 Elean S. V. Tay
Elean S. V. Tay Yi T. Ting
Yi T. Ting Poh-Yi Gan
Poh-Yi Gan Joshua D. Ooi
Joshua D. Ooi- Center for Inflammatory Diseases, School of Clinical Sciences at Monash Health, Monash University, Melbourne, VIC, Australia
Anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV) is a rare autoimmune disease characterized by the inflammation of small vessels. It is most commonly caused by ANCA targeting proteinase 3 (PR3) and myeloperoxidase (MPO) which are found in neutrophil lysosomes. The most common affected organs are respiratory tracts and kidneys, though other organs can be involved too. Although the cause of disease between PR3-AAV and MPO-AAV is similar, they vary in pathogenesis. Epigenetic and genetic factors may play a role in the disease development as certain population such as Chinese with HLA-DRB1*04:05 are more prevalent in MPO-AAV patient population. The prognosis for them is usually poor, often resulting in end-stage renal failure even with existing treatment. Current treatment for AAV relies heavily on corticosteroids which are toxic for long-term usage. Hence, there is a strong need to develop new, less toxic and targeted therapy for this disease. Regulatory T cell (Treg) therapy is a new type of therapy with the potential to specifically re-establish tolerance to the target autoantigen (MPO or PR3). This review will delve into the pathogeneses of AAV and discuss the potential of using genetically engineered Tregs to treat the cause of disease.
Anti-neutrophil cytoplasmic antibodies-associated vasculitis
AAV is a rare autoimmune disease that causes inflammation and subsequently damage to the small vessels (1–3). The main characteristic of the disease is the pauci-immune state of deposition of immunoglobulin within the small vessels of the glomeruli, which stands out as a subgroup of the small vessel vasculitides (1–3). The common affected organs include the respiratory tract, lungs and kidney. The underlying causes are autoantibodies to the neutrophil proteins; particularly proteinase 3 (PR3) and myeloperoxidase (MPO), however, other neutrophil proteins including lysosome-associated membrane protein 2 (LAMP-2) and elastase are involved to a lesser extent.
Classification and clinical presentation
AAV can be subclassified into granulomatosis with polyangiitis (GPA) (previously known as Wegener’s granulomatosis), microscopic polyangiitis (MPA) and eosinophilic GPA (EGPA) (previously known as Churg-Strauss syndrome) according to the 2012 Revised Chapel Hill Consensus Conference (4). GPA is presented with necrotizing vasculitis with extravascular granulomatosis that usually involves respiratory tract and kidney whereas MPA is like GPA, but in the absence of granulomatosis. EGPA is characterized with necrotizing granulomatous vasculitis in the presence of eosinophilia, and it usually involves the respiratory tract with an association with asthma. A famous disease manifestation is the detection of little or no deposition of immune complexes in the affected tissue area. This results in an interesting disease presentation of pauci-immune necrotizing crescentic glomerulonephritis (NCGN). The absence of the immune complexes is due to the release of elastase from the dead neutrophils, digesting the immunoglobulin (5).
Furthermore, PR3-ANCA is usually associated with GPA (around 85% of patients) whereas MPO-ANCA is more frequently associated with MPA (around 60% of patients) (6). The titres of ANCA do not correlate with the severity of the disease as ANCA can be found in asymptomatic or healthy individuals (1, 7). The pathogenic T cell epitopes of MPO-AAV have been delineated in experimental models of disease (8).
Epigenetic and genetic factors of MPO-AAV
The prevalence of AAV has increased in recent years from 48–184 cases to 300–421 cases per million individuals, indicating improved survival of patients (6). Efforts have been put into genome wide association studies (GWAS) to investigate genetic variants and subsets occurring in patients that might be the key towards disease pathogenesis. PR3 is mostly found in Caucasian population whereas Asians such as Chinese and Japanese dominates MPA, indicating the role of different genetic subsets. Studies have supported an association of the disease with non-major histocompatibility complex (MHC) and MHC, though MPO-AAV is mostly associated with MHC II alleles (9). Japanese patients with MPO-AAV are skewed towards DRB1*09:01 being the risk allele (10) whereas Chinese patients are frequently carrying DQA1*03:02, DQB1*03:03 and DRB1*04:05 (11, 12). In particular, MPO-AAV patients carrying DRB1*04:05 exhibit the worst prognosis, with ~50% of patients progressing to end-stage renal failure (ESRF) within 1 year of diagnosis (11).
Other factors including infections, drugs and silica exposure have been associated with disease progression. Patients who are carriers of S. aureus are more susceptible to relapse as S. aureus infection triggers the disease onset for PR3-AAV. This is because S. aureus demonstrates a molecular mimicry to the complementary peptide of PR3105-201, and the B cells responds by producing antibodies against this cPR3105–201 peptide (13). However, the resulting antibodies were also found to be reactive towards the PR3105-201, contributing towards the autoimmunity in patients. Whereas in MPO-AAV, Ooi et al. demonstrated that certain S. aureus strain carries a plasmid-encoded 6-phosphogluconate dehydrogenase amino acid sequence (6PGD391-410) can trigger anti-MPO immunity due to molecular mimicry (14). Certain drugs such as propylthiouracil (PTU), an antithyroid drug is commonly associated with MPO-AAV even though its exact mechanism is unknown, further contributing towards the risk of certain population to develop AAV (6). Intensity of silica exposure impacts on the development of AAV as well, though its exact mechanism is unknown (15).
Previous studies have identified the immunodominant region of MPO, which tends to be in a ‘hotspot’ region in the heavy chain of MPO. Patients’ sera were used to map these immunogenic epitopes, with identification of shared epitopes between T and B cells further emphasize the importance of the hotspot region through their persistence presence in remission patients. The human chimera to the identified immunogenic MPO epitope in a mouse study by Ooi et. al., MPO435–453 lies within the hotspot region (8), whereas clinical studies had identified MPO393-402, MPO437-446, MPO447-461, MPO479-488, MPO717–726 to be immunogenic (7, 16, 17).
Diagnosis
Currently, patients are diagnosed based on clinical symptoms; pathology tests (urine and blood) and tissue biopsy (6). However, it must be noted that having a positive ANCA does not necessarily mean the patient has AAV, rather it could be other autoimmune diseases such as lupus nephritis (LN), inflammatory bowel disease (IBD), ulcerative colitis (UC) and autoimmune hepatitis (AIH). Birmingham Vasculitis Activity Score (BVAS) is a well-developed tool that is used by clinicians to quantify the disease activity in patients (18–20). Histologic examination of tissue biopsy remains to be the gold standard in diagnosing AAV. It is crucial to differentiate them with immune complex small vessel vasculitis because it determines the type of treatment and prognosis of patients (6).
Pathogenesis of MPO-AAV
Innate immune response
The pathogenesis of AAV is not completely elucidated and the suggested pathways are based on patient and experimental observations (Figure 1). The autoantigen, MPO is a sequestered antigen, found in the lysosomes of neutrophils. Hence, a stimulus is required to activate neutrophils to cause an upregulation of membrane bound MPO in neutrophils. Proinflammatory cytokines such as tumour necrosis factor alpha (TNF-α) are usually the trigger for disease onset, which commonly happens in infection. The presence of infectious agent stimulates the release of transforming growth factor beta (TGFβ) and interleukin 6 (IL-6) by dendritic cells (21, 22). ANCAs produced by B cells bind to membrane bound MPO on activated neutrophils. This in turn triggers the naïve T cells to differentiate into T helper 17 (Th17) cells and produce IL-17. IL-17 is a potent proinflammatory cytokine that stimulates the immune system, including the monocytes and macrophages to release TNFα and IL-1β, which further prime the neutrophils during the acute phase of the disease (21, 22). Renal neutrophils are found to be capable of producing proinflammatory cytokines TNFα and IL-17 as well, contributing to the differentiation of Th17 cells. The innate γδ T cells migrate to the site of inflammation and produce IL-17 that contributes to disease development (23).
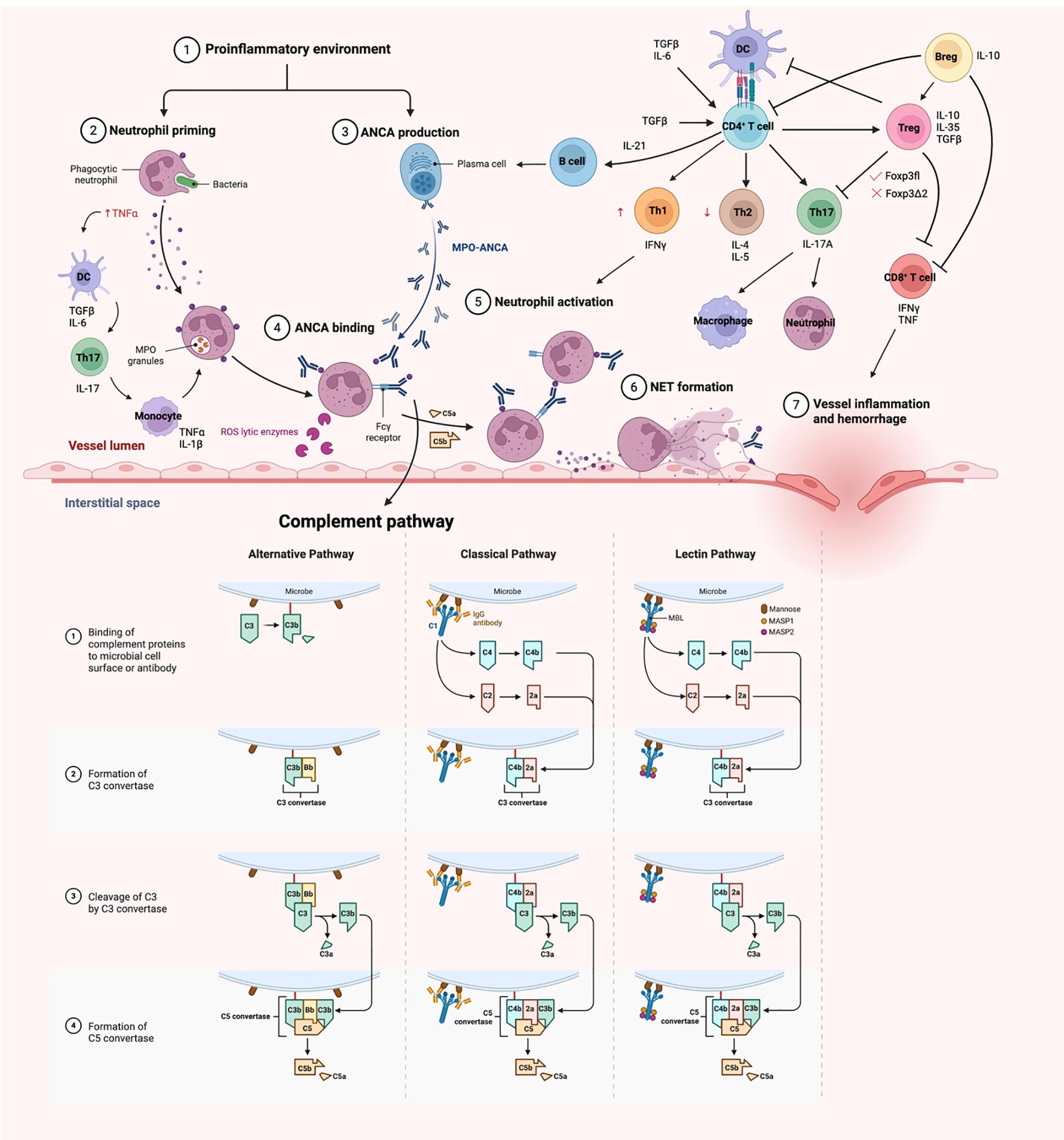
Figure 1. A schematic overview of pathogenesis of MPO-AAV. Neutrophils are primed with TNFα in a proinflammatory environment, resulting in an upregulation of membrane MPO. These neutrophils are targeted by ANCA, hence destroying them in the process, resulting in damage of endothelium lining. The adaptive immune system comes in later and act in a delayed-type hypersensitivity (DTH) fashion, enhancing the disease activity. Diagrams created with Biorender.com.
Studies have shown that TNF-α primed neutrophils upregulates membrane expression of MPO, allowing the interaction between MPO and MPO-ANCA, in which the MPO-ANCA acts as a bridge between the Fcγ receptors and MPO. The FcγR of the adjacent neutrophils can then recognize the Fc portion of these ANCAs, resulting in neutrophil activation, hence releasing reactive oxygen species (ROS) that kills the ‘victim’ neutrophils. This causes the neutrophils to undergo respiratory burst, degranulation, releasing neutrophil extracellular traps (NETosis), apoptosis and necrosis (3).
Furthermore, the FcγR becomes the target of complement, activating the classical and alternate complement pathways, encouraging the destruction of neutrophils. In particular, C5a is the central complement in both pathways and studies had shown an increase in circulating levels of C5a in patients (24). The ablation of the C5a receptor on neutrophils also attenuate the disease phenotype, indicating a probable therapeutic target (25, 26). Despite the importance of FcγR being stated, FcγRIIB plays a role as the only inhibitory Fc receptor that regulates and maintains peripheral tolerance (27). An in vivo mouse model showed that the FcγRIIB deficient mice developed more severe glomerular injury. The whole process releases proinflammatory cytokines which attract more neutrophils to the site by adhering to the vessel wall and undergo diapedesis (1–3, 6). Spillage of plasma then happens, and the presence of the coagulation factor encourages the formation of fibrinoid necrosis, ultimately damaging the fragile monolayer endothelium.
Involvement of the adaptive immune response
B cells
B cells are responsible for generating autoantibodies, and some studies suggests that this arises from a deficiency or dysfunction of regulatory T cells (Treg), leading to a loss of immune tolerance. Neutrophils also contribute by releasing B-cell activating factors (BAFF)/B lymphocyte stimulator (BLyS) (28) which stimulate B cell differentiation, proliferation and immunoglobulins (Igs) production. Regulatory B cells (Breg) that express CD5 and is important for IL-10 production, are downregulated in disease. These Bregs play a key role in suppressing activated T cells and supporting Treg differentiation (29, 30). Research has shown that CD5+CD24hiCD38+ B cells are reduced during active disease but return to levels comparable levels to healthy controls (HC) during remission (31). Additionally, CD4+ T cells contribute to B cells stimulation through the production of IL-21 (30).
T cells
T cells play a critical, non-redundant role in disease pathogenesis (6, 32. 1, 33). They mediate delayed-type hypersensitivity (DTH) responses, perpetuating inflammation and ultimately leading to tissue destruction (Figure 1).
CD4+ T helper (Th) cells are central to adaptive immunity, activating B cells, recruiting macrophages and promoting cytotoxic T cell responses, all of which drive autoimmune response. Gan et al. demonstrated that the depletion of MPO-specific CD4+ T cells ameliorates GN, highlighting their importance in disease activity (33). Specifically, Th1 cell contributes to the nephritogenic immune responses, where IL-12p40 guides Th1 cells in inducing crescentic GN (34). The balance between Th1 and Th2 cells is critical for maintaining self-immune tolerance (21). Th1 cells drive the severity and progression of autoimmune disease through IFNγ production, while Th2 cells act in opposition (35). Th17 cell is another subset linked to autoimmunity due to its pro-inflammatory nature. Retinoic acid receptor-related orphan nuclear receptor γt (RORγt) is an important transcription factor essential for Th17 cells differentiation, and its absence attenuates GN (36). Both TGFβ and IL-6 encourages the Th17 lineage differentiation, but TGFβ alone supports Treg differentiation (37). Th17 cells produce IL-17 which significantly contributes to disease manifestation, with elevated IL-17 levels exacerbating crescentic GN formation (38). IL-17A not only activates Th17 cells but also enhances neutrophils and macrophages recruitment (39).
In experimental anti-MPO GN mouse models, depletion of MPO-specific CD8+ T cells ameliorate kidney injury, as evidenced by reduction in albuminuria, blood urea nitrogen (BUN) and proteinuria (40). Notably, CD8+ T cells can mediate glomerular injury in an MPO+ environment even in the absence of CD4+ T cells, highlighting their independent pathogenic potential (40). These CD8+ T cells are major sources of proinflammatory cytokines such as interferon gamma (IFNγ) and TNF. Furthermore, IL7R (CD127) signalling is critical for Teff function, as demonstrated by increased expression in a transcriptome analysis (41).
Besides, toll-like receptors (TLRs) which function to recognize pathogen associated molecular patterns are found to engage in disease pathogenesis. The activation of TLR2 stimulates IL-17A production, promoting Th17 cells activity, while TLR9 enhances anti-MPO driven autoimmunity through Th1 committed lineage pathway (42). In addition, TLR4 is constitutively expressed in glomeruli and its expression is upregulated during GN (43), drive the production of chemokines, CXCL1 and CXCL2 where they serve as the major chemoattractant for neutrophils (43). An experimental murine anti-MPO model further showed that lipopolysaccharide (LPS) can synergize with anti-MPO autoimmunity, exacerbating the NCGN disease phenotype (44). Collectively, these findings suggest that infection and innate immune activation can amplify the anti-MPO driven autoimmunity.
Regulatory T cells
A subset of T cells, known as the regulatory T cells (Tregs) are key mediators of self-tolerance and immune homeostasis. The critical role of Tregs was first elucidated in the 1990s by Sakaguchi et. al, where adoptive transfer of CD4+CD25+ but not CD4+CD25- T cells could prevent autoimmune disease in athymic mice (45). The scurfy phenotype observed in mice is linked to mutations in the Forkhead box protein p3 (Foxp3) gene, which is also implicated in the human immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome. This syndrome arises from recessive mutations in Foxp3 and results in severe autoimmune manifestations, underscoring the essential role of Foxp3 in Treg function (45).
Foxp3 is a transcription factor uniquely expressed in Treg, but its precise regulatory mechanisms remain incompletely understood. At the genomic level, Foxp3 controls the expression of genes critical for T cell function, including nuclear factor of activated T cells (NFAT) and AML1/RunX1, which are required for effector T cells (Teff) differentiation (46). Foxp3 also interacts with RORγt, inhibiting the differentiation of naïve T cells into Th17, further clarifying its role in MPO-AAV disease mechanism. Notably, only the full length Foxp3 isoform (Foxp3fl) can interact with RORγt, while the exon 2-spliced variant (Foxp3Δ2) cannot (32).
Given their potent immunosuppressive capacity, Tregs are a great therapeutic tool to treat autoimmune diseases. In patients with MPO-AAV, Treg numbers are often elevated, but their suppressive function is frequently imparied (47, 48), potentially due to a reduction in activated Treg (aTreg) characterized by CD45RA-Foxp3high51. This impairment may also relate to increased expression of the Foxp3Δ2 isoform, which is associated with reduced suppressive function and a higher proportion of resistant CD4+ Teff cells within the patient population (47). A recent discovery demonstrated that co-expression of Foxp3fl and Foxp3Δ2 are essential for optimal Treg suppressive capacity (49).
Autoimmune regulator (Aire) is a transcription factor found in lymphoid organs, particularly in medullary thymic epithelial cells (mTECs) of the thymus, where it plays an inevitable role in immune regulation and the establishment of central tolerance. Aire enables mTECs to express a broad array of tissue-specific antigens, facilitating the presentation of self-peptides on major histocompatibility complex (pMHC) molecules to developing T cells. This process ensures that T cells with high affinity TCRs for self-antigens are either deleted or diverted to differentiate into Treg, resulting in a Treg population with a diverse and high affinity T-cell receptor (TCR) repertoire. The importance of Aire is further highlighted through an experimental anti-MPO murine model where Aire deficient mice exhibit more severe disease phenotype due to the escape of autoreactive anti-MPO T cells (50).
Tregs naturally develop in the thymus (nTreg), but they can also be peripherally induced (pTreg) from naïve T cells. While pTreg can be induced transiently in the presence of anti-inflammatory cytokines such as TGFβ and IL-10, they may revert to Teff cells under proinflammatory conditions. TGFβ is crucial for pTreg induction, as it phosphorylates the Smad transcription factors, Smad2 and Smad3, which interact with conserved non-coding sequence (CNS) 1 region in the Foxp3 gene locus, driving Foxp3 expression and thus Treg differentiation. Stable Treg identity is maintained through epigenetic mechanisms. In nTreg, the CpG island in the CNS2 region of the Foxp3 gene which is known as the Treg-specific demethylated region (TSDR) remains hypomethylated, supporting sustained Foxp3 expression and Treg lineage stability even in inflammatory environments. which constitutes towards the promoter (51). The TSDR methylation status distinguishes nTreg from pTreg and is crucial for long-term suppressive function, as TSDR demethylation enables the binding of key transcription factors and preserves cell memory. TSDR comprises of a few functionally crucial genes for Treg differentiation and function, including Foxp3, Ctla4, Il2ra, Ikzf4 and Tnfrs18 (51). On top of the suggested phenotype, CD127 is found to be inversely correlated with Foxp3+ cells, and the isolated Treg population is highly purified if included with this marker (52). Therefore, the current best phenotype of Treg is CD4+CD25highCD127lowFoxp3+55.
Previous pioneer work revealed how Treg functions although its exact working mechanism is still unknown. A primary mode of suppression is the secretion of inhibitory cytokines IL-10, IL-35 and TGFβ, which collectively act to inhibit the activity of effector immune cells (32, 53). IL-35 is a heterodimer formed through the pairing of Epstein-Barr virus-induced gene 3 (Ebi3) and interleukin-12 alpha (Il12a), which is proficient in suppressing T cell proliferation (53, 54). Treg also mediate cytotoxicity through the production of granzymes A and B, which are involved in the direct killing of target cells. The high expression of CD25 on Treg enable them to mop up IL-2, a cytokine essential for T cells survival, thereby depriving Teffs of this growth factor, although IL-2 depletion alone does not fully account for Treg-mediated suppression (53). Another key mechanism involves the generation of pericellular adenosine via the CD73/CD39 ectonucleotidase pathway on Treg (55). This adenosine acts on the adenosine 2A (A2A) receptor on activated T cells, potently inhibiting their activity. Activation of A2A receptor also supresses IL-6 production by Teff while promoting TGFβ generation (56), further shifting the immune response towards regulation rather than inflammation. Furthermore, Tregs interact with dendritic cells (DC) via lymphocyte activation gene (LAG) 3 and cytotoxic T lymphocyte-associated Ag (CTLA)-4 (32, 53). These interactions promote the development of tolerogenic DC, which can produce immunoregulatory enzymes like indoleamine 2, 3-dioxygenase (IDO) that suppresses T cell responses. Odobasic et al. have demonstrated the importance of these tolerogenic DCs in an experimental murine anti-MPO GN model (57).
Current animal model
Animal models are essential for studying diseases like MPO-AAV because they allow researchers to identify therapeutic targets and evaluate potential treatments in vivo. However, most current MPO-AAV models primarily replicate the acute phase of disease rather than full remission, highlighting the need for improved models that better capture the complexity of human disease. To summarize, there are currently 4 ways to induce MPO-AAV in mice (refer to Figure 2).
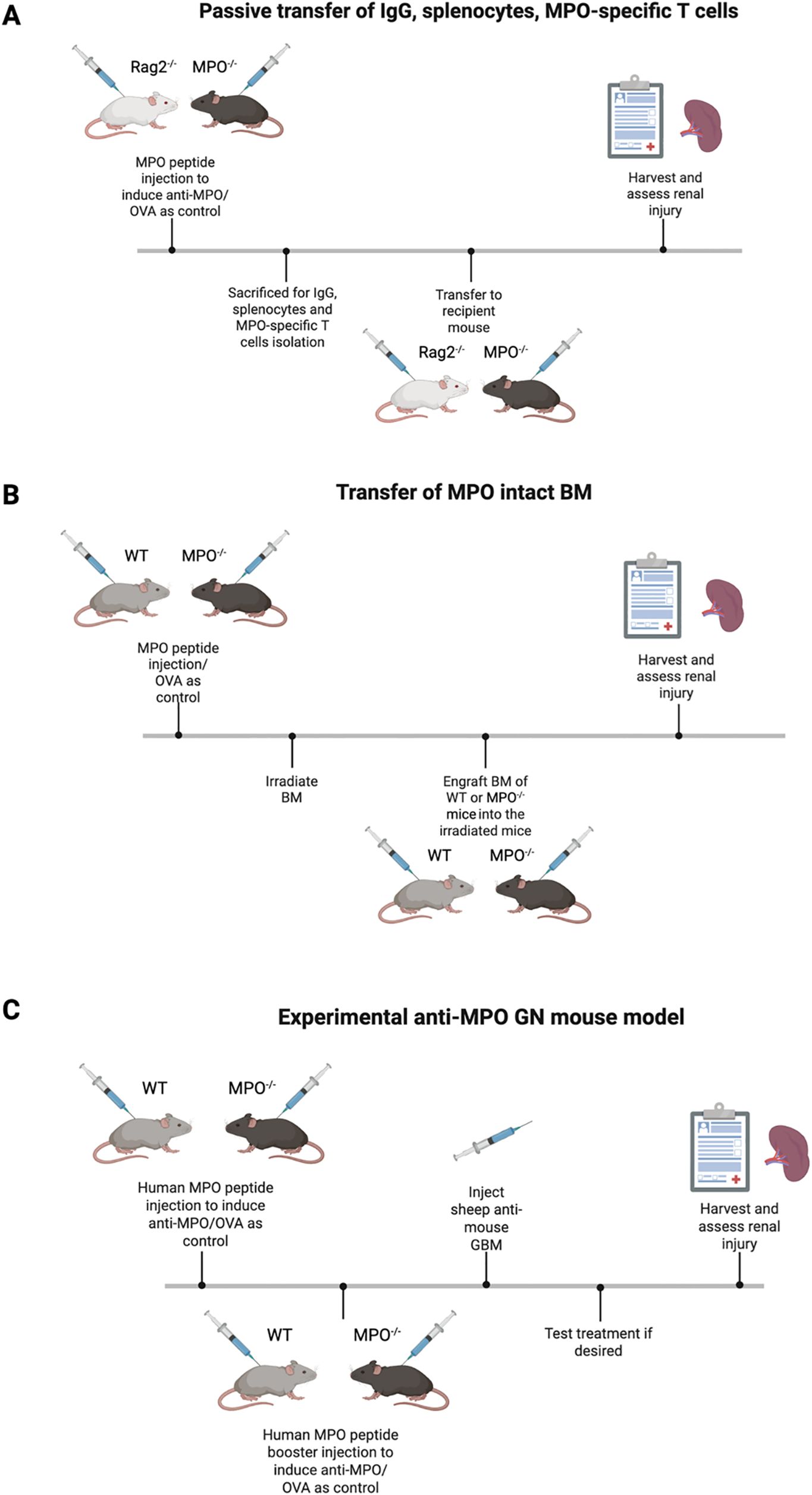
Figure 2. An overview of the current most used mouse models. (A) Passive transfer mouse model that showed MPO-ANCA initiates the hallmark of renal injury. (B) BM engraftment model further digs into the role of adaptive immune system where MPO-specific T cells enhance renal injury. (C) Experimental anti-MPO GN model allows the investigation of MPO-AAV pathogenesis and testing new therapies. Diagrams created with Biorender.com.
Passive transfer of IgG, splenocytes, MPO-specific T cells into mice
The foundational anti-MPO GN mouse model was developed by Xiao et al. and has been instrumental in understanding MPO-AAV pathogenesis (58, 59). In this model, MPO knockout (MPO-/-) and recombinase-activating gene-2-deficient (Rag2-/-) mice, where the Rag2-/- mice lack both T and B cells are first immunized with MPO to induce anti-MPO immune responses. IgG is then isolated from these immunized mice and injected into wildtype (WT) or Rag2-/- mice. This transfer reliably induces focal necrotizing GN, providing direct evidence that anti-MPO IgG alone can cause renal injury. Disease severity increases when splenocytes of the MPO-immunized MPO-/- mice are transferred into WT and Rag2-/- mice (58). These recipient mice develop severe NCGN, with marked elevation of BUN and serum creatinine, indicating significant kidney dysfunction. The disease can be further exacerbated by introducing proinflammatory stimuli such as LPS, resulting in even more severe pathology. Despite the utility of this model in establishing the pathogenicity of anti-MPO antibodies and immune cell, they have limitations. Notably, glomerular immune complex deposition is commonly observed, which differs from the pauci-immune pattern in human cases of MPO-AAV. Therefore, while these models are invaluable for dissecting disease mechanisms and testing therapies, further refinement is necessary to more accurately reflect human disease.
Transfer of MPO intact bone marrow
Another strategy involves first immunizing MPO-/- and WT mice with MPO, followed by irradiation (59, 60). Bone marrow from naïve WT or MPO-/- mice is then transplanted into these immunized recipients to repopulate their immune cells. Only mice receiving WT bone marrow but not those receiving MPO-/- bone marrow develop NCGN after transfer. This finding underscores the necessity of MPO+ neutrophils in peripheral blood of recipient mice for disease development post engraftment. These results suggest that immune cells, particularly neutrophils are the pathogenic targets for anti-MPO IgG (60).
Active immunization of MPO inducing experimental GN
The experimental anti-MPO GN murine model is particularly valuable as it enables the study of loss of tolerance and the development of active autoimmunity. This model also facilitates the identification of potential therapeutic targets by allowing reestablishment of immune tolerance, which is not possible in the passive transfer model. In this approach, WT and MPO-/- mice are sensitized with human MPO to induce anti-MPO antibodies (59, 61). These mice are then injected with sheep anti-mouse glomerular basement membrane (GBM) ten days later to activate neutrophils and deposit MPO in the glomeruli, thereby inducing experimental MPO-ANCA. The use of a minimal dose of sheep anti-mouse GBM is important as it triggers neutrophils influx into the glomeruli without causing significant anti-GBM disease, which would confound the results. Importantly, only MPO-sensitized mice develop diseases, confirming the requirement for both anti-MPO immunity and MPO deposition in disease pathogenesis. Ovalbumin (OVA) was also included as control as an irrelevant antigen confirm the specificity of the model (61). This model advances our knowledge in characterizing the role of CD4+ and CD8+ T cells as well as B cells in disease pathogenesis.
Although the above-mentioned animal models are well-established for the study of MPO-AAV, exploring options beyond them is necessary to facilitate with the understanding of later phase of disease. Humanized mouse model and organoids are promising next steps for the advancement of preclinical models (62, 63). Harnessing immunodeficient mice to allow engraftment of human cells creates a 3D environment for the study of cells and tissues interactions, however, it is usually expensive and is met with the common limitations of cross-species difference. On the other hand, the cost-effective organoids-based approaches is attractive as it aligns with the reduction usage of animals but retaining the capability for inter-species translation. Kidney is a complex organ and the development of 3D organoids is still on-going with efforts being put into mimicking the kidney environment with reproducible conditions (64). Nevertheless, more research is still required for the development of appropriate preclinical models to assist with the study of chronic disease pathogenesis and treatment targets.
Treatment
Current therapy
Treatment for AAV is segregated into two phases, that are induction of remission and maintenance therapy. The current standard treatment for severe AAV is the combination of glucocorticoids; either prednisone with cyclophosphamide (CYC) or with rituximab (RTX) (65, 66). Oral CYC is associated with greater toxicity due to prolonged drug exposure, thus pulse intravenous (i.v.) CYC is preferred, despite a higher relapse risk. This preference is supported by evidence showing that patients receiving i.v. CYC experience less renal impairment and fewer cases of leucopaenia (67, 68). CYC toxicity arises from its broad immunosuppressive effects, often lead to opportunistic infections such as Pneumocystis jiroveci pneumonia, haemorrhagic cystitis, malignancy, and gonadal failure resulting in infertility (68). Meanwhile, RTX is an anti-CD20 monoclonal antibody drug that depletes B cells, thereby preventing ANCA production. RTX is preferred if CYC overdose is a concern, or in patients prone to relapse. Additionally, RTX had been shown to enhance Treg immunomodulatory capacity by inducing B cell apoptosis, though B cell depletion can result in hypogammaglobulinaemia that leads to immune suppression (69).
To minimize cumulative CYC exposure, alternate therapies such as methotrexate (MTX) are considered in early disease, although longer treatment is required and relapse rates are higher compared to CYC (70, 71). Maintenance therapy may involve MTX or azathioprine (AZA), both of which are noninferior to CYC and associated with lesser adverse events (66, 72). Leflunomide and mycophenolate mofetil (MMF) are less commonly used as leflunomide is associated with higher frequency of adverse events (73, 74), whereas in the case of MMF, showed lower efficacy than AZA except in MPO-AAV patients experience less severe renal disease (75, 76).
Given the toxicity of long-term immunosuppressive therapy, biologic agents are being explored. TNFα blocker like etanercept is tested in GPA patients though not proven to be effective in MPO-AAV patients (66, 77). On the contrary, plasma exchange is considered adjunctive for severe cases with pulmonary and renal involvement (78, 79), though recent study shows no superiority over standard treatment (80). Long-term outcome on these patients, however, have shown promising results as they require lower steroid dosage for maintenance therapy, reducing toxicity. Avacopan (CCX168), a C5a inhibitor, has shown promise in replacing glucocorticoids for maintenance therapy (81, 82), potentially reducing morbidity and mortality in AAV patients (83). Patients are spared from higher dose of steroids and safer for renal recovery. Low dose IL-2 is another emerging option, selectively stimulating Treg cells to modulate immune response (84). A recent study employing spatial transcriptomics and digital pharmacology had identified ustekinumab, a human monoclonal antibody which target IL-12 and IL-23 to be effective in disease remission (85). It potently inhibits the dominated Th1/Tc1 and Th17/Tc17 cells in inflamed kidneys as seen in patients, suggesting the use of personalized therapy through combination of current available drug with rapid immune profiling.
Future therapy
While current standard treatment is well-defined with mature clinical trials results supporting in place (Table 1), retaining kidney function is the top priority and with several events of relapse, patients eventually develop end-stage renal failure. Therefore, exploring alternative therapeutic options is important, and recent success in emerging cell-based therapies that offer personalized treatment with fewer side effects through direct target of disease mechanism.
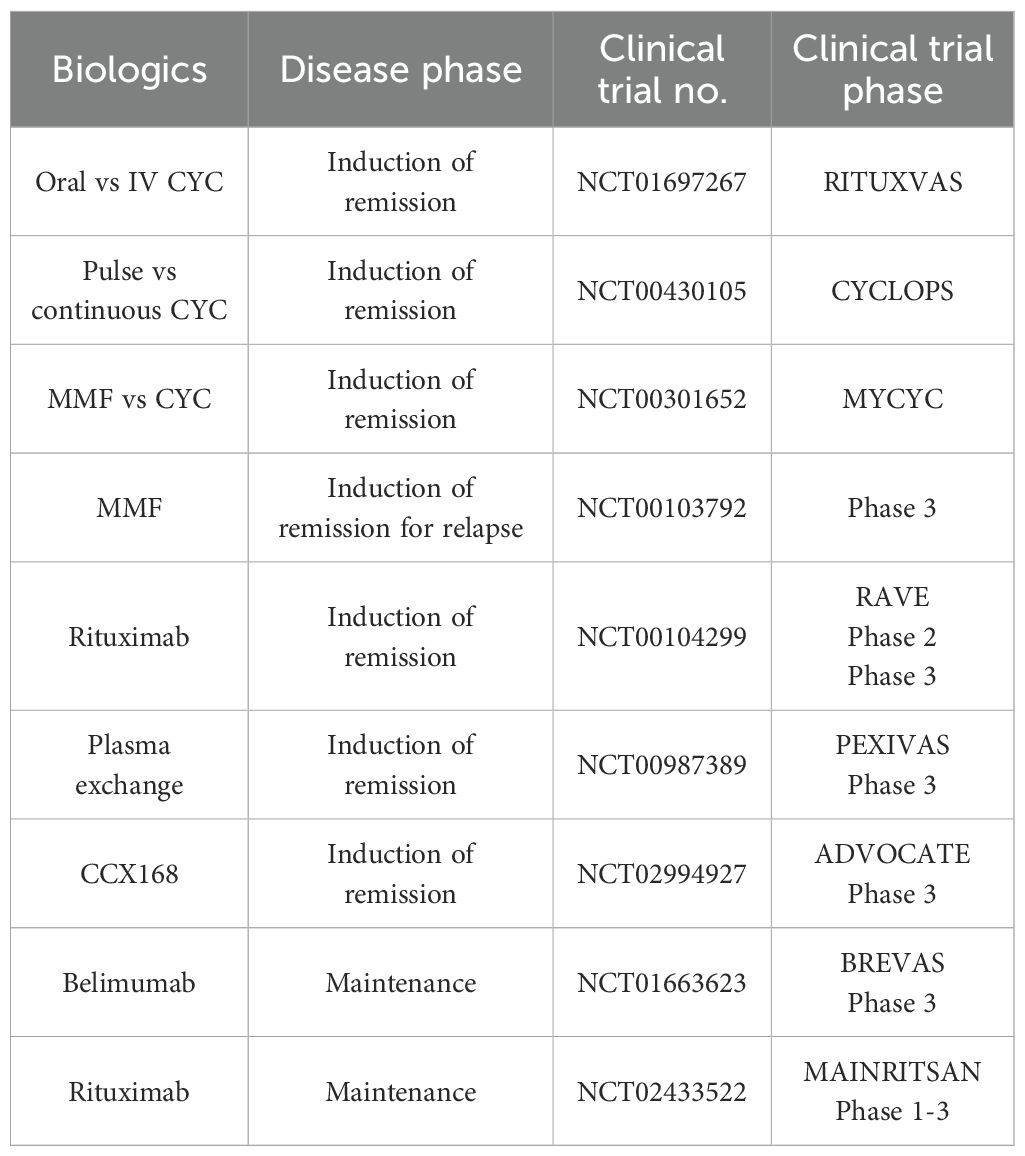
Table 1. Summary of current available therapy associated with their clinical trials studies.
Adoptive cell transfer
Previous attempts on harnessing the potent immunosuppressive capacity of Treg have led to various studies evaluating their efficacy in treating autoimmune diseases and preventing graft rejection. Ex vivo expanded polyclonal Treg have been tested in multiple clinical settings and have shown promise in inducing immune tolerance. This process involves isolating Tregs from patients’ peripheral blood mononuclear cells (PBMCs), expanding them in vitro, and then reinfusing them into the patients.
Multiple studies have demonstrated that adoptive transfer of Tregs attenuates symptoms of immune-mediated conditions, such as asthma (86), graft versus host disease (GvHD) (87) and several autoimmune diseases including type 1 diabetes (T1D) (88), multiple sclerosis (MS) (89) and autoimmune hepatitis (AIH) (90). For example, a phase 1 clinical trial evaluated the polyclonal Treg therapy in paediatric T1D patients, with results indicating therapeutic effectiveness following the infusion of two doses of polyclonal Tregs (91). However, similar benefit was not observed in adult trials, where no significant efficacy was detected (88). This discrepancy may be due to differences in immune system maturity between children and adults, leading to varied responses to therapy.
Another possible explanation is that polyclonal Treg may function differently in inducing peripheral immune tolerance. One study suggested that polyclonal Treg might inhibit the migration of Teff cells to specific tissue sites, whereas antigen-specific iTreg can act directly at the target site, potentially providing more precise immune regulation (92). This distinction highlights the potential advantages of using antigen-specific Tregs, especially since autoimmune diseases are often diagnosed only after tissue damage has occurred. In such cases, Teff cells may already have infiltrated the target site, making site-specific intervention by engineered Tregs a more effective therapeutic approach.
CAR-T cell therapy
Chimeric antigen receptor (CAR)-T cell therapy is an FDA-approved treatment that specifically targets CD19, a B cell antigen, and has demonstrated significant efficacy in the management of B cell leukaemias and lymphomas (93, 94). However, the application of CAR-T cell therapy to solid tumours has met with limited success, primarily due to the unique challenges posed by the tumour microenvironment and tumour biology (95, 96).
The engineering of CAR-T cells involves the introduction of a synthetic receptor, the chimeric antigen receptor, which is composed of several key domains. The extracellular portion consists of a single-chain variable fragment (scFv), derived from the variable region of antibody heavy and light chains (refer to Figure 3), which confers antigen specificity (97, 98). A flexible spacer region links the scFv to a transmembrane domain, allowing for optimal antigen binding. The transmembrane domain anchors the receptor within the T cell membrane and connects to intracellular signalling domains. The first-generation CARs utilized only the CD3ζ signalling domain, resulting in weak T cell activation and limited therapeutic persistence. Subsequent generations incorporated additional costimulatory molecules, such as CD28 or 4-1BB (CD137), which markedly enhance CAR-T cell activation, persistence, and overall therapeutic efficacy.
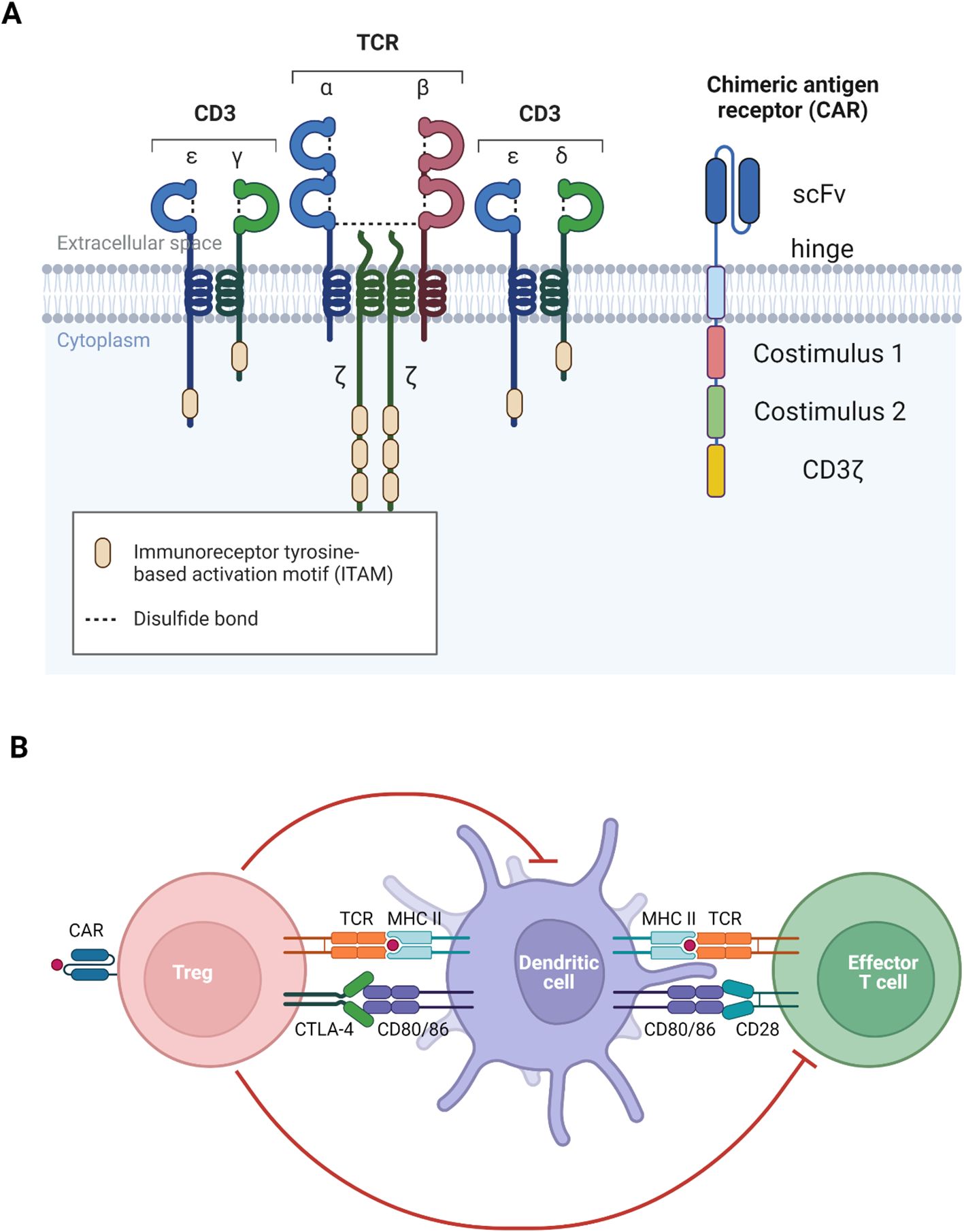
Figure 3. Visualization of TCR and CAR. (A) The main difference between TCR and CAR is that TCR recognizes MHC-bound peptide but CAR recognizes free antigen. (B) Both TCR and CAR function to activate the generated antigen-specific Treg to recognize self-antigen to induce immune tolerance. Diagrams created with Biorender.com.
Despite these advances, several barriers hinder the effectiveness of CAR-T cell therapy in solid tumours. A major challenge is the limited infiltration of CAR-T cells into the tumour site, which is influenced by the tumour’s structural complexity and the surrounding extracellular matrix. Additionally, the tumour microenvironment is often immunosuppressive, characterized by low oxygen levels, high acidity, and nutrient scarcity, all of which impair CAR-T cell function and survival. Tumour heterogeneity, whereby cancer cells exhibit diverse antigen expression profiles, further complicates the targeting of all malignant cells by CAR-T therapy.
The success of CAR-T cell therapy in haematologic malignancies has spurred interest in extending its application to other disease areas (98). For example, preclinical and early clinical studies are exploring the use of CAR-T cells for the treatment of autoimmune diseases, such as systemic lupus erythematosus (SLE) (NCT03030976) and carcinoembryonic antigen (CEA)-specific CAR-Treg to treat ulcerative colitis (UC) (99). Additionally, CAR-T cell approaches are being investigated to induce immune tolerance in patients with haemophilia A, with the aim of enabling sustained recombinant Factor VIII therapy without the risk of inhibitor formation. Other autoimmune diseases including myelin-oligodendrocyte glycoprotein (MOG)-specific CAR-Treg to treat MS (100) and Dsg-3 chimeric autoantibody receptor (CAAR)-T to treat pemphigus vulgaris (PV) (101). This review will only cover the aspects of autoimmune diseases for the sake of comparison between treatments in MPO-AAV. CAR-Treg is easier to produce and convenient to use as it is independent of MHC antigen recognition, hence it does not select patient population for a specific HLA to use (Table 2).
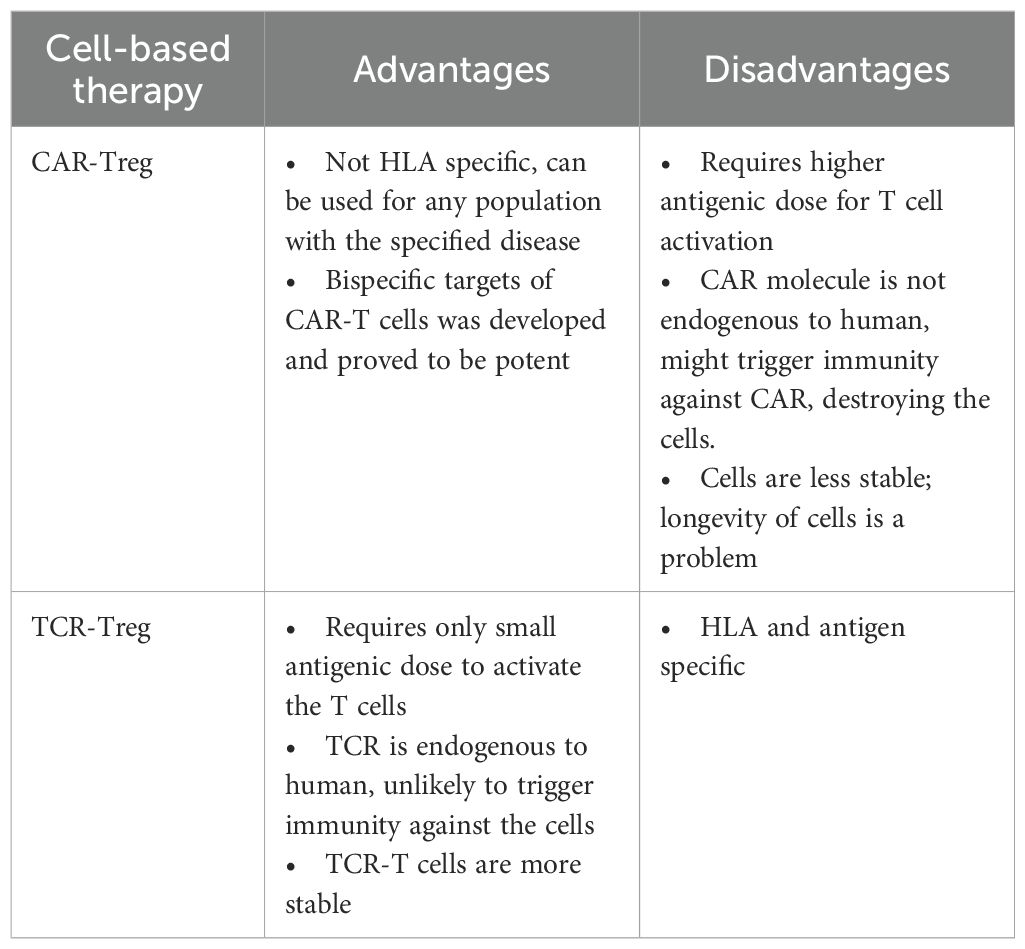
Table 2. Comparison between the cell-based therapies.
TCR-Treg therapy
Since the manifestation of MPO-AAV also involves the autoantigen recognition of immune cells presenting processed MPO on MHC II (refer to Figure 3), TCR-Treg is suited to use to target these autoreactive cells. The approach involves directing Treg to recognize MHC II-bound MPO, which can be achieved through gene transfer of T cell receptor (TCR) specific to the epitope of interest.
Although further studies are required to confirm the efficacy of TCR-Treg, several in vitro studies provide foundational insights. T1D, a highly prevalent autoimmune disease, serves as a notable reference point for such research. An in vitro study on T1D had employed TCR-Treg specific for islet antigen 2 (IA2) and insulin, with influenza-specific TCR-Tregs included as controls to assess specificty (102). The suppressive potency of these TCR-Tregs was evaluated by co-culturing them with either wild-type or antigen-presenting cells loaded with their respective peptides. Results demonstrated that the generated TCR-Tregs are highly immunosuppressive in the presence, but not absence of their cognate peptides (102). Conversely, patients with Haemophilia A require constant intake of recombinant FVIII (rFVIII) as they lack the critical coagulation factor FVIII, however, rFVIII is not native to patients’ immune systems, thus causing gradual destruction of rFVIII (103, 104). To address this, studies have introduced FVIII-specific TCR-Treg into patients, showing successful suppression of immune responses against FVIII-specific Teff cells (103). Additionally, another study had explored the use of myelin-basic protein (MBP)-specific TCR-Treg to investigate the efficacy in treating MS (105). Despite the need for further validation, these findings collectively highlight the therapeutic potential of antigen-specific TCR-Treg in a range of autoimmune conditions.
Previous studies have successfully determined the immunodominant epitope of MPO that triggers autoimmune activation, with disease severity and prevalence are HLA-linked. The importance of HLA-DRB1*04:05 and HLA-DQA1*03:02, DQB1*03:03 in Chinese patients whereas HLA-DRB1*09:01 in Japanese patients were well elucidated. This attracts molecular studies to be performed on these disease-linked HLAs, which could possibly open the doors to enhance the development of better cell-based therapy. HLA-DRB1*04:05 is the most severe related HLA class II molecule found in patients that usually progress into ESRF within six months even with existing treatment. Thus, initiating the alternative cell-based therapy with HLA-DRB1*04:05 would be a major step towards the development of next-generation personalized therapy (Table 2).
Challenges in TCR-Treg production
Cell-based therapy represents a promising frontier in individualized immunotherapy, yet the high production cost remains a significant barrier to widespread clinical adoption. The process is resource-intensive, requiring the collection of blood, isolation of Treg, transduction and expansion of these cells, and ultimately their infusion into patients. To address scalability and cost, one proposed solution is the use of platforms such as Lonza, which can be adopted by laboratories that mimic clean room environments, thereby commercializing production while maintaining the Good Manufacturing Practice (GMP) standards. A major safety concern in TCR-Treg therapy is the potential for the introduced transgenic TCR to mispair with the endogenous TCR, leading to the formation of hybrid receptors that may trigger harmful autoimmune responses. To mitigate this risk, clustered regularly interspaced short palindromic repeats (CRISPR)-based knockout of the endogenous TCR prior to gene transfer has been explored. Studies have shown that this approach enhances the expression of the transduced TCR, resulting in improved function of engineered T cells (106). By also knocking out endogenous HLA presenting on the Tregs, the product could be possibly made off-shelf, significantly improving the feasibility of the therapy. Although autologous T cell therapy is more favourable due to unlikely event of graft-versus-host disease (GvHD) in patients, it is less available to rural area and is often required to be operated at a more centred venue for product production, storage and delivery. This makes off-shelf product to be more convenient as it does not require patients’ cells and with proper management, they can be delivered to more rural areas for infusion. Knocking in the TCR of interest using the same method can therefore create super-Tregs. These super-Tregs can be further produced through induction from peripheral T cells to iTreg; and through CRISPR-based demethylation of the TSDR region of T cells (107, 108), can create a more stable Treg phenotype, overcoming the limit on the Tregs number since they occur in small amount naturally. The pipeline for Treg cell therapy production includes isolation, expansion, storage, transport, infusion into the patients. The toughest part is the scalability and cost as the naturally existing Tregs occur in small numbers in human body. But with the above-mentioned gene editing method, CD4+ T cells can be isolated in larger numbers and expanded. The study of Tregs stability is crucial as Tregs can still convert to Teffs under pro-inflammatory environment, hence, in vivo humanized model is required for not only to study the functionality of the engineered Tregs, but also to determine their longevity and stability.
There are several current Treg manufacturing protocols, and no method is proven to be superior to one another. A standardized protocol is yet to be developed as little is known about how the different manufacturing options can cause different patients’ outcome, though they are not supposed to differ significantly to each other. Briefly, the enriched Treg population can be expanded in presence of rapamycin to maintain Treg phenotype and prevent Teff expansion. Other methods including bead-based enrichment and flow cytometry-based selection to expand then isolate is considered as well. A recent GMP certified protocol approved by the Spanish authority can be considered as well (109), and using this as a foundation to further modify the product can help with the establishment of Treg product. As the study only used natural Treg as the source for amplification and subsequent product development, the iTreg product is yet to be investigated.
There are several challenges regarding cell-based therapies, especially the optimal treatment regimen for patients. Given the nature of complexity of autoimmune diseases, current landscape involves infusing the engineered Tregs into patients for induction of remission. However, the longevity of these infused cells remains unknown, as a recent CAR-T study in an autoimmune disease, idiopathic inflammatory myositis indicate patients may relapse within a year (110). It is still unknown whether the cell-based therapies can be given for maintenance therapy. Options for therapy include one-time induction until disease recurrence or utilizing a higher initial dose followed by lower, regular doses for maintenance. Patient outcomes are still measured using standards like the BVAS score, which provides clear criteria for diagnosis and disease activity. A key advance would be the elimination of steroids as part of therapy, greatly improving quality of life and disease manageability. Autoimmune diseases are challenging and long-term, therefore, robust and comprehensive studies including long-term clinical trials spanning 5–10 years are necessary to fully understand patient outcomes and refine future treatment strategies.
Future directions
MPO-AAV with HLA-DRB1*04:05 garners particular interest because, despite its rarity, it is associated with the poorest 5-year survival among MPO-AAV. Cell-based therapies using engineered Treg are attractive for MPO-AAV as they may circumvent the broad immunosuppression and associated side effects observed with conventional treatments such as CYC or RTX combined with glucocorticoids. Two main strategies are under investigation, which are CAR-Treg and TCR-Treg therapies. Of these, TCR-Treg which recognize antigen-MHC complexes, may offer superior efficacy in MPO-AAV due to their MHC-dependence in ensuring the specificity of the developed therapy to minimize possible side effects. Although clinical trials using CAR-Treg and TCR-Treg therapies in MPO-AAV have not yet commenced, both in vitro and in vivo studies in other autoimmune diseases support their feasibility and therapeutic potential. This is further underscored by promising results in related models, such as MOG-specific CAR-Treg and MBP-specific TCR-Treg for MS as well as the FVIII-specific CAR-Treg and FVIII-specific TCR-Treg to tolerate rFVIII in Haemophilia A patients. In summary, while TCR-Treg therapy holds great promise for the treatment in refractory autoimmune diseases, its clinical translation will require overcoming challenges related to manufacturing scalability, cost, and safety. Advances in gene editing technologies and optimized manufacturing protocols are critical next steps for bridging these therapies into routine clinical practice.
This also allows exploration of other similar diseases of vasculitis in the broader terms for the application of the Treg-based therapy. Since other vasculitis involving big and medium vessels like giant cell arthritis involves a more systemic disease rather than organ-specific, a potent alternate therapy to the corticosteroid’s regimen is required for the patients’ quality of lives and prognosis (111). The involvement of adaptive immune system points to the vital role of Tregs to be developed as a possible next-generation personalized therapy.
Literature searches and article selection
Clinicaltrials.gov (Pubmed) was used for clinical trial searches on current treatment in ANCA-AAV using the keyword ANCA associated vasculitis. The search was further filtered to include only interventional, completed and active, but not recruiting participant studies. 73 studies were filtered out and only studies covering MPA were selected. The Google search engine was used to select top 30 articles with keywords including pathogenesis of MPO-AAV, Treg function, therapy for MPO-AAV in separate occasions. Related references within the selected articles were further studied and included as part of review writing.
Author contributions
ET: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization. YT: Supervision, Writing – review & editing, Conceptualization, Data curation, Project administration. PG: Conceptualization, Supervision, Writing – review & editing, Data curation, Methodology. JO: Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Data curation, Methodology, Project administration, Resources.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jennette JC and Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. (2014) 10:463–73. doi: 10.1038/nrrheum.2014.103
2. Jennette JC, Falk RJ, and Gasim AH. Pathogenesis of ANCA vasculitis. Curr Opin Nephrol Hyperten. (2011) 20:263–70. doi: 10.1097/MNH.0b013e3283456731
3. Jennette JC, Xiao H, and Falk RJ. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. (2006) 17:1235–42. doi: 10.1681/ASN.2005101048
4. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
5. Futamata E, Masuda S, Nishibata Y, Tanaka S, Tomaru U, and Ishizu A. Vanishing immunoglobulins: the formation of pauci-immune lesions in myeloperoxidase-antineutrophil cytoplasmic antibody-associated vasculitis. Nephron. (2018) 138:328–30. doi: 10.1159/000485902
6. Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, et al. ANCA-associated vasculitis. Nat Rev Dis Primers. (2020) 6:71. doi: 10.1038/s41572-020-0204-y
7. Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, Poulton CE, et al. Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. J Clin Invest. (2013) 123:1773–83. doi: 10.1172/JCI65292
8. Ooi JD, Chang J, Hickey MJ, Borza DB, Fugger L, Holdsworth SR, et al. The immunodominant myeloperoxidase T-cell epitope induces local cell-mediated injury in antimyeloperoxidase glomerulonephritis. Proc Natl Acad Sci United States America. (2012) 109:E2615–2624. doi: 10.1073/pnas.1210147109
9. Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DRW, et al. Genetically distinct subsets within ANCA-Associated vasculitis. New Engl J Med. (2012) 367:214–23. doi: 10.1056/NEJMoa1108735
10. Hiwa R, Ohmura K, Arase N, Jin H, Hirayasu K, Kohyama M, et al. Myeloperoxidase/HLA class II complexes recognized by autoantibodies in microscopic polyangiitis. Arthritis Rheumatol. (2017) 69:2069–80. doi: 10.1002/art.40170
11. Chang DY, Luo H, Zhou XJ, Chen M, and Zhao MH. Association of HLA genes with clinical outcomes of ANCA-associated vasculitis. Clin J Am Soc Nephrol. (2012) 7:1293–9. doi: 10.2215/CJN.13071211
12. Wang HY, Cui Z, Pei ZY, Fang SB, Chen SF, Zhu L, et al. Risk HLA class II alleles and amino acid residues in myeloperoxidase-ANCA-associated vasculitis. Kidney Int. (2019) 96:1010–9. doi: 10.1016/j.kint.2019.06.015
13. Pendergraft WF 3rd, Preston GA, Shah RR, Tropsha A, Carter CW Jr, Jennette JC, et al. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. (2004) 10:72–9. doi: 10.1038/nm968
14. Ooi JD, Jiang JH, Eggenhuizen PJ, Chua LL, van Timmeren M, Loh KL, et al. A plasmid-encoded peptide from Staphylococcus aureus induces anti-myeloperoxidase nephritogenic autoimmunity. Nat Commun. (2019) 10:3392. doi: 10.1038/s41467-019-11255-0
15. Hogan SL, Cooper GS, Savitz DA, Nylander-French LA, Parks CG, Chin H, et al. Association of silica exposure with anti-neutrophil cytoplasmic autoantibody small-vessel vasculitis: a population-based, case-control study. Clin J Am Soc Nephrol. (2007) 2:290–9. doi: 10.2215/CJN.03501006
16. Bruner BF, Vista ES, Wynn DM, and James JA. Epitope specificity of myeloperoxidase antibodies: identification of candidate human immunodominant epitopes. Clin Exp Immunol. (2011) 164:330–6. doi: 10.1111/j.1365-2249.2011.04372.x
17. Free ME, Stember KG, Hess JJ, McInnis EA, Lardinois O, Hogan SL, et al. Restricted myeloperoxidase epitopes drive the adaptive immune response in MPO-ANCA vasculitis. J Autoimmun. (2020) 106:102306. doi: 10.1016/j.jaut.2019.102306
18. Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM Int J Med. (1994) 87:671–8. doi: 10.1093/qjmed/87.11.671
19. Mukhtyar C, Lee R, Brown D, Carruthers D, Dasgupta B, Dubey S, et al. Modification and validation of the birmingham vasculitis activity score (version 3). Ann Rheum Dis. (2009) 68:1827–32. doi: 10.1136/ard.2008.101279
20. Suppiah R, Mukhtyar C, Flossmann O, Alberici F, Baslund B, Batra R, et al. A cross-sectional study of the Birmingham Vasculitis Activity Score version 3 in systemic vasculitis. Rheumatology. (2011) 50:899–905. doi: 10.1093/rheumatology/keq400
22. Nakazawa D, Masuda S, Tomaru U, and Ishizu A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol. (2019) 15:91–101. doi: 10.1038/s41584-018-0145-y
23. Gan PY, Fujita T, Ooi JD, Alikhan MA, Dick J, Shim R, et al. Pathogenic role for γδ T Cells in autoimmune anti-myeloperoxidase glomerulonephritis. J Immunol. (2017) 199:3042–50. doi: 10.4049/jimmunol.1602025
24. Yuan J, Gou SJ, Huang J, Hao J, Chen M, and Zhao MH. C5a and its receptors in human anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Arthritis Res Ther. (2012) 14:R140. doi: 10.1186/ar3873
25. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, and Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. (2009) 20:289–98. doi: 10.1681/ASN.2008050497
26. Dick J, Gan PY, Ford SL, Odobasic D, Alikhan MA, Loosen SH, et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental anti-myeloperoxidase glomerulonephritis. Kidney Int. (2018) 93:615–25. doi: 10.1016/j.kint.2017.09.018
27. Ooi JD, Gan PY, Chen T, Eggenhuizen PJ, Chang J, Alikhan MA, et al. FcγRIIB regulates T-cell autoreactivity, ANCA production, and neutrophil activation to suppress anti-myeloperoxidase glomerulonephritis. Kidney Int. (2014) 86:1140–9. doi: 10.1038/ki.2014.189
28. Scapini P, Bazzoni F, and Cassatella MA. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett. (2008) 116:1–6. doi: 10.1016/j.imlet.2007.11.009
29. Bunch DO, McGregor JG, Khandoobhai NB, Aybar LT, Burkart ME, Hu Y, et al. Decreased CD5(+) B cells in active ANCA vasculitis and relapse after rituximab. Clin J Am Soc Nephrol. (2013) 8:382–91. doi: 10.2215/CJN.03950412
30. Dumoitier N, Terrier B, London J, Lofek S, and Mouthon L. Implication of B lymphocytes in the pathogenesis of ANCA-associated vasculitides. Autoimmun Rev. (2015) 14:996–1004. doi: 10.1016/j.autrev.2015.06.008
31. Aybar LT, McGregor JG, Hogan SL, Hu Y, Mendoza CE, Brant EJ, et al. Reduced CD5(+) CD24(hi) CD38(hi) and interleukin-10(+) regulatory B cells in active anti-neutrophil cytoplasmic autoantibody-associated vasculitis permit increased circulating autoantibodies. Clin Exp Immunol. (2015) 180:178–88. doi: 10.1111/cei.12483
32. von Borstel A, Sanders JS, Rutgers A, Stegeman CA, Heeringa P, and Abdulahad WH. Cellular immune regulation in the pathogenesis of ANCA-associated vasculitides. Autoimmun Rev. (2018) 17:413–21. doi: 10.1016/j.autrev.2017.12.002
33. Gan PY, Holdsworth SR, Kitching AR, and Ooi JD. Myeloperoxidase (MPO)-specific CD4+ T cells contribute to MPO-antineutrophil cytoplasmic antibody (ANCA) associated glomerulonephritis. Cell Immunol. (2013) 282:21–7. doi: 10.1016/j.cellimm.2013.04.007
34. Kitching AR, Turner AL, Wilson GR, Semple T, Odobasic D, Timoshanko JR, et al. IL-12p40 and IL-18 in crescentic glomerulonephritis: IL-12p40 is the key Th1-defining cytokine chain, whereas IL-18 promotes local inflammation and leukocyte recruitment. J Am Soc Nephrol. (2005) 16:2023–33. doi: 10.1681/ASN.2004121075
35. Nicholson LB and Kuchroo VK. Manipulation of the Th1/Th2 balance in autoimmune disease. Curr Opin Immunol. (1996) 8:837–42. doi: 10.1016/s0952-7915(96)80013-6
36. Steinmetz OM, Summers SA, Gan PY, Semple T, Holdsworth SR, and Kitching AR. The Th17-defining transcription factor RORgammat promotes glomerulonephritis. J Am Soc Nephrol. (2011) 22:472–83. doi: 10.1681/ASN.2010040435
37. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
38. Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele KM, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. (2010) 25:2209–17. doi: 10.1093/ndt/gfp783
39. Gan PY, Steinmetz OM, Tan DS, O'Sullivan KM, Ooi JD, Iwakura Y, et al. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol. (2010) 21:925–31. doi: 10.1681/ASN.2009070763
40. Chang J, Eggenhuizen P, O'Sullivan KM, Alikhan MA, Holdsworth SR, Ooi JD, et al. CD8+ T Cells effect glomerular injury in experimental anti-myeloperoxidase GN. J Am Soc Nephrol. (2017) 28:47–55. doi: 10.1681/ASN.2015121356
41. McKinney EF, Lyons PA, Carr EJ, Hollis JL, Jayne DR, Willcocks LC, et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. (2010) 16:586–91. doi: 10.1038/nm.2130
42. Summers SA, Steinmetz OM, Gan PY, Ooi JD, Odobasic D, Kitching AR, et al. Toll-like receptor 2 induces Th17 myeloperoxidase autoimmunity while Toll-like receptor 9 drives Th1 autoimmunity in murine vasculitis. Arthritis Rheumatol. (2011) 63:1124–35. doi: 10.1002/art.30208
43. Summers SA, van der Veen BS, O'Sullivan KM, Gan P-Y, Ooi JD, Heeringa P, et al Intrinsic renal cell and leukocyte-derived TLR4 aggravate experimental anti-MPO glomerulonephritis. Kidney Int. (2010) 78:1263–74. doi: 10.1038/ki.2010.327
44. Huugen D, Xiao H, van Esch A, Falk RJ, Peutz-Kootstra CJ, Buurman WA, et al. Aggravation of anti-myeloperoxidase antibody-induced glomerulonephritis by bacterial lipopolysaccharide: role of tumour necrosis factor-alpha. Am J Pathol. (2005) 167:47–58. doi: 10.1016/s0002-9440(10)62952-5
45. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, and Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64. doi: 10.4049/jimmunol.155.3.1151
46. Sakaguchi S, Yamaguchi T, Nomura T, and Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
47. Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, et al. ANCA-associated vasculitis patients have defective Treg function exacerbated by presence of a suppression-resistant effector population. Arthritis Rheum. (2013) 65:1922–33. doi: 10.1002/art.37959
48. Wang Y, Zhang S, Zhang N, Feng M, Liang Z, Zhao X, et al, et al. Reduced activated regulatory T cells and imbalance of Th17/activated Treg cells marks renal involvement in ANCA-associated vasculitis. Mol Immunol. (2020) 118:19–29. doi: 10.1016/j.molimm.2019.11.010
49. Sato Y, Liu J, Lee E, Perriman R, Roncarolo MG, and Bacchetta R. Co-expression of FOXP3FL and FOXP3Delta2 isoforms is required for optimal treg-like cell phenotypes and suppressive function. Front Immunol. (2021) 12:752394. doi: 10.3389/fimmu.2021.752394
50. Tan DS, Gan PS, O'Sullivan KM, Hammett MV, Summers SA, Ooi JD, et al. Thymic deletion and regulatory T cells prevent antimyeloperoxidase GN. J Am Soc Nephrol. (2013) 24:573–85. doi: 10.1681/ASN.2012090898
51. Kanamori M, Nakatsukasa H, Okada M, Lu Q, and Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol. (2016) 37:803–11. doi: 10.1016/j.it.2016.08.012
52. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. (2006) 203:1701–11. doi: 10.1084/jem.20060772
53. Vignali DA, Collison LW, and Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
54. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
55. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalysed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
56. Zarek PE, Huang C-T, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signalling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. (2008) 111:251–9. doi: 10.1182/blood-2007-03-081646
57. Odobasic D, Oudin V, Ito K, Gan P-Y, Kitching AR, and Holdsworth S. Tolerogenic dendritic cells attenuate experimental autoimmune antimyeloperoxidase glomerulonephritis. J Am Soc Nephrol. (2019) 30:2140–57. doi: 10.1681/ASN.2019030236
58. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. (2002) 110:955–63. doi: 10.1172/JCI15918
59. Salama AD and Little MA. Animal models of antineutrophil cytoplasm antibody-associated vasculitis. Curr Opin Rheumatol. (2012) 24:1–7. doi: 10.1097/BOR.0b013e32834d2d52
60. Schreiber A, Xiao H, Falk RJ, and Jennette JC. Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol. (2006) 17:3355–64. doi: 10.1681/ASN.2006070718
61. Ruth AJ, Kitching AR, Kwan RYQ, Odobasic D, Ooi JDK, and Timoshanko JR. Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play nonredundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J Am Soc Nephrol. (2006) 17:1940–9. doi: 10.1681/ASN.2006020108
62. Chen J, Liao S, Xiao Z, Pan Q, Wang X, Shen K, et al. The development and improvement of immunodeficient mice and humanized immune system mouse models. Front Immunol. (2022) 13:1007579. doi: 10.3389/fimmu.2022.1007579
63. Park G, Rim YA, Sohn Y, Nam Y, and Ju JH. Replacing animal testing with stem cell-organoids: advantages and limitations. Stem Cell Rev Rep. (2024) 20:1375–86. doi: 10.1007/s12015-024-10723-5
64. Liu Q, Yue L, Deng J, Tan Y, and Wu C. Progress and breakthroughs in human kidney organoid research. Biochem Biophys Rep. (2024) 39:101736. doi: 10.1016/j.bbrep.2024.101736
65. Byng-Maddick R and Ehrenstein MR. The impact of biological therapy on regulatory T cells in rheumatoid arthritis. Rheumatology. (2015) 54:768–75. doi: 10.1093/rheumatology/keu487
66. Lally L and Spiera R. Current therapies for ANCA-associated vasculitis. Annu Rev Med. (2015) 66:227–40. doi: 10.1146/annurev-med-011514-023051
67. de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. (2009) 150:670–80. doi: 10.7326/0003-4819-150-10-200905190-00004
68. Harper L, Morgan MD, Walsh M, Hoglund P, Westman K, Flossmann O, et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis. (2012) 71:955–60. doi: 10.1136/annrheumdis-2011-200477
69. Gan PY, Dick J, O'Sullivan KM, Oudin V, Cao Le A, Koo Yuk Cheong D, et al. Anti-CD20 mAb-induced B cell apoptosis generates T cell regulation of experimental myeloperoxidase ANCA-associated vasculitis. J Am Soc Nephrol. (2021) 32:1071–83. doi: 10.1681/ASN.2020060834
70. De Groot K, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. (2005) 52:2461–9. doi: 10.1002/art.21142
71. Faurschou M, Westman K, Rasmussen N, de Groot K, Flossmann O, Hoglund P, et al. Brief report: long-term outcome of a randomized clinical trial comparing methotrexate to cyclophosphamide for remission induction in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. (2012) 64:3472–7. doi: 10.1002/art.34547
72. Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. (2008) 359:2790–803. doi: 10.1056/NEJMoa0802311
73. Bremer JP, Ullrich S, Laudien M, Gross WL, and Lamprecht P. Methotrexate plus leflunomide for the treatment of relapsing Wegener’s granulomatosis. A retrospective uncontrolled study. Clin Exp Rheumatol. (2010) 28:67–71. doi: 10.1002/art.21142
74. Metzler C, Miehle N, Manger K, Iking-Konert C, de Groot K, Hellmich B, et al. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener’s granulomatosis. Rheumatol (Oxford). (2007) 46:1087–91. doi: 10.1093/rheumatology/kem029
75. Hiemstra TF, Walsh M, Mahr A, Savage CO, de Groot K, Harper L, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA. (2010) 304:2381–8. doi: 10.1001/jama.2010.1658
76. Silva F, Specks U, Kalra S, Hogan MC, Leung N, Sethi S, et al. Mycophenolate mofetil for induction and maintenance of remission in microscopic polyangiitis with mild to moderate renal involvement–a prospective, open-label pilot trial. Clin J Am Soc Nephrol. (2010) 5:445–53. doi: 10.2215/CJN.06010809
77. Wegener’s Granulomatosis Etanercept Trial Research G. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. (2005) 352:351–61. doi: 10.1056/NEJMoa041884
78. Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. (2007) 18:2180–8. doi: 10.1681/ASN.2007010090
79. Walsh M, Catapano F, Szpirt W, Peh CA, Thorlund K, Bruchfeld A, et al. Plasma exchange for renal vasculitis and idiopathic rapidly progressive glomerulonephritis: a meta-analysis. Am J Kidney Dis. (2011) 57:566–74. doi: 10.1053/j.ajkd.2010.10.049
80. Walsh M, Merkel PA, Peh CA, Szpirt WM, Puechal X, Fujimoto S, et al. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med. (2020) 382:622–31. doi: 10.1056/NEJMoa1803537
81. Jayne DRW, Merkel PA, Schall TJ, Bekker P, and Group AS. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. (2021) 384:599–609. doi: 10.1056/NEJMoa2023386
82. Merkel PA, Jayne DR, Wang C, Hillson J, and Bekker P. Evaluation of the safety and efficacy of avacopan, a C5a receptor inhibitor, in patients with antineutrophil cytoplasmic antibody-associated vasculitis treated concomitantly with rituximab or cyclophosphamide/azathioprine: protocol for a randomized, double-blind, active-controlled, phase 3 trial. JMIR Res Protoc. (2020) 9:e16664. doi: 10.2196/16664
83. Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. (2017) 28:2756–67. doi: 10.1681/ASN.2016111179
84. Wu R, Su R, Ding T, Xue H, An J, Jiang L, et al. Low-dose IL-2 restores Treg-mediated immune tolerance in patients with ANCA-associated vasculitis. Ann Rheum Dis 79. (2020) 384. doi: 10.1136/annrheumdis-2020-eular.4346
85. Engesser J, Khatri R, Schaub DP, Zhao Y, Paust HJ, Sultana Z, et al. Immune profiling-based targeting of pathogenic T cells with ustekinumab in ANCA-associated glomerulonephritis. Nat Commun. (2024) 15:8220. doi: 10.1038/s41467-024-52525-w
86. Xu W, Lan Q, Chen M, Chen H, Zhu N, Zhou X, et al. Adoptive transfer of induced-Treg cells effectively attenuates murine airway allergic inflammation. PloS One. (2012) 7:e40314. doi: 10.1371/journal.pone.0040314
87. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal Regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant. (2017) 17:2945–54. doi: 10.1111/ajt.14415
88. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Trans Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
89. Duffy SS, Keating BA, and Moalem-Taylor G. Adoptive transfer of regulatory T Cells as a promising immunotherapy for the treatment of multiple sclerosis. Front Neurosci. (2019) 13:1107. doi: 10.3389/fnins.2019.01107
90. Lapierre P, Beland K, Yang R, and Alvarez F. Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology. (2013) 57:217–27. doi: 10.1002/hep.26023
91. Marek-Trzonkowska N, Myśliwiec M, Iwaszkiewicz-Grześ D, Gliwiński M, Derkowska I, Żalińska M, et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Transl Med. (2016) 14:332. doi: 10.1186/s12967-016-1090-7
92. Shevach EM and Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. (2014) 259:88–102. doi: 10.1111/imr.12160
93. June CH and Sadelain M. Chimeric antigen receptor therapy. New Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
94. Park JH, Geyer MB, and Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic Malignancies: interpreting clinical outcomes to date. Blood. (2016) 127:3312–20. doi: 10.1182/blood-2016-02-629063
95. Zhang H, Ye ZL, Yuan ZG, Luo ZQ, Jin HJ, and Qian QJ. New strategies for the treatment of solid tumours with CAR-T cells. Int J Biol Sci. (2016) 12:718–29. doi: 10.7150/ijbs.14405
96. Beavis PA, et al. Reprogramming the tumour microenvironment to enhance adoptive cellular therapy. Semin Immunol. (2016) 28:64–72. doi: 10.1016/j.smim.2015.11.003
97. Sadeqi Nezhad M, Seifalian A, Bagheri N, Yaghoubi S, Karimi MH, and Adbollahpour-Alitappeh M. Chimeric antigen receptor based therapy as a potential approach in autoimmune diseases: how close are we to the treatment? Front Immunol. (2020) 11:603237. doi: 10.3389/fimmu.2020.603237
98. Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, et al. Chimeric antigen receptor (CAR) Treg: a promising approach to inducing immunological tolerance. Front Immunol. (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
99. Blat D, Zigmond E, Alteber Z, Waks T, and Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T Cells. Mol Ther. (2014) 22:1018–28. doi: 10.1038/mt.2014.41
100. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflamm. (2012) 9:1–12. doi: 10.1186/1742-2094-9-112
101. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
102. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun. (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
103. Kim YC, Zhang A-H, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. (2015) 125:1107–15. doi: 10.1182/blood-2014-04-566786
104. Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW, et al. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. (2017) 129:238–45. doi: 10.1182/blood-2016-07-727834
105. Kim YC, Zhang A-H, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun. (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
106. Legut M, Dolton G, Mian AA, Ottmann OG, and Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. (2018) 131:311–22. doi: 10.1182/blood-2017-05-787598
107. Amini L, Greig J, Schmueck-Henneresse M, Volk HD, Bezie S, Reinke P, et al. Super-treg: toward a new era of adoptive treg therapy enabled by genetic modifications. Front Immunol. (2020) 11:611638. doi: 10.3389/fimmu.2020.611638
108. Stoops J, Morton T, Powell J, Pace AL, and Bluestone JA. Treg cell therapy manufacturability: current state of the art, challenges and new opportunities. Front Immunol. (2025) 16:1604483. doi: 10.3389/fimmu.2025.1604483
109. Bernaldo-de-Quirós E, Cózar B, López-Esteban R, Clemente M, Gil-Jaurena JM, Pardo C, et al. A novel GMP protocol to produce high-quality treg cells from the paediatric thymic tissue to be employed as cellular therapy. Front Immunol. (2022) 13:893576. doi: 10.3389/fimmu.2022.893576
110. Muller F, Wirsching A, Hagen M, Völkl S, Tur C, Raimondo MG, et al. BCMA CAR T cells in a patient with relapsing idiopathic inflammatory myositis after initial and repeat therapy with CD19 CAR T cells. Nat Med. (2025) 31:1793–7. doi: 10.1038/s41591-025-03718-3
Keywords: MPO-AAV, HLA, preclinical model, TCR-T and CAR-T therapy, ANCA
Citation: Tay ESV, Ting YT, Gan P-Y and Ooi JD (2025) Regulatory T cell therapy for myeloperoxidase-specific anti-neutrophil cytoplasmic antibody associated vasculitis. Front. Immunol. 16:1675251. doi: 10.3389/fimmu.2025.1675251
Received: 29 July 2025; Accepted: 20 October 2025;
Published: 03 November 2025.
Edited by:
Maria Giovanna Danieli, Università Politecnica delle Marche, ItalyReviewed by:
Davide Firinu, University of Cagliari, ItalyAntonio Giovanni Solimando, University of Bari Aldo Moro, Italy
Mario Andrea Piga, Università Politecnica delle Marche, Italy
Copyright © 2025 Tay, Ting, Gan and Ooi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elean S. V. Tay, ZWxlYW4udGF5QG1vbmFzaC5lZHU=