 Kengo Nishino1
Kengo Nishino1 Takumi Kiwamoto1*
Takumi Kiwamoto1* Masashi Matsuyama1
Masashi Matsuyama1 Zhenting Wei1Sosuke Matsumura1Kenya Kuramoto1
Zhenting Wei1Sosuke Matsumura1Kenya Kuramoto1 Yuki Yabuuchi1Kai Yazaki1
Yuki Yabuuchi1Kai Yazaki1 Kazufumi Yoshida1Yosuke Matsuno2
Kazufumi Yoshida1Yosuke Matsuno2 Yuko Morishima1Nobuyuki Hizawa1
Yuko Morishima1Nobuyuki Hizawa1- 1Department of Pulmonary Medicine, Institute of Medicine, University of Tsukuba, Tsukuba, Japan
- 2Department of Pulmonary Medicine, Nikko Memorial Hospital, Hitachi, Japan
Chronic obstructive pulmonary disease (COPD) is a progressive inflammatory lung disorder primarily caused by prolonged exposure to harmful substances, such as cigarette smoke. Hyaluronic acid synthase 2 (HAS2) synthesizes high-molecular-weight hyaluronic acid (HMW-HA), which has anti-inflammatory properties. Previous studies have revealed that HAS2 dysfunction may exacerbate COPD. However, the specific impact of HAS2 on pulmonary emphysema progression remains unclear. Therefore, this study examined whether HAS2 dysfunction worsens airway inflammation and emphysema in a mouse COPD model. Has2 heterozygous-deficient (Has2+/−) mice and their wild-type (WT) littermates were evaluated using a porcine pancreatic elastase (PPE)-induced COPD model. After the administration of PPE, the Has2+/− mice exhibited a significant increase in total cell and neutrophil counts in the bronchoalveolar lavage fluid samples compared with the WT mice. Further, the Has2+/− mice presented with enhanced emphysema development on histological analyses, with higher mean linear intercept values relative to the WT mice. The PPE-stimulated Has2+/− mice also had increased G-CSF levels and tumor growth factor-beta (TGF-β) attenuation in the lungs. RNA-sequencing analysis revealed that PPE stimulation promoted the synthesis of HMW-HA and TGF-β signaling. Gene Ontology analysis using Has2+/− mice-specific differentially expressed genes showed that the genes associated with the pathways that promote the negative regulation of the TGF-β receptor signaling pathway were activated after the administration of PPE. Therefore, Has2 dysfunction exacerbates neutrophilic airway inflammation and emphysema, thereby underscoring the protective role of HAS2 in a PPE-induced emphysema model. The exacerbated response may involve G-CSF- and TGF-β-related signaling pathways. These findings may contribute to the development of novel therapeutic strategies for COPD management.
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous lung condition characterized by chronic respiratory symptoms attributed to abnormalities of the airways and/or alveoli that cause persistent airflow obstruction (1). Further, it is one of the leading causes of mortality worldwide (2–4). The financial implications of COPD management are projected to increase further on a global scale (5). Therefore, the early identification of patients at risk for rapid COPD progression is important in reducing the disease-related costs. COPD is characterized anatomically by small airway remodeling and emphysema. In pulmonary emphysema, elastin is the major target of decomposition leading to loss of lung elasticity (6, 7). A previous study has shown that hyaluronan (HA) binds to elastic fibers and protects them from elastase-induced injury (8, 9). Another study found that patients with asthma or COPD had lower levels of HA in their airway smooth muscle cells (10). This study also revealed a significant decrease in the HA synthase 1 (HAS1) and HAS2 levels in the same tissue (10). HA synthases synthesize HA polymers of various sizes. HAS1 and HAS2 produce high-molecular-weight HA (HMW-HA, 2 × 104 kDa). Meanwhile, HAS3 synthesizes low-molecular-weight HA (2 × 102 kDa) (11). Our previous genome-wide association study has found that the HAS2 gene is a potential susceptibility factor for adult asthma (12). Using HAS2 gene (Has2) heterozygous-deficient (Has2+/−) mice, we also found that the attenuation of Has2 exacerbates eosinophilic airway inflammation, increases airway hyperresponsiveness, and promotes airway remodeling (13, 14). HMW-HA has anti-inflammatory effects. However, it remains unclear whether the dysfunction of Has2, which produces HMW-HA, contributes to the severity and intractability of other inflammatory lung diseases such as COPD. To validate the role of Has2 in the pathogenesis of COPD, this study analyzed the development of porcine pancreatic elastase-induced pulmonary emphysema in Has2+/− mice. Results showed that the Has2+/− mice exhibited a more severe neutrophilic airway inflammatory reaction, higher degree of emphysema formation, and increased granulocyte-colony stimulating factor (G-CSF) with transforming growth factor beta 1 (TGF-β1) attenuation.
Methods
Animals
The experiments used 8–12-week-old female C57BL/6 background wild-type (WT) mice and Has2 heterozygous (Has2+/-) mice. Breeding sets of Has2+/− mice (Jackson Laboratory, Bar Harbor, ME) were backcrossed to the C57BL/6 background for at least 10 generations (13–15). The Institutional Review Board of the University of Tsukuba approved all animal studies (approval numbers: 21-028, 23-004, and 24-057).
Elastase inoculation
Mice were injected intraperitoneally with triple mixed anesthesia of medetomidine hydrochloride (0.30 mg/kg; Nippon Zenyaku Kogyo, Fukushima, Japan), midazolam (4 mg/kg; Fujifilm Wako Pure Chemical, Osaka, Japan), and butorphanol tartrate (5 mg/kg; Meiji Animal Health, Kumamoto, Japan). After induction of anesthesia, the mice were inoculated with 3.75 U of porcine pancreatic elastase (PPE; Elastin Products Company, Owensville, MO) in 100 μl of saline intratracheally. After the administering PPE, atipamezole hydrochloride (0.30 mg/kg; Nippon Zenyaku Kogyo) was injected intraperitoneally to awaken the mice. The mice were sacrificed by exsanguination through the abdominal aorta under anesthesia on days 1, 4, and 21. An age-matched PPE non-treated mice group was used as the control group.
Experimental groups
Animals were randomly divided into four groups:
WT-Control: WT mice maintained under filtered air without the administration of PPE (n = 8).
Has2+/−-Control: Has2+/− mice maintained under filtered air without the administration of PPE (n = 8).
WT-PPE: WT mice maintained under filtered air that received PPE administration (n = 8 for each time point).
Has2+/−-PPE: Has2+/− mice maintained under filtered air that received PPE administration (n = 8 for each time point).
To avoid the influence of each measurement value during specimen collection, three experimental setups were necessary for the protocol. In the first, lungs (right and left) were used to conduct morphometric analysis. The second setup was required to perform bronchoalveolar lavage fluid cell analysis. The left lungs from the third set up were required to obtain lung homogenates for cytokine analysis. RNA-seq analysis was performed on the right lung samples from the third setup.
Lung histology
Lung paraffin sections were stained with hematoxylin and eosin (HE) staining to assess air space enlargement and airway inflammatory cell infiltration. The mean linear intercept (MLI) was quantified, as described in a previous study (16). Briefly, we prepared at least four slides from both the side lung and performed MLI analysis in the distal regions of the parenchymal tissue. For each prepared section, ten fields of view (500 μm × 373.5 μm each) of peripheral airspaces that were free of blood vessels and bronchioles were selected. The MLI was then calculated based on the average number of intersections between ten horizontally spaced lines, which were placed equidistant from each other and the alveolar walls within all field of view.
Bronchoalveolar lavage fluid cell counting
Mouse lungs were lavaged using five repeated instillations of 0.6 mL of saline each via the tracheal cannula. The recovery rate was between 75 and 85%. The cell count was determined using a hemocytometer, and a differential cell count was subsequently performed based on count of 300 cells. The cells were morphologically classified using staining with Diff-Quik (Polysciences, Inc., Warrington, PA), using standard light microscopic techniques.
Enzyme-linked immunosorbent assay
Quantification of TGF-β1 in the lung homogenates was performed using commercially available ELISA kits (R&D Systems, Minneapolis, MN).
Multiplex cytokine assay
Cytokines and chemokines in the lung homogenates were measured using the MILLIPLEX MAP Kit (MilliporeSigma, Burlington, MA), according to the manufacturer’s instructions.
Lung RNA extraction and RNA sequencing
Total RNA was extracted from the mouse lungs using the TRIzol® reagent (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer’s instructions (n = 6–7, each group). The concentration and purity of the RNA samples were determined via automated optical density evaluation (OD 260/OD 280 ≥ 1.8 and OD 260/OD 230 ≥ 1.8) using the NanoDrop spectrophotometer (Thermo Fisher Scientific). RNA sequencing (RNA-seq) libraries were prepared using the NEBNext rRNA Depletion Kit (New England Biolabs, Ipswich, MA) and the ENBNext Ultra Directional RNA Library Prep Kit (New England Biolabs), according to the manufacturer’s instructions, using 500 ng of total RNA samples. Next, 2 × 36 base paired-end sequencing was performed using the NextSeq 500 sequencer (Illumina, San Diego, CA) by Tsukuba i-Laboratory LLP (Tsukuba, Japan). Sequences were mapped to the mm10 mouse genome and quantified using CLC Genomics Workbench version 10.1.1 (QIAGEN, Hilden, Germany). Differentially expressed genes were identified by filtering according to the P-values obtained via analysis of variance (Gaussian Statistical Analysis in CLC). The data are available under GEO series accession number GSE 304417.
Pathway analysis of DEGs and CIBERSORT analysis
The biosynthesis and catabolism of the hyaluronan glycosylation pathway were visualized using GlycoMaple (https://glycosmos.org/glycomaple/Human) (Supplementary Figure 1). Upstream analysis was performed on RNA-seq data using the Ingenuity Pathway Analysis software (QIAGEN) with the Fisher’s exact test (P < 0.05, indicates statistically significant differences). Unique differentially expressed genes (DEGs) between WT-Control vs WT-PPE and WT-Control vs Has2+/–PPE were identified using Venny (v2.0; http://bioinfogp.cnb.csic.es/tools/venny/index.html). The Gene Ontology (GO) terms enriched in the DE genes were identified using Metascape (http://metascape.org). CIBERSORT analysis was performed on the RNA-seq data using the analytical tool (https://cibersort.stanford.edu/) (17). A previously published mouse reference signature matrix, comprising 511 distinguishing genes for 25 immune cell types, was used as a reference profile (18).
Statistical analysis
Data were presented as means ± standard error of the means or individual dot plots with means ± standard error of the means. Between-group differences were evaluated using the Mann–Whitney U test or analysis of variance with the Tukey’s multiple comparison test. P values < 0.05 indicated statistically significant differences.
Results
The development of elastase-induced pulmonary emphysema was enhanced in Has2+/− mice
To elucidate the protective role of HAS2 against emphysema, we initially evaluated the development of emphysema 1, 4, and 21 days after the administration of PPE (Figure 1A). Pathological differences were not observed between WT and Has2+/− mice before PPE treatment upon microscopic examination (Figure 1B, panels a and e). Air space enlargement and alveolar wall disruption worsened over time after PPE treatment in both WT (Figure 1B, upper panel) and Has2+/− mice (Figure 1B, lower panel). However, these pathological changes were more severe in Has2+/− mice than in WT mice. To quantify morphologic changes, MLI was then evaluated. Morphometric analysis revealed that the Has2+/− mice had significantly higher MLI values than the WT mice on days 1, 4, and 21 after the administration of PPE (Figure 1C). Taken together, these histological evaluations have revealed that Has2+/− mice are more susceptible to PPE-induced emphysema.
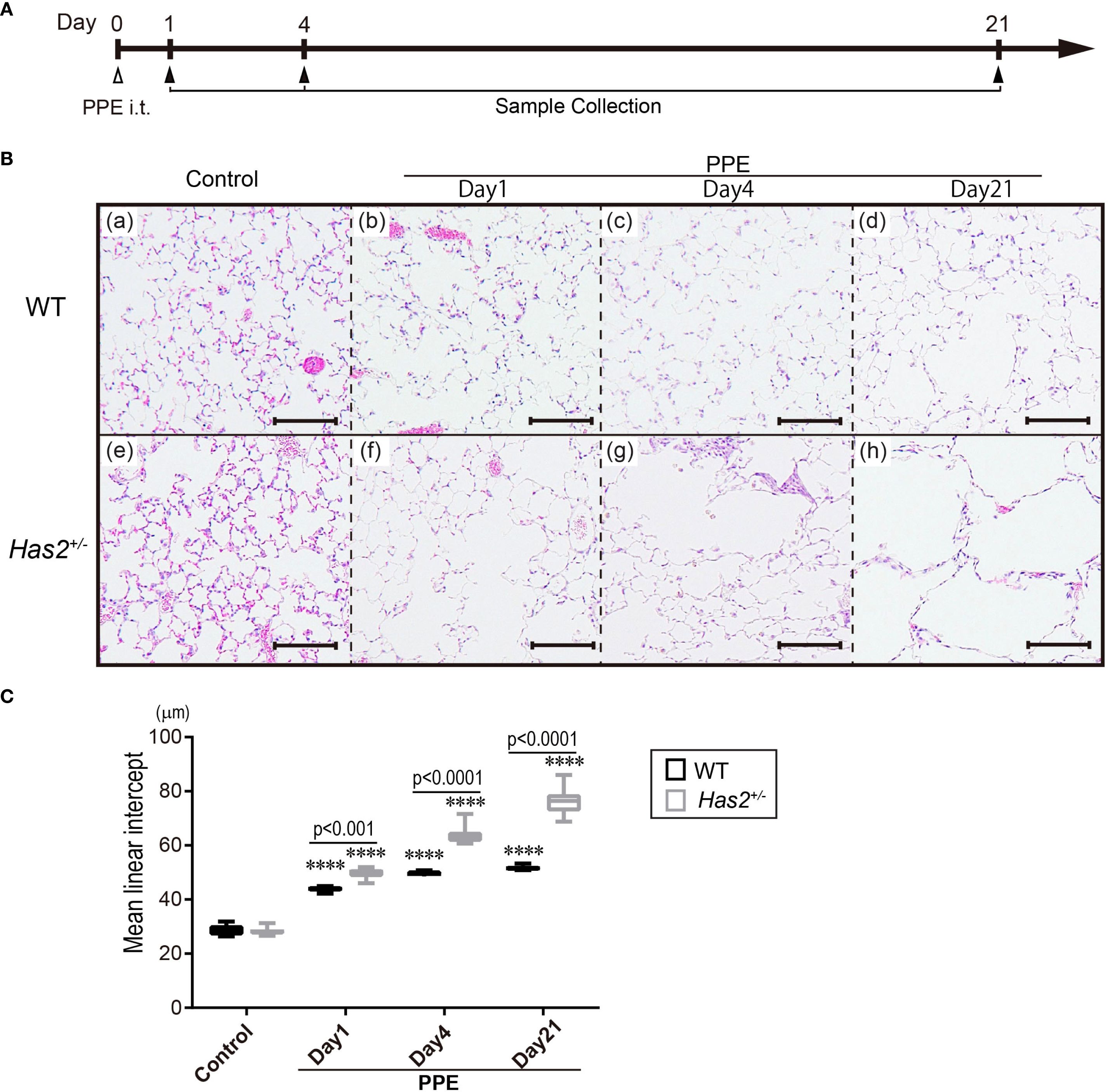
Figure 1. Has2 attenuation exacerbated lung emphysema in a PPE-induced mouse model of COPD. (A) Schematic illustration of the experimental design. (B) Lung tissue HE staining in the WT mice (upper lane, panels a–d) and the Has2+/− mice (lower lane, panels e–h). Scale bar: 100 μm. (C) Mean linear intercept (MLI) values of the alveoli (n = 8). Statistical significance was determined using the Tukey’s multiple comparison test. ****P < 0.0001 relative to the WT-Control mice. Horizontal bars indicate direct statistical comparisons between WT and Has2+/− mice.
The severity and duration of elastase-induced pulmonary inflammation were enhanced in Has2+/− mice
To examine the severity of pulmonary inflammation induced by elastase, the number of inflammatory cells in the bronchoalveolar lavage fluid (BALF) was determined. Both the WT-PPE and Has2+/−-PPE mice had a significantly higher number of total cells and macrophages than the WT-Control mice (Figures 2A, B). Leukocyte subsets were analyzed to characterize inflammation in WT-PPE and Has2+/−-PPE mice. The number of BALF neutrophils on day 1 was significantly higher in the Has2+/− mice than in the WT mice (Figure 2D). In the Has2+/− mice, the significant increase in the neutrophil counts persisted 4 days after the administration of PPE (Figure 2D). The WT-PPE mice had a higher number of lymphocytes and eosinophils on day 1 than the Has2+/−-PPE mice (Figures 2C, E). This phenomenon could be attributed to the fact that the absolute numbers of lymphocytes and eosinophils were considerably lower than the absolute number of neutrophils. These results indicate that Has2 attenuation worsens PPE-induced neutrophilic airway inflammation.
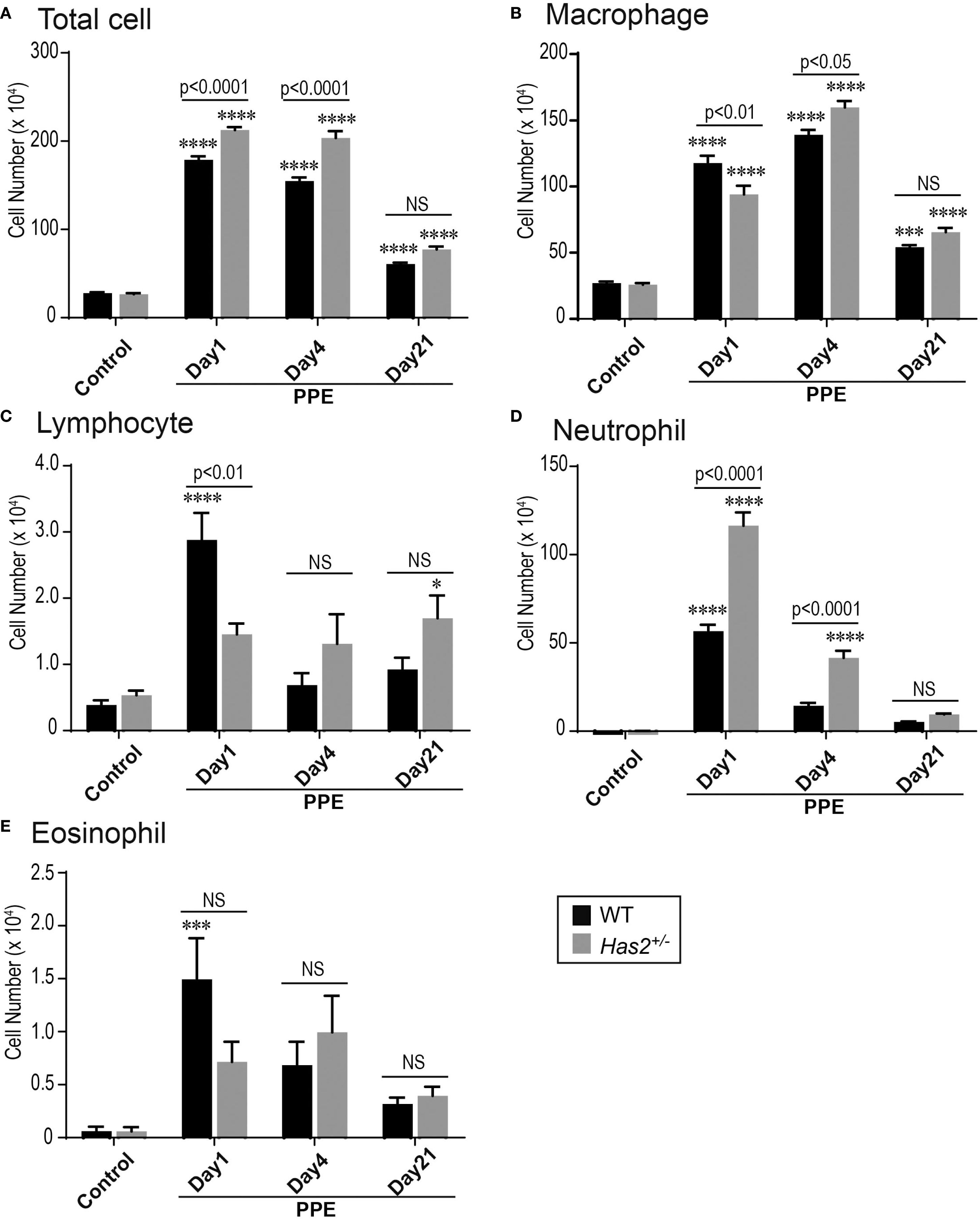
Figure 2. Has2 attenuation exacerbated acute neutrophilic airway inflammation in a PPE-induced mouse model of COPD. BALF cytology of each indicated cell type (n = 8). (A) Total cell. (B) Macrophage. (C) Lymphocyte. (D) Neutrophil. (E) Eosinophil. Statistical significance was determined using the Tukey’s multiple comparison test. * P < 0.05, *** P < 0.001, **** P < 0.0001 relative to the WT-Control mice. Horizontal bars indicate direct statistical comparisons between WT and Has2+/− mice. NS, not significant.
Has2 attenuation results in the downregulation of TGF-β1
TGF-β is known to be regulated by CD44, a HA-binding protein (19). To determine whether Has2 attenuation affects the expression of lung TGF-β1, the TGF-β1 levels in the lung homogenate were evaluated. As shown in Figure 3, the protein-adjusted levels of TGF-β1 in the lung homogenates of Has2+/− mice were more likely to be lower than those of WT mice. These levels decreased significantly 21 days after the administration of PPE (Figure 3D), as determined using the Mann–Whitney U test. These results may support the hypothesis that Has2 attenuation prolongs the inhibitory state of HA-binding protein expression and TGF-β signaling.
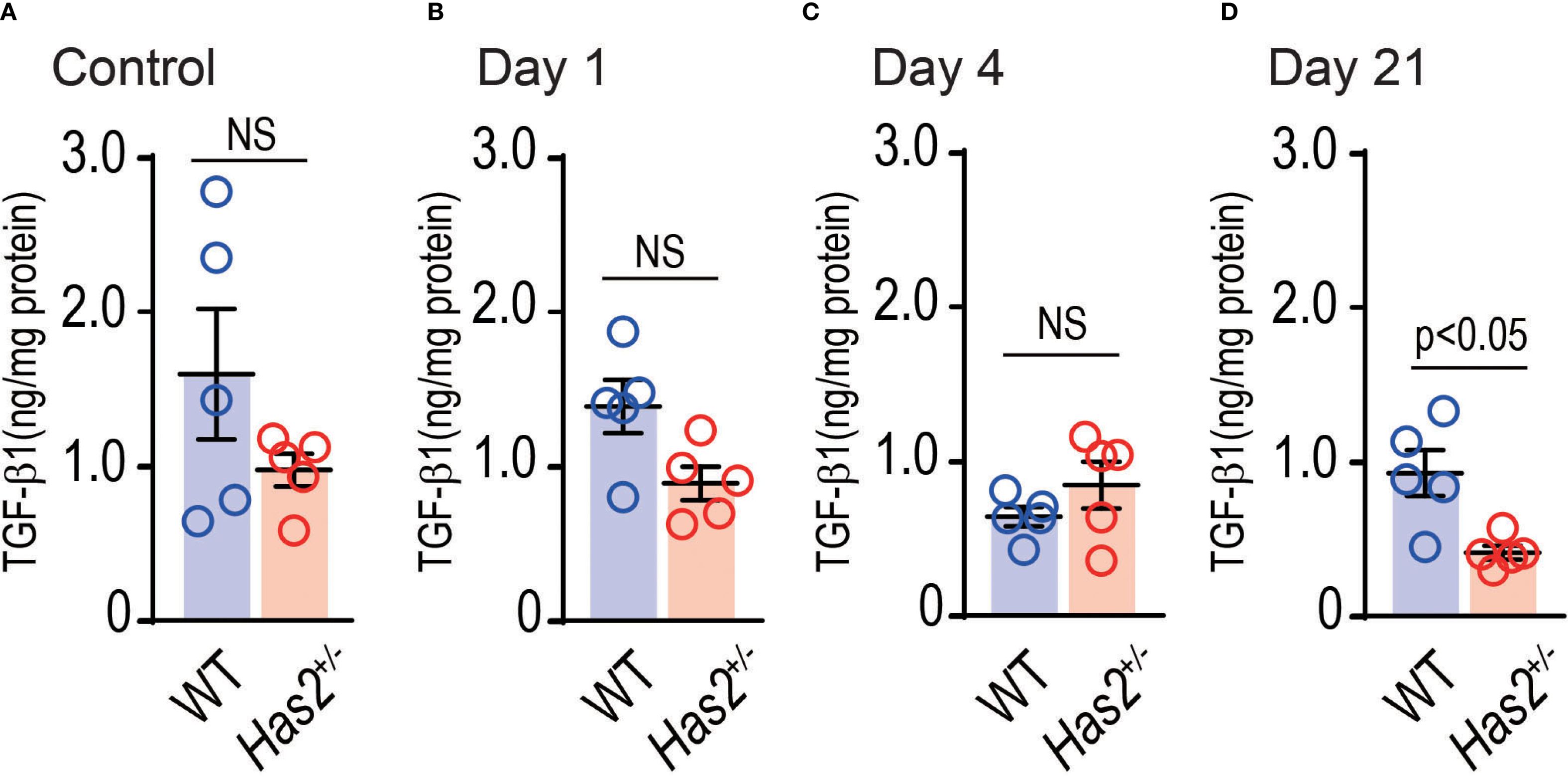
Figure 3. Effects of Has2 attenuation on the TGF-β1 levels in the lung homogenates. Protein adjusted levels of TGF-β1 (n = 5). (A) Control. (B) Day 1. (C) Day4. (D) Day 21. Statistical significance was determined using the Mann–Whitney U test. Horizontal bars indicate direct statistical comparisons between WT and Has2+/−− mice. NS, not significant.
Elastase-stimulated Has2+/− mice exhibit increased G-CSF levels in the lung
To examine the hypothesis that significant lung neutrophilia and emphysema progression in Has2+/− mice was attributed to an altered inflammatory cytokine and chemokine response, lung homogenate samples were tested for a variety of potentially relevant mediators (Figure 4, Supplementary Figures 2, 3). The Has2+/−-PPE mice had significantly higher G-CSF levels in the lung homogenates than the WT-PPE mice on day 1 (Figure 4A, panel a). Conversely, the Has2+/−-PPE mice had significantly lower levels of GM-CSF, IL-5, IL-7, IL-10, IL-12(p70), IL-13, and IL-15 than the WT-PPE mice on day 1 (Figure 4, Supplementary Figures 2, 3). The Has2+/−-PPE group had significantly higher protein concentrations than the WT-PPE group on day 1. Therefore, this difference might have affected the results of the protein adjusted cytokine and chemokine measurements (Supplementary Figures 4). Interestingly, on day 4, the Has2+/−-PPE group had significantly lower levels of IFN-γ, and IL-13 than the WT-PPE group (Figure 4B, panels a and f). In addition, there were no significant differences between the WT-PPE and Has2+/−-PPE mice in terms of the levels of IL-1β, IL-6, or IL-17, all of which are associated with neutrophilic inflammation (Figure 4A, panels d–f). Based on these results, Has2 attenuation induced severe neutrophilic airway inflammation due to increased G-CSF after PPE stimulation.

Figure 4. Effects of Has2 attenuation on various cytokine and chemokine levels in the lung homogenates. (A) Protein adjusted levels of inflammatory cytokines in the lung homogenates (n = 4–5). (B) Protein adjusted levels of the indicated Th1-/Th2-related cytokines in the lung homogenates (n = 4–5). Statistical significance was determined using the Tukey’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 relative to the WT-Control mice. Horizontal bars indicate direct statistical comparisons between WT and Has2+/− mice. NS, not significant.
Elastase stimulation induced high-molecular-weight hyaluronan via Has2, and the TGF-β1 pathway activation
RNA-seq was conducted to elucidate the alterations in gene expression that occurred 4 days after the administration of PPE in the WT and Has2+/− mice. Initially, a comprehensive analysis of the glycosylation pathway was conducted to visualize the impact of PPE administration on the hyaluronic acid synthesis pathway using Glycomaple. The results of the analysis, which were based on the transfer product number per million (TPM), showed that the synthesis pathway of HMW-HA was suppressed in the non-stimulated state (Figure 5A, panel a). However, it was activated after the administration of PPE (Figure 5A, panel b). Among the 16 genes implicated in the biosynthesis and catabolism of hyaluronan pathway that were examined, only Has2 has been implicated in the synthesis of HMW-HA (Figures 5A, B). The WT mice exhibited an increase in the Has2 gene expression after the administration of PPE (Figure 5B). However, the Has2 gene expression did not differ between the WT-PPE and Has2+/− -PPE mice (Supplementary Figure 5). The generation of Has2-deficient mice was established with 60-codon deletion in exon 4. This short deletion could potentially explain the absence of differences in the gene expression that were not detected via RNA-seq. Further, based on the results of an upstream analysis using the Ingenuity Pathway Analysis software, the response to the administration of PPE was significantly regulated by TGFB1 gene activation (Figure 5C, Supplementary Table 1). These findings indicate that the elastase-induced emphysema model led to the production of HMW-HA via Has2, which is then regulated via the TGF-β1 pathway.
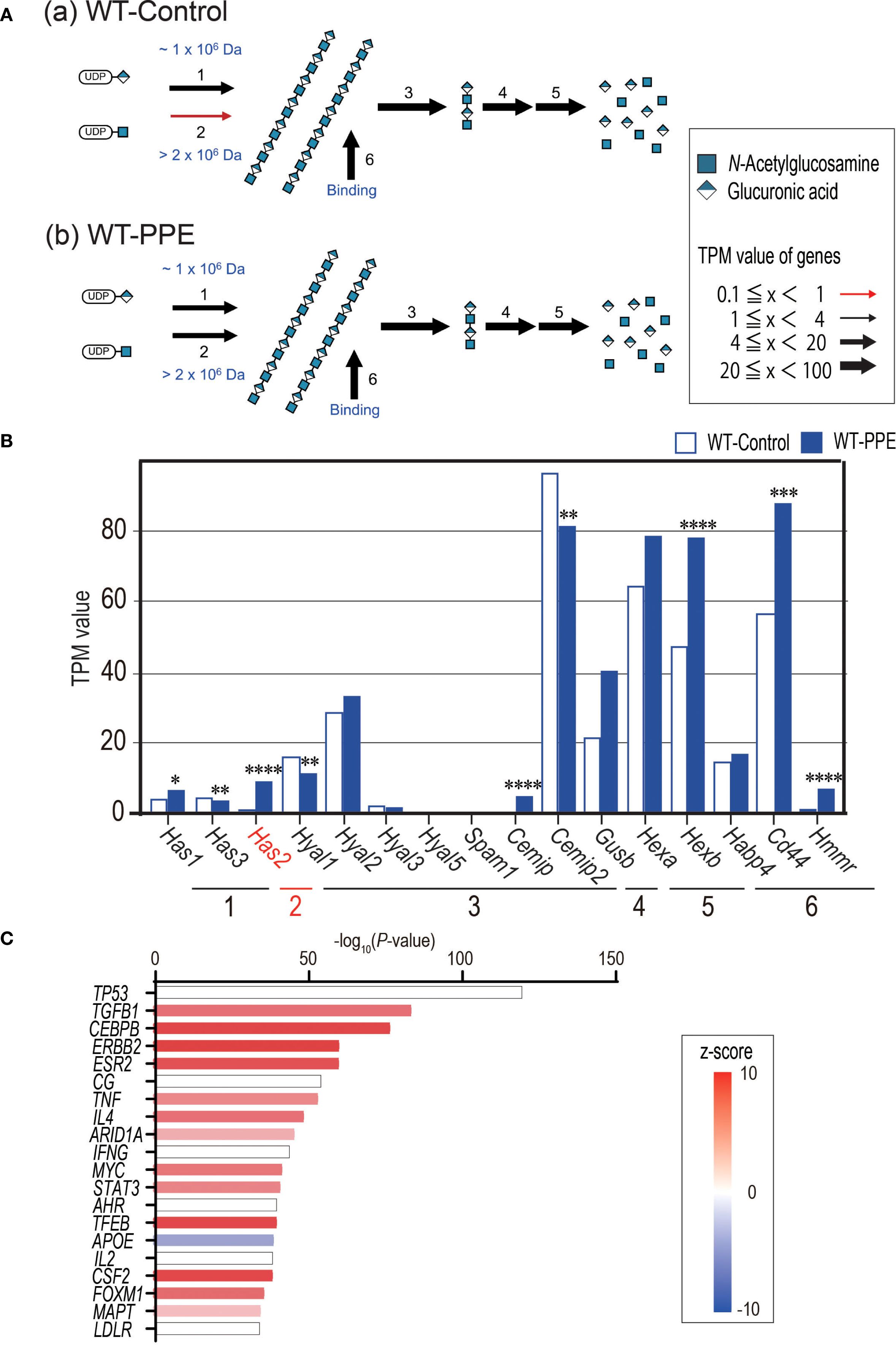
Figure 5. PPE administration affects the HMW-HA synthesis and activates the TGF-b1-related pathway. (A) Biosynthesis and catabolism of hyaluronan pathways in the lungs of WT-Control mice (A) and WT-PPE mice (B) were visualized using GlycoMaple. (B) The TPM value of hyaluronan pathway related genes between the lungs of WT-Control and WT-PPE mice (n = 7). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 relative to the WT-Control mice. (C) The top 20 significant genes detected via upstream analysis of the DEGs in the lungs of WT-Control and WT-PPE mice (n = 7). DEGs, differentially expressed genes.
Has2 attenuation affected inflammatory phenotype, macrophage differentiation, and impaired the TGF-β-related signaling
Five genes had significantly different expressions in the lungs of WT-PPE mice and Has2+/−-PPE mice (Figure 6A). GO analyses identified myeloid cell differentiation (GO: 0030099) (Figure 6B). Next, our analysis focused on specifically altered genes in WT-PPE and Has2+/−-PPE mice compared with WT-Control mice (Figure 6C). In total, 960 genes were uniquely altered in the WT-PPE mice (WT-PPE unique genes, Figure 6C). Meanwhile, 664 genes were uniquely altered in the Has2+/−-PPE mice (Has2+/−-PPE unique genes, Figure 6C). GO analyses revealed several significant biological processes, including ribonucleoprotein complex biogenesis (GO: 0022613), neutrophil degranulation (R-MMU-6798695), leukocyte migration (GO: 0050900), innate immune response (GO: 0045087), and comprehensive IL-17A signaling (WP5242) among the uniquely upregulated genes in WT-PPE (Figure 6D, panel a, right lane, and Supplementary Table 2). Conversely, the pathways associated with adaptive immune response (GO: 0002250, GO: 0002706, GO: 0051249, GO: 0042110, GO: 0002253, GO: 0031295, and mmu04658) was identified among the uniquely downregulated genes in the WT-PPE mice (Figure 6D, panel a, left lane, Supplementary Table 3). These results might be related to the increase in the lymphocyte counts in the BALF of the WT-PPE group during the acute phase, and the significant increase in the neutrophil counts in the Has2+/−-PPE group. The pathways associated with ciliary movement (GO: 0035082 and GO: 0070286) and heart development (GO: 0007507) were also identified among the uniquely downregulated genes in the WT-PPE mice (Figure 6D, panel a, left lane, Supplementary Table 3). Further, the pathways related to cargo concentration in ER (R-MMU-5694530) and the negative regulation of transforming growth factor beta receptor signaling pathway (GO: 0030512) were detected in the uniquely upregulated genes in the Has2+/−-PPE mice (Figure 6D, panel b, right lane, Supplementary Table 4). These pathways, detected in Has2+/−-PPE unique genes, are associated with HA or Has2 (14, 19).
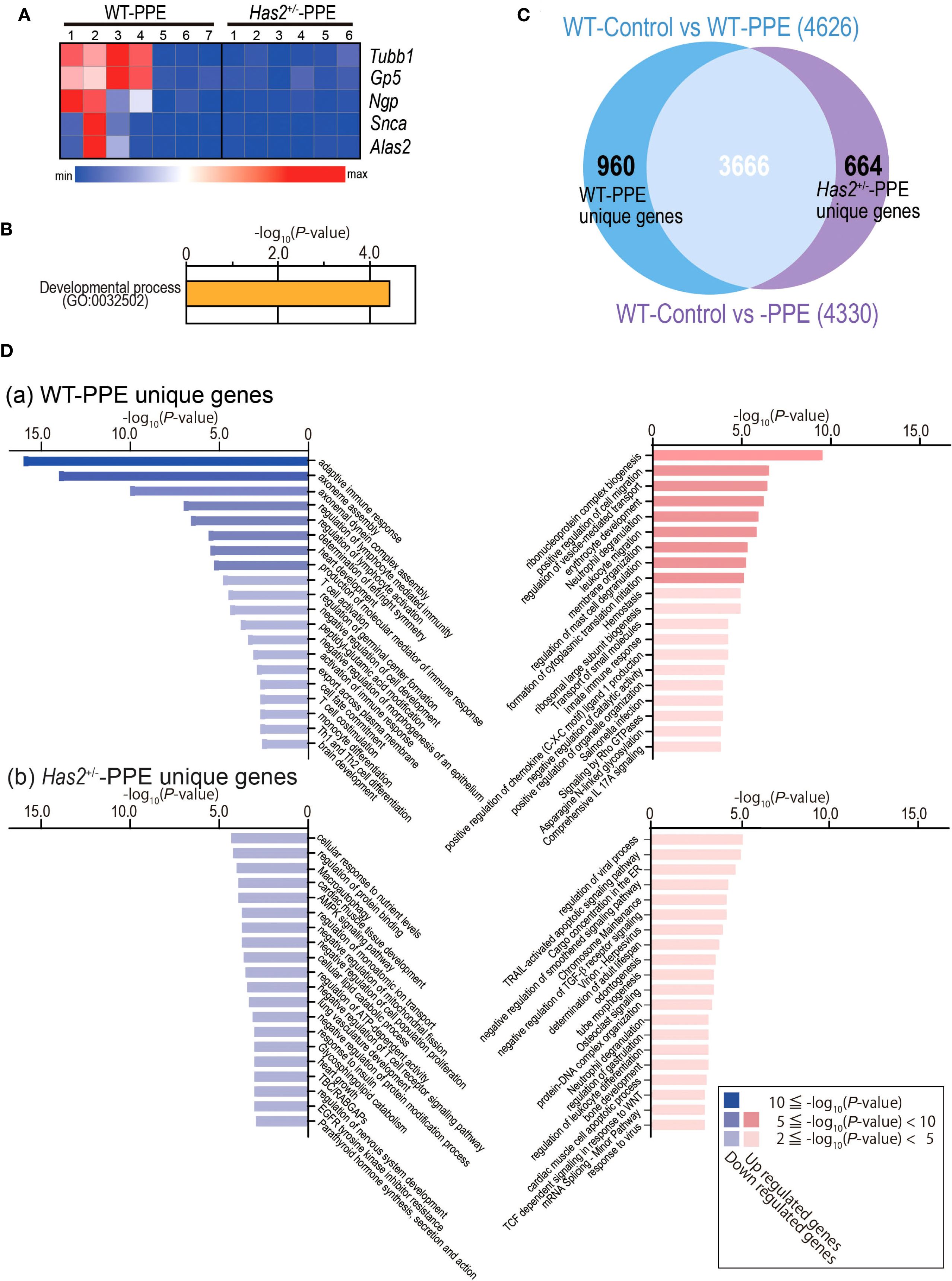
Figure 6. DEG analysis between the lungs of the WT-Control, WT-PPE, and Has2+/−-PPE mice. (A) Heatmap of the DEGs in the lungs of WT-PPE and Has2+/−-PPE mice (n = 6–7, cutoff: adjusted P < 0.01; log2 fold change > 1.2.). Hierarchical clustering was performed based on the mean log2 fold change. (B) Significant biological process terms detected via GO analysis in WT-PPE and Has2+/−-PPE mice. (C) Identification of the unique DEGs between WT-Control vs. WT-PPE mice and WT-Control vs. Has2+/−-PPE mice using the Venn diagram. (D) The top 20 significant terms detected via GO analysis among the unique genes in the WT-PPE mice (a) and Has2+/−-PPE mice (b).
To understand the significant changes in immune cell fractions and cytokines in the lungs of WT-PPE and Has2+/−-PPE mice, a CIBERSORT analysis was performed. In this analysis, the proportion of M1 macrophage cells was significantly high in the Has2+/−-PPE mice (Figure 7). However, there were no significant differences in the proportion of Treg cells, Th17 cells, and γδT cells affecting the production of TGF-β or IL-17 between the two groups (Figure 7). Based on these results, the differentiation of M1 macrophages in the Has2+/−-PPE group may be affected by the dysfunction of Has2 under these conditions.
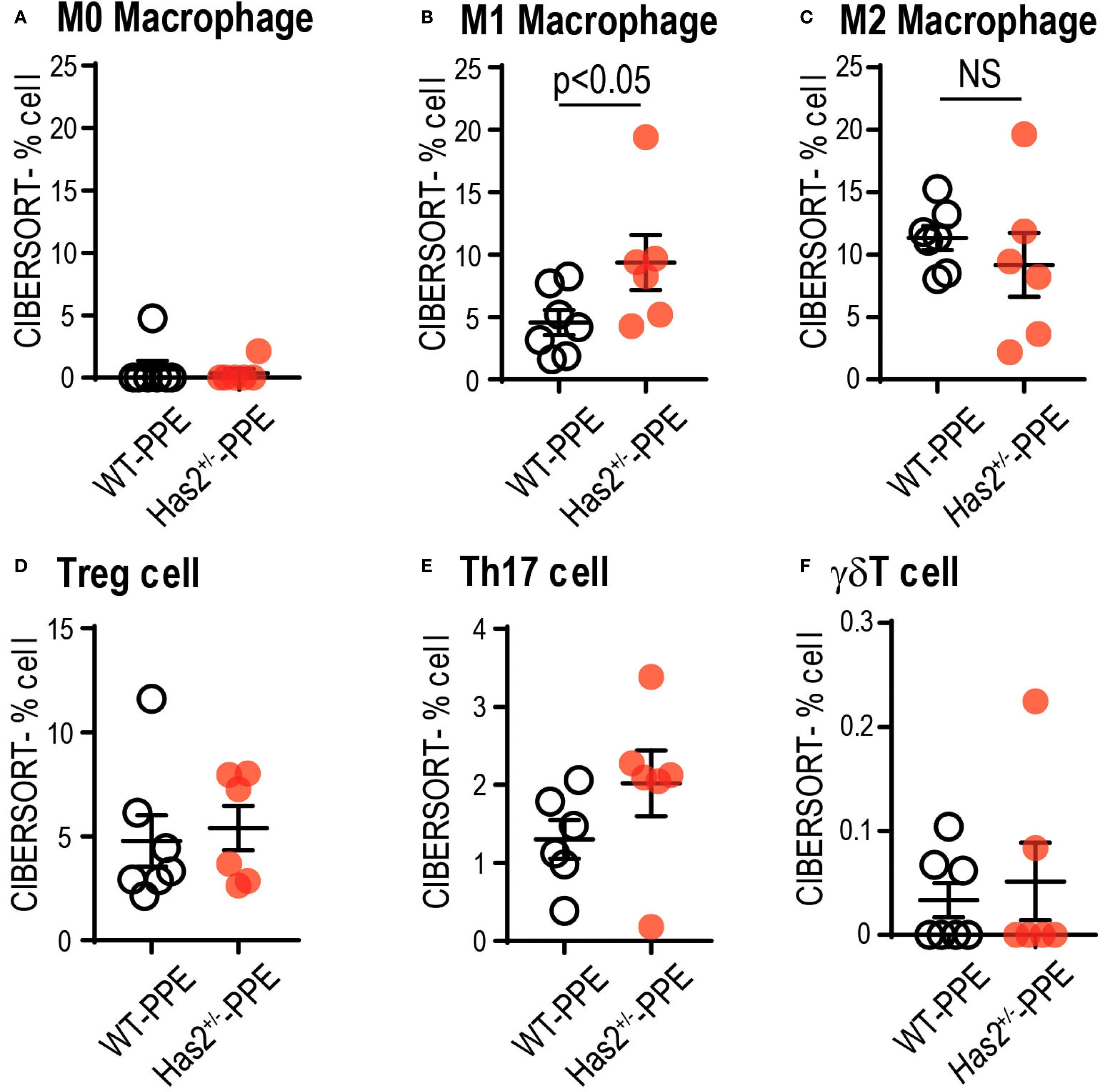
Figure 7. Attenuation of Has2 affected macrophage differentiation. Semi-quantitative evaluation of immune cell infiltrates after PPE stimulation, as determined via CIBERSORT analysis on whole-lung RNA-seq data (n = 6–7). (A) M0 Macrophage. (B) M1 Macrophage. (C) M2 Macrophage. (D) Treg cell. (E) Th17 cell. (F) γδT cell. All samples were obtained 4 days after the PPE stimulation. Statistical significance was determined using the Mann–Whitney U test. Horizontal bars indicate direct statistical comparisons between WT and Has2+/− mice. NS, not significant.
Discussion
The protease–antiprotease imbalance hypothesis was proposed after the discovery of alpha-1 antitrypsin deficiency, a disease susceptibility gene for COPD (20, 21). Currently, this hypothesis is applicable to COPD that is not associated with alpha-1 antitrypsin deficiency (21). A previous study reported that smoking also causes a reduction in the ability of alpha-1 protein to inhibit elastase activity (21). HA is relevant the vulnerability of matrix elastin to the effects of elastases (21, 22). The loss of HA was observed in human emphysema lungs (23, 24). Meanwhile, aerosolized HA significantly reduced elastase- or cigarette smoke-induced airspace enlargement (21, 25). Based on these data, the design of the current study was established.
To the best of our knowledge, this study first report that the attenuation of Has2 mRNA affected the severity of PPE-induced pulmonary emphysema and airway inflammation in the Has2+/− mice. A quantitative analysis using MLI revealed that the Has2+/− mice exhibited more severe emphysema at least 1 day after the administration of PPE compared with the WT mice (Figure 1C). Has2+/− mice showed a significant increase and prolongation in the number of BALF neutrophils after PPE stimulation (Figure 2D). Consistent with previous studies, an increase in the lymphocyte and eosinophil counts was observed during the acute phase in the WT mice (26, 27). However, the Has2+/− mice had a lower increase in the lymphocyte and eosinophil counts than the WT mice. The Has2+/− mice had significantly lower lymphocyte counts on day 1 after the administration of PPE than the WT mice (Figure 2C). This phenotypic difference may be attributed to the significantly higher number of BALF neutrophils compared with other cells in the Has2+/−-PPE mice.
In the cytokine and chemokine analyses, a significantly increase in the G-CSF levels was observed in the lungs of the Has2+/− mice on day 1 after the administration of PPE (Figure 4A, panel a). G-CSF is an important factor in the survival, proliferation, and differentiation of progenitor cells that differentiate into neutrophils (28). Previous studies have reported an association between genetic variation in the G-CSF and a decline in the forced expiratory volume in 1 second (FEV1) in smokers (29), and the elevated levels of G-CSF in the BALF of patients with COPD (30). In addition, high serum G-CSF levels promote COPD exacerbations characterized by neutrophilic inflammation with underlying bacterial dysbiosis (31). Further, the deletion of G-CSF reduces airway inflammation, lung tissue destruction, and the severity of comorbidities in mice (30). Based on these results, the increase in the G-CSF levels associated with Has2 dysfunction worsens emphysema formation and airway inflammation during the acute phase after PPE administration in the Has2+/−-KO mice. Conversely, there were no significant increases in the levels of other neutrophil- or Th1-related cytokines in the lung homogenates of the Has2+/−-PPE mice (Figure 4, Supplementaary Figures 2, 3). In addition, the expression of Th2-type cytokines, including IL-4, IL-5, and IL-13, decreased in the lung homogenates of the Has2+/−-PPE mice compared with the WT mice (Figure 4B). Similar results were observed in our previous chronic Ovalbumin (OVA)-stimulated asthma model (14). Based on these findings, the exacerbation of airway inflammation due to Has2 dysfunction is influenced by a pathway that is different from the Th1/Th2 imbalance pathway.
HA, CD44, and TGF-β1 are involved in the resolution of acute inflammation of tissue damage occurs (32–34). In the bleomycin-induced acute lung injury model, 75% of CD44-deficient mice die within a 2-week period. The underlying cause of this condition is believed to be abnormalities in the inflammatory resolution mechanism, characterized by low TGF-β1 activity, impaired removal of apoptotic neutrophils, and accumulation of HA (32). Moreover, TGF-β has a protective role for airway inflammation and AHR (35, 36). However, it exacerbates airway fibrosis and airway muscle hyperplasia (37). In the current study, RNA-seq analysis revealed high levels of Has2 mRNA expression in the lungs 4 days after the administration of PPE (Figure 5B). Using GlycoMaple to elucidate the alterations in the synthetic metabolic pathway of HA, an enhancement in the HMW-HA synthetic pathway was observed following the administration of PPE (Figure 5A). An upstream analysis revealed that the differentially expressed genes that underwent alterations subsequent to the administration of PPE in the WT mouse lungs were under the regulatory influence of TGF-β1 (Figure 5C). Further, GO analysis revealed that the negative regulation of the TGF-β receptor signaling pathway was significantly enriched among the uniquely upregulated genes in the Has2+/−-PPE mice (Figure 6D, panel b, right lane, Supplementary Table 4). A statistically significant difference in the TGF-β concentrations was observed between the WT mice and Has2+/− mice groups only 21 days after the administration of PPE (Figure 4). However, the Has2+/− mice were more likely to have lower protein-adjusted TGF-β concentrations. Based on these results suggest that Has2 inhibition leads to the sustained suppression of HA-binding protein expression and TGF-β signaling.
Regardless of the TGF-β pathway results, the GO analysis results using the unique genes in the WT-PPE or Has2+/−-PPE mice are supported by the findings of this study. Several significant biological processes, which are related to neutrophilic airway inflammation, were observed among the uniquely upregulated genes in the WT-PPE mice (Figure 6D, panel a, right lane, Supplementary Table 2). Meanwhile, the pathways associated with the adaptive immune response were identified among the unique downregulated genes in the WT-PPE mice (Figure 6D, panel a, left lane, Supplementary Table 3). These results are consistent with the BALF results (Figure 2). In particular, the airway inflammation phenotype in the Has2+/−-PPE mice is predominantly neutrophil-dominant, with lymphocyte-mediated airway inflammation being relatively suppressed compared with that in the WT mice. In our previous RNA-seq analysis conducted under chronic OVA stimulation, the downregulation of EIF2 signaling pathways, which are associated with ER stress response, and the TGF-β signaling pathways, and Th17 bias were observed in the BALB/c background Has2+/− mice (14). In the current study, RNA-seq analysis identified comprehensive IL-17A signaling among the uniquely upregulated genes in the WT-PPE mice. Further, cargo concentration in the ER was detected among the uniquely upregulated genes in the Has2+/−-PPE mice (Figure 6D, panel b, right lane, Supplementary Table 4). However, the roles of IL-17 and endoplasmic reticulum stress in this process are not yet completely understood. This may be attributed to the fact that PPE is a more potent stimulus than OVA in inducing G-CSF and neutrophilic inflammation, which complicates the detection of CD4+ T cells compared with OVA. Furthermore, previous studies have reported that C57BL/6 mice exhibit weaker increases in IL-17 than BALB/c mice in PPE administration models (24). Therefore, although significant increases in IL-6 and G-CSF were observed in the lung homogenate samples from this experiment, changes in IL-17 were likely difficult to detect.
The findings of this study do not provide a comprehensive explanation for the observed association between Has2 dysfunction and the subsequent development of acute-phase G-CSF and neutrophil increase, accompanied by severe emphysema formation. However, based on previous research results, there are several hypotheses that must be considered. In a bleomycin-induced pulmonary fibrosis model, Has2 induces and regulates cellular senescence in fibroblasts via the p27-CDK2-SKP2 pathway (38). Further, recent studies have revealed that fibroblasts responsive to FGF promote the production of G-CSF, HA secretion, and neutrophilic inflammation, thereby contributing to steroid-resistant airway inflammation in patients with severe asthma (39). In light of the aforementioned data, it can be hypothesized that Has2 dysfunction in fibroblasts may influence G-CSF production and HA secretion, thereby exacerbating the pathogenesis of inflammatory lung diseases. Considering that the proportion of M1 macrophages in the Has2+/− mice was significantly higher than that of the WT mice in the CIBERSORT analysis (Figure 7) and that alveolar destruction progressed after PPE administration, it can be hypothesized that Has2 expression abnormalities directly affect M1 macrophages and epithelial cells. Live E. coli instilled into the airways of mice induced rapid and selective increases in the mRNA expression of Has1 and Has2 in the whole lung (40).Primary cultures of murine airway epithelial cells showed significant increases in the Has2 expression (40). However, the upregulation of Has1 expression, but not Has2 expression, in the macrophages was found to be a selective response to LPS, the M1 classical activation via TLR4 in vitro (40). These results indicate that the Has2 expression in epithelial cells and fibroblasts may directly influence this disease state.
The current study had several limitations that should be considered before translating the findings into therapies for airway diseases. For instance, because the present study used lung bulk RNA sequencing, cell-type-specific data could not be obtained. Further research is needed to validate these mechanisms using methods such as single-cell RNA sequencing, spatial transcriptomics, and lineage-specific Has2 knockouts in fibroblast, epithelial, and macrophage cells. Additionally, the airway glycan ligand that acts on CD44 or regulates G-CSF secretion may have decreased in the Has2+/− mice. However, this decrease has not been confirmed. Therefore, future experiments should be performed to confirm treatment efficacy. In particular, whether the supply of HAS2 enzymes, HMW-HA, and anti-G-CSF Ab into the airway inhibits lung neutrophilic airway inflammation and emphysema should be examined. Furthermore, determining whether the administration of TGF-β signaling modulators exacerbates COPD pathology could be useful in evaluating potential therapeutic targets. Recent clinical studies have shown that the inhalation of HMW-HA decreases the duration of respiratory failure and the need for noninvasive ventilation therapy in patients experiencing acute exacerbation of COPD (41). Further, the inhibition of G-CSF receptors reduces neutrophilic inflammation, mucosal injury, and airways fibrosis (42, 43). These results show the clinical efficacy of treatments targeting hyaluronic acid abnormalities and support our results. Additionally, some experiments, such as cytokine assays and RNA-seq, have small sample sizes, which limits their power and generalizability. By increasing the sample size, it may be possible to detect cytokine changes and DEGs under Has2 functional suppression that were not detected in this study. Previous studies have reported that the progression of emphysematous changes persists for up to two to three months (26). A longer observation period is required because TGF-β is significantly reduced in Has2+/- mice 21 days after PPE administration. However, this study did not evaluate TGF-β-related structural changes including small airway remodeling such as airway wall thickness, collagen deposition, airway smooth muscle hypertrophy. Further investigation, including a morphological assessment of small airway remodeling at late onset, is necessary to elucidate the characteristic phenotypes associated with the mechanistic relationship between TGF-β and extracellular matrix homeostasis under HA dysfunction. Furthermore, this study did not validate the use of data from COPD patients or human lung tissue. Further validation using human specimens is necessary for clinical application. For instance, evaluating the activity of HAS2, the expression levels and fractions of HMW-HA and LMW-HA, and the concentrations of G-CSF and TGF-β in lung specimens from COPD patients, as well as examining their correlation with FEV1 decline, the frequency of acute exacerbations, and prognosis, is expected to enhance their clinical value.
Nevertheless, the observation that Has2+/− mice exhibited more severe emphysema progression, neutrophilic airway inflammation accompanied by a significant increase in the G-CSF levels, and reduced TGF-β levels in the lungs strongly indicate that low Has2 levels impair extracellular matrix homeostasis for controlling PPE-induced lung injury. Further, disorders associated with reduced HAS2 function in the lungs may result in persistent airway inflammation and remodeling. The serum HA concentrations of patients with COPD increased during exacerbations and remained high. Thus, serum HA can be an independent predictor of overall survival (44). Elucidating the effects of HAS2 and HA could help identify patients with COPD who have poor prognosis. Moreover, this research could support the efficacy of HA inhalation therapy, anti-G-CSF antibody therapy, and G-CSF receptor-targeted antagonistic therapy in patients with COPD. Considering these factors, the results of this study could help improve the mortality rates of COPD patients.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSE304417 (GEO). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE304417.
Ethics statement
The animal study was approved by The Animal Experiment Committee of Tsukuba University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
KN: Software, Writing – original draft, Formal analysis, Data curation. TK: Visualization, Validation, Supervision, Formal analysis, Funding acquisition, Project administration, Writing – original draft, Software, Investigation, Methodology, Data curation, Resources, Writing – review & editing, Conceptualization. MM: Writing – review & editing, Methodology, Supervision, Software, Data curation. ZW: Formal analysis, Writing – review & editing, Methodology, Validation, Software, Data curation, Resources. SM: Writing – review & editing, Methodology. KK: Methodology, Writing – review & editing. YY: Writing – review & editing, Methodology. KYa: Writing – review & editing, Supervision, Methodology. KYo: Methodology, Writing – review & editing, Supervision. YoM: Methodology, Supervision, Writing – review & editing. YuM: Writing – review & editing, Supervision. NH: Supervision, Writing – review & editing, Investigation.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported in part by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (21K08150, 24K11310 to TK).
Acknowledgments
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author. This work was performed in part at One-stop Sharing Facility Center for Future Drug Discoveries in Graduate School of Pharmaceutical Sciences, the University of Tokyo.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1683385/full#supplementary-material
References
1. Celli B, Fabbri L, Criner G, Martinez FJ, Mannino D, Vogelmeier C, et al. Definition and nomenclature of chronic obstructive pulmonary disease: time for its revision. Am J Respir Crit Care Med. (2022) 206:1317–25. doi: 10.1164/rccm.202204-0671PP
2. Smith LA, Oakden-Rayner L, Bird A, Zeng M, To MS, Mukherjee S, et al. Machine learning and deep learning predictive models for long-term prognosis in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Digit Health. (2023) 5:e872–e81. doi: 10.1016/S2589-7500(23)00177-2
3. Stolz D, Mkorombindo T, Schumann DM, Agusti A, Ash SY, Bafadhel M, et al. Towards the elimination of chronic obstructive pulmonary disease: a Lancet Commission. Lancet. (2022) 400:921–72. doi: 10.1016/S0140-6736(22)01273-9
4. Christenson SA, Smith BM, Bafadhel M, and Putcha N. Chronic obstructive pulmonary disease. Lancet. (2022) 399:2227–42. doi: 10.1016/S0140-6736(22)00470-6
5. Zafari Z, Li S, Eakin MN, Bellanger M, and Reed RM. Projecting long-term health and economic burden of COPD in the United States. Chest. (2021) 159:1400–10. doi: 10.1016/j.chest.2020.09.255
6. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, and Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J. (2017) 50. doi: 10.1183/13993003.01805-2016
7. Karakioulaki M, Papakonstantinou E, and Stolz D. Extracellular matrix remodelling in COPD. Eur Respir Rev. (2020) 29. doi: 10.1183/16000617.0124-2019
8. Cantor JO, Shteyngart B, Cerreta JM, Liu M, Armand G, and Turino GM. The effect of hyaluronan on elastic fiber injury in vitro and elastase-induced airspace enlargement in vivo. Proc Soc Exp Biol Med. (2000) 225:65–71. doi: 10.1046/j.1525-1373.2000.22508.x
9. Cantor JO, Shteyngart B, Cerreta JM, and Turino GM. The effect of lysozyme on elastase-mediated injury. Exp Biol Med (Maywood). (2002) 227:108–13. doi: 10.1177/153537020222700205
10. Klagas I, Goulet S, Karakiulakis G, Zhong J, Baraket M, Black JL, et al. Decreased hyaluronan in airway smooth muscle cells from patients with asthma and COPD. Eur Respir J. (2009) 34:616–28. doi: 10.1183/09031936.00070808
11. Karamanos NK, Piperigkou Z, Theocharis AD, Watanabe H, Franchi M, Baud S, et al. Proteoglycan chemical diversity drives multifunctional cell regulation and therapeutics. Chem Rev. (2018) 118:9152–232. doi: 10.1021/acs.chemrev.8b00354
12. Yatagai Y, Sakamoto T, Yamada H, Masuko H, Kaneko Y, Iijima H, et al. Genomewide association study identifies HAS2 as a novel susceptibility gene for adult asthma in a Japanese population. Clin Exp Allergy. (2014) 44:1327–34. doi: 10.1111/cea.12415
13. Tsunoda Y, Sherpa MT, Kiwamoto T, Matsuyama M, Osawa H, Sakurai H, et al. Has2 deficiency enhances OVA-induced airway inflammation and hyperresponsiveness in mice. Allergy. (2021) 76:2214–8. doi: 10.1111/all.14715
14. Sherpa MT, Kiwamoto T, Matsuyama M, Tsunoda Y, Yazaki K, Yoshida K, et al. Has2 regulates the development of ovalbumin-induced airway remodeling and steroid insensitivity in mice. Front Immunol. (2021) 12:770305. doi: 10.3389/fimmu.2021.770305
15. Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A Jr., et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. (2000) 106:349–60. doi: 10.1172/JCI10272
16. Hayashi S, Matsuno Y, Tsunoda Y, Sakurai H, Kiwamoto T, Morishima Y, et al. Transcription factor T-bet attenuates the development of elastase-induced emphysema in mice. Am J Respir Cell Mol Biol. (2019) 61:525–36. doi: 10.1165/rcmb.2018-0109OC
17. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. (2019) 37:773–82. doi: 10.1038/s41587-019-0114-2
18. Chen Z, Huang A, Sun J, Jiang T, Qin FX, and Wu A. Inference of immune cell composition on the expression profiles of mouse tissue. Sci Rep. (2017) 7:40508. doi: 10.1038/srep40508
19. Liang J, Jiang D, and Noble PW. Hyaluronan as a therapeutic target in human diseases. Adv Drug Delivery Rev. (2016) 97:186–203. doi: 10.1016/j.addr.2015.10.017
20. Laurell CB and Eriksson S. The electrophoretic α;1-globulin pattern of serum in α;1-antitrypsin deficiency. Scandinavian J Clin Lab Invest. (1963) 15:132–40. doi: 10.1080/00365516309051324
21. Turino GM, Ma S, Lin YY, and Cantor JO. The therapeutic potential of hyaluronan in COPD. Chest. (2018) 153:792–8. doi: 10.1016/j.chest.2017.12.016
22. Garantziotis S, Brezina M, Castelnuovo P, and Drago L. The role of hyaluronan in the pathobiology and treatment of respiratory disease. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L785–95. doi: 10.1152/ajplung.00168.2015
23. Cantor J, Armand G, and Turino G. Lung hyaluronan levels are decreased in alpha-1 antiprotease deficiency COPD. Respir Med. (2015) 109:656–9. doi: 10.1016/j.rmed.2015.03.006
24. Konno K, Arai H, Motomiya M, Nagai H, Ito M, Sato H, et al. A biochemical study on glycosaminoglycans (mucopolysaccharides) in emphysematous and in aged lungs. Am Rev Respir Dis. (1982) 126:797–801. doi: 10.1164/arrd.1982.126.5.797
25. Cantor JO, Cerreta JM, Ochoa M, Ma S, Chow T, Grunig G, et al. Aerosolized hyaluronan limits airspace enlargement in a mouse model of cigarette smoke-induced pulmonary emphysema. Exp Lung Res. (2005) 31:417–30. doi: 10.1080/01902140590918669
26. Limjunyawong N, Craig JM, Lagasse HA, Scott AL, and Mitzner W. Experimental progressive emphysema in BALB/cJ mice as a model for chronic alveolar destruction in humans. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L662–76. doi: 10.1152/ajplung.00214.2015
27. Craig JM, Scott AL, and Mitzner W. Immune-mediated inflammation in the pathogenesis of emphysema: insights from mouse models. Cell Tissue Res. (2017) 367:591–605. doi: 10.1007/s00441-016-2567-7
28. Tsioumpekou M, Krijgsman D, Leusen JHW, and Olofsen PA. The role of cytokines in neutrophil development, tissue homing, function and plasticity in health and disease. Cells. (2023) 12. doi: 10.3390/cells12151981
29. He JQ, Shumansky K, Connett JE, Anthonisen NR, Pare PD, and Sandford AJ. Association of genetic variations in the CSF2 and CSF3 genes with lung function in smoking-induced COPD. Eur Respir J. (2008) 32:25–34. doi: 10.1183/09031936.00040307
30. Tsantikos E, Lau M, Castelino CM, Maxwell MJ, Passey SL, Hansen MJ, et al. Granulocyte-CSF links destructive inflammation and comorbidities in obstructive lung disease. J Clin Invest. (2018) 128:2406–18. doi: 10.1172/JCI98224
31. Chakrabarti A, Mar JS, Choy DF, Cao Y, Rathore N, Yang X, et al. High serum granulocyte-colony stimulating factor characterises neutrophilic COPD exacerbations associated with dysbiosis. ERJ Open Res. (2021) 7. doi: 10.1183/23120541.00836-2020
32. Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, et al. Resolution of lung inflammation by CD44. Sci (New York NY). (2002) 296:155–8. doi: 10.1126/science.1069659
33. DeGrendele HC, Estess P, and Siegelman MH. Requirement for CD44 in activated T cell extravasation into an inflammatory site. Sci (New York NY). (1997) 278:672–5. doi: 10.1126/science.278.5338.672
34. McDonald B and Kubes P. Interactions between CD44 and hyaluronan in leukocyte trafficking. Front Immunol. (2015) 6:68. doi: 10.3389/fimmu.2015.00068
35. Nakao A, Miike S, Hatano M, Okumura K, Tokuhisa T, Ra C, et al. Blockade of transforming growth factor beta/Smad signaling in T cells by overexpression of Smad7 enhances antigen-induced airway inflammation and airway reactivity. J Exp Med. (2000) 192:151–8. doi: 10.1084/jem.192.2.151
36. Hansen G, McIntire JJ, Yeung VP, Berry G, Thorbecke GJ, Chen L, et al. CD4(+) T helper cells engineered to produce latent TGF-beta1 reverse allergen-induced airway hyperreactivity and inflammation. J Clin Invest. (2000) 105:61–70. doi: 10.1172/JCI7589
37. Liu G, Philp AM, Corte T, Travis MA, Schilter H, Hansbro NG, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. (2021) 225:107839. doi: 10.1016/j.pharmthera.2021.107839
38. Li Y, Liang J, Yang T, Monterrosa Mena J, Huan C, Xie T, et al. Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix biology: J Int Soc Matrix Biol. (2016) 55:35–48. doi: 10.1016/j.matbio.2016.03.004
39. Guidi R, Xu D, Choy DF, Ramalingam TR, Lee WP, Modrusan Z, et al. Steroid-induced fibroblast growth factors drive an epithelial-mesenchymal inflammatory axis in severe asthma. Sci Transl Med. (2022) 14:eabl8146. doi: 10.1126/scitranslmed.abl8146
40. Chang MY, Tanino Y, Vidova V, Kinsella MG, Chan CK, Johnson PY, et al. A rapid increase in macrophage-derived versican and hyaluronan in infectious lung disease. Matrix biology: J Int Soc Matrix Biol. (2014) 34:1–12. doi: 10.1016/j.matbio.2014.01.011
41. Galdi F, Pedone C, McGee CA, George M, Rice AB, Hussain SS, et al. Inhaled high molecular weight hyaluronan ameliorates respiratory failure in acute COPD exacerbation: a pilot study. Respir Res. (2021) 22:30. doi: 10.1186/s12931-020-01610-x
42. Wang H, Aloe C, Wilson N, and Bozinovski S. G-CSFR antagonism reduces neutrophilic inflammation during pneumococcal and influenza respiratory infections without compromising clearance. Sci Rep. (2019) 9:17732. doi: 10.1038/s41598-019-54053-w
43. Wang H, Aloe C, McQualter J, Papanicolaou A, Vlahos R, Wilson N, et al. G-CSFR antagonism reduces mucosal injury and airways fibrosis in a virus-dependent model of severe asthma. Br J Pharmacol. (2021) 178:1869–85. doi: 10.1111/bph.15415
Keywords: HAS2, chronic obstructive pulmonary disease, emphysema, TGF-β1, G-CSF, neutrophil
Citation: Nishino K, Kiwamoto T, Matsuyama M, Wei Z, Matsumura S, Kuramoto K, Yabuuchi Y, Yazaki K, Yoshida K, Matsuno Y, Morishima Y and Hizawa N (2025) Hyaluronic acid synthase 2 dysfunction exacerbates elastase-induced neutrophilic airway inflammation and emphysema in mice. Front. Immunol. 16:1683385. doi: 10.3389/fimmu.2025.1683385
Received: 11 August 2025; Accepted: 22 September 2025;
Published: 14 October 2025.
Edited by:
Fernanda Degobbi Tenorio Quirino dos Santos Lopes, University of São Paulo, BrazilReviewed by:
Leandro do Nascimento Camargo, University of São Paulo, BrazilCarlos Rio, Fundació Institut d’Investigació Sanitaria Illes Balear, Spain
Copyright © 2025 Nishino, Kiwamoto, Matsuyama, Wei, Matsumura, Kuramoto, Yabuuchi, Yazaki, Yoshida, Matsuno, Morishima and Hizawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takumi Kiwamoto, dC1raXdhbW90b0BtZC50c3VrdWJhLmFjLmpw