 Kameron B. Rodrigues
Kameron B. Rodrigues Peter J. Eggenhuizen
Peter J. Eggenhuizen Rosa Bacchetta
Rosa Bacchetta Zinaida Good
Zinaida Good- 1Division of Immunology and Rheumatology, Department of Medicine, Stanford University, Stanford, CA, United States
- 2Center for Biomedical Informatics Research, Department of Medicine, Stanford University, Stanford, CA, United States
- 3Center for Inflammatory Diseases, Department of Medicine, School of Clinical Sciences, Monash University, Clayton, VIC, Australia
- 4Division of Hematology, Oncology, Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford University, Stanford, CA, United States
- 5Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, Stanford, CA, United States
- 6Center for Definitive and Curative Medicine, Stanford University, Stanford, CA, United States
- 7Parker Institute for Cancer Immunotherapy, Stanford University, Stanford, CA, United States
- 8Weill Cancer Hub West, Stanford University, Stanford, CA, United States
Regulatory T cell (Treg) therapies are emerging as powerful tools for treating autoimmune and inflammatory diseases, preventing graft-versus-host disease (GvHD), and promoting organ transplant tolerance. Building on the identification of chimeric antigen receptor (CAR)-expressing Tregs as a correlate of poor patient outcomes in CD19-CAR T cell therapy, this review examines strategies for learning from clinical samples and data to improve Treg therapies. We highlight current and next-generation Treg modalities, including polyclonal, antigen-specific, converted, TCR-engineered, and CAR-engineered Tregs, provide a comprehensive overview of Treg clinical trials, and evaluate the evolving toolkit for in vivo Treg monitoring. Emphasis is placed on advanced immunomonitoring technologies, such as single-cell multi-omic profiling, epigenetic analysis, and spatial transcriptomics, which enable precise characterization of Treg persistence, function, and lineage stability. By integrating insights from adoptive T cell therapies and cutting-edge multi-omic platforms, this review outlines how Treg therapies can be optimized as “living drugs” capable of establishing immune tolerance across diverse clinical contexts.
1 Introduction
Regulatory T cells (Tregs) represent a specialized subset of CD4+ T lymphocytes crucial for maintaining immune homeostasis and preventing autoimmunity. Originally characterized by their high expression of CD25 (the IL-2 receptor α-chain) and the transcription factor FOXP3 (1–4), Tregs play an essential role in dampening excessive immune responses and promoting tolerance to self-antigens (5). Although detrimental in cancer, immunosuppressive functions have positioned Tregs as attractive candidates for cell-based therapies aimed at controlling unwanted immune reactions in autoimmune and inflammatory diseases, graft-versus-host disease (GvHD), and solid organ transplantation (6). Recent clinical observations from adoptive T cell therapy trials have underscored the potential for uncovering correlates of therapeutic outcomes and understanding the mechanism of failure in the context of cancer T cell therapies. Notably, the identification of chimeric antigen receptor (CAR)-expressing Tregs as negative correlates of patient outcomes in CD19-CAR T cell therapy for large B-cell lymphoma has provided insights on how engineered Tregs can be monitored in clinical settings and provided evidence for function of engineered Tregs in humans (7, 8). In this review, we examine the evolving landscape of clinical trials for Treg cell therapies, from non-engineered polyclonal Tregs to antigen-specific, T cell receptor (TCR)-engineered, and CAR-engineered Tregs. We discuss cutting-edge technologies for tracking and characterizing Tregs in patients and highlight operational considerations for maximizing insights from clinical trials. By drawing lessons from other adoptive transfer approaches, we aim to provide a framework for optimizing Treg therapies and expanding their clinical applications.
2 Treg cell therapies in the clinic
2.1 Polyclonal Tregs
The earliest clinical applications of Treg therapy employed non-engineered, polyclonal Tregs isolated from peripheral blood (9) (Figure 1). These approaches typically involved isolation of CD4+CD127low T cells through fluorescence-activated cell sorting (FACS) and/or magnetic bead-based methods (often CliniMACS Plus System, Miltenyi Biotec, for CD25+ selection), followed by cryopreservation or direct administration (10–12). The CliniMACS bead enrichment approach, while practical, often results in around 80% Tregs mixed with other cell types (13–16) (NCT02371434, NCT02385019). Given how rare Tregs are in peripheral blood — comprising only 5-10% of CD4+ T cells (17, 18) (2-8% in our hands) — the field sought for additional clinical sources of Tregs with goals to improve purity. Ex vivo expansion methods were developed and implemented in clinical trials (19), specifically with anti-CD3/CD28 stimulation in the presence of high-dose interleukin-2 (IL-2) alone (20, 21), or with rapamycin — an inhibitor of mammalian target of rapamycin (mTOR) — resulting in improved Treg purity of about 90% (14, 22). Other sources of Tregs are also implemented in clinical trials, such as cryopreserved umbilical cord blood (NCT05027815, NCT05349591) or discarded thymus tissue from pediatric heart transplants (NCT04924491, NCT06052436). Polyclonal Tregs have shown promising safety profiles in allogeneic settings such as GvHD prevention and solid organ transplantation (22, 23), and also in autologous settings such as type 1 diabetes (T1D) (24). One important example of polyclonal non-engineered Treg therapy is Orca-T, where allogeneic (graft-matched) Tregs were freshly administered at a 1:1 ratio with T conventional cells (Tconv) along with CD34+ hematopoietic stem cells to prevent GvHD (10), leading to positive phase 2 trial results (25, 26).
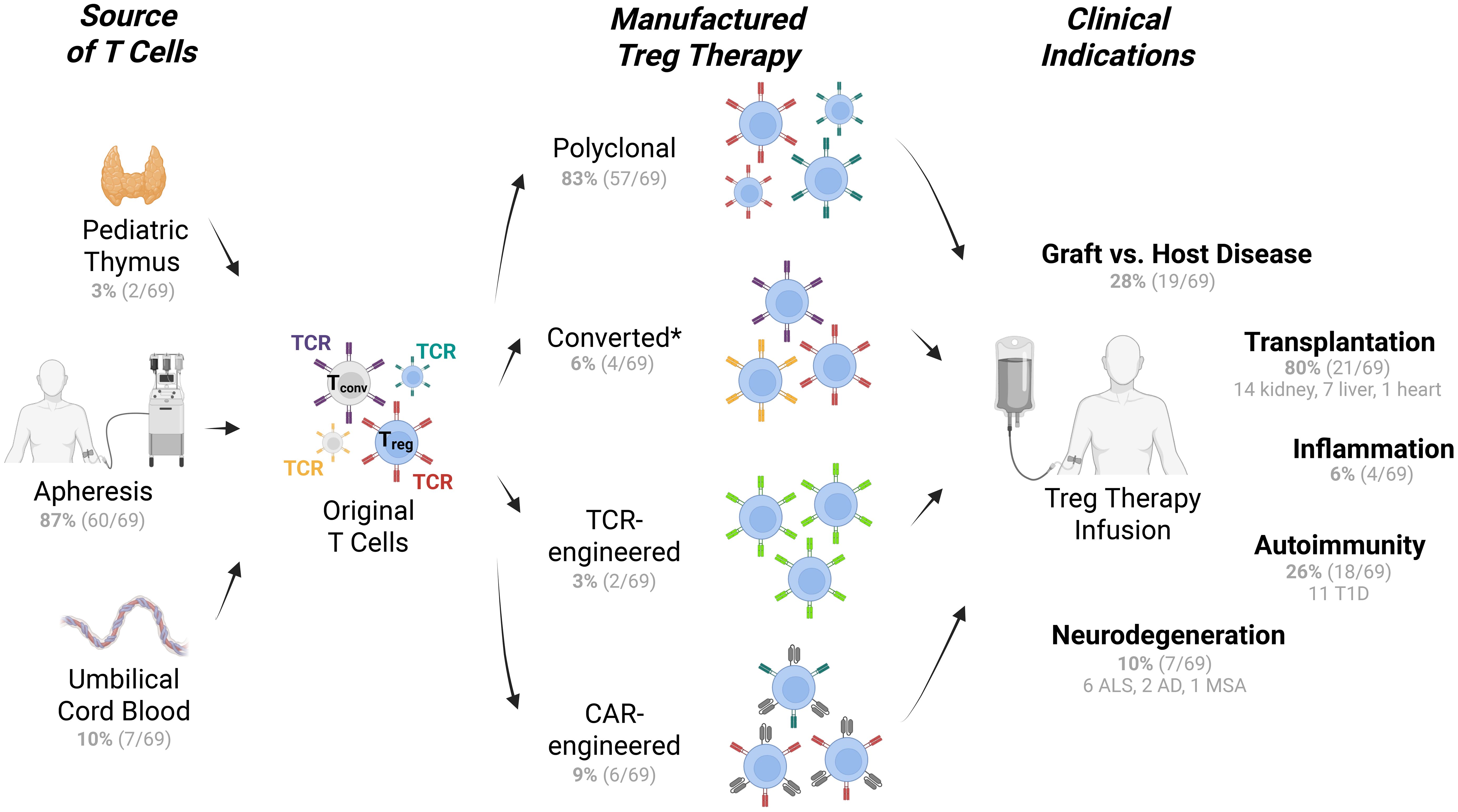
Figure 1. Types of Treg therapies, T cell sources, and indications in clinical trials. Treg therapies from multiple sources of T cells (left) are manufactured via multiple approaches (middle) for multiple applications under evaluation in clinical trials (right). Tconv cells are represented with grey cytoplasm and nucleus, while Tregs are represented in blue. Colored receptors denote TCR or CAR where relevant. The number of clinical trials are listed in parentheses. See Treg therapy clinical trial details in Supplementary Table S1. *Converted Treg products may originate from CD4+ T conventional cells. Trials that were withdrawn, terminated, or suspended were excluded. T1D, type 1 diabetes; ALS, amyotrophic lateral sclerosis; AD, Alzheimer's disease; MSA, multiple system atrophy (108).
Antigen-specific Tregs can be enriched from purified polyclonal Tregs through ex vivo expansion. In the context of allogeneic setting, host Tregs can be isolated then exposed to donor cells ex vivo to expand donor-alloantigen reactive Tregs. This approach has been used in clinical trials to prevent transplantation rejection for either kidney (NCT02091232, NCT02244801) (15, 16) or liver (NCT02188719, NCT02474199, NCT03577431, NCT03654040). Enriched antigen-specific Tregs have also been applied in autologous settings. In a trial for Alzheimer’s disease (AD), Tregs were expanded ex vivo in the presence of amyloid beta antigen to enrich for amyloid beta reactive Tregs (27). An interesting extension of this approach is ‘CRANE’ technology from Cellenkos that expands Tregs from cord blood while enriching for a specific population that has high levels of specific homing receptors. For example, Tregs expressing integrin protein CD11a, in the case of CK0803 for trial NCT05695521, have been used to target the CXCR3/CXCL10 axis with the aim of engaging the inflamed microglia in patients with amyotrophic lateral sclerosis (ALS). Additionally, CD49d is targeted in CK0802 for trial NCT04468971 (28).
Although approaches for ex vivo polyclonal Treg expansion faced several limitations, the early clinical experiences with polyclonal Tregs provided valuable insights into dosing, safety, and monitoring strategies. Polyclonal Treg trials have advanced into late-stage trials despite the limitations of restricted antigen specificity, possible in vivo instability, variable purity, and limited persistence. In summary, the trials for polyclonal Tregs have paved the way for next-generation Treg therapies by expanding Treg sources, improving cell isolation and expansion technologies, establishing the importance of antigen specificity, and providing initial evidence of clinical benefit in both autologous and allogeneic settings.
2.2 Converted Tregs
By 2025, approaches of polyclonal T cell products included the reprogramming of conventional T cells (Tconv) to acquire regulatory function, generating induced Treg (iTreg) or converted Treg cells (Figure 1). Generally, iTregs are produced in clinical trials by culturing CD4+CD25– T cells with IL-2, rapamycin, transforming growth factor β (TGF-β), and anti-CD3 monoclonal antibody-loaded artificial antigen-presenting cells to generate FOXP3+ iTregs with potent suppressive function (NCT01634217 for GvHD) (29). A similar protocol leveraging rapamycin has been used to reprogram Tconv cells in the context of ALS and COVID-19 related acute respiratory distress syndrome. For example, NCT06169176, NCT04220190, and NCT04482699 utilize Rapa-501, a two-step, 7-day culture process. First, T cells are de-differentiated using rapamycin with media starvation, which drives T cells towards a T stem cell memory phenotype. The final step is to re-differentiate the T cells into Treg and Th2 programs with IL-2, IL-4, and TGF-β. As of 2025, NCT04220190 which used this rapamycin reprograming approach was the most progressed clinical trial of the converted Treg class and is in phase 3 for treatment of ALS. Cell engineering approaches for converted Treg products have begun clinical trials, where high FOXP3 expression is induced by lentiviral transduction of CD4+ T cells, together with a surface marker gene tNGFR. This autologous converted Treg-like cell product (CD4LVFOXP3) is administered to patients who genetically lack functional Tregs in a first-in-human trial for conditions including immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome (NCT05241444) (30). In a similar gene-transfer approach, high IL-10 production is induced by lentiviral transduction into CD4+ T cells (CD4LV-IL10) to produce allogenic type 1 regulatory T cells (Tr1 cells) for “off-the shelf” GvHD and inflammatory bowel disease (IBD) treatment in clinical trials led by Tr1X Bio (31, 32). Type 1 Tregs are important for peripheral tolerance, with suppressive functions mediated by IL-10, TGF-β, and CTLA4, independent of FOXP3 (33, 34). These approaches, which still produce polyclonal Treg-like cells, differ from expanded polyclonal Tregs in that they do not enrich for Tregs prior to differentiation/expansion, and rather utilize all CD4+ T cells as the starting material, overcoming the issues of reaching sufficient Treg number and purity. Converted Tregs are particularly valuable in settings where functional Tregs cannot be obtained in sufficient numbers as a starting material (e.g. IPEX), or where inflammation is naturally controlled by iTreg or Tr1 cells (e.g. IBD), with additional settings under investigation.
2.3 TCR-engineered Tregs
T cell receptor (TCR) engineering of Tregs renders them antigen-specific for a particular disease target and is an emerging approach yet to fully transition to the clinic (6). Preclinical models of transplantation tolerance demonstrate that TCR-engineered Tregs exhibit enhanced potency compared to polyclonal Treg populations and can mediate “linked suppression” of responses against other antigens present in the same microenvironment (35–37). To enhance antigen specificity and potentially improve therapeutic efficacy, the field is developing TCR-engineered Tregs. By introducing TCRs specific for relevant disease antigens (e.g., alloantigens in transplantation or self-antigens in autoimmunity), TCR-engineered Tregs are expected to exert targeted immunosuppression at pathogenic sites (Figure 1). In the context of autoimmunity, preclinical models of TCR-engineered Tregs include the following targets: (i) myelin basic protein in multiple sclerosis (38), (ii) Smith autoantigen in lupus nephritis (39), (iii) factor VIII in hemophilia A (40), (iv) type IV collagen in anti-glomerular basement membrane disease (41), and (v) glutamic acid decarboxylase in type 1 diabetes (42). These studies demonstrated the potential of TCR-engineered Tregs in restoring immune tolerance. Key potential advantages of TCR-engineered Tregs, if successful, would include improved localization reachable with lower cell doses, enhanced persistence, superior antigen specificity, and reduced risk of undesired immunosuppression via lowered bystander suppression. However, challenges remain in selecting optimal target antigens, which are still largely unknown for the majority of autoimmune diseases (43), managing potential off-target effects and genotoxicity, and ensuring reliable and safe manufacturing of these more complex cellular products. As of late-2025, only two clinical trials existed for TCR-engineered Tregs. Abata therapeutics engineered autologous Tregs to express a TCR that specifically recognizes immunogenic myelin fragments in the CNS (ABA-101, NCT06566261). In late 2025 a phase 1 clinical trial begun for GENTI-122, a converted Treg product from Gentibio to treat T1D where CD4+ T cells are engineered to express FOXP3, a chemically inducible signaling complex (CISC) that provides IL-2 signaling support in response to rapamycin, and IGRP305-TCR that recognizes the pancreatic islet-specific glucose-6-phosphatase catalytic subunit–related protein (IGRP) peptide (NCT06919354) (44). More work is needed for progressing preclinical results of TCR-engineered Tregs into clinical trials, whereas the first TCR-engineered Treg trials will provide crucial data for future product optimization.
2.4 CAR-engineered Tregs
Chimeric antigen receptor (CAR) technology, which has revolutionized cancer immunotherapy (45), has been adapted to engineer Tregs with enhanced specificity and function (6). CAR Tregs express synthetic receptors that recognize cell surface antigens independent of MHC presentation, combining the specificity of an antibody with intracellular signaling and leading to Treg activation and regulatory function. Initial preclinical studies demonstrated that CAR Tregs targeting HLA-A2, factor VIII, or myelin oligodendrocyte glycoprotein (MOG) could suppress alloimmunity, autoimmunity against factor VIII in hemophilia, or experimental autoimmune encephalomyelitis, respectively (46–48). The first clinical trials of CAR Tregs are now underway, including a phase 1/2a trial of HLA-A2-specific CAR Tregs for prevention of kidney transplant rejection (Sangamo Therapeutics, NCT04817774) (49, 50) or liver transplant rejection (Quell Therapeutics, NCT05234190), citrullinated vimentin-specific CAR Tregs for rheumatoid arthritis (Sonoma Therapeutics, NCT06201416) (51) and hidradenitis suppurativa (NCT06361836), and CD6-specific CAR Tregs for GvHD (NCT05993611) (Figure 1). Although not yet in clinical trials, Tr1X Bio is developing TRX319, an allogeneic polyclonal CAR Treg therapy for the treatment of multiple B cell mediated autoimmune diseases. The finding that CD19-CAR Tregs correlate with poor outcomes in cancer immunotherapy further underscores the potential of CAR Treg therapies for in vivo suppression in clinical settings (7, 8). CAR Treg approaches offer several potential advantages, including MHC-independent recognition, tunable signaling domains, and diverse targeting options. However, the complexity of CAR Treg biology presents unique challenges, such as balancing activation and stability, preventing exhaustion or plasticity due to CAR tonic signaling, and addressing manufacturing considerations.
Leveraging our recently published list of Treg therapy clinical trials (52), we have retrieved data for each interventional trial and presented results in Supplementary Table S1, as summarized in Figure 1. Polyclonal Treg approaches are the most established, representing 83% of trials as of late 2025. Converted Tregs are emerging (representing 6% of all Treg trials). Among engineered Treg cell therapies, CAR Treg approaches are more mature than TCR-engineered Tregs (representing 9% vs. 3% of all Treg trials, respectively). It remains to be determined whether other forms of engineered or “modified” Tregs can be exploited in the clinic, for example to leverage Treg metabolism or enhance inflammatory activity in cancer settings (53–56).
3 Technologies for Treg immunomonitoring in clinical trials
Comprehensive monitoring of Treg therapies requires sophisticated technologies that can track cell persistence, phenotype, stability, immune rejection, tissue distribution, and function over time. In this process, it is also critical to monitor disease state, potential for infectious tolerance, and the overall immune state in the relevant tissues and blood. Several complementary approaches have emerged as essential tools for understanding Treg behavior in vivo (Figure 2).
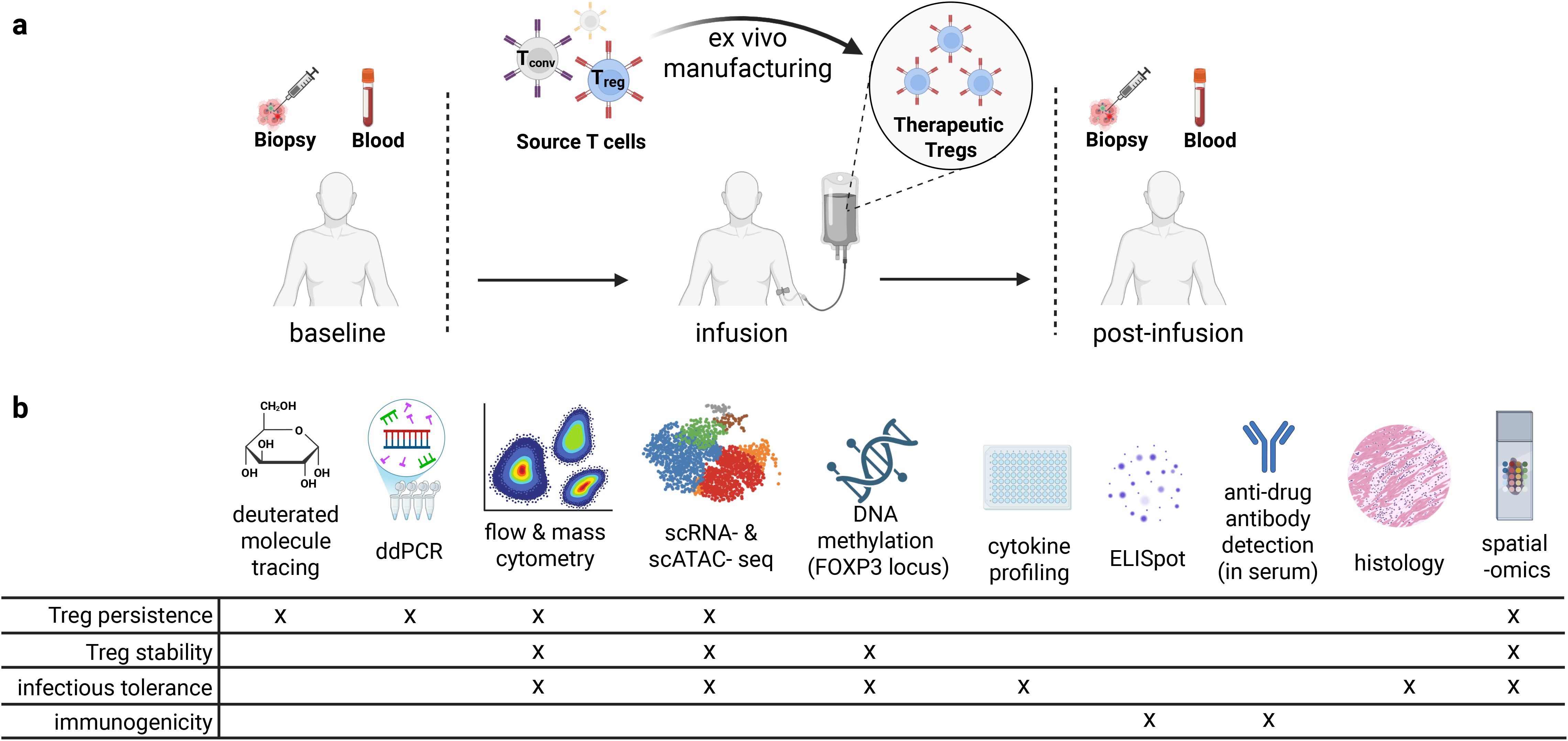
Figure 2. Experimental approaches for immune monitoring of Treg cell therapies in clinical trials. (a) Timepoints and sample types that can be valuable for correlative studies in clinical trials. (b) Assays and specific insights into properties of Treg cell therapies that can be obtained from relevant clinical samples (109).
3.1 Treg tracking methods
Historically, Jeffrey Bluestone’s group pioneered the first approach to tracking Tregs in vivo. The technique involves labeling Tregs with deuterium (²H) prior to infusion to enable long-term tracking of cell persistence and proliferation. By culturing Tregs in deuterated glucose (57) or media containing deuterated water during ex vivo expansion, Tregs incorporate the stable isotope into newly synthesized DNA and proteins (44, 58). After transfer, deuterium-labeled Tregs can be detected in blood and tissue samples through mass spectrometry, allowing assessment of persistence, proliferation, and tissue distribution. A phase I trial of deuterium-labelled polyclonal Tregs for type 1 diabetes showed safety and Treg detection for up to one year post-transfer (22). This technique provides unique insights into Treg kinetics in vivo without genetic manipulation, offering advantages for clinical studies where genetic tracking methods are not feasible.
Genetic tracking of engineered Tregs in clinical trials can be achieved by encoding an inert and non-immunogenic human cell surface transgenic protein that is detectable by flow cytometry (59) and immunohistochemistry. One example is truncated epidermal growth factor receptor (EGFRt), which can be detected by the antibody cetuximab that is available with good manufacturing practices (GMP) certification (60). EGFRt is utilized in CD19-CAR T cell trials (NCT05625594). It is also often used in pre-clinical testing of CAR Tregs (61). In addition to being an engineered Treg tracking tool, EGFRt can enable enrichment of engineered cells pre-transfer, as well as function as a ‘kill switch’ in case of toxicity or malignancy through cetuximab-mediated in vivo elimination. Another example of cell surface transgenic protein for tracking Tregs is truncated nerve growth factor receptor (tNGFR), also known as LNGFR or CD271, and utilized for GMP-compatible pre-transfer enrichment, post-transfer tracking, and quantification of engineered Tregs (30). tNGFR-transduced cells were shown to be safe (62) and were generated in the phase I clinical trial of CD4LVFOXP3 cells for IPEX (NCT05241444; see above). Alternatively, antibodies can be used to detect a functional engineered Treg protein, such as CAR or TCR (7). For example, CAR idiotype antibodies are specific to the scFv binding pocket of the CAR construct (59). Antibody-mediated detection of genetic Treg markers can be combined with single-cell technologies through CITE-seq (63) or spatial technologies, such as immunohistochemistry (IHC) (59) or CODEX (64), although CAR idiotype antibodies often have excessive background signal in spatial applications. DNA or RNA transcripts encoding engineered proteins remain detectable and can be traced to identify infused Tregs using quantitative real-time PCR, highly sensitive digital droplet PCR (ddPCR) (65, 66), single-cell sequencing, or spatial transcriptomics. Overall, engineered cell tracking technologies provide data on persistence and biodistribution of engineered Tregs in humans.
3.2 Assessing Treg phenotype, stability, and function
3.2.1 Flow cytometry and mass cytometry
Flow cytometry remains the cornerstone of Treg identification and characterization in clinical samples. Conventional panels typically include markers such as CD4, CD25, CD127, and FOXP3, along with activation markers (e.g. CD39), homing receptors (e.g. CCR4), and functional markers (e.g. Ki-67) (67). Spectral flow cytometry panels enable practical quantification of 30–40 markers. Mass cytometry (CyTOF) extends this capability by enabling practical detection of 40–50 parameters using metal-tagged antibodies, allowing more comprehensive phenotyping with minimal spectral overlap (7, 68–70). This approach is generally applied to batched cryopreserved samples, revealing previously unappreciated heterogeneity within the Treg compartment and distinct Treg subpopulations associated with clinical outcomes (71). Key considerations for flow-based monitoring include thoughtful antibody panel development and validation, standardization of staining procedure (e.g. Using lyophilized, pre-mixed antibody panels formatted as single-bead aliquots), and including batch controls that express all markers up to maximum level in the test samples. These methods can provide critical information on Treg persistence and stability, functional and homing marker assessment, trafficking patterns, and the overall state of the immune system.
3.2.2 Single-cell sequencing
Single-cell RNA-sequencing (scRNA-seq) has enabled deep characterization of the in vivo cellular heterogeneity and proven crucial for tracking Tregs during reconstitution post-stem cell transplantation (72). The comprehensive transcriptional profiles of individual cells provided by scRNA-seq reveals functional states, activation status, and potential loss of phenotypic and functional stability that may be missed by protein-based methods. In the context of Treg therapies, scRNA-seq is useful in identifying transcriptional signatures and pathways associated with therapeutic efficacy or toxicity, tracking clonal dynamics of transferred Tregs through integration with single-cell TCR sequencing (scTCR-seq) (73), detecting lineage instability through expression of non-Treg lineage genes, incorporating expression of key proteins through CITE-seq (63), and mapping interactions between Tregs and other immune or tissue cells through interactome analyses (74, 75).
Assay for transposase-accessible chromatin using sequencing (ATAC-seq) provides insights into the epigenetic landscape of cells by quantifying regions of open chromatin (76). When applied to Tregs, this technique reveals regulatory elements controlling Treg identity and function, including those associated with FOXP3 expression and stability (77, 78). Single-cell ATAC-seq (scATAC-seq) can identify epigenetic changes occurring in Treg subpopulations during therapy, potentially predicting functional alterations before they become apparent at the transcriptional or protein level (79, 80). Further, scATAC-seq can assess the extent that engineered Tregs recapitulate natural Tregs epigenetically (including at the FOXP3 locus), providing information on cell stability, enhancer activity, and the extent that Tregs are ‘primed’ for future cell states. Integration of scATAC-seq with scRNA-seq data through multi-omic approaches (81–84) enables trajectory inferencing (85, 86), while providing a more complete picture of the Treg cellular states and kinetics.
The main limitations of single-cell sequencing technologies for Treg clinical trials are the cost and limited cell numbers. Thus, correlative studies often leverage fluorescence-activated cell sorting (FACS) to enrich a population of interest – such as infused Tregs from blood – prior to scRNA-seq or scATAC-seq. Costs can be further reduced through selecting the most informative samples, barcoding and pooling samples in batched analyses, and leveraging rapidly evolving technologies (e.g. 10x Genomics GEM-X, BD Rhapsody, Parse Evecode, Illumina Single Cell) and kits for single-cell sequencing (e.g. 48-sample kit is more cost effective than a 16-sample kit).
3.2.3 Spatial omics
Spatial omics technologies build on the original spatial analysis methods, including hematoxylin and eosin (H&E) stain, IHC, and immunofluorescence. Single-cell spatial transcriptomic tools, including Nanostring CosMx, 10x Genomics Xenium, and Vizgen MERSCOPE, utilize probes to detect a preset panel of up to 6,000 genes and support custom probes for engineered proteins, such as CAR or TCR. Spatial proteomics technologies, including MIBI (87) and CODEX (64, 88), can be used instead of or in parallel with spatial transcriptomics methods to learn insights from the relevant tissue biopsies. Already applied in studies on Treg therapy for kidney transplantation (89–92), spatial omics methods could be essential to comprehensively profile the immune state within the relevant tissue biopsies in Treg trials, detect Tregs in tissues and define their phenotype, and examine Treg-rich organized lymphoid structures (TOLS) (93), if present. In addition to assessing Treg persistence, phenotype, and microenvironment, spatial transcriptomics can define spatial cell-cell communication (94, 95). Important advantages of spatial omic technologies are spatial information and more accurate cell proportions when compared to single-cell analyses of dissociated tissues. Limitations of spatial omics include lower precision in cell type separation due to imperfect cell segmentation and spillover effects, higher background in spatial proteomics compared to flow cytometry, and higher dropout in spatial transcriptomics compared to single-cell sequencing. Constructing tissue microarrays (TMAs) from serial tissue biopsies can reduce costs of spatial omics analyses.
3.2.4 TSDR demethylation
The biological instability of Tregs represents a concern for Treg cell therapies, as infused cells could lose their identity when exposed to inflammatory environments in vivo. This instability manifests as FOXP3 downregulation, phenotypic conversion, proinflammatory cytokine production, and unpredictable therapeutic performance (67). While single-cell technologies can assess Treg phenotype for inference of stability, DNA methylation information is considered gold standard. Treg identity and stability are closely linked to demethylation of specific regulatory regions of the FOXP3 locus, particularly the Treg-specific demethylated region (TSDR) (96–99). Quantitative analysis of TSDR demethylation serves as a reliable measure of bona fide Tregs and can be used to track the stability of transferred Treg cell products over time.
3.2.5 Treg functional assays
Assessing function of therapeutic Tregs in clinical samples remains an active area of methodological development. In addition to antigen-specific suppression, Tregs can induce bystander suppression to antigens that are distinct from their original antigenic specificity (100–103). Infectious tolerance is a phenomenon that could occur in the context of Treg therapies where Tregs induce tolerance in Tconv and other immune cells, effectively ‘spreading’ their regulatory function beyond their direct or bystander suppressive effects and potentially lasting even if the therapeutic Tregs wane (104, 105). Although in vitro suppression or antigen-specific suppression assays can provide evidence of Treg function in blood samples, the quality of clinical samples collected, stored, and transported over years may not always be sufficient for functional assays. Pathway activity or proliferation markers of therapeutic Tregs based on flow cytometry or scRNA-seq analysis can provide evidence of function (7). Further, measuring changes in regulatory plasma cytokines (e.g. IL-10, TGF-β) versus inflammatory cytokines can provide evidence of function and indirect evidence of infectious tolerance. Infectious tolerance and bystander suppression can be assessed using flow cytometry, single-cell sequencing, TSDR demethylation, and spatial omics analysis of tolerogenic features and reduction in inflammatory features among non-therapeutic cells, respectively (e.g. increase in FOXP3+Helios– T cells may indicate de novo Treg induction). Finally, examining relevant tissue histology for a reduction in disease-specific features, tissue structures associated with tolerance (e.g. TOLS), and tolerogenic state of non-therapeutic cells using a combination of H&E, IHC, and spatial omics methods can provide vital information on therapeutic Treg suppression and infectious tolerance in situ. Clinical efficacy is ultimately the most important metric of therapeutic Treg function that is assessed through disease-specific metrics.
3.3 Monitoring Treg rejection
The immune system may recognize foreign antigens in allogeneic Tregs or in genetically modified Tregs, such as scFv in CAR or junction sequences in TCR, leading humoral (antibody-mediated) and cellular (T cell-mediated) rejection of the therapeutic Treg cells (106). Humoral rejection can be measured by ELISA of serum samples to detect anti-drug antibodies targeting the engineered protein. Cellular rejection can be tested in patient-derived peripheral blood mononuclear cells (PBMCs) (e.g. against peptides spanning the engineered protein) via IFN-γ production by ELISpot. Monitoring Treg rejection can provide valuable information when therapeutic Tregs do not persist.
4 Operational considerations and future directions
4.1 Maximizing insights from Treg correlative studies
To maximize biological insights from Treg clinical trials, we recommend a standardized yet flexible approach to sample collection and correlative assays (Figure 2). Longitudinal peripheral blood samples should be collected at baseline (ideally at the time of apheresis, if applicable), immediate (days 1-2), early (days 7-14), mid (weeks 4-8), and late (months 3-6) post-infusion; precise timing would be driven by the biology of disease and Treg therapy. At each timepoint, PBMCs, plasma, and serum should be processed and cryopreserved. Tissue biopsies — if clinically justified — should be collected at baseline and at matched post-infusion timepoints (e.g. 4–8 weeks and 3–6 months). Treg infusion products and pre-manufacture cell products should also be banked. We recommend spectral flow cytometry or mass cytometry analysis of all batched cryopreserved PBMCs, pre-manufacture cells, and Treg infusion products to assess Treg persistence, stability, and function and examine correlates of patient outcomes. Single-cell RNA-seq and paired scTCR-seq should ideally be performed on pre-manufacture cells, Treg infusion products, and FACS-enriched blood CD4+ T cells that are positive for a genetically encoded surface marker, if available (e.g. EGFRt, tNGFR, CAR). When performed at high-quality timepoints, these approaches enable deep profiling of infused Treg cell state and lineage stability. FOXP3 TSDR demethylation assessed by bisulfite sequencing can provide gold standard information on Treg lineage stability and potential infectious tolerance. In tissue, immunohistochemistry on full slides and spatial transcriptomics on TMAs should be used to define Treg localization, phenotype, and tissue microenvironment. For engineered Tregs, ddPCR should assess persistence, whereas tracking non-engineered Tregs is limited to deuterium labeling and may be difficult to implement. Cytokine profiling (e.g. Luminex) of plasma and anti-drug antibody ELISA of serum provide functional and rejection data, respectively. When rejection is suspected, ELISpot for T cell responses against engineered domains is recommended. These harmonized protocols should be combined with monitoring relevant disease biomarkers to enhance biological insights across Treg therapy trials.
4.2 Future landscape of Treg cell therapy
The field of Treg cell therapy stands at an inflection point, with fundamental insights from preclinical studies and lessons from early clinical experiences converging to guide next-generation approaches (6). Future Treg cell therapies will likely be shaped by several emerging trends: engineered antigen specificity, allogeneic approaches for off-the-shelf availability, induced/converted Tregs to overcome natural Treg limitations, and controlled expansion in vivo to enhance persistence of therapeutically relevant cells. By learning from both successes and challenges of adoptive T cell therapies (107), the field can accelerate the development of Treg-based approaches that harness their full potential as ‘living drugs’ using cutting-edge technologies for engineering and monitoring Tregs, coupled with thoughtful trial design and data analysis strategies. With at least 69 Treg clinical trials across autoimmune and inflammatory diseases and transplantation as of 2025 (Supplementary Table S1), Treg cell therapies have demonstrated their potential for precise immune regulation that could transform treatment paradigms.
Author contributions
KR: Data curation, Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. PE: Writing – original draft, Writing – review & editing. RB: Data curation, Writing – review & editing. ZG: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. ZG was supported by a Parker Bridge Award (C-02895) and a Kona Innovation Challenge Award (C-04134) from the Parker Institute for Cancer Immunotherapy, an NIH/NCI Pathway to Independence Award (1K99CA293149-01, 4R00CA293149-02), an NIH Office of the Director Award (1OT2OD038101), and Weill Cancer Hub West. ZG is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program. RB was supported as an Anne T. and Robert M. Bass Faculty Scholar and through the Maternal and Child Research Institute (MCHRI) and the Department of Pediatrics at Stanford University. Major funding to RB included a California Institute for Regenerative Medicine Award (CLIN2 13259) and an FDA Award (R01FD007540).
Acknowledgments
We thank all members of the Stanford Division on Immunology and Rheumatology and the Stanford Cellular Immune Tolerance Program for their insights into advances in Treg cell therapies. We also thank all members of the Stanford Center for Cancer Cell Therapy for guidance on innovations in correlative studies for T cell therapies. Figures were created in BioRender (108, 109).
Conflict of interest
ZG is an inventor on a patent in the T cell immunotherapy space; holds equity and advises Boom Capital Ventures on T cell therapies; received reagents, technical support, and/or speaker fees from 10x Genomics, Standard Biotools, AstraZeneca, and Sangamo Therapeutics; is a part of a sponsored research agreement with Kite Pharma, a subsidiary of Gilead Sciences; and has a non-sponsored research agreement with Sangamo Therapeutics that is related to HLA-A2 CAR Treg therapy. None of the above interests were directly related to nor aided the writing of this manuscript. RB and PE hold multiple patents in the Treg cell therapy field.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1675114/full#supplementary-material
Supplementary Table 1 | Treg therapy clinical trials and associated details. Only interventional trials included.
References
1. Hori S, Nomura T, and Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
2. Khattri R, Cox T, Yasayko S-A, and Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. (2003) 4:337–42. doi: 10.1038/ni909
3. Fontenot JD, Gavin MA, and Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
4. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
5. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, and Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
6. Wardell CM, Boardman DA, and Levings MK. Harnessing the biology of regulatory T cells to treat disease. Nat Rev Drug Discov. (2025) 24:93–111. doi: 10.1038/s41573-024-01089-x
7. Good Z, Spiegel JY, Sahaf B, Malipatlolla MB, Ehlinger ZJ, Kurra S, et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat Med. (2022) 28:1860–71. doi: 10.1038/s41591-022-01960-7
8. Haradhvala NJ, Leick MB, Maurer K, Gohil SH, Larson RC, Yao N, et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat Med. (2022) 28:1848–59. doi: 10.1038/s41591-022-01959-0
9. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. (2011) 117:3921–8. doi: 10.1182/blood-2010-10-311894
10. Meyer EH, Laport G, Xie BJ, MacDonald K, Heydari K, Sahaf B, et al. Transplantation of donor grafts with defined ratio of conventional and regulatory T cells in HLA-matched recipients. JCI Insight. (2019) 4(10):e127244. doi: 10.1172/jci.insight.127244
11. Gladstone DE, Howard C, Lyu M-A, Mock J, Adams D, Gibbs K, et al. Randomized, multi-center, double-blinded, placebo controlled safety and early efficacy trial of cryopreserved cord blood derived T-regulatory cell infusions (CK0802) in the treatment of COVID-19 induced ARDS. (RESOLVE Trial) Blood. (2021) 138:828. doi: 10.1182/blood-2021-153616
12. Bader CS, Pavlova A, Lowsky R, Muffly LS, Shiraz P, Arai S, et al. Single-center randomized trial of T-reg graft alone vs T-reg graft plus tacrolimus for the prevention of acute GVHD. Blood Adv. (2024) 8(5):1105–15. doi: 10.1182/bloodadvances.2023011625
13. Sánchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant. (2020) 20:1125. doi: 10.1111/ajt.15700
14. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget. (2016) 7:7563–77. doi: 10.18632/oncotarget.6927
15. Roemhild A, Otto NM, Moll G, Abou-El-Enein M, Kaiser D, Bold G, et al. Regulatory T cells for minimising immune suppression in kidney transplantation: phase I/IIa clinical trial. BMJ. (2020) 371:m3734. doi: 10.1136/bmj.m3734
16. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. (2020) 395:1627–39. doi: 10.1016/S0140-6736(20)30167-7
17. Seddiki N, Santner-Nanan B, Tangye SG, Alexander SI, Solomon M, Lee S, et al. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood. (2006) 107:2830–8. doi: 10.1182/blood-2005-06-2403
18. Kleinewietfeld M, Starke M, Di Mitri D, Borsellino G, Battistini L, Rötzschke O, et al. CD49d provides access to “untouched” human Foxp3+ Treg free of contaminating effector cells. Blood. (2009) 113:827–36. doi: 10.1182/blood-2008-04-150524
19. Mathew JM, H.-Voss J, LeFever A, Konieczna I, Stratton C, He J, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. (2018) 8:1–12. doi: 10.1038/s41598-018-25574-7
20. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
21. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
22. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Trans Med. (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
23. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatol (Baltimore Md). (2016) 64(2):632–43. doi: 10.1002/hep.28459
24. Rosenzwajg M, Churlaud G, Mallone R, Six A, Dérian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun. (2015) 58:48–58. doi: 10.1016/j.jaut.2015.01.001
25. Muffly L, Ananth S, Danley L, Wagner C, Kordek D, DeNoble K, et al. Safe and effective combination of donor-derived, allogeneic CD19/CD22-CAR T cells with myeloablative graft-engineered allo-HCT for high-risk B-ALL. Blood. (2024) 144:679. doi: 10.1182/blood-2024-211959
26. Meyer EH, Pavlova A, Villar-Prados A, Bader C, Xie B, Muffly L, et al. Donor regulatory T-cell therapy to prevent graft-versus-host disease. Blood. (2025) 145:2012–24. doi: 10.1182/blood.2024026446
27. Yang H, Byun MS, Ha NY, Yang J, Park SY, Park JE, et al. A preclinical and phase I clinical study of ex vivo-expanded amyloid beta-specific human regulatory T cells in Alzheimer’s disease. Biomed Pharmacother = Biomed Pharmacother. (2024) 181:117721. doi: 10.1016/j.biopha.2024.117721
28. Huang M, Ke Z, Lyu MA, Masarova L, Sadeghi T, Flowers CR, et al. CXCR4-enriched T regulatory cells preferentially home to bone marrow and resolve inflammation. iScience. (2024) 27(9):110830. doi: 10.1016/j.isci.2024.110830
29. MacMillan ML, Hippen KL, McKenna DH, Kadidlo D, Sumstad D, DeFor TE, et al. First-in-human phase 1 trial of induced regulatory T cells for graft-versus-host disease prophylaxis in HLA-matched siblings. Blood Adv. (2021) 5(5):1425–36. doi: 10.1182/bloodadvances.2020003219
30. Sato Y, Passerini L, Piening BD, Uyeda MJ, Goodwin M, Gregori S, et al. Human-engineered Treg-like cells suppress FOXP3-deficient T cells but preserve adaptive immune responses. vivo. Clin Trans Immunol. (2020) 9:e1214. doi: 10.1002/cti2.1214
31. Andolfi G, Fousteri G, Rossetti M, Magnani CF, Jofra T, Locafaro G, et al. Enforced IL-10 expression confers type 1 regulatory T cell (Tr1) phenotype and function to human CD4(+) T cells. Mol Ther. (2012) 20:1778–90. doi: 10.1038/mt.2012.71
32. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, and Gagliani N. The biology of T regulatory type 1 cells and their therapeutic application in immune-mediated diseases. Immunity. (2018) 49(6):1004–19. doi: 10.1016/j.immuni.2018.12.001
33. Freeborn RA, Strubbe S, and Roncarolo MG. Type 1 regulatory T cell-mediated tolerance in health and disease. Front Immunol. (2022) 13:1032575. doi: 10.3389/fimmu.2022.1032575
34. Sayitoglu EC, Freeborn RA, and Roncarolo MG. The yin and yang of type 1 regulatory T cells: from discovery to clinical application. Front Immunol. (2021) 12:693105. doi: 10.3389/fimmu.2021.693105
35. Tsang JY, Tanriver Y, Jiang S, Xue SA, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest. (2008) 118(11):3619–28. doi: 10.1172/JCI33185
36. Wardell CM, Fung VCW, Chen E, Haque M, Gillies J, Spanier JA, et al. Short Report: CAR Tregs mediate linked suppression and infectious tolerance in islet transplantation. bioRxivorg. (2024) [Preprint]. doi: 10.1101/2024.04.06.588414
37. Durgam SS, Rosado-Sánchez I, Yin D, Speck M, Mojibian M, Sayin I, et al. CAR Treg synergy with anti-CD154 promotes infectious tolerance and dictates allogeneic heart transplant acceptance. JCI Insight. (2025) 10(7):e188624. doi: 10.1172/jci.insight.188624
38. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun. (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
39. Eggenhuizen PJ, Cheong RMY, Lo C, Chang J, Ng BH, Ting YT, et al. Smith-specific regulatory T cells halt the progression of lupus nephritis. Nat Commun. (2024) 15:1–14. doi: 10.1038/s41467-024-45056-x
40. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. (2015) 125(7):1107–15. doi: 10.1182/blood-2014-04-566786
41. Eggenhuizen PJ, Ng BH, Lo C, Chang J, Snelgrove SL, Cheong RMY, et al. Engineered antigen-specific T regulatory cells suppress autoreactivity to the anti-glomerular basement membrane disease antigen. Kidney Int. (2025) 107(4):751–6. doi: 10.1016/j.kint.2025.01.005
42. Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, et al. Avidity and bystander suppressive capacity of human regulatory T cells expressing de novo autoreactive T-cell receptors in type 1 diabetes. Front Immunol. (2017) 8:1313. doi: 10.3389/fimmu.2017.01313
43. Arshad S, Cameron B, and Joglekar AV. Immunopeptidomics for autoimmunity: unlocking the chamber of immune secrets. NPJ Syst Biol Appl. (2025) 11:10. doi: 10.1038/s41540-024-00482-x
44. Uenishi GI, Repic M, Yam JY, Landuyt A, Saikumar-Lakshmi P, Guo T, et al. GNTI-122: an autologous antigen-specific engineered Treg cell therapy for type 1 diabetes. JCI Insight. (2024) 9(6):e171844. doi: 10.1172/jci.insight.171844
45. Labanieh L and Mackall CL. Author Correction: CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 619:E26. doi: 10.1038/s41586-023-06088-3
46. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant: Off J Am Soc Transplant Am Soc Transplant Surg. (2017) 17(4):931–43. doi: 10.1111/ajt.14185
47. Yoon J, Schmidt A, Zhang AH, Königs C, Kim YC, and Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. (2017) 129(2):238–45. doi: 10.1182/blood-2016-07-727834
48. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflamm. (2012) 9:1–12. doi: 10.1186/1742-2094-9-112
49. Schreeb K, Culme-Seymour E, Ridha E, Dumont C, Atkinson G, Hsu B, et al. Study design: human leukocyte antigen class I molecule A∗02-chimeric antigen receptor regulatory T cells in renal transplantation. Kidney Int Rep. (2022) 7:1258. doi: 10.1016/j.ekir.2022.03.030
50. Clinicaltrials.gov . Available online at: https://clinicaltrials.gov/study/NCT04817774?cond=NCT04817774&rank=1 (Accessed March 24, 2025).
51. Development of a novel regulatory T cell-based therapy for patients with rheumatoid arthritis (2022). ACR Meeting Abstracts. Available online at: https://acrabstracts.org/abstract/development-of-a-novel-regulatory-t-cell-based-therapy-for-patients-with-rheumatoid-arthritis/ (Accessed March 24, 2025).
52. Bluestone JA, Burnett BK, Crute CE, Fellows MD, Levings M, Lebrec H, et al. Regulatory T cell therapies to treat autoimmune diseases and transplant rejection. Nat Immunol. (2025) 26:819–24. doi: 10.1038/s41590-025-02154-2
53. Cieniewicz B, Uyeda MJ, Chen PP, Sayitoglu EC, Liu JM-H, Andolfi G, et al. Engineered type 1 regulatory T cells designed for clinical use kill primary pediatric acute myeloid leukemia cells. Haematologica. (2021) 106:2588–97. doi: 10.3324/haematol.2020.263129
54. Niu C, Wei H, Pan X, Wang Y, Song H, Li C, et al. Foxp3 confers long-term efficacy of chimeric antigen receptor-T cells via metabolic reprogramming. Cell Metab. (2025) 37:1426–1441.e7. doi: 10.1016/j.cmet.2025.04.008
55. Cochrane RW, Robino RA, Granger B, Allen E, Vaena S, Romeo MJ, et al. High-affinity chimeric antigen receptor signaling induces an inflammatory program in human regulatory T cells. Mol Ther Methods Clin Dev. (2024) 32:101385. doi: 10.1016/j.omtm.2024.101385
56. Li Y, Singh N, Dong C, Sharma S, Zhou Z, Du J, et al. Reprogramming intratumoral Treg cells by morpholino-mediated splicing of FOXP3 for cancer immunotherapy. Sci Immunol. (2025) 10:eadr9933.
57. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant. (2017) 17:2945–54. doi: 10.1111/ajt.14415
58. Orozco G, Gupta M, Gedaly R, and Marti F. Untangling the knots of regulatory T cell therapy in solid organ transplantation. Front Immunol. (2022) 13:883855. doi: 10.3389/fimmu.2022.883855
59. Jena B, Maiti S, Huls H, Singh H, Lee DA, Champlin RE, et al. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PloS One. (2013) 8:e57838. doi: 10.1371/journal.pone.0057838
60. Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. (2011) 118(5):1255–63. doi: 10.1182/blood-2011-02-337360
61. Lamarthée B, Marchal A, Charbonnier S, Blein T, Leon J, Martin E, et al. Transient mTOR inhibition rescues 4-1BB CAR-Tregs from tonic signal-induced dysfunction. Nat Commun. (2021) 12:1–19. doi: 10.1038/s41467-021-26844-1
62. Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med. (2003) 9:367–9. doi: 10.1038/nm0403-367
63. Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. (2017) 14:865–8. doi: 10.1038/nmeth.4380
64. Goltsev Y and Nolan G. CODEX multiplexed tissue imaging. Nat Rev Immunol. (2023) 23:613. doi: 10.1038/s41577-023-00936-z
65. Mika T, Maghnouj A, Klein-Scory S, Ladigan-Badura S, Baraniskin A, Thomson J, et al. Digital-droplet PCR for quantification of CD19-directed CAR T-cells. Front Mol Biosci. (2020) 7:84. doi: 10.3389/fmolb.2020.00084
66. Lu A, Liu H, Shi R, Cai Y, Ma J, Shao L, et al. Application of droplet digital PCR for the detection of vector copy number in clinical CAR/TCR T cell products. J Transl Med. (2020) 18:191. doi: 10.1186/s12967-020-02358-0
67. Vignali DAA, Collison LW, and Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
68. Mason GM, Lowe K, Melchiotti R, Ellis R, de Rinaldis E, Peakman M, et al. Phenotypic complexity of the human regulatory T cell compartment revealed by mass cytometry. J Immunol (Baltimore Md: 1950). (2015) 195(5):2030–7. doi: 10.4049/jimmunol.1500703
69. Barcenilla H, Åkerman L, Pihl M, Ludvigsson J, and Casas R. Mass cytometry identifies distinct subsets of regulatory T cells and natural killer cells associated with high risk for type 1 diabetes. Front Immunol. (2019) 10:422932. doi: 10.3389/fimmu.2019.00982
70. Garcia Castillo J, DeBarge R, Mende A, Tenvooren I, Marquez DM, Straub A, et al. A mass cytometry method pairing T cell receptor and differentiation state analysis. Nat Immunol. (2024) 25(9):1754–63. doi: 10.1038/s41590-024-01937-3
71. Kordasti S, Costantini B, Seidl T, Perez AP, Martinez LM, McLornan D, et al. Deep phenotyping of Tregs identifies an immune signature for idiopathic aplastic anemia and predicts response to treatment. Blood. (2016) 128(9):1193–205. doi: 10.1182/blood-2016-03-703702
72. Luo Y, Xu C, Wang B, Niu Q, Su X, Bai Y, et al. Single-cell transcriptomic analysis reveals disparate effector differentiation pathways in human Treg compartment. Nat Commun. (2021) 12:1–14. doi: 10.1038/s41467-021-24213-6
73. Pai JA and Satpathy AT. High-throughput and single-cell T cell receptor sequencing technologies. Nat Methods. (2021) 18:881–92. doi: 10.1038/s41592-021-01201-8
74. Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. (2021) 12:1088. doi: 10.1038/s41467-021-21246-9
75. Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature. (2018) 563:347–53. doi: 10.1038/s41586-018-0698-6
76. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. (2013) 10:1213–8. doi: 10.1038/nmeth.2688
77. van der Veeken J, Glasner A, Zhong Y, Hu W, Wang ZM, Bou-Puerto R, et al. The transcription factor foxp3 shapes regulatory T cell identity by tuning the activity of trans-acting intermediaries. Immunity. (2020) 53:971–984.e5. doi: 10.1016/j.immuni.2020.10.010
78. Chowdhary K, Léon J, Mathis D, and Benoist C. An integrated transcription factor framework for Treg identity and diversity. Proc Natl Acad Sci. (2024) 121:e2411301121. doi: 10.1073/pnas.2411301121
79. Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol. (2019) 37:925–36. doi: 10.1038/s41587-019-0206-z
80. Reed KSM, Davis ES, Bond ML, Cabrera A, Thulson E, Quiroga IY, et al. Temporal analysis suggests a reciprocal relationship between 3D chromatin structure and transcription. Cell Rep. (2022) 41(5):111567. doi: 10.1016/j.celrep.2022.111567
81. Lee MYY, Kaestner KH, and Li M. Benchmarking algorithms for joint integration of unpaired and paired single-cell RNA-seq and ATAC-seq data. Genome Biol. (2023) 24:1–33. doi: 10.1186/s13059-023-03073-x
82. Cai L, Ma X, and Ma J. Integrating scRNA-seq and scATAC-seq with inter-type attention heterogeneous graph neural networks. Briefings Bioinf. (2024) 26(1):bbae711. doi: 10.1093/bib/bbae711
83. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell. (2019) 177(7):1888–902.e21. doi: 10.1016/j.cell.2019.05.031
84. Mitra S, Malik R, Wong W, Rahman A, Hartemink AJ, Pritykin Y, et al. Single-cell multi-ome regression models identify functional and disease-associated enhancers and enable chromatin potential analysis. Nat Genet. (2024) 56:627–36. doi: 10.1038/s41588-024-01689-8
85. Sha Y, Qiu Y, Zhou P, and Nie Q. Reconstructing growth and dynamic trajectories from single-cell transcriptomics data. Nat Mach Intell. (2024) 6:25–39. doi: 10.1038/s42256-023-00763-w
86. Stassen SV, Yip GGK, Wong KKY, Ho JWK, and Tsia KK. Generalized and scalable trajectory inference in single-cell omics data with VIA. Nat Commun. (2021) 12:5528. doi: 10.1038/s41467-021-25773-3
87. Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. (2014) 20:436–42. doi: 10.1038/nm.3488
88. Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. (2018) 174:968–981.e15. doi: 10.1016/j.cell.2018.07.010
89. Ma M, Luo Q, Chen L, Liu F, Yin L, and Guan B. Novel insights into kidney disease: the scRNA-seq and spatial transcriptomics approaches: a literature review. BMC Nephrol. (2025) 26:1–19. doi: 10.1186/s12882-025-04103-5
90. McCallion O, Cross AR, Brook MO, Hennessy C, Ferreira R, Trzupek D, et al. Regulatory T cell therapy is associated with distinct immune regulatory lymphocytic infiltrates in kidney transplants. Med (New York NY). (2024) 6(5):100561. doi: 10.1016/j.medj.2024.11.014
91. Lamarthée B, Callemeyn J, Van Herck Y, Antoranz A, Anglicheau D, Boada P, et al. Transcriptional and spatial profiling of the kidney allograft unravels a central role for FcyRIII+ innate immune cells in rejection. Nat Commun. (2023) 14:1–22. doi: 10.1038/s41467-023-39859-7
92. Azim S, Zubair H, Rousselle T, McDaniels JM, Shetty AC, Kuscu C, et al. Single-cell RNA sequencing reveals peripheral blood mononuclear immune cell landscape associated with operational tolerance in a kidney transplant recipient. Am J Transplant. (2023) 23:1434–45. doi: 10.1016/j.ajt.2023.04.035
93. Rosales IA, Yang C, Farkash EA, Ashry T, Ge J, Aljabban I, et al. Novel intragraft regulatory lymphoid structures in kidney allograft tolerance. Am J Transplant. (2022) 22:705–16. doi: 10.1111/ajt.16880
94. Cang Z, Zhao Y, Almet AA, Stabell A, Ramos R, Plikus MV, et al. Screening cell-cell communication in spatial transcriptomics via collective optimal transport. Nat Methods. (2023) 20:218–28. doi: 10.1038/s41592-022-01728-4
95. Zhu J, Wang Y, Chang WY, Malewska A, Napolitano F, Gahan JC, et al. Mapping cellular interactions from spatially resolved transcriptomics data. Nat Methods. (2024) 21:1830–42. doi: 10.1038/s41592-024-02408-1
96. Huehn J, Polansky JK, and Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. (2009) 9(2):83–9. doi: 10.1038/nri2474
97. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, and Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the foxp3 locus. Cell. (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
98. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, and Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. (2010) 463:808–12. doi: 10.1038/nature08750
99. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PloS Biol. (2007) 5:e38. doi: 10.1371/journal.pbio.0050038
100. Thornton AM and Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. (1998) 188:287–96. doi: 10.1084/jem.188.2.287
101. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. (2000) 192:303–10. doi: 10.1084/jem.192.2.303
102. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
103. Yang F, Zhang D, Jiang H, Ye J, Zhang L, Bagley SJ, et al. Small-molecule toosendanin reverses macrophage-mediated immunosuppression to overcome glioblastoma resistance to immunotherapy. Sci Transl Med. (2023) 15:eabq3558. doi: 10.1126/scitranslmed.abq3558
104. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. Infectious” transplantation tolerance. Science. (1993) 259:974–7. doi: 10.1126/science.8094901
105. Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, and Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med. (2002) 196:255–60. doi: 10.1084/jem.20020394
106. Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. (2021) 18:379–93. doi: 10.1038/s41571-021-00476-2
107. Labanieh L and Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
108. Rodrigues K. Created in BioRender. (2025). https://BioRender.com/r2ueosz.
109. Rodrigues K. Created in BioRender. (2025). https://BioRender.com/bjd32dh.
Keywords: Treg, regulatory T cell, autoimmunity, transplantation, GvHD, T cell therapy, immune tolerance, immunomonitoring
Citation: Rodrigues KB, Eggenhuizen PJ, Bacchetta R and Good Z (2025) Regulatory T cell therapies: from patient data to biological insights. Front. Immunol. 16:1675114. doi: 10.3389/fimmu.2025.1675114
Received: 28 July 2025; Accepted: 10 October 2025;
Published: 31 October 2025.
Edited by:
Fabien Depis, TregShield Bio, United StatesReviewed by:
Subhash Kumar Tripathi, Seattle Children’s Research Institute, United StatesCopyright © 2025 Rodrigues, Eggenhuizen, Bacchetta and Good. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zinaida Good, emluYWlkYUBzdGFuZm9yZC5lZHU=