Malandrakis P, Kostopoulos IV, Ntanasis-Stathopoulos I, Solia I, Theodorakakou F, Spiliopoulou V, et al. Circulating tumor cells by next generation flow cytometry may be a new prognostic biomarkers among patients with asymptomatic monoclonal gammopathies. Blood. (2024) 144:1020. doi: 10.1182/blood-2024-203751
103. Termini R, Žihala D, Terpos E, Perez-Montaña A, Jelínek T, Raab M, et al. Circulating tumor and immune cells for minimally invasive risk stratification of smoldering multiple myeloma. Clin Cancer Res. (2022) 28:4771–81. doi: 10.1158/1078-0432.CCR-22-1594
104. Bianchi G, Kyle RA, Larson DR, Witzig TE, Kumar S, Dispenzieri A, et al. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia. (2013) 27:680–5. doi: 10.1038/leu.2012.237
105. Fernández de Larrea C, Kyle RA, Durie BG, Ludwig H, Usmani S, Vesole DH, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia. (2013) 27:780–91. doi: 10.1038/leu.2012.336
106. Jin X, Jiang X, Li H, Shen K, Liu S, Chen M, et al. Prognostic implications of circulating plasma cell percentage in multiple myeloma and primary plasma cell leukemia defined by new criteria. Acta Haematol. (2025) 148:48–57. doi: 10.1159/000538658
107. Jelinek T, Bezdekova R, Zihala D, Sevcikova T, Anilkumar Sithara A, Pospisilova L, et al. More than 2% of circulating tumor plasma cells defines plasma cell leukemia-like multiple myeloma. J Clin Oncol. (2023) 41:1383–92. doi: 10.1200/JCO.22.01226
108. Chu B, Wang YT, Gao S, Shi L, Lu MQ, Fang LJ, et al. R2-ISS staging combined with circulating plasma cells improves risk stratification for newly diagnosed multiple myeloma: a single-center real-world study. Ann Hematol. (2024) 103:3677–90. doi: 10.1007/s00277-024-05806-9
109. Ntanasis-Stathopoulos L, Kostopoulos LV, Malandrakis P, Rousakis P, Panteli C, Eleutherakis-Papaiakovou E, et al. A novel prognostic system based on circulating tumor cells for newly diagnosed multiple myeloma. Blood. (2024) 144:490. doi: 10.1182/blood-2024-194090
110. Han W, Jin Y, Xu M, Zhao SS, Shi Q, Qu X, et al. Prognostic value of circulating clonal plasma cells in newly diagnosed multiple myeloma. Hematology. (2021) 26:510–7. doi: 10.1080/16078454.2021.1948208
111. Yao W, Yang H, You H, Shang J, Zhai Y, Yan Z, et al. The prognostic significance of circulating plasma cells in newly diagnosed multiple myeloma patients. Front Oncol. (2023) 13:1266868. doi: 10.3389/fonc.2023.1266868
112. Cheng Q, Cai L, Zhang Y, Chen L, Hu Y, and Sun C. Circulating plasma cells as a biomarker to predict newly diagnosed multiple myeloma prognosis: developing nomogram prognostic models. Front Oncol. (2021) 11:639528. doi: 10.3389/fonc.2021.639528
113. Soloviev MV, Mendeleeva L, Samoilova O, Davydkin I, Kaplanov K, Kaporskaya T, et al. The impact of circulating plasma cells on overall survival (OS) in patients with newly diagnosed multiple myeloma (MM) - results of a multicenter study. Blood. (2024) 144:3332. doi: 10.1182/blood-2024-204591
114. Liang D, Yan Y, Bai S, Xu W, Wang Q, Feng D, et al. Clinical outcome of ≥2% circulating tumor cells in newly diagnosed multiple myeloma: insights from a multicenter study. Ann Med. (2025) 57:2496796. doi: 10.1080/07853890
115. Kostopoulos IV, Ntanasis-Stathopoulos I, Rousakis P, Malandrakis P, Panteli C, Eleutherakis-Papaiakovou E, et al. Low circulating tumor cell levels correlate with favorable outcomes and distinct biological features in multiple myeloma. Am J Hematol. (2024) 99:1887–96. doi: 10.1002/ajh.27414
116. Bezdekova R, Jelinek T, Kralova R, Stork M, Polackova P, Vsianska P, et al. Necessity of flow cytometry assessment of circulating plasma cells and its connection with clinical characteristics of primary and secondary plasma cell leukaemia. Br J Haematol. (2021) 195:95–107. doi: 10.1111/bjh.17713
117. Tembhare PR, Sriram H, Khanka T, Gawai S, Ghogale S, Deshpande N, et al. Circulating clonal plasma cells at diagnosis and peripheral blood measurable residual disease assessment provide powerful prognostication biomarkers in newly-diagnosed multiple myeloma patients treated without autologous transplant. Blood. (2022) 140:1135–6. doi: 10.1182/blood-2022-165715
118. Dingli D, Nowakowski GS, Dispenzieri A, Lacy MQ, Hayman SR, Rajkumar SV, et al. Flow cytometric detection of circulating myeloma cells before transplantation in patients with multiple myeloma: a simple risk stratification system. Blood. (2006) 107:3384–8. doi: 10.1182/blood-2005-08-3398
119. Chakraborty R, Muchtar E, Kumar SK, Jevremovic D, Buadi FK, Dingli D, et al. Risk stratification in myeloma by detection of circulating plasma cells prior to autologous stem cell transplantation in the novel agent era. Blood Cancer J. (2016) 6:e512. doi: 10.1038/bcj.2016.117
120. Chakraborty R, Muchtar E, Kumar SK, Jevremovic D, Buadi FK, Dingli D, et al. Serial measurements of circulating plasma cells before and after induction therapy have an independent prognostic impact in patients with multiple myeloma undergoing upfront autologous transplantation. Haematologica. (2017) 102:1439–45. doi: 10.3324/haematol.2017.166629
121. Tanenbaum B, Miett T, and Patel SA. The emerging therapeutic landscape of relapsed/refractory multiple myeloma. Ann Hematol. (2023) 102:1–11. doi: 10.1007/s00277-022-05058-5
122. Chen Q, Zhang M, Zheng S, Tong Y, and Tan Y. Therapeutic progress in relapsed/refractory multiple myeloma. Ann Hematol. (2024) 103:1833–41. doi: 10.1007/s00277-024-05730-y
123. Boussi LS, Avigan ZM, and Rosenblatt J. Immunotherapy for the treatment of multiple myeloma. Front Immunol. (2022) 13:1027385. doi: 10.3389/fimmu.2022.1027385
124. Zhao JH, Xu QL, Ma S, Li CY, Zhang HC, Zhao LJ, et al. Recent advance of small-molecule drugs for clinical treatment of multiple myeloma. Eur J Med Chem. (2023) 257:115492. doi: 10.1016/j.ejmech.2023.115492
125. Bertamini L, Fokkema C, Rodríguez-Otero P, van Duin M, van de Donk NWCJ, D’Agostino M, et al. Circulating tumor cells as a biomarker to identify high-risk transplant eligible myeloma patients treated with bortezomib, lenalidomide and dexamethasone with or without daratumumab during induction/consolidation, and lenalidomide with or without daratumumab during maintenance: results from the Perseus Study. Blood. (2024) 144:487–9. doi: 10.1182/blood-2024-199550
126. Xiang Y, Zhang L, Xiang P, and Zhang J. Circulating miRNAs as auxiliary diagnostic biomarkers for multiple myeloma: a systematic review, meta-analysis, and recommendations. Front Oncol. (2021) 11:698197. doi: 10.3389/fonc.2021.698197
127. Ferreira B, Caetano J, Barahona F, Lopes R, Carneiro E, Costa-Silva B, et al. Liquid biopsies for multiple myeloma in a time of precision medicine. J Mol Med (Berl). (2020) 98:513–25. doi: 10.1007/s00109-020-01897-9
128. Marivel AJ, Ma Y, Becker TM, Verma A, Trieu S, Roberts TL, et al. The use of cell free DNA (cfDNA) for mutational screening of multiple myeloma. Leuk Res Rep. (2023) 20:100393. doi: 10.1016/j.lrr.2023.100393
129. Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun. (2017) 8:268. doi: 10.1038/s41467-017-00296-y
130. Lohr JG, Kim S, Gould J, Knoechel B, Drier Y, Cotton MJ, et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci Transl Med. (2016) 8:363ra147. doi: 10.1126/scitranslmed.aac7037
131. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Multiple Myeloma. Available online at: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1445 (Accessed May 10, 2025). Version 1. 2025.
132. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. (2014) 15:e538–48. doi: 10.1016/S1470-2045(14)70442-5
133. Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. (2022) 97:1086–107. doi: 10.1002/ajh.26590
134. Rajkumar SV. Multiple myeloma: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. (2024) 99:1802–24. doi: 10.1002/ajh.27422
135. Musto P, Engelhardt M, Caers J, Bolli N, Kaiser M, Van de Donk N, et al. 2021 European Myeloma Network review and consensus statement on smoldering multiple myeloma: how to distinguish (and manage) Dr. Jekyll and Mr. Hyde. Haematologica. (2021) 106:2799–812. doi: 10.3324/haematol.2021.278519
136. Hiepe F, Dörner T, Hauser AE, Hoyer BF, Mei H, and Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. (2011) 7:170–8. doi: 10.1038/nrrheum.2011.1
137. Dang VD, Stefanski AL, Lino AC, and Dörner T. B- and plasma cell subsets in autoimmune diseases: translational perspectives. J Invest Dermatol. (2022) 142:811–22. doi: 10.1016/j.jid.2021.05.038
138. Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. (2004) 199:1577–84. doi: 10.1084/jem.20040168
139. Streicher K, Morehouse CA, Groves CJ, Rajan B, Pilataxi F, Lehmann KP, et al. The plasma cell signature in autoimmune disease. Arthritis Rheumatol. (2014) 66:173–84. doi: 10.1002/art.38194
140. Parodis I, Gatto M, and Sjöwall C. B cells in systemic lupus erythematosus: targets of new therapies and surveillance tools. Front Med (Lausanne). (2022) 9:952304. doi: 10.3389/fmed.2022.952304
141. Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, et al. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis. (2010) 69:305–8. doi: 10.1136/ard.2008.096495
142. Vital EM, Dass S, Buch MH, Henshaw K, Pease CT, Martin MF, et al. B cell biomarkers of rituximab responses in systemic lupus erythematosus. Arthritis Rheumatol. (2011) 63:3038–47. doi: 10.1002/art.30466
143. Md Yusof MY, Shaw D, El-Sherbiny YM, Dunn E, Rawstron AC, Emery P, et al. Predicting and managing primary and secondary non-response to rituximab using B-cell biomarkers in systemic lupus erythematosus. Ann Rheum Dis. (2017) 76:1829–36. doi: 10.1136/annrheumdis-2017-211191
144. Yang DH, Chang DM, Lai JH, Lin FH, and Chen CH. Significantly higher percentage of circulating CD27(high) plasma cells in systemic lupus erythematosus patients with infection than with disease flare-up. Yonsei Med J. (2010) 51:924–31. doi: 10.3349/ymj.2010.51.6.924
145. Parodis I, Gomez A, Lindblom J, Chow JW, Doria A, and Gatto M. Early changes in B and plasma cell subsets and traditional serological markers as predictors of SRI-4 response to therapy in systemic lupus erythematosus. Front Med (Lausanne). (2022) 9:852162. doi: 10.3389/fmed.2022.852162
146. Parodis I, Gomez A, Chow JW, Borg A, Lindblom J, and Gatto M. Early B cell and plasma cell kinetics upon treatment initiation portend flares in systemic lupus erythematosus: a post-hoc analysis of three phase III clinical trials of belimumab. Front Immunol. (2022) 13:796508. doi: 10.3389/fimmu.2022.796508
Keywords: plasma cells, circulating plasma cells, plasma cell disorders, multiple myeloma, flow cytometry
Citation: Wang P, Su N, Yan X, Xu F and Zhang Y (2025) Circulating plasma cells: from basic mechanisms to clinical applications. Front. Immunol. 16:1674088. doi: 10.3389/fimmu.2025.1674088
Received: 27 July 2025; Accepted: 16 October 2025;
Published: 30 October 2025.
Edited by:
Doan C. Nguyen, Emory University, United StatesReviewed by:
Ioannis V. Kostopoulos, National and Kapodistrian University of Athens, GreeceGodhev Kumar Manakkat Vijay, University of Pittsburgh, United States
Copyright © 2025 Wang, Su, Yan, Xu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zhang, MzYwMDc2NzMyQHFxLmNvbQ==; Feng Xu, eWV6aXh1ZmVuZ0AxNjMuY29t; Xiaojing Yan, eWFueGlhb2ppbmdfcHBAaG90bWFpbC5jb20=
†These authors have contributed equally to this work