 Ruike Fan
Ruike Fan Zihan Sun2
Zihan Sun2 Lishang Dai
Lishang Dai- 1Department of Pharmacy, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China
- 2School of Traditional Chinese Medicine, Wenzhou Medical University, Wenzhou, China
This study investigates the immunomodulatory effects of Lonicera japonica Thunb. extract, a perennial semi-evergreen vine of the Caprifoliaceae family, on lipopolysaccharide (LPS)-induced immune responses in loach (Misgurnus anguillicaudatus) through miRNA regulatory mechanisms. Small RNA libraries constructed from hepatic tissues of LPS-challenged (CK) and Lonicera japonica-treated (LJ) groups yielded 139.6 million clean reads with characteristic 21–23 nucleotide length distribution. Abundance analysis revealed significant differential expression patterns within the let-7 family: miR-let-7-9, miR-let-7-6, and miR-let-7–18 exhibited higher abundance in the infection group, whereas miR-let-7-1, miR-let-7-17, and miR-let-7–16 showed elevated abundance in the treatment group. Comparative profiling identified 55 differentially expressed miRNAs (41 upregulated, 14 downregulated), with animal-undef-351, animal-mir-21-6, and animal-undef-603 demonstrating the most significant expression differences (P<0.01). KEGG enrichment analysis highlighted predominant involvement in sphingolipid signaling pathway, glycerophospholipid metabolism, T cell receptor signaling pathway, and TNF signaling pathway. GO analysis revealed significant enrichment in biological processes related to glycosylation, cellular components of transcription regulator complexes, and molecular functions associated with double-stranded DNA binding. These findings demonstrate that L. japonica alleviates LPS-induced inflammation by modulating miRNA expression networks, particularly through sphingolipid metabolism and TNF signaling pathways, providing novel molecular evolutionary insights into the immunoregulatory mechanisms of traditional Chinese medicine.
1 Introduction
The loach (Misgurnus anguillicaudatus), a benthic fish species of the family Cobitidae, derives its scientific name from its ecological niche in lacustrine and pond sediments rich in humic substrates (1). This species is widely distributed across freshwater ecosystems in Asia, with primary habitats concentrated in the Yangtze River Basin of China, as well as in Korea and Japan (2–4). Recognized as a high-value aquaculture species, the loach is not only rich in high-quality proteins, vitamins, and essential amino acids (5), but also exhibits remarkable environmental adaptability, surviving under extreme conditions such as elevated temperatures (>30 °C), hypoxic environments, and intermittent drought (6, 7). Recent studies have revealed that bioactive components in loach tissues, including mucous polysaccharides and antioxidant enzymes, possess anti-inflammatory, hepatoprotective, and antitumor potential (8), driving its transition from traditional culinary use to medicinal applications. Owing to these attributes, the loach has emerged as a cornerstone species in China’s freshwater aquaculture industry (8). However, intensive farming practices are frequently challenged by pathogenic microbial infections (e.g., Aeromonas hydrophila and Edwardsiella tarda) and environmental stressors such as ammonia nitrogen accumulation, which trigger outbreaks of inflammatory diseases, thereby posing significant threats to aquaculture productivity and industry sustainability (9, 10).
Lipopolysaccharide (LPS), a major component of Gram-negative bacterial outer membranes, triggers systemic inflammation in fish by activating the Toll-like receptor 4 (TLR4)/NF-κB signaling pathway, leading to excessive production of pro-inflammatory cytokines such as TNF-α and IL-1β (11–14). Honeysuckle (L. japonica), a traditional Chinese herb, contains bioactive compounds including luteolin (LUT), chlorogenic acid, and caffeic acid, which exhibit broad-spectrum anti-inflammatory, immunomodulatory, and antioxidant properties (15–17). Previous studies have demonstrated that honeysuckle extracts alleviate mammalian inflammatory injuries via TLR4 pathway inhibition (18), yet their regulatory effects on LPS-induced inflammation in fish remain unexplored.
MicroRNAs (miRNAs), a class of endogenous non-coding RNAs (~22 nt in length), regulate gene expression by binding to the 3’ untranslated region (UTR) of target mRNAs, playing pivotal roles in immune responses and cellular homeostasis (19). Since the discovery of the first miRNA (lin-4) in Caenorhabditis elegans in 1993 (20), high-throughput sequencing technologies have identified hundreds of conserved miRNAs in model fish species such as zebrafish (Danio rerio) (21, 22). Recent advances further highlight the critical functions of miRNAs in stress adaptation and disease resistance of non-model aquaculture species (23, 24), providing novel insights into deciphering anti-pathogenic mechanisms.
Although existing studies have focused on loach immunity (8, 25, 26), the molecular mechanisms underlying herbal intervention in LPS-induced inflammatory responses are largely unknown. This study pioneers the integration of small RNA sequencing and molecular biology approaches to systematically analyze the miRNA expression profiles in loach muscle tissues following LPS challenge and honeysuckle treatment, which contributed to further understanding of the anti-inflammatory mechanism of Honeysuckle, and thus provided a basis for further optimization of artificial breeding methods of loach.
2 Materials and methods
2.1 Experimental animal and LPS treatment
A total of 300 healthy loaches of similar size were purchased from aquatic animal breeding base of Zhejiang Province and randomly divided into 6 groups with an average of 50 in each group. Normal feeding for 3 days, 20 g feed per day. On the fourth day, three groups were given Chinese herbal powder, that is, 20 g of feed and 10g of Chinese herbal medicine, and they were set as the experimental group, and the other three groups were given the same amount of normal feeding and were set as the control group. After 12 h, each loach in both the control and treatment groups was intraperitoneally injected with 100 μL of LPS solution (60 mg/100 μL, dissolved in phosphate-buffered saline (PBS)). The LPS used in this study was purchased from Beijing Solarbio Science & Technology Co., Ltd. The PBS solution preparation method is as follows: 8.00 g NaCl, 0.20 g KCl, 1.44 g Na2HPO4, and 0.24 g KH2PO4 and dissolved in 800 mL of distilled water. Adjust the HCl solution to pH 7.4, and supplementary distilled water to make a constant volume of 1 liter from then on. Two sets of samples were observed for 3,6,12 and 24 h. The hepatic tissue of the sample was ground and quickly frozen with liquid nitrogen and stored in the refrigerator at -80 °C.
2.2 Total RNA extraction, cDNA library preparation
Total RNA was extracted from loach tissues using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) followed by DNase I (RNase-free) treatment to eliminate genomic DNA contamination. RNA purity and concentration were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA), while RNA integrity was evaluated via an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples meeting the following criteria were selected for subsequent analysis: 28S/18S rRNA ratio ≥1.0, RNA Integrity Number (RIN) ≥7.0, OD260/280 = 1.8-2.2, and OD260/230 ≥2.0. To enrich small RNA molecules (18–30 nt), RNA fragments were size-selected using 15% denaturing polyacrylamide gel electrophoresis (PAGE). The purified small RNAs were ligated with 3′ and 5′ adapters using T4 RNA ligase (New England Biolabs, Ipswich, MA, USA). Reverse transcription was performed with adapter-specific primers, followed by PCR amplification to generate cDNA libraries. PCR products were purified via 3.5% agarose gel electrophoresis to isolate fragments of the desired size (140–160 bp), thereby removing unincorporated primers, primer dimers, and adapter contaminants. The final cDNA libraries were quantified by qPCR and subjected to paired-end sequencing (PE150) on an Illumina HiSeq platform (Shanghai Personal Biotechnology Co., Ltd., Shanghai, China).
The original sequence was sequenced by using the script independently developed by Shanghai Personal Biotechnology Co., Ltd. to remove the joint, and then mass cutting was performed according to the quality of the sequence. Specifically, the original sequence was searched with 5 base lengths as a window. When the average sequencing quality of bases in the window was lower than 20, the part starting from the front of the window was truncated and discarded. The filter sequences, namely clean reads, were obtained. The number of Clean Reads (Total Reads) whose sequence length was more than 18 nt and less than 36 nt was analyzed. The identical sequences in a single sample were de-processed and sequence abundance was counted to obtain Unique Reads for subsequent analysis.
2.3 Small RNA annotation
Using Blast (27), Unique Reads were compared with the Rfam13 database to screen four known classes of ncRNAs, including rRNA, tRNA, snRNA and snoRNA. The screening criteria are no more than 2 perfect matches or mismatches.
Reads that were not annotated to the above four types of ncRNAs were screened from Blast and all mature miRNA sequences in miRBase22 (http://www.mirbase.org/, Griffths -Jones S, 2004) for conserved miRNAs. The screening criteria are perfect match or mismatch number no more than 2.
we summarized the comparison and annotation of all small RNAs with other types of RNAs. Small RNA sequences are short, and it is generally easy to match different fragments, so there are multiple annotation results. In order to make each small RNA have a unique comment, the existing comment results are sorted according to the priority of known miRNA > rRNA > tRNA > snRNA > snoRNA > novel miRNA. At the same time, because different base positions of miRNAs may have different base preferences, base preference analysis was performed for known miRNAs.
2.4 Differentially expressed miRNAs analysis
In order to observe the conserved miRNA among species, miRNA of Loach was Blast compared with the mature miRNA sequences of related species in the miRBase database, and the Reads Count value of miRNA was calculated according to the number of conserved miRNA sequences. Then, the conservative miRNA expression data of the control group and the treatment group were collated, and DESeq (version 1.18.0, Anders S and Huber W, 2010) was used to analyze the miRNA with differential expression. They were mainly screened according to the expression multiple difference (|fold change| > 2) and the significance of expression difference (p-value < 0.05), and finally the results were obtained for statistics. At the same time, in order to judge the expression patterns of the obtained differentially expressed genes under different experimental conditions, we used R language Pheatmap software package to conduct bidirectional cluster analysis of genes and samples. The distance was calculated by Euclidean method and the Complete Linkage method was used for hierarchical clustering.
2.5 Target gene prediction and functional analysis of miRNAs
Because miRNAs bind to target sites mainly through complementary pairing. Therefore, we used miranda to predict miRNA target genes in animals and psRobot_tar to predict miRNA target genes in plants. Then, GO functional enrichment analysis was performed on the target genes of different miRNAs to obtain GO functional items that were significantly enriched in the target genes of different miRNAs, thus displaying relevant biological functions. (Map all genes to each Term in the Gene Ontology database, and calculate the number of target genes for each Term. Hypergeometric distribution was used to calculate the Term of significant enrichment of target genes against the background of the whole genome. The number of target genes contained in different levels of each KEGG Pathway was then counted to determine the metabolic pathways and signaling pathways that the target genes were mainly involved in (hypergeometric distribution was used to calculate the pathways with significant enrichment of target genes against the background of the whole genome).
2.6 Quantitative Real-Time PCR Validation
In order to verify the accuracy and reliability of the sequencing results, we selected qRT-PCR based on the principle of │log2(fc)│>1 and fpkm > 10. A total of 7 genes (5 down-regulated and 2 up-regulated) were selected for verification, and primers were designed by DNAstar (primer design is shown in Table 1). The qPCR was verified by SYBR®Green Supermix (TransGen Biotech) method with primer length of 18–24 bp as standard.

Table 1. Information of RT-qPCR primers.
3 Results
3.1 Analysis of small RNA sequencing results
To investigate the expression dynamics of miRNAs in loach (M. anguillicaudatus) under pathogenic infection and Honeysuckle (L. japonica) treatment, total RNA was extracted from hepatic tissues of both experimental (Honeysuckle-fed) and control groups, followed by the construction of six independent small RNA sequencing libraries. Deep sequencing generated 144.7 million raw reads across all libraries. After rigorous quality control procedures including adapter trimming, removal of contaminated reads, and elimination of low-quality sequences, 139.6 million high-quality clean reads were retained for subsequent analysis. Notably, the proportion of high-quality clean reads exceeded 95.6% in all six libraries (Table 2). Subsequently, we performed length distribution analysis on high-quality clean reads (total reads) ranging from 18 to 36 nucleotides (nt). Following deduplication to eliminate redundant sequences within individual samples, sequence abundance profiling revealed that small RNAs were predominantly enriched in the 21–23 nt range, with 22 nt sequences constituting the most abundant population (Figure 1 and Figure 2). Notably, this size distribution pattern aligns with canonical characteristics of animal miRNAs, thereby substantiating the biological relevance of our sequencing data.
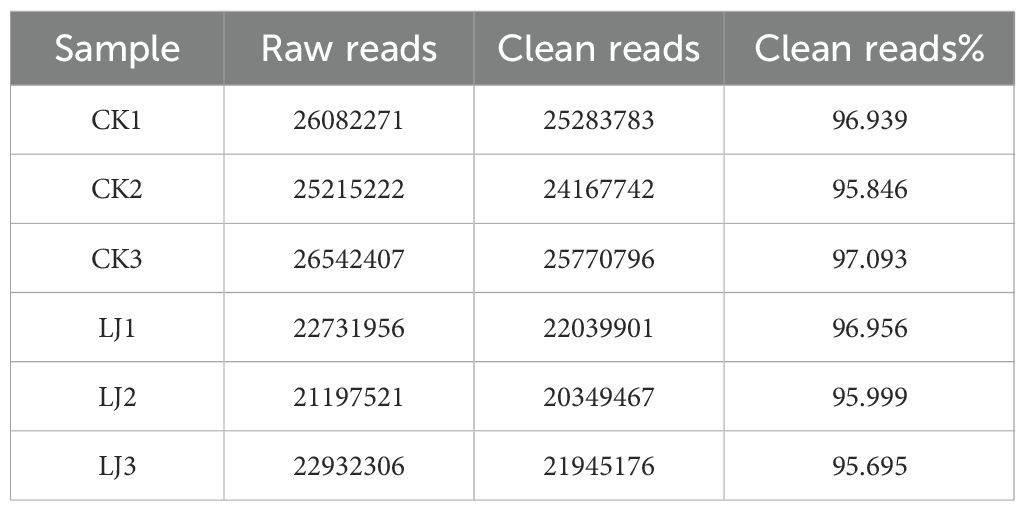
Table 2. Raw data statistics.
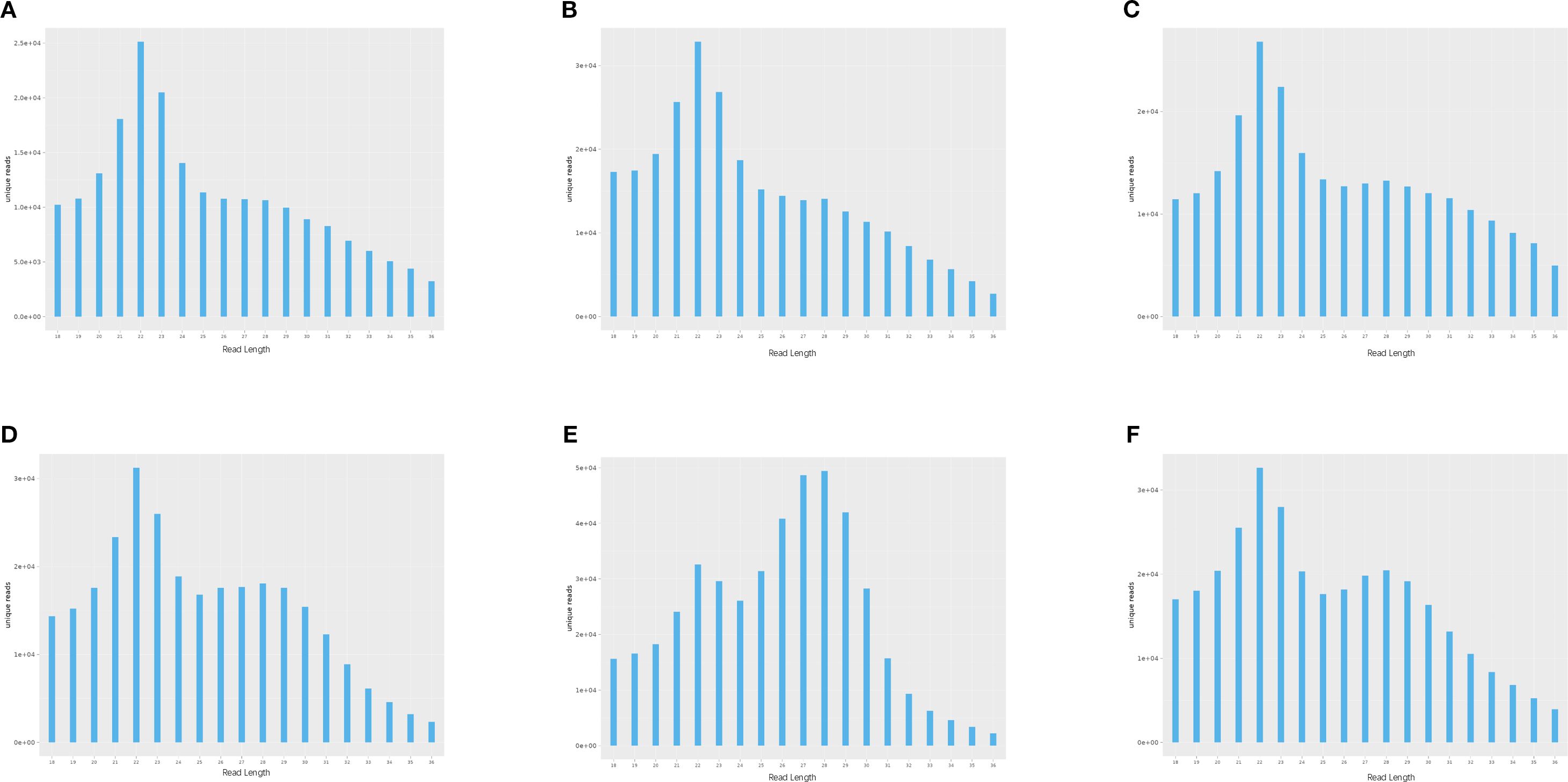
Figure 1. Length-specific distribution of deduplicated unique reads. (X: Read Length [nt]; Y: Abundance [×104]): (A) CK1, (B) CK2, (C) CK3, (D) LJ1, (E) LJ2, (F) LJ3.
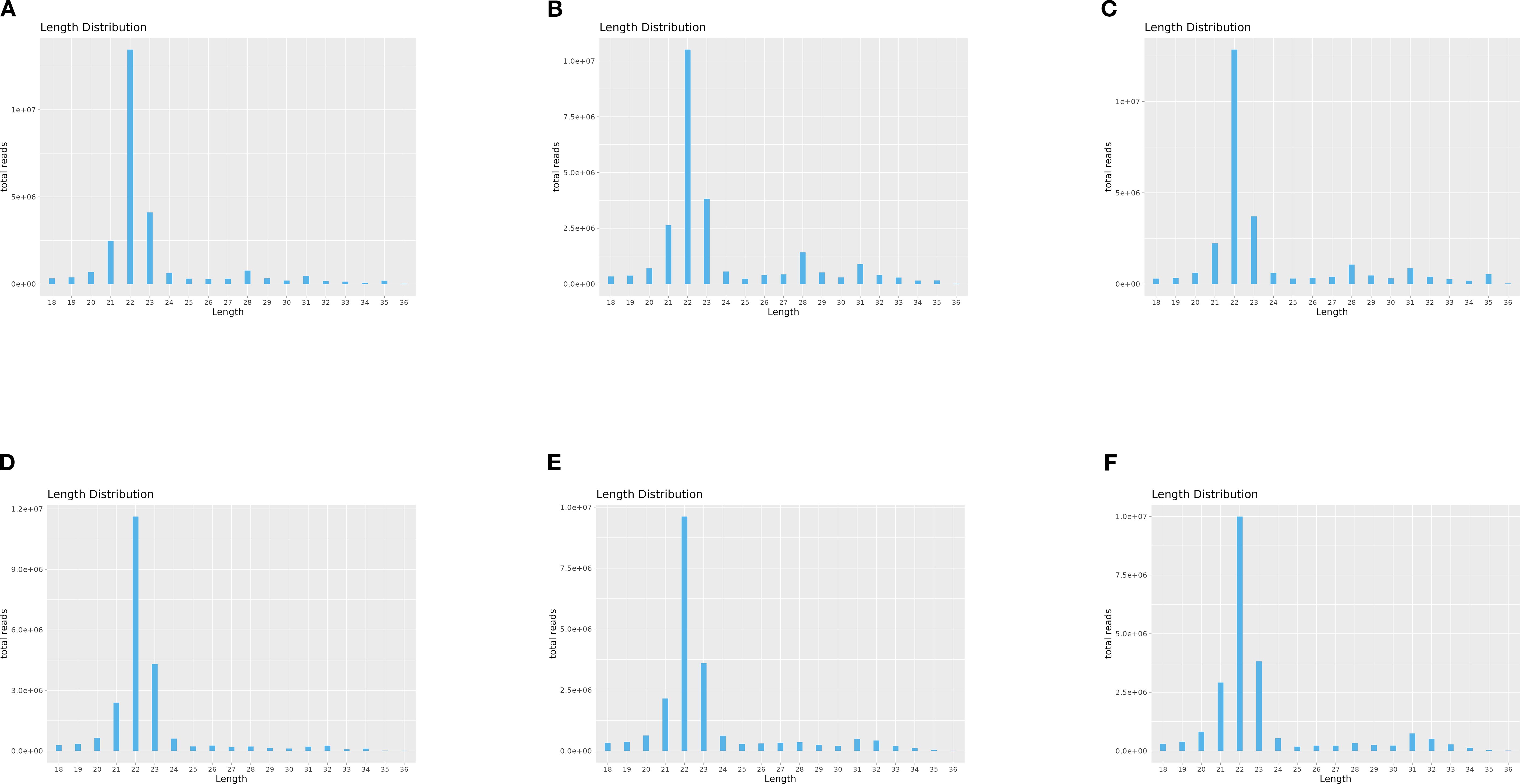
Figure 2. Length-specific distribution of deduplicated total reads. (X: Read Length [nt]; Y: Abundance [×104]): (A) CK1, (B) CK2, (C) CK3, (D) LJ1, (E) LJ2, (F) LJ3.
3.2 Identification of miRNAs in hepatic tissues of loach
The clean reads derived from both infected and control groups were systematically classified and annotated into three categories: known RNAs, unannotated RNAs, and non-coding RNAs (ncRNAs) including ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA). Bioinformatic analysis revealed that the proportion of known RNAs in deduplicated clean reads ranged between 4% and 8% across groups (Table 3), whereas this proportion exceeded 68% prior to deduplication (Table 4). This striking reduction in known RNA abundance post-deduplication (from >68% to <10%) demonstrates significant dynamic changes in sequence complexity, suggesting effective removal of redundant transcriptional fragments during miRNA enrichment (Figure 3).
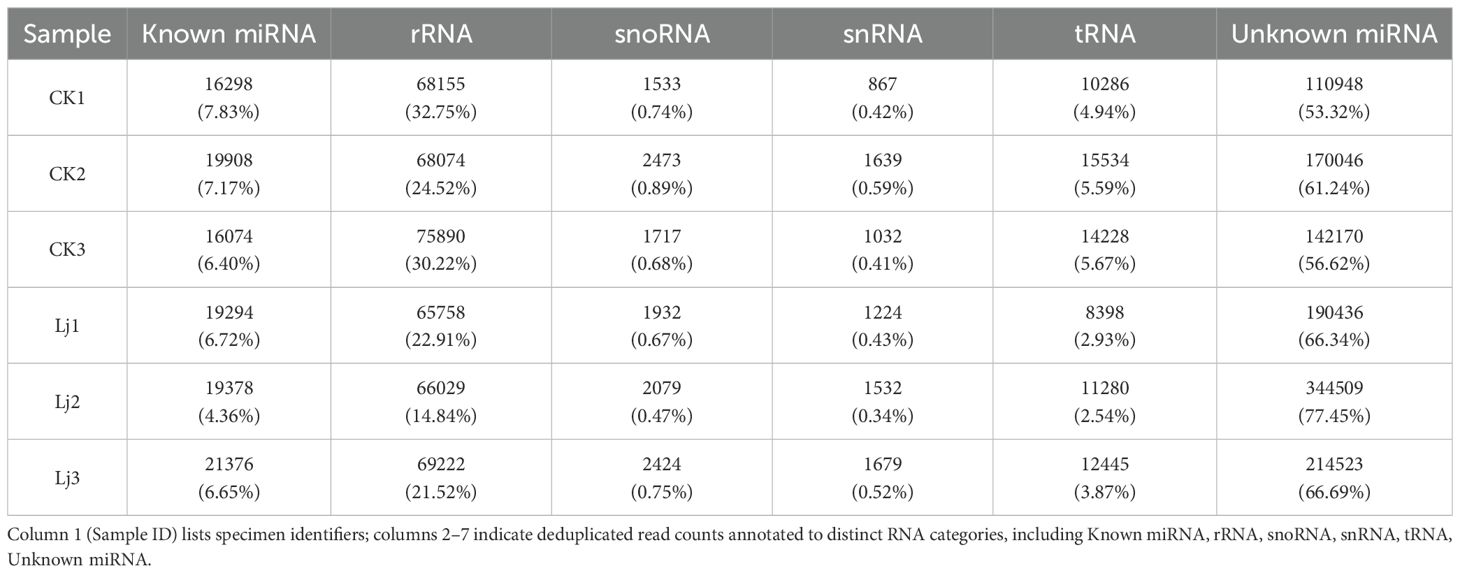
Table 3. Statistical results of miRNA in each sample (Unique).
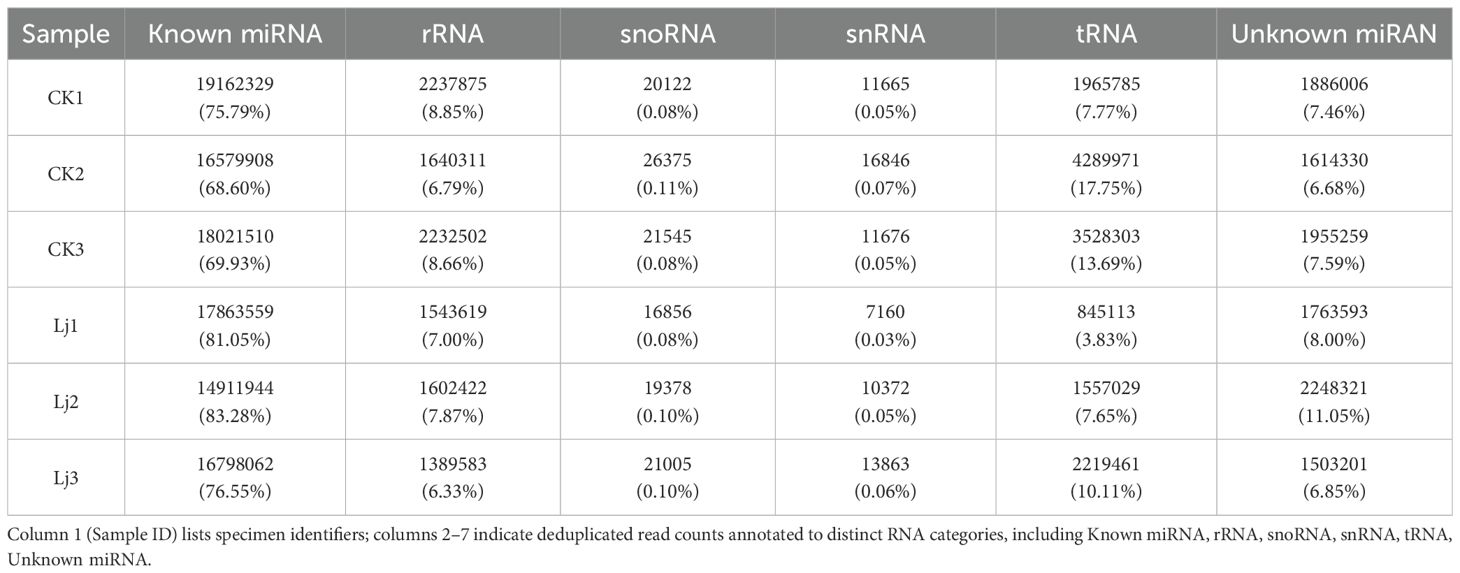
Table 4. Statistical results of miRNA in each sample (Total).
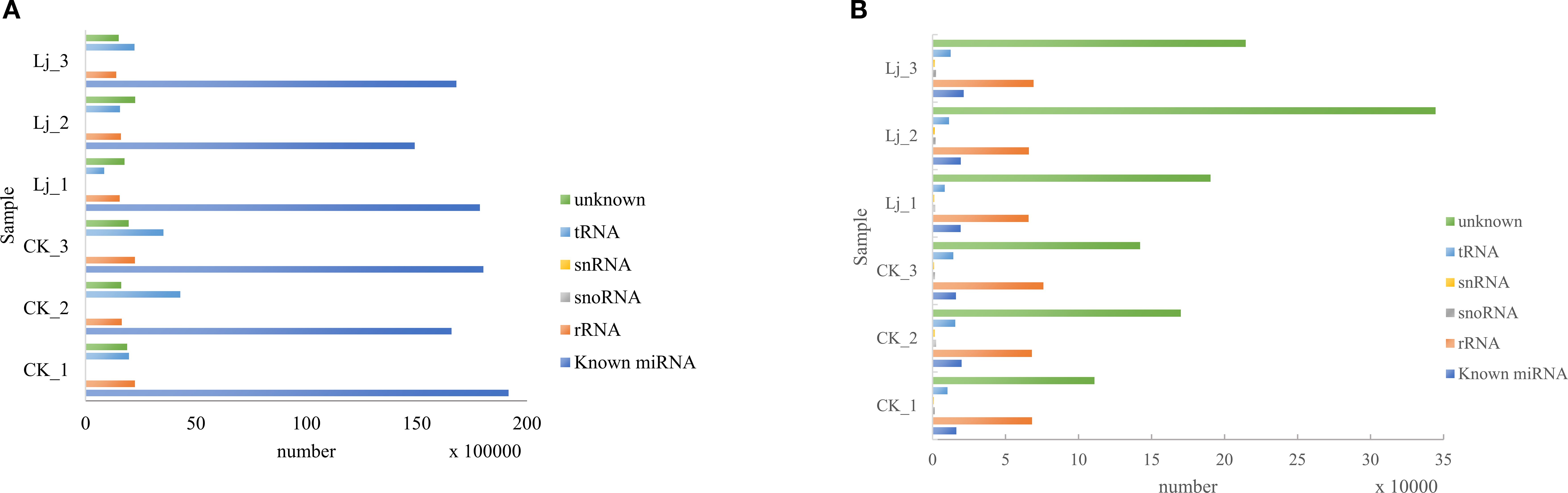
Figure 3. Small RNA Annotation: (A) Total Reads, (B) Deduplicated Reads.
Different base locations of miRNAs may have different base preferences. Therefore, base preference analysis of known miRNAs found that the 5 ‘end had a strong U tendency, that is, the typical miRNA base ratio caused by Dicer enzymes and DCL enzymes recognizing and cutting precursor miRNAs. Dicer and DCL enzymes recognize and cut the precursor miRNA, and its 5’ end has a strong U tendency. Through base preference analysis of miRNA, the typical miRNA base ratio was obtained (Figure 4).
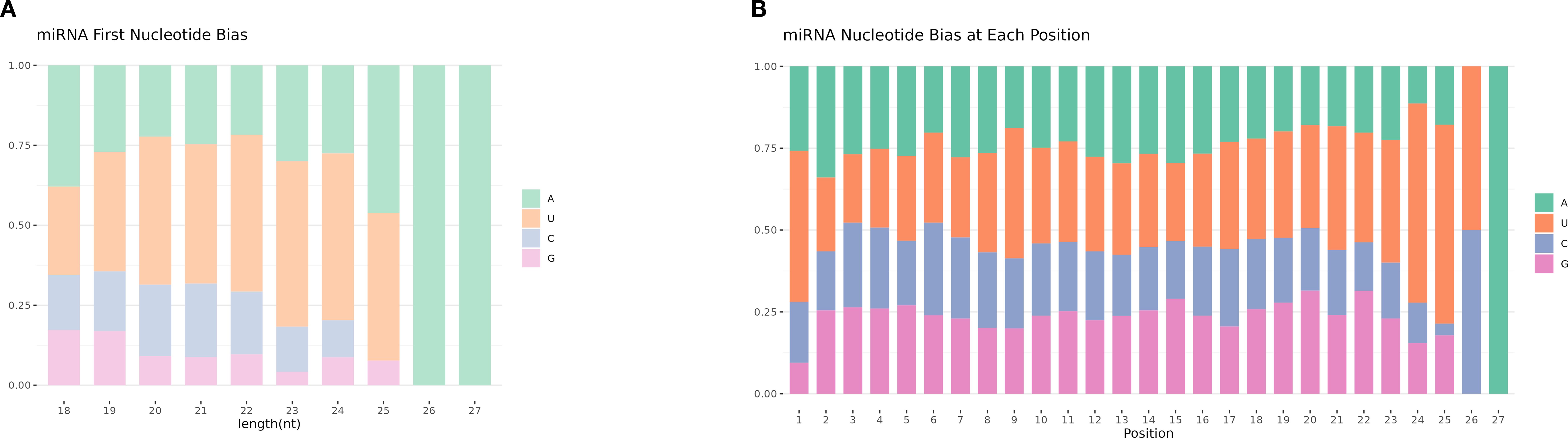
Figure 4. miRNA base preference. (A) The first base distribution of miRNAs of different lengths. The abscissa indicates sequences of different lengths; the ordinate indicates the percentage of the first base of each length of miRNA. (B) miRNA site base distribution.
3.3 Differentially expressed miRNAs identification
Through alignment with evolutionarily conserved miRNAs, we quantified miRNA read counts across experimental groups (Table 5). Comparative analysis revealed that animal-let-7-9, animal-let-7-6, animal-let-7–18 exhibited statistically significant upregulation in the infected group compared to the honeysuckle-treated group. Conversely, animal-let-7-1, animal-let-7-17, and animal-let-7–16 demonstrated marked transcriptional activation following honeysuckle intervention, with fold-change values exceeding 2.5 (Figure 5). These differential expression patterns suggest pathogen-responsive and phytotherapeutic regulatory roles within the let-7 miRNA family.
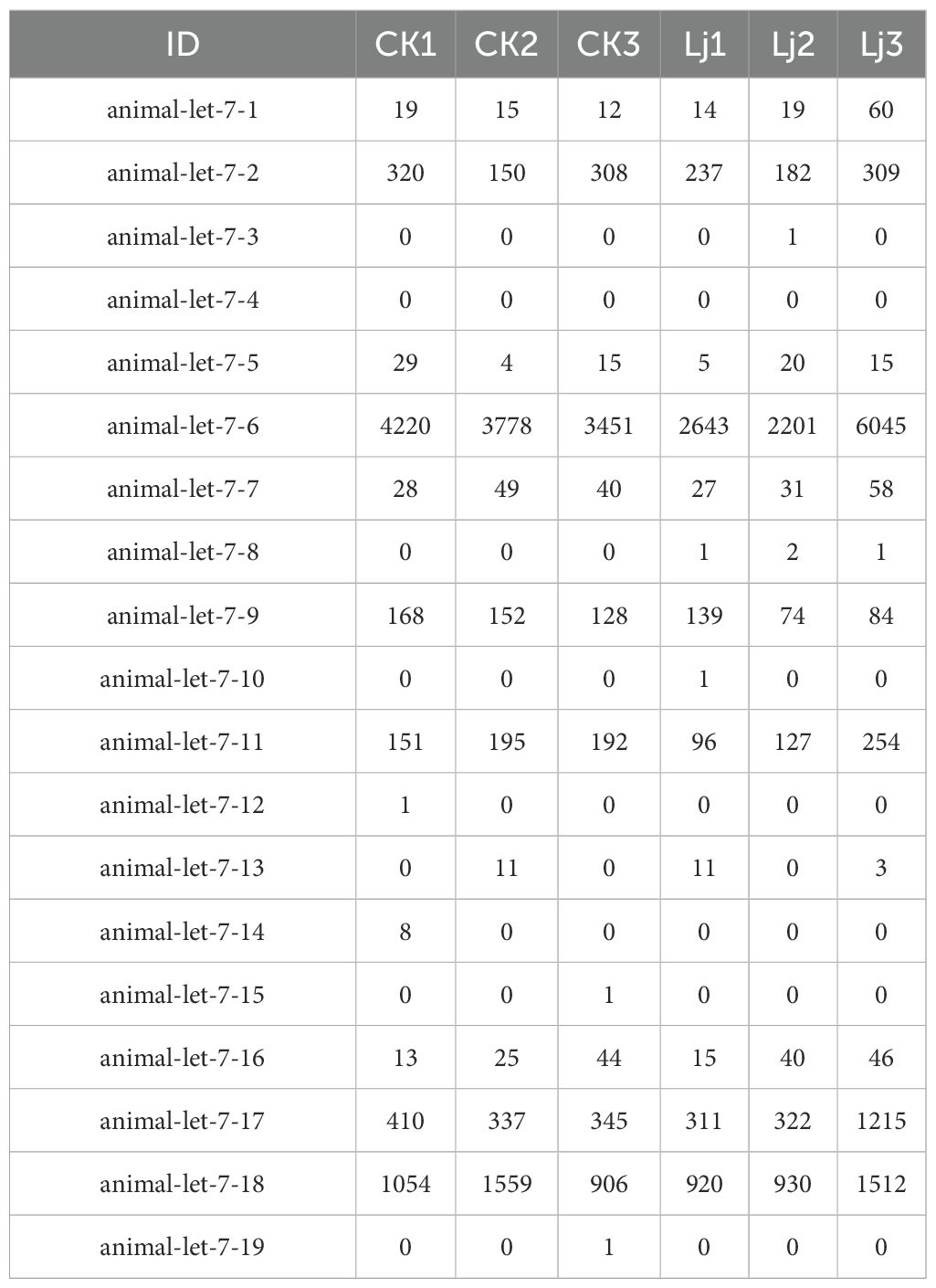
Table 5. miRNA Expression table.
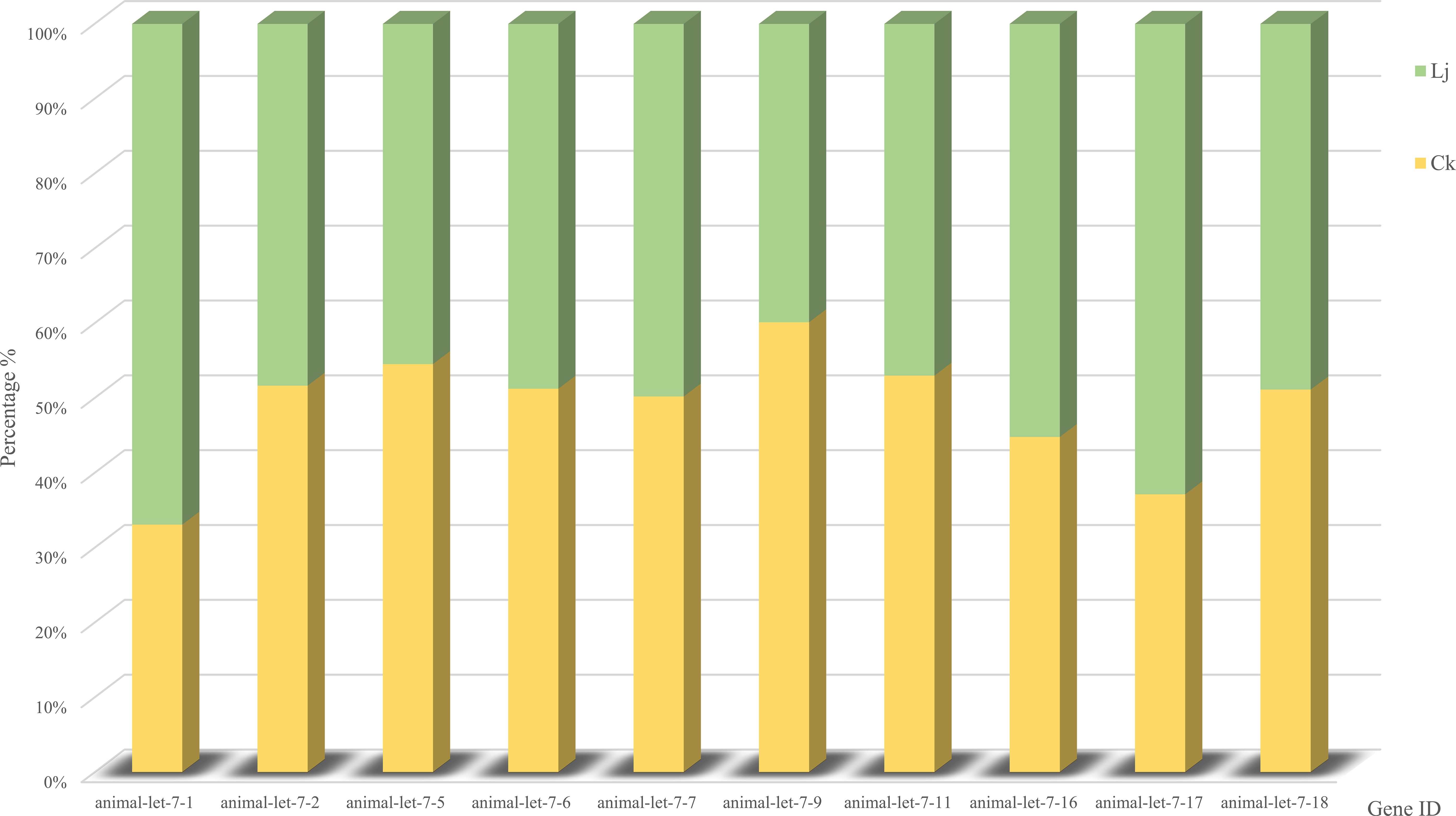
Figure 5. Comparative miRNA expression abundance profiles: infection group vs. control group.
Comparing the expression differences of miRNA between LJ group and CK group, 55 differentially expressed genes were found (Figure 6), of which 41 were significantly up-regulated and 14 were significantly down-regulated. The expression profiles of all miRNA in group LJ and group CK are shown in the heatmap in Figure 5. Notably, among these differentially expressed miRNAs, animal-undef-351, animal-mir-21-6, and animal-undef-603 exhibited the most pronounced expression differences (P<0.01). Also, we found animal-mir-22-2, animal-mir-181-4, animal-mir-122-2, animal-mir-454-2, animal-mir-10-13, animal-mir-29-11, animal-mir-129–3 Down-regulated trends were observed in all miRNAs, suggesting that the transcription levels of these miRNAs may be regulated by common factors in LJ (Table 6).
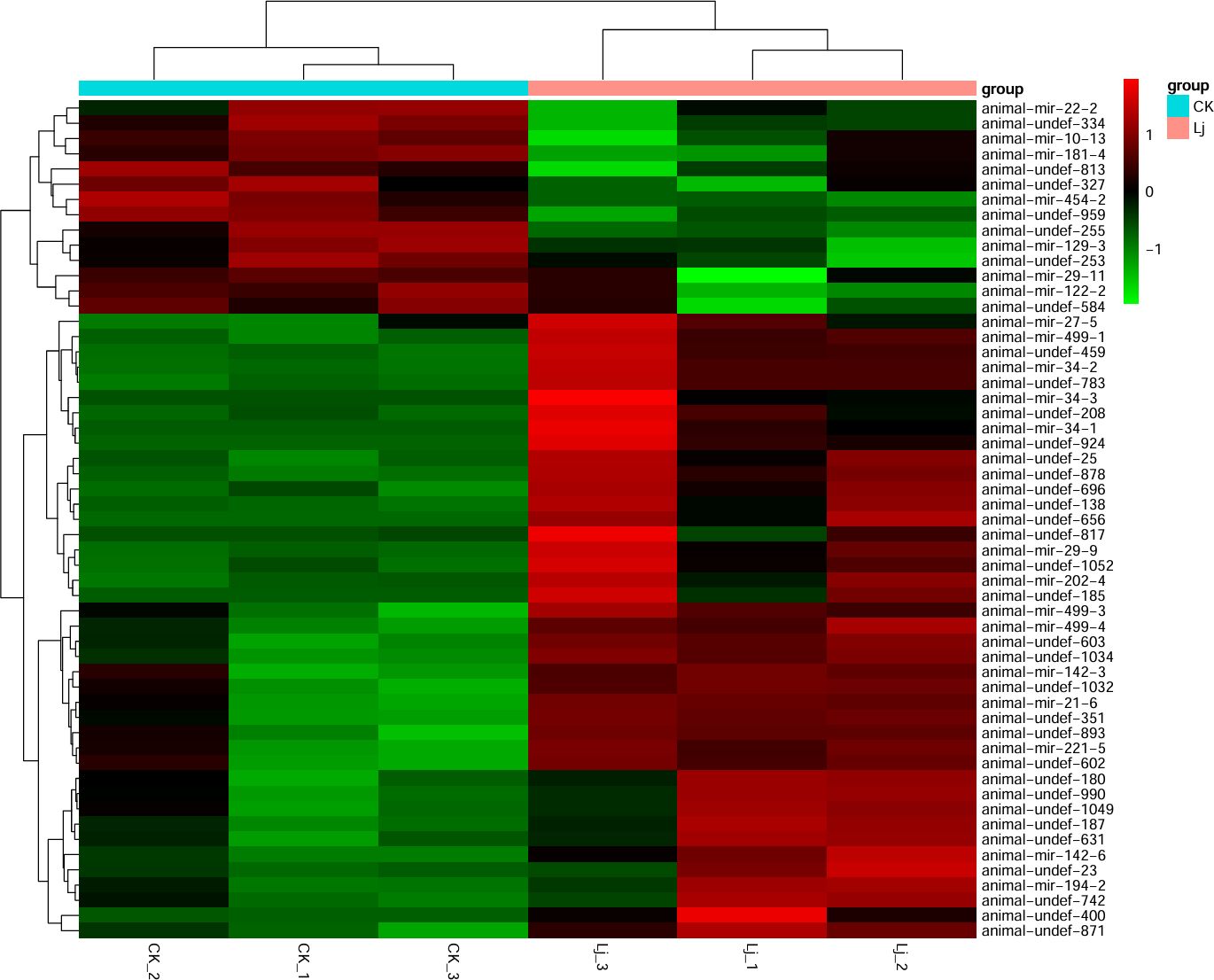
Figure 6. Cluster map of differentially expressed miRNAs. Red color indicates elevated expression and green color indicates decreased expression.
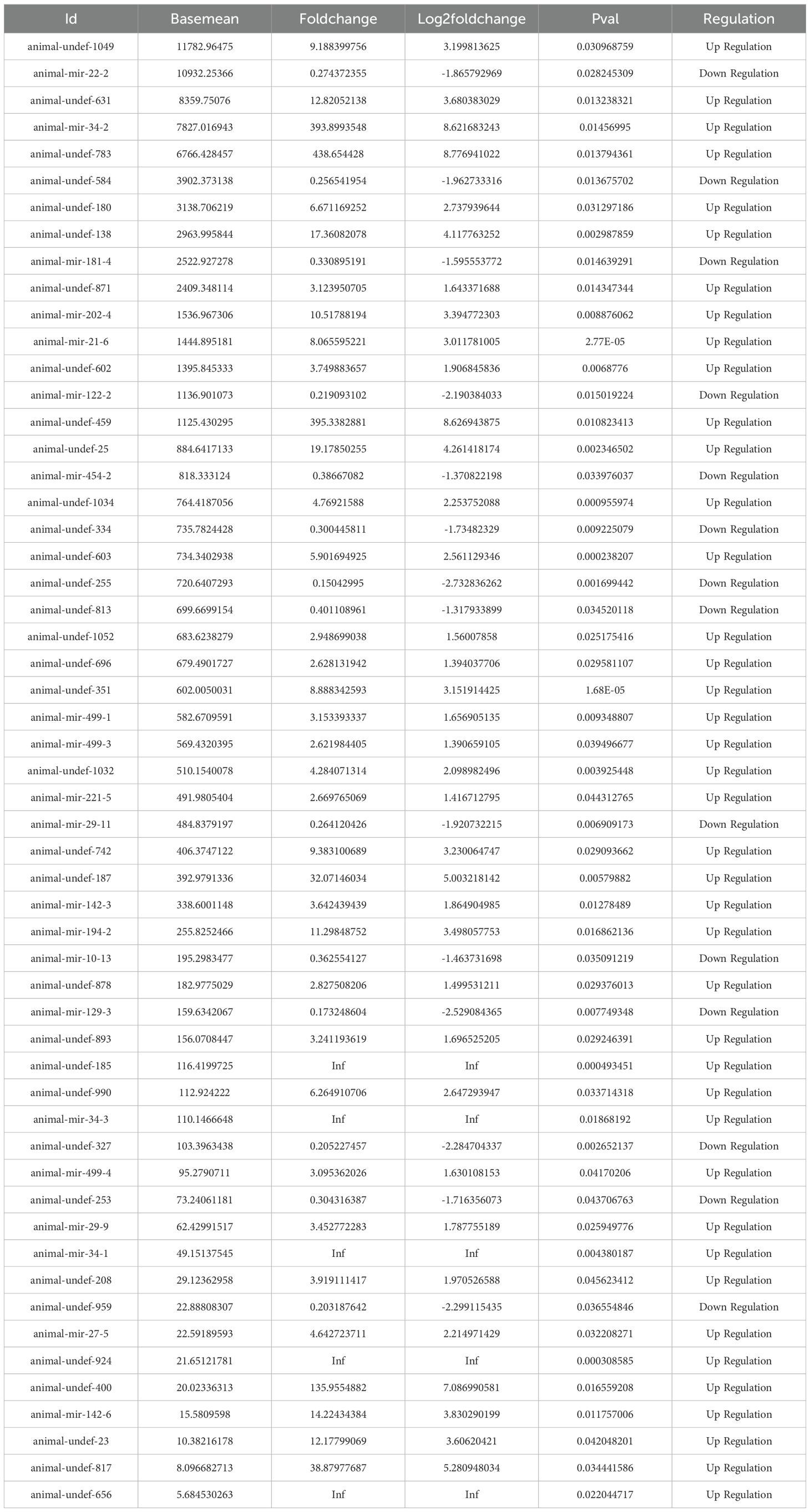
Table 6. Details of the DE miRNA.
3.4 Enrichment analysis of GO and KEGG pathways of target genes predicted by miRNA
In this experiment, a total of 91,596 target genes were predicted, among which 150 miRNAs had target genes. At the same time, a target gene might correspond to multiple targets, so a total of 913,690 target sites were predicted. Then in the GO enrichment, we selected the top 10 biological process (BP), cellular component (CC), and molecular function (MF) directories. In the category of biological processes, most of the noted targets involve glycosylation and Glycoprotein biosynthetic processes. Within the cell component category, most targets were assigned to transcription regulator complex and intracellular membrane-bounded organelle. In the molecular function category, most targets are associated with double-stranded DNA binding and transcription cis-regulatory region binding (Figure 7).
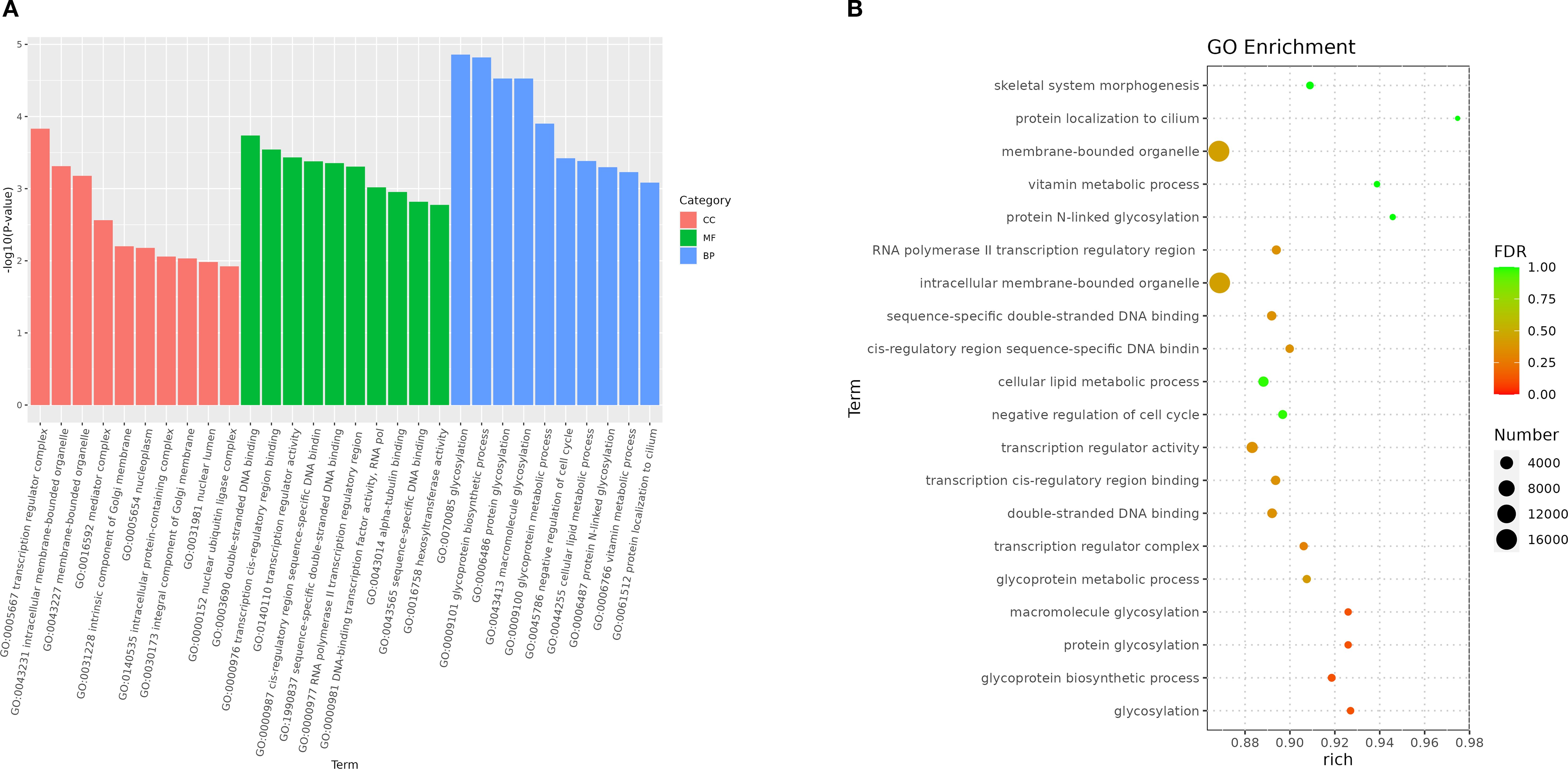
Figure 7. Enriched GO of genes targeted by DEM in 2 groups. (A) GO enrichment bar plot of differentially expressed miRNA target genes. (B) Bubble Plot of GO Enrichment Analysis for Target Genes of Differentially Expressed miRNAs.
The Gene Ontology (GO) enrichment analysis revealed distinct hierarchical patterns across functional categories: Biological Process (BP): A functionally coherent hierarchy spanning from ubiquitin-dependent endocytosis (GO:0048497) through macromolecule modification (GO:0043412) to protein glycosylation (GO:0006486) exhibited statistically significant enrichment, indicating systematic activation of post-translational modification pathways (Figure 8A). Cellular Component (CC): The parent-child relationship between membrane-bounded organelle (GO:0043227) and its descendant term intracellular membrane-bounded organelle (GO:0043231) showed pronounced enrichment, highlighting compartment-specific regulatory mechanisms (Figure 8B). Molecular Function (MF): Enriched GO terms within the DNA binding (GO:0003677) and extracellular vesicle biogenesis (GO:0140115), suggesting coordinated regulation of genomic integrity maintenance and intercellular communication processes (Figure 8C).
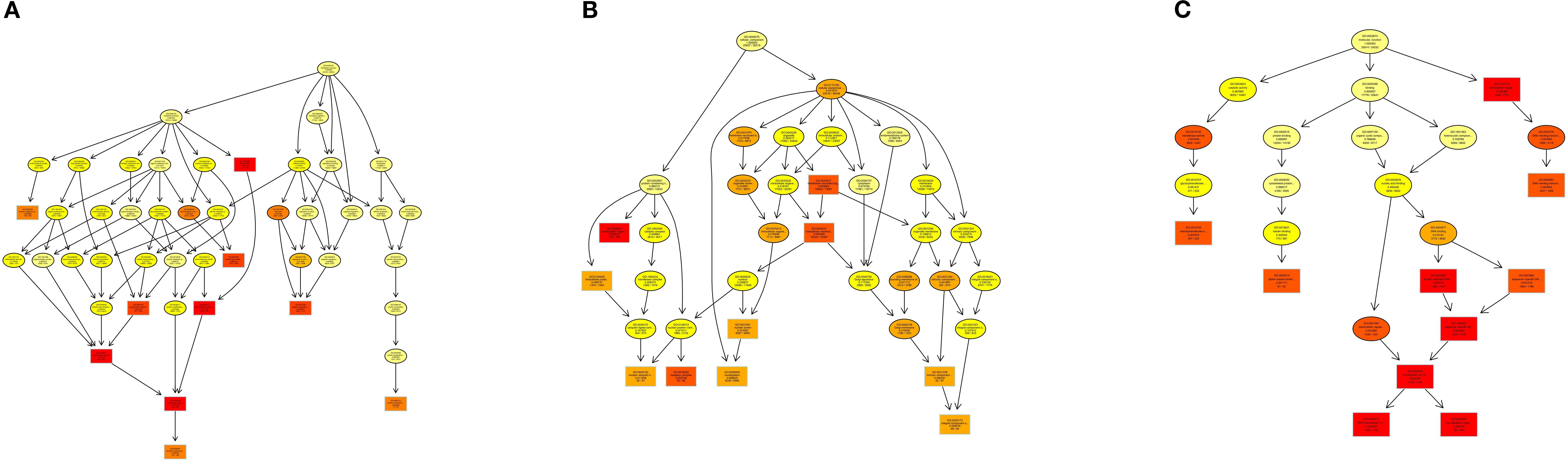
Figure 8. DAG visualization of GO enrichment analysis: (A) Biological Process (BP), (B) Cellular Component (CC), (C) Molecular Function (MF).
In addition, In the KEGG rich concentration (Figure 9 and Figure 10), we found sphingolipid signaling pathway and glycerophospholipid metabolism involved in cell recognition and signal transduction, and T cell receptor involved in anti-inflammatory signaling pathway and TNF signaling pathway.
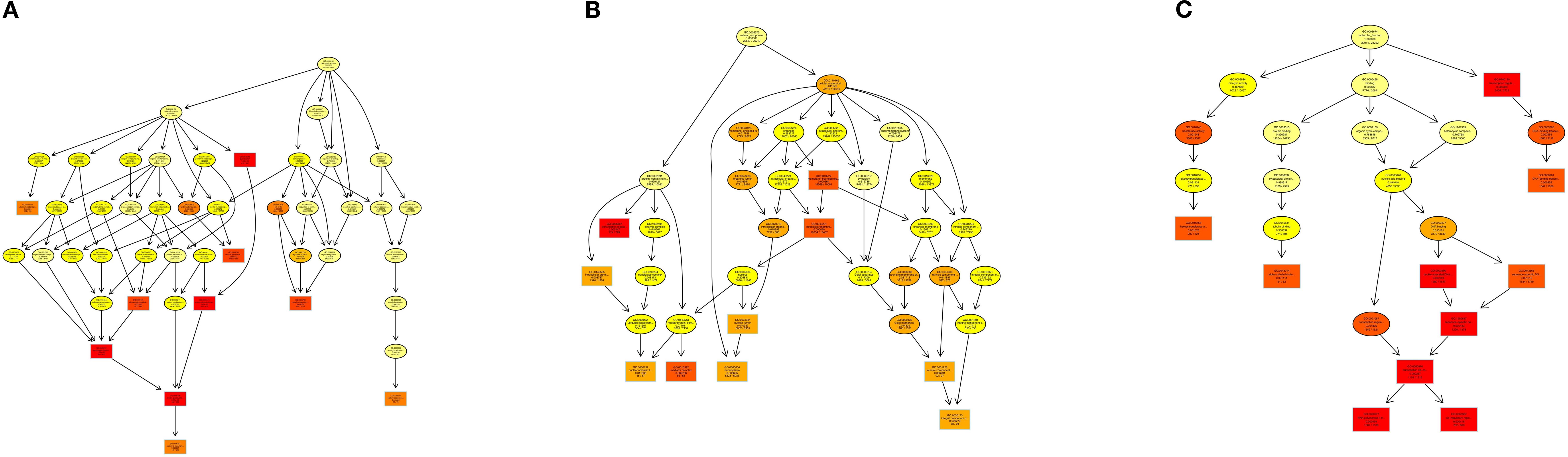
Figure 9. Enriched KEGG of genes targeted by DEM in 2 groups. (A) KEGG enrichment bar plot of differentially expressed miRNA target genes. (B) Bubble Plot of KEGG Enrichment Analysis for Target Genes of Differentially Expressed miRNAs.
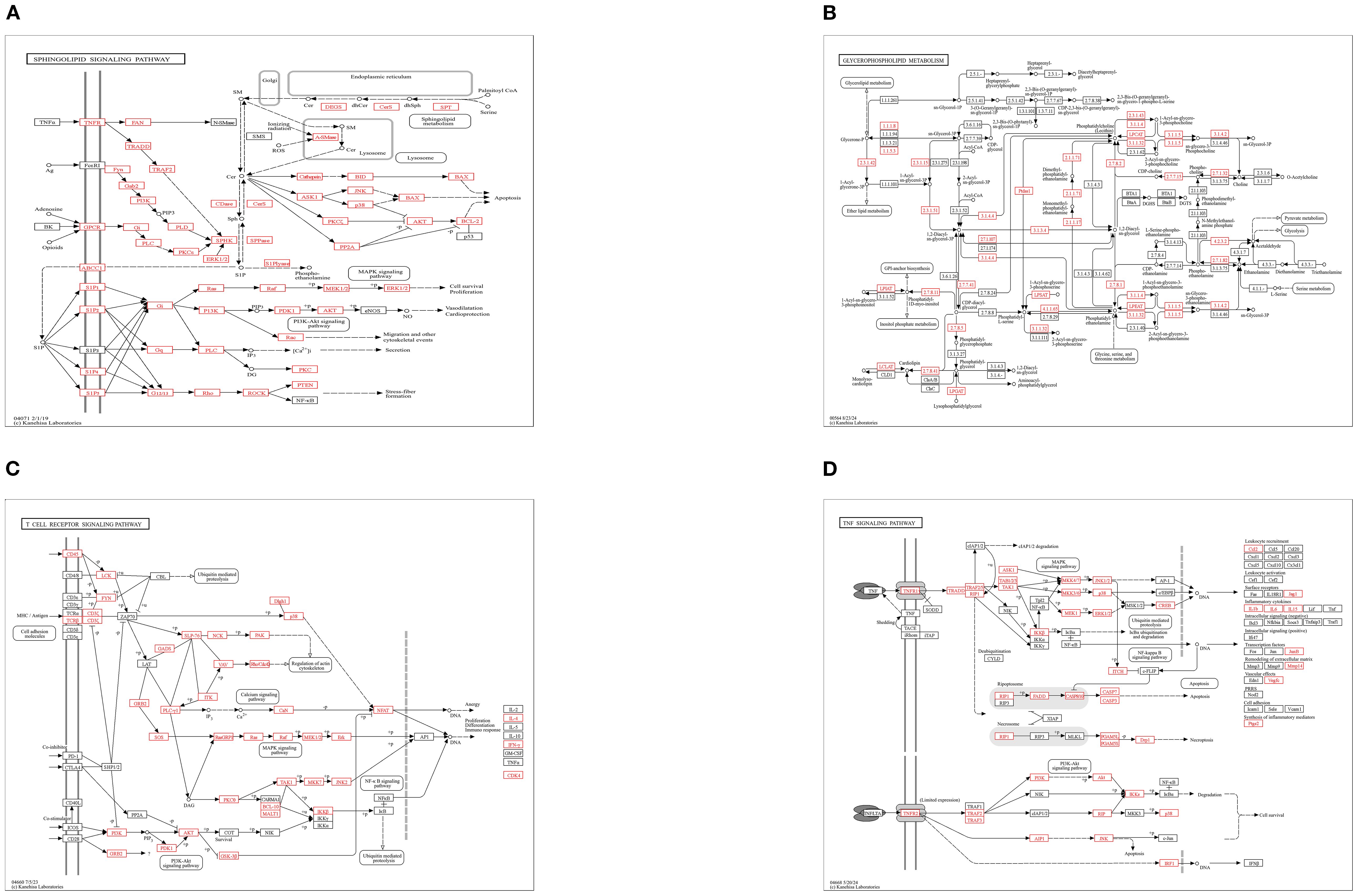
Figure 10. KEGG pathway enrichment analysis: (A) Sphingolipid Signaling Pathway, (B) Glycerophospholipid Metabolism, (C) T-cell Receptor (TCR) Signaling Pathway, (D) TNF Signaling Pathway.
3.5 qRT-PCR consistency for selected miRNAs
To further verify the differentially expressed miRNAs identified by RNA-seq, we used qRT-PCR for confirmation. Seven miRNAs (animal-undef-180, animal-undef-459, animal-mir-122-2, animal-mir-181-4, animal-mir-22-2, animal-mir-29-11, and animal-u) were selected ndef-334 performed qRT-PCR, and it was found that 2 up-regulated genes and 5 down-regulated genes showed the same regulatory trend with qRT-PCR compared with the control group. This indicated that the expression pattern of differentially expressed miRNA was consistent with the results of real-time PCR detection (Figure 11).
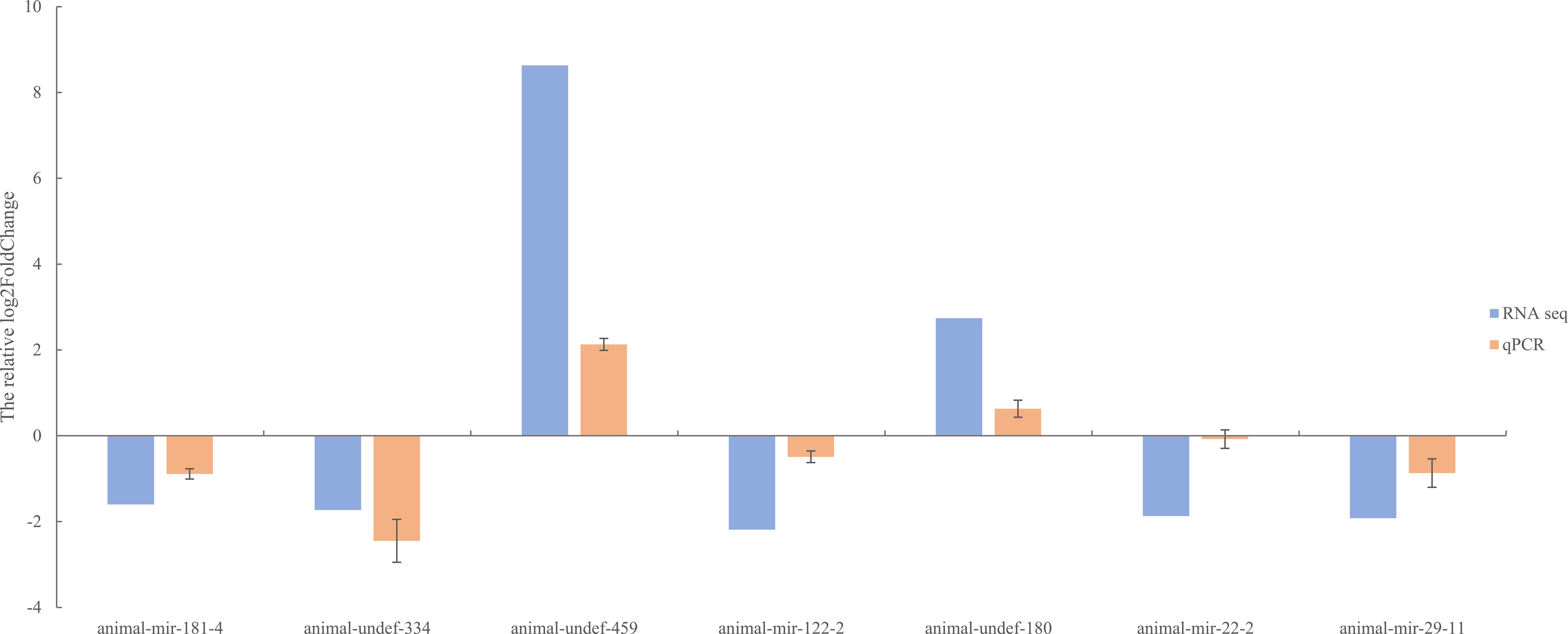
Figure 11. QPCR validation figure of differentially expressed miRNAs.
4 Discussion
The loach, a commercially vital freshwater aquaculture species in China, faces critical challenges in sustainable production due to inflammation-related diseases. Enhancing its growth performance and disease resistance remains a priority for aquaculture optimization. Honeysuckle (L. japonica), a traditional Chinese herb, contains bioactive compounds (e.g., chlorogenic acid and luteoloside) with demonstrated anti-inflammatory, immunomodulatory, and antimicrobial properties (28–30). Previous studies in lipopolysaccharide (LPS)-induced acute lung injury (ALI) murine models revealed that honeysuckle extracts significantly suppressed pro-inflammatory cytokines (TNF-α, IL-6) via modulation of the NF-κB pathway, concurrently improving survival rates (31). Nevertheless, the molecular mechanisms underlying herbal immunoregulation in aquatic species remain underexplored. Therefore, this study employed an integrated experimental design combining lipopolysaccharide (LPS) challenge with L. japonica-supplemented dietary intervention in loach, to systematically characterize miRNA expression dynamics.
In this study, a total of 20,349,467-25,283,783 clean reads were identified by sequencing technology. with clean read ratios exceeding 95%, confirming robust data integrity that meets established sequencing quality benchmarks for subsequent bioinformatic analyses. The six groups of clean reads were distributed in the range of 18–30 bp in length, mainly 21–23 bp, and clustered most at 22 bp. This is consistent with the typical sequence length distribution of Dicer derivatives.
Integrated analysis of small RNA sequencing data from Aeromonas hydrophila-infected and honeysuckle-treated loach groups identified 55 differentially expressed miRNAs (DEMs), comprising 41 upregulated and 14 downregulated species. Notably, several DEMs have been previously implicated in inflammatory regulation and muscle development: miR-21, miR-29, mir-142 and miR-122 were differentially modulated. Specifically, miR-21 was shown to preserve intestinal tight junction (TJ) barrier integrity via ROCK1-dependent upregulation of occludin expression, with its knockout exacerbating barrier dysfunction and inflammatory cascades (32–34). In A. hydrophila-infected grass carp, miR-142 exerted anti-apoptotic and anti-inflammatory effects by directly targeting TNFAIP-2 and GLUT-3 (35, 36). While miR-122 attenuated A. hydrophila-induced inflammation via suppression of IL-6, IL-15, and IL-1β (37), it concurrently disrupted TJ barrier proteins (occludin, claudin-2, claudin-5) and EGFR signaling, potentiating gastrointestinal hyperpermeability and secondary inflammation (38–42). Significant downregulation of miR-29 in stenotic mucosal regions of Castleman’s disease patients suggests its role in suppressing pathological hyperplasia-driven inflammation (43, 44). These findings collectively suggest that honeysuckle intervention may alleviate inflammatory phenotypes and enhance muscle quality in loach by orchestrating DEM expression networks (e.g., miR-21/miR-142) to suppress pro-inflammatory cascades while optimizing myogenic pathways.
KEGG pathway enrichment analysis revealed that target genes of differentially expressed miRNAs (DEMs) were significantly enriched in four pivotal signaling pathways: sphingolipid signaling, glycerophospholipid metabolism, T-cell receptor (TCR) signaling, and TNF signaling. As central regulators of lipid metabolism, sphingolipid and glycerophospholipid pathways orchestrate cellular proliferation, apoptosis, stress responses, and inflammatory cascades through bioactive lipid mediators such as sphingosine-1-phosphate (S1P) and lysophosphatidic acid (LPA) (45, 46). Pharmacological evidence demonstrates that gastrodin and gallic acid ameliorate sphingolipid dysregulation by upregulating S1P receptor 1 (S1PR1) expression, consequently suppressing pro-inflammatory cytokines (e.g., IL-1β and TNF-α) (47). Concurrently, glycerophospholipid metabolic perturbations elevate systemic inflammatory markers (e.g., CRP, IL-6), correlating with metabolic disorder progression (48). The TCR pathway reinforces immune tolerance via IL-10 secretion from regulatory T cells (Tregs), effectively curbing excessive inflammation (49), while TNF-α, a key effector of the TNF pathway, drives inflammatory cascades through NF-κB and MAPK activation. Notably, herbal interventions attenuate these pathways to mitigate inflammatory damage (50, 51). GO enrichment analysis revealed that the majority of differentially expressed genes were significantly enriched in the biological process of glycosylation, the cellular component of transcription regulator complexes, and the molecular function of double-stranded DNA binding-all of which are critically involved in inflammatory regulation. We propose that glycosylation modifications regulate inflammatory pathways (e.g., NF-κB) by modulating transcription factor stability and facilitating transcriptional condensate formation. Furthermore, the functional activity of double-stranded DNA-binding proteins is influenced by their glycosylation status, while the assembly of transcriptional condensates depends on DNA scaffolding. These three mechanisms synergistically orchestrate inflammatory resolution through coordinated regulation of gene expression, chromatin remodeling, and signal transduction cascades (52–58). Collectively, integrated KEGG and GO evidence suggests that honeysuckle dietary intervention exerts synergistic anti-inflammatory effects in loach by multi-target modulation of lipid metabolic reprogramming, immune tolerance establishment, and epigenetic remodeling.
5 Conclusion
In this study, based on miRNA transcriptome sequencing technology, the mechanism of LPS-mediated effects of honeysuckle on the immune function of Loach was investigated. The results showed that honeysuckle had certain effects on the immune function of loach. This study provides a new idea for the rational utilization of honeysuckle, and provides theoretical guidance for the healthy feeding of loach.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, SRX25348514.
Ethics statement
The animal study was approved by Animal experiments were approved by the Research and Animal Ethics Committee of Wenzhou Medical University in accordance with the “Experimental Animal Law”. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
RF: Writing – original draft, Conceptualization. ZS: Data curation, Writing – original draft. LD: Resources, Project administration, Writing – review & editing, Writing – original draft. XJ: Writing – original draft, Visualization, Resources, Conceptualization, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We appreciate the funding for the present study by Wenzhou Key Laboratory of Research and Transformation of Chinese Medicine.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sun B, Huang Y, Castro LFC, Yang S, Huang S, Jin W, et al. The chromosome-level genome and key genes associated with mud-dwelling behavior and adaptations of hypoxia and noxious environments in loach (Misgurnus anguillicaudatus). BMC Biol. (2023) 21:18. doi: 10.1186/s12915-023-01517-1
2. Zeng L, Wang J, Sheng J, Gu Q, and Hong Y. Molecular characteristics of mitochondrial DNA and phylogenetic analysis of the loach (Misgurnus anguillicaudatus) from the Poyang Lake. Mitochondrial DNA. (2012) 23:187–200. doi: 10.3109/19401736.2012.668893
3. Zhong J, Yi S, Ma L, and Wang W. Evolution and phylogeography analysis of diploid and polyploid Misgurnus anguillicaudatus populations across China. Proc Biol Sci. (2019) 286:20190076. doi: 10.1098/rspb.2019.0076
4. Yun L, Han C, He X, Li Q, Fersht V, and Zhang M. Structure characterization and immunomodulatory activity of misgurnus anguillicaudatus carbohydrates. Molecules (Basel Switzerland). (2023) 28:5771. doi: 10.3390/molecules28155771
5. Kim H, Shin J, Yang J, Sim Y, and Yang JY. Biomarker development for identifying mud loach (Misgurnus mizolepis) origin country using untargeted metabolite profiling. Life (Basel Switzerland). (2023) 13:2149. doi: 10.3390/life13112149
6. Ip YK and Chew SF. Air-breathing and excretory nitrogen metabolism in fishes. Acta histochemica. (2018) 120:680–90. doi: 10.1016/j.acthis.2018.08.013
7. Qin C, Huang K, and Xu H. Protective effect of polysaccharide from the loach on the in vitro and in vivo peroxidative damage of hepatocyte. J Nutr Biochem. (2002) 13:592–7. doi: 10.1016/S0955-2863(02)00193-6
8. Wang M, Xu W, Zou J, Li S, Song Z, Zheng F, et al. The programming of antioxidant capacity, immunity, and lipid metabolism in dojo loach (Misgurnus anguillicaudatus) larvae linked to sodium chloride and hydrogen peroxide pre-treatment during egg hatching. Front Physiol. (2021) 12:768907. doi: 10.3389/fphys.2021.768907
9. Shamsi S, Steller E, and Zhu X. The occurrence and clinical importance of infectious stage of Echinocephalus (Nematoda: Gnathostomidae) larvae in selected Australian edible fish. Parasitol Int. (2021) 83:102333. doi: 10.1016/j.parint.2021.102333
10. Kuroda M, Fujimoto T, Murakami M, Yamaha E, and Arai K. Aberrant meiotic configurations cause sterility in clone-origin triploid and inter-group hybrid males of the dojo loach, misgurnus anguillicaudatus. Cytogenetic Genome Res. (2019) 158:46–54. doi: 10.1159/000500303
11. Ciesielska A, Matyjek M, and Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life sciences: CMLS. (2021) 78:1233–61. doi: 10.1007/s00018-020-03656-y
12. Chen Y, Chen Y, Yan X, Li Q, Wang P, Sun Y, et al. hnRNPub inhibits LPS-induced NF-κB pathway by targeting TRAF6 for K48-linked ubiquitination in miiuy croaker (Miichthys miiuy). Fish shellfish Immunol. (2022) 121:498–504. doi: 10.1016/j.fsi.2022.01.029
13. Hwang JH, Kim KJ, Ryu SJ, and Lee BY. Caffeine prevents LPS-induced inflammatory responses in RAW264.7 cells and zebrafish. Chemico-biological Interact. (2016) 248:1–7. doi: 10.1016/j.cbi.2016.01.020
14. Chen YT, Chen SJ, Yin LJ, Hu CY, Dong CD, Singhania RR, et al. Anti-inflammatory effects of fish bone fermented using Monascus purpureus in LPS-induced RAW264.7 cells by regulating NF-κB pathway. J Food Sci Technol. (2023) 60:958–65. doi: 10.1007/s13197-022-05413-4
15. Ling X, Yan W, Yang F, Jiang S, Chen F, and Li N. Research progress of chlorogenic acid in improving inflammatory diseases, Zhong nan da xue xue bao. Yi xue ban = J Cent South University. Med Sci. (2023) 48:1611–20. doi: 10.11817/j.issn.1672-7347.2023.230146
16. Jeong SH, Park MY, Bhosale PB, Abusaliya A, Won CK, Park KI, et al. Potential antioxidant and anti-inflammatory effects of lonicera japonica and citri reticulatae pericarpium polyphenolic extract (LCPE). Antioxidants (Basel Switzerland). (2023) 12:1582. doi: 10.3390/antiox12081582
17. Tang X, Liu X, Zhong J, and Fang R. Potential application of lonicera japonica extracts in animal production: from the perspective of intestinal health. Front Microbiol. (2021) 12:719877. doi: 10.3389/fmicb.2021.719877
18. Shang X, Pan H, Li M, Miao X, and Ding H. Lonicera japonica Thunb.: ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J ethnopharmacology. (2011) 138:1–21. doi: 10.1016/j.jep.2011.08.016
19. Ahirwar SS, Rizwan R, Sethi S, Shahid Z, Malviya S, Khandia R, et al. Comparative analysis of published database predicting microRNA binding in 3’UTR of mRNA in diverse species. MicroRNA (Shariqah United Arab Emirates). (2024) 13:2–13. doi: 10.2174/0122115366261005231018070640
20. Lee RC, Feinbaum RL, Ambros V, and The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-Y
21. Ribeiro AO, de Oliveira AC, Costa JM, Nachtigall PG, Herkenhoff ME, Campos VF, et al. MicroRNA roles in regeneration: Multiple lessons from zebrafish. Dev dynamics: an Off Publ Am Assoc Anatomists. (2022) 251:556–76. doi: 10.1002/dvdy.421
22. Brown W, Bardhan A, Darrah K, Tsang M, and Deiters A. Optical control of microRNA function in zebrafish embryos. J Am Chem Soc. (2022) 144:16819–26. doi: 10.1021/jacs.2c04479
23. Ren L, Zhang H, Luo M, Gao X, Cui J, Zhang X, et al. Heterosis of growth trait regulated by DNA methylation and miRNA in allotriploid fish. Epigenet chromatin. (2022) 15:19. doi: 10.1186/s13072-022-00455-6
24. Nie M, Ni W, Wang L, Gao Q, Liu D, Tian F, et al. Insights Into miRNA-mRNA Regulatory Mechanisms of Cold Adaptation in Gymnocypris eckloni: Ubiquitin-Mediated Proteolysis Is Pivotal for Adaptive Energy Metabolism. Front Genet. (2022) 13:903995. doi: 10.3389/fgene.2022.903995
25. Lee JW, Kim JE, Goo IB, Hwang JA, Im JH, Choi HS, et al. Expression of Immune-Related Genes during Loach (Misgurnus anguillicaudatus) Embryonic and Early Larval Development. Dev Reprod. (2015) 19:181–7. doi: 10.12717/DR.2015.19.4.181
26. Kim CH, Kim EJ, Seo C, and Nam YK. Transcriptome analysis of maternal gene transcripts in unfertilized eggs of misgurnus anguillicaudatus and identification of immune-related maternal genes. Int J Mol Sci. (2020) 21:3872. doi: 10.3390/ijms21113872
27. Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
28. Zhang J, Wu M, Li W, and Bai G. Regulation of chlorogenic acid biosynthesis by hydroxycinnamoyl CoA quinate hydroxycinnamoyl transferase in Lonicera japonica. Plant Physiol biochemistry: PPB. (2017) 121:74–9. doi: 10.1016/j.plaphy.2017.10.017
29. Guan R, Guo F, Guo R, Wang S, Sun X, Zhao Q, et al. Integrated metabolic profiling and transcriptome analysis of Lonicera japonica flowers for chlorogenic acid, luteolin and endogenous hormone syntheses. Gene. (2023) 888:147739. doi: 10.1016/j.gene.2023.147739
30. Zhao YX, Wang JJ, Zhang L, Zhang S, Su SL, Duan JA, et al. Bioactive luteoloside produced by Myroides odoratimimus, solvent-tolerant bacterium form the rhizosphere of Lonicera japonica. Natural product Res. (2019) 33:3559–62. doi: 10.1080/14786419.2018.1481838
31. Xiong L, Liu Y, Wang Y, Zhao H, Song X, Fan W, et al. The protective effect of Lonicera japonica Thunb. against lipopolysaccharide-induced acute lung injury in mice: Modulation of inflammation, oxidative stress, and ferroptosis. J ethnopharmacology. (2024) 331:118333. doi: 10.1016/j.jep.2024.118333
32. Liu Z, Li C, Chen S, Lin H, Zhao H, Liu M, et al. MicroRNA-21 increases the expression level of occludin through regulating ROCK1 in prevention of intestinal barrier dysfunction. J Cell Biochem. (2019) 120:4545–54. doi: 10.1002/jcb.27742
33. Lu X, Yu Y, and Tan S. The role of the miR-21-5p-mediated inflammatory pathway in ulcerative colitis. Exp Ther Med. (2020) 19:981–9. doi: 10.3892/etm.2019.8277
34. Zhang L, Shen J, Cheng J, and Fan X. MicroRNA-21 regulates intestinal epithelial tight junction permeability. Cell Biochem Funct. (2015) 33:235–40. doi: 10.1002/cbf.3109
35. Tao L, Pang Y, Wang A, Li L, Shen Y, Xu X, et al. Functional miR-142a-3p Induces Apoptosis and Macrophage Polarization by Targeting tnfaip2 and glut3 in Grass Carp (Ctenopharyngodon idella). Front Immunol. (2021) 12:633324. doi: 10.3389/fimmu.2021.633324
36. Xu XY, Shen YB, Fu JJ, Yu HY, Huang WJ, Lu LQ, et al. MicroRNA-induced negative regulation of TLR-5 in grass carp. Ctenopharyngodon idella Sci Rep. (2016) 6:18595. doi: 10.1038/srep18595
37. Liu X, Hao Y, Peng L, Liu Y, Wei N, and Liang Q. MiR-122 is involved in immune response by regulating Interleukin-15 in the orange-spotted grouper (Epinephelus coioides). Fish shellfish Immunol. (2020) 106:404–9. doi: 10.1016/j.fsi.2020.08.021
38. Sadeghi H, Karimi Dermani F, Gheibi N, Afshar D, Heidarzadeh S, Datta I, et al. Potential role of microRNAs in response to aeromonas infection in fish. Arch Razi Institute. (2023) 78:1668–79. doi: 10.32592/ARI.2023.78.6.1668
39. Shi L, Zheng X, Fan Y, Yang X, Li A, and Qian J. The contribution of miR-122 to the innate immunity by regulating toll-like receptor 4 in hepatoma cells. BMC Gastroenterol. (2019) 19:130. doi: 10.1186/s12876-019-1048-3
40. Zhang B, Tian Y, Jiang P, Jiang Y, Li C, Liu T, et al. MicroRNA-122a regulates zonulin by targeting EGFR in intestinal epithelial dysfunction. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol. (2017) 42:848–58. doi: 10.1159/000478629
41. Al-Sadi R, Engers J, and Abdulqadir R. Talk about micromanaging! Role of microRNAs in intestinal barrier function. Am J Physiol Gastrointestinal liver Physiol. (2020) 319:G170–g174. doi: 10.1152/ajpgi.00214.2020
42. Ye D, Guo S, Al-Sadi R, and Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. (2011) 141:1323–33. doi: 10.1053/j.gastro.2011.07.005
43. Fukata T, Mizushima T, Nishimura J, Okuzaki D, Wu X, Hirose H, et al. The supercarbonate apatite-microRNA complex inhibits dextran sodium sulfate-induced colitis, molecular therapy. Nucleic Acids. (2018) 12:658–71. doi: 10.1016/j.omtn.2018.07.007
44. Nijhuis A, Biancheri P, Lewis A, Bishop CL, Giuffrida P, Chan C, et al. In Crohn’s disease fibrosis-reduced expression of the miR-29 family enhances collagen expression in intestinal fibroblasts. Clin Sci (London England: 1979). (2014) 127:341–50. doi: 10.1042/CS20140048
45. Du X, Liu Y, He X, Tao L, Fang M, and Chu M. Identification and expression profile analysis of circRNAs associated with goat uterus with different fecundity during estrous cycle. BMC Genomics. (2025) 26:349. doi: 10.1186/s12864-025-11489-x
46. Zhang Y, Xi Y, Yang C, Gong W, Wang C, Wu L, et al. Short-Chain Fatty Acids Attenuate 5-Fluorouracil-Induced THP-1 Cell Inflammation through Inhibiting NF-κB/NLRP3 Signaling via Glycerolphospholipid and Sphingolipid Metabolism. Molecules (Basel Switzerland). (2023) 28:494. doi: 10.3390/molecules28020494
47. Wang S, Zhu C, Zhang S, Ma S, Li B, Zhao S, et al. The combination of gastrodin and gallic acid synergistically attenuates angII-induced apoptosis and inflammation via regulation of sphingolipid metabolism. J Inflammation Res. (2024) 17:6971–88. doi: 10.2147/JIR.S477554
48. Siddiqui MK, Smith G, St Jean P, Dawed AY, Bell S, Soto-Pedre E, et al. Diabetes status modifies the long-term effect of lipoprotein-associated phospholipase A2 on major coronary events. Diabetologia. (2022) 65:101–12. doi: 10.1007/s00125-021-05574-5
49. Dikiy S, Ghelani AP, Levine AG, Martis S, Giovanelli P, Wang ZM, et al. Terminal differentiation and persistence of effector regulatory T cells essential for preventing intestinal inflammation. Nat Immunol. (2025) 26:444–58. doi: 10.1038/s41590-024-02075-6
50. Wang H, Xi Y, Gu F, Peng L, and Li J. Protective effects of a polyherbal mixture on intestinal injury via the NF-κB signaling pathway and gut microbiota modulation in hyperuricemic mice. Foods (Basel Switzerland). (2025) 14:1118. doi: 10.3390/foods14071118
51. Zhang Y, Deng Z, Li H, and Jiang Z. A spermidine derivative ameliorates dextran sulfate sodium-induced colitis in mice by inhibiting the MAPK4/AKT signaling pathway. Foods (Basel Switzerland). (2025) 14:1110. doi: 10.3390/foods14071110
52. Biersteker R, Larsen OF, Wuhrer M, Huizinga TWJ, Toes REM, and Hafkenscheid L. Variable domain glycosylation as a marker and modulator of immune responses: Insights into autoimmunity and B-cell Malignancies. Semin Immunol. (2025) 78:101946. doi: 10.1016/j.smim.2025.101946
53. Trzos S, Link-Lenczowski P, and Pocheć E. The role of N-glycosylation in B-cell biology and IgG activity. The aspects of autoimmunity and anti-inflammatory therapy. Front Immunol. (2023) 14:1188838. doi: 10.3389/fimmu.2023.1188838
54. Park SH, Hong H, Han YM, Kangwan N, Kim SJ, Kim EH, et al. Nonsteroidal anti-inflammatory drugs (NSAID) sparing effects of glucosamine hydrochloride through N-glycosylation inhibition; strategy to rescue stomach from NSAID damage. J Physiol Pharmacol. (2013) 64:157–65.
55. Kato Y, Adachi Y, and Ohno N. Contribution of N-linked oligosaccharides to the expression and functions of beta-glucan receptor. Dectin-1 Biol Pharm Bull. (2006) 29:1580–6. doi: 10.1248/bpb.29.1580
56. Xu R, Zhang J, Hu X, Xu P, Huang S, Cui S, et al. Yi-shen-hua-shi granules modulate immune and inflammatory damage via the ALG3/PPARγ/NF-κB pathway in the treatment of immunoglobulin a nephropathy. J ethnopharmacology. (2024) 319:117204. doi: 10.1016/j.jep.2023.117204
57. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. (2021) 592:296–301. doi: 10.1038/s41586-021-03341-5
Keywords: Misgurnus anguillicaudatus, Lonicera japonica, anti-inflammatory, lipopolysaccharide, microRNA
Citation: Fan R, Sun Z, Dai L and Jiang X (2025) Deciphering the regulatory mechanisms of honeysuckle (Lonicera japonica) on lipopolysaccharide-induced inflammatory responses in loach (Misgurnus anguillicaudatus) through small RNA sequencing. Front. Immunol. 16:1683771. doi: 10.3389/fimmu.2025.1683771
Received: 11 August 2025; Accepted: 05 September 2025;
Published: 23 September 2025.
Edited by:
Saima Kausar, University of Agriculture, PakistanReviewed by:
Xu-Dong Zhou, Hunan University of Chinese Medicine, ChinaCongfen Zhang, Chengdu University, China
Copyright © 2025 Fan, Sun, Dai and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lishang Dai, bGlzaGFuZzIwMTZAd211LmVkdS5jbg==; Xiajuan Jiang, NDIxNzE3MDQxQHFxLmNvbQ==