 Hanjun Xu1†
Hanjun Xu1† Baojun Tu
Baojun Tu- 1Department of Urology, Taizhou Second People’s Hospital Affiliated to Yangzhou University, Taizhou, Jiangsu, China
- 2The First Clinical Medical College, Nanjing Medical University, Nanjing, Jiangsu, China
- 3Department of Nephrology, Taizhou Jiangyan Traditional Chinese Medicine Hospital, Jiangyan Affiliated Hospital of Nanjing University of Chinese Medicine, Taizhou, Jiangsu, China
Renal cell carcinoma (RCC) evolves within a chronic inflammatory renal niche, where angiogenesis, metabolism, and immune programs synergize to reshape the tumor immune microenvironment (TIME). Recurrent renal damage and tissue hypoxia sustain NF-κB/STAT3 and HIF-VEGF signaling, while a sustained IFN-γ response enhances antigen presentation while inducing inhibitory checkpoints, promoting a state of “inflammation but constrained.” Single-cell and spatial studies reveal early microenvironment heterogeneity and the chemokine-checkpoint paradox: regions enriched in CXCL9/10 coexist with endothelial inertia, cancer-associated fibroblasts (CAFs)-mediated stromal barriers, and metabolic stress, which collectively exclude functional CD8+T cells. In advanced clear cell RCC, immuno-inflammatory, immuno-excluded, and immuno-desert phenotypes often coexist and undergo transitions during treatment, leading to heterogeneity in response to immune checkpoint blockade (ICB). We propose a modular perspective-the NF-κB/STAT3, HIF-VEGF, IFN-γ circuits and auxiliary regulatory factors-to link stage-specific biology with treatment matching. Integrative biomarkers couple IFN-γ characteristics with angiogenesis/stromal modules and spatial indicators, offering superior predictive power compared to single tests. These insights support the adoption of a combined strategy: integrating vascular normalization or stromal/myeloid cell reprogramming on the basis of ICB, and encouraging the use of longitudinal “immune snapshots” to guide intervention and precision immunotherapy for renal cancer.
1 Introduction
Renal cell carcinoma (RCC) accounts for the vast majority of kidney malignancies and remains one of the leading causes of death from genitourinary system cancers globally. Its incidence rate is influenced by the increased exposure to metabolic and vascular risk factors, as well as the widespread use of cross-sectional imaging (1, 2). From a biological perspective, RCC exhibits significant inter- and intratumoral heterogeneity. Clear cell RCC (ccRCC) is the predominant subtype, characterized by the loss of the VHL gene and activation of the HIF pathway. Equally decisive is the ability of RCC to remodel and exploit the tumor immune microenvironment (TIME): tumor cells, stromal cells, and endothelial cell populations jointly regulate cytokine and chemokine programs, thereby refining lymphocyte migration, antigen presentation, and effector functions, gradually driving the transition from immune surveillance to immune evasion. The resulting immune ecology includes: immune infiltration regions accompanied by interferon response programs; immune exclusion zones where lymphocytes are sequestered at the edge due to fibrovascular and stromal barriers; and immune deserts with almost no leukocyte presence. These phenotypes are closely correlated with the heterogeneity of clinical behavior and differential responses to combined treatment with immune checkpoint blockade (ICB) and VEGF targeting (3, 4).
Under physiological conditions, the kidney maintains a relatively immune-tolerant and low-inflammatory environment to safeguard filtration and tubular transport, balancing constant antigen exposure with the need to avoid collateral tissue damage. This homeostasis is maintained by tissue-resident immune cells, such as macrophages, dendritic cells, and natural killer cells, which interact with tubular epithelial and endothelial cells to regulate antigen clearance and prevent excessive immune responses (5). However, this balance can be disrupted by persistent tissue injury, including chronic kidney disease, autoimmune nephritis, ischemia–reperfusion injury, metabolic stress, and smoldering microinflammation. These insults activate inflammatory pathways such as NF-κB, STAT3, and interferon-related signaling, leading to elevated chemokine expression, immune cell infiltration, and stromal remodeling. Together, these processes create an immune environment that supports tumor development (6). RCC originates and progresses within this altered immune context, gradually shifting from immune surveillance toward immune evasion.
During early tumorigenesis and the preneoplastic phase, interferon-γ (IFN-γ) signaling enhances antigen presentation components, such as MHC I/II, TAP1, and PSMB9. At the same time, it induces upregulation of inhibitory immune checkpoints, including PD-L1, VISTA, and B7-H3. These molecular changes are accompanied by the expansion of regulatory T cells and the emergence of dysfunctional CD8+T cells, collectively forming an “immune rewiring” state characterized by simultaneous immune activation and suppression (7). This immune remodeling shows considerable spatial heterogeneity, with immune-infiltrated regions coexisting with immune-excluded areas, where T cell access is limited by fibroblast-mediated matrix barriers, endothelial dysfunction, and metabolic constraints (8). As the tumor advances, the TIME evolves into distinct immune phenotypes such as T cell-inflamed, immune-excluded, and immune-desert. These phenotypes are not fixed but change dynamically under therapeutic pressure, which may explain the variable responses to ICB and VEGF-targeted therapies observed in RCC. Recognizing the spatiotemporal evolution of these immune states is crucial for identifying reliable biomarkers and guiding the development of adaptive immunotherapeutic strategies.
2 Chronic inflammation drives renal immune landscape remodeling
The healthy kidney maintains a low-inflammatory, immune-tolerant milieu to protect filtration and tubular transport. Persistent tissue injury, including chronic kidney disease, autoimmune nephritis, ischemia–reperfusion, metabolic stress, toxins, and smoldering micro-inflammation, can disrupt this balance and is associated with progressive rewiring of the renal immune niche (9, 10). These changes elevate chemokine production, recruit myeloid populations, and remodel stroma and endothelium, pre-positioning the renal TIME for immune editing (11, 12).
2.1 Core inflammatory modules and effector programs
Three interacting modules organize this transition and reinforce one another. First, innate sensors and alarmins sustain NF-κB/STAT3 signaling; complement and inflammasome pathways further amplify myeloid-biased inflammation and promote tolerogenic dendritic-cell states with low CCR7/CD86, limiting effective T-cell priming and egress (13, 14). Second, capillary rarefaction and fibrosis drive tubulointerstitial hypoxia, stabilizing HIF-1/2 and amplifying VEGF-dependent angiogenic programs that reshape vascular structure and trafficking cues (15–17). Third, IFN-γ signaling is a double-edged module: it enhances antigen-presentation machinery while concurrently inducing inhibitory checkpoints and fostering dysfunctional/exhausted T-cell states under chronic exposure (18, 19). In parallel, cancer-associated fibroblasts (CAFs) and activated endothelium remodel matrix and vasculature to limit lymphocyte access, consolidating an inflamed-yet-restrained program (20).
2.2 Cellular and stromal remodeling in chronically inflamed kidneys
Chronically inflamed kidneys undergo coordinated remodeling of immune and stromal compartments. Neutrophils accumulate and release elastase and neutrophil extracellular traps (NETs), injuring epithelium and potentially unmasking neoantigens (21). Monocyte-derived macrophages polarize toward M2-like states that secrete IL-10, TGF-β, and prostaglandins. Dendritic cells remain functionally immature, limiting effective T-cell priming and egress, while natural killer cells exhibit reduced cytotoxicity (22). CD4+T cells skew toward Th17 and Treg subsets, and CD8+T cells display early dysfunction/exhaustion markers despite increased antigen exposure (23). In parallel, fibroblasts acquire CAF-like phenotypes, produce CXCL12, and reorganize collagen, creating physical and chemotactic barriers; the vasculature becomes abnormal and anergic, with reduced expression of adhesion molecules such as ICAM-1/VCAM-1 that impair leukocyte trafficking. Animal models, including ischemia–reperfusion injury, adenine or folate nephropathy, and chemical carcinogen exposure, recapitulate these features, with progressive vascular dysfunction, checkpoint induction, and myeloid dominance preceding overt tumorigenesis (24, 25). Human bulk and single-cell datasets from diseased kidneys and early RCC similarly capture these transitions, linking chronic inflammation to tumor development and suggesting a tractable pre-malignant window in which anti-cytokine, vascular-normalizing, and stroma- or myeloid-targeted interventions may help restore immune surveillance and delay or prevent RCC initiation (26). Therapeutic options aligned to these modules are outlined in Table 1.
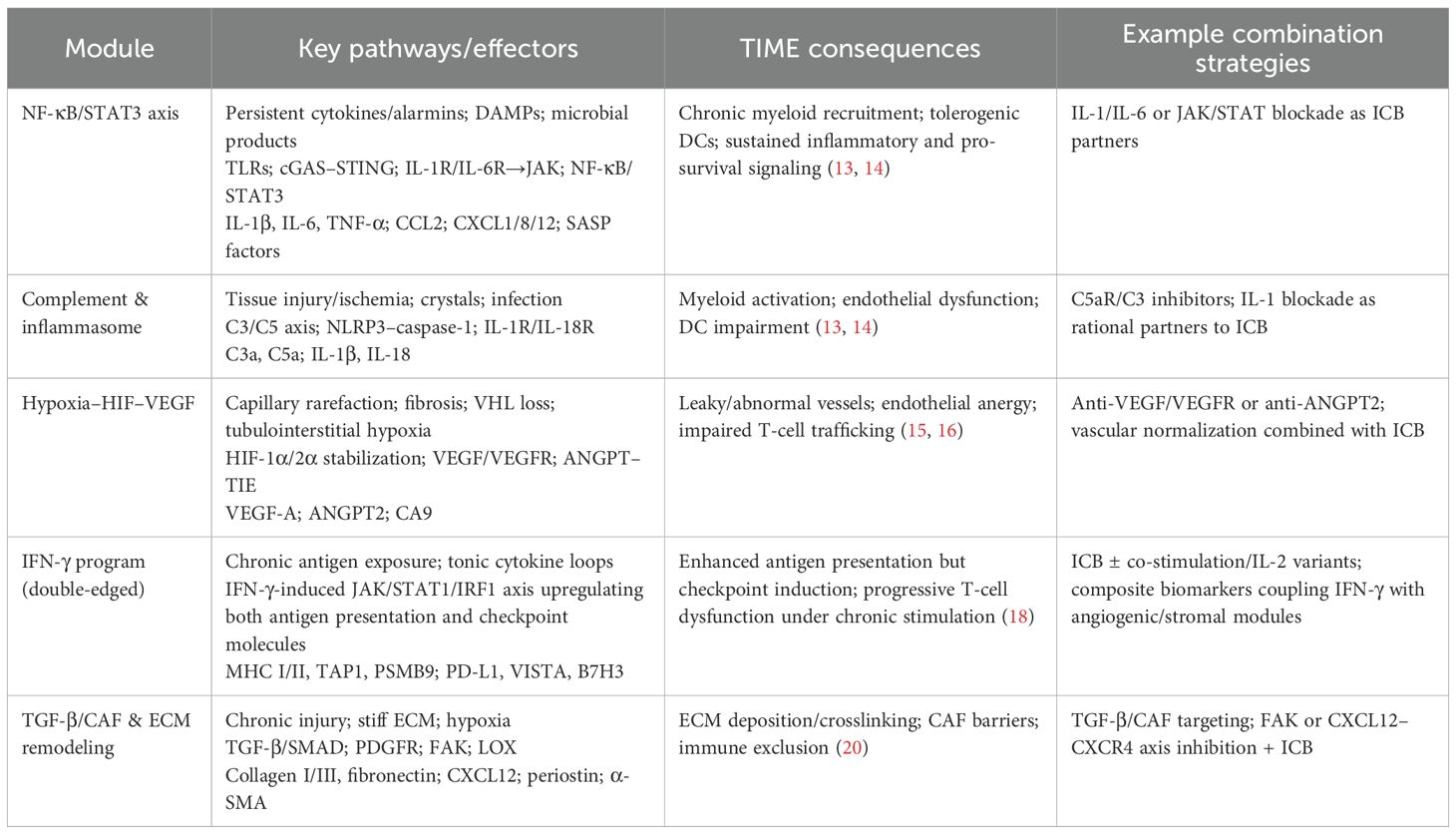
Table 1. Modular view of chronic-inflammation–driven remodeling of the renal tumor immune microenvironment (TIME) and therapeutic implications in RCC.
3 Immune rewiring in preneoplastic/early RCC
3.1 IFN-γ–driven epithelial responses and early checkpoint induction
In the preneoplastic and early stages of RCC, persistent IFN-γ signaling—mainly derived from tissue-resident and infiltrating T lymphocytes, amplifies antigen processing and presentation on the one hand, and installs “molecular brakes” for effector activities on the other hand. IFN-γ activates the STAT1/IRF1 pathway, which upregulates classical MHC-I molecules and key regulators such as NLRC5 and B2M. It also induces MHC-II expression through CIITA, enhancing tumor antigen presentation to CD4+ T cells (27). Pattern-recognition pathways, including RIG-I/MDA5 and the cGAS-STING axis, can via type I/II interferon signaling, reinforce antigen-processing and presentation programs.
However, this enhanced immunogenic state is accompanied by early and coordinated expression of inhibitory immune checkpoints including PD-L1, VISTA, B7-H3, and occasionally B7-H4, across epithelial, endothelial, and myeloid cell populations. These molecules together form a suppressive network that limits effective T cell co-stimulation (28). In parallel, the tumor epithelium undergoes metabolic reprogramming that dampens immune responses. For example, IDO1 and TDO2 facilitate kynurenine production, activating the aryl hydrocarbon receptor (AHR); extracellular adenosine accumulates via CD39/CD73; and elevated glycolytic activity leads to lactic acid buildup—all of which impair cytotoxic T cell function and antigen processing (29). Although these early changes may serve as a protective adaptation, they gradually favor the survival of epithelial cell clones resistant to IFN signaling, promoting immune editing. With tumor progression, temporary regulatory adaptations, such as PD-L1 promoter acetylation or inducible MHC-II expression, are often replaced by irreversible genetic alterations, including loss of HLA heterozygosity, disruption of B2M, and mutations in JAK-STAT signaling components, leading to a fixed immune escape phenotype (30). This “inflamed but restrained” immune state represents a common bottleneck in the early evolution of RCC and is increasingly recognized as a window for therapeutic intervention.
3.2 Spatial and single-cell insights: heterogeneity emerges early
Recent advances in spatial transcriptomics and single-cell RNA sequencing have revealed that, even at early stages of ccRCC, the TIME is highly heterogeneous at the tissue level. Immune-active “inflammatory islands” can be found next to immunosuppressive regions within the same tumor. The former are characterized by IRF1/STAT1 signaling, secretion of CXCL9/10, and inducible expression of HLA class II, while the latter show features such as TGF-β signaling, EMT activation, checkpoint ligand expression, and impaired antigen processing (31). This spatial pattern is partly related to epithelial lineage, where proximal tubule-like cells retain stronger immune visibility, whereas distal or collecting duct-like phenotypes tend toward immune evasion (32). The immune cells reflect this organization: Tregs accumulate near active niches, while CD8+ T cells divide into precursor exhausted and terminally exhausted subsets, the latter enriched in metabolically stressed areas with high lactate and adenosine (33). The number of conventional dendritic cells was maintained but the function was immature, which limited the effective T-cell priming and egress. Although plasmacytoid and monocyte-derived DCs express interferon-stimulating genes in varying degrees, they do not perform well in cross presentation. Meanwhile, macrophages polarize toward an M2-like state, producing IL-10 and TGF-β, which promote immune suppression and activate CAFs. These CAFs further diversify into inflammatory and myofibroblastic types, with the latter forming aligned collagen that directs but restricts CD8+T cell entry. Tumor-associated endothelial cells also vary, with stalk-like cells lacking HEV features and expressing low ICAM-1/VCAM-1, disrupting immune cell trafficking (34). TLS may form near inflammatory zones, containing B and T cells with germinal center markers. Their function depends on context—supportive when antigen presentation is active and matrix barriers are limited, but potentially tolerogenic when CAFs and suppressive myeloid cells dominate. These high-resolution data explain why the overall detection can show “inflammatory type”, while the lesions are “unresponsive”: the inflammation and immune escape microregions are staggered on the sub millimeter scale, and their relative proportion rather than simple existence determines the function.
3.3 Chemokine–checkpoint paradox and early risk stratification
A recurring feature of early RCC evolution is the chemokine checkpoint paradox: Although the level of chemokine that attracts T cells is very high, the degree of truly effective CD8+ entering the epithelial “nest” is limited. This leads to a pattern of spatial immune exclusion despite a seemingly inflamed transcriptional profile. Several mechanisms contribute to this paradox: (i) Matrix gating: CXCL12 produced by CAF forms gradients that trap CXCR4+ T cells in perivascular regions, while ECM remodeling through collagen I/III deposition increases tissue stiffness and pressure, limiting T-cell movement. (II) Endothelium unresponsive: high VEGF, HIF-dependent vascular down-regulation of adhesion molecules, abnormal pericyte coating, and the expression of FasL or PD-L1, collectively scavenge or inactivate effector T cells during transendothelial migration. (III) Metabolism/ion barrier: lactate accumulation, hypoxia and adenosine signaling reduce T cell motility, cytokine production and survival; Potassium leakage from dying cells disrupts T cell translation and biosynthesis. (IV) Premature checkpoint tone: epithelial and myeloid cells express PD-L1/TIM-3 ligand, and myeloid/endothelial cells express VISTA, which inactivates the effector function just at the “entrance” indicated by chemotactic signals. As a result, the TIME appears immunologically active at the transcript level but functionally excludes effector T cells (35). For clinical application, single biomarkers like PD-L1 staining are often insufficient. Instead, integrated models that combine IFN-γ response signatures, co-expression of inhibitory receptors, myeloid/CAF-derived suppressive signals, and spatial parameters offer a more accurate basis for risk assessment (8). These multidimensional approaches can help identify lesions with high progression risk and support early intervention strategies—such as vascular or ECM normalization to facilitate immune entry, tailored checkpoint blockade to prevent early tolerance, and myeloid reprogramming to restore responsiveness—before RCC becomes clinically evident.
4 Established RCC: from immune dysfunction to clinical translation
In established ccRCC, the TIME typically presents as one of three immune phenotypes, including immune-infiltrated, immune-excluded, or immune-desert, each defined by distinct immune cell profiles, spatial patterns, and functional states. Immune-infiltrated tumors contain abundant intratumoral CD8+T cells, activated DCs, and elevated interferon signaling, reflecting an engaged, though often exhausted, immune response. In immune-excluded tumors, immune cells are present but confined to the tumor periphery or fibrotic stromal zones. Infiltration is limited by endothelial dysfunction, ECM barriers, and suppressive cytokines like TGF-β and VEGF, commonly shaped by CAF activity and hypoxia (36). Desert tumors, by contrast, show minimal immune infiltration, low MHC expression, and weak chemokine activity. These are often associated with low tumor mutation burden and are generally resistant to ICB therapies. Spatial transcriptomics and multiplex imaging have revealed that these phenotypes can coexist within the same tumor, with sharp transitions between inflamed and excluded areas at sub-millimeter scales (37). This spatial diversity helps explain the frequent presence of mixed immune regions in a single lesion. Moreover, TIME phenotypes are not fixed. They shift over time in response to tumor-intrinsic factors like hypoxia or loss of BAP1 and CDKN2A/B, as well as external pressures from therapy. Longitudinal studies have shown, for example, that inflamed tumors may transition to excluded phenotypes after ICB due to immune exhaustion or stromal remodeling, while anti-VEGF therapy may restore vascular function and allow T cell entry (38). These insights support the view that TIME phenotypes are dynamic and modifiable, reinforcing the need for flexible, time-adapted immunotherapy strategies in RCC treatment. Collectively, Figure 1 integrates epithelial IFN-γ responses, vascular/stromal constraints, and early spatial heterogeneity, providing a mechanistic basis for response prediction and for combination strategies pairing ICB with vascular normalization and stromal/myeloid reprogramming.
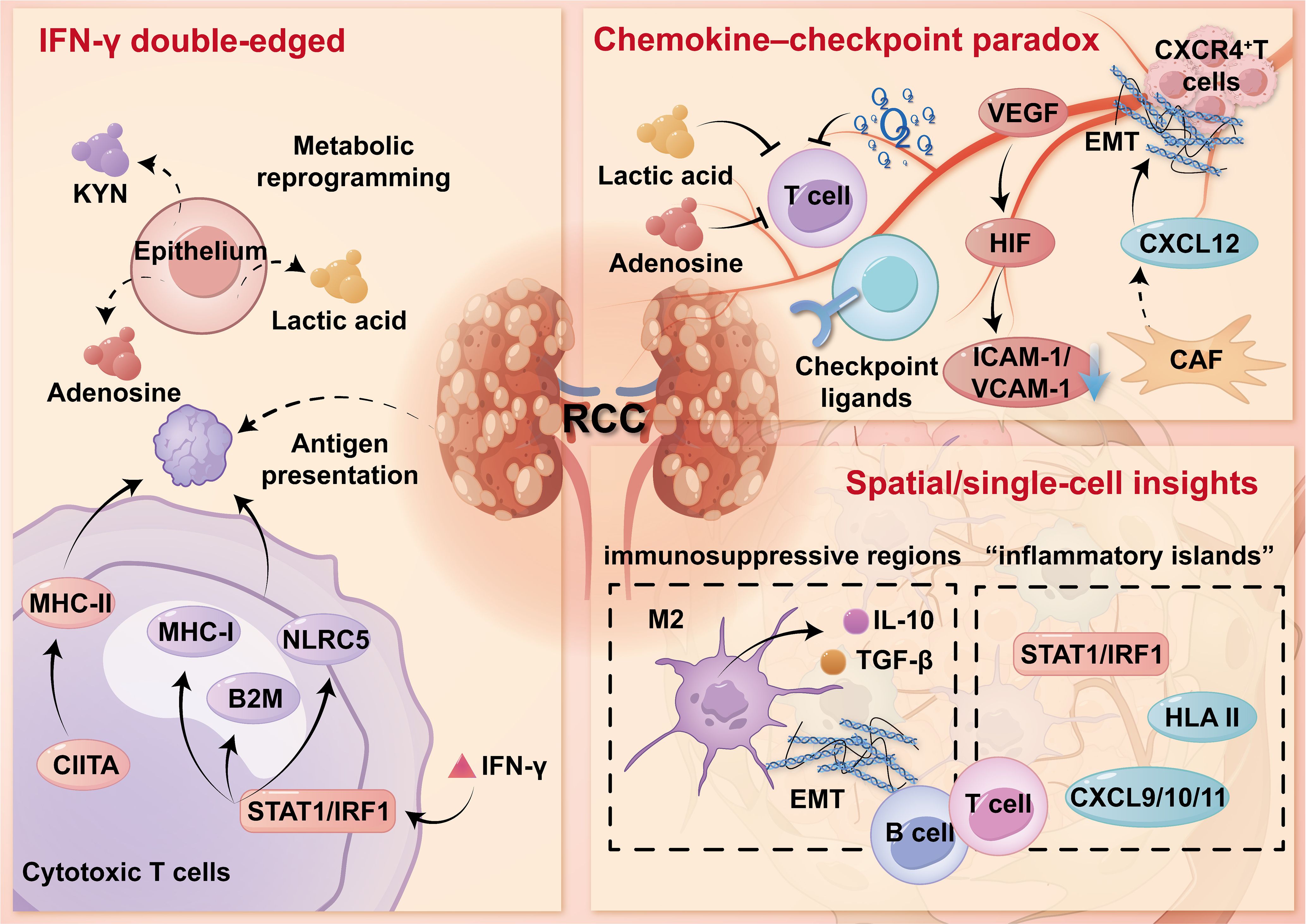
Figure 1. Chronic inflammation primes RCC for an “inflamed-yet-restrained” TIME.
4.1 T-cell dysfunction, myeloid/CAF suppression, and hypoxia coupling
In the TIME of established ccRCC, CD8+T cells often remain dysfunctional despite their presence in adequate numbers, reflecting progression along an exhaustion spectrum. These cells range from progenitor-like to terminally exhausted states, characterized by diminished cytotoxic potential, reduced granzyme and perforin production, impaired mitochondrial biogenesis, and metabolic stress driven by lactate accumulation and lipid overload (39). Persistent antigen stimulation activates NFAT-dependent TOX expression, reinforcing epigenetic programs that lock in the exhausted phenotype. At the same time, immunosuppressive myeloid populations—including M2-polarized macrophages and both granulocytic and monocytic MDSCs—produce suppressive mediators such as NO, ROS, arginase, IL-10, and TGF-β. These factors inhibit T cell priming and cytotoxicity while impairing DC maturation and cross-priming (40). CAFs contribute further by depositing aligned collagen fibers and releasing CXCL12, which traps CXCR4+T cells in perivascular zones, forming both structural and chemotactic barriers to infiltration. In parallel, VEGF and hypoxia induce endothelial anergy by reducing ICAM-1/VCAM-1 expression and altering pericyte coverage. Some endothelial cells additionally express FasL and PD-L1, directly eliminating incoming T cells. Hypoxia also stabilizes HIF-1α/2α, promoting VEGF-driven but inefficient angiogenesis that sustains acidosis and nutrient deprivation, ultimately limiting T cell survival and motility. Notably, recent studies show that chronic antigen exposure under hypoxic conditions induces rapid terminal exhaustion of CD8+T cells via mitochondrial ROS accumulation and suppression of PGC-1α signaling, linking metabolic stress to immunotherapy resistance. Taken together, the interplay between intrinsic T cell exhaustion, suppressive myeloid and stromal elements, and hypoxia-induced metabolic dysfunction creates a TIME that strongly predicts poor response to immune checkpoint blockade, unless these barriers—particularly stromal and vascular components—are actively reprogrammed (41).
4.2 Clinical translation and therapeutic matching
Around the dominant TIME phenotype matching therapy, the response probability and persistence can be improved at the same time. Immune-infiltrated tumors, marked by pre-existing TILs, intact antigen presentation, and interferon signaling, respond well to PD-1/PD-L1 inhibitors, either alone or combined with CTLA-4 blockade. In contrast, immune-excluded tumors—often shaped by abnormal angiogenesis and dysfunctional endothelium—may benefit more from combining ICB with VEGF/VEGFR inhibitors. These agents can restore vascular integrity, upregulate ICAM-1/VCAM-1, reduce myeloid infiltration, and enhance T cell entry. Immune-desert tumors, or those dominated by suppressive myeloid cells and CAFs, may require additional interventions targeting pathways, alongside strategies to reduce ECM stiffness or metabolic constraints, in order to create a more immune-permissive environment prior to or together with checkpoint inhibition (42). Biomarker strategies that combine indices of T cell inflammation with angiogenic and stromal signatures offer better predictive power than single markers like PD-L1 IHC. In cases with unclear immune profiles, genomic alterations in chromatin remodeling or DNA repair genes may further refine patient selection. Clinical management should also be adaptive, with immune phenotype shifts tracked by serial biopsy or validated surrogates like imaging-based assessments of perfusion or immune activity. For example, adding anti-angiogenic or myeloid-targeting agents may help shift immune-excluded tumors toward inflamed states, while inflamed tumors may remain responsive to ICB alone. Current RCC trials increasingly stratify patients by TIME phenotype and dominant resistance mechanism and define success not only by traditional endpoints but also by biological transitions such as reversal of immune exclusion, aiming to reprogram the tumor microenvironment and restore cytotoxic immunity.
5 Discussion
Despite major advances in immune profiling technologies, turning static immune “snapshots” into clinically useful biomarkers in RCC remains challenging. Technical factors—such as biopsy location, ischemia time, and fixation protocols—can introduce variability, while differences between profiling platforms and downstream analysis pipelines often result in inconsistent interpretations of immune cell states (43). RCC is also highly spatially heterogeneous: a single core can miss key features of the TIME, from inflamed pockets to immune-excluded zones or myeloid/CAF-dominated areas. Time compounds the problem. Therapy, intercurrent infection, or ischemia-reperfusion can quickly remodel the TIME, so a baseline sample rarely captures true dynamics. Cross-platform integration remains immature: scRNA-seq, spatial transcriptomics, multiplex IHC/IMC, and digital pathology each show a different slice of biology, and we lack robust reference atlases and external validation to fuse them into stable, generalizable classifiers. Clinical qualification is another hurdle: assays must show analytical validity, clinical validity, and real utility, within the constraints of trial timelines, budgets, and regulators. Meaningful progress will require harmonized SOPs, multi-region and longitudinal sampling, built-in QC, and prospective replication in diverse cohorts before immune snapshots can routinely guide RCC care (26).
Looking ahead, progress in RCC immunotherapy will depend on adopting a longitudinal, multi-modal approach that integrates spatial, single-cell, and histopathologic data to monitor transitions between inflamed, excluded, and desert TIME states. Trial stratification should consider both immune phenotype and dominant resistance pathways—whether checkpoint-driven, angiogenic, or myeloid/CAF-mediated—to guide rational combinations. Adaptive trial designs with dynamic treatment adjustment based on TIME remodeling could improve therapeutic sequencing. Endpoints should move beyond traditional metrics like ORR and PFS to capture meaningful biological transitions, such as exclusion-to-inflamed conversion or restoration of myeloid–lymphoid balance, supported by real-time biomarkers. Developing standardized immune atlases, validated surrogate markers, and accessible high-fidelity assays will be essential to enable clinical translation. Ultimately, refining immune classification tools and matching therapies to the underlying resistance mechanisms will allow immune snapshots to serve not just as static readouts but as real-time guides for interception, sequencing, and combination treatment in RCC.
Author contributions
HX: Visualization, Data curation, Writing – review & editing, Conceptualization, Writing – original draft. BT: Conceptualization, Writing – original draft. HL: Writing – review & editing. YS: Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bukavina L, Bensalah K, Bray F, Carlo M, Challacombe B, Karam JA, et al. Epidemiology of renal cell carcinoma: 2022 update. Eur Urol. (2022) 82:529–42. doi: 10.1016/j.eururo.2022.08.019
2. Makino T, Kadomoto S, Izumi K, and Mizokami A. Epidemiology and prevention of renal cell carcinoma. Cancers (Basel). (2022) 14(16):4059. doi: 10.3390/cancers14164059
3. Zheng W, Zhang S, Guo H, Chen X, Huang Z, Jiang S, et al. Multi-omics analysis of tumor angiogenesis characteristics and potential epigenetic regulation mechanisms in renal clear cell carcinoma. Cell communication signaling: CCS. (2021) 19:39. doi: 10.1186/s12964-021-00728-9
4. Chappell JC, Payne LB, and Rathmell WK. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J Clin Invest. (2019) 129:442–51. doi: 10.1172/jci120855
5. Qu Z, Chu J, Jin S, Yang C, Zang J, Zhang J, et al. Tissue-resident macrophages and renal diseases: landscapes and treatment directions. Front Immunol. (2025) 16:1548053. doi: 10.3389/fimmu.2025.1548053
6. Kruk L, Mamtimin M, Braun A, Anders HJ, Andrassy J, Gudermann T, et al. Inflammatory networks in renal cell carcinoma. Cancers (Basel). (2023) 15(8):2212. doi: 10.3390/cancers15082212
7. Qin S, Jiang J, Lu Y, Nice EC, Huang C, Zhang J, et al. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal transduction targeted Ther. (2020) 5:228. doi: 10.1038/s41392-020-00313-5
8. Bruni S, Mercogliano MF, Mauro FL, Cordo Russo RI, and Schillaci R. Cancer immune exclusion: breaking the barricade for a successful immunotherapy. Front Oncol. (2023) 13:1135456. doi: 10.3389/fonc.2023.1135456
9. Arabi T, Shafqat A, Sabbah BN, Ashraf N, Shah H, Abdulkader H, et al. Obesity-related kidney disease: beyond hypertension and insulin-resistance. Front Endocrinol. (2022) 13:1095211. doi: 10.3389/fendo.2022.1095211
10. Oda Y, Nishi H, and Nangaku M. Role of inflammation in progression of chronic kidney disease in type 2 diabetes mellitus: clinical implications. Semin Nephrol. (2023) 43:151431. doi: 10.1016/j.semnephrol.2023.151431
11. Kim J, Ha S, Son M, Kim D, Kim MJ, Kim B, et al. Tlr7 activation by Mir-21 promotes renal fibrosis by activating the pro-inflammatory signaling pathway in tubule epithelial cells. Cell communication signaling: CCS. (2023) 21:215. doi: 10.1186/s12964-023-01234-w
12. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the sting pathway lead to renal inflammation and fibrosis. Cell Metab. (2019) 30:784–99.e5. doi: 10.1016/j.cmet.2019.08.003
13. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal transduction targeted Ther. (2021) 6:263. doi: 10.1038/s41392-021-00658-5
14. Kourtis N, Wang Q, Wang B, Oswald E, Adler C, Cherravuru S, et al. A single-cell map of dynamic chromatin landscapes of immune cells in renal cell carcinoma. Nat Cancer. (2022) 3:885–98. doi: 10.1038/s43018-022-00391-0
15. Liu ZJ, Semenza GL, and Zhang HF. Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang Univ Sci B. (2015) 16:32–43. doi: 10.1631/jzus.B1400221
16. Hoefflin R, Harlander S, Schäfer S, Metzger P, Kuo F, Schönenberger D, et al. Hif-1α and Hif-2α Differently regulate tumor development and inflammation of clear cell renal cell carcinoma in mice. Nat Commun. (2020) 11:4111. doi: 10.1038/s41467-020-17873-3
17. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium fasl establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
18. Jorgovanovic D, Song M, Wang L, and Zhang Y. Roles of ifn-Γ in tumor progression and regression: A review. biomark Res. (2020) 8:49. doi: 10.1186/s40364-020-00228-x
19. Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, et al. Ifn-Γ from lymphocytes induces Pd-L1 expression and promotes progression of ovarian cancer. Br J Cancer. (2015) 112:1501–9. doi: 10.1038/bjc.2015.101
20. Salminen A, Kaarniranta K, and Kauppinen A. Tissue fibroblasts are versatile immune regulators: an evaluation of their impact on the aging process. Ageing Res Rev. (2024) 97:102296. doi: 10.1016/j.arr.2024.102296
21. Xie R, Shang B, Shi H, Bi X, Song Y, Qu W, et al. Neutrophil extracellular traps in relationship to efficacy of systemic therapy for metastatic renal cell carcinoma. Cancer Med. (2023) 12:21807–19. doi: 10.1002/cam4.6748
22. Li J, Yang Y, Wang Y, Li Q, and He F. Metabolic signatures of immune cells in chronic kidney disease. Expert Rev Mol Med. (2022) 24:e40. doi: 10.1017/erm.2022.35
23. Li L, Yang C, Zhao Z, Xu B, Zheng M, Zhang C, et al. Skewed T-helper (Th)1/2- and Th17/T regulatory−Cell balances in patients with renal cell carcinoma. Mol Med Rep. (2015) 11:947–53. doi: 10.3892/mmr.2014.2778
24. Guzzi F, Cirillo L, Roperto RM, Romagnani P, and Lazzeri E. Molecular mechanisms of the acute kidney injury to chronic kidney disease transition: an updated view. Int J Mol Sci. (2019) 20(19):4941. doi: 10.3390/ijms20194941
25. Jaworska K, Ratajczak J, Huang L, Whalen K, Yang M, Stevens BK, et al. Both Pd-1 ligands protect the kidney from ischemia reperfusion injury. J Immunol. (2015) 194:325–33. doi: 10.4049/jimmunol.1400497
26. Borcherding N, Vishwakarma A, Voigt AP, Bellizzi A, Kaplan J, Nepple K, et al. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun Biol. (2021) 4:122. doi: 10.1038/s42003-020-01625-6
27. Shukla A, Cloutier M, Appiya Santharam M, Ramanathan S, and Ilangumaran S. The Mhc class-I transactivator Nlrc5: implications to cancer immunology and potential applications to cancer immunotherapy. Int J Mol Sci. (2021) 22(4):1964. doi: 10.3390/ijms22041964
28. Mielcarska S, Kot A, Dawidowicz M, Kula A, Sobków P, Kłaczka D, et al. B7-H3 in cancer immunotherapy-prospects and challenges: A review of the literature. Cells. (2025) 14(15):1209. doi: 10.3390/cells14151209
29. Labadie BW, Bao R, and Luke JJ. Reimagining Ido pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin Cancer Res. (2019) 25:1462–71. doi: 10.1158/1078-0432.Ccr-18-2882
30. Wang H, Fu C, Du J, Wang H, He R, Yin X, et al. Enhanced histone H3 acetylation of the Pd-L1 promoter via the Cop1/C-Jun/Hdac3 axis is required for Pd-L1 expression in drug-resistant cancer cells. J Exp Clin Cancer Res. (2020) 39:29. doi: 10.1186/s13046-020-1536-x
31. Song X, Zhu Y, Geng W, Jiao J, Liu H, Chen R, et al. Spatial and single-cell transcriptomics reveal cellular heterogeneity and a novel cancer-promoting Treg cell subset in human clear-cell renal cell carcinoma. J immunotherapy Cancer. (2025) 13(1):e010183. doi: 10.1136/jitc-2024-010183
32. Yu Z, Lv Y, Su C, Lu W, Zhang R, Li J, et al. Integrative single-cell analysis reveals transcriptional and epigenetic regulatory features of clear cell renal cell carcinoma. Cancer Res. (2023) 83:700–19. doi: 10.1158/0008-5472.Can-22-2224
33. Yang G, Cheng J, Xu J, Shen C, Lu X, He C, et al. Metabolic heterogeneity in clear cell renal cell carcinoma revealed by single-cell Rna sequencing and spatial transcriptomics. J Transl Med. (2024) 22:210. doi: 10.1186/s12967-024-04848-x
34. Krishna C, DiNatale RG, Kuo F, Srivastava RM, Vuong L, Chowell D, et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccrcc to tumor topology and therapy efficacy. Cancer Cell. (2021) 39:662–77.e6. doi: 10.1016/j.ccell.2021.03.007
35. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. (2018) 175:984–97.e24. doi: 10.1016/j.cell.2018.09.006
36. Zheng S, Wang W, Shen L, Yao Y, Xia W, and Ni C. Tumor battlefield within inflamed, excluded or desert immune phenotypes: the mechanisms and strategies. Exp Hematol Oncol. (2024) 13:80. doi: 10.1186/s40164-024-00543-1
37. Sobottka B, Vetter V, Banaei-Esfahani A, Nowak M, Lorch A, Sirek A, et al. Immune phenotype-genotype associations in primary clear cell renal cell carcinoma and matched metastatic tissue. Modern Pathol. (2024) 37:100558. doi: 10.1016/j.modpat.2024.100558
38. Tsuzuki T, Ohe C, Osawa T, Yasuda Y, Tanaka T, Anai S, et al. Prognostic value of immune phenotype and Pd-L1 status in recurrent or metastatic renal cell carcinoma: an exploratory analysis of the archery study. Pathology. (2023) 55:31–9. doi: 10.1016/j.pathol.2022.07.013
39. Pichler R, Siska PJ, Tymoszuk P, Martowicz A, Untergasser G, Mayr R, et al. A chemokine network of T cell exhaustion and metabolic reprogramming in renal cell carcinoma. Front Immunol. (2023) 14:1095195. doi: 10.3389/fimmu.2023.1095195
40. Annels NE, Denyer M, Nicol D, Hazell S, Silvanto A, Crockett M, et al. The dysfunctional immune response in renal cell carcinoma correlates with changes in the metabolic landscape of ccrcc during disease progression. Cancer Immunol Immunother. (2023) 72:4221–34. doi: 10.1007/s00262-023-03558-5
41. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. (2021) 22:205–15. doi: 10.1038/s41590-020-00834-9
42. Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal transduction targeted Ther. (2021) 6:218. doi: 10.1038/s41392-021-00641-0
Keywords: renal cell carcinoma, tumor immune microenvironment, chronic inflammation, IFN-γ signaling, immune exclusion, immunotherapy
Citation: Xu H, Tu B, Li H and Shan Y (2025) From chronic inflammation to immune escape: mapping the tumor microenvironment evolution in renal cell carcinoma. Front. Immunol. 16:1683844. doi: 10.3389/fimmu.2025.1683844
Received: 11 August 2025; Accepted: 17 October 2025;
Published: 04 November 2025.
Edited by:
Cristiana Tanase, Victor Babes National Institute of Pathology (INCDVB), RomaniaReviewed by:
Lingsong Tao, Second People’s Hospital of Wuhu, ChinaCopyright © 2025 Xu, Tu, Li and Shan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Shan, RHIuU2hhbllvbmdAb3V0bG9vay5jb20=
†These authors have contributed equally to this work