Abstract
Introduction:
Ducks rank among the most important sources of animal protein globally, yet hepatic and splenic hemorrhage and necrosis in Muscovy ducks present a critical challenge to the poultry industry. The causes behind such diseases are often multifaceted, involving both established and newly emerging pathogens.
Methods:
In this study, we leveraged metatranscriptomic sequencing to profile the intestinal viral communities of healthy and diseased Muscovy ducks from a Guangdong Province farm that experienced a hepatic and splenic hemorrhage in June 2024.
Results:
Our findings revealed marked differences in viral community profiles between the two groups, with the diseased cohort exhibiting higher α-diversity. Taxonomic analyses across multiple levels uncovered significant variations in viral composition, including shifts in phylums like Uroviricota and families such as Demerecviridae. At the genus and species levels, several bacteriophages and eukaryotic viruses displayed differential abundance. Notably, Avian orthoreovirus was detected exclusively in diseased ducks, with a specific novel duck reovirus (NDRV) validated via RT-qPCR as a potential contributor to hepatic and splenic pathogenesis. In contrast, known pathogens such as Duck hepatitis A virus (DHAV) and Fowl adenovirus serotype 4 (FAdV-4) were not detected.
Discussion:
This study constitutes the first comprehensive analysis of the Muscovy duck gut virome, highlighting NDRV as a potential causative agent and emphasizing the utility of metatranscriptomics in pathogen discovery.
1 Introduction
Ducks represent an important source of animal protein and are widely consumed around the world, especially in Asia (1). However, hemorrhage and necrosis of the liver and spleen, particularly in Muscovy ducks (Cairina moschata), pose a serious threat to the poultry industry (2). These pathological manifestations could be caused by a variety of pathogens, and novel causative agents keep emerging over time. Previous studies have identified Duck Hepatitis A Virus (DHAV) as one of the primary etiological agents responsible for hepatic hemorrhage in ducks, leading to liver necrosis and hemorrhagic lesions that severely impact animal health and farming profitability (3, 4). In addition to DHAV, fowl adenovirus serotype 4 (FAdV-4) has also been recognized as a significant pathogen causing hepatic hemorrhage in ducks (5, 6). More recently, novel duck reovirus (NDRV) has been identified as another causative agent. NDRV infection has also been associated with severe splenic necrosis, which might result in immunosuppression and secondary infections (7, 8). Moreover, NDRV can be transmitted vertically, further exacerbating its potential for widespread dissemination within duck populations (9). Collectively, these findings underscore the complex etiology of hepatic and splenic hemorrhagic disease in ducks, which often involves co-infection or emergence of novel pathogens. Such infections not only compromise duck health but also lead to substantial economic losses in poultry production. Therefore, early detection and accurate diagnosis of both known and emerging pathogens are critical for the development of effective prevention and control strategies.
Viral metagenomics is an emerging and powerful tool that has grown increasingly valuable for the identification of novel pathogens (10). By enabling the comprehensive characterization of complex microbial communities without prior knowledge of the target organisms, metaranscriptomic or metagenomic approaches and newly developed deep-learning based algorithms allow the unbiased detection of both known and previously unrecognized viruses (11–14). This technology has already demonstrated great promise in the fields of public health surveillance and infectious disease diagnostics (15). In contrast, the application of viral metagenomics in poultry remains limited. Nevertheless, several studies have shown its utility in avian virus monitoring. For instance, metagenomic analysis of samples from six poultry farms, including chicken, duck, and goose farms, in eastern China enabled the simultaneous detection of multiple avian viruses, including avian influenza virus and Newcastle disease virus (16). Similarly, metatranscriptome sequencing has been employed to profile the diversity and abundance of avian viruses in live poultry markets in Guangdong Province, further demonstrating its diagnostic and epidemiological potential (17). In addition to known pathogens, this approach has proven invaluable in the discovery of novel viruses from environmental samples that would otherwise evade detection by conventional techniques (18). It has also been successfully applied to wildlife studies for virus identification and characterization (19). Despite the growing interest and impact in viral metagenomics, its application in domestic poultry—particularly in ducks—remains underexplored.
In June 2024, a Muscovy duck farm in Guangdong Province reported severe cases of hepatic and splenic hemorrhage and necrosis, the etiology of which remained unknown. To date, no studies have characterized the intestinal virome of Muscovy ducks. To address this gap and investigate potential causative pathogens, we applied metatranscriptomic sequencing to characterize and compare the intestinal viral communities of diseased and healthy ducks. Our comparative analysis found that Avian orthoreovirus was significantly enriched in the intestines of diseased individuals, suggesting its potential role as a causative agent. This finding was further supported by RT-qPCR validation, which confirmed the presence of novel duck reovirus (NDRV) in affected ducks. This study represents the first comprehensive analysis of the intestinal virome in Muscovy ducks and provides critical foundational data for future research into duck gut health. Furthermore, our identification of NDRV as a potential etiological agent demonstrates the practical utility of metatranscriptomics in pathogen discovery, offering a useful model for rapid diagnosis and detection of unknown pathogens in poultry farming.
2 Materials and methods
2.1 Ethics statement
The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC), Sun Yat-Sen University, Guangzhou, China (Approval ID: SYXK-2023-0112). All procedures involving animals were conducted in line with institutional guidelines for animal welfare and ethical research practices.
2.2 Sample collection
In June 2024, a severe outbreak characterized by marked hepatic and splenic hemorrhage occurred in 1–2-week-old Muscovy ducks (Cairina moschata) at a commercial farm in Guangdong Province, China. Fresh cecal contents were collected from diseased ducks immediately after necropsy. For sample pooling, contents from every three diseased ducks were combined into one biological replicate, yielding 10 pooled samples for the diseased group. Age-matched healthy Muscovy ducks from the same farm served as controls, with cecal contents collected using the same pooling strategy. All samples were immediately flash-frozen in liquid nitrogen immediately after collection, transported to the laboratory under cryogenic conditions, and stored at -80 °C until subsequent processing.
2.3 Total RNA extraction for sequencing
Total RNA was extracted from cecal content samples using the RNAPowerSoil Total RNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA; Cat. No. 12866-25) according to the manufacturer’s protocol. RNA concentration and purity were then assessed using a Qubit Fluorometer and a Nanodrop 2000 UV spectrophotometer. Only samples meeting the following criteria were used for library construction: minimum concentration ≥ 80 ng/μL, A260/A280 ratio ≥ 1.8, and total RNA amount ≥ 2 μg per sample.
2.4 Viral metatranscriptomic library construction
Ribosomal RNA (rRNA) was depleted by hybridizing rRNA-specific biotinylated probes to total RNA, followed by capturing of rRNA-probe complexes using streptavidin-coated magnetic beads (Illumina, San Diego, CA, USA). The remaining mRNA was purified via ethanol precipitation. cDNA library construction was carried out using the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina) according to the manufacturer’s instructions, including reverse transcription, adaptor ligation, and PCR amplification. Library integrity was validated using an Agilent Bioanalyzer 2100 with the Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA), with qualification requiring a single distinct peak without adapter dimers. Library quantification was performed using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific) on a Promega QuantiFluor fluorometer, with only libraries achieving a concentration ≥ 2 nM deemed qualified for sequencing.
2.5 Metatranscriptomic sequencing
Individual qualified libraries, each bearing unique indices, were diluted to 2 nM and pooled in equimolar ratios based on target sequencing depth. Pooled libraries were denatured into single-stranded DNA using 0.2 N NaOH and sequenced on an Illumina NovaSeq 6000 platform, generating 2 × 150 bp paired-end reads.
2.6 Metatranscriptomic data analysis
Raw sequencing data were processed using the Hecatomb pipeline (20), a specialized, high-performance tool optimized for virome analysis of metatranscriptomic datasets. The workflow included the following steps: (1) removal of adapter sequences, primers, and low-quality reads using Trimmomatic (21); (2) depletion of host genomic and transcriptomic sequences via alignment to the Muscovy duck reference genome (GenBank: GCA_048319975.1) utilizing Bowtie2 (22); (3) reduction of sequence redundancy via clustering with CD-HIT-EST (23); (4) generation of sequence count tables. Taxonomic classification was performed using an iterative search strategy implemented in MMSeqs2 (24). Initially, processed sequences were aligned to a virus-specific protein database (RefSeq Viral Proteins) using a translated nucleotide-to-protein (tblastx) approach. Sequences assigned to a viral taxonomy were subjected to secondary validation against a comprehensive reference database containing sequences from bacteria, plants, vertebrates, fungi, and other domains to eliminate false positives. This two-step strategy balanced computational efficiency with classification accuracy, avoiding the prohibitive cost of direct searches against large general databases.
2.7 Real-time quantitative PCR validation
For RT-qPCR validation, 2 g aliquots of cecal samples were homogenized in 500 μL ice-cold phosphate-buffered saline (PBS) by vortexing vigorously. After centrifugation at 12,000 × g for 10 min at 4°C, the supernatant was collected for viral nucleic acid extraction. Total viral DNA/RNA was isolated using the Vazyme FastPure Viral DNA/RNA Mini Kit V2 (Vazyme Biotech, Nanjing, China; Cat. No. RC313) following the manufacturer’s protocol, with all procedures performed on ice to maintain nucleic acid integrity. RNA concentration and purity were determined spectrophotometrically (NanoDrop 2000; Thermo Fisher Scientific, Wilmington, DE, USA), with samples exhibiting A260/A280 ratios between 1.8 and 2.0 considered suitable for downstream analysis. Reverse transcription was performed using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions, incorporating a genomic DNA elimination step prior to cDNA synthesis. Quantitative PCR amplification was conducted using SYBR® Premix Ex Taq II (Tli RNaseH Plus; Takara Bio) on a LightCycler 480 II Real-Time PCR System (Roche Diagnostics, Basel, Switzerland). The thermal cycling protocol consisted of an initial incubation at 50°C for 2 min, followed by denaturation at 95°C for 10 min, and 40 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 30 s. All reactions were performed in triplicate, with appropriate negative controls included in each run. Primer sequences targeting specific viral genes are provided in Supplementary Table S1 (25–27).
2.8 Statistical analysis and visualization
Partial least squares discriminant analysis (PLS-DA) was performed using the package mixOmics (28) to visualize differences in viral community composition between groups. Heatmaps of viral abundance were generated using the ComplexHeatmap (29) package, and scatter plots were constructed with ggplot2. Statistical comparisons of viral abundance between healthy and diseased groups were conducted using Student’s t-test, with p-values < 0.05 considered statistically significant. All analyses were performed in R version 4.2.1.
3 Results
3.1 Increased viral diversity characterizes the gut virome of diseased ducklings
In June 2024, a sudden surge in mortality occurred at a Muscovy duck farm in Guangdong Province, triggering an urgent pathological investigation. Necropsies revealed extensive punctate hepatic hemorrhages and diffuse splenic bleeding (Figure 1A), raising suspicion of a viral etiology. However, the exact causative virus remained undetermined. To identify potential pathogens, we extracted cecal contents from both symptomatic and asymptomatic ducks and conducted metatranscriptomic sequencing. Following the removal of host and bacterial sequences, the remaining reads were annotated using viral databases (Figure 1B). Partial least squares discriminant analysis (PLS-DA) revealed clear clustering that separated diseased from healthy samples, reflecting strong within-group similarity and substantial inter-group differences in viral composition (Figure 1C). Moreover, alpha-diversity indices were significantly elevated in diseased ducks, suggesting greater viral richness and complexity (Figures 1D–G). These findings provide a comparative dataset of the gut virome in healthy versus diseased Muscovy ducks, forming a basis for pathogen discovery.
Figure 1

Overview of clinical sample collection and intestinal metatranscriptomic sequencing. (A) Representative gross pathological features of both healthy and diseased Muscovy ducks; (B) Schematic workflow illustrating the steps involved in intestinal metatranscriptomic sequencing; (C) Partial least squares discriminant analysis (PLS-DA) of virome profiles; (D–G) Alpha diversity indices of the duck gut virome, including Shannon (D), Simpson (E), ACE (F), and Chao1 (G).
3.2 Distinct viral taxonomic patterns emerge in diseased ducklings across all levels of classification
To further explore virome alterations, we annotated viral taxa from phylum to family, covering both bacteriophages and eukaryotic viruses. At the phylum level, Uroviricota dominated both groups but was substantially more abundant in diseased ducks (63.5%) compared to healthy ones (49.9%). Negarnaviricota and Pisuviricota were also more enriched in diseased ducklings, while Phixviricota appeared more prevalent in healthy individuals (Figures 2A, E). At the class level, Caudoviricetes was the most enriched taxon in diseased samples (63.5% vs. 49.9%). Additional enriched classes in the diseased group included Leviviricetes, Insthoviricetes, and Malgrandaviricetes, whereas Amabiliviricetes and Revtraviricetes were more common in the healthy group (Figures 2B, F). At the order level, unclassified Caudoviricetes were dominant in both groups, aligning with class-level patterns. Diseased ducks showed increased representation of Articulavirales and Norzivirales, while Petitvirales and Ortervirales were relatively more abundant in healthy individuals (Figures 2C, G). Family-level comparisons further highlighted marked differences in viral composition. Demerecviridae (12.3%), Aliceevansviridae (5.9%), and Herelleviridae (3.2%) were enriched in diseased ducks, whereas Aliceevansviridae (8.1%), Fiersviridae (1.6%), and Ackermannviridae (1.3%) were more abundant in healthy ducks. Notably, unclassified Caudoviricetes families were 7.5% more abundant in the diseased group (Figures 2D, H). A scatter plot depicting the relative abundances of the top 30 viral families across groups further corroborates these differential patterns, underscoring the distinct family-level viral profiles that distinguish healthy from diseased ducklings (Supplementary Figure S1).
Figure 2

Taxonomic profiling of the duck intestinal virome. (A–D) Bar charts showing relative taxonomic abundances at the phylum to family levels, comparing healthy and diseased groups; (E–H) Sample-wise relative abundance plots at the phylum to family levels.
3.3 Bacteriophage composition reveals gut virome shifts in diseased ducks
To gain deeper insight into virome structure and uncover potential pathogenic phages, we classified viruses based on host specificity—bacteriophages versus eukaryotic viruses—and performed family, genus, and species-level annotations of bacteriophages.
At the family level, Demerecviridae, Straboviridae, and Autographiviridae showed marked enrichment in diseased ducks (log2 FC > 3), while Microviridae and Ackermannviridae were relatively more abundant in healthy individuals (Supplementary Figure S2, Supplementary Table S2).
At the genus level, Brussowvirus, Vansinderenvirus, Tequintavirus, Schiekvirus, and Moineauvirus represented dominant taxa in the duck gut. While Brussowvirus, Vansinderenvirus, and Moineauvirus were present at relatively low abundance in some healthy samples, no significant overall differences were observed between groups. However, Tequintavirus and Schiekvirus were markedly elevated in the diseased group, with approximate fold increases of 9,024 (log2 FC = 13.1) and 4.3 (log2 FC = 2.1), respectively, compared to the healthy group (Figure 3A, Supplementary Table S3). Several unclassified genera, such as unclassified Aliceevansviridae, unclassified Demerecviridae, unclassified Ackermannviridae, and unclassified Herelleviridae, were also prevalent in both groups. Notably, unclassified Demerecviridae showed a significant enrichment in the diseased group (483-fold; log2 FC = 8.9), whereas unclassified Fiersviridae was more abundant in the healthy group (2.8-fold; log2 FC = 1.5) (Figure 3A, Supplementary Table S3).
Figure 3

Characterization of gut-associated bacteriophages in ducks. (A, B) Heatmaps displaying the top 30 most abundant bacteriophage taxa at the genus (A) and species (B) levels across all samples.
At the species level, the most abundant phages included Brussowvirus 20617, Vansinderenvirus SW4, unclassified Tequintavirus species, Schiekvirus EFP01, and unclassified Demerecviridae species, representing the core bacteriophage constituents of the Muscovy duck gut virome (Figure 3B, Supplementary Table S4). Schiekvirus EFP01 was significantly more abundant in diseased ducks (4.3-fold; log2 FC = 2.1), while Pseudotevenvirus margaery also showed elevated abundance in the diseased group (2.8-fold; log2 FC = 1.5). In contrast, Leviviridae sp. was depleted in diseased ducks, being 2.8-fold more abundant (log2 FC = 1.5) in healthy individuals (Figure 3B, Supplementary Table S4).
3.4 Differentially abundant phages highlight microbial shifts in disease
To identify differentially abundant phage species, we performed a statistical comparison of relative abundances between groups and highlighted the top 10 significantly altered bacteriophages (Figure 4). These included Pseudotevenvirus margaery (p = 0.04; Figure 4A), unclassified Herelleviridae species (p = 0.10; Figure 4B), Fischettivirus C1 (p = 0.12; Figure 4C), Schiekvirus EFP01 (p = 0.13; Figure 4D), Citexvirus dobby (p = 0.14; Figure 4E), Shalavirus Shbh1 (p = 0.15; Figure 4F), unclassified Rountreeviridae species (p = 0.16; Figure 4G), Kayvirus S253 (p = 0.17; Figure 4H), unclassified Straboviridae species (p = 0.19; Figure 4I), and Kayvirus SA12 (p = 0.20; Figure 4J). These data indicate substantial shifts in phage community composition between healthy and diseased ducks, suggesting a possible role of phage dynamics in disease progression or microbiome imbalance.
Figure 4

Differential abundance analysis of bacteriophages. (A–J) Top 10 bacteriophage taxa showing the most significant differences in abundance between healthy and diseased ducks.
3.5 Eukaryotic viral community structure varies between healthy and diseased ducks
To investigate the eukaryotic vertebrate-infecting viral community, we performed annotations at both genus and species levels. A heatmap of eukaryotic viral families highlights key abundance shifts: Sedoreoviridae and Orthomyxoviridae were notably enriched in diseased ducks (log2 FC > 1.9), while Picobirnaviridae and Retroviridae showed higher representation in healthy individuals(Supplementary Figure S3, Supplementary Table S5).
At the genus level, dominant taxa included unclassified Picobirnaviridae, Ludopivirus, Sapelovirus, unclassified Caliciviridae, and Picobirnavirus. Among these, Sapelovirus was significantly enriched in diseased ducks (18.4-fold; log2 FC = 4.2), while unclassified Picobirnaviridae, Ludopivirus, and Picobirnavirus were more prevalent in the healthy group (Figure 5A, Supplementary Table S6).
Figure 5
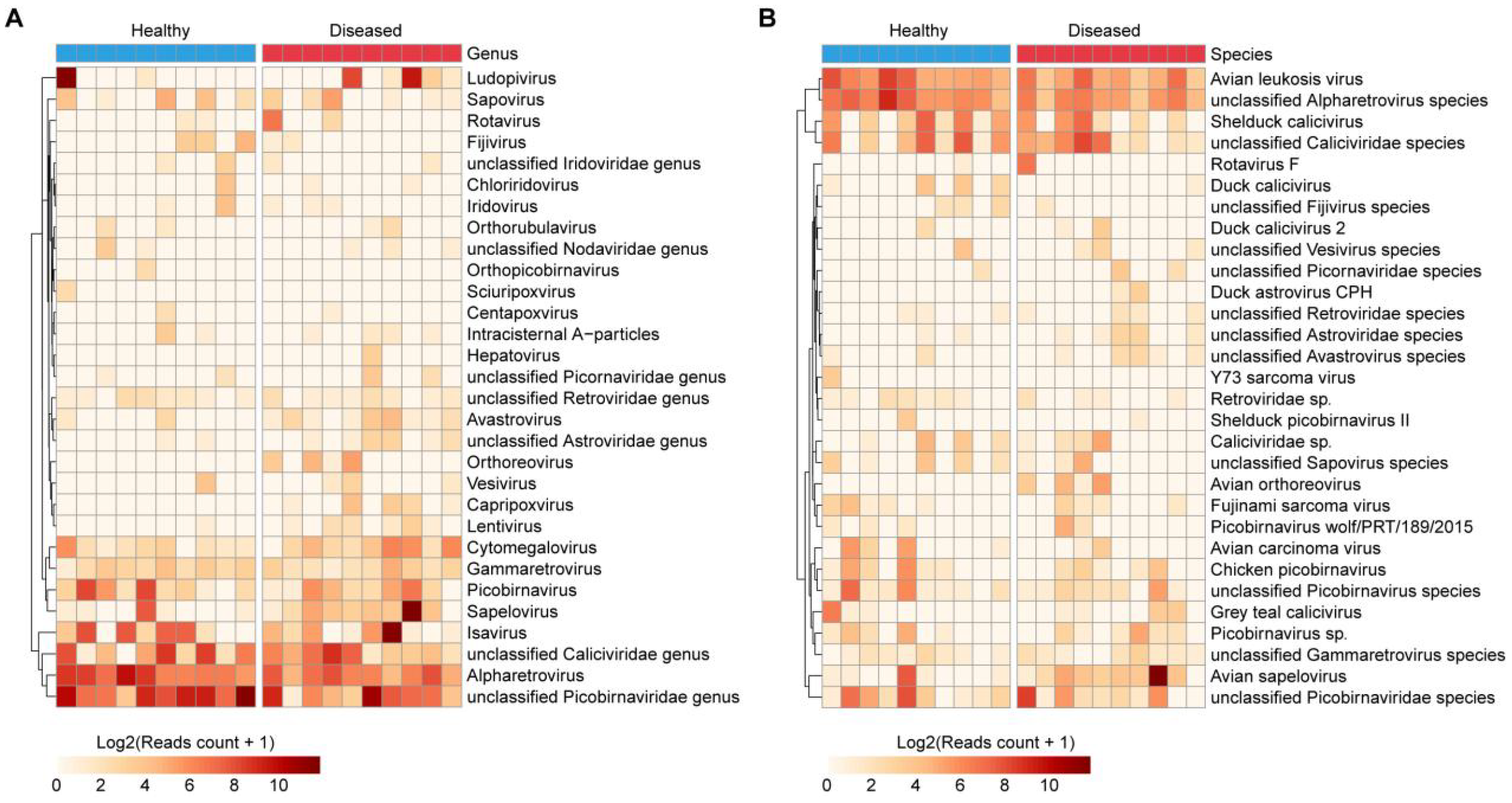
Composition of eukaryotic viruses in the duck gut. (A, B) Heatmaps illustrating the 30 most abundant eukaryotic viral taxa at the genus (A) and species (B) levels among all samples.
At the species level, the most abundant annotated viruses included Avian sapelovirus, unclassified Caliciviridae species, unclassified Picobirnaviridae species, Shelduck calicivirus, unclassified Picobirnavirus species, Chicken picobirnavirus, and Grey teal calicivirus (Figure 5B, Supplementary Table S7). Notably, Avian sapelovirus was significantly more abundant in diseased ducks (18.4-fold; log2 FC = 4.2). Furthermore, several viruses were detected exclusively in the diseased group but not in healthy individuals, such as Avian orthoreovirus and Duck astrovirus CPH (Figure 5B, Supplementary Table S7).
3.6 Differential abundance analyses reveal eukaryotic virus associations with disease
To identify significantly different eukaryotic viral species, differential abundance analysis was conducted, highlighting the top 10 differentially enriched viruses (Figure 5). These included Duck calicivirus (p = 0.10; Figure 6A), unclassified Astroviridae species (p = 0.10; Figure 6B), Avian orthoreovirus (p = 0.12; Figure 6C), unclassified Alpharetrovirus species (p = 0.13; Figure 6D), Avian carcinoma virus (p = 0.18; Figure 6E), unclassified Avastrovirus species (p = 0.18; Figure 6F), unclassified Fijivirus species (p = 0.18; Figure 6G), Retroviridae sp. (p = 0.20; Figure 6H), unclassified Retroviridae species (p = 0.22; Figure 6I), and Duck astrovirus CPH (p = 0.24; Figure 6J). These results further emphasize the distinct viral signatures associated with diseased ducks.
Figure 6

Differentially enriched eukaryotic viruses. (A–J) Top 10 eukaryotic viral taxa exhibiting the most significant abundance changes between healthy and diseased groups.
3.7 Novel duck reovirus is the primary candidate causative agent of the outbreak
Importantly, several viruses, including Avian orthoreovirus, were exclusively detected in the diseased group (Figures 5B, 6C). Further analysis of Avian orthoreovirus sequences revealed annotations corresponding primarily to Duck reovirus, Muscovy duck reovirus, and Goose orthoreovirus. At the genomic level, annotated fragments included M3, M1, L1, and L2 segments (Figure 7A). Among these, Novel duck reovirus (NDRV)—a subtype of Avian orthoreovirus—has previously been associated with splenic and hepatic hemorrhages and necrosis in ducks (30). Based on our findings, we hypothesize that NDRV is the causative pathogen of the observed outbreak.
Figure 7

Avian reovirus identification and validation. (A) Taxonomic and gene-level annotations of Avian reovirus detected via metatranscriptomic sequencing; (B) Quantitative RT-PCR results confirming the presence of Avian reovirus.
To validate this hypothesis, we performed RT-qPCR using primers specific for Novel duck reovirus, Duck hepatitis A virus (DHAV) and Fowl adenovirus serotype 4 (FAdV-4). All diseased samples tested positive for Novel duck reovirus, while the other two viruses were not detected (Figure 7B). These findings provide strong support that Novel duck reovirus is the probable cause of the observed outbreak.
4 Discussion
In this study, we employed a metatranscriptomic approach for the first time to investigate the gut virome of healthy and diseased Muscovy ducks (Cairina moschata). To our knowledge, this is the first metatranscriptomic study of the Muscovy duck gut virome. Through differential analysis and RT-qPCR validation, we identified Avian orthoreovirus as a potential etiological agent responsible for hemorrhagic necrosis in the liver and spleen of diseased ducklings. This strategy presents a promising approach for rapid pathogen detection and the discovery of unknown infectious agents in poultry.
Accurate and timely diagnosis is essential for disease prevention and control not only in Muscovy duck farming but also across the broader livestock, wildlife industries and public health (31–33). This is particularly important when the potential etiological candidates are unknown. Advanced technologies such as high-throughput sequencing (HTS) have become indispensable tools for identifying potential pathogens under such circumstances, as demonstrated in uncovering the causes of diseases like Human Febrile Illness and Chapare Hemorrhagic Fever (34, 35). HTS has transformed molecular diagnostics, enabling comprehensive, scalable, and precise identification of pathogens within a short timeframe (36, 37). For instance, a nationwide study involving 1,941 mammalian samples from China identified 102 virus species from 13 families capable of infecting mammals, significantly broadening our understanding of wildlife viral diversity and strengthening surveillance strategies (19). In the context of livestock and poultry farming, HTS has already been applied in pathogen detection, diagnosis, and zoonotic disease surveillance. It has demonstrated significant advantages in identifying both known and emerging infectious agents, thereby enhancing the early warning capacity against animal-derived threats (38). While previous virome studies have explored the viral composition in passerine birds using metaviromics (39, 40), they have primarily focused on wild birds. Our investigation represents the first application of metatranscriptomics to characterize the gut virome of Muscovy ducks, offering a powerful diagnostic tool that can improve the efficiency, accuracy, and responsiveness of poultry disease management. Moreover, the adoption of such techniques contributes to the sustainable development of the farming industry and bolsters efforts to prevent zoonotic transmission.
Ducks, particularly wild waterfowl such as mallards (Anas platyrhynchos), are widely recognized as natural reservoirs or intermediate hosts for a variety of pathogens, including influenza viruses, coronaviruses, and Escherichia coli (41–43). These viruses circulate among wild waterfowl and frequently spill over into domestic poultry populations (44). In addition to influenza viruses, ducks can also carry other significant avian pathogens such as Newcastle disease virus (NDV) (45). Ducks, as natural hosts for multiple viruses, play a critical role in cross-species transmission, which holds significant public health implications (46). For instance, both gamma- and delta-coronaviruses have been detected in waterfowl, and ducks appear to be asymptomatic carriers. Ducks also contributed to the interspecies transmission of delta-coronaviruses from birds to pigs (47). In our study, only minimal reads were assigned to coronaviruses and influenza viruses, suggesting that Muscovy ducks were unlikely infected with these pathogens. This observation may also reflect the success of current vaccination and biosecurity measures implemented in Guangdong’s duck farming systems. Therefore, characterizing the virome of ducks not only has implications for their health management but also plays a vital role in public health. In-depth virome studies in ducks could inform effective disease control strategies and help anticipate and prevent cross-species transmission events.
Previous metatranscriptomic analyses of duck fecal viromes have identified a range of viral families, including Paramyxoviridae, Narnaviridae, Bunyaviridae, Birnaviridae, and Reoviridae (48). Viral metatranscriptomics has also enabled the discovery of novel viruses in species like ruddy shelducks, underscoring the importance of continuous monitoring for emerging threats to both wildlife and public health (49). Some viruses once thought to be restricted to Southeast Asia have since been detected in northern Eurasia, indicating wider geographic distribution than previously assumed (50). These findings reinforce the role of ducks as viral reservoirs and potential conduits for pathogen persistence and transmission. Our study revealed significant restructuring of the bacteriophage community in diseased ducks (Figures 3, 4). Notably, lytic bacteriophage families typically associated with pathogenic bacteria (Demerecviridae and Straboviridae) were markedly enriched in the diseased group, whereas temperate phage taxa implicated in microbiome homeostasis maintenance (Microviridae and Ackermannviridae) showed relative attenuation. This bidirectional imbalance pattern—characterized by lytic phage proliferation alongside temperate phage suppression—may synergize with intestinal microbial dysbiosis in diseased ducks (51, 52), ultimately contributing to microecological collapse. Future work should investigate the contributions of these viral groups to disease processes in Muscovy ducks. In addition to novel virus discovery, virome sequencing provides insights into viral evolution, recombination, and transmission patterns (40). Additionally, Genome assembly can uncover recombination events that may generate new viral strains, potentially impacting vaccine efficacy and posing economic risks to poultry production (53). Therefore, a comprehensive and integrative approach to studying the Muscovy duck virome is vital for understanding the interplay between viruses, hosts, and the environment, ultimately promoting sustainable poultry development.
Novel duck reoviruses (NDRV) have been associated with more severe clinical presentations than classical Muscovy duck reoviruses, particularly extensive hemorrhagic necrosis in the spleen and liver, highlighting their high pathogenic potential (54). NDRV infection has also been linked to immunosuppression, increasing susceptibility to secondary bacterial or viral infections (55). In our study, we observed marked hemorrhagic necrosis in the liver and spleen (Figure 1A). Metatranscriptomic analysis revealed a clear increase in Avian orthoreovirus abundance in diseased ducks, whereas no such signal was detected in healthy individuals. Avian orthoreoviruses in ducks primarily include Muscovy duck reovirus (MDRV) and NDRV. MDRV typically manifests with disseminated white necrotic foci in the liver and intestinal mucosal damage (56, 57), whereas NDRV is characterized by splenic necrosis, swelling, and hepatic lesions (58). Integrating clinical symptoms with multi-omics detection results, we conclude that these pathological changes were likely caused by NDRV, a conclusion further supported by RT-qPCR validation. While NDRV is a strong candidate, causal confirmation requires future isolation and challenge studies. It should be noted that the universal NDRV positivity in diseased samples by RT-qPCR appears discordant with the relatively low abundance of Avian orthoreovirus in some samples (Figure 6C). This apparent discrepancy is closely associated with technical limitations in sequencing depth. Our selection of moderate depth was driven by practical application considerations for future field deployment—excessive depth increase would substantially raise per-sample costs, hindering large-scale implementation. Crucially, the complete absence of NDRV detection in healthy ducks (Figures 5B, 6C) confirms assay specificity. While our study provides new insights into the diagnosis and identification of viral pathogens in Muscovy ducks, further direct evidence, such as virus isolation and animal challenge experiments, is necessary to definitively confirm the pathogenic role of NDRV. Although metatranscriptomic genome assembly and evolutionary analysis are valuable tools, the relatively low abundance of reovirus sequences in this study precluded full genome assembly or phylogenetic characterization. Future research will focus on improving sampling strategies, sequencing depth, and assembly algorithms, potentially incorporating artificial intelligence tools (12) maximize the diagnostic power of metatranscriptomics in uncovering novel pathogens.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The sequencing data that support the findings of this study has been deposited in the National Center of Biotechnology Information (NCBI) Sequence Read Archive (SRA) and is accessible via the accession number SRR34855210–SRR34855229.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee (IACUC), Sun Yat-Sen University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
CL: Writing – review & editing, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft. QK: Writing – review & editing, Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft. XW: Data curation, Formal Analysis, Writing – review & editing, Methodology, Software, Visualization. FZ: Data curation, Writing – review & editing, Resources, Validation. YH: Data curation, Writing – review & editing, Methodology, Software. YY: Investigation, Methodology, Resources, Writing – review & editing. RG: Investigation, Methodology, Resources, Writing – review & editing. JL: Data curation, Investigation, Writing – review & editing. XL: Data curation, Investigation, Writing – review & editing. ZY: Methodology, Resources, Writing – review & editing. LY: Resources, Writing – review & editing. SC: Resources, Writing – review & editing. FC: Resources, Writing – review & editing. HZ: Conceptualization, Resources, Writing – review & editing. OP: Conceptualization, Formal Analysis, Writing – review & editing. YC: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Guangdong Provincial Project for Agricultural Science and Technology Development and Resource-Environmental Protection Management (Grant No. 2019KJ137).
Conflict of interest
FZ, ZY, LY, and SC were employed by Wen’s Foodstuff Group Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1680275/full#supplementary-material
Supplementary Figure 1Scatter plot showing the relative abundances of the top 30 viral families across different experimental groups.
Supplementary Figure 2Heatmap presenting sample-wise total abundance rankings for the top 30 phage families at the family level.
Supplementary Figure 3Heatmap depicting the total abundance rankings of the top 30 eukaryotic viral families in each sample at the family level.
References
1
Fouad AM Ruan D Wang S Chen W Xia W Zheng C . Nutritional requirements of meat-type and egg-type ducks: what do we know? J Anim Sci Biotechnol. (2018) 9:1. doi: 10.1186/s40104-017-0217-x
2
Wang W Liang J Shi M Chen G Huang Y Zhang Y et al . The diagnosis and successful replication of a clinical case of Duck Spleen Necrosis Disease: An experimental co-infection of an emerging unique reovirus and Salmonella Indiana reveals the roles of each of the pathogens. Vet Microbiol. (2020) 246:108723. doi: 10.1016/j.vetmic.2020.108723
3
Gu CQ Xie CQ Hu XY Zhang WP Bi DR Cheng GF . Cytokine gene expression in the livers of ducklings infected with duck hepatitis virus-1 JX strain. Poult Sci. (2012) 91:583–91. doi: 10.3382/ps.2011-01743
4
Niu Y Ma H Ding Y Li Z Sun Y Li M et al . The pathogenicity of duck hepatitis A virus types 1 and 3 on ducklings. Poult Sci. (2019) 98:6333–9. doi: 10.3382/ps/pez455
5
Tang Z Liu M Gao Z Li M Cao J Ye H et al . Pathogenicity and virus shedding ability of fowl adenovirus serotype 4 to ducks. Vet Microbiol. (2022) 264:109302. doi: 10.1016/j.vetmic.2021.109302
6
Wu B Yang B He D Tang Y Diao Y . Genetic evolution of fowl adenovirus serotype 4 and its pathogenicity to Cherry Valley ducks in China. Vet Microbiol. (2022) 274:109578. doi: 10.1016/j.vetmic.2022.109578
7
Wang H Jiang C Xu B Lei D Fang R Tang Y . Transcriptomic analysis revealed ferroptosis in ducklings with splenic necrosis induced by NDRV infection. Vet Res. (2025) 56:54. doi: 10.1186/s13567-025-01479-y
8
Wang H Gao B Chen H Diao Y Tang Y . Isolation and characterization of a variant duck orthoreovirus causing spleen necrosis in Peking ducks, China. Transbound Emerg Dis. (2019) 66:2033–44. doi: 10.1111/tbed.13252
9
Wang H Wang Y Gao B Zhang S Diao Y Tang Y . Evidence of vertical transmission of novel duck orthoreovirus in ducks. Vet Microbiol. (2020) 251:108861. doi: 10.1016/j.vetmic.2020.108861
10
Marais G Hardie D Brink A . A case for investment in clinical metagenomics in low-income and middle-income countries. Lancet Microbe. (2023) 4:e192–9. doi: 10.1016/s2666-5247(22)00328-7
11
Oguzie JU Petros BA Oluniyi PE Mehta SB Eromon PE Nair P et al . Metagenomic surveillance uncovers diverse and novel viral taxa in febrile patients from Nigeria. Nat Commun. (2023) 14:4693. doi: 10.1038/s41467-023-40247-4
12
Hou X He Y Fang P Mei SQ Xu Z Wu WC et al . Using artificial intelligence to document the hidden RNA virosphere. Cell. (2024) 187:6929–6942.e6916. doi: 10.1016/j.cell.2024.09.027
13
Zárate A Díaz-González L Taboada B . VirDetect-AI: a residual and convolutional neural network-based metagenomic tool for eukaryotic viral protein identification. Brief Bioinform. (2024) 26(1):bbaf001. doi: 10.1093/bib/bbaf001
14
Fu Y Yu S Li J Lao Z Yang X Lin Z . DeepMineLys: Deep mining of phage lysins from human microbiome. Cell Rep. (2024) 43:114583. doi: 10.1016/j.celrep.2024.114583
15
Miller RR Montoya V Gardy JL Patrick DM Tang P . Metagenomics for pathogen detection in public health. Genome Med. (2013) 5:81. doi: 10.1186/gm485
16
Qiu Y Wang S Huang B Zhong H Pan Z Zhuang Q et al . Viral infection detection using metagenomics technology in six poultry farms of eastern China. PloS One. (2019) 14:e0211553. doi: 10.1371/journal.pone.0211553
17
Zhou Y Zhang Y Jia W . Next-generation sequencing technology reveals the viruses carried by poultry in the live poultry market of Guangdong, China. Vet Microbiol. (2024) 295:110136. doi: 10.1016/j.vetmic.2024.110136
18
Calayag AM Priest T Oldenburg E Muschiol J Popa O Wietz M et al . Arctic Ocean virus communities and their seasonality, bipolarity, and prokaryotic associations. Nat Commun. (2025) 16:6427. doi: 10.1038/s41467-025-61568-6
19
He WT Hou X Zhao J Sun J He H Si W et al . Virome characterization of game animals in China reveals a spectrum of emerging pathogens. Cell. (2022) 185:1117–1129.e1118. doi: 10.1016/j.cell.2022.02.014
20
Roach MJ Beecroft SJ Mihindukulasuriya KA Wang L Paredes A Cárdenas LAC et al . Hecatomb: an integrated software platform for viral metagenomics. Gigascience. (2024) 13:giae020. doi: 10.1093/gigascience/giae020
21
Bolger AM Lohse M Usadel B . Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
22
Langmead B Salzberg SL . Fast gapped-read alignment with Bowtie 2. Nat Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
23
Li W Godzik A . Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. (2006) 22:1658–9. doi: 10.1093/bioinformatics/btl158
24
Steinegger M Söding J . MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. (2017) 35:1026–8. doi: 10.1038/nbt.3988
25
Wang J Wang J Chen P Liu L Yuan W . Development of a TaqMan-based real-time PCR assay for rapid and specific detection of fowl aviadenovirus serotype 4. Avian Pathol. (2017) 46:338–43. doi: 10.1080/03079457.2016.1278428
26
Qiu Q Hu R Liu Z Yan L Dai X Xing C et al . Taqman-probe-based multiplex real-time RT-QPCR for simultaneous detection of duck tembusu virus, novel duck reovirus, duck hepatitis a type 1 virus and duck hepatitis a type 3 virus. Poult Sci. (2025) 104:105508. doi: 10.1016/j.psj.2025.105508
27
Zhang S Li W Liu X Li X Gao B Diao Y et al . A TaqMan-based real-time PCR assay for specific detection of novel duck reovirus in China. BMC Vet Res. (2020) 16:306. doi: 10.1186/s12917-020-02523-z
28
Rohart F Gautier B Singh A KA LC . mixOmics: An R package for ‘omics feature selection and multiple data integration. PloS Comput Biol. (2017) 13:e1005752. doi: 10.1371/journal.pcbi.1005752
29
Gu Z . Complex heatmap visualization. Imeta. (2022) 1:e43. doi: 10.1002/imt2.43
30
Li B Mao M Li H Man X Wu M Lu C et al . Synergistic pathogenicity of novel duck Orthoreovirus and salmonella typhimurium in ducks. Poult Sci. (2025) 104:104929. doi: 10.1016/j.psj.2025.104929
31
Wang H Chen J An T Chen H Wang Y Zhu L et al . Development and application of quadruplex real time quantitative PCR method for differentiation of Muscovy duck parvovirus, Goose parvovirus, Duck circovirus, and Duck adenovirus 3. Front Cell Infect Microbiol. (2024) 14:1448480. doi: 10.3389/fcimb.2024.1448480
32
You D Xu T Huang BZ Wu F Deng LS Liu ZY et al . Rapid, sensitive, and visual detection of pseudorabies virus with an RPA-CRISPR/EsCas13d-based dual-readout portable platform. Anal Chim Acta. (2024) 1318:342918. doi: 10.1016/j.aca.2024.342918
33
Leifels M Khalilur Rahman O Sam IC Cheng D Chua FJD Nainani D et al . The one health perspective to improve environmental surveillance of zoonotic viruses: lessons from COVID-19 and outlook beyond. ISME Commun. (2022) 2:107. doi: 10.1038/s43705-022-00191-8
34
Zhang XA Ma YD Zhang YF Hu ZY Zhang JT Han S et al . A new orthonairovirus associated with human febrile illness. N Engl J Med. (2024) 391:821–31. doi: 10.1056/NEJMoa2313722
35
Loayza Mafayle R Morales-Betoulle ME Romero C Cossaboom CM Whitmer S Alvarez Aguilera CE et al . Chapare hemorrhagic fever and virus detection in rodents in Bolivia in 2019. N Engl J Med. (2022) 386:2283–94. doi: 10.1056/NEJMoa2110339
36
Maggi F Pistello M Antonelli G . Future management of viral diseases: role of new technologies and new approaches in microbial interactions. Clin Microbiol Infect. (2019) 25:136–41. doi: 10.1016/j.cmi.2018.11.015
37
Ni PX Ding X Zhang YX Yao X Sun RX Wang P et al . Rapid detection and identification of infectious pathogens based on high-throughput sequencing. Chin Med J (Engl). (2015) 128:877–83. doi: 10.4103/0366-6999.154281
38
Suminda GGD Bhandari S Won Y Goutam U Kanth Pulicherla K Son YO et al . High-throughput sequencing technologies in the detection of livestock pathogens, diagnosis, and zoonotic surveillance. Comput Struct Biotechnol J. (2022) 20:5378–92. doi: 10.1016/j.csbj.2022.09.028
39
Williams RAJ Sánchez-Llatas CJ Doménech A Madrid R Fandiño S Cea-Callejo P et al . Emerging and novel viruses in passerine birds. Microorganisms. (2023) 11(9):2355. doi: 10.3390/microorganisms11092355
40
Wille M Shi M Klaassen M Hurt AC Holmes EC . Virome heterogeneity and connectivity in waterfowl and shorebird communities. Isme J. (2019) 13:2603–16. doi: 10.1038/s41396-019-0458-0
41
Huang Y Li Y Burt DW Chen H Zhang Y Qian W et al . The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet. (2013) 45:776–83. doi: 10.1038/ng.2657
42
Xu Y Han Y Xu P Zhou S Zhao P Wang Y et al . Avian migration-mediated transmission and recombination driving the diversity of gammacoronaviruses and deltacoronaviruses. Mol Biol Evol. (2025) 42(3):msaf045. doi: 10.1093/molbev/msaf045
43
Zhang S Chen S Rehman MU Yang H Yang Z Wang M et al . Distribution and association of antimicrobial resistance and virulence traits in Escherichia coli isolates from healthy waterfowls in Hainan, China. Ecotoxicol Environ Saf. (2021) 220:112317. doi: 10.1016/j.ecoenv.2021.112317
44
Pepin KM Leach CB Barrett NL Ellis JW VanDalen KK Webb CT et al . Environmental transmission of influenza A virus in mallards. mBio. (2023) 14:e0086223. doi: 10.1128/mbio.00862-23
45
Habib M Yaqub T Nazir J Shehzad W Aziz Ul R Sohail T et al . Genomic and biological characterization of Newcastle disease viruses isolated from migratory mallards (Anas platyrhynchos). Arch Virol. (2018) 163:2179–88. doi: 10.1007/s00705-018-3840-8
46
Peiris JS Cowling BJ Wu JT Feng L Guan Y Yu H et al . Interventions to reduce zoonotic and pandemic risks from avian influenza in Asia. Lancet Infect Dis. (2016) 16:252–8. doi: 10.1016/s1473-3099(15)00502-2
47
Guo J He J Liang Z Huang S Wen F . Birds as reservoirs: unraveling the global spread of Gamma- and Deltacoronaviruses. mBio. (2024) 15:e0232424. doi: 10.1128/mbio.02324-24
48
Zhao L Niu Y Lu T Yin H Zhang Y Xu L et al . Metagenomic analysis of the jinding duck fecal virome. Curr Microbiol. (2018) 75:658–65. doi: 10.1007/s00284-018-1430-3
49
Ji L Wang Y Sun Y Ji L Wang X Liu Y et al . Identification and characterization of multiple novel viruses in fecal samples of ruddy shelducks using viral metagenomics methods. Heliyon. (2024) 10:e38338. doi: 10.1016/j.heliyon.2024.e38338
50
Dubovitskiy N Derko A Loginova A Khozyainova A Denisov E Apanasevich M et al . Viral metagenomics in wild ducks reveals the presence of seadornaviruses in Siberia. Arch Virol. (2025) 170:41. doi: 10.1007/s00705-025-06226-4
51
Agapé L Menanteau P Kempf F Schouler C Boulesteix O Riou M et al . Prophylactic phage administration reduces Salmonella Enteritidis infection in newly hatched chicks. Microbiologyopen. (2024) 13:e70002. doi: 10.1002/mbo3.70002
52
Gao SM Fei HL Li Q Lan LY Huang LN Fan PF . Eco-evolutionary dynamics of gut phageome in wild gibbons (Hoolock tianxing) with seasonal diet variations. Nat Commun. (2024) 15:1254. doi: 10.1038/s41467-024-45663-8
53
Vibin J Chamings A Klaassen M Alexandersen S . Metagenomic characterisation of additional and novel avian viruses from Australian wild ducks. Sci Rep. (2020) 10:22284. doi: 10.1038/s41598-020-79413-9
54
Yang H Zhang W Wang M Yuan S Zhang X Wen F et al . Characterization and pathogenicity evaluation of recombinant novel duck reovirus isolated from Southeast China. Front Vet Sci. (2023) 10:1124999. doi: 10.3389/fvets.2023.1124999
55
Yang Y Li L Liu X Jiang M Zhao J Li X et al . Quantitative proteomic analysis of duck embryo fibroblasts infected with novel duck reovirus. Front Vet Sci. (2020) 7:577370. doi: 10.3389/fvets.2020.577370
56
Chen X Zheng M Huang M Xiao S Lin F Chen S et al . Muscovy duck reovirus infection disrupts the composition of intestinal microbiota in muscovy ducklings. Curr Microbiol. (2020) 77:769–78. doi: 10.1007/s00284-019-01865-8
57
Wu Y Jiang H Zhu E Li J Wang Q Zhou W et al . Hericium erinaceus polysaccharide facilitates restoration of injured intestinal mucosal immunity in Muscovy duck reovirus-infected Muscovy ducklings. Int J Biol Macromol. (2018) 107:1151–61. doi: 10.1016/j.ijbiomac.2017.09.092
58
Yan H Xu G Zhu Y Xie Z Zhang R Jiang S . Isolation and characterization of a naturally attenuated novel duck reovirus strain as a live vaccine candidate. Vet Microbiol. (2021) 261:109214. doi: 10.1016/j.vetmic.2021.109214
Summary
Keywords
Muscovy ducks, metatranscriptomic sequencing, intestinal viral communities, novel duck reovirus, hepatic and splenic hemorrhage
Citation
Liu C, Kang Q, Wang X, Zeng F, Huang Y, Yang Y, Geng R, Liao J, Luo X, Yan Z, Yin L, Cao S, Chen F, Zhang H, Peng O and Cao Y (2025) Metatranscriptomic analysis of the gut virome in Muscovy ducks reveals a novel duck reovirus potentially associated with hepatic and splenic hemorrhage. Front. Immunol. 16:1680275. doi: 10.3389/fimmu.2025.1680275
Received
05 August 2025
Accepted
06 October 2025
Published
23 October 2025
Volume
16 - 2025
Edited by
Qi Zhao, University of Macau, China
Reviewed by
Qingtao Liu, Jiangsu Academy of Agricultural Sciences, China
Linlin Li, Guangdong Academy of Agricultural Sciences, China
Xinwei Wang, Henan Agricultural University, China
Jing Zhao, China Agricultural University, China
Updates
Copyright
© 2025 Liu, Kang, Wang, Zeng, Huang, Yang, Geng, Liao, Luo, Yan, Yin, Cao, Chen, Zhang, Peng and Cao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongchang Cao, caoych@mail.sysu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.