 Xiaojing Guo1,2,3†
Xiaojing Guo1,2,3† Siwei Wang1,2,3†
Siwei Wang1,2,3† Jiuli Ding3†Weiwei Liu1,2,3Jiaqi Xu1,2,3Mutian Wang4Hongyuan Sun1,2Yuening Ma1,2
Jiuli Ding3†Weiwei Liu1,2,3Jiaqi Xu1,2,3Mutian Wang4Hongyuan Sun1,2Yuening Ma1,2 Wei Liu1,2*Lei Zhang5*Min Liu1,2*
Wei Liu1,2*Lei Zhang5*Min Liu1,2*- 1Department of Infectious Diseases, First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 2Department of Infectious Diseases, National Clinical Research Center for Chinese Medicine Acupuncture and Moxibustion, Tianjin, China
- 3Graduate School, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 4Department of Respiratory Medicine, Shanxi Provincial Academy of Traditional Chinese Medicine, Shanxi, China
- 5Office of the President,Tianjin Academy of Traditional Chinese Medicine Affiliated Hospital, Tianjin, China
Periodontitis (PD) is a chronic inflammatory disease linked to microbial dysbiosis, while rheumatoid arthritis (RA) is an autoimmune disorder characterized by anti-citrullinated protein antibodies (ACPA). Despite their distinct etiologies, a clinical and serological association between PD and RA has been observed. Oral microorganisms, especially Porphyromonas gingivalis (P. gingivalis), may contribute to RA onset or progression through dissemination to joints or systemic inflammation. This review explores a: the role of oral microbiota and immune responses in RA b; clinical pathogenic pathways from oral pathogens to the joints c; mechanistic studies on the impact of periodontal pathogens on RA; and d. preventive and therapeutic strategies. P. gingivalis and other periodontal pathogens have been detected in synovial tissues and fluids of RA patients. Microbiome analyses show a more diverse oral microbiota with elevated periodontal disease-associated bacteria in RA patients. Studies demonstrate that P. gingivalis can induce citrullination, autoantibody production, and inflammation, exacerbating joint damage. Future research should investigate the impact of periodontal therapy and RA treatments on the oral microbiota, while large-scale clinical trials are needed to validate the causal relationship between periodontal pathogens and RA.
1 Introduction
Rheumatoid arthritis (RA) has attracted widespread attention due to its substantial impact on human health and the considerable socioeconomic burden it imposes. It is commonly associated with progressive disability, premature mortality, and elevated healthcare and societal costs. The global prevalence of RA varies substantially, ranging from 0.25% to 1% (1), and is projected to continue rising through 2040. This trend poses a significant challenge to global public health (2). RA primarily affects individuals over the age of 40. The prevalence is particularly higher in women, who are two to three times more likely to be affected than men (3). RA is a systemic autoimmune inflammatory disease that primarily affects the joints. The pathological hallmark of RA is persistent synovitis, which leads to joint pain, cartilage degradation, joint deformity, and functional impairment. In advanced stages, RA can result in severe disability (4).
1.1 Risk factors for rheumatoid arthritis
The development of RA is influenced by both genetic and environmental risk factors (Figure 1). Genetic predisposition is estimated to account for approximately 60 percent of RA risk (5, 6). First-degree relatives of RA patients have a two- to fivefold higher risk compared to the general population. The genetic susceptibility to RA is strongly associated with specific human leukocyte antigen (HLA) haplotypes, particularly the presence of the shared epitope (SE), a five–amino acid motif encoded by certain HLA-DRB1 alleles (7). Compelling evidence supports HLA-DRB1 as the primary genetic determinant in anti-citrullinated protein antibodies(ACPA)-positive RA (8). Genome-wide association studies (GWAS) have further revealed the contribution of non-HLA genes to RA pathogenesis. The genetic risk of seropositive RA, defined by the presence of ACPA and/or rheumatoid factor, is primarily linked to signaling networks involving interferon alpha/beta and interleukin-12/23. Variants in genes within the JAK-STAT pathway, including STAT4, TYK2, and FLT3, have shown significant associations with disease susceptibility (9). Individuals who are ACPA-positive have a substantially increased risk of progressing to classified RA and are considered to be in an at-risk state (10), which is marked by immune dysregulation. Epigenetic studies have revealed nonspecific regulatory abnormalities in peripheral B cells, as well as in naïve and memory T cells of ACPA-positive individuals. Studies of antigen-specific T cells have identified a higher frequency of CD4+ T cells that recognize citrullinated epitopes, especially citrullinated cartilage intermediate layer protein I (cit-CILP), in at-risk individuals. These T cells tend to exhibit pro-inflammatory phenotypes, such as T helper cell (Th) 1, Th17, and stem-like memory characteristics. Additionally, antibody–antigen microarray analysis has detected increased levels of autoantibodies against citrullinated clusterin, fibrinogen, and histone H4 in both at-risk individuals and patients with early RA, with the highest levels found in the latter (10). These findings suggest that widespread early epigenetic alterations may disrupt immune tolerance and lead to the activation of cit-CILP–specific T cells, which in turn support B cell responses and drive the production of autoantibodies against a broader array of citrullinated antigens. This multifaceted immune imbalance promotes the progression from asymptomatic autoimmunity to clinically evident arthritis.
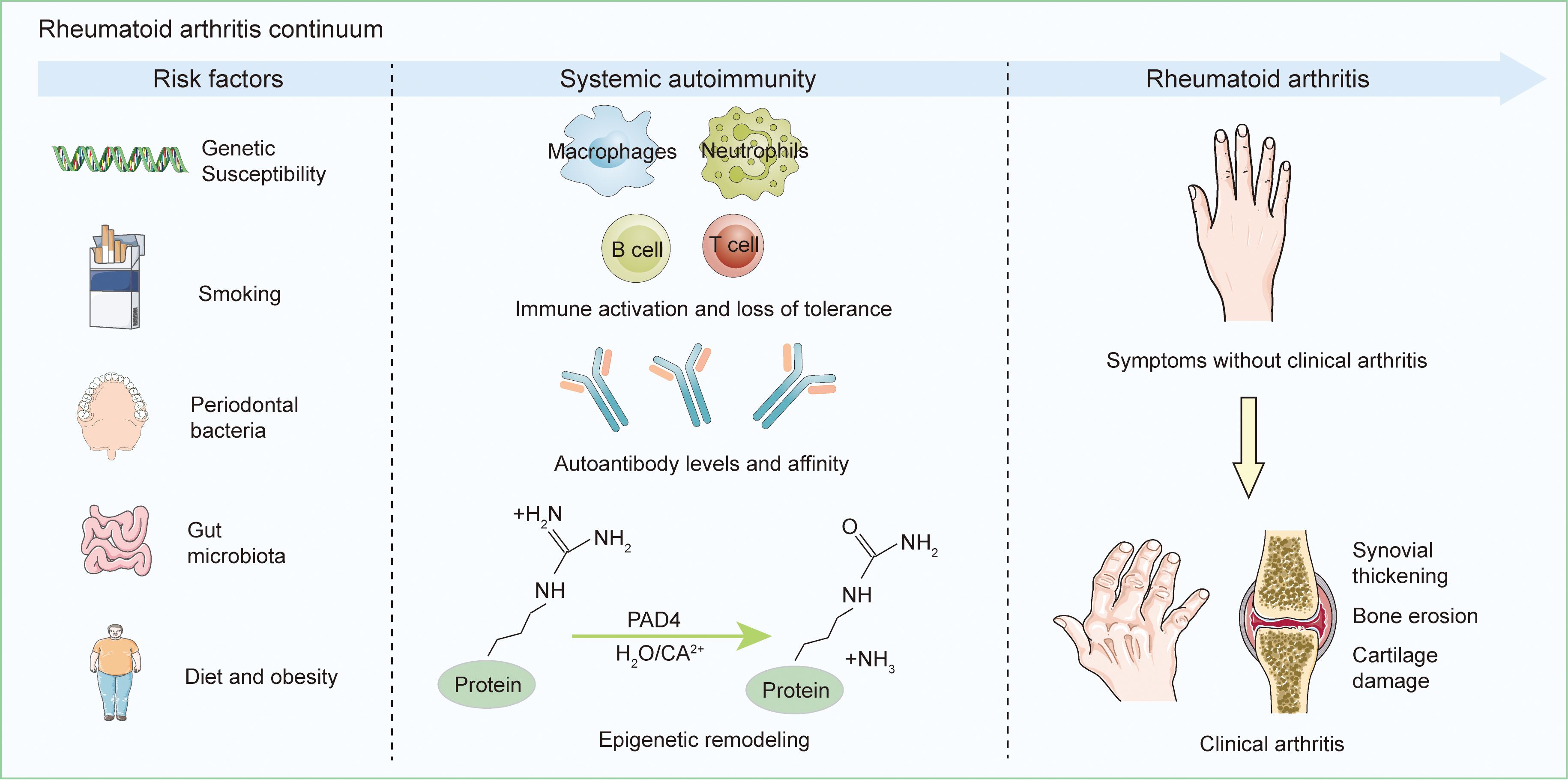
Figure 1. Initiation and Advancement of RA. A combination of individual genetic predispositions and environmental exposures contributes to systemic autoimmunity in those susceptible to RA, resulting in the localized synthesis of RA-related antibodies, predominantly in the oral, lung, or gut mucosal tissues. Many patients then progress through a stage of subclinical synovitis before developing RA. This process is not inevitable; ACPA positivity before synovitis may convert to ACPA negativity, and some ACPA-positive individuals may not develop RA.
An increasing number of environmental factors have been identified as contributing to the risk of developing RA. Among them, cigarette smoking is currently recognized as the most strongly associated environmental risk factor for seropositive RA, with a clear dose-dependent effect (11). Compared to non-smokers, RA patients who carry SE alleles and smoke exhibit elevated ACPA production by peripheral B cells (12). Further studies indicate that smoking and HLA-SE contribute at distinct stages of RA development. Smoking promotes the generation of ACPA and the onset of symptoms, while HLA-SE primarily facilitates the emergence of clinical manifestations and progression to classified RA (13). The role of the microbiome in RA has also garnered growing interest, and the involvement of oral and gut microbial communities is now well recognized. Studies suggest that mucosal exposure and/or dysbiosis in patients with early RA (diagnosed within one year) or long-standing active disease may play a causal role in the at-risk phase of RA progression (14). Periodontal dysbiosis may induce local inflammation, disrupt mucosal barriers, and allow microbial components to translocate into the bloodstream. Certain bacterial elements may also mimic host autoantigens at the molecular level, potentially triggering joint-specific immune responses (15). Overweight and obesity, defined as a body mass index (BMI) of ≥25 kg/m2 and ≥30 kg/m2, respectively, have also been linked to increased RA risk (16). Studies have shown that individuals with a BMI ≥25 kg/m2 and ≥30 kg/m2 experience a 15% and 21–31% greater risk of developing RA, respectively (17). This association may be mediated by the pro-inflammatory activity of white adipose tissue and its immunomodulatory effects (18). Nevertheless, the role of obesity in RA onset remains controversial. Other potential environmental risk factors, such as dietary habits and estrogen levels, have been proposed but require further investigation to confirm their relevance (1). Overall, most known environmental contributors to RA point toward the involvement of chronic mucosal inflammation in disease initiation, although additional research is required to clarify the underlying mechanisms (1).
1.2 The pivotal role of citrullination in RA pathogenesis
The pathogenesis of RA is complex. Immune dysregulation may begin years before the appearance of joint symptoms, a period commonly referred to as the preclinical phase of RA (19). The formation and recognition of citrullinated self-antigens span the entire course of the disease, from the subclinical phase to the clinical manifestation (20). This process is primarily attributed to the loss of immune tolerance, resulting in aberrant immune responses against citrullinated self-antigens (21). Citrullination is a key post-translational modification in which arginine residues in specific self-antigens, such as immunoglobulin G (IgG), type II collagen, and vimentin, are converted into citrulline by peptidyl arginine deiminases (PADs) (22). Among the five calcium-dependent PAD isoenzymes in humans (23), PAD2 and PAD4 are most strongly associated with RA. These enzymes are highly expressed in synovial tissue, synovial fluid, and infiltrating mononuclear cells, with expression levels positively correlated with the severity of inflammation (24, 25). Citrullinated proteins have also been abundantly detected in synovial fluid, where they serve as key targets for local immune responses (26–28). Citrullination exposes cryptic epitopes that are normally hidden from immune surveillance, leading to their misrecognition as non-self antigens. This is achieved by altering protease cleavage sites and destabilizing protein structures, which unmasks peptides derived from native sequences (29). In the presence of susceptibility alleles such as HLA-DR1 and HLA-DR4, the positively charged P4 pocket of the SE binding groove selectively binds citrullinated peptides through electrostatic complementarity. These complexes are misrecognized as foreign, triggering downstream immune cascades (30). Citrullinated antigens are taken up by antigen-presenting cells, which initiate dendritic cell-mediated responses and activate both T and B cells in a process known as costimulation. B cells undergo somatic hypermutation and class-switch recombination, eventually differentiating into plasma cells that produce specific autoantibodies against citrullinated epitopes, known as ACPAs (31). ACPAs can be detected in the bloodstream approximately four to five years before the clinical onset of arthritis. As genetic and environmental risk factors interact, ACPA levels increase, the range of recognized protein epitopes expands, and circulating pro-inflammatory proteins rise (20). These changes collectively contribute to the breakdown of immune tolerance and the eventual transition to clinically evident arthritis, whether classified or undifferentiated (29).
Increasing evidence supports the mucosal origin hypothesis in the pathogenesis of RA, suggesting that autoimmune processes may begin at extra-articular mucosal sites—such as the lungs, oral cavity, and gut—where local immunological changes lead to both local and systemic autoantibody production (14). Among these sites, the oral mucosa, particularly in the setting of PD, has garnered significant attention. A meta-analysis demonstrated that individuals with PD lasting more than five years are significantly more likely to develop RA, with a 69% higher risk than healthy controls (32). Citrullination may initiate autoimmune responses at periodontal sites before the clinical onset of RA (33, 34). PAD2, PAD4, and ACPAs have been detected in inflamed gingival tissues and gingival crevicular fluid in individuals with PD (35). ACPAs targeting citrullinated peptides are among the most extensively studied autoantibodies in RA. Their targets include intracellular proteins such as α-enolase and vimentin, as well as extracellular citrullinated proteins such as fibrinogen, type II collagen, and fibronectin (36, 37). These antigen–antibody complexes accumulate in synovial fluid and contribute to RA pathogenesis (38). Porphyromonas gingivalis (P. gingivalis), a key periodontal pathogen, produces peptidyl arginine deiminase (PPAD), an enzyme that citrullinates arginine residues in host periodontal proteins and its own bacterial proteins. Arginine-specific gingipains (Rgps), another major virulence factor of P. gingivalis, cleaves fibrinogen and α-enolase to generate C-terminal arginine-containing peptides, which serve as preferred substrates for PPAD (39, 40). Co-incubation of these proteins with PPAD and RgpB has confirmed the formation of novel citrullinated peptides, including 37 derived from fibrinogen and 11 from α-enolase (41).Rheumatoid factors (RFs), another class of autoantibodies in RA, target the Fc region of host IgG (42). Rgps cleave lysine and arginine residues within the CH2 and CH3 domains of IgG, producing fragments that enhance Rheumatoid factor (RF) binding. Moreover, P. gingivalis generates short-chain fatty acids that attract neutrophils to periodontal tissues (43), where they activate and release reactive oxygen species (ROS) such as superoxide, hydrogen peroxide, and hydroxyl radicals. These ROS enhance the reactivity of IgG, facilitating RF-mediated immune complex formation (44). The resulting immune complexes involving ACPAs and RFs accumulate in synovial fluid, where they drive persistent synovitis and joint destruction—the pathological hallmarks of RA (38, 42). Other oral pathogens that may contribute to citrullination include Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans) (45) and Prevotella intermedia (P. intermedia) (46), which will be further discussed in subsequent sections.
1.3 Epidemiological association RA– periodontal disease
Periodontal disease affects the gums, supporting connective tissues, and alveolar bone, ranking sixth in disease burden among chronic non-communicable diseases. Between 2011 and 2020, its prevalence reached nearly 62% among adults who had received dental treatment (47). In 2021, over 1 billion people worldwide suffered from severe periodontitis, accounting for approximately 11% of the adult population (48). It is broadly classified into gingivitis and PD. Gingivitis is characterized by inflammation limited to the gingival epithelium and adjacent soft connective tissues, whereas periodontitis is a chronic, non-communicable, and multifactorial inflammatory condition primarily initiated by bacterial infection, leading to inflammation of the tooth-supporting structures (49). This process results in the loss of connective tissue attachment and alveolar bone destruction, with an overall population prevalence estimated at 45 to 50 percent. The pathogenesis of periodontitis involves the invasion of periodontal tissues by pathogenic bacteria and their products within the dental biofilm, which stimulate the production of pro-inflammatory mediators. Recurrent disruption of the oral mucosal barrier facilitates the translocation of oral pathogens and their virulence factors into the bloodstream (50). This leads to elevated systemic levels of inflammatory cytokines and a sustained immune-inflammatory response, which may serve as a persistent driver of systemic immune activation.
A growing body of clinical and epidemiological evidence supports a bidirectional relationship between RA and PD (51). The prevalence of PD among individuals with RA is at least twice as high as that observed in healthy populations (52). A study by Eriksson et al. reported that approximately 75 percent of RA patients exhibited moderate to severe periodontitis, while the remaining patients had either no periodontitis or only mild forms (53). A meta-analysis further revealed that individuals with RA display significantly worse periodontal conditions, characterized by greater clinical attachment loss (CAL), higher rates of tooth loss, elevated plaque index scores, and increased probing depth (54). These differences appear to be independent of age, sex, ethnicity, and smoking status (55). Notably, RA patients with severe periodontitis tend to exhibit higher levels of disease activity, highlighting a potential link between periodontal burden and RA progression (56).
2 Oral microorganisms in RA
The oral microbiota is a highly diverse and structured microbial community comprising over 700 bacterial species, including more than 250 recognized pathogens, which can influence host pathophysiology on a systemic level (57). Factors such as aging, poor oral hygiene, systemic comorbidities, and the use of medications can disrupt the composition of the oral microbiota, contributing to the onset of oral diseases (58). Over the past two decades, a growing number of clinical studies have examined the oral microbial characteristics of individuals at high risk of developing RA and of patients already diagnosed with the condition (summary provided in Table 1), revealing a potential link between oral microbial dysbiosis and RA pathogenesis.
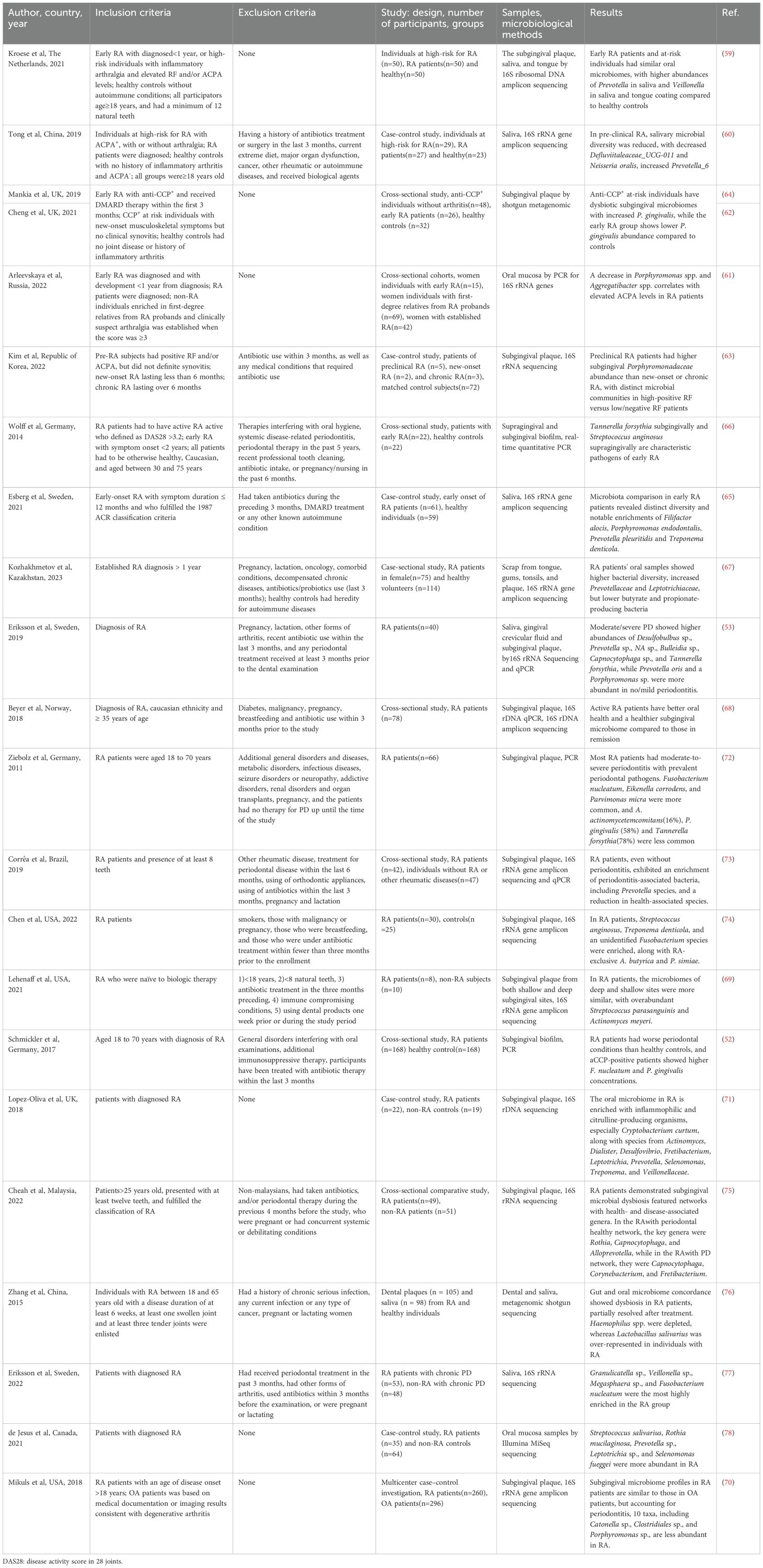
Table 1. Studies on oral microbiota in RA patients.
2.1 Oral microbiota
Individuals at high risk of RA are characterized by seropositivity for autoantibodies and inflammatory arthralgia in the absence of clinically evident synovitis (59–64). Oral microbiota perturbations have been observed during this “preclinical” phase of RA. Studies have demonstrated a reduction in salivary microbial diversity among these high-risk individuals, particularly a decrease in Defluviitaleaceae_UCG-011 and Neisseria oralis, along with an increase in Prevotella_6 (60). Dysbiosis of the subgingival microbiota has also been detected in anti-cyclic citrullinated peptide (CCP)+ individuals without arthritis, who show higher prevalence of both periodontitis and gingivitis (62, 64). Furthermore, the oral microbiota composition in at-risk individuals closely resembles that of early RA patients, with no significant differences observed in the microbial profiles of saliva and tongue coating. Compared to healthy controls, these groups exhibit increased abundance of Veillonella in both saliva and tongue coating, and Prevotella in saliva (59). This similarity suggests that specific shifts in the oral microbiota may constitute early pathogenic events in RA development. Additionally, studies have identified enrichment of Porphyromonadaceae family bacteria and Saccharimonas species in subgingival plaque samples from at-risk individuals prior to RA diagnosis (63). These microbial signatures may hold predictive potential but require further confirmation in prospective investigations.
To investigate the oral microbiota across different stages of RA, patients are commonly categorized into early-stage and chronic RA, with healthy individuals serving as controls (61–66). Early RA is typically defined as meeting the 2010 ACR/EULAR classification criteria, with disease duration under 6 months (63), less than one year since diagnosis (61, 65), initiation of disease-modifying antirheumatic drugs (DMARDs) therapy within 3 months (62, 64), or symptom onset within two years (66). Compared to healthy individuals, early RA patients tend to experience more severe periodontitis, including greater loss of periodontal attachment and alveolar bone. Subgingival Tannerella forsythia and supragingival Streptococcus anginosus are markedly elevated—by sixfold and threefold, respectively—indicating their potential as characteristic pathogens in early RA (66). The composition of the oral microbiota in RA patients shifts in accordance with the severity of coexisting periodontitis. In individuals with no or mild periodontitis, Prevotella oris and Porphyromonas species are more abundant, whereas in moderate to severe cases, the dominant species include Desulfobulbus sp., Prevotella sp., NA sp., Bulleidia sp., and Capnocytophaga sp., along with higher levels of Tannerella forsythia (53). This indicates that the hypoxic and inflamed periodontal microenvironment remains a critical determinant of subgingival microbial composition in the context of RA. Distinct oral microbial signatures have also been identified based on RA disease activity. Staphylococcus and Porphyromonas are associated with higher disease activity, while Treponema sp. and Absconditabacteriales are more frequently found in patients during remission (67). A seemingly paradoxical yet significant observation is that patients with active RA sometimes exhibit better periodontal health and a more “eubiotic” subgingival microbiota compared to those in remission (68). This strongly suggests that anti-rheumatic treatments may substantially alter the oral microbial landscape and inflammatory status, necessitating careful consideration of therapy as a major confounding factor when interpreting microbiota–disease activity relationships. Oral microbiota may also correlate with the presence and titers of RA-specific autoantibodies. Marked differences in subgingival microbial communities have been observed between RF-high and RF-negative/low patients, with enrichment of Fusobacteria, Saccharibacteria, and Spirochaetes, as well as the families Saccharimonas, Porphyromonadaceae, and Spirochaetaceae, associated with elevated RF levels (63). Similarly, ACPA positivity has been linked to more severe PD (53). Certain taxa, such as Prevotella_9, show a positive correlation with both ACPA and RF levels (67), whereas reductions in Porphyromonas and Aggregatibacter have been associated with higher ACPA titers, independent of RF status, HLA-DRB1 SE alleles, or methotrexate usage (61). This may reflect a unique host–microbe interaction in which ACPA targets citrullinated bacterial membrane antigens, potentially facilitating the clearance of specific pathogens both locally and systemically. Notably, no significant microbial differences have been found between superficial and deep gingival sites in RA patients (69). Moreover, subgingival microbiota profiles in RA patients resemble those of individuals with osteoarthritis (OA), and microbial diversity indices do not correlate with RA disease activity, suggesting that microbial dysbiosis in the subgingival niche is not exclusive to RA (70). Overall, RA patients exhibit higher total bacterial loads and a more diverse oral microbiota, with increased prevalence of proinflammatory and citrulline-producing organisms (71), while the abundance of health-associated commensals is concurrently reduced (67, 72, 73).
2.2 Periodontal pathogen distribution in oral niches and clinical correlates in RA patients
Although numerous studies have highlighted the alterations in oral microbiota associated with different stages of RA, disease activity levels, and autoantibody profiles, it is important to consider the complexity of the oral environment. The oral cavity consists of distinct ecological niches that support heterogeneous microbial communities. Microbial composition differs significantly between sites such as dental plaque, the dorsal tongue, and keratinized gingiva (79). Within this complex microenvironment, P. gingivalis has become a central focus of mechanistic research due to its unique ability to generate citrullinated proteins and its strong epidemiological association with RA, as discussed in Section 1.3. As a Gram-negative anaerobe, P. gingivalis is commonly found in individuals with poor oral hygiene and has also been implicated in several systemic diseases. Meta-analyses have demonstrated that individuals exposed to P. gingivalis have a significantly increased risk of developing RA (80). In ACPA+ individuals at high risk of RA, P. gingivalis shows increased abundance in subgingival plaque (62, 64), but reduced levels in oral mucosal surfaces and saliva (60, 61). A large cross-sectional study involving 600 participants found that elevated P. gingivalis DNA levels in saliva were associated with increased serum ACPA titers (81), suggesting the potential for systemic dissemination. Furthermore, in participants with concurrently elevated salivary P. gingivalis DNA and C-reactive protein (CRP), ACPA reactivity against citrullinated α-enolase and vimentin was commonly observed (82). Interestingly, other studies have reported no significant difference in the prevalence of P. gingivalis in subgingival plaque between RA patients and healthy controls (83–85). Additionally, P. gingivalis abundance has shown no consistent association with RA disease activity, ACPA, or RF levels (68, 70, 81, 86), but instead correlates more strongly with PD severity. These findings suggest that PD, rather than P. gingivalis per se, may be the primary factor modulating RA outcomes. Nonetheless, this does not negate the potential involvement of P. gingivalis in RA. In tongue biofilm samples, the proportion of P. gingivalis has been positively correlated with RA disease activity, with higher detection rates in non-remission patients compared to those in remission (83). Although findings vary depending on the sample type, existing evidence supports the participation of P. gingivalis in the pathogenesis of RA. Beyond P. gingivalis, other periodontal pathogens have also been implicated in RA. A. actinomycetemcomitans is more frequently detected in the gingival crevicular fluid of RA patients (87), and its subgingival presence is positively correlated with DAS28, RF, anti-CCP, and ACPA levels (88). In addition, Staphylococcus aureus has been found at higher prevalence in the oral cavities of RA patients compared to controls (89, 90), indicating that multiple oral microbes may contribute to RA-related immune dysregulation.
Substantial evidence indicates that individuals at high risk for RA and RA patients often show characteristic oral microbiome dysbiosis, including reduced microbial diversity and altered abundance of various bacterial taxa. These changes have been linked to autoantibody presence, especially ACPA, as well as disease stage and activity. However, most studies are cross-sectional, limiting conclusions about causality and temporal sequence. Small sample sizes (n < 100), limited geographic scope (mainly Europe and East Asia), and inconsistent sampling sites further constrain generalizability. Although some studies control for confounders like smoking, age, sex, and medication use, fully eliminating these factors is difficult, particularly given the frequent co-occurrence of periodontitis and RA. Thus, observed microbial changes may partly reflect confounding rather than RA-specific patterns. Identifying reliable microbial biomarkers for RA diagnosis or prediction remains challenging. Future research should adopt large-scale, standardized, prospective designs and evaluate microbiome-targeted interventions for RA prevention and treatment. The next section discusses the role of periodontal pathogens in RA pathogenesis.
3 Pathogenic translocation of oral microbiota to synovial compartments
Oral pathogens possess the capacity to translocate from their primary infection sites to distant local tissues, including the joints. Several clinical studies in patients with RA (Table 2) have identified nucleic acids from periodontal pathogens within synovial fluid and synovial tissues, lending support to the hypothesis that oral bacteria may reach the joint environment and participate in the initiation and progression of arthritis. In a pioneering study, Moen et al. reported a greater diversity and higher concentration of oral bacterial DNA in synovial fluid compared to serum samples from patients with active RA. Notably, multiple periodontal pathogens such as Tannerella forsythensis, P. intermedia, Streptococcus intermedius, P. gingivalis, A. actinomycetemcomitans, and Fusobacterium nucleatum were detected (91), preliminarily confirming that oral bacteria can disseminate into the joint cavity. Subsequent studies have concentrated on specific pathogenic species. Reichert et al. demonstrated that the detection rate of P. gingivalis DNA was significantly higher in both the synovial fluid and oral plaque of RA patients compared to non-RA controls (92), suggesting preferential enrichment of this pathogen within the RA-affected joint environment. Further investigations revealed that P. gingivalis DNA was present not only in synovial fluid but also in synovial tissue, with a significantly higher positivity rate in RA patients (33.3%) than in those with undifferentiated polyarthritis (5.9%) (93). Moreover, the positivity rate in synovial tissue was particularly elevated in patients carrying the HLA-DRB1*04 allele, implying a potential interaction between bacterial localization and genetic susceptibility (93). However, it is important to note that the detection rates of P. gingivalis in subgingival plaque, peripheral blood, and synovial fluid did not significantly differ between patients with RA and those with undifferentiated arthritis (93). Furthermore, the presence of P. gingivalis was not significantly correlated with disease activity, disability levels, or autoantibody status in RA patients. In patients with refractory RA complicated by periodontitis, P. intermedia and P. gingivalis DNA were frequently detected in subgingival plaque and synovial fluid, with detection rates of 73.6% and 42.1%, respectively (94). These findings further support the hypothesis that specific periodontal bacteria may colonize the joint space, particularly in patients with both systemic and oral inflammatory conditions. Further insights into the joint microbiome have been gained from a study conducted in China, which found abundant bacterial nucleic acids in both synovial fluid and synovial tissue samples from RA patients. Among periodontal species, Porphyromonas was consistently detected in all samples, while Veillonella dispar, Haemophilus parainfluenzae, Prevotella copri, and Treponema amylovorum were more prevalent in synovial fluid. In contrast, genera such as Agrobacterium, Comamonas, Kocuria, Meiothermus, and Rhodoplanes were more abundant in synovial tissue (95), revealing the complex microbial composition within the RA joint microenvironment. Oral streptococci are dominant commensals in the human oral and pharyngeal cavities, and their bacterial fragments have also been identified in the joint spaces of RA patients (96). Animal models further corroborate the dissemination of periodontal pathogens. P. gingivalis, Treponema denticola, and Tannerella forsythia are recognized as periodontal pathogens that typically emerge in the mature stages of biofilm development. This microbial consortium, known as the red complex, constitutes a keystone polybacterial community within the subgingival biofilm of progressive periodontitis sites (97). Among members of the red complex, only P. gingivalis was found to disseminate hematogenously to synovial joints. Fluorescence in situ hybridization confirmed its presence in the perinuclear region of ankle joint cells in mice (98). Additionally, streptococcal cell walls extracted from the oral cavity of RA patients have been shown to induce arthritis in murine models. Local gingival inflammation and barrier dysfunction may facilitate the translocation of pro-inflammatory streptococcal components into the synovium, especially given the limited degradation capacity for such pathogen-associated molecular patterns (99).
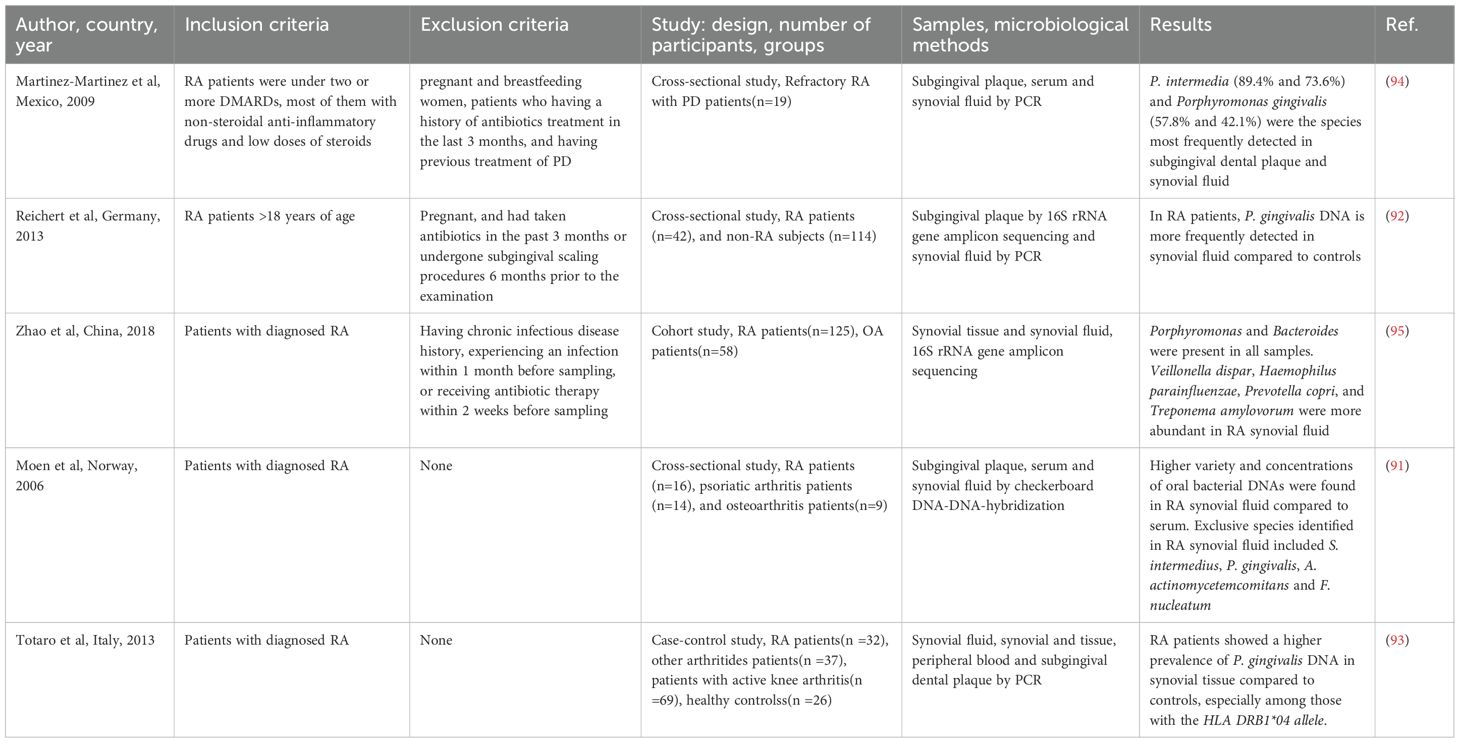
Table 2. Studies detecting oral bacteria in synovial fluid/tissue of RA patients.
These studies have established a chain of evidence for the translocation of oral bacteria to the joints, with particular emphasis on the colonization of P. gingivalis in synovial tissues, its intracellular localization potential, and its association with genetic susceptibility. These findings provide strong clues for its direct involvement in local joint pathology. However, the specific routes through which oral bacteria reach the joints, the extent of their contribution to arthritis pathogenesis, and the active components involved, such as intact bacteria, DNA, or metabolic products, remain to be fully elucidated.
4 Effects of periodontopathogen on experimental arthritis
Although many oral bacteria have been linked to RA, P. gingivalis is the most extensively studied species in terms of its impact on arthritis progression in animal models. As the predominant pathogen in subgingival polymicrobial communities associated with PD, P. gingivalis has been the focus of numerous experimental investigations in this field. Various arthritis models incorporating P. gingivalis infection have been employed to simulate its differential effects on RA. In these models, variations in immunization protocols, bacterial dosages, and routes of administration can markedly influence experimental outcomes. Moreover, the timing of arthritis induction relative to periodontal infection introduces additional complexity to experimental design, making reproducibility and interpretation particularly challenging. Twenty-three studies have examined the effect of P. gingivalis infection on the progression of RA using animal models (Table 3). Seventeen studies reported that P. gingivalis exacerbated arthritis, while four studies investigated the role of P. gingivalis combined with other oral pathogens, with three showing synergistic effects and one showing no significant effect on pristane-induced arthritis (98, 100, 101). Three reported synergistic effects, while one found no significant effect on pristane-induced arthritis (102). One study showed that oral inoculation with P. gingivalis led to the onset of seropositive arthritis, systemic inflammation, and bone erosion over an 8-month observation period (103). Additional study suggested that P. gingivalis failed to worsen arthritis but altered synovial proteome characteristics, suggesting that oral pathogens may contribute to the formation of particular disease subtypes (104). These studies had highly variable designs, using different P. gingivalis strains, models, and time points.
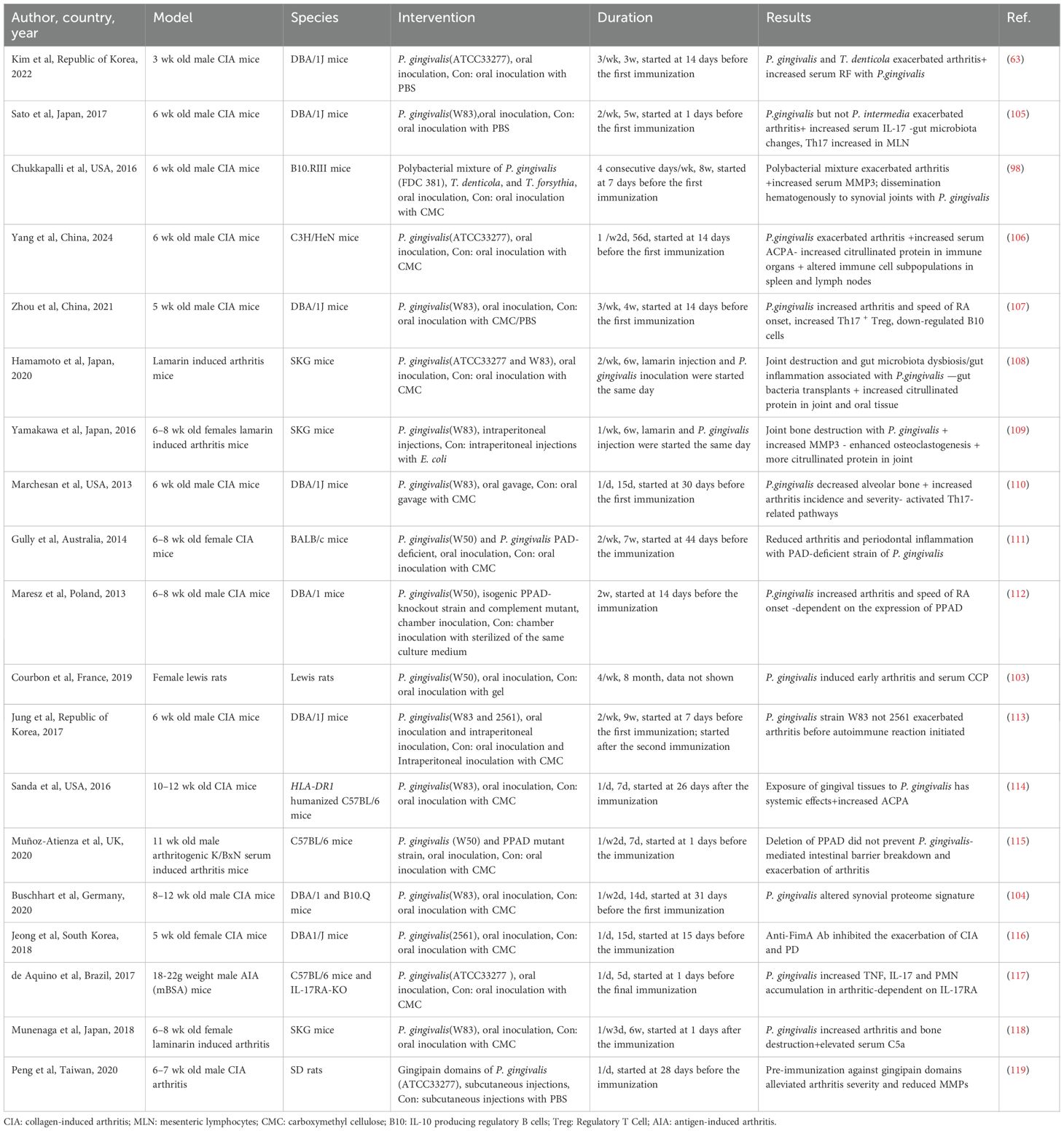
Table 3. Studies using animals on the association P. gingivalis – RA.
Polymicrobial oral infections have been shown to accelerate experimental arthritis, leading to earlier disease onset and increased severity characterized by enhanced inflammatory cell infiltration and pannus formation. In addition to the well-established role of the red complex, a major pathogenic group in periodontitis (98), the combined infection with P. gingivalis, Fusobacterium nucleatum, and A. actinomycetemcomitans similarly promotes the onset and progression of RA (100). Interestingly, while this bacterial consortium exacerbates arthritis, it results in comparatively less alveolar bone loss in mice than individual pathogens, suggesting a complex interplay between local and systemic immune responses (100). Repeated oral inoculation with P. gingivalis and Prevotella nigrescens promotes RA through Th17-mediated immune responses, and their effect is directly correlated with the severity of joint bone erosion (101). However, not all oral bacteria aggravate experimental arthritis. For instance, periodontitis induced by A. actinomycetemcomitans does not affect the course of antigen-induced arthritis (AIA) in mice, whereas arthritis aggravates alveolar bone loss caused by this bacterium, primarily due to enhanced systemic inflammation and lymphocyte polarization (120). Notably, both anti-TNF-α and antimicrobial therapies can alleviate periodontitis in this context (120).
5 Systemic humoral immune responses to periodontopathogens in RA patients
5.1 Circulating antibodies against P. gingivalis and related bacterial components
Recent studies suggest that humoral immune responses targeting oral bacteria may play a role in the pathogenesis of RA (121). Infection with P. gingivalis elicits a host immune response characterized by the generation of specific antibodies, primarily targeting its bacterial components and major virulence factors, including arginine-specific gingipains (RgpA and RgpB) and PPAD (122). Multiple serological studies have consistently reported that individuals with RA exhibit significantly elevated levels of P. gingivalis-specific antibodies compared to healthy controls or first-degree relatives (123–126). The seroprevalence of these antibodies is also comparable between patients with RA (48.9%) and those with PD (52.7%) (83). Moreover, antibody titers have been shown to correlate with the severity of PD (127).
Antibodies targeting P. gingivalis are closely associated with key biomarkers of RA (Table 4). Among these, the association between serum P. gingivalis antibody titers and ACPAs has been one of the most robust and consistently observed findings across multiple studies (124, 128–132). Mikuls et al. reported that elevated P. gingivalis antibody titers were positively correlated with specific ACPA subtypes, particularly anti-CCP IgM and IgG2 (124). Similarly, another study observed that patients with subgingival P. gingivalis colonization and elevated antibody titers exhibited increased expression of selective ACPAs, including antibodies to citrullinated filaggrin (85). Additionally, P. gingivalis antibody titers have been shown to correlate with RF levels in patients with RA, particularly among those with moderate-to-high disease activity (126). Notably, individuals with RF-IgA levels exceeding 75 IU/mL exhibited a five-fold increase in P. gingivalis antibody titers (133). However, some studies did not observe a direct association between P. gingivalis antibody levels and RA disease activity (123, 126, 127), suggesting that these antibodies may play a more specific role in autoimmune mechanisms rather than contributing directly to systemic inflammation.
Table 4. Studies in humans on the association periodontal microorganisms Porphyromonas gingivalis and RA.
Serum antibodies against P. gingivalis demonstrate potential in the diagnosis of RA, as well as in predicting disease progression and treatment outcomes (Table 4). Double positivity for anti-RgpA and anti-PPAD antibodies in peripheral blood significantly enhances the diagnostic accuracy for RA, with a specificity of 93.7% and a positive predictive value of 82.5% (134). Two independent studies have implicated P. gingivalis in accelerating the severity and progression of joint damage in RA. In patients experiencing rapid radiographic progression (RRP), the IgA/IgG ratio of anti-P. gingivalis lipopolysaccharide (LPS) antibodies was closely correlated with disease activity parameters, including DAS28-ESR, RF, erythrocyte sedimentation rate (ESR), and CRP levels (135). Similarly, a large early RA cohort study found that non-smoking patients with elevated P. gingivalis antibody titers had a higher rate of bone erosion (47.5%) (136). Beyond diagnosis and progression, P. gingivalis antibodies may serve as prognostic indicators of suboptimal therapeutic response. Early RA patients who are seropositive for these antibodies tend to exhibit higher disease activity, impaired joint function, and elevated levels of anti-CCP, RF, and systemic inflammation. Even after 12 months of standard treatment, this subgroup continues to display a trend toward increased disease activity (130). Convergent evidence from a separate study involving 82 treatment-naïve RA patients revealed that those with higher P. gingivalis antibody titers showed a less favorable response to therapy after three months (129).
Although existing evidence suggests a role for P. gingivalis antibodies in RA, growing evidence indicates that these antibodies are more strongly associated with PD. This suggests that the observed link to RA may reflect the broader pathophysiological state of PD rather than the effect of a specific pathogen.
5.2 Circulating antibodies against other periodontal pathogens and related bacterial components
Beyond P. gingivalis, circulating antibodies targeting other periodontal pathogens and their virulence factors have also been implicated in the pathogenesis and clinical manifestations of RA, expanding our understanding of how humoral immune responses to periodontal infections may contribute to RA development.
Serological studies have identified a significant association between elevated anti-A. actinomycetemcomitans IgG titers and RA (126). Notably, all individuals infected with A. actinomycetemcomitans were found to be anti-CCP+ (144). The influence of HLA-DRB1 SE alleles on autoantibody positivity appears to be restricted to RA patients exposed to A. actinomycetemcomitans (145). Leukotoxin A (LtxA), a key virulence factor secreted by A. actinomycetemcomitans, has recently been identified as a potential risk factor for the progression from arthralgia to RA (146). Anti-LtxA antibodies, including IgM, IgG, and IgA isotypes, can be detected across different stages of RA development, with significantly elevated IgM levels observed in both early and established RA patients (146). These antibodies are more prevalent among ACPA+ arthralgia patients and are associated with an increased likelihood of progression to RA (147). LtxA is thought to mimic membranolytic mechanisms involved in sustaining self-antigen citrullination in RA joints, inducing neutrophil morphological alterations and promoting neutrophil extracellular trap (NET) formation, which facilitates the release of highly citrullinated proteins in the oral cavity (145). Additionally, the N-terminal region of the DnaJ protein from A. actinomycetemcomitans elicits a specific immune response in RA patients, offering further insights into the disease’s etiology (148).
P. intermedia is a key periodontal pathogen implicated in chronic oral infection. Musculoskeletal ultrasound examinations have revealed that more than half of RA patients in clinical remission exhibit persistent synovitis. Notably, IgG titers against P. intermedia are significantly higher in patients with active RA and in those with ultrasound-detected synovitis (149). These findings suggest that P. intermedia may contribute to the chronic inflammatory state observed in RA. Through its enzymatic capacity to induce protein citrullination, P. intermedia may play a role in the generation of specific ACPA subtypes during the preclinical phase of RA. Prior to diagnosis, anti-P. intermedia antibodies are positively correlated with anti-CCP2, ACPA targeting vimentin, histones, and α-enolase, as well as with RF isotypes including IgA, IgG, and IgM (46). Further research has shown that P. intermedia-specific antibodies in gingival crevicular fluid are closely associated with ACPAs against citrullinated cytokeratin 13, a novel antigenic target with heightened antibody responses in RA patients that appears to lack cross-reactivity with antibodies directed against P. gingivalis gingipains (150). Interestingly, a large cross-sectional study (n = 33,994) found that elevated IgG responses to P. intermedia and Capnocytophaga ochracea were inversely associated with RF seropositivity (151). These findings imply that humoral immune responses to different periodontal pathogens may differentially influence RA-associated serological markers.
Circulating antibodies targeting other periodontal bacterial components, as well as the presence of these bacteria in the oral cavity, have also been linked to RA serological markers, although current evidence remains limited. Haemophilus species have been found to be depleted in both dental and salivary samples from RA patients and are negatively correlated with anti-CCP antibody levels (76). Subgingival colonization by Aminipila butyrica and Peptococcus simiae has been proposed to contribute to the generation of ACPAs (74). Additionally, Lactobacillus salivarius has been associated with elevated RA disease activity (74), further suggesting a potential role of specific oral microbiota in modulating systemic inflammatory states.
6 Molecular mimicry and immune cross-reactivity as core mechanisms of periodontopathogen-induced autoimmunity
6.1 Induction of citrullination and breaking immune tolerance
One of the central pathogenic mechanisms of RA is the breakdown of immune tolerance to citrullinated proteins, which involves increased production of citrullinated antigens and enhanced autoimmune responses against these antigens, particularly ACPA. Among various periodontal pathogens, P. gingivalis has drawn considerable attention due to its unique ability to induce citrullination. In animal models, oral administration of P. gingivalis has been shown to significantly elevate serum ACPA levels, accompanied by the detection of abundant P. gingivalis components and citrullinated proteins in the ankle joints (109). Upregulation of citrullinated protein expression has also been observed in multiple organs (108, 114). Moreover, a marked increase in the proportion of Tfh-gcB cell subsets responsible for autoantibody generation has been reported in lymphoid tissues (106). The contribution of P. gingivalis PPAD to citrullination has been demonstrated using gene manipulation approaches. Notably, wild-type P. gingivalis, but not PAD-deficient strains, induced the development of ACPA (108, 111) and production of citrullinated proteins (108, 112, 113), underscoring the mechanistic role of PPAD in linking P. gingivalis infection to RA pathogenesis. However, opposing evidence suggests that PPAD may not serve as the initial cross-reactive target for ACPA induction, as its absence does not prevent P. gingivalis-induced intestinal barrier impairment or exacerbation of arthritis in certain models (115).
A key mechanism by which PPAD induces host immune responses is enzymatic mimicry. This hypothesis, first proposed in 2009, suggests that P. gingivalis infection and PPAD-mediated citrullination of human antigens can trigger the ACPA response (39). Subsequent studies in RA patients have provided supporting evidence. It has been observed that peripheral plasmablasts in RA patients can produce ACPA, and approximately 63% of these antibodies cross-react with P. gingivalis outer membrane antigens and/or citrullinated enolase (152). Notably, in addition to C-terminal citrullination, P. gingivalis PAD exhibits prominent endocitrullination, targeting internal arginine residues (152). A 2020 study cloned and purified the PPAD enzyme (RACH2007-PPAD) from P. gingivalis strain CH2007 isolated from an RA patient and found that it could citrullinate internal arginine residues of major RA autoantigens such as fibrinogen and vimentin. Moreover, anti-RACH2007-PPAD antibody levels correlated with ACPA titers (153). More recently, a 2023 study proposed a possible mechanism by which persistent P. gingivalis exposure disrupts immune tolerance in RA. Repeated episodes of oral bacteremia in RA patients may result in widespread citrullination of oral bacteria, with subsequent translocation of citrullinated microbial antigens into circulation due to mucosal barrier breaches. This process activates proinflammatory monocyte subsets and inflamed synovial tissue, and further stimulates ACPA-producing B cells, thereby promoting affinity maturation and epitope spreading toward citrullinated human proteins (154).
Subsequent research has indicated that P. gingivalis primarily serves as an exogenous source of citrullinated antigens rather than modulating host-derived citrullination processes. Experimental evidence shows that when P. gingivalis is co-cultured with human peripheral blood mononuclear cells and macrophages, PPAD activity is detected exclusively in mononuclear cells exposed to the bacteria, leading to increased extracellular citrullination. In contrast, the expression of endogenous PAD enzymes and intracellular citrullination within monocytes and macrophages remains unaffected by P. gingivalis exposure (155).
6.2 Immune evasion
P. gingivalis employs various mechanisms to evade the host’s innate immune defense. P. gingivalis evades the immune response through PPAD-mediated citrullination by: 1) decreasing levels of neutrophil-derived proteins associated with phagocytosis and the antimicrobial activity of histone H2; 2) citrullinating histone H3, which facilitates P. gingivalis in evading NETs; 3) citrullinating the lysozyme-derived cationic antimicrobial peptide LP9, thereby limiting its antimicrobial activity (156). However, this immune evasion has been primarily observed in the oral cavity, and further confirmation in the context of RA is needed. Additionally, P. gingivalis facilitates immune evasion through A-type lipopolysaccharide (A-LPS), which assists in the localization of PPAD to Outer membrane vesicles (OMVs). OMVs are secreted by P. gingivalis as spherical bilayer structures made up of outer membrane proteins, phospholipids, DNA, lipopolysaccharides and segments of the periplasm (157). These vesicles package and transport bacterial virulence factors, including PPAD, to various host tissues. A-LPS modification not only localizes PPAD to OMVs, but also protects PPAD from proteolytic degradation, thus facilitating its “protective secretion” (158). Although PPAD is not essential for bacterial biofilm formation, or for the attachment and penetration of gingival epithelial cells by P. gingivalis, it functions as a powerful regulator of the immune response by enhancing the expression of genes related to cytokines (159).
6.3 Modulation of host immune responses
Exposure to P. gingivalis in gingival tissue exerts systemic effects by activating the host immune response. It may accelerate arthritis progression in CIA mice by mobilizing dendritic cells, macrophages, and neutrophils to the joints (116). Oral infection with P. gingivalis in CIA mice not only increases arthritis scores but also elevates serum RF levels (63). Chronic oral colonization with P. gingivalis prior to arthritis induction enhances immune activation that favors Th17 cell responses (107, 110, 114). Studies using IL-17RA knockout mice have confirmed that the exacerbating effect of P. gingivalis on arthritis is dependent on the Th17/IL-17 signaling pathway (117). Interestingly, P. gingivalis can also compromise the intestinal barrier and alter gut microbiota, thereby contributing to systemic inflammation and further aggravating arthritis (105, 108). These effects are partially mediated by increased serum IL-17 levels and a higher proportion of Th17 cells in mesenteric lymph nodes (105). In addition, downregulation of IL-10–producing regulatory B cells has been proposed as a key mechanism by which P. gingivalis promotes RA, since other immunosuppressive cells such as Tregs and myeloid-derived suppressor cells (MDSCs) are upregulated rather than suppressed (107). It is noteworthy that experimental arthritis does not affect the degree of P. gingivalis-induced alveolar bone loss (117).
6.4 Effects on promotes bone destruction and cartilage damage
Osteoclast overactivation and differentiation are the primary mechanisms underlying articular bone erosion in RA. P. gingivalis is a key contributor to bone loss associated with inflammation. Toll-like receptor (TLR) signaling modulates both inflammation and bone metabolism, whereas the receptor activators of nuclear factor kappa-B(NF-κB) ligand (RANKL) and its receptor, RANK, serves as a crucial regulator of osteoclast activation and bone remodeling processes. In isolated cultures of mouse parietal bone, P. gingivalis-LPS, via TLR2, promotes the formation of differentiated, mature osteoclasts, leading to bone loss. This process is primarily dependent on elevated RANKL levels (160). Subsequent studies revealed novel crosstalk between TLR and RANKL signaling cascades in the process of osteoclastogenesis. P. gingivalis modulates differently RANKL-induced nuclear factor of activated T-cells 1 (NFATc1) and cellular proto-oncogene Fos (c-Fos) expression, contingent upon the differentiation stage of osteoclast precursors. It inhibits RANKL-driven osteoclastogenesis at initial stages but promotes osteoclast maturation at later phases. TLR2/MyD88 is a central regulator of osteoclast differentiation by P. gingivalis, while RANKL reduces cytokine production triggered by P. gingivalis by down-regulating TLR/NF-κB and up-regulating NFATc1 (161). Mycorrhizal hairs, a major virulence factor of P. gingivalis, also contribute to osteoclast activation. The long-type FimA and short-type Mfa1 filaments are proteinaceous structures originating from P. gingivalis (162). The study found that both Mfa1 and FimA hyphae promote the differentiation and activation of osteoclasts. Significantly, the effect of Mfa1 on osteoclast differentiation was intense than that of FimA, and Mfa1 promoted osteoclast differentiation and activation in osteoclast precursor cells stimulated by RANKL (163).
Infection of macrophages by bacteria triggers the release of pro-inflammatory cytokines, with TNF-α stimulating osteoclasts that differentiate bone marrow macrophages both directly in vitro and indirectly via osteoblasts. A study in an environment exposed to P. gingivalis highlights the critical role of TLR2 signaling in osteoclastogenesis. P. gingivalis promotes the increased expression of TLR2 on macrophage surfaces, which in turn increases TNF-α production and enables a functional response of these macrophages to reinfection. The macrophage reaction to P. gingivalis stimulation depends on TNF-α and is independent of RANKL, IL-6, and IL-1β (164). Conversely, an alternative investigation proposed that P. gingivalis directly promotes RANKL-triggered osteoclastogenesis in RAW264 subclonal mouse macrophages, with TNF-α not involved in this process (165).
Chondrocyte apoptosis can lead to cartilage damage, resulting in tissue degeneration and joint deformity. A study found that infection with P. gingivalis significantly increased both early and late apoptosis of human primary chondrocytes (166). This finding was corroborated by Pischon et al., who showed that P. gingivalis can localize within cells (167).
In conclusion, numerous in vitro experiments and animal studies have highlighted the potential of P. gingivalis to elicit changes associated with RA. In animal studies, P. gingivalis is often introduced into the oral cavity to observe changes in distal joints, thereby mimicking the clinical situation. Moreover, the experimental design, in which the control group is typically sham or bacteria-free, should be reconsidered to better assess the specific role of P. gingivalis compared to other periodontal pathogens.
7 Preventive aspects from the microbiological point of view
The essence of establishing proof-of-concept hinges on tackling the underlying causative factor with treatments like nonsurgical periodontal treatment (NSPT), which involves providing oral hygiene guidance and mechanically removing microbes from tooth surfaces both above and below the gingival margin. Currently, there is no consensus on the common management strategies for periodontitis and rheumatoid arthritis. Studies have found that NSPT relieves inflammation for 6 weeks to 6 months and reduces DAS28, VAS, SJC, TJC and improves clinical periodontal parameters (168, 169). The meta-analysis showed that, beyond enhancing the management of disease activity in RA, NSPT alleviated the systemic biomarker serum CRP among individuals with RA. It also exhibited a tendency of decreased ESR, morning stiffness, RF, TNF-α, and IL-6 (170). Therefore, promoting and developing NSPT for RA patients should be prioritized. However, due to issues such as selection bias and frequent changes in RA medication, there remains a pressing need for well-designed studies to further investigate this topic. At present, it remains challenging to definitively assess the effect of periodontal treatment on RA. Nonetheless, the promotion and development of NSPT for RA patients should not be overlooked.
Another approach targets the virulence factors of microorganisms. FimA, an important bacterial protein of P. gingivalis, demonstrated the ability to minimize alveolar bone loss and arthritic bone destruction when preincubated with FimA antibody. It also lowered the adhesion and aggregation of bacteria on gingival and synovial fibroblasts derived from humans (116). A distinct amino acid sequence within the catalytic domain of RgpA exhibits homology to the sequence of type II collagen. Pre-immunization rats with the purified recombinant structural region of RgpA similarly reduced joint inflammation and bone erosion (119). Targeting key virulence factors of P. gingivalis through preimmunization or small molecule inhibitors have the potential to diminish the likelihood of pathogens spreading to distal tissues and decrease their ability to trigger or exacerbate pathological changes in RA.
Recent studies have shown that DMARDs can improve clinical parameters of PD while treating RA. To eliminate the potential confounding influence of patients adopting proper oral hygiene habits, the investigators selected a 4-week follow-up period. Following 4 weeks of treatment with methotrexate (MTX), hydroxychloroquine (HCQ), and sulfasalazine (SSZ), patients diagnosed with RA and comorbid periodontitis exhibited improvements in CAL and probing depths when compared to periodontally healthy subjects. Moreover, MTX alone reduced probing depth after 3 months in contrast to the combination of MTX with SSZ and HCQ. Other drugs, including the JAK-inhibitor baricitinib, tocilizumab, rituximab, and anti-TNF-α monoclonal antibodies, also provide similar remission in PD (171). Paradoxically, some studies have questioned the treatment effectiveness of DMARDs in PD. Treatment with synthetic DMARDs, alone or in combination with another synthetic or biologic DMARD, for 6 months did not significantly improve periodontal parameters, nor did it affect the presence of P. gingivalis (172). In contrast, no improvement in the periodontal inflammatory surface area or in anti-P. gingivalis antibody levels was observed after 2 months of treatment with MTX or 3–6 months of treatment with anti-TNF-α antibody (173). The heterogeneity of periodontal parameters, variations in monitoring period duration, and the small quantity of randomized controlled trials with diverse DMARDs make it difficult to identify the most effective DMARD for treating PD. This requires further investigation.
Plant-derived products have been proposed as potential modulators of both microbiota and host responses. Curcumin, the primary curcuminoid in turmeric, is an effective antimicrobial, antioxidant, and anti-inflammatory agent against P. gingivalis infections and biofilm formation. It suppresses the pro-inflammatory response of Th17 cells and enhances the immunoregulatory functions of Tregs, yielding a dual therapeutic effect (174). Clinically, topical curcumin has been shown to reduce microbial load in patients with chronic PD (175). Curcumin can inhibit P. gingivalis biofilm formation by more than 80%, even at very low concentrations (20 μg/mL) (176).
8 Conclusion and outlook
An increasing body of preclinical animal studies and epidemiological research supports a close association between RA and the oral microbiome. This review explores the role of oral pathogens in the initiation and progression of RA, and highlights the presence and potential effects of various pathogenic species in the oral microbiota of RA patients. Most clinical studies have found that dysbiosis of the oral microbiome is already evident during the preclinical phase of RA. Pathogenic bacteria and their virulence factors may enter systemic circulation, elicit immune responses, and be detectable through the identification of bacterial DNA in peripheral samples. The review also analyzes key virulence factors of oral microbes, their pathogenic mechanisms, and their impact on host immune responses. Notably, P. gingivalis infection appears to increase the risk of developing RA and may reduce the therapeutic efficacy of DMARDs. However, most clinical studies are cross-sectional in nature and are subject to heterogeneity and confounding factors, making it difficult to establish definitive causal relationships. Identifying reliable microbial biomarkers for RA diagnosis or prediction remains a major challenge. In contrast, animal studies have provided stronger evidence that P. gingivalis and its major virulence factors accelerate the progression of experimental arthritis. These pathogenic effects are primarily mediated through four mechanisms (1): protein citrullination, (2) immune evasion, (3) modulation of host immune responses, and (4) promotion of bone destruction. It is important to note that current studies predominantly focus on the role of a single bacterial species in RA pathogenesis. Given that periodontitis is a polymicrobial and chronic inflammatory condition, future research should also investigate how microbial consortia contribute to RA development.
The unique pathogenicity of P. gingivalis remains a subject of ongoing debate. Although experimental animal models have demonstrated RA-related alterations following P. gingivalis exposure, the complexity of human disease makes the available evidence inconsistent, and the observed associations do not establish causality. Since RA primarily affects the joints, human studies, due to ethical and humanitarian considerations, are largely limited to analyses of the oral cavity and peripheral blood. As a result, there are limited opportunities to directly investigate changes at the primary site of disease. Although several studies have detected periodontal pathogens in synovial fluid and tissue, the specific pathogenic features, migration pathways from the oral cavity to the joints, and key molecular mediators involved in this process remain to be fully elucidated.
In conclusion, the roles of PD, P. gingivalis, and other periodontal pathogens in the pathogenesis of RA remain incompletely understood. The underlying mechanisms by which non-surgical periodontal therapy improves RA outcomes also require further clarification. Well-designed clinical trials with large sample sizes and extended follow-up periods are needed to provide more robust evidence. In the future, rigorous, standardized, and large-scale prospective studies, along with in-depth mechanistic investigations, are essential to clarify the causal relationship between oral pathogens and RA. Moreover, exploring interventions targeting the oral microbiome may offer valuable insights for RA prevention and management, potentially contributing to personalized therapeutic strategies.
Author contributions
XG: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. SW: Investigation, Writing – original draft, Writing – review & editing. JD: Formal Analysis, Writing – original draft. WWL: Methodology, Resources, Writing – review & editing. JX: Methodology, Resources, Writing – review & editing. MW: Data curation, Formal Analysis, Resources, Writing – review & editing. HS: Project administration, Software, Visualization, Writing – review & editing. YM: Project administration, Software, Visualization, Writing – review & editing. WL: Project administration, Supervision, Writing – review & editing. LZ: Project administration, Supervision, Writing – review & editing. ML: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Finckh A, Gilbert B, Hodkinson B, Bae S, Thomas R, Deane KD, et al. Global epidemiology of rheumatoid arthritis. Nat Rev Rheumatol. (2022) 18:591–602. doi: 10.1038/s41584-022-00827-y
2. Shi G, Liao X, Lin Z, Liu W, Luo X, Zhan H, et al. Estimation of the global prevalence, incidence, years lived with disability of rheumatoid arthritis in 2019 and forecasted incidence in 2040: results from the Global Burden of Disease Study 2019. Clin Rheumatol. (2023) 42:2297–309. doi: 10.1007/s10067-023-06628-2
3. Scott IC, Whittle R, Bailey J, Twohig H, Hider SL, Mallen CD, et al. Rheumatoid arthritis, psoriatic arthritis, and axial spondyloarthritis epidemiology in England from 2004 to 2020: An observational study using primary care electronic health record data. Lancet Reg Health Eur. (2022) 23:100519. doi: 10.1016/j.lanepe.2022.100519
4. McInnes IB and Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
5. MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. (2000) 43:30–7. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B
6. Van Der Woude D, Houwing-Duistermaat JJ, Toes RE, Huizinga TW, Thomson W, Worthington J, et al. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. (2009) 60:916–23. doi: 10.1002/art.24385
7. Scherer HU, Haupl T, and Burmester GR. The etiology of rheumatoid arthritis. J Autoimmun. (2020) 110:102400. doi: 10.1016/j.jaut.2019.102400
8. Di Matteo A, Bathon JM, and Emery P. Rheumatoid arthritis. Lancet. (2023) 402:2019–33. doi: 10.1016/S0140-6736(23)01525-8
9. Saevarsdottir S, Stefansdottir L, Sulem P, Thorleifsson G, Ferkingstad E, Rutsdottir G, et al. Multiomics analysis of rheumatoid arthritis yields sequence variants that have large effects on risk of the seropositive subset. Ann Rheum Dis. (2022) 81:1085–95. doi: 10.1136/annrheumdis-2021-221754
10. James EA, Holers VM, Iyer R, Prideaux EB, Rao NL, Rims C, et al. Multifaceted immune dysregulation characterizes individuals at-risk for rheumatoid arthritis. Nat Commun. (2023) 14:7637. doi: 10.1038/s41467-023-43091-8
11. Liu X, Tedeschi SK, Barbhaiya M, Leatherwood CL, Speyer CB, Lu B, et al. Impact and timing of smoking cessation on reducing risk of rheumatoid arthritis among women in the nurses' Health studies. Arthritis Care Res (Hoboken). (2019) 71:914–24. doi: 10.1002/acr.23837
12. Bellatin MF, Han M, Fallena M, Fan L, Xia D, Olsen N, et al. Production of autoantibodies against citrullinated antigens/peptides by human B cells. J Immunol. (2012) 188:3542–50. doi: 10.4049/jimmunol.1100577
13. Wouters F, Maurits MP, Van Boheemen L, Verstappen M, Mankia K, Matthijssen X, et al. Determining in which pre-arthritis stage HLA-shared epitope alleles and smoking exert their effect on the development of rheumatoid arthritis. Ann Rheum Dis. (2022) 81:48–55. doi: 10.1136/annrheumdis-2021-220546
14. Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol. (2018) 14:542–57. doi: 10.1038/s41584-018-0070-0
15. Lin L, Zhang K, Xiong Q, Zhang J, Cai B, Huang Z, et al. Gut microbiota in pre-clinical rheumatoid arthritis: From pathogenesis to preventing progression. J Autoimmun. (2023) 141:103001. doi: 10.1016/j.jaut.2023.103001
16. Crowson CS, Matteson EL, Davis JR, and Gabriel SE. Contribution of obesity to the rise in incidence of rheumatoid arthritis. Arthritis Care Res (Hoboken). (2013) 65:71–7. doi: 10.1002/acr.21660
17. Tedeschi SK, Cui J, Arkema EV, Robinson WH, Sokolove J, Lingampalli N, et al. Elevated BMI and antibodies to citrullinated proteins interact to increase rheumatoid arthritis risk and shorten time to diagnosis: A nested case-control study of women in the Nurses' Health Studies. Semin Arthritis Rheum. (2017) 46:692–98. doi: 10.1016/j.semarthrit.2016.09.001
18. Petrovska N, Prajzlerova K, Vencovsky J, Senolt L, and Filkova M. The pre-clinical phase of rheumatoid arthritis: From risk factors to prevention of arthritis. Autoimmun Rev. (2021) 20:102797. doi: 10.1016/j.autrev.2021.102797
19. Firestein GS and McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. (2017) 46:183–96. doi: 10.1016/j.immuni.2017.02.006
20. Sokolove J, Bromberg R, Deane KD, Lahey LJ, Derber LA, Chandra PE, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PloS One. (2012) 7:e35296. doi: 10.1371/journal.pone.0035296
21. Gravallese EM and Firestein GS. Rheumatoid arthritis - common origins, divergent mechanisms. N Engl J Med. (2023) 388:529–42. doi: 10.1056/NEJMra2103726
22. Curran AM, Naik P, Giles JT, and Darrah E. PAD enzymes in rheumatoid arthritis: pathogenic effectors and autoimmune targets. Nat Rev Rheumatol. (2020) 16:301–15. doi: 10.1038/s41584-020-0409-1
23. Vossenaar ER, Zendman AJ, Van Venrooij WJ, and Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays. (2003) 25:1106–18. doi: 10.1002/bies.10357
24. Foulquier C, Sebbag M, Clavel C, Chapuy-Regaud S, Al Badine R, Méchin M, et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheumatism. (2007) 56:3541–53. doi: 10.1002/art.22983
25. Kinloch A, Lundberg K, Wait R, Wegner N, Lim NH, Zendman AJ, et al. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. (2008) 58:2287–95. doi: 10.1002/art.23618
26. Van Beers JJ, Schwarte CM, Stammen-Vogelzangs J, Oosterink E, Bozic B, and Pruijn GJ. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and beta-actin. Arthritis Rheum. (2013) 65:69–80. doi: 10.1002/art.37720
27. Tilvawala R, Nguyen SH, Maurais AJ, Nemmara VV, Nagar M, Salinger AJ, et al. The rheumatoid arthritis-associated citrullinome. Cell Chem Biol. (2018) 25:691–704. doi: 10.1016/j.chembiol.2018.03.002
28. Wang F, Chen FF, Gao WB, Wang HY, Zhao NW, Xu M, et al. Identification of citrullinated peptides in the synovial fluid of patients with rheumatoid arthritis using LC-MALDI-TOF/TOF. Clin Rheumatol. (2016) 35:2185–94. doi: 10.1007/s10067-016-3247-4
29. Curran AM, Girgis AA, Jang Y, Crawford JD, Thomas MA, Kawalerski R, et al. Citrullination modulates antigen processing and presentation by revealing cryptic epitopes in rheumatoid arthritis. Nat Commun. (2023) 14:1061. doi: 10.1038/s41467-023-36620-y
30. McElwee MK, Dileepan T, Mahmud SA, and Jenkins MK. The CD4+ T cell repertoire specific for citrullinated peptides shows evidence of immune tolerance. J Exp Med. (2023) 220:e20230209. doi: 10.1084/jem.20230209
31. Stavnezer J, Guikema JEJ, and Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
32. Qiao Y, Wang Z, Li Y, Han Y, Zhou Y, and Cao X. Rheumatoid arthritis risk in periodontitis patients: A systematic review and meta-analysis. Joint Bone Spine. (2020) 87:556–64. doi: 10.1016/j.jbspin.2020.04.024
33. Maldonado A, Pirracchio L, Imber JC, Burgin W, Moller B, Sculean A, et al. Citrullination in periodontium is associated with Porphyromonas gingivalis. Arch Oral Biol. (2020) 114:104695. doi: 10.1016/j.archoralbio.2020.104695
34. Potempa J, Mydel P, and Koziel J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13:606–20. doi: 10.1038/nrrheum.2017.132
35. Harvey GP, Fitzsimmons TR, Dhamarpatni AA, Marchant C, Haynes DR, and Bartold PM. Expression of peptidylarginine deiminase-2 and -4, citrullinated proteins and anti-citrullinated protein antibodies in human gingiva. J Periodontal Res. (2013) 48:252–61. doi: 10.1111/jre.12002
36. Darrah E and Andrade F. Rheumatoid arthritis and citrullination. Curr Opin Rheumatol. (2018) 30:72–8. doi: 10.1097/BOR.0000000000000452
37. Ge C, Tong D, Liang B, Lonnblom E, Schneider N, Hagert C, et al. Anti-citrullinated protein antibodies cause arthritis by cross-reactivity to joint cartilage. JCI Insight. (2017) 2:e93688. doi: 10.1172/jci.insight.93688
38. Yu HC and Lu MC. The roles of anti-citrullinated protein antibodies in the immunopathogenesis of rheumatoid arthritis. Tzu Chi Med J. (2019) 31:5–10. doi: 10.4103/tcmj.tcmj_116_18
39. Wegner N, Wait R, Sroka A, Eick S, Nguyen K, Lundberg K, et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheumatism. (2010) 62:2662–72. doi: 10.1002/art.27552
40. Chow YC, Yam HC, Gunasekaran B, Lai WY, Wo WY, Agarwal T, et al. Implications of Porphyromonas gingivalis peptidyl arginine deiminase and gingipain R in human health and diseases. Front Cell Infect Microbiol. (2022) 12:987683. doi: 10.3389/fcimb.2022.987683
41. Montgomery AB, Kopec J, Shrestha L, Thezenas ML, Burgess-Brown NA, Fischer R, et al. Crystal structure of Porphyromonas gingivalis peptidylarginine deiminase: implications for autoimmunity in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1255–61. doi: 10.1136/annrheumdis-2015-207656
42. Ingegnoli F, Castelli R, and Gualtierotti R. Rheumatoid factors: clinical applications. Dis Markers. (2013) 35:727–34. doi: 10.1155/2013/726598
43. Dahlstrand RA, Khamzeh A, Venkatakrishnan V, Persson T, Gabl M, Savolainen O, et al. Porphyromonas gingivalis produce neutrophil specific chemoattractants including short chain fatty acids. Front Cell Infect Microbiol. (2020) 10:620681. doi: 10.3389/fcimb.2020.620681
44. Olsen I and Hajishengallis G. Major neutrophil functions subverted by Porphyromonas gingivalis. J Oral Microbiol. (2016) 8:30936. doi: 10.3402/jom.v8.30936
45. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
46. Lee JA, Mikuls TR, Deane KD, Sayles HR, Thiele GM, Edison JD, et al. Serum antibodies to periodontal pathogens prior to rheumatoid arthritis diagnosis: A case-control study. Semin Arthritis Rheum. (2023) 59:152176. doi: 10.1016/j.semarthrit.2023.152176
47. Trindade D, Carvalho R, MaChado V, Chambrone L, Mendes JJ, and Botelho J. Prevalence of periodontitis in dentate people between 2011 and 2020: A systematic review and meta-analysis of epidemiological studies. J Clin Periodontol. (2023) 50:604–26. doi: 10.1111/jcpe.13769
48. Nascimento GG, Alves-Costa S, and Romandini M. Burden of severe periodontitis and edentulism in 2021, with projections up to 2050: The Global Burden of Disease 2021 study. J Periodontal Res. (2024) 59:823–67. doi: 10.1111/jre.13337
49. Sanz M, Marco DCA, Jepsen S, Gonzalez-Juanatey JR, D'Aiuto F, Bouchard P, et al. Periodontitis and cardiovascular diseases: Consensus report. J Clin Periodontol. (2020) 47:268–88. doi: 10.1111/jcpe.13189
50. Di Stefano M, Polizzi A, Santonocito S, Romano A, Lombardi T, and Isola G. Impact of oral microbiome in periodontal health and periodontitis: A critical review on prevention and treatment. Int J Mol Sci. (2022) 23:5142. doi: 10.3390/ijms23095142
51. Rodríguez-Lozano B, González-Febles J, Garnier-Rodríguez JL, Dadlani S, Bustabad-Reyes S, Sanz M, et al. Association between severity of periodontitis and clinical activity in rheumatoid arthritis patients: a case-control study. Arthritis Res Ther. (2019) 21:27. doi: 10.1186/s13075-019-1808-z
52. Schmickler J, Rupprecht A, Patschan S, Patschan D, Müller GA, Haak R, et al. Cross-sectional evaluation of periodontal status and microbiologic and rheumatoid parameters in a large cohort of patients with rheumatoid arthritis. J Periodontol. (2017) 88:368–79. doi: 10.1902/jop.2016.160355
53. Eriksson K, Fei G, Lundmark A, Benchimol D, Lee L, Hu Y, et al. Periodontal health and oral microbiota in patients with rheumatoid arthritis. J Clin Med. (2019) 8:630. doi: 10.3390/jcm8050630
54. Rak D, Kulloli AM, Shetty SK, Tripathy S, Mathur A, Mehta V, et al. Correlation between rheumatoid arthritis and chronic periodontitis: a systematic review and meta-analysis. Minerva Dent Oral Sci. (2024) 73:294–302. doi: 10.23736/S2724-6329.23.04891-X
55. Ortiz P, Bissada NF, Palomo L, Han YW, Al-Zahrani MS, Panneerselvam A, et al. Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J Periodontol. (2009) 80:535–40. doi: 10.1902/jop.2009.080447
56. Silva DS, De Vries C, Rovisco J, Serra S, Kaminska M, Mydel P, et al. The impact of periodontitis and periodontal treatment on rheumatoid arthritis outcomes: an exploratory clinical trial. Rheumatol (Oxford). (2025) 64:1679–88. doi: 10.1093/rheumatology/keae358
57. Baker JL, Mark WJ, Kauffman KM, McLean JS, and He X. The oral microbiome: diversity, biogeography and human health. Nat Rev Microbiol. (2024) 22:89–104. doi: 10.1038/s41579-023-00963-6
58. Sedghi L, DiMassa V, Harrington A, Lynch SV, and Kapila YL. The oral microbiome: Role of key organisms and complex networks in oral health and disease. Periodontol 2000. (2021) 87:107–31. doi: 10.1111/prd.12393
59. Kroese JM, Brandt BW, Buijs MJ, Crielaard W, Lobbezoo F, Loos BG, et al. Differences in the oral microbiome in patients with early rheumatoid arthritis and individuals at risk of rheumatoid arthritis compared to healthy individuals. Arthritis Rheumatol. (2021) 73:1986–93. doi: 10.1002/art.41780
60. Tong Y, Zheng L, Qing P, Zhao H, Li Y, Su L, et al. Oral microbiota perturbations are linked to high risk for rheumatoid arthritis. Front Cell Infect Microbiol. (2020) 9:475. doi: 10.3389/fcimb.2019.00475
61. Arleevskaya MI, Boulygina EA, Larionova R, Validov S, Kravtsova O, Shagimardanova EI, et al. Anti-citrullinated peptide antibodies control oral porphyromonas and aggregatibacter species in patients with rheumatoid arthritis. Int J Mol Sci. (2022) 23:12599. doi: 10.3390/ijms232012599
62. Cheng Z, Do T, Mankia K, Meade J, Hunt L, Clerehugh V, et al. Dysbiosis in the oral microbiomes of anti-CCP positive individuals at risk of developing rheumatoid arthritis. Ann Rheum Dis. (2021) 80:162–68. doi: 10.1136/annrheumdis-2020-216972
63. Kim J, Jung H, Baek I, Nam Y, Kang J, Chung MK, et al. Differential effects of periodontal microbiome on the rheumatoid factor induction during rheumatoid arthritis pathogenesis. Sci Rep. (2022) 12:19636. doi: 10.1038/s41598-022-21788-y
64. Mankia K, Cheng Z, Do T, Hunt L, Meade J, Kang J, et al. Prevalence of periodontal disease and periodontopathic bacteria in anti–cyclic citrullinated protein antibody–positive at-risk adults without arthritis. JAMA Netw Open. (2019) 2:e195394. doi: 10.1001/jamanetworkopen.2019.5394
65. Esberg A, Johansson L, Johansson I, and Dahlqvist SR. Oral microbiota identifies patients in early onset rheumatoid arthritis. Microorganisms. (2021) 9:1657. doi: 10.3390/microorganisms9081657
66. Wolff B, Berger T, Frese C, Max R, Blank N, Lorenz HM, et al. Oral status in patients with early rheumatoid arthritis: a prospective, case-control study. Rheumatol (Oxford). (2014) 53:526–31. doi: 10.1093/rheumatology/ket362
67. Kozhakhmetov S, Babenko D, Issilbayeva A, Nurgaziyev M, Kozhakhmetova S, Meiramova A, et al. Oral microbial signature of rheumatoid arthritis in female patients. J Clin Med. (2023) 12:3694. doi: 10.3390/jcm12113694
68. Beyer K, Zaura E, Brandt BW, Buijs MJ, Brun JG, Crielaard W, et al. Subgingival microbiome of rheumatoid arthritis patients in relation to their disease status and periodontal health. PloS One. (2018) 13:e202278. doi: 10.1371/journal.pone.0202278
69. Lehenaff R, Tamashiro R, Nascimento MM, Lee K, Jenkins R, Whitlock J, et al. Subgingival microbiome of deep and shallow periodontal sites in patients with rheumatoid arthritis: a pilot study. BMC Oral Health. (2021) 21:248. doi: 10.1186/s12903-021-01597-x
70. Mikuls TR, Walker C, Qiu F, Yu F, Thiele GM, Alfant B, et al. The subgingival microbiome in patients with established rheumatoid arthritis. Rheumatol (Oxford). (2018) 57:1162–72. doi: 10.1093/rheumatology/key052
71. Lopez Oliva I, Paropkari AD, Saraswat S, Serban S, Yonel Z, Sharma P, et al. Dysbiotic subgingival microbial communities in periodontally healthy patients with rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:1008–13. doi: 10.1002/art.40485
72. Ziebolz D, Pabel SO, Lange K, Krohn Grimberghe B, Hornecker E, and Mausberg RF. Clinical periodontal and microbiologic parameters in patients with rheumatoid arthritis. J Periodontol. (2011) 82:1424–32. doi: 10.1902/jop.2011.100481
73. Corrêa JD, Fernandes GR, Calderaro DC, Mendonça SMS, Silva JM, Albiero ML, et al. Oral microbial dysbiosis linked to worsened periodontal condition in rheumatoid arthritis patients. Sci Rep. (2019) 9:8379. doi: 10.1038/s41598-019-44674-6
74. Chen Y, Hung W, Chou Y, Lai C, Peng P, Jhou P, et al. Subgingival microbiome in rheumatoid arthritis patients with periodontitis. Int J Mol Sci. (2022) 23:9883. doi: 10.3390/ijms23179883
75. Cheah CW, Al-Maleki AR, Vaithilingam RD, Vadivelu J, Sockalingam S, Baharuddin NA, et al. Associations between inflammation-related LL-37 with subgingival microbial dysbiosis in rheumatoid arthritis patients. Clin Oral Investig. (2022) 26:4161–72. doi: 10.1007/s00784-022-04388-y
76. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. (2015) 21:895–905. doi: 10.1038/nm.3914
77. Eriksson K, Lundmark A, Delgado LF, Hu YOO, Fei G, Lee L, et al. Salivary microbiota and host-inflammatory responses in periodontitis affected individuals with and without rheumatoid arthritis. Front Cell Infect Microbiol. (2022) 12:841139. doi: 10.3389/fcimb.2022.841139
78. De Jesus VC, Singh M, Schroth RJ, Chelikani P, and Hitchon CA. Association of bitter taste receptor T2R38 polymorphisms, oral microbiota, and rheumatoid arthritis. Curr Issues Mol Biol. (2021) 43:1460–72. doi: 10.3390/cimb43030103
79. Eren AM, Borisy GG, Huse SM, and Mark Welch JL. Oligotyping analysis of the human oral microbiome. Proc Natl Acad Sci. (2014) 111:E2875–84. doi: 10.1073/pnas.1409644111
80. Li Y, Guo R, Oduro PK, Sun T, Chen H, Yi Y, et al. The relationship between porphyromonas gingivalis and rheumatoid arthritis: A meta-analysis. Front Cell Infect Microbiol. (2022) 12:956417. doi: 10.3389/fcimb.2022.956417
81. Hashimoto M, Yamazaki T, Hamaguchi M, Morimoto T, Yamori M, Asai K, et al. Periodontitis and porphyromonas gingivalis in preclinical stage of arthritis patients. PloS One. (2015) 10:e122121. doi: 10.1371/journal.pone.0122121
82. Oluwagbemigun K, Yucel-Lindberg T, Dietrich T, Tour G, Sherina N, Hansson M, et al. A cross-sectional investigation into the association betweenPorphyromonas gingivalis and autoantibodies to citrullinated proteins in a German population. Ther Adv Musculoskelet Dis. (2019) 11:1759720X19883152. doi: 10.1177/1759720X19883152
83. Ceccarelli F, Orru G, Pilloni A, Bartosiewicz I, Perricone C, Martino E, et al. Porphyromonas gingivalis in the tongue biofilm is associated with clinical outcome in rheumatoid arthritis patients. Clin Exp Immunol. (2018) 194:244–52. doi: 10.1111/cei.13184
84. De Smit M, Westra J, Vissink A, Doornbos-Van DMB, Brouwer E, and Van Winkelhoff AJ. Periodontitis in established rheumatoid arthritis patients: a cross-sectional clinical, microbiological and serological study. Arthritis Res Ther. (2012) 14:R222. doi: 10.1186/ar4061
85. Mikuls TR, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, et al. Periodontitis and porphyromonas gingivalisin patients with rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:1090–100. doi: 10.1002/art.38348
86. Wan JT, Taib H, Majdiah WMW, Mohamad S, and Syamimee WGW. Periodontal health status, Porphyromonas gingivalis and anti-cyclic citrullinated peptide antibodies among rheumatoid arthritis patients. Int Immunopharmacol. (2023) 124:110940. doi: 10.1016/j.intimp.2023.110940
87. Laugisch O, Wong A, Sroka A, Kantyka T, Koziel J, Neuhaus K, et al. Citrullination in the periodontium—a possible link between periodontitis and rheumatoid arthritis. Clin Oral Investig. (2016) 20:675–83. doi: 10.1007/s00784-015-1556-7
88. Huang Y and Ni S. Aggregatibacter actinomycetemcomitans with periodontitis and rheumatoid arthritis. Int Dent J. (2024) 74:58–65. doi: 10.1016/j.identj.2023.06.011
89. Jacobson JJ, Patel B, Asher G, Woolliscroft JO, and Schaberg D. Oral staphylococcus in older subjects with rheumatoid arthritis. J Am Geriatr Soc. (1997) 45:590–93. doi: 10.1111/j.1532-5415.1997.tb03092.x
90. Jackson MS, Bagg J, Gupta MN, and Sturrock RD. Oral carriage of staphylococci in patients with rheumatoid arthritis. Rheumatol (Oxford). (1999) 38:572–75. doi: 10.1093/rheumatology/38.6.572
91. Moen K, Brun JG, Valen M, Skartveit L, Eribe EKR, Olsen I, et al. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin Exp Rheumatol. (2006) 24:656. doi: 10.1007/s10725-008-9267-6
92. Reichert S, Haffner M, Keyßer G, Schäfer C, Stein JM, Schaller HG, et al. Detection of oral bacterialDNAin synovial fluid. J Clin Periodontol. (2013) 40:591–98. doi: 10.1111/jcpe.12102
93. Totaro MC, Cattani P, Ria F, Tolusso B, Gremese E, Fedele AL, et al. Porphyromonas gingivalis and the pathogenesis of rheumatoid arthritis: analysis of various compartments including the synovial tissue. Arthritis Res Ther. (2013) 15:R66. doi: 10.1186/ar4243
94. Martinez Martinez RE, Abud Mendoza C, Patiño Marin N, Rizo Rodríguez JC, Little JW, and Loyola Rodríguez JP. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol. (2009) 36:1004–10. doi: 10.1111/j.1600-051X.2009.01496.x
95. Zhao Y, Chen B, Li S, Yang L, Zhu D, Wang Y, et al. Detection and characterization of bacterial nucleic acids in culture-negative synovial tissue and fluid samples from rheumatoid arthritis or osteoarthritis patients. Sci Rep. (2018) 8:14305. doi: 10.1038/s41598-018-32675-w
96. Zeisel MB, Neff LA, Randle J, Klein J, Sibilia J, and Wachsmann D. Impaired release of IL-18 from fibroblast-like synoviocytes activated with protein I/II, a pathogen-associated molecular pattern from oral streptococci, results from defective translation of IL-18 mRNA in pro-IL-18. Cell Microbiol. (2004) 6:593–98. doi: 10.1111/j.1462-5822.2004.00385.x
97. Holt SC and Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the "red complex", a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. (2005) 38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x
98. Chukkapalli S, Rivera-Kweh M, Gehlot P, Velsko I, Bhattacharyya I, Calise SJ, et al. Periodontal bacterial colonization in synovial tissues exacerbates collagen-induced arthritis in B10.RIII mice. Arthritis Res Ther. (2016) 18:161. doi: 10.1186/s13075-016-1056-4
99. Moentadj R, Wang Y, Bowerman K, Rehaume L, Nel H, O Cuiv P, et al. Streptococcus species enriched in the oral cavity of patients with RA are a source of peptidoglycan-polysaccharide polymers that can induce arthritis in mice. Ann Rheum Dis. (2021) 80:573–81. doi: 10.1136/annrheumdis-2020-219009
100. Ebbers M, Lübcke PM, Volzke J, Kriebel K, Hieke C, Engelmann R, et al. Interplay between P. gingivalis, F. nucleatum and A. actinomycetemcomitans in murine alveolar bone loss, arthritis onset and progression. Sci Rep. (2018) 8:15129. doi: 10.1038/s41598-018-33129-z
101. De Aquino SG, Abdollahi-Roodsaz S, Koenders MI, Van De Loo FA, Pruijn GJ, Marijnissen RJ, et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J Immunol. (2014) 192:4103–11. doi: 10.4049/jimmunol.1301970
102. Eriksson K, Lönnblom E, Tour G, Kats A, Mydel P, Georgsson P, et al. Effects by periodontitis on pristane-induced arthritis in rats. J Transl Med. (2016) 14:311. doi: 10.1186/s12967-016-1067-6
103. Courbon G, Rinaudo-Gaujous M, Blasco-Baque V, Auger I, Caire R, Mijola L, et al. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann Rheum Dis. (2019) 78:594–99. doi: 10.1136/annrheumdis-2018-213697
104. Buschhart A, Bolten L, Volzke J, Ekat K, Kneitz S, Mikkat S, et al. Periodontal pathogens alter the synovial proteome. Periodontal pathogens do not exacerbate macroscopic arthritis but alter the synovial proteome in mice. PloS One. (2020) 15:e242868. doi: 10.1371/journal.pone.0242868
105. Sato K, Takahashi N, Kato T, Matsuda Y, Yokoji M, Yamada M, et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci Rep. (2017) 7:6955. doi: 10.1038/s41598-017-07196-7
106. Yang C, Hu Z, Wang L, Fang L, Wang X, Li Q, et al. Porphyromonas gingivalis with collagen immunization induces ACPA-positive rheumatoid arthritis in C3H mice. Clin Immunol. (2024) 258:109859. doi: 10.1016/j.clim.2023.109859
107. Zhou N, Zou F, Cheng X, Huang Y, Zou H, Niu Q, et al. Porphyromonas gingivalis induces periodontitis, causes immune imbalance, and promotes rheumatoid arthritis. J Leukoc Biol. (2021) 110:461–73. doi: 10.1002/JLB.3MA0121-045R
108. Hamamoto Y, Ouhara K, Munenaga S, Shoji M, Ozawa T, Hisatsune J, et al. Effect of Porphyromonas gingivalis infection on gut dysbiosis and resultant arthritis exacerbation in mouse model. Arthritis Res Ther. (2020) 22:249. doi: 10.1186/s13075-020-02348-z
109. Yamakawa M, Ouhara K, Kajiya M, Munenaga S, Kittaka M, Yamasaki S, et al. Porphyromonas gingivalis infection exacerbates the onset of rheumatoid arthritis in SKG mice. Clin Exp Immunol. (2016) 186:177–89. doi: 10.1111/cei.12847
110. Marchesan JT, Gerow EA, Schaff R, Taut AD, Shin SY, Sugai J, et al. Porphyromonas gingivalis oral infection exacerbates the development and severity of collagen-induced arthritis. Arthritis Res Ther. (2013) 15:R186. doi: 10.1186/ar4376
111. Gully N, Bright R, Marino V, Marchant C, Cantley M, Haynes D, et al. Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PloS One. (2014) 9:e100838. doi: 10.1371/journal.pone.0100838
112. Maresz KJ, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PloS Pathog. (2013) 9:e1003627. doi: 10.1371/journal.ppat.1003627
113. Jung H, Jung SM, Rim YA, Park N, Nam Y, Lee J, et al. Arthritic role of Porphyromonas gingivalis in collagen-induced arthritis mice. PloS One. (2017) 12:e188698. doi: 10.1371/journal.pone.0188698
114. Sandal I, Karydis A, Luo J, Prislovsky A, Whittington KB, Rosloniec EF, et al. Bone loss and aggravated autoimmune arthritis in HLA-DRβ1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res Ther. (2016) 18:249. doi: 10.1186/s13075-016-1143-6
115. Muñoz-Atienza E, Flak MB, Sirr J, Paramonov NA, Aduse-Opoku J, Pitzalis C, et al. TheP. gingivalis autocitrullinome is not a target for ACPA in early rheumatoid arthritis. J Dent Res. (2020) 99:456–62. doi: 10.1177/0022034519898144
116. Jeong SH, Nam Y, Jung H, Kim J, Rim YA, Park N, et al. Interrupting oral infection of Porphyromonas gingivalis with anti-FimA antibody attenuates bacterial dissemination to the arthritic joint and improves experimental arthritis. Exp Mol Med. (2018) 50:e460. doi: 10.1038/emm.2017.301
117. De Aquino SG, Talbot J, Sônego F, Turato WM, Grespan R, Avila-Campos MJ, et al. The aggravation of arthritis by periodontitis is dependent of IL-17 receptor A activation. J Clin Periodontol. (2017) 44:881–91. doi: 10.1111/jcpe.12743
118. Munenaga S, Ouhara K, Hamamoto Y, Kajiya M, Takeda K, Yamasaki S, et al. The involvement of C5a in the progression of experimental arthritis with Porphyromonas gingivalis infection in SKG mice. Arthritis Res Ther. (2018) 20:247. doi: 10.1186/s13075-018-1744-3
119. Peng H, Chen S, Siao S, Chang JT, Xue T, Lee Y, et al. Targeting a cysteine protease from a pathobiont alleviates experimental arthritis. Arthritis Res Ther. (2020) 22:114. doi: 10.1186/s13075-020-02205-z
120. Queiroz Junior CM, Madeira MFM, Coelho FM, De Oliveira CR, Cândido LCM, Garlet GP, et al. Experimental arthritis exacerbatesA ggregatibacter actinomycetemcomitans -induced periodontitis in mice. J Clin Periodontol. (2012) 39:608–16. doi: 10.1111/j.1600-051X.2012.01886.x
121. Rosenstein ED, Greenwald RA, Kushner LJ, and Weissmann G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation. (2004) 28:311–18. doi: 10.1007/s10753-004-6641-z
122. Bender P, Bürgin WB, Sculean A, and Eick S. Serum antibody levels against Porphyromonas gingivalis in patients with and without rheumatoid arthritis – a systematic review and meta-analysis. Clin Oral Investig. (2017) 21:33–42. doi: 10.1007/s00784-016-1938-5
123. Lee JY, Choi IA, Kim J, Kim K, Lee EY, Lee EB, et al. Association between anti-Porphyromonas gingivalis or anti-α-enolase antibody and severity of periodontitis or rheumatoid arthritis (RA) disease activity in RA. BMC Musculoskelet Disord. (2015) 16:190. doi: 10.1186/s12891-015-0647-6
124. Mikuls TR, Payne JB, Reinhardt RA, Thiele GM, Maziarz E, Cannella AC, et al. Antibody responses to Porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int Immunopharmacol. (2009) 9:38–42. doi: 10.1016/j.intimp.2008.09.008
125. Hitchon CA, Chandad F, Ferucci ED, Willemze A, Ioan-Facsinay A, Van Der Woude D, et al. Antibodies toporphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J Rheumatol. (2010) 37:1105–12. doi: 10.3899/jrheum.091323
126. Okada M, Kobayashi T, Ito S, Yokoyama T, Komatsu Y, Abe A, et al. Antibody responses to periodontopathic bacteria in relation to rheumatoid arthritis in Japanese adults. J Periodontol. (2011) 82:1433–41. doi: 10.1902/jop.2011.110020
127. Kim J, Choi IA, Lee JY, Kim K, Kim S, Koo K, et al. Periodontal pathogens and the association between periodontitis and rheumatoid arthritis in Korean adults. J Periodontal Implant Sci. (2018) 48:347. doi: 10.5051/jpis.2018.48.6.347
128. Kononoff A, Arstila L, Pussinen P, Kautiainen H, Elfving P, Savolainen E, et al. Incidence of inflammatory joint diseases in Finland: results from a population-based epidemiological study. Rheumatol Int. (2017) 37:1693–700. doi: 10.1007/s00296-017-3779-1
129. Takeuchi-Hatanaka K, Koyama Y, Okamoto K, Sakaida K, Yamamoto T, and Takashiba S. Treatment resistance of rheumatoid arthritis relates to infection of periodontal pathogenic bacteria: a case–control cross-sectional study. Sci Rep. (2022) 12:12353. doi: 10.1038/s41598-022-16279-z
130. Arvikar SL, Collier DS, Fisher MC, Unizony S, Cohen GL, McHugh G, et al. Clinical correlations with Porphyromonas gingivalis antibody responses in patients with early rheumatoid arthritis. Arthritis Res Ther. (2013) 15:R109. doi: 10.1186/ar4289
131. Bello Gualtero JM, Lafaurie GI, Hoyos LX, Castillo DM, De Avila J, Munevar JC, et al. Periodontal disease in individuals with a genetic risk of developing arthritis and early rheumatoid arthritis: A cross-sectional study. J Periodontol. (2016) 87:346–56. doi: 10.1902/jop.2015.150455
132. Rinaudo-Gaujous M, Blasco-Baque V, Miossec P, Gaudin P, Farge P, Roblin X, et al. Infliximab induced a dissociated response of severe periodontal biomarkers in rheumatoid arthritis patients. J Clin Med. (2019) 8:751. doi: 10.3390/jcm8050751
133. Manoil D, Bostanci N, Mumcu G, Inanc N, Can M, Direskeneli H, et al. Novel and known periodontal pathogens residing in gingival crevicular fluid are associated with rheumatoid arthritis. J Periodontol. (2021) 92:359–70. doi: 10.1002/JPER.20-0295
134. Castillo DM, Lafaurie GI, Romero-Sánchez C, Delgadillo NA, Castillo Y, Bautista-Molano W, et al. The interaction effect of anti-rgpA and anti-PPAD antibody titers: an indicator for rheumatoid arthritis diagnosis. J Clin Med. (2023) 12:3027. doi: 10.3390/jcm12083027
135. Terato K, Waritani T, Fukai R, Shionoya H, Itoh H, and Katayama K. Contribution of bacterial pathogens to evoking serological disease markers and aggravating disease activity in rheumatoid arthritis. PloS One. (2018) 13:e190588. doi: 10.1371/journal.pone.0190588
136. Seror R, Le Gall-David S, Bonnaure-Mallet M, Schaeverbeke T, Cantagrel A, Minet J, et al. Anti-Porphyromonas gingivalis antibodies titres are associated with non-smoking status in early rheumatoid arthritis: Results from the ESPOIR cohort. Arthritis Rheumatol (Hoboken N.J.). (2015) 67:1729–37. doi: 10.1002/art.39118
137. Svärd A, Kastbom A, Ljungberg KR, Potempa B, Potempa J, Persson GR, et al. Antibodies against Porphyromonas gingivalis in serum and saliva and their association with rheumatoid arthritis and periodontitis. Data from two rheumatoid arthritis cohorts in Sweden. Front Immunol. (2023) 14:1183194. doi: 10.3389/fimmu.2023.1183194
138. Kharlamova N, Jiang X, Sherina N, Potempa B, Israelsson L, Quirke AM, et al. Antibodies toPorphyromonas gingivalis Indicate Interaction Between Oral Infection, Smoking, and Risk Genes in Rheumatoid Arthritis Etiology. Arthritis Rheumatol. (2016) 68:604–13. doi: 10.1002/art.39491
139. Davison E, Johnston W, Piela K, Rosier BT, Paterson M, Mira A, et al. The Subgingival Plaque Microbiome, Systemic Antibodies against Bacteria and Citrullinated Proteins following Periodontal Therapy. Pathogens. (2021) 10:193. doi: 10.3390/pathogens10020193
140. Quirke A, Lugli EB, Wegner N, Hamilton BC, Charles P, Chowdhury M, et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: a potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann Rheum Dis. (2014) 73:263–69. doi: 10.1136/annrheumdis-2012-202726
141. Sherina N, De Vries C, Kharlamova N, Sippl N, Jiang X, Brynedal B, et al. Antibodies to a citrullinated porphyromonas gingivalis epitope are increased in early rheumatoid arthritis, and can be produced by gingival tissue B cells: implications for a bacterial origin in RA etiology. Front Immunol. (2022) 13:804822. doi: 10.3389/fimmu.2022.804822
142. Konig MF, Paracha AS, Moni M, Bingham CO, and Andrade F. Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann Rheum Dis. (2015) 74:2054–61. doi: 10.1136/annrheumdis-2014-205385
143. Shimada A, Kobayashi T, Ito S, Okada M, Murasawa A, Nakazono K, et al. Expression of anti-Porphyromonas gingivalis peptidylarginine deiminase immunoglobulin G and peptidylarginine deiminase-4 in patients with rheumatoid arthritis and periodontitis. J Periodontal Res. (2016) 51:103–11. doi: 10.1111/jre.12288
144. Reichert S, Jurianz E, Natalie P, Schlumberger W, Dähnrich C, Johannsen N, et al. Is periodontitis a prognostic factor in order to indicate antibodies against citrullinated peptides in patients with rheumatoid arthritis? Clin Exp Rheumatol. (2020) 38:227–38. doi: 10.55563/clinexprheumatol/9p1bcm
145. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans –induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
146. Gomez-Bañuelos E, Johansson L, Konig M, Lundquist A, Paz M, Buhlin K, et al. Exposure to Aggregatibacter actinomycetemcomitans before Symptom Onset and the Risk of Evolving to Rheumatoid Arthritis. J Clin Med. (2020) 9:1906. doi: 10.3390/jcm9061906
147. Martinsson K, Di Matteo A, Öhman C, Johansson A, Svärd A, Mankia K, et al. Antibodies to leukotoxin A from the periodontal pathogen Aggregatibacter actinomycetemcomitans in patients at an increased risk of rheumatoid arthritis. Front Med (Lausanne). (2023) 10:1176165. doi: 10.3389/fmed.2023.1176165
148. Yoshida A, Nakano Y, Yamashita Y, Oho T, Ito H, Kondo M, et al. Data from: Immunodominant region of Actinobacillus actinomycetemcomitans 40-kilodalton heat shock protein in patients with rheumatoid arthritis. J Dent Res. (2001) 80:346–50. doi: 10.1177/00220345010800010901
149. Kimura Y, Yoshida S, Takeuchi T, Kimura M, Yoshikawa A, Hiramatsu Y, et al. Periodontal pathogens participate in synovitis in patients with rheumatoid arthritis in clinical remission: a retrospective case–control study. Rheumatol (Oxford). (2015) 54:2257–63. doi: 10.1093/rheumatology/kev274
150. Schwenzer A, Quirke AM, Marzeda AM, Wong A, Montgomery AB, Sayles HR, et al. Association of Distinct Fine Specificities of Anti–Citrullinated Peptide Antibodies With Elevated Immune Responses toPrevotella intermediain a Subgroup of Patients With Rheumatoid Arthritis and Periodontitis. Arthritis Rheumatol. (2017) 69:2303–13. doi: 10.1002/art.40227
151. Goh CE, Kopp J, Papapanou PN, Molitor JA, and Demmer RT. Association between serum antibodies to periodontal bacteria and rheumatoid factor in the third national health and nutrition examination survey. Arthritis Rheumatol. (2016) 68:2384–93. doi: 10.1002/art.39724
152. Li S, Yu Y, Yue Y, Liao H, Xie W, Thai J, et al. Autoantibodies From Single Circulating Plasmablasts React With Citrullinated Antigens andPorphyromonas gingivalisin Rheumatoid Arthritis. Arthritis Rheumatol. (2016) 68:614–26. doi: 10.1002/art.39455
153. Jenning M, Marklein B, Ytterberg J, Zubarev RA, Joshua V, Van Schaardenburg D, et al. Bacterial citrullinated epitopes generated by Porphyromonas gingivalis infection—a missing link for ACPA production. Ann Rheum Dis. (2020) 79:1194–202. doi: 10.1136/annrheumdis-2019-216919
154. Brewer RC, Lanz TV, Hale CR, Sepich-Poore GD, Martino C, Swafford AD, et al. Oral mucosal breaks trigger anti-citrullinated bacterial and human protein antibody responses in rheumatoid arthritis. Sci Transl Med. (2023) 15:eabq8476. doi: 10.1126/scitranslmed.abq8476
155. Marchant C, Smith MD, Proudman S, Haynes DR, and Bartold PM. Effect ofPorphyromonas gingivalis on Citrullination of Proteins by Macrophages In Vitro. J Periodontol. (2013) 84:1272–80. doi: 10.1902/jop.2012.120103
156. Stobernack T, Du Teil Espina M, Mulder LM, Palma Medina LM, Piebenga DR, Gabarrini G, et al. A secreted bacterial peptidylarginine deiminase can neutralize human innate immune defenses. Mbio. (2018) 9:e01704–18. doi: 10.1128/mBio.01704-18
157. Kulp A and Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. (2010) 64:163–84. doi: 10.1146/annurev.micro.091208.073413
158. Gabarrini G, Heida R, Van Ieperen N, Curtis MA, Van Winkelhoff AJ, and Van Dijl JM. Dropping anchor: attachment of peptidylarginine deiminase via A-LPS to secreted outer membrane vesicles of Porphyromonas gingivalis. Sci Rep. (2018) 8:8949. doi: 10.1038/s41598-018-27223-5
159. Aliko A, Kamińska M, Bergum B, Gawron K, Benedyk M, Lamont RJ, et al. Impact of porphyromonas gingivalis peptidylarginine deiminase on bacterial biofilm formation, epithelial cell invasion, and epithelial cell transcriptional landscape. Sci Rep. (2018) 8:14144. doi: 10.1038/s41598-018-32603-y
160. Kassem A, Henning P, Lundberg P, Souza PPC, Lindholm C, and Lerner UH. Porphyromonas gingivalis Stimulates Bone Resorption by Enhancing RANKL (Receptor Activator of NF-κB Ligand) through Activation of Toll-like Receptor 2 in Osteoblasts. J Biol Chem. (2015) 290:20147–58. doi: 10.1074/jbc.M115.655787
161. Zhang P, Liu J, Xu Q, Harber G, Feng X, Michalek SM, et al. TLR2-dependent Modulation of Osteoclastogenesis by Porphyromonas gingivalis through Differential Induction of NFATc1 and NF-κB. J Biol Chem. (2011) 286:24159–69. doi: 10.1074/jbc.M110.198085
162. Yoshimura F, Murakami Y, Nishikawa K, Hasegawa Y, and Kawaminami S. Surface components of Porphyromonas gingivalis. J Periodontal Res. (2009) 44:1–12. doi: 10.1111/j.1600-0765.2008.01135.x
163. Suzuki Y, Kikuchi T, Goto H, Takayanagi Y, Kawamura S, Sawada N, et al. Porphyromonas gingivalis Fimbriae Induce Osteoclastogenesis via Toll-like Receptors in RAW264 Cells. Int J Mol Sci. (2022) 23:15293. doi: 10.3390/ijms232315293
164. Ukai T, Yumoto H, Gibson FC, and Genco CA. Macrophage-elicited osteoclastogenesis in response to bacterial stimulation requires toll-like receptor 2-dependent tumor necrosis factor-alpha production. Infect Immun. (2008) 76:812–19. doi: 10.1128/IAI.01241-07
165. Kukita A, Ichigi Y, Takigawa I, Watanabe T, Kukita T, and Miyamoto H. Infection of RANKL-primed RAW-D macrophages with Porphyromonas gingivalis promotes osteoclastogenesis in a TNF-alpha-independent manner. PloS One. (2012) 7:e38500. doi: 10.1371/journal.pone.0038500
166. Röhner E, Detert J, Kolar P, Hocke A, N Guessan P, Matziolis G, et al. Induced apoptosis of chondrocytes by porphyromonas gingivalis as a possible pathway for cartilage loss in rheumatoid arthritis. Calcif Tissue Int. (2010) 87:333–40. doi: 10.1007/s00223-010-9389-5
167. Pischon N, Röhner E, Hocke A, N'Guessan P, Müller HC, Matziolis G, et al. Effects of Porphyromonas gingivalis on cell cycle progression and apoptosis of primary human chondrocytes. Ann Rheum Dis. (2009) 68:1902–07. doi: 10.1136/ard.2008.102392
168. Lopez-Oliva I, Malcolm J, and Culshaw S. Periodontitis and rheumatoid arthritis-Global efforts to untangle two complex diseases. Periodontol 2000. (2024) 00:1–19. doi: 10.1111/prd.12530
169. De Molon RS, Rossa CJ, Thurlings RM, Cirelli JA, and Koenders MI. Linkage of periodontitis and rheumatoid arthritis: current evidence and potential biological interactions. Int J Mol Sci. (2019) 20:45410. doi: 10.3390/ijms20184541
170. Sun J, Zheng Y, Bian X, Ge H, Wang J, and Zhang Z. Non-surgical periodontal treatment improves rheumatoid arthritis disease activity: a meta-analysis. Clin Oral Investig. (2021) 25:4975–85. doi: 10.1007/s00784-021-03807-w
171. Inchingolo F, Inchingolo AM, Avantario P, Settanni V, Fatone MC, Piras F, et al. Data from: the effects of periodontal treatment on rheumatoid arthritis and of anti-rheumatic drugs on periodontitis: A systematic review. Int J Mol Sci, (2023) 24:17228. doi: 10.3390/ijms242417228
172. Ayravainen L, Heikkinen AM, Kuuliala A, Ahola K, Koivuniemi R, Moilanen E, et al. Anti-rheumatic medication and salivary MMP-8, a biomarker for periodontal disease. Oral Dis. (2018) 24:1562–71. doi: 10.1111/odi.12930
173. De Smit MJ, Westra J, Posthumus MD, Springer G, Van Winkelhoff AJ, Vissink A, et al. Effect of anti-rheumatic treatment on the periodontal condition of rheumatoid arthritis patients. Int J Environ Res Public Health. (2021) 18:2529. doi: 10.3390/ijerph18052529
174. Asteriou E, Gkoutzourelas A, Mavropoulos A, Katsiari C, and Sakkas LI. Bogdanos DP data from: curcumin for the management of periodontitis and early ACPA-positive rheumatoid arthritis: killing two birds with one stone. Nutrients, (2018) 10:908. doi: 10.3390/nu10070908
175. Anitha V, Rajesh P, Shanmugam M, Priya BM, Prabhu S, and Shivakumar V. Comparative evaluation of natural curcumin and synthetic chlorhexidine in the management of chronic periodontitis as a local drug delivery: a clinical and microbiological study. Indian J Dent Res. (2015) 26:53–6. doi: 10.4103/0970-9290.156806
Keywords: rheumatoid arthritis, periodontitis, porphyromonas gingivalis, citrullinated antigens, oral pathogens
Citation: Guo X, Wang S, Ding J, Liu W, Xu J, Wang M, Sun H, Ma Y, Liu W, Zhang L and Liu M (2025) Periodontopathic bacteria in rheumatoid arthritis pathogenesis: bridging clinical associations to molecular mechanisms. Front. Immunol. 16:1681037. doi: 10.3389/fimmu.2025.1681037
Received: 06 August 2025; Accepted: 30 September 2025;
Published: 17 October 2025.
Edited by:
Javier Pascual, Darwin Bioprospecting Excellence, SpainReviewed by:
Adriana Sant’Ana, University of São Paulo, Bauru, BrazilMuhammad Annurdin Sabarudin, Universiti Sains Islam Malaysia, Malaysia
Copyright © 2025 Guo, Wang, Ding, Liu, Xu, Wang, Sun, Ma, Liu, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Liu, bGl1bWludGNtQDE2My5jb20=; Lei Zhang, emhhbmdsZWl0ajIwMDhAMTYzLmNvbQ==; Wei Liu, ZmVuZ3NoaWxpdXdlaUAxNjMuY29t
†These authors have contributed equally to this work