 Yeying Jin1,2
Yeying Jin1,2 Rui Yang
Rui Yang- 1Department of Breast Surgery, Shanxi Cancer Hospital/Chinese Academy of Medical Sciences Cancer Hospital Shanxi Hospital/Affiliated Cancer Hospital of Shanxi Medical University, Taiyuan, China
- 2School of Basic Medical Sciences, Shanxi Medical University, Jinzhong, China
Chemokine ligand 2 (CCL2) is a key regulatory molecule in the tumor microenvironment (TME) participating in the occurrence, progression, and metastasis of tumors through complex mechanisms. This paper systematically reviews the production and regulation of CCL2 in tumors and its pleiotropic effects. CCL2 can be continuously produced by tumor cells, stromal cells, and host–tumor interactions through constitutive secretion, microenvironmental stimulation response, and interaction network. Its expression is regulated by transcription factors such as Nuclear factor-kappa B (NF-κB), signal transducer and activator of transcription 3 (STAT3), and activator protein 1 (AP-1); single nucleotide polymorphisms (SNPs); and epigenetic modifications such as DNA methylation and noncoding RNA. Inflammatory factors (such as tumor necrosis factor [TNF]-α, interleukin [IL]-1β, and IL-6) and hypoxia signal in the TME further amplify CCL2 secretion through the activation of NF-κB, MAPK, and other pathways, forming a positive feedback loop. CCL2 directly promotes the proliferation, migration, and epithelial–mesenchymal transition of cancer cells by activating CCR2 receptor and its downstream PI3K/AKT, MAPK, and other signaling pathways and remodels the immunosuppressive microenvironment by recruiting tumor-associated macrophages and myeloid-derived suppressor cells. Furthermore, CCL2 drives tumor invasion and distant metastasis by inducing angiogenesis, enhancing matrix metalloproteinase activity, and promoting premetastatic niche formation. Although clinical trials targeting the CCL2–CCR2 axis have been carried out, the efficacy is limited by the redundancy of CCL2 expression and its crosstalk with other factors. Given our incomplete understanding of its mechanism, the development of combined strategies or miRNA, epigenetic intervention, and other source regulation methods is necessary. This study provides a theoretical basis for understanding the tumor regulatory network of CCL2 and the development of precise targeted therapy.
1 Introduction
The occurrence and progression of cancer are far from a simple cell-autonomous event, but a complex ecosystem driven by gene mutation, microenvironmental remodeling, and immune evasion. From the initial accumulation of genetic abnormalities to malignant transformation, tumor cells gradually get rid of growth regulation, recruit and reprogram surrounding stromal cells by secreting a variety of cytokines, thereby jointly constructing a tumor microenvironment (TME) that supports their survival and spread (1). This dynamic network includes immune cells, fibroblasts, endothelial cells and extracellular matrix (ECM), especially tumor-associated macrophages (TAM) (2). The interaction of cytokines, chemokines and growth factors promotes the growth of vascular tissues and the metastasis of tumor cells. It also prompts the host immune response to gradually change from the initial immune attack to immune evasion, distorting the local inflammatory response into a cancer-promoting engine. In addition, cytokines secreted by the primary tumor also lay the foundation for the formation of the premetastatic niche by “preconditioning” the organs that will be metastasized in the future (3).
Among the many molecules regulating TME, chemokines have become a key bridge connecting inflammation and tumor progression due to their core functions of mediating immune cell migration and activation (4). Among them, chemokine ligand 2 (CCL2) has attracted much attention owing to its pleiotropic and contradictory functions. CCL2, also known as monocyte chemoattractant protein-1 (MCP-1), was first isolated and purified from the culture supernatant of peripheral blood monocytes and tumor cell lines in 1989 (5). Early studies have emphasized its anti-tumor effect by recruiting monocytes/macrophages (6). In the study of non-intracranial tumors, CCL2 gene transduction has been regarded as an effective vaccine strategy to enhance the host cell defense ability against tumor cell attack (7). Furthermore, studies have shown that CCL2 synergistically with lipopolysaccharide (LPS) can activate the tumoricidal properties of macrophages, thereby reducing lung metastasis of colon cancer (8). However, with a deeper understanding of the complexity of TME, studies have found that the pro-tumor properties of CCL2 far outweigh the anti-tumor effects (9). On the one hand, its abnormally high expression attracts immunosuppressive cells such as CCR2+ TAMs through chemotaxis, activates pro-cancer pathways such as PI3K/AKT and NF-κB, and induces EMT, directly promoting tumor invasion and metastasis[ (10). On the other hand, CCL2 is closely related to angiogenesis, matrix degradation and the up-regulation of immune checkpoint molecules, and forms a positive feedback loop with inflammatory factors such as TNF-α to further consolidate the immunosuppressive microenvironment (11, 12). This transformation from “guardian” to “traitor” highlights the hijacking of the host physiological mechanism by tumors, and also makes CCL2 one of the core molecules to understand the malignant progression of tumors.
As cytokines involved in cancer progression, neutralizing inhibitors targeting IL-1, IL-4, TGFβ, and IL-10 have shown the potential to improve cancer (13). However, due to the pleiotropic, dual-action nature of cytokines in cancer and the redundant mechanisms of multicellular receptors, the results of relevant clinical trials are often not ideal, and many trials are abandoned (14). Blocking the CCL2-CCR2 pathway in cancer has also been investigated in a variety of clinical trials. Some trials have reported positive responses, but the efficacy of anti-CCL2 antibodies or CCR2 antagonists as monotherapies is not ideal (15). Therefore, understanding the production mechanism and downstream effects of CCL2 in TME is not only the key to reveal the logic of tumor progression, but also provides a theoretical fulcrum for the development of CCL2 therapeutic strategies.
This article systematically reviews the biological characteristics of CCL2-CCR2, as well as the constitutive secretion of CCL2 in the TME, its microenvironment-responsive expression, and the multi-dimensional regulation resulting from tumor-stroma interactions. It focuses on the pleiotropic roles of CCL2 in the EMT, angiogenesis, immune escape, and metastatic cascades. Finally, we discuss the limitations and directions of therapeutic interventions targeting CCL2. The aim is to provide a theoretical basis for precise anti-tumor strategies targeting CCL2. Please refer to Figure 1 for further details. Created in https://BioRender.com.
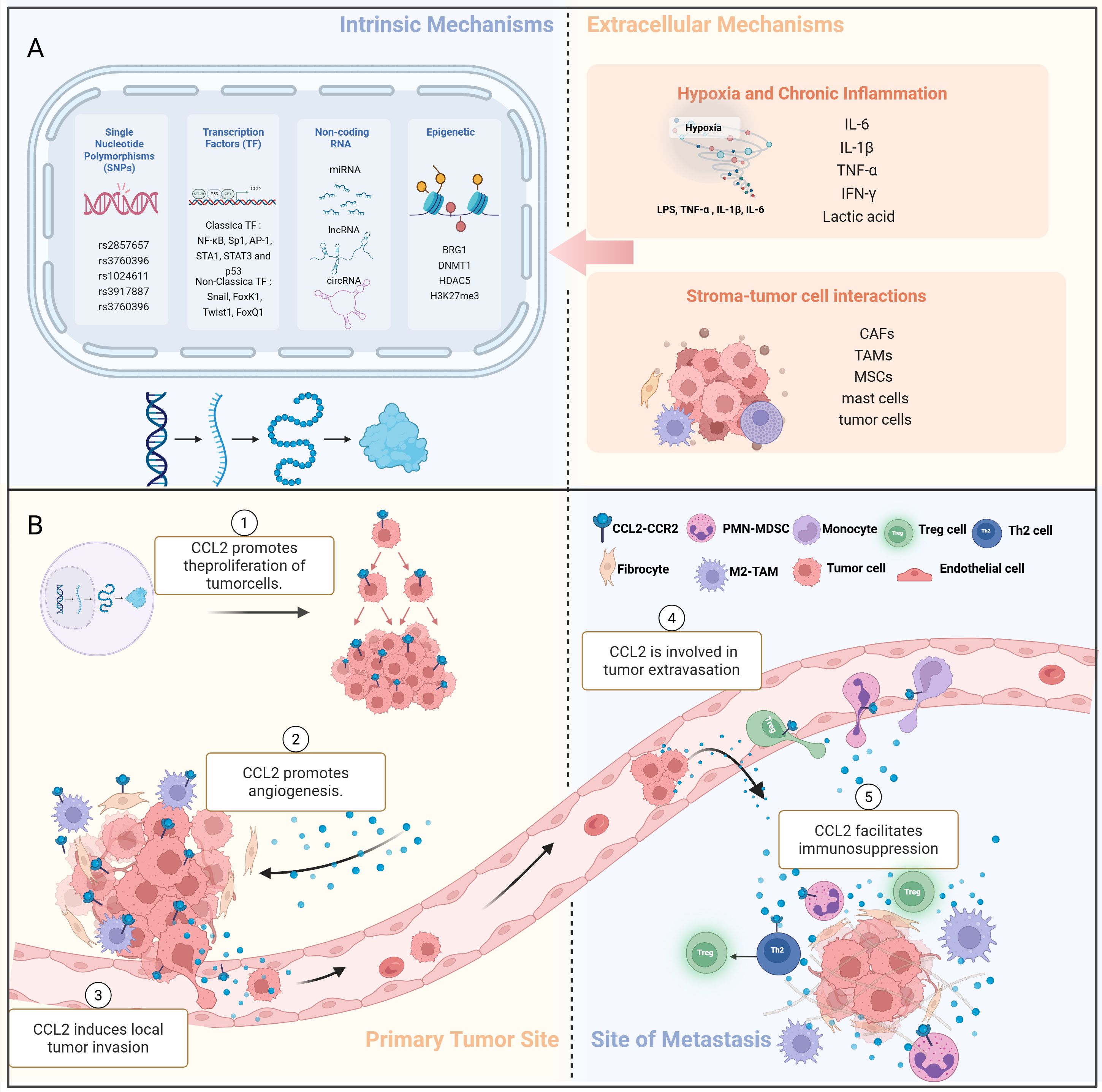
Figure 1. Production and pleiotropic regulation of CCL2 in the TME. (A) (1) The intrinsic cellular mechanism of CCL2 production:single nucleotide polymorphisms (SNPs), epigenetic regulation, non-coding RNA regulation. (2) The extrinsic cellular mechanism of CCL2 production:hypoxia and chronic inflammation; stromal tumor-cell interactions. (B) (1) Regulation of CCL2 production in TME. (2) CCL2 promotes the proliferation of tumor cells, enhancing their growth and survival. (3) CCL2 directly or indirectly promotes angiogenesis, the formation of new blood vessels, which supports tumor growth. (4) CCL2 induces local tumor invasion, allowing cancer cells to spread beyond the primary tumor site. (5) CCL2 is involved in the process of tumor extravasation. (6) CCL2 facilitates immunosuppression, inhibiting the immune response against cancer cells, thereby promoting tumor growth and metastasis. M2-TAM, M2 tumor-associated macrophages;Treg cell, Regulatory T cells;Th2 cell, Helper T cells; PMN-MDSC, Polymorphonuclear myeloid-derived suppressor cells; SNP, single nucleotide polymorphism.
2 Biological characteristics and functions of CCL2
CCL2, also known as MCP-1, is one of the earliest discovered human CC chemokines (16). It is a 76-amino acid basic protein encoded by the CCL2 gene located on chromosome 17q11.2-q12 (16).In the TME, monocytes and macrophages serve as the primary producers of this molecule (17). As a small-molecule cytokine, CCL2 shows remarkable pro-inflammatory chemotactic activity. Under normal physiological conditions, CCL2 recruits immune cells such as monocytes, macrophages, memory T cells, natural killer (NK) cells, and dendritic cells from the blood to the inflammation site through the chemokine gradient, which helps maintain the normal distribution and function of immune cells (18). In addition to its chemotactic function, CCL2 has a multifaceted role in the regulation of immune cell function. In the inflammatory response, CCL2 can enhance the adhesion of monocytes to vascular endothelium by increasing the expression of integrin β2 family and the release of arachidonic acid, thereby strengthening the retention capacity of monocytes (19, 20). It also regulates the secretion of effector molecules and the autophagy, killing, and survival of immune cells (21). Moreover, CCL2 participates in the development, differentiation, maturation, homing, and interaction of immune cells (18, 22, 23); initiates and maintains inflammatory responses; and balances the intensity of inflammation by regulating the polarization state of macrophages and cytokine secretion (24). In tissue repair, CCL2 promotes the migration and polarization of monocytes and macrophages for tissue remodeling and angiogenesis (25–27).
The main receptor of CCL2 is C-C chemokine receptor type 2 (CCR2), a seven-transmembrane G protein-coupled receptor with extremely strong affinity (Kd = 0.77 nM) (28, 29). In terms of binding mechanism, the N-terminus of CCL2 forms a key hydrogen bond with the third transmembrane domain (TM3) and the second extracellular loop (ECL2) of CCR2. These interactions make CCL2 the most potent ligand to activate CCR2 signaling (30). The binding of CCL2 and CCR2 is essential for initiating signal transduction pathways and stimulating cell migration. Upon binding to CCR2, CCL2 activates the G protein-coupled receptor signaling pathway to initiate an intracellular signaling cascade, which includes PI3K/AKT, mitogen-activated protein kinase (MAPK)/p38, protein kinase C (PKC), calmodulin-dependent protein kinase II, and JAK/STAT3 pathways (31). By activating these signaling pathways, CCL2 regulates cytoskeleton reorganization and phospholipase C-dependent calcium release, which plays a key role in anti-apoptosis, angiogenesis, and cell invasion and migration. CCL2 can also bind to other G protein-coupled receptors. For example, the CCL2–CCR4 signaling axis is a key mechanism to promote brain metastasis in breast cancer (32) and mediates cell migration in head and neck squamous cell carcinoma (33). Furthermore, CCL2 can bind to atypical receptors (ACKR1 and ACKR2). Although they have a sevenfold transmembrane structure similar to classical chemokine receptors, atypical receptors are not involved in G-protein-mediated signal transduction and do not affect intracellular calcium concentration. They also do not trigger cell migration (34, 35). Although they do not generate signals through G proteins, they are involved in the formation of chemokine gradients and regulate chemokine bioavailability and signaling by internalizing and degrading or redirecting CCL2, acting as regulatory components of the chemokine network (36, 37). The biological function of CCL2 is shown in Figure 2.
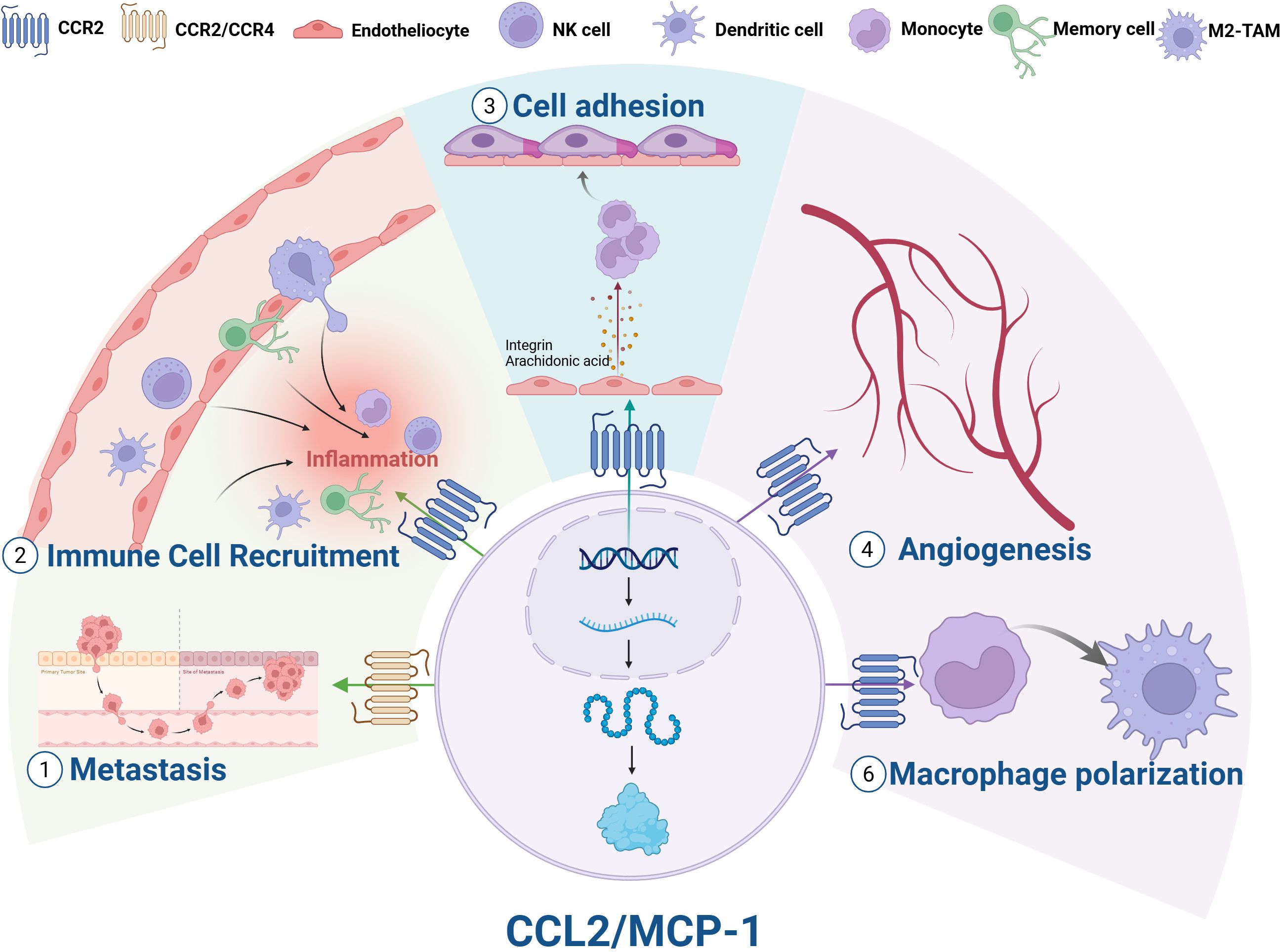
Figure 2. Biological functions of CCL2. (1) Metastasis; (2) Immune cell recruitment; (3) Cell adhesion; (4) Angiogenesis; (5) Macrophage polarization; (6) Enhanced tumor growth and metastasis. NK cell, natural killer cell; M2-TAMs, M2 tumor-associated macrophages.
3 Mechanisms of CCL2 production in the tumor microenvironment
In the TME, CCL2 is synthesized by various cell types such as cancer cells and stromal cells in response to various stimuli. The production pathways of CCL2 can be summarized into the following three categories: (1) Cells constitutively produce CCL2; (2) The responsive expression of CCL2 to the stimulatory factors in TME; (3) Stromal tumor-cell interactions produce CCL2. These three mechanisms interact to construct a complex regulatory network, which leads to the high level of CCL2 expression in the TME (Figure 3), and also provides a large number of precise targets for CCL2-targeted therapy.
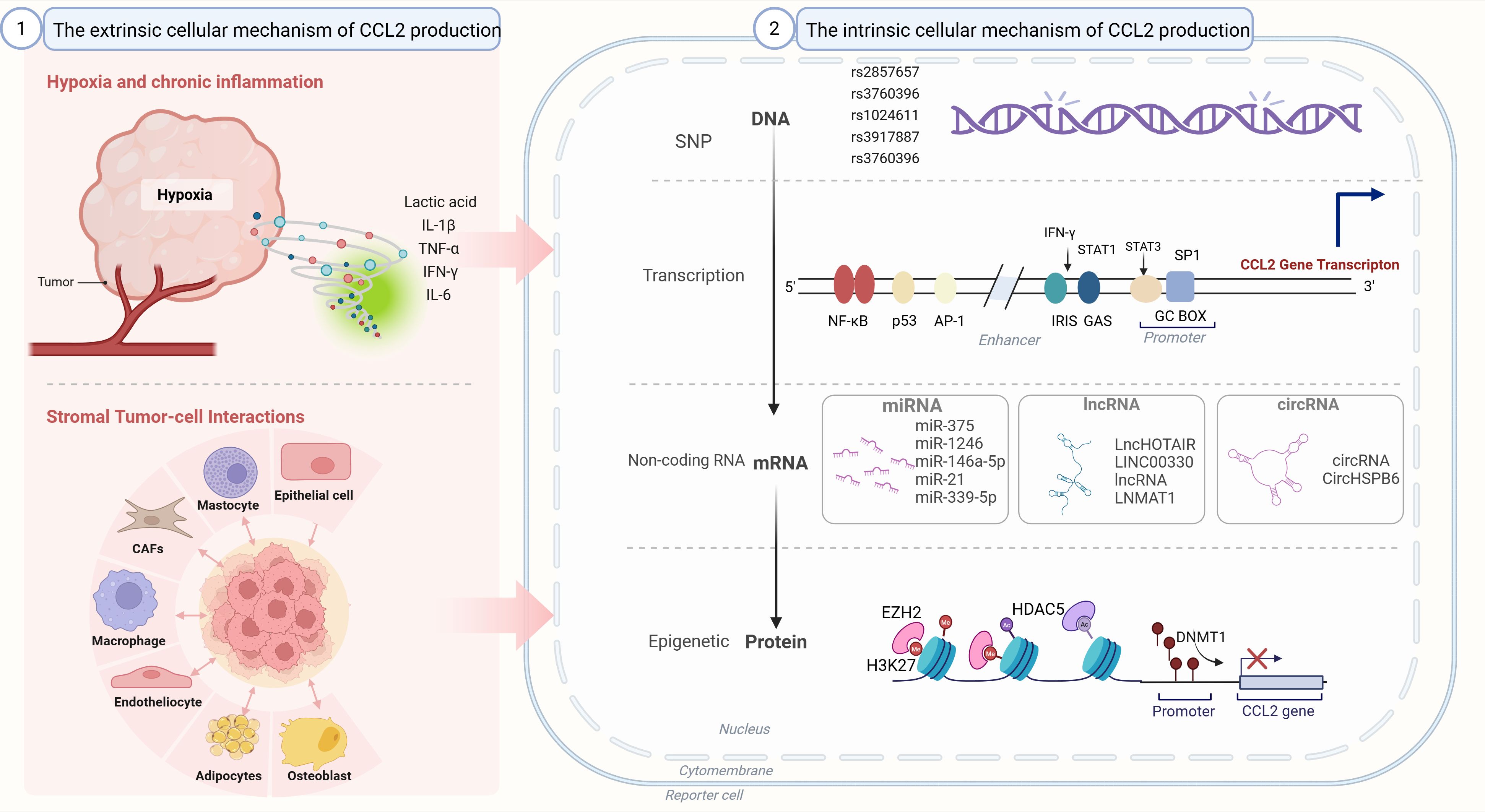
Figure 3. Regulation of CCL2 production. The intrinsic cellular mechanism of CCL2 production: single nucleotide polymorphisms (SNPs), epigenetic regulation, non-coding RNA regulation. (2) The extrinsic cellular mechanism of CCL2 production:hypoxia and chronic inflammation; stromal tumor-cell interactions. SNP: single nucleotide polymorphism. CAFs, Cancer-associated fibroblasts; miRNA, MicroRNA; lncRNA, Long non-coding RNA; circRNA, Circular RNA; SNP, Single nucleotide polymorphism; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; p53, Tumor protein p53; AP-1, Activator protein 1; IRIS, Inflammation and response sensor; GAS, Gamma interferon activation site; STAT1, Signal transducer and activator of transcription 1; STAT3, Signal transducer and activator of transcription 3; GC BOX, Guanine cytosine box; EZH2, Enhancer of zeste homolog 2; HDACs, Histone deacetylases; H3K27, Histone H3 lysine 27; DNMT1, DNA methyltransferase 1.
3.1 Cells constitutively produce CCL2
Several studies have confirmed that overexpression of various transcription factors in cancer cells can affect the transcription process of CCL2. Tumor cells can continuously and autonomously secrete CCL2 through intrinsic mechanisms due to intrinsic gene or mutation or epigenetic changes without external stimulation.
2.1.1 Single nucleotide polymorphisms (SNPs) regulate CCL2 expression
Single nucleotide polymorphisms (SNPs) in the regulatory regions of cytokine genes may affect CCL2 transcription and alter the expression levels of these genes in specific populations (38). Among CCL2 SNPs, +764 C/G (rs2857657), −927 G/C (rs3760396), −2518 A/G, and −2835C/A are associated with high CCL2 expression; −2518 A/G (rs1024611), −927 G/C (rs3760396), and −2835 C/A (rs2857654) are located in the promoter regulatory region; and +764 C/G and CCL2 14bp I/D (rs3917887) are located in the intronic region (39, 40). Meanwhile, −2518A/G (rs1024611) is the most widely studied CCL2 regulatory SNP and has the most well-defined function in tumors; its G allele rs1024611G is associated with high CCL2 expression and poor clinical prognosis in a variety of cancers (41). By contrast, rs1024611A is associated with low CCL2 expression (42). rs2857656 (−362 G/C), rs1024611(−2518 A/G), and rs4586 (Cys35Cys) of CCL2/MCP1 are positively associated with elevated circulating levels of CCL2 and significantly associated with an increased risk of breast cancer (43). CCL2 rs1024611 SNP may also be associated with the occurrence and development of cervical cancer, deep stromal invasion, large tumor diameter, and parametrial invasion (44). The effects of other SNPs on CCL2 expression and their diagnostic values in different cancers are detailed in Table 1.
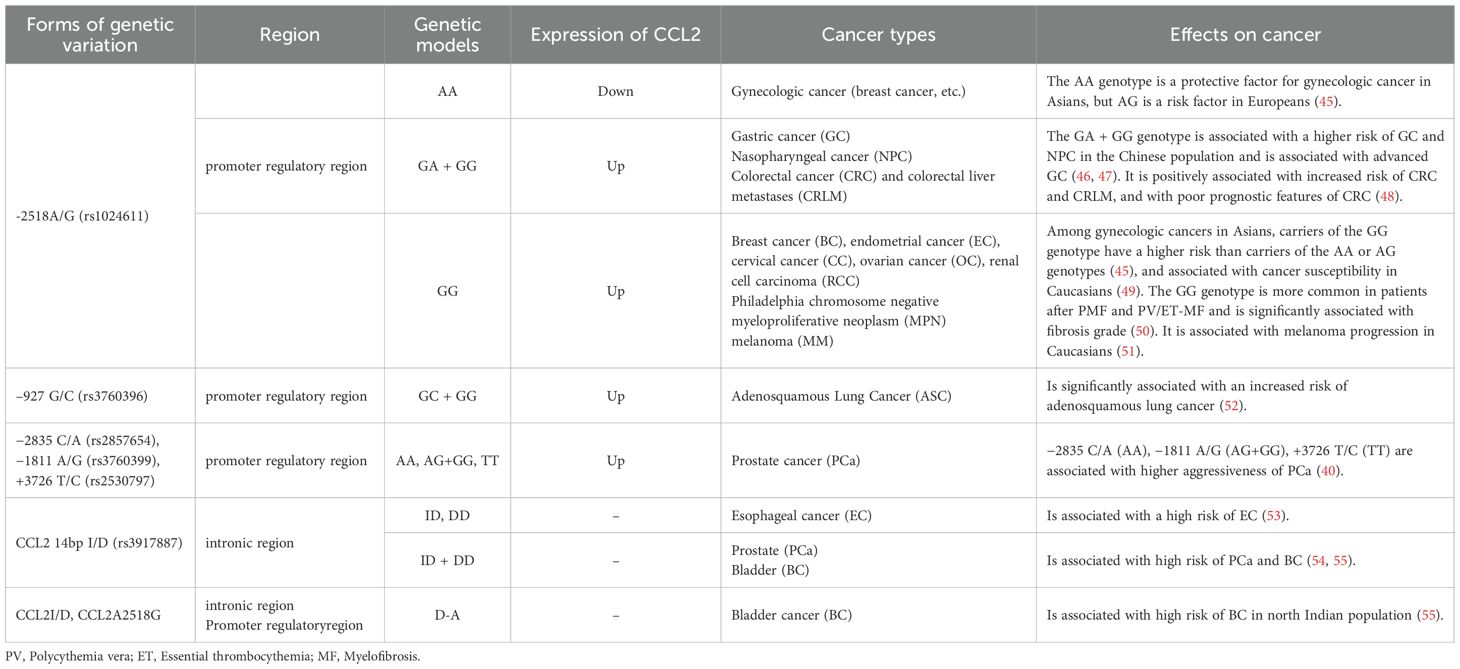
Table 1. SNPs associated with different cancer types in CCL2.
2.1.2 Epigenetic modifications regulate CCL2 expression
Epigenetic alteration refers to the process of regulating gene expression through chemical modification or chromatin structure changes without altering the DNA sequence. CCL2 expression is affected by a variety of epigenetic regulatory mechanisms, including DNA methylation, histone modification, chromatin remodeling.
Posttranslational modifications of histone N-terminal tails, including phosphorylation, acetylation, ubiquitination, and methylation, can regulate nucleosome structure (56, 57). Among them, acetylation usually activates gene expression by promoting the interaction between transcription complexes and DNA (57). Methylation is closely related to the activation or repression of genes (58). In small cell lung cancer, DNA methyltransferase 1 (DNMT1) inhibits CCL2 expression by adding methyl groups to CpG islands in the promoter region of CCL2 gene, preventing TF binding (59). As a histone methyltransferase, Enhancer of Zeste Homolog 2 (Enhancer of Zeste Homolog 2) catalyzes the trimethylation of histone Histone H3 Lysine 27 (H3K27) and represses gene expression. Its binding to the enhancer region of CCL2 leads to an increase in the trimethylation level of H3K27 and ultimately inhibits the transcription of CCL2 (59). In breast cancer, EZH2 and DNMT1 act synergistically to regulate CCL2 expression. EZH2 can recruit DNMT1 to the promoter region of CCL2 gene, leading to DNA hypermethylation, and then up-regulate miR-124-3p to further inhibit CCL2 expression (60). miR-124-3p regulates CCL2 expression at the posttranscriptional level by targeting the 3′ untranslated region (3′ UTR) of CCL2 and degrading its mRNA or inhibiting its translation (61). In macrophage inflammatory activation, transcription repressors G protein pathway suppressor 2 (GPS2) and silencing mediator for retinoid and thyroid hormone receptors (SMRT) regulate enhancer and silencer remodeling via the transcription of long noncoding enhancer RNAs, consequently suppressing CCL2 transcription (62). In an iKPC mouse model, histone deacetylase 5 (HDAC5) was found to be a potential downstream target of the proto-oncogene KRAS. Its inhibition can lead to the down-regulation of negative regulator Socs3, which in turn increases CCL2 expression and promotes the infiltration of macrophages recruited by CCL2 (63). Brahma-related gene 1 (BRG1) promotes the recruitment of acetylated histone H3 and H4 and nuclear factor NF-κB to the CCL2 promoter region by regulating chromatin structure and TF binding. This process consequently up-regulates CCL2 (64).
2.1.3 Noncoding RNAs regulate CCL2 expression
Noncoding RNA Regulation (ncRNA) play a key role in the development and course of many diseases, especially cancer. The family of ncRNAs encompasses various molecular species, including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs), among other type (65). In vitro, long noncoding RNA H19 (H19-IRP) promotes glioblastoma multiforme (GBM) immunosuppression by binding to CCL2 and galectin-9 promoters to activate its transcription and recruiting myeloid-derived suppressor cells (MDSCs) and TAMs to induce T cell exhaustion and an immunosuppressive GBM-TME. Traditional GBM vaccines such as Rindopepimut targeting EGFRvIII and dendritic cell (DC) vaccine ICT-107 rely on T cell activation, but have limited improvement on immunosuppressed GBM-TME. However, the circular RNA vaccine against human H19-IRP activates anti-tumor T cells and modifies GBM–TME to enhance the antitumor effect (66). In the TME, miRNAs also regulate CCL2 and thus have become effective prognostic markers (67). Compared with lentivirus or protein-based gene therapy, ncRNAs have more potential in clinical application because of their simple structure and lower immunogenicity and toxicity (68). Therefore, CCL2-regulated ncRNAs serve as novel cancer biomarker and therapeutic target for cancer patients. Other ncRNAs that regulate CCL2 expression are listed in Table 2.
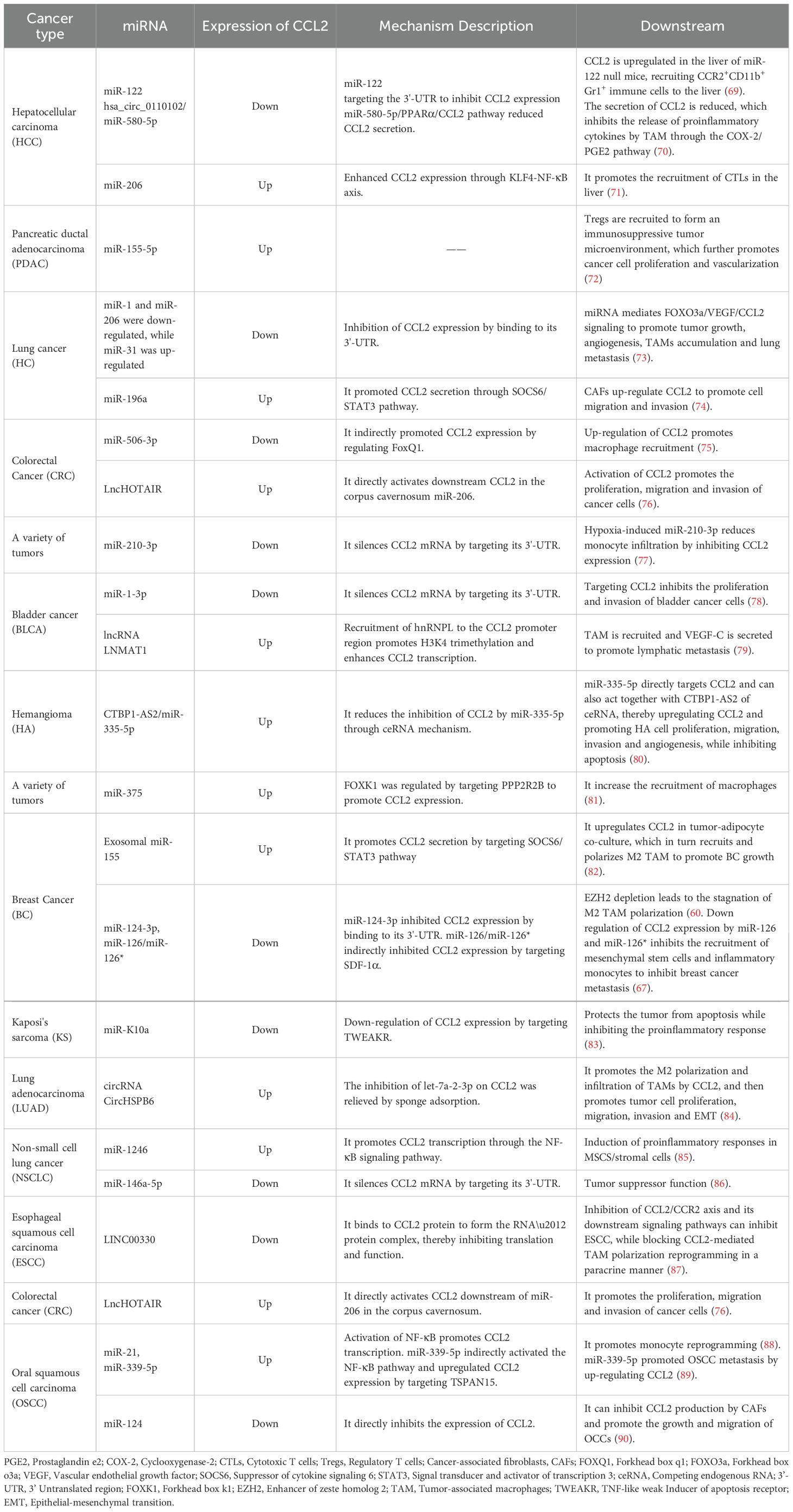
Table 2. Noncoding RNAs regulate CCL2 expression.
3.2 Microenvironmental stimulation regulate CCL2 expression
In addition to the constitutive expression of CCL2 driven by intrinsic genetic variations in cells, the chronic inflammatory state and hypoxic conditions within the TME further promote abnormal CCL2 expression (91). It is worth noting that these factors usually do not act independently but rather precisely regulate the expression profile of chemokines in the TME through synergistic effects. For instance, in papillary thyroid carcinoma (PTC), the combined stimulation of IFN-γ and TNF-α can significantly synergistically upregulate the expression of CXCL9 and CXCL11, activating the anti-tumor immune pathway (92). Therefore, the synergy among inflammatory factors and the positive feedback network form the basis for constructing a complex regulatory network.
3.2.1 Chronic inflammation regulate CCL2 expression
Cytokines including, IL-6, and IL-1β engage in positive feedback interactions with CCL2 in the TME—a phenomenon with important implications for tumor therapy.
TNF-α, a key pro-inflammatory cytokine in tumors (93), is constitutively overexpressed via its receptor on tumor cells, correlating with sustained NF-κB activation and elevated MCP-1/CCL2 expression. In breast and ovarian cancers, the TNF-α secreted by malignant cells potently induces CCL2/MCP-1 expression, with nearly a twofold increase upon TNF-α stimulation compared with its inhibition (94, 95). Moreover, the TNF-α secreted by CCL2-recruited TAMs further amplifies CCL2 production, establishing a reinforcing cycle that perpetuates CCL2 activation (93).
IL-1β, another proinflammatory cytokine belonging to the IL-1 family (96), promotes CCL2 expression in macrophages and tumor cells within the TME (97). The coordinated action of TNF-α and IL-1β leads to a sustained CCL2 release from tumor and endothelial cells (98, 99). Furthermore, IL-1β and CCL2 mutually enhance each other’s production, forming a feedback loop that amplifies macrophage infiltration into tumor tissues (100).
IL-6, a pivotal tumor-promoting cytokine, induces CCL2 secretion via STAT3 activation; the two engage in macrophage-mediated feedback amplification (101, 102). Functionally, IL-6 and CCL2 exhibit synergistic effects: certain p53 protein isoforms enhance cancer cell migration and metastasis by upregulating both cytokines (103). The compound N-2 suppresses tumor growth through its concurrent inhibition of IL-6 and CCL2 via p53 and NF-κB pathways (104). IL-6 and CCL2 induced by CMTM4 upregulate MDSC expansion in the TME, and silencing CMTM4 enhances anti-PD-1 efficacy (105). Oligonucleotide–chitosan complexes suppress IL-6 and CCL2 production and cooperate with p53 to inhibit ATM signaling and tumor progression (106). These findings underscore the therapeutic potential of targeting the IL-6/CCL2 axis to overcome resistance to immunotherapy.
2.2.2 Hypoxia regulates CCL2 expression
Hypoxia is a common feature of solid tumors and a key regulator of the TME (107). Chronic (continuous, uninterrupted) and periodic (intermittent, transient) hypoxia both occur in malignant tumors. In the early stages of tumor development, the inability of blood vessels to provide oxygen to the tumor interior leads to the formation of chronic hypoxic regions (108). In the later stages of tumor development, the abnormal structure of tumor-generated blood vessels leads to periodic hypoxia in different regions inside the tumor (109). Under chronic hypoxia, oxygen-dependent Jumonji histone demethylase activity is reduced, which increases histone methylation in the CCL2 gene promoter, thereby reducing CCL2 expression (110). In addition, hypoxia induces histone deacetylase 1 (HDAC1) activation via CK2 (111). The activated HDAC1 forms a complex with p65 NF-κB and acts as a transcriptional repressor through histone deacetylation to reduce CCL2 expression (112). HIF-1α is activated as a major TF in chronic hypoxia; it can bind to the hypoxia response element (HRE) in the promoter region of CCL2 gene and directly promote CCL2 transcription (113). Hypoxia can also indirectly regulate CCL2 expression. Solid tumor-associated hypoxia can up-regulate the gene expression of the regulator of G protein signaling 2 (Rgs2) in MDSCs, leading to a significant increase in CCL2 (114).
The main pathway of CCL2 production is cyclic hypoxia, where NF-κB activation is the most important mechanism to increase CCL2 expression (91). The transcriptional expression of CCL2 is induced by increasing NF-κB p65/p50 binding activity (115). Hypoxia/reoxygenation (H/R) also induces CCL2 expression in melanoma cells through the synergistic effect of TFs NF-κB and SP1; this process involves the MAPK signaling pathway (91). Cyclic hypoxia can also indirectly regulate CCL2 expression. In THP-1 monocytes, hypoxia indirectly enhances CCL2/MCP-1 expression by up-regulating the receptor for advanced glycation end products through the activation of HIF-1α and NF-κB (116).
In addition to the above common tumor environmental factors, lactate accumulation and growth factors can regulate CCL2 expression in the TME. The binding of lactate to HCAR1 could activate scaffold protein 14-3-3, which in turn activates the STAT3 signaling pathway. The activated STAT3 enhances CCL2 transcription, resulting in increased CCL2 expression (117). In breast fibroblasts, TGF-β signaling inhibits CCL2 expression and negatively regulates TAM recruitment and tumor metastasis (118).
2.2.3 Integrated role of classical transcription factors in CCL2 regulation
By conducting extensive literature review, it has been found that inflammation and hypoxia signals can eventually activate a series of classical TFs, including NF-κB, Sp1, AP-1, STAT3, and p53, which jointly regulate the basal expression of CCL2 gene. Taking these TFs as the core, we systematically sorted out their upstream stimulating factors, downstream effects, and synergistic effects as detailed in Table 3.
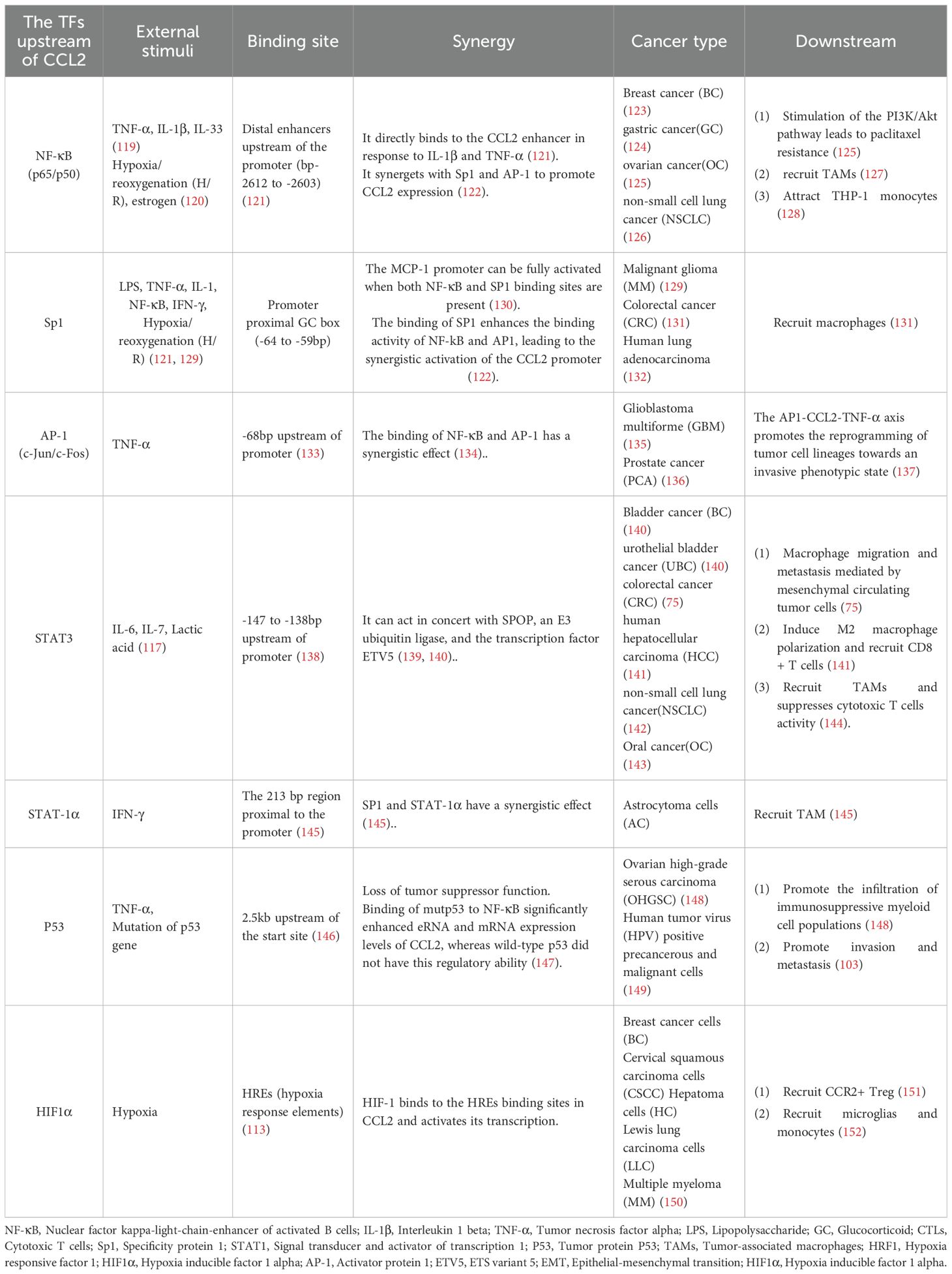
Table 3. The external cellular environment influences CCL2 expression through transcription factors.
Among these TFs, NF-κB plays the most crucial role in CCL2 regulation. NF-κB is located in a distal enhancer approximately 2.5 kbp upstream of the basic promoter. Two specific binding sites can be found in the distal region of the CCL2 gene promoter (153). The far upstream κB binding site (bp −2612 to −2603) is essential for the enhancer activity induced by cytokines such as IL-1β and TNF-α, and its mutation completely abolishes this activity (121).
A complex, bidirectional, positive-feedback regulatory relationship exists between NF-κB and CCL2. On the one hand, NF-κB is a key upstream regulatory factor for CCL2 expression; on the other hand, CCL2 can inhibit the M1 phenotype of TAMs through ZC3H12A, and this process depends on the NF-κB signaling pathway (154). The drug degreen can significantly inhibit the activation of the PI3K/Akt/NF-κB signaling pathway in GBM cells, thereby hindering the disease progression; by contrast, exogenous CCL2 addition can activate the NF-κB signaling and reverse the inhibitory effect of degreen on DBTRG cells (155). In hepatocellular carcinoma (HCC), CCL2 elevated PD-L1 expression by activating the NF-κB pathway (156).
2.2.4 Non-classical transcription factors regulate CCL2 expression
In addition to the classic transcription factor pathways, CCL2 is also regulated by some non-transcription factor factors. PPARγ belongs to the ligand-activated transcription factor of the nuclear receptor superfamily. In anaplastic thyroid carcinoma (ATC) in humans, PPARγ agonists can inhibit the secretion of CCL2 in some tumor cells and simultaneously suppress the activation of NF-κB and ERK1/2 signaling pathways (157). Creb-binding protein (CBP) can interact with the transcription repressor Snail, which increases the promoter activity of CCL2 through the acetylation modification of Snail (158). Insulin-induced activation of the mammalian target of rapamycin complex 1 induces the dephosphorylation of transcription factor FOXK1, which directly binds to the promoter region of the CCL2 gene and promotes its transcriptional activation. This process is independent of the classical NF-κB signaling pathway (159). Twist1 up-regulation in cancer cells activates the transcription of CCL2 mRNA, thus promoting CCL2 expression (160). In breast cancer, estrogen enhances Twist expression by activating ERα and PI3K/AKT/NF-κB signaling, which in turn induces CCL2 autocrine (120). Forkhead box Q1 (FoxQ1) is a major regulator of tumor metastasis. It can bind to the promoter region of Versican V1 gene and induce its expression, ultimately promoting CCL2 secretion (161). FoxQ1 also enhances CCL2 expression by activating Twist1 (162). CCAAT/enhancer binding protein β (C/EBPβ) may interact with AP-1 family members to drive CCL2 transcription (163). Fli-1 is a key member of the Ets TF family and can directly regulate the expression of CCL2 gene by binding to its promoter. The interaction between Fli-1 and p65, a member of the NF-κB family, can enhance the transcriptional activity of CCL2 promoter (164). KRAS gene mutation is one of the most common mutations in human cancers, especially in metastatic cancers such as pancreatic cancer, lung cancer, and colorectal cancer (165). KRAS activation promotes CCL2 expression. In the mouse model of pancreatic ductal adenocarcinoma, KRAS indirectly promotes CCL2 expression by regulating HDAC5 and negative regulator Socs3, thereby shaping the immune cell composition in the TME (63).
3.2 Stromal tumor-cell interactions regulate CCL2 expression
Cells do not act in isolation. They coordinate to complete pathological tasks through interactions and the release of cytokines. Some studies have revealed the regulation of CCL2 production by the interaction between tumor cells and stromal cells, but the mechanisms are mostly unknown and deserve further investigation. In hepatocellular carcinoma (HCC), The indirect co-culture system revealed that TAMs released more CCL2 into the culture medium when they were cultured with HCC cells than when they were cultured alone. NF-κB was also enhanced in the co-culture system, which might be related to the upregulation of CCL2 expression (166). In oral squamous cell carcinoma (OSCC), CCL2 release was significantly increased in the coculture system compared with that in the conditioned medium of OSCC cell lines PCI-13 and LUVA (MCs). But the mechanism of this process remains unknown (23). The interaction between epithelial cells and BC cells significantly elevated CCL2 expression levels by up to 8 folds, possibly by influencing mRNA and post-translational modifications (167). In prostate cancer, tumor-derived CXCL8 induced CCL2 gene expression in WPMY-1 fibroblasts and THP-1 cells (168). TNF-α and IL-1β secreted by stromal cells and BC cells synergistically stimulate mesenchymal stem cells and CAFs to release CCL2 (169). The levels of CCL2 detected in the culture supernatants of cancer-associated adipocytes cocultured with MCF-7 or MDA-MB-231 cells were higher than those in the normal breast adipocytes cocultured with BC cells (170).
4 The role of CCL2 in tumors
Cancer cells establish a microenvironment that facilitates tumor progression by recruiting and reprogramming non-cancerous host cells, as well as remodeling the vasculature and ECM. This dynamic process relies on heterotypic interactions between cancer cells and resident or recruited non-cancer cells in the TME (171). CCL2 plays an important role in the survival, proliferation, migration and colonization of distant organs of cancer cells through paracrine and autocrine mechanisms, which runs through all stages of cancer development (172, 173). A large number of pan-cancer analyses have shown that high levels of CCL2 are associated with more aggressive malignancies, higher risk of metastasis, and poor cancer prognosis (174). The high expression of CCL2 may also be used as a potential diagnostic marker for early detection and prognosis evaluation of tumors (175).
4.1 CCL2 acts in a paracrine and autocrine manner to stimulate cancer cell proliferation and migration
In the TME, CCL2 secreted by cancer cells and stromal cells can activate downstream pathways, up-regulate the expression of anti-apoptotic proteins and cell cycle regulatory proteins to promote the survival and anti-apoptotic ability of tumor cells (Figure 4).
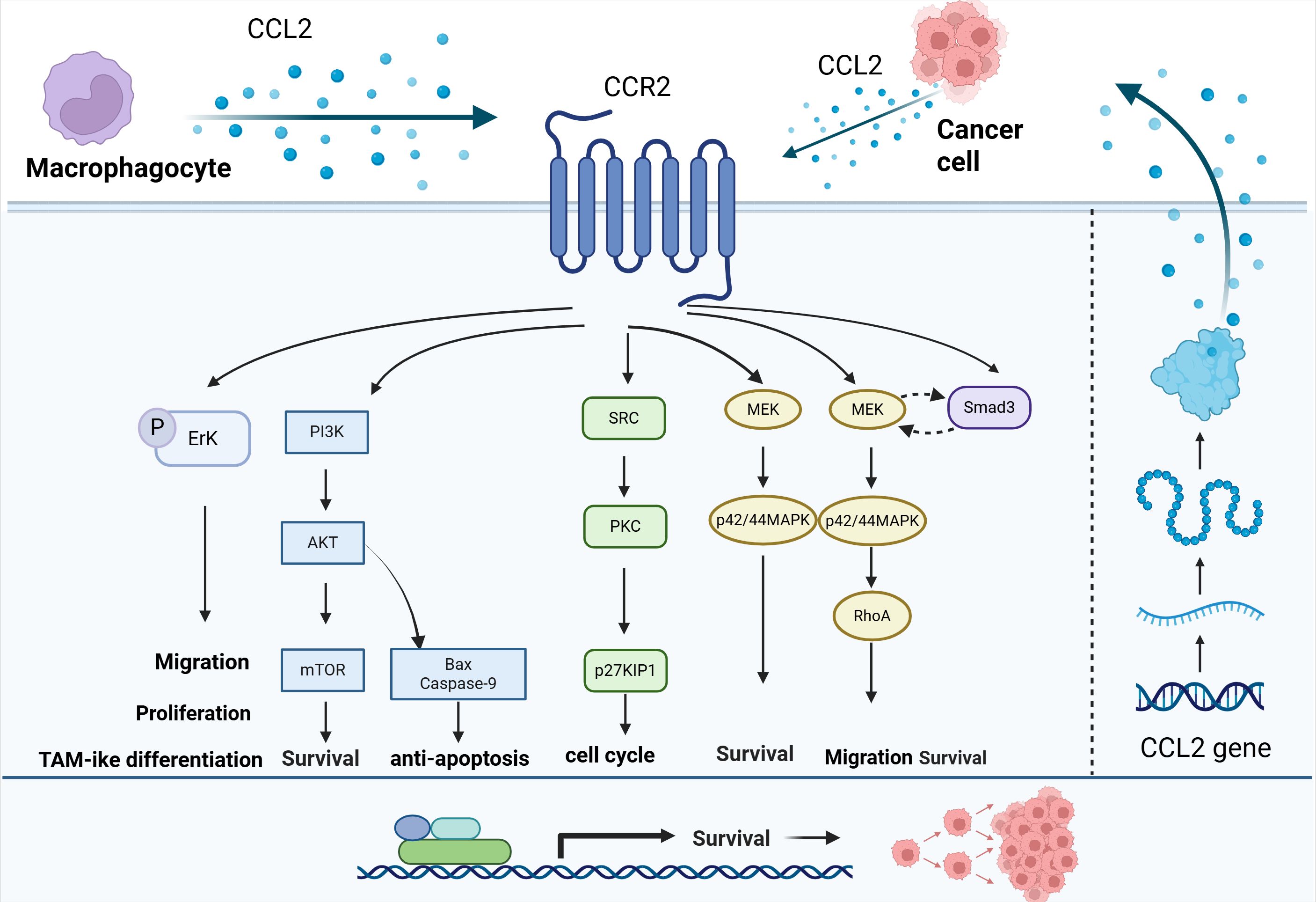
Figure 4. Effect of CCL2 on the proliferation of tumor cells. CCL2 can activate downstream pathways such as PI3K/AKT, SRC/PKC, and MEK-p42/44MAPK through CCR2, up-regulate the expression of anti-apoptotic proteins and cell cycle regulatory proteins, and promote the survival and anti-apoptotic ability of tumor cells.
In the indirect co-culture system of HCC and macrophages, CCL2 secretion was observed to be significantly increased, and the proliferation and migration ability of the two types of cells were promoted via autocrine and paracrine mechanisms. The CCL2-CCR2 axis also regulated the phenotypes of HCC cells and macrophages through downstream Erk (176). Through PI3K/AKT, CCL2 can inhibit the expression of pro-apoptotic proteins Bax and Caspase-9, leading to docetaxel (DTX) resistance in lung cancer (177). CCL2 regulates autophagy in a PI3K/AKT-dependent manner through the mTOR pathway and upregulates the expression of Survivin, conferring a significant survival advantage to cells (178). In BC cells, CCL2 binds to CCR2 receptors to activate Smad3 and p42/44MAPK signaling pathways. Specifically, CCL2 induces MEK signaling independently of Smad3 in the p42/44MAPK pathway, thereby promoting cell survival. In addition, CCL2 also regulates cell motility and survival through a RhoA-dependent mechanism by activating Smad3, allowing it to act synergistically with the MEK-p42/44MAPK pathway (179). In BC, CCL2 activates SRC and PKC signaling pathways through its receptor CCR2, which in turn negatively regulates the expression of cell cycle inhibitory protein p27KIP1 and releases cell cycle inhibition (180).
4.2 CCL2 directly or indirectly promotes angiogenesis
Angiogenesis, the process of developing new blood vessels, is essential for tumorigenesis. Tumor blood vessels are constantly exposed to factors such as vascular endothelial growth factor (VEGF) and angiopoietin (Ang), leading to vascular system disorder and leakage. This affects tumor oxygenation, alters immune cell kinetics, and reduces drug penetration into tumors, thus promoting high proliferation rates and drug resistance in tumors (181).
Recent studies have shown that CCL2 forms a complex pro-angiogenic network by directly acting on vascular endothelial cells or indirectly regulating the functions of immune cells and stromal cells (Figure 5). In vitro experiments, CCL2 can directly promote the tubular structure of human umbilical vein endothelial cells (HUVECs) to form blood vessels without the involvement of inflammatory cells (182). CCL2 also promotes the proliferation and migration of HUVECs through MAPK/ERK1/2/MMP9, PI3K/AKT and Wnt/β-catenin signaling pathways, degrades the ECM and provides space for angiogenesis. At the same time, CCL2 can up-regulate VEGF in endothelial cells to promote angiogenesis (183).
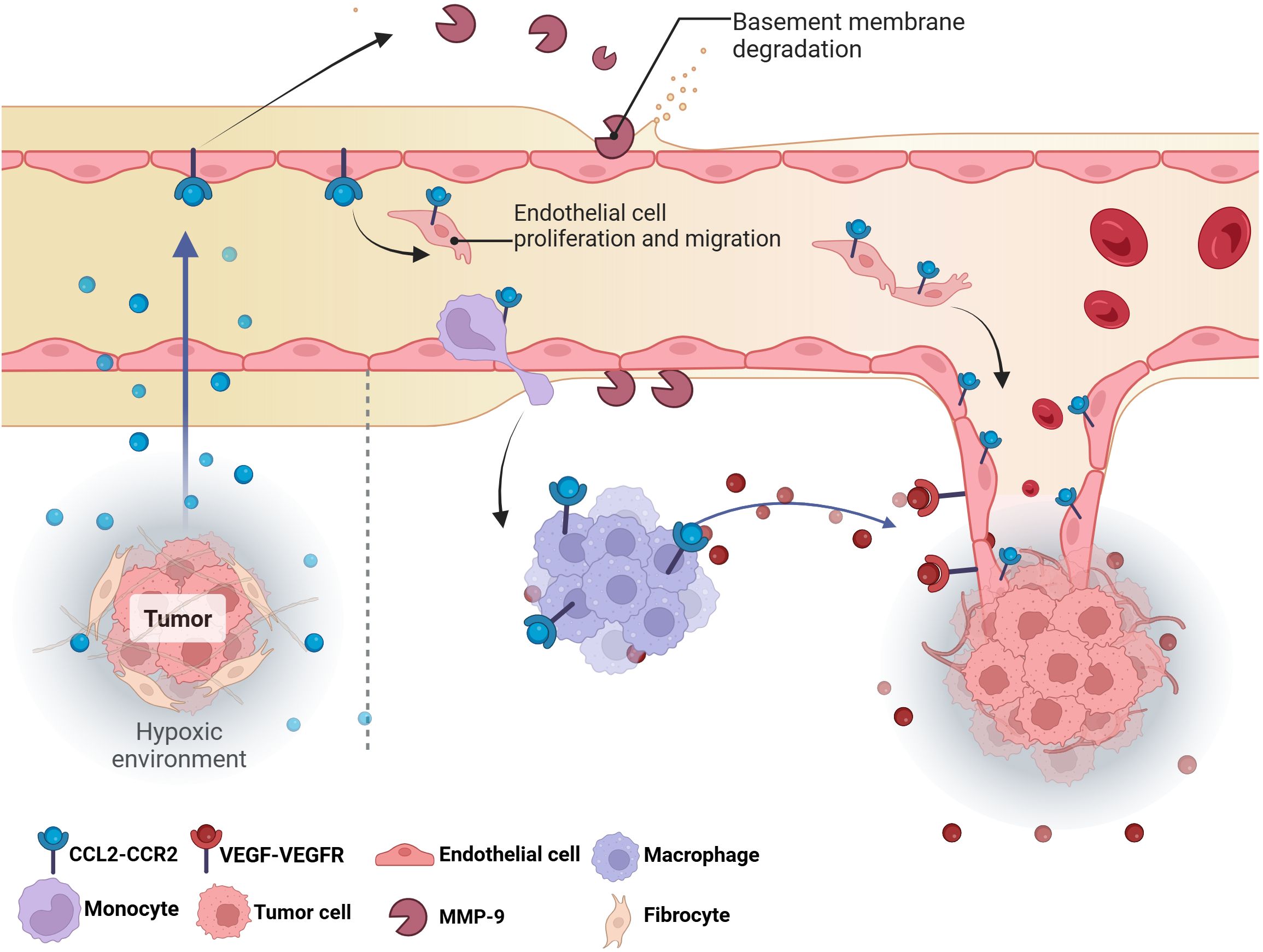
Figure 5. The effect of CCL2 on tumor angiogenesis. The hypoxic environment in tumors triggers the release of CCL2, directly stimulating the proliferation and migration of endothelial cells to form new blood vessels. Meanwhile, macrophages and monocytes are recruited to the tumor site by CCL2, which release MMP-9 and VEGF to degrade the basement membrane and create space for angiogenesis, thereby stimulating angiogenesis.VEGF, Vascular endothelial growth factor; MMP-9; Matrix metalloproteinase-9.
In addition to directly acting on endothelial cells, CCL2 can also act on helper cells in the TME to indirectly regulate angiogenesis. Most of the stromal cells in tumor tissue are fibroblasts. These cells are the main source of CCL2 (184). Additionally, immune cells, especially macrophages and neutrophils, are also key sources of angiogenesis related chemokines, growth factors and proteases (185). CCL2 is a key chemotactic factor for monocytes/macrophages (185). In vitro experiments showed that upregulation of Twist1 in cancer cells activated CCL2 mRNA transcription; A chemotactic gradient is generated by the secretion of CCL2 protein to promote macrophage infiltration and subsequent angiogenesis stimulation (160). These TAMs amplify pro-angiogenic signals by secreting VEGF, FGF, IL-8 and other pro-angiogenic factors, releasing MMP-9 to reconstitute the extracellular matrix (186).
Tissue hypoxia is a characteristic of tumor tissue and the main trigger of angiogenesis (107). Many molecules such as VEGF that respond to hypoxia can promote angiogenesis. As mentioned above, both chronic hypoxia and cyclic hypoxia can significantly up-regulate the expression of CCL2, and a large amount of CCL2 will escort the angiogenesis, forming a vicious cycle. Therefore, CCL2 constructs a multi-layered pro-angiogenic network by directly activating endothelial cell signaling pathways and indirectly regulating TAMs and pro-angiogenic factors.
4.3 CCL2 is involved in tumor metastasis
Metastasis is the leading cause of cancer-related death in humans (187). Tumor metastasis begins with local invasion. Tumor cells alter their intrinsic characteristics, degrade the extracellular matrix, modulate the immune microenvironment, and acquire invasive capabilities. Subsequently, tumor cells intravasate into blood vessels or lymphatic vessels and inhibit anoikis to survive in the circulation. They then prepare for colonization by forming a premetastatic niche (PMN) in distal organs. Tumor cells then extravasate from blood vessels, proliferate in distant organs, form metastases, and harvest nutrients through angiogenesis. Eventually, micrometastases continue to grow, evade immune attack, and develop into detectable metastases (15). The role of CCL2 in metastasis is pleiotropic, and it is associated with many metastatic steps, such as local invasion, intravasation, PMN formation, anoikis, extravasation, chemotaxis, and colonization (188).
3.3.1 Epithelial-mesenchymal transition (EMT) and local invasion
Once the tumor has successfully formed blood vessels, it can enter the next stage of disease progression, namely local invasion. Aggressive growth is one of the important characteristics of cancer, separating from neighboring cancer cells by breaking the adhesion between epithelial cells and tumor cells, accompanied by an EMT-like state (15).
CCL2 regulates the expression of EMT markers and promotes the expression and activity of matrix metalloproteinases through downstream signaling pathways. This contributes to increased tumor cell motility and invasiveness, ultimately facilitating local tumor invasion and distant metastasis (Figure 6). Recent studies have shown that CCL2 expression can predict clinical outcomes, activate the EMT pathway and angiogenesis in pituitary tumors (189). CCL2 secreted by TAMs can activate the PI3K/Akt signaling pathway, increase the expression of β-catenin and bind to TCF/LEF transcription factors, resulting in the down-regulation of epithelial markers E-cadherin and up-regulation of interstitial markers such as N-cadherin and Vimentin, which endow cancer cells with the ability of migration and invasion (190). CCL2 also induces ERK/GSK-3β/Snail signaling to promote EMT and the migration of MCF-7 human breast cancer cells (191). M2-like TAMs activate the MEK/ERK/ELK1/Snail signaling pathway in gallbladder cancer (GBC) cells by secreting CCL2, thereby promoting EMT, stemness and metastasis of GBC (192). In vitro experiments, CCL2-CCR2 axis was found to induce invasion and EMT of hepatocellular carcinoma in vitro by activating Hedgehog signaling pathway (193). In the pre-metastatic microenvironment, CCL2 secreted by cancer cells and bone marrow cells attracts and induces M2-TAM and other cells to activate Wnt/β-catenin signaling pathway, which destroys the E-cadherin junction between early cancer cells. Prior to tumor formation, these processes facilitate the establishment of an early disseminated microenvironment, enhance the active invagination of tumor cells and their entry into the circulatory system, and ultimately facilitate metastasis to distant organs (194). In addition, pharmacological inhibition of CCL2/CCR4/Vav2/Rac1/MLC signaling pathway can effectively reduce the motility of cancer cells, thereby inhibiting local invasion and distant lymph node metastasis in head and neck squamous cell carcinoma (HNSCC) cells (33). These results suggest that CCL2-CCR4 may serve as a potential therapeutic molecular target by inhibiting tumor invasion and metastasis.
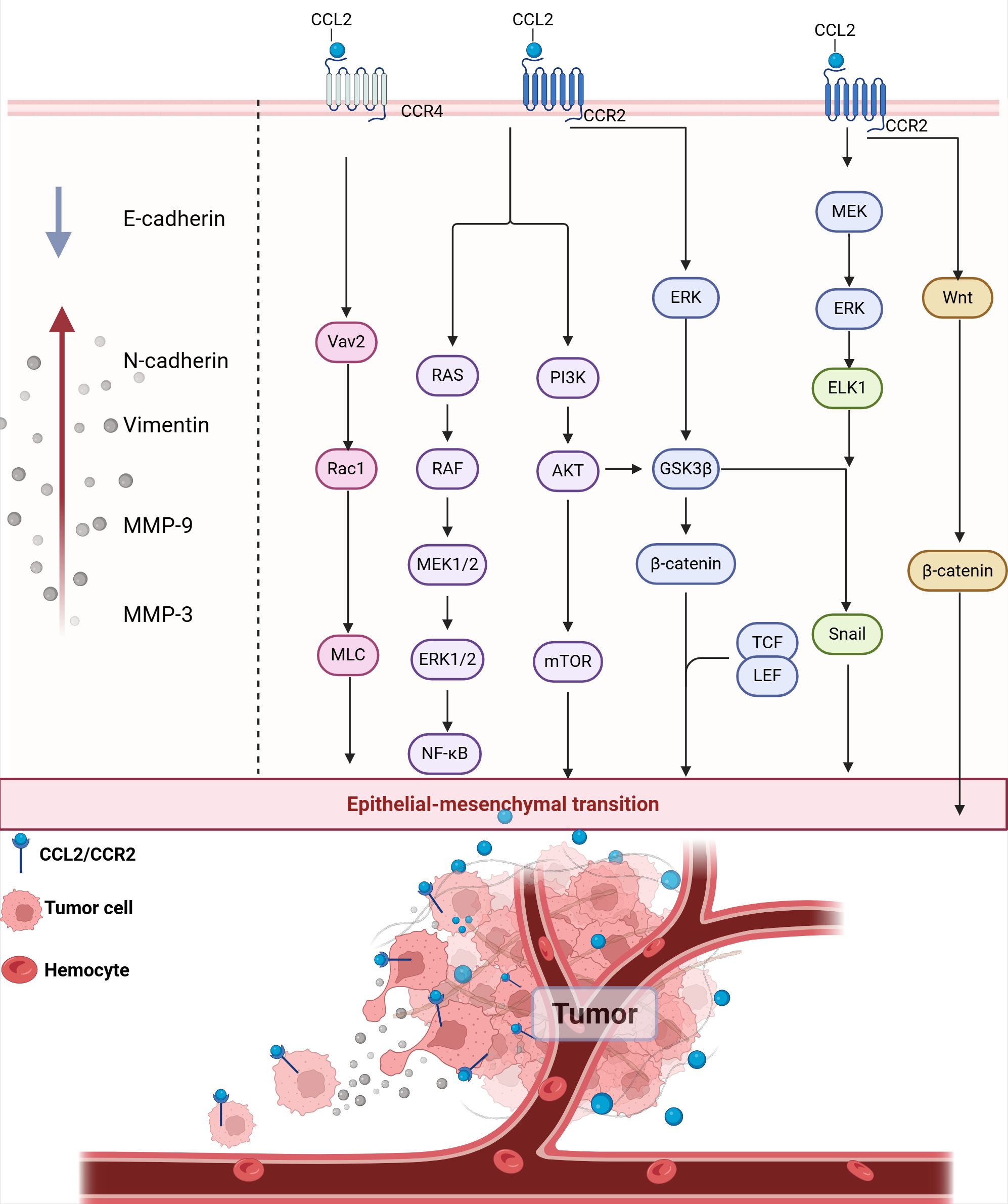
Figure 6. CCL2 promotes EMT and local invasion. CCL2 binding to CCR2 activates multiple signaling pathways, including the RAS/RAF/MEK1/2, PI3K/AKT/mTOR, and ERK pathways. These pathways converge to activate transcription factors such as NF-κB, Snail, TCF/LEF, and β-catenin, which promote the expression of EMT markers. E-cadherin, Epithelial Cadherin; N-cadherin, Neural cadherin; MMP-3, Matrix metallopeptidase 3; MMP-9, Matrix metallopeptidase 9.
Matrix metalloproteinase (MMP) can degrade the extracellular matrix and basement membrane through its proteolytic activity, make cancer cells transition from epithelial to mesenchymal state, and enhance cell motility and invasion ability. MMP also creates conditions for tumor cells to enter blood vessels or lymphatic vessels by destroying the vascular endothelial cells layer (195). CCL2-CCR2 axis can induce EMT by promoting the recruitment of MMP in the extracellular matrix, which leads to the degradation of tight junction proteins and basement membranes. In non-small cell lung cancer (NSCLC), CCL2 reduces E-cadherin levels and upregulates vimentin, MMP-2 and MMP-9 protein levels through PI3K/Akt/mTOR to activate EMT (196). In GBM induced by the mitochondrial pro-apoptotic protein BV6, NF-κB activation induces the upregulation of CCL2 and triggers the expression of MMP-9 in an autocrine/paracrine manner, promoting the migration and invasion of GBM cells (197). CCL2 also increases the level of MMP-9 in human chondrosarcoma cells via the Ras/Raf/MEK/ERK/NF-κB signaling pathway to promote cell motility (198). In vivo and in vitro experiments with pulmonary metastatic osteosarcoma showed that CCL2 enhances MMP-3 dependent cell movement by inhibiting miR-3659 synthesis (199).
3.3.2 Immunosuppression and Pre-metastatic Niche formation
The PMN refers to a specialized microenvironment that facilitates the colonization and growth of tumor cells in the distal organ. This is achieved through the secretion of factors and extracellular vesicles by the primary tumor prior to the arrival of tumor cells at the distal site. This concept was first proposed by Kaplan et al., in 2005 (200).
CCL2 provides support for tumor cell metastasis by not only activating and recruiting immunosuppressive cells, but also by directly inhibiting immune cell activity, upregulate the expression of immune checkpoints, acting together on the formation of the PMN (Figure 7). As a regulator of T cells, CCL2 regulates the differentiation of T helper 2 cells (Th2 cells) into more immunosuppressive regulatory T cells (Treg cells) (201). CCL2 can also recruit bone MDSCs and regulatory Treg cells through CCR2 and CCR4 receptors, and the two synergistically inhibit the clearance of glioma cells by cytotoxic T cells (202). In the lung metastasis model of renal cell carcinoma, CCL2 secreted by lung macrophages can attract TAM and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC), and the aggregation of these immunosuppressive cells forms an immunosuppressive and metastatic microenvironment (203). Under hypoxic conditions, activation of the lactate receptor HCAR1 signaling pathway induces the expression of CCL2 and CCL7 in colorectal tumor cells, which then attracts immunosuppressive CCR2+ PMN-MDSCs into the TME (117). CCL2 also activates the STAT3 signaling pathway by binding to the receptors on the surface of PMN-MDSCs, thereby regulating the expression of calcium-binding proteins gp91phox, S100A8 and S100A9, enhancing the production of ROS and inhibiting the ability of T cells (204). Furthermore, CCL2 can also suppress cytotoxic NK cells in the pre-metastatic niche, helping triple-negative breast cancer cells evade immune surveillance and thereby promoting their metastasis (205). In gastric cancer, CCL2 + fibroblasts activate M2-TAM via STAT3 while inhibiting T cell activation and also inducing the production of inhibitory TME (206).
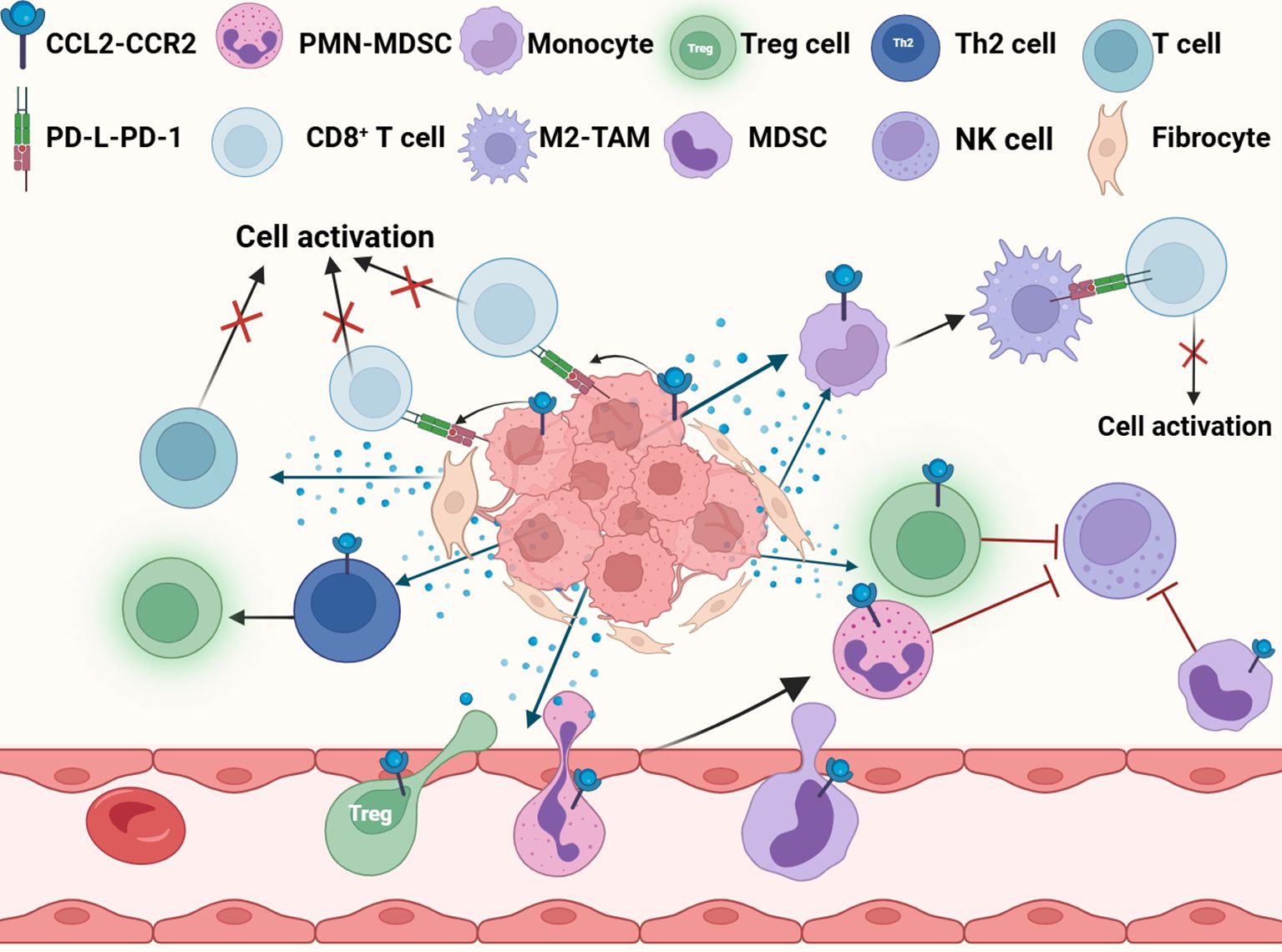
Figure 7. CCL2 promotes immunosuppression. CCL2 fosters the establishment of an immunosuppressive microenvironment by recruiting immunosuppressive cells, suppressing immune cell activity, and upregulating PD-L1 expression in tumor cells. M2-TAM, M2-tumor associated macrophage; Treg cell, Regulatory T cell; MDSC, Myeloid-derived suppressor cell; PMN-MDSC, Polymorphonuclear myeloid-derived suppressor Cells; NK cell, Natural killer cell.
Additionally, CCL2 induces the upregulation of programmed death ligand 1 (PD-L1) and programmed death ligand 2 (PD-L2) in tumor cells and immune cells, and inhibit the activation of CD8+ T cells mediated by PD-1 antibodies. CCl2-induced M2 polarization increases the expression of PD-L1 and PD-L2 in TAMs, which transmit inhibitory signals to CD8 + T cells through PD-1 signaling and helps tumor cells escape the attack of the immune system (207, 208). Even when PD-L1 is completely blocked, CCL2 release can still counteract the effect of PD-1/PD-L1 inhibitors by recruiting immunosuppressive cells into the TME (209).
3.3.3 Tumor cell extravasation
Tumor Cell Extravasation refers to the process by which circulating tumor cells (CTCs) pass through the endothelial cell layer from blood vessels or lymphatic vessels and enter distant organs or tissues (210). CCL2 secreted by tumor cells promotes cancer cell extravasation through a dual mechanism (Figure 8) (1): CCL2 attracts CCR2+ Ly6C high expressing monocytes, which are used by tumor cells as carriers across the vessel wall (2); CCL2 enhances the permeability of local blood vessels through endothelial cells (210).
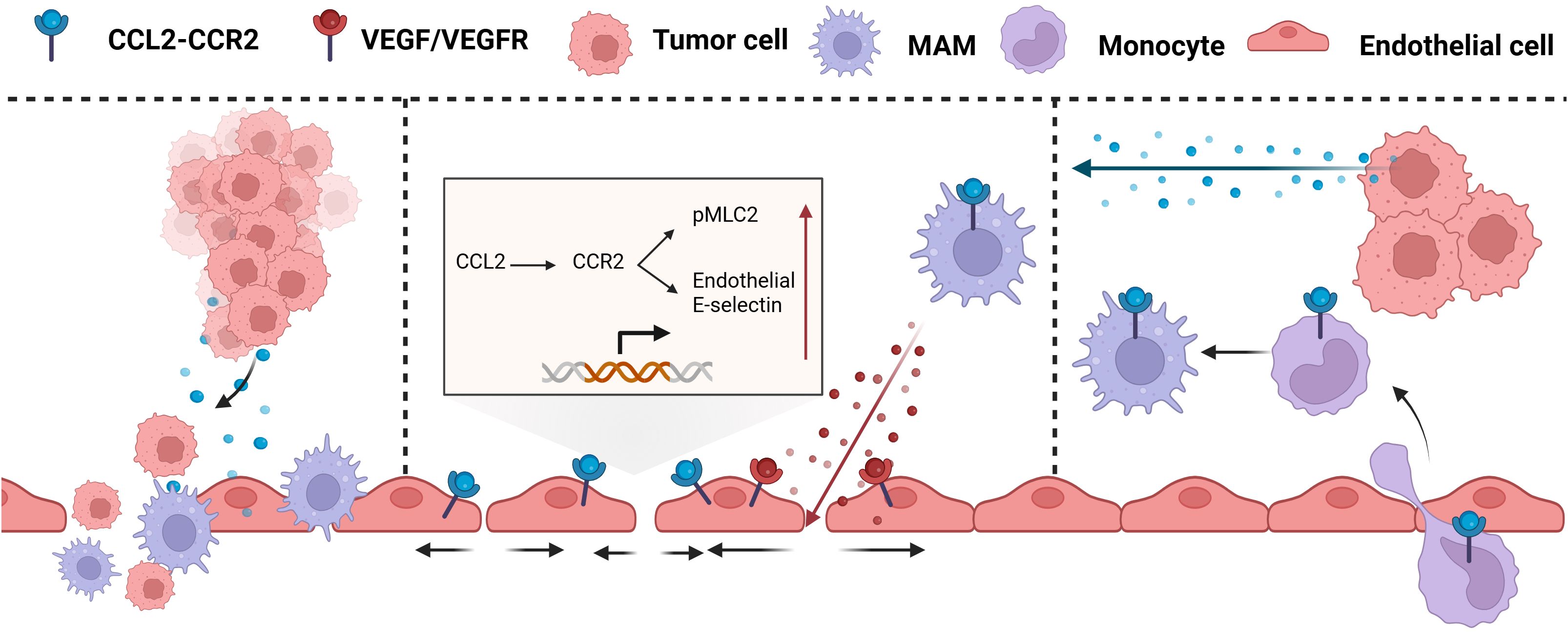
Figure 8. CCL2 promotes tumor extravasation. CCL2 chemotactic CCR2+ Ly6C high expression monocytes act as carriers through the vascular wall and enhance local vascular permeability through endothelial cells. MAM, Mammalian Target of Rapamycin; VEGF, Vascular Endothelial growth factor; VEGFR, Vascular endothelial growth factor receptor.
At the site of metastasis, inflammatory monocytes, their derivatives, and metastasis-associated macrophages (MAMs) are critical in facilitating the extravasation and proliferation of metastatic tumor cells. Inflammatory monocytes, which highly express CCR2 and respond to CCL2 produced by tumor and stromal cells, are recruited to metastatic sites such as the lungs and liver before tumor cells and differentiate into MAMs, secreting VEGF and promoting tumor cell metastasis (211, 212). The CCL2-induced CCL3 cascade also prolongs the retention time of MAMs at the tumor site, thereby accelerating the extravasation of tumor cells (213). However, it has been demonstrated in mouse models of colon cancer lung metastasis that CCL2-induced high expression of Ly6C monocytes is a necessary but not sufficient condition for effective metastasis. The loss of CCR2 in monocytes reduces but does not prevent metastasis, while the absence of CCR2 in endothelial cells prevents lung metastasis (214).
Endothelial retraction is a key step for tumors to undergo intravasation and extravasation, which promotes tumor metastasis by enhancing the contraction of vascular endothelial cells and intercellular space. In the lung metastasis model, CCL2 stimulates pulmonary endothelial cells, leading to the phosphorylation of myosin light chain (MLC2) and the upregulation of E-selectin expression on endothelial cells. This process triggers cytoskeletal rearrangement and facilitates chemotactic recruitment, adhesion, and activation of monocytes, thereby shaping the TME. Additionally, it enhances vascular permeability, which is essential for initiating metastasis, and promotes the tranendothelial migration of tumor cells (215). Furthermore, CCL2 derived from tumor cells can activate the CCR2 receptor on vascular endothelial cells, inducing the JAK2/STAT5 and p38MAPK signaling pathways. The two pathways act in concert to increase (214).
In conclusion, CCL2 acts as a core driver of tumor metastasis by regulating EMT, ECM degradation, vascular leakage, and premetastatic niche formation. CCL2 plays a crucial role throughout the entire process of metastasis, including initiation, progression, and colonization, forming a complex interaction network with immune cells and stromal cells within the TME.
5 The clinical efficacy and potential breakthrough areas of CCL2
Previous literature has systematically described the current status of clinical treatment of CCL2 in tumors (18, 216). this article will focus on the clinical efficacy and extension of targeting CCL2.
5.1 The clinical efficacy and limitations of CCL2
Immunomodulatory antibodies and small molecule inhibitors targeting cancer cells have shown definite efficacy in a variety of solid tumors such as breast cancer and lung cancer (217). Among them, neutralizing antibodies are still the main means to interfere with CCL2/CCR2 signaling axis. In theory, CCL2 blockade can simultaneously inhibit the recruitment of immunosuppressive cells and enhance the infiltration of killer T cells to achieve tumor suppression, but the clinical benefit of targeting the CCL2/CCR2 axis is still pending.
In the case of S0916 (MLN1202; prozolizumab; TAK-202), a humanized IgG1 monoclonal antibody targeting CCR2, the results of a phase II study (NCT01015560) in bone metastases or solid tumors have not been published, and its anticancer activity or potential toxicity cannot be confirmed at this time (218). Another phase 1 study in advanced melanoma (NCT02723006) was stopped early because of a 58% incidence of serious adverse events (219). In contrast, the CCR2 inhibitor CCX872 showed benefit over single-agent chemotherapy in combination with FOLFIRINOX in a phase Ib trial of pancreatic cancer (220).
Carlumab (CNTO888), the first human recombinant monoclonal antibody (IgG1κ) to enter the clinic, significantly reduced the infiltration of tumor-associated macrophages and inhibited tumor growth and metastasis in preclinical models. However, the clinical efficacy of Carlumab is far from expected. In a phase I trial in patients with advanced solid tumors (NCT00537368), free CCL2 was only transiently inhibited after the first dose, followed by a rapid rebound and above baseline levels (221); Total CCL2 (including complexes) can increase up to thousfold, and no clear clinical efficacy has been observed (221). In another phase I study (NCT01204996), Carlumab combined with chemotherapy failed to enhance the efficacy of chemotherapy and did not prolong the inhibition of free CCL2 (222). In a phase II trial in metastatic castration-resistant prostate cancer (NCT00992186), free CCL2 showed a trend of rapid rebound despite a manageable safety profile (223). Pharmacokinetic analysis further revealed that Carlumab could not achieve sustained CCL2 neutralization due to its large fluctuation in blood concentration and short half-life (about 2.4 days) (224). In addition, studies in breast cancer models have shown that withdrawal of CCL2 inhibitors triggers massive monocyte mobilization, cancer cell activation, and IL-6/VEGF-A-driven metastatic recurrence, whereas concurrent blockade of CCL2 and IL-6 significantly inhibits metastasis and extends survival (225). In summary, the reasons why CCL2 neutralizing antibodies have been frustrated in clinical trials can be summarized as follows:
1. Poor pharmacokinetics and rebound effect: the antibody has insufficient affinity with CCL2 and the complex is easy to dissociate; At the same time, the body’s synthesis rate of CCL2 is extremely fast, which together lead to the unstable maintenance of blood drug concentration, and the level of CCL2 often rebound sharply after drug administration.
2. Limitations of the mechanism of action: Neutralization strategies can only temporarily block extracellular CCL2 but cannot silence CCL2 at its source, thus failing to restore the homeostasis of CCL2 in the tumor microenvironment.
3. Signaling compensatory network: A single CCL2 blockade triggers a wide range of compensatory mechanisms. This involves not only the compensatory upregulation of CCL2, but is frequently accompanied by increased levels of other key inflammatory factors, such as IL-6 and TNF-α, leading to the establishment of one or more alternative inflammatory pathways that bypass the initial therapeutic targets.
This is the common problem of all neutralizing antibody therapy, and it is also the breakthrough point of precision medicine.
5.2 Future development direction
Although the current strategies to target CCL2 face challenges, emerging studies are attempting from the aspects of source intervention and synergistic blockade, which provide new directions for overcoming the existing limitations.
5.2.1 Interfering with CCL2/CCR2 signaling axis at the source
Source inhibition at the level of gene expression is a feasible strategy to achieve long-term blockade. For example, the Ca-TAT nanocomplexes are formed through non-covalent cross-linking mediated by calcium ions, which enable the complexation of siRNA with TAT cell-penetrating peptides. Topical delivery of CCL2 siRNA via Ca-TAT nanocomplexes can achieve efficient and durable gene silencing in tumor cells with minimal effect on normal tissues. Similarly, siRNA delivery using polyethylenimide-coated mesoporous silica nanoparticles effectively silenced TWIST1 and inhibited CCL2 expression, thereby inhibiting tumor growth in an in vivo model. In addition to nucleic acid drugs, small molecule drugs such as Bindarit can reduce the synthesis of CCL2 by inhibiting AKT and NF-κB pathways, showing potential inhibitory effects on tumor progression and metastasis (226). The sicCR2-encapsulated nanoparticles (CNP/siCCR2) can effectively silence the CCR2 expression of macrophages, inhibit the secretion of IL-10 and TGF-β immunosuppressive factors, and improve the cytotoxic efficacy of adriamycin. It can significantly inhibit the function of CCL2 and modify the immunosuppressive tumor microenvironment (227). In addition, targeted regulation of upstream signaling nodes is also an important direction. TNF-α inhibitors such as etanercept and adalimumab can inhibit the expression of CCL2 in monocytes through MAPK, NF-κB and epigenetic pathways. microRNA-206 can inhibit the induced expression of CCL2 in tumor-associated macrophages and cytotoxic T lymphocytes by regulating the Kruppel-like factor 4 (Klf4) transcription factor.
5.2.2 Synergistic blocking and combined treatment strategies
In view of the redundancy and compensatory mechanism of CCL2 signaling network, combined targeting of its cofactors has become the key to improve the therapeutic effect. In the pancreatic cancer model, although inhibition of CCR2 alone can reduce TAM infiltration, it leads to a compensatory accumulation of CXCR2+ TAN. Meanwhile, simultaneous blockade of CCR2 and CXCR2 can synergistically inhibit tumor and enhance chemotherapy response (228). The synergistic axis formed by IL-6 and CCL2 is of particular interest: both of them are often up-regulated functionally by p53 isoforms to promote metastasis (103). Compound N-2 could inhibit tumor growth by synergistically inhibiting the p53 and NF-κB pathways and simultaneously reducing the levels of IL-6 and CCL2 (104). CMTM4 could also induce both CCL2 and IL-6 to promote the expansion of MDSCs, while its knockdown enhanced the anti-PD-1 efficacy (106). In recurrent head and neck squamous cell carcinoma, combined blockade of IL-6 and CCR2 can significantly enhance NK cell activity, and the effect is better than that of single pathway inhibition (229), suggesting that this combined strategy has the potential to reverse the mask transformation of immunotherapy resistance.
In addition, CCL2 also synergetic with HGF/MET pathway to promote the progression and metabolic reprogramming of breast cancer, and the combination of CCR2 knockout and MET inhibitor meritinib can more effectively inhibit tumor growth and stromal response (230). At the same time, CCL2 is also involved in the resistance mechanism of PD-1/PD-L1 inhibitors: when PI3K/AKT and NF-κB pathways jointly activate CCL2 and PD-L1, even if PD-L1 is blocked, the release of CCL2 may still lead to treatment failure. Dual inhibition of PD-L1 and CCL2 could reverse the drug resistance phenotype. On the basis of this, a phase I clinical trial (NCT02723006) evaluating the combination of CCR2 inhibitors and nivolumab in the treatment of advanced melanoma is ongoing to verify the synergistic effect of combined blockade of CCR2 and immune checkpoints in activating CD8+ T cells and overcoming immunosuppression (209).
It must be pointed out that the majority of the aforementioned therapies are still in the preclinical research stage. Section 2 of this article attempts to systematically explain the upstream regulatory mechanisms of CCL2 in tumors, from gene transcription, mRNA regulation to protein expression and secretion, and even the complex downstream synergistic effects. For this reason, each link in this pathway constitutes a potential intervention target. Future research should avoid the limitations of single therapy: first, to deeply analyze the upstream generation mechanism of CCL2 to develop more fundamental intervention methods; second, to clarify its downstream compensatory and synergistic networks, laying a theoretical foundation for the design of effective combination therapies.
6 Conclusions and future perspectives
This work systematically reviews the production mechanism of CCL2 in a variety of tumors and its multifaceted role in the TME, focusing on its upstream regulation and downstream effects. The aim is to comprehensively clarify the production, regulation, and key biological processes of CCL2 in the TME and provide theoretical basis and potential targets for the development of CCL2 targeted therapy.
CCL2 is continuously produced by tumor cells and stromal cells through three main pathways: constitutive secretion, microenvironmental stimulation response and tumor-stroma interaction. Among them, SNPs, epigenetic regulation and noncoding RNA regulation are the core mechanisms of CCL2 constitutive expression. External inflammatory factors in the TME (such as TNF-α, IL-1β, and IL-6) and hypoxia signals further amplify the secretion of CCL2 by activating NF-κB and other transcription factor pathways. This continuous release of CCL2 reshapes the immune microenvironment by recruiting TAMs and MDSCs and forms a positive feedback loop through autocrine and paracrine signals. These interesting crosstalks are the reasons for breaking CCL2 homeostasis and should be considered for future treatments.
As a “good soldier” of tumor cells, the activation of the CCL2–CCR2 axis directly induces the expression of EMT-related transcription factors and enhances the invasion ability of cancer cells by regulating PI3K/Akt, MAPK, NF-κB, and other signaling pathways. CCL2 also promotes the secretion of MMPs and the release of VEGF, accelerates the degradation of ECM and angiogenesis, and creates conditions for the intravasation of tumor cells and distal colonization of CTCs. In PMN formation, CCL2 recruits immunosuppressive cells and inhibits the activity of NK and other immune cells, providing a “protective umbrella” for the immune escape of metastases.
To date, the downstream effects mediated by CCL2 have been clearly elucidated. In clinical intervention strategies, small-molecule inhibitors and neutralizing antibodies are mainly used to block the CCL2–CCR2 signaling axis (221, 231). However, these monotherapies have not significantly improved the prognosis of patients. In preclinical studies and clinical trials, the use of CNTO888 against CCL2 or MLN1202 antihuman antibodies against CCR2 in different cancer types have only achieved short-term and hardly predictable anticancer responses (18). These negative outcomes can be attributed to its complex upstream regulatory network and compensatory mechanisms.
Recent studies have shown that targeting the upstream signals of CCL2 modified by lactic acid and the STAT3 axis overcomes the immunotherapy resistance of pancreatic ductal adenocarcinoma (144). microRNA-206 regulates CCL2 expression through mRNA and promotes the recruitment of cytotoxic T lymphocytes (CTLS) via CCR2 in the liver. Targeting microRNA-206 inhibits its CCL2 induction in macrophages (71). Moreover, the combination of CCL2 inhibition with an immunomodulator targeting PD-1/PD-L1 has shown more favorable results than each monotherapy (32). These studies suggest that silencing CCL2 at its source (such as mRNA interference, siRNA, epigenetic modulation, or promoter inhibition), and combined with immunomodulatory agents to disrupt its positive feedback loop with PD-L1, TNF-α, IL-1β, and IL-6, may constitute a potentially effective cancer treatment strategy.
In summary, the mechanism of CCL2 in the TME is complex and multifaceted. Therapeutic strategies targeting CCL2 still require a deep understanding of its upstream and downstream mechanisms in tumors, and effective intervention can be achieved only by fully grasping the complexity of its function. The development of combined strategies or source regulation of CCL2 may be the direction of future research.
Author contributions
YJ: Writing – original draft. RY: Writing – review & editing. YW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by National Natural Science Foundation of China (NO.82205132). Fundamental Research Program of Shanxi Province (NO.202203021212065). Cancer Hospital of Chinese Academy of Medical Sciences, Shanxi Hospital, Doctoral Talents Foundation(NO.QH2023006). Shanxi Province Traditional Chinese Medicine Research (NO.2024ZYY2D025). Shanxi Cancer Hospital Doctoral Research Foundation (NO.Dr202312). College Students’ Innovation Training Program Project (NO: 20250408).
Acknowledgments
All Figures were Created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
AP-1: Activator protein 1
Ang: Angiopoietin
BC: Breast cancer
BLCA: Bladder cancer
CCR2: C-C chemokine receptor type 2
CAF: Cancer-associated fibroblasts
CBP: Creb-binding protein
CCL2: Chemokine ligand 2
CRC: Colorectal cancer
CTCs: Circulating tumor cells
CNP/siCCR2: sicCR2-encapsulated nanoparticles
DADS: Diallyl disulfide
DNMT1: DNA methyltransferase 1
DTX: Docetaxel
EMT: Epithelial-mesenchymal transition
ECL2: The second extracellular loop
ESCC: Esophageal squamous cell carcinoma
FAP: Fibrocell-activating protein
FoxQ1: Forkhead box Q1
GBC: Gallbladder cancer
HA: Hemangioma
HCC: Hepatocellular carcinoma
HDAC1: Histone deacetylase 1
HDAC5: Histone deacetylase 5
hFOB: Human fetal osteoblasts
HBMECs: Human bone marrow endothelial cells
HUVECs: Human umbilical vein endothelial cells
HNSCC: Head and neck squamous cell carcinoma
HRE: Hypoxia response element
Il-1β: Interleukin-1β
IL-6: Interleukin-6
JHDM: Jumonji histone demethylase
KS: Kaposi’s sarcoma
LC: Lung cancer
LUAD: Lung adenocarcinoma
LncRNA: Long non-coding RNA
LPS: Lipopolysaccharide
MAPK: Mitogen-activated protein kinase
MAMs: Metastasis-associated macrophages
MCP-1: Monocyte chemoattractant protein-1
MDSCs: Myeloid-derived suppressor cells
MCs: Mast cells
MLC2: Myosin light chain
MMP: Matrix metalloproteinase
NBAs: Normal breast adipocytes
NF-κB: Nuclear factor kappa-B
NK: Natural killer
NSCLC: Non-small cell lung cancer
OSCC: Oral squamous cell carcinoma
PDAC: Pancreatic ductal adenocarcinoma
PD-L1: Programmed death ligand 1
PD-L2: Programmed death ligand 2
PMN: Premetastatic niche
PMN-MDSC: Polymorphonuclear myeloid-derived suppressor cells
PTHrP: Parathyroid hormone-related protein
RAGE: Receptor for advanced glycation end
Rgs2: Regulator of G protein signaling 2
SNPs: Single nucleotide polymorphisms
Sp1: Specific Sequence Protein 1
STAT: Signal transducer and activator of transcription
TAM: Tumor-associated macrophages
TME: Tumor microenvironment
TNF-α: Tumor necrosis factor-α
TM3: Third transmembrane domain
Treg cells: T cells
VEGF: Vascular endothelial growth factor
VSMCs: Vascular smooth muscle cells
3’UTR: 3’
References
1. De Visser KE and Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
2. Hanahan D and Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
3. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
5. Matsushima K, Larsen CG, DuBois GC, and Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med. (1989) 169:1485–90. doi: 10.1084/jem.169.4.1485
6. Zachariae CO, Anderson AO, Thompson HL, Appella E, Mantovani A, Oppenheim JJ, et al. Properties of monocyte chemotactic and activating factor (MCAF) purified from a human fibrosarcoma cell line. J Exp Med. (1990) 171:2177–82. doi: 10.1084/jem.171.6.2177
7. Manome Y, Wen PY, Hershowitz A, Tanaka T, Rollins BJ, Kufe DW, et al. Monocyte chemoattractant protein-1 (MCP-1) gene transduction: an effective tumor vaccine strategy for non-intracranial tumors. Cancer Immunol Immunother. (1995) 41:227–35. doi: 10.1007/BF01516997
8. Huang S, Singh RK, Xie K, Gutman M, Berry KK, Bucana CD, et al. Expression of the JE/MCP-1 gene suppresses metastatic potential in murine colon carcinoma cells. Cancer Immunol Immunother. (1994) 39:231–8. doi: 10.1007/BF01525986
9. Conti I and Rollins BJ. CCL2 (monocyte chemoattractant protein-1) and cancer. Semin Cancer Biol. (2004) 14:149–54. doi: 10.1016/j.semcancer.2003.10.009
10. Yoshimura T, Li C, Wang Y, and Matsukawa A. The chemokine monocyte chemoattractant protein-1/CCL2 is a promoter of breast cancer metastasis. Cell Mol Immunol. (2023) 20:714–38. doi: 10.1038/s41423-023-01013-0
11. Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, et al. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy. J Neuroinflamm. (2018) 15:290. doi: 10.1186/s12974-018-1330-2
12. Xu M, Wang Y, Xia R, Wei Y, and Wei X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. (2021) 54:e13115. doi: 10.1111/cpr.13115
13. Weckmann AL and Alcocer-Varela J. Cytokine inhibitors in autoimmune disease. Semin Arthritis Rheum. (1996) 26:539–57. doi: 10.1016/S0049-0172(96)80042-4
14. Donnelly RP, Young HA, and Rosenberg AS. An overview of cytokines and cytokine antagonists as therapeutic agents. Ann N Y Acad Sci. (2009) 1182:1–13. doi: 10.1111/j.1749-6632.2009.05382.x
15. Lim SY, Yuzhalin AE, Gordon-Weeks AN, and Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. (2016) 7:28697–710. doi: 10.18632/oncotarget.7376
16. Zlotnik A and Yoshie O. Chemokines: A new classification system and their role in immunity. Immunity. (2000) 12:121–7. doi: 10.1016/S1074-7613(00)80165-X
17. Naruse K, Ueno M, Satoh T, Nomiyama H, Tei H, Takeda M, et al. A YAC contig of the human CC chemokine genes clustered on chromosome 17q11.2. Genomics. (1996) 34:236–40. doi: 10.1006/geno.1996.0274
18. Pozzi S and Satchi-Fainaro R. The role of CCL2/CCR2 axis in cancer and inflammation: The next frontier in nanomedicine. Adv Drug Deliv Rev. (2024) 209:115318. doi: 10.1016/j.addr.2024.115318
19. Maus U, Henning S, Wenschuh H, Mayer K, Seeger W, and Lohmeyer J. Role of endothelial MCP-1 in monocyte adhesion to inflamed human endothelium under physiological flow. Am J Physiol Heart Circ Physiol. (2002) 283:H2584–91. doi: 10.1152/ajpheart.00349.2002
20. Locati M, Zhou D, Luini W, Evangelista V, Mantovani A, and Sozzani S. Rapid induction of arachidonic acid release by monocyte chemotactic protein-1 and related chemokines. Role of Ca2+ influx, synergism with platelet-activating factor and significance for chemotaxis. J Biol Chem. (1994) 269:4746–53. doi: 10.1016/S0021-9258(17)37607-X
21. Gschwandtner M, Derler R, and Midwood KS. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol. (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
22. Kwak M, Erdag G, Leick KM, Bekiranov S, Engelhard VH, and Slingluff CL. Associations of immune cell homing gene signatures and infiltrates of lymphocyte subsets in human melanomas: discordance with CD163(+) myeloid cell infiltrates. J Transl Med. (2021) 19:371. doi: 10.1186/s12967-021-03044-5
23. Hemmerlein B, Reinhardt L, Wiechens B, Khromov T, Schliephake H, and Brockmeyer P. Is CCL2 an important mediator of mast cell-tumor cell interactions in oral squamous cell carcinoma? Int J Mol Sci. (2023) 24:3641. doi: 10.3390/ijms24043641
24. Wang R, Liang Q, Zhang Q, Zhao S, Lin Y, Liu B, et al. Ccl2-induced regulatory T cells balance inflammation through macrophage polarization during liver reconstitution. Adv Sci. (2024) 11:2403849. doi: 10.1002/advs.202403849
25. Whelan DS, Caplice NM, and Clover AJP. Mesenchymal stromal cell derived CCL2 is required for accelerated wound healing. Sci Rep. (2020) 10:2642. doi: 10.1038/s41598-020-59174-1
26. Mulholland BS, Forwood MR, and Morrison NA. Monocyte chemoattractant protein-1 (MCP-1/CCL2) drives activation of bone remodelling and skeletal metastasis. Curr Osteoporos Rep. (2019) 17:538–47. doi: 10.1007/s11914-019-00545-7
27. Ishida Y, Kuninaka Y, Nosaka M, Furuta M, Kimura A, Taruya A, et al. CCL2-mediated reversal of impaired skin wound healing in diabetic mice by normalization of neovascularization and collagen accumulation. J Invest Dermatol. (2019) 139:2517–27.e5. doi: 10.1016/j.jid.2019.05.022
28. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun Biol. (2020) 3:720. doi: 10.1038/s42003-020-01441-y
29. Ernst CA, Zhang YJ, Hancock PR, Rutledge BJ, Corless CL, and Rollins BJ. Biochemical and biologic characterization of murine monocyte chemoattractant protein-1. Identification of two functional domains. J Immunol. (1994) 152:3541–9. doi: 10.4049/jimmunol.152.7.3541
30. Shao Z, Tan Y, Shen Q, Hou L, Yao B, Qin J, et al. Molecular insights into ligand recognition and activation of chemokine receptors CCR2 and CCR3. Cell Discov. (2022) 8:44. doi: 10.1038/s41421-022-00403-4
31. Mir MA, Jan U, and Ishfaq. CCL2–CCR2 signaling axis in cancer. In: Mir MA, editor. Cytokine and chemokine networks in cancer. Springer Nature Singapore, Singapore (2023). p. 241–70.
32. Israeli Dangoor S, Khoury R, Salomon K, Pozzi S, Shahar S, Miari A, et al. CCL2 blockade combined with PD-1/P-selectin immunomodulators impedes breast cancer brain metastasis. Brain. (2024) 148:awae347. doi: 10.1093/brain/awae347
33. Ling Z, Li W, Hu J, Li Y, Deng M, Zhang S, et al. Targeting CCL2-CCR4 axis suppress cell migration of head and neck squamous cell carcinoma. Cell Death Dis. (2022) 13:158. doi: 10.1038/s41419-022-04610-5
34. Comerford I and McColl SR. Atypical chemokine receptors in the immune system. Nat Rev Immunol. (2024) 24:753–69. doi: 10.1038/s41577-024-01025-5
35. Benoit A, Lequeux A, Harter P, Berchem G, and Janji B. Atypical chemokine receptor 2 expression is directly regulated by hypoxia inducible factor-1 alpha in cancer cells under hypoxia. Sci Rep. (2024) 14:26589. doi: 10.1038/s41598-024-77628-8
36. Darbonne WC, Rice GC, Mohler MA, Apple T, Hébert CA, Valente AJ, et al. Red blood cells are a sink for interleukin 8, a leukocyte chemotaxin. J Clin Invest. (1991) 88:1362–9. doi: 10.1172/JCI115442
37. Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol. (2009) 10:101–8. doi: 10.1038/ni.1675
38. Shao LN, Zhou SH, Wang N, Zhang ST, and Liu M. Association between the genetic polymorphisms of CCL2, CCL5, CCL8, CCR2, and CCR5 with chronic hepatitis C virus infection in the chinese han population. Immunol Invest. (2022) 51:1182–97. doi: 10.1080/08820139.2021.1916524
39. Chinoy H, Salway F, Fertig N, Tait BD, Oddis CV, Ollier WE, et al. Monocyte chemotactic protein-1 single nucleotide polymorphisms do not confer susceptibility for the development of adult onset polymyositis/dermatomyositis in UK Caucasians. Rheumatol (Oxford). (2007) 46:604–7. doi: 10.1093/rheumatology/kel359
40. Sun T, Mary LG, Oh WK, Freedman ML, Pomerantz M, Pienta KJ, et al. Inherited variants in the chemokine CCL2 gene and prostate cancer aggressiveness in a Caucasian cohort. Clin Cancer Res. (2011) 17:1546–52. doi: 10.1158/1078-0432.CCR-10-2015
41. Colobran R, Pujol-Borrell R, Armengol MP, and Juan M. The chemokine network. II. On how polymorphisms and alternative splicing increase the number of molecular species and configure intricate patterns of disease susceptibility. Clin Exp Immunol. (2007) 150:1–12. doi: 10.1111/j.1365-2249.2007.03489.x
42. Pham MH, Bonello GB, Castiblanco J, Le T, Sigala J, He W, et al. The rs1024611 regulatory region polymorphism is associated with CCL2 allelic expression imbalance. PloS One. (2012) 7:e49498. doi: 10.1371/journal.pone.0049498
43. Miao KK, Li J, Wu LN, Zhang B, and Li MQ. Genetic variants, circulating levels of monocyte chemoattractant protein-1 with risk of breast cancer: a case-control study and Mendelian randomization analysis. Zhonghua Yu Fang Yi Xue Za Zhi. (2022) 56:590–4. doi: 10.3760/cma.j.cn112150-20211107-01030
44. Wu HH, Lee TH, Tee YT, Chen SC, Yang SF, Lee SK, et al. Relationships of single nucleotide polymorphisms of monocyte chemoattractant protein 1 and chemokine receptor 2 with susceptibility and clinicopathologic characteristics of neoplasia of uterine cervix in Taiwan women. Reprod Sci. (2013) 20:1175–83. doi: 10.1177/1933719113477481
45. He S and Zhang X. The rs1024611 in the CCL2 gene and risk of gynecological cancer in Asians: a meta-analysis. World J Surg Oncol. (2018) 16:34. doi: 10.1186/s12957-018-1335-4
46. Gu H, Ni M, Guo X, Feng P, Xu Y, Gu X, et al. The functional polymorphism in monocyte chemoattractant protein-1 gene increases susceptibility to gastric cancer. Med Oncol. (2011) 28:S280–5. doi: 10.1007/s12032-010-9748-0
47. Niu Y, Zhou G, Wang Y, Qin J, Ping J, Zhang Q, et al. Association of MCP-1 promoter polymorphism with susceptibility to nasopharyngeal carcinoma. J Cell Biochem. (2019) 120:6661–70. doi: 10.1002/jcb.27962
48. Boughriba R, Sahraoui G, Chaar I, Weslati M, Ayed K, Ounissi D, et al. Significant association of MCP1 rs1024611 and CCR2 rs1799864 polymorphisms with colorectal cancer and liver metastases susceptibility and aggressiveness: A case-control study. Cytokine. (2023) 167:156193. doi: 10.1016/j.cyto.2023.156193
49. Chen Z, Yin S, Zheng L, Tang W, Kang M, Wei W, et al. Relationship between the Monocyte Chemo-attractant Protein-1 gene rs1024611 A>G Polymorphism and Cancer Susceptibility: A Meta-analysis Involving 14,617 Subjects. Immunol Investigations. (2021) 50:461–77. doi: 10.1080/08820139.2020.1776726
50. Hodeib H, Abd El Hai D, Tawfik MA, Allam AA, Selim A, Elsawy AA, et al. CCL2 rs1024611Gene polymorphism in philadelphia-negative myeloproliferative neoplasms. Genes (Basel). (2022) 13:492. doi: 10.3390/genes13030492
51. Rodero M, Rodero P, Descamps V, Lebbe C, Wolkenstein P, Aegerter P, et al. Melanoma susceptibility and progression: Association study between polymorphisms of the chemokine (CCL2) and chemokine receptors (CX3CR1, CCR5). J Dermatol Sci. (2007) 46:72–6. doi: 10.1016/j.jdermsci.2006.11.007
52. Li X, Lin F, and Zhou H. Genetic polymorphism rs3760396 of the chemokine (C-C motif) ligand 2 gene (CCL2) associated with the susceptibility of lung cancer in a pathological subtype-specific manner in Han-ancestry Chinese: a case control study. BMC Cancer. (2016) 16:298. doi: 10.1186/s12885-016-2328-8
53. Mahajan D, Sambyal V, Uppal MS, Sudan M, and Guleria K. Association of insertion/deletion polymorphisms of MDM2, MCP-1 and VEGF with esophageal cancer risk in north-west Indians: A case - control study. Asian Pac J Cancer Prev. (2025) 26:1623–31. doi: 10.31557/APJCP.2025.26.5.1623
54. Mandal RK, Agrawal T, and Mittal RD. Genetic variants of chemokine CCL2 and chemokine receptor CCR2 genes and risk of prostate cancer. Tumour Biol. (2015) 36:375–81. doi: 10.1007/s13277-014-2646-x
55. Singh V, Srivastava P, Srivastava N, Kapoor R, and Mittal RD. Association of inflammatory chemokine gene CCL2I/D with bladder cancer risk in North Indian population. Mol Biol Rep. (2012) 39:9827–34. doi: 10.1007/s11033-012-1849-8
56. Imhof A. Histone modifications: an assembly line for active chromatin? Curr Biol. (2003) 13:R22–R4. doi: 10.1016/S0960-9822(02)01383-0
57. Lusser A. Acetylated, methylated, remodeled: chromatin states for gene regulation. Curr Opin Plant Biol. (2002) 5:437–43. doi: 10.1016/S1369-5266(02)00287-X
58. Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. (2002) 12:198–209. doi: 10.1016/S0959-437X(02)00287-3
59. Zheng Y, Wang Z, Wei S, Liu Z, and Chen G. Epigenetic silencing of chemokine CCL2 represses macrophage infiltration to potentiate tumor development in small cell lung cancer. Cancer Lett. (2021) 499:148–63. doi: 10.1016/j.canlet.2020.11.034
60. Wang Y-F, Yu L, Hu Z-L, Fang Y-F, Shen Y-Y, Song M-F, et al. Regulation of CCL2 by EZH2 affects tumor-associated macrophages polarization and infiltration in breast cancer. Cell Death Dis. (2022) 13:748. doi: 10.1038/s41419-022-05169-x
61. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
62. Huang Z, Liang N, Goñi S, Damdimopoulos A, Wang C, Ballaire R, et al. The corepressors GPS2 and SMRT control enhancer and silencer remodeling via eRNA transcription during inflammatory activation of macrophages. Mol Cell. (2021) 81:953–68.e9. doi: 10.1016/j.molcel.2020.12.040
63. Hou P, Kapoor A, Zhang Q, Li J, Wu CJ, Li J, et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov. (2020) 10:1058–77. doi: 10.1158/2159-8290.CD-19-0597
64. Xu M, Lu X, Zhu F, Sun X, Yao H, Zhang J, et al. BRG1 mediates epigenetic regulation of TNFα-induced CCL2 expression in oral tongue squamous cell carcinoma cells. J Cell Biochem. (2024) 125:e30535. doi: 10.1002/jcb.30535
65. Ambros V. microRNAs: tiny regulators with great potential. Cell. (2001) 107:823–6. doi: 10.1016/S0092-8674(01)00616-X
66. Chen J, Gao Y, Zhong J, Wu X, Leng Z, Liu M, et al. Lnc-H19-derived protein shapes the immunosuppressive microenvironment of glioblastoma. Cell Rep Med. (2024) 5:101806. doi: 10.1016/j.xcrm.2024.101806
67. Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, et al. miR-126 and miR-126* repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat Cell Biol. (2013) 15:284–94. doi: 10.1038/ncb2690
68. Bertoli G, Cava C, and Castiglioni I. MicroRNAs: new biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics. (2015) 5:1122–43. doi: 10.7150/thno.11543
69. Teng KY, Han J, Zhang X, Hsu SH, He S, Wani NA, et al. Blocking the CCL2-CCR2 axis using CCL2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol Cancer Ther. (2017) 16:312–22. doi: 10.1158/1535-7163.MCT-16-0124
70. Wang X, Sheng W, Xu T, Xu J, Gao R, and Zhang Z. CircRNA hsa_circ_0110102 inhibited macrophage activation and hepatocellular carcinoma progression via miR-580-5p/PPARα/CCL2 pathway. Aging (Albany NY). (2021) 13:11969–87. doi: 10.18632/aging.202900
71. Liu N, Wang X, Steer CJ, and Song G. MicroRNA-206 promotes the recruitment of CD8(+) T cells by driving M1 polarisation of Kupffer cells. Gut. (2022) 71:1642–55. doi: 10.1136/gutjnl-2021-324170
72. Sun H, Zhao L, Pan K, Zhang Z, Zhou M, and Cao G. Integrated analysis of mRNA and miRNA expression profiles in pancreatic ductal adenocarcinoma. Oncol Rep. (2017) 37:2779–86. doi: 10.3892/or.2017.5526
73. Shen H, Yu X, Yang F, Zhang Z, Shen J, Sun J, et al. Reprogramming of Normal Fibroblasts into Cancer-Associated Fibroblasts by miRNAs-Mediated CCL2/VEGFA Signaling. PloS Genet. (2016) 12:e1006244. doi: 10.1371/journal.pgen.1006244
74. Lee S, Hong JH, Kim JS, Yoon JS, Chun SH, Hong SA, et al. Cancer-associated fibroblasts activated by miR-196a promote the migration and invasion of lung cancer cells. Cancer Lett. (2021) 508:92–103. doi: 10.1016/j.canlet.2021.03.021
75. Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. (2019) 18:64. doi: 10.1186/s12943-019-0976-4
76. Shengnan J, Dafei X, Hua J, Sunfu F, Xiaowei W, and Liang X. Long non-coding RNA HOTAIR as a competitive endogenous RNA to sponge miR-206 to promote colorectal cancer progression by activating CCL2. J Cancer. (2020) 11:4431–41. doi: 10.7150/jca.42308
77. Arora L, Patra D, Roy S, Nanda S, Singh N, Verma AK, et al. Hypoxia-induced miR-210-3p expression in lung adenocarcinoma potentiates tumor development by regulating CCL2 mediated monocyte infiltration. Mol Oncol. (2024) 18:1278–300. doi: 10.1002/1878-0261.13260
78. Wang W, Shen F, Wang C, Lu W, Wei J, Shang A, et al. MiR-1-3p inhibits the proliferation and invasion of bladder cancer cells by suppressing CCL2 expression. Tumour Biol. (2017) 39:1010428317698383. doi: 10.1177/1010428317698383
79. Chen C, He W, Huang J, Wang B, Li H, Cai Q, et al. LNMAT1 promotes lymphatic metastasis of bladder cancer via CCL2 dependent macrophage recruitment. Nat Commun. (2018) 9:3826. doi: 10.1038/s41467-018-06152-x
80. Li R, Liu Y, Liu J, Chen B, Ji Z, Xu A, et al. CCL2 regulated by the CTBP1-AS2/miR-335-5p axis promotes hemangioma progression and angiogenesis. Immunopharmacol Immunotoxicol. (2024) 46:385–94. doi: 10.1080/08923973.2024.2330651
81. Frank A-C, Ebersberger S, Fink AF, Lampe S, Weigert A, Schmid T, et al. Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype. Nat Commun. (2019) 10:1135. doi: 10.1038/s41467-019-08989-2
82. Li B, Liu S, Yang Q, Li Z, Li J, Wu J, et al. Macrophages in tumor-associated adipose microenvironment accelerate tumor progression. Adv Biol (Weinh). (2023) 7:e2200161. doi: 10.1002/adbi.202200161
83. Abend JR, Uldrick T, and Ziegelbauer JM. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol. (2010) 84:12139–51. doi: 10.1128/JVI.00884-10
84. Li D, Du F, Jiao H, Zhang F, Wang X, and Zhang S. CircHSPB6 promotes tumor-associated macrophages M2 polarization and infiltration to accelerate cell Malignant properties in lung adenocarcinoma by CCL2. Biochem Genet. (2024) 62:1379–95. doi: 10.1007/s10528-023-10482-x
85. Bott A, Erdem N, Lerrer S, Hotz-Wagenblatt A, Breunig C, Abnaof K, et al. miRNA-1246 induces pro-inflammatory responses in mesenchymal stem/stromal cells by regulating PKA and PP2A. Oncotarget. (2017) 8:43897–914. doi: 10.18632/oncotarget.14915
86. Iacona JR, Monteleone NJ, Lemenze AD, Cornett AL, and Lutz CS. Transcriptomic studies provide insights into the tumor suppressive role of miR-146a-5p in non-small cell lung cancer (NSCLC) cells. RNA Biol. (2019) 16:1721–32. doi: 10.1080/15476286.2019.1657351
87. Zhao L, Wang G, Qi H, Yu L, Yin H, Sun R, et al. LINC00330/CCL2 axis-mediated ESCC TAM reprogramming affects tumor progression. Cell Mol Biol Lett. (2024) 29:77. doi: 10.1186/s11658-024-00592-8
88. Momen-Heravi F and Bala S. Extracellular vesicles in oral squamous carcinoma carry oncogenic miRNA profile and reprogram monocytes via NF-κB pathway. Oncotarget. (2018) 9:34838–54. doi: 10.18632/oncotarget.26208
89. Zhang B, Zhang Z, Li L, Qin YR, Liu H, Jiang C, et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-κB signaling. Nat Commun. (2018) 9:1423. doi: 10.1038/s41467-018-03716-9
90. Li X, Fan Q, Li J, Song J, and Gu Y. MiR-124 down-regulation is critical for cancer associated fibroblasts-enhanced tumor growth of oral carcinoma. Exp Cell Res. (2017) 351:100–8. doi: 10.1016/j.yexcr.2017.01.001
91. Korbecki J, Simińska D, Gąssowska-Dobrowolska M, Listos J, Gutowska I, Chlubek D, et al. Chronic and cycling hypoxia: drivers of cancer chronic inflammation through HIF-1 and NF-κB activation: A review of the molecular mechanisms. Int J Mol Sci. (2021) 22:10701. doi: 10.3390/ijms221910701
92. Fallahi P, Ferrari SM, Piaggi S, Luconi M, Cantini G, Gelmini S, et al. The paramount role of cytokines and chemokines in papillary thyroid cancer: a review and experimental results. Immunol Res. (2018) 66:710–22. doi: 10.1007/s12026-018-9056-x
93. Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. (2006) 25:409–16. doi: 10.1007/s10555-006-9005-3
94. Kanga KJW, Mendonca P, Soliman KFA, Ferguson DT, and Darling-Reed SF. Effect of diallyl trisulfide on TNF-α-induced CCL2/MCP-1 release in genetically different triple-negative breast cancer cells. Anticancer Res. (2021) 41:5919–33. doi: 10.21873/anticanres.15411
95. Thaklaewphan P, Ruttanapattanakul J, Monkaew S, Buatoom M, Sookkhee S, Nimlamool W, et al. Kaempferia parviflora extract inhibits TNF-α-induced release of MCP-1 in ovarian cancer cells through the suppression of NF-κB signaling. Biomed Pharmacother. (2021) 141:111911. doi: 10.1016/j.biopha.2021.111911
96. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. (1996) 87:2095–147. doi: 10.1182/blood.V87.6.2095.bloodjournal8762095
97. Guo B, Fu S, Zhang J, Liu B, and Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. (2016) 6:36107. doi: 10.1038/srep36107
98. Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, et al. Inflammatory mediators in breast cancer: coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer. (2011) 11:130. doi: 10.1186/1471-2407-11-130
99. Won Jun H, Kyung Lee H, Ho Na I, Jeong Lee S, Kim K, Park G, et al. The role of CCL2, CCL7, ICAM-1, and VCAM-1 in interaction of endothelial cells and natural killer cells. Int Immunopharmacol. (2022) 113:109332. doi: 10.1016/j.intimp.2022.109332
100. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc Natl Acad Sci. (2019) 116:1361–9. doi: 10.1073/pnas.1812266115
101. Wu CY, Shu LH, Liu HT, Cheng YC, Wu YH, and Wu YH. The role of MRE11 in the IL-6/STAT3 pathway of lung cancer cells. Curr Issues Mol Biol. (2022) 44:6132–44. doi: 10.3390/cimb44120418
102. Zhou B, Mo Z, Lai G, Chen X, Li R, Wu R, et al. Targeting tumor exosomal circular RNA cSERPINE2 suppresses breast cancer progression by modulating MALT1-NF-?B-IL-6 axis of tumor-associated macrophages. J Exp Clin Cancer Res. (2023) 42:48. doi: 10.1186/s13046-023-02620-5
103. Roth I, Campbell H, Rubio C, Vennin C, Wilson M, Wiles A, et al. The Δ133p53 isoform and its mouse analogue Δ122p53 promote invasion and metastasis involving pro-inflammatory molecules interleukin-6 and CCL2. Oncogene. (2016) 35:4981–9. doi: 10.1038/onc.2016.45
104. Hwang SG, Park J, Park JY, Park CH, Lee KH, Cho JW, et al. Anti-cancer activity of a novel small molecule compound that simultaneously activates p53 and inhibits NF-κB signaling. PloS One. (2012) 7:e44259. doi: 10.1371/journal.pone.0044259
105. Ding J, Hu H, Zhu Y, Xu X, Yin B, Yang M, et al. Inhibiting CMTM4 reverses the immunosuppressive function of myeloid-derived suppressor cells and augments immunotherapy response in cervical cancer. J Immunother Cancer. (2025) 13. doi: 10.1136/jitc-2025-011776
106. Chen LM, Liu PY, Chen YA, Tseng HY, Shen PC, Hwang PA, et al. Oligo-Fucoidan prevents IL-6 and CCL2 production and cooperates with p53 to suppress ATM signaling and tumor progression. Sci Rep. (2017) 7:11864. doi: 10.1038/s41598-017-12111-1
107. Suvac A, Ashton J, and Bristow RG. Tumour hypoxia in driving genomic instability and tumour evolution. Nat Rev Cancer. (2025) 25:167–88. doi: 10.1038/s41568-024-00781-9
108. Licausi F and Giuntoli B. Synthetic biology of hypoxia. New Phytol. (2021) 229:50–6. doi: 10.1111/nph.16441
109. Lanzen J, Braun RD, Klitzman B, Brizel D, Secomb TW, and Dewhirst MW. Direct demonstration of instabilities in oxygen concentrations within the extravascular compartment of an experimental tumor. Cancer Res. (2006) 66:2219–23. doi: 10.1158/0008-5472.CAN-03-2958
110. Tausendschön M, Dehne N, and Brüne B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine. (2011) 53:256–62. doi: 10.1016/j.cyto.2010.11.002
111. Pluemsampant S, Safronova OS, Nakahama K, and Morita I. Protein kinase CK2 is a key activator of histone deacetylase in hypoxia-associated tumors. Int J Cancer. (2008) 122:333–41. doi: 10.1002/ijc.23094
112. Safronova O, Pluemsampant S, Nakahama K, and Morita I. Regulation of chemokine gene expression by hypoxia via cooperative activation of NF-kappaB and histone deacetylase. Int J Biochem Cell Biol. (2009) 41:2270–80. doi: 10.1016/j.biocel.2009.05.003
113. Mojsilovic-Petrovic J, Callaghan D, Cui H, Dean C, Stanimirovic DB, and Zhang W. Hypoxia-inducible factor-1 (HIF-1) is involved in the regulation of hypoxia-stimulated expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) and MCP-5 (Ccl12) in astrocytes. J Neuroinflamm. (2007) 4:12. doi: 10.1186/1742-2094-4-12
114. Boelte KC, Gordy LE, Joyce S, Thompson MA, Yang L, and Lin PC. Rgs2 mediates pro-angiogenic function of myeloid derived suppressor cells in the tumor microenvironment via upregulation of MCP-1. PloS One. (2011) 6:e18534. doi: 10.1371/journal.pone.0018534
115. Galindo M, Santiago B, Alcami J, Rivero M, Martín-Serrano J, and Pablos JL. Hypoxia induces expression of the chemokines monocyte chemoattractant protein-1 (MCP-1) and IL-8 in human dermal fibroblasts. Clin Exp Immunol. (2001) 123:36–41. doi: 10.1046/j.1365-2249.2001.01412.x
116. Bai W, Zhou J, Zhou N, Liu Q, Cui J, Zou W, et al. Hypoxia-increased RAGE expression regulates chemotaxis and pro-inflammatory cytokines release through nuclear translocation of NF-κ B and HIF1α in THP-1 cells. Biochem Biophys Res Commun. (2018) 495:2282–8. doi: 10.1016/j.bbrc.2017.12.084
117. He J, Chai X, Zhang Q, Wang Y, Wang Y, Yang X, et al. The lactate receptor HCAR1 drives the recruitment of immunosuppressive PMN-MDSCs in colorectal cancer. Nat Immunol. (2025) 26:391–403. doi: 10.1038/s41590-024-02068-5
118. Hembruff SL, Jokar I, Yang L, and Cheng N. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia. (2010) 12:425–33. doi: 10.1593/neo.10200
119. Yue Y, Lian J, Wang T, Luo C, Yuan Y, Qin G, et al. Interleukin-33-nuclear factor-κB-CCL2 signaling pathway promotes progression of esophageal squamous cell carcinoma by directing regulatory T cells. Cancer Sci. (2020) 111:795–806. doi: 10.1111/cas.14293
120. Han R, Gu S, Zhang Y, Luo A, Jing X, Zhao L, et al. Estrogen promotes progression of hormone-dependent breast cancer through CCL2-CCR2 axis by upregulation of Twist via PI3K/AKT/NF-κB signaling. Sci Rep. (2018) 8:9575. doi: 10.1038/s41598-018-27810-6
121. Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. (1994) 153:2052–63. doi: 10.4049/jimmunol.153.5.2052
122. Lim SP and Garzino-Demo A. The human immunodeficiency virus type 1 Tat protein up-regulates the promoter activity of the beta-chemokine monocyte chemoattractant protein 1 in the human astrocytoma cell line U-87 MG: role of SP-1, AP-1, and NF-kappaB consensus sites. J Virol. (2000) 74:1632–40. doi: 10.1128/JVI.74.4.1632-1640.2000
123. Bauer D, Redmon N, Mazzio E, Taka E, Reuben JS, Day A, et al. Diallyl disulfide inhibits TNFα induced CCL2 release through MAPK/ERK and NF-Kappa-B signaling. Cytokine. (2015) 75:117–26. doi: 10.1016/j.cyto.2014.12.007
124. Zhao Z, Sun H, Liu Y, Zhang Y, Wang X, Wang X, et al. PDPN+ cancer-associated fibroblasts enhance gastric cancer angiogenesis via AKT/NF-κB activation and the CCL2-ACKR1 axis. MedComm. (2025) 6:e70037. doi: 10.1002/mco2.70037
125. Yang YI, Wang YY, Ahn JH, Kim BH, and Choi JH. CCL2 overexpression is associated with paclitaxel resistance in ovarian cancer cells via autocrine signaling and macrophage recruitment. BioMed Pharmacother. (2022) 153:113474. doi: 10.1016/j.biopha.2022.113474
126. Chen X, Zhou J, Wang Y, Wang X, Chen K, Chen Q, et al. PIM1/NF-κB/CCL2 blockade enhances anti-PD-1 therapy response by modulating macrophage infiltration and polarization in tumor microenvironment of NSCLC. Oncogene. (2024) 43:2517–30. doi: 10.1038/s41388-024-03100-6
127. Wolfsberger J, Sakil HAM, Zhou L, Van Bree N, Baldisseri E, De Souza Ferreira S, et al. TAp73 represses NF-κB-mediated recruitment of tumor-associated macrophages in breast cancer. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2017089118
128. Liu Z, Huang C, Mao X, Mi J, Zhang Q, Xie Y, et al. Single cell RNA-sequencing reveals mast cells enhance mononuclear phagocytes infiltration in bladder cancer microenvironment. J Cancer. (2024) 15:5672–90. doi: 10.7150/jca.99554
129. Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. (2018) 15:335–45. doi: 10.1038/cmi.2017.135
130. Kunz M, Bloss G, Gillitzer R, Gross G, Goebeler M, Rapp UR, et al. Hypoxia/reoxygenation induction of monocyte chemoattractant protein-1 in melanoma cells: involvement of nuclear factor-kappaB, stimulatory protein-1 transcription factors and mitogen-activated protein kinase pathways. Biochem J. (2002) 366:299–306. doi: 10.1042/bj20011749
131. Shi M, An G, Chen N, Jia J, Cui X, Zhan T, et al. UVRAG promotes tumor progression through regulating SP1 in colorectal cancer. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15092502
132. Li X and Tai HH. Activation of thromboxane A2 receptor (TP) increases the expression of monocyte chemoattractant protein -1 (MCP-1)/chemokine (C-C motif) ligand 2 (CCL2) and recruits macrophages to promote invasion of lung cancer cells. PloS One. (2013) 8:e54073. doi: 10.1371/journal.pone.0054073
133. Martin T, Cardarelli PM, Parry GC, Felts KA, and Cobb RR. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur J Immunol. (1997) 27:1091–7. doi: 10.1002/eji.1830270508
134. Martin T, Cardarelli PM, Parry GCN, Felts KA, and Cobb RR. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-χB and AP-1. Eur J Immunol. (1997) 27:1091–7. doi: 10.1002/eji.1830270508
135. Jung Y, Ahn SH, Park H, Park SH, Choi K, Choi C, et al. MCP-1 and MIP-3α Secreted from necrotic cell-treated glioblastoma cells promote migration/infiltration of microglia. Cell Physiol Biochem. (2018) 48:1332–46. doi: 10.1159/000492092
136. Ting H, Deep G, Kumar S, Jain AK, Agarwal C, and Agarwal R. Beneficial effects of the naturally occurring flavonoid silibinin on the prostate cancer microenvironment: role of monocyte chemotactic protein-1 and immune cell recruitment. Carcinogenesis. (2016) 37:589–99. doi: 10.1093/carcin/bgw039
137. Tu M, Klein L, Espinet E, Georgomanolis T, Wegwitz F, Li X, et al. TNF-α-producing macrophages determine subtype identity and prognosis via AP1 enhancer reprogramming in pancreatic cancer. Nat Cancer. (2021) 2:1185–203. doi: 10.1038/s43018-021-00258-w
138. Xue J, Ge X, Zhao W, Xue L, Dai C, Lin F, et al. PIPKIγ Regulates CCL2 expression in colorectal cancer by activating AKT-STAT3 signaling. J Immunol Res. (2019) 2019:3690561. doi: 10.1155/2019/3690561
139. Feng H, Liu K, Shen X, Liang J, Wang C, Qiu W, et al. Targeting tumor cell-derived CCL2 as a strategy to overcome Bevacizumab resistance in ETV5+ colorectal cancer. Cell Death Dis. (2020) 11:916. doi: 10.1038/s41419-020-03111-7
140. Li M, Cui Y, Qi Q, Liu J, Li J, Huang G, et al. SPOP downregulation promotes bladder cancer progression based on cancer cell-macrophage crosstalk via STAT3/CCL2/IL-6 axis and is regulated by VEZF1. Theranostics. (2024) 14:6543–59. doi: 10.7150/thno.101575
141. Tang B, Zhu J, Shi Y, Wang Y, Zhang X, Chen B, et al. Tumor cell-intrinsic MELK enhanced CCL2-dependent immunosuppression to exacerbate hepatocarcinogenesis and confer resistance of HCC to radiotherapy. Mol Cancer. (2024) 23:137. doi: 10.1186/s12943-024-02049-0
142. Cheng H, Shao Y, Zhang A, Li C, Li X, Fu Y, et al. Interleukin-7 enhances recruitment of MDSCs by regulating MCP-1 via JAK1/STAT3 signaling pathway in non-small cell lung cancer. Sci Rep. (2025) 15:16869. doi: 10.1038/s41598-025-01868-5
143. Wang L, Wang YJ, Wang R, Gong FL, Chen JY, Yu YT, et al. Fasting-mimicking diet enhances EGFR-TKI efficacy in oral cancer through dual mechanisms: direct cancer cell sensitization and tumor-associated macrophage crosstalk. Front Pharmacol. (2025) 16:1641024. doi: 10.3389/fphar.2025.1641024
144. Chen Q, Yuan H, Bronze MS, and Li M. Targeting lactylation and the STAT3/CCL2 axis to overcome immunotherapy resistance in pancreatic ductal adenocarcinoma. J Clin Invest. (2025) 135. doi: 10.1172/JCI191422
145. Zhou ZH, Chaturvedi P, Han YL, Aras S, Li YS, Kolattukudy PE, et al. IFN-gamma induction of the human monocyte chemoattractant protein (hMCP)-1 gene in astrocytoma cells: functional interaction between an IFN-gamma-activated site and a GC-rich element. J Immunol. (1998) 160:3908–16. doi: 10.4049/jimmunol.160.8.3908
146. Hacke K, Rincon-Orozco B, Buchwalter G, Siehler SY, Wasylyk B, Wiesmüller L, et al. Regulation of MCP-1 chemokine transcription by p53. Mol Cancer. (2010) 9:82. doi: 10.1186/1476-4598-9-82
147. Rahnamoun H, Lu H, Duttke SH, Benner C, Glass CK, and Lauberth SM. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun. (2017) 8:754. doi: 10.1038/s41467-017-01117-y
148. Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, et al. CRISPR/cas9-mediated trp53 and brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res. (2016) 76:6118–29. doi: 10.1158/0008-5472.CAN-16-1272
149. Carlsen L, Zhang S, Tian X, De La Cruz A, George A, Arnoff TE, et al. The role of p53 in anti-tumor immunity and response to immunotherapy. Front Mol Biosci. (2023) 10:1148389. doi: 10.3389/fmolb.2023.1148389
150. Korbecki J, Kojder K, Barczak K, Simińska D, Gutowska I, Chlubek D, et al. Hypoxia alters the expression of CC chemokines and CC chemokine receptors in a tumor–A literature review. Int J Mol Sci. (2020) 21:5647. doi: 10.3390/ijms21165647
151. Boutet M, Nishitani K, Couturier N, Erler P, Zhang Z, Militello AM, et al. Mutations in MLL3 promote breast cancer progression via HIF1α-dependent intratumoral recruitment and differentiation of regulatory T cells. Immunity. (2025) 58:2035–53.e9. doi: 10.1016/j.immuni.2025.07.008
152. Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK, Shin YJ, et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. (2019) 26:409–25. doi: 10.1038/s41418-018-0126-3
153. Khan A, Zhang Y, Ma N, Shi J, and Hou Y. NF-κB role on tumor proliferation, migration, invasion and immune escape. Cancer Gene Ther. (2024) 31:1599–610. doi: 10.1038/s41417-024-00811-6
154. Sun W, Wang X, Wang D, Lu L, Lin H, Zhang Z, et al. CD40×HER2 bispecific antibody overcomes the CCL2-induced trastuzumab resistance in HER2-positive gastric cancer. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-005063
155. Qian Y, Dong J, Zhang W, Xue X, Xiong Z, Zeng W, et al. Deguelin inhibits the glioblastoma progression through suppressing CCL2/NFκB signaling pathway. Neuropharmacology. (2024) 259:110109. doi: 10.1016/j.neuropharm.2024.110109
156. Zhou C, Weng J, Liu C, Liu S, Hu Z, Xie X, et al. Disruption of SLFN11 deficiency-induced CCL2 signaling and macrophage M2 polarization potentiates anti-PD-1 therapy efficacy in hepatocellular carcinoma. Gastroenterology. (2023) 164:1261–78. doi: 10.1053/j.gastro.2023.02.005
157. Ferrari SM, Elia G, Piaggi S, Baldini E, Ulisse S, Miccoli M, et al. CCL2 is modulated by cytokines and PPAR-γ in anaplastic thyroid cancer. Anticancer Agents Med Chem. (2018) 18:458–66. doi: 10.2174/1871520617666170719152349
158. Hsu DS, Wang HJ, Tai SK, Chou CH, Hsieh CH, Chiu PH, et al. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell. (2014) 26:534–48. doi: 10.1016/j.ccell.2014.09.002
159. Nakatsumi H, Matsumoto M, and Nakayama KI. Noncanonical pathway for regulation of CCL2 expression by an mTORC1-FOXK1 axis promotes recruitment of tumor-associated macrophages. Cell Rep. (2017) 21:2471–86. doi: 10.1016/j.celrep.2017.11.014
160. Low-Marchelli JM, Ardi VC, Vizcarra EA, Van Rooijen N, Quigley JP, and Yang J. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. (2013) 73:662–71. doi: 10.1158/0008-5472.CAN-12-0653
161. Xia L, Huang W, Tian D, Zhang L, Qi X, Chen Z, et al. Forkhead box Q1 promotes hepatocellular carcinoma metastasis by transactivating ZEB2 and VersicanV1 expression. Hepatology. (2014) 59:958–73. doi: 10.1002/hep.26735
162. Tang H, Zheng J, Bai X, Yue KL, Liang JH, Li DY, et al. Forkhead box Q1 is critical to angiogenesis and macrophage recruitment of colorectal cancer. Front Oncol. (2020) 10:564298. doi: 10.3389/fonc.2020.564298
163. Abraham S, Sweet T, Sawaya BE, Rappaport J, Khalili K, and Amini S. Cooperative interaction of C/EBPβ and Tat modulates MCP-1 gene transcription in astrocytes. J Neuroimmunol. (2005) 160:219–27. doi: 10.1016/j.jneuroim.2004.11.009
164. Lennard Richard ML, Nowling TK, Brandon D, Watson DK, and Zhang XK. Fli-1 controls transcription from the MCP-1 gene promoter, which may provide a novel mechanism for chemokine and cytokine activation. Mol Immunol. (2015) 63:566–73. doi: 10.1016/j.molimm.2014.07.013
165. Choi DH, Jang HL, Lim SH, Kim ST, Hong JY, Park SH, et al. Prevalence of KRAS amplification in patients with metastatic cancer: Real-world next-generation sequencing analysis. Pathol Res Pract. (2024) 261:155473. doi: 10.1016/j.prp.2024.155473
166. Ishihara N, Koma Y-I, Omori M, Komatsu S, Torigoe R, Yokoo H, et al. CCL2/CCR2/erk signal induced through cancer cell-macrophage interaction contributes to hepatocellular carcinoma progression. Am J Pathol. (2025) 195:589–608. doi: 10.1016/j.ajpath.2024.12.007
167. Potter SM, Dwyer RM, Hartmann MC, Khan S, Boyle MP, and Curran CE. Influence of stromal-epithelial interactions on breast cancer in vitro and in vivo. Breast Cancer Res Treat. (2012) 131:401–11. doi: 10.1007/s10549-011-1410-9
168. Maxwell PJ, Neisen J, Messenger J, and Waugh DJ. Tumor-derived CXCL8 signaling augments stroma-derived CCL2-promoted proliferation and CXCL12-mediated invasion of PTEN-deficient prostate cancer cells. Oncotarget. (2014) 5:4895–908. doi: 10.18632/oncotarget.2052
169. Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, and Leibovich-Rivkin T. Inflammatory mediators in breast cancer: coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition.BMC Cancer. (2011) 11:130. doi: 10.1186/1471-2407-11-130
170. Fujisaki K, Fujimoto H, Sangai T, Nagashima T, Sakakibara M, Shiina N, et al. Cancer-mediated adipose reversion promotes cancer cell migration via IL-6 and MCP-1. Breast Cancer Res Treat. (2015) 150:255–63. doi: 10.1007/s10549-015-3318-2
171. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. (2017) 8:761–73. doi: 10.7150/jca.17648
172. Zhang J, Lu Y, and Pienta KJ. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. (2010) 102:522–8. doi: 10.1093/jnci/djq044
173. Lu Y, Cai Z, Xiao G, Liu Y, Keller ET, Yao Z, et al. CCR2 expression correlates with prostate cancer progression. J Cell Biochem. (2007) 101:676–85. doi: 10.1002/jcb.21220
174. O'Connor T and Heikenwalder M. CCL2 in the tumor microenvironment. Adv Exp Med Biol. (2021) 1302:1–14. doi: 10.1007/978-3-030-62658-7_1
175. Li L, Liu YD, Zhan YT, Zhu YH, Li Y, Xie D, et al. High levels of CCL2 or CCL4 in the tumor microenvironment predict unfavorable survival in lung adenocarcinoma. Thorac Cancer. (2018) 9:775–84. doi: 10.1111/1759-7714.12643
176. Ishihara N, Koma Y-I, Omori M, Komatsu S, Torigoe R, Yokoo H, et al. Chemokine (C-C motif) ligand 2/CCR2/extracellular signal-regulated kinase signal induced through cancer cell–macrophage interaction contributes to hepatocellular carcinoma progression. Am J Pathol. (2025) 195:589–608. doi: 10.1016/j.ajpath.2024.12.007
177. Wang T, Zhan Q, Peng X, Qiu Z, and Zhao T. CCL2 influences the sensitivity of lung cancer A549 cells to docetaxel. Oncol Lett. (2018) 16:1267–74. doi: 10.3892/ol.2018.8769
178. Roca H, Varsos Z, and Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem. (2008) 283:25057–73. doi: 10.1074/jbc.M801073200
179. Fang WB, Sofia Acevedo D, Smart C, Zinda B, Alissa N, Warren K, et al. Expression of CCL2/CCR2 signaling proteins in breast carcinoma cells is associated with invasive progression. Sci Rep. (2021) 11:8708. doi: 10.1038/s41598-021-88229-0
180. Yao M, Fang W, Smart C, Hu Q, Huang S, Alvarez N, et al. CCR2 chemokine receptors enhance growth and cell-cycle progression of breast cancer cells through SRC and PKC activation. Mol Cancer Res. (2019) 17:604–17. doi: 10.1158/1541-7786.MCR-18-0750
181. Acharya SS and Kundu CN. Havoc in harmony: Unravelling the intricacies of angiogenesis orchestrated by the tumor microenvironment. Cancer Treat Rev. (2024) 127:102749. doi: 10.1016/j.ctrv.2024.102749
182. Zhu Z, Guo L, Yeltai N, Xu H, and Zhang Y. Chemokine (C-C motif) ligand 2-enhanced adipogenesis and angiogenesis of human adipose-derived stem cell and human umbilical vein endothelial cell co-culture system in adipose tissue engineering. J Tissue Eng Regenerative Med. (2022) 16:163–76. doi: 10.1002/term.3264
183. Peng Z, Pang H, Wu H, Peng X, Tan Q, Lin S, et al. CCL2 promotes proliferation, migration and angiogenesis through the MAPK/ERK1/2/MMP9, PI3K/AKT, Wnt/β−catenin signaling pathways in HUVECs. Exp Ther Med. (2023) 25:77. doi: 10.3892/etm.2022.11776
184. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3–CCL2 signaling. Cancer Res. (2016) 76:4124–35. doi: 10.1158/0008-5472.CAN-15-2973
185. Nyberg P, Salo T, and Kalluri R. Tumor microenvironment and angiogenesis. Front Biosci. (2008) 13:6537–53. doi: 10.2741/3173
186. Kzhyshkowska J, Shen J, and Larionova I. Targeting of TAMs: can we be more clever than cancer cells? Cell Mol Immunol. (2024) 21:1376–409. doi: 10.1038/s41423-024-01232-z
187. Gupta GP and Massagué J. Cancer metastasis: building a framework. Cell. (2006) 127:679–95. doi: 10.1016/j.cell.2006.11.001
188. Mukaida N and Baba T. Chemokines in tumor development and progression. Exp Cell Res. (2012) 318:95–102. doi: 10.1016/j.yexcr.2011.10.012
189. Silva AL, Barry S, Lopes-Pinto M, Joaquim R, Miranda C, Reis F, et al. CCL2 expression predicts clinical outcomes, and regulates E-cadherin and angiogenesis in pituitary tumours. Endocrine-Related Cancer. (2025) 32:ERC-24-0293. doi: 10.1530/ERC-24-0293
190. Chen X, Yang M, Yin J, Li P, Zeng S, Zheng G, et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/β-catenin signaling. Cell Commun Signal. (2022) 20:92. doi: 10.1186/s12964-022-00888-2
191. Li S, Lu J, Chen Y, Xiong N, Li L, Zhang J, et al. MCP-1-induced ERK/GSK-3β/Snail signaling facilitates the epithelial-mesenchymal transition and promotes the migration of MCF-7 human breast carcinoma cells. Cell Mol Immunol. (2017) 14:621–30. doi: 10.1038/cmi.2015.106
192. Chen W, Chen M, Hong L, Xiahenazi A, Huang M, Tang N, et al. M2-like tumor-associated macrophage-secreted CCL2 facilitates gallbladder cancer stemness and metastasis. Exp Hematol Oncol. (2024) 13:83. doi: 10.1186/s40164-024-00550-2
193. Zhuang H, Cao G, Kou C, and Liu T. CCL2/CCR2 axis induces hepatocellular carcinoma invasion and epithelial-mesenchymal transition in vitro through activation of the Hedgehog pathway. Oncol Rep. (2018) 39:21–30. doi: 10.3892/or.2017.6069
194. Linde N, Casanova-Acebes M, Sosa MS, Mortha A, Rahman A, Farias E, et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun. (2018) 9:21. doi: 10.1038/s41467-017-02481-5
195. Azevedo Martins JM, Rabelo-Santos SH, Do Amaral Westin MC, and Zeferino LC. Tumoral and stromal expression of MMP-2, MMP-9, MMP-14, TIMP-1, TIMP-2, and VEGF-A in cervical cancer patient survival: a competing risk analysis. BMC Cancer. (2020) 20:660. doi: 10.1186/s12885-020-07150-3
196. Xu H, Wang J, Al-Nusaif M, Ma H, and Le W. CCL2 promotes metastasis and epithelial-mesenchymal transition of non-small cell lung cancer via PI3K/Akt/mTOR and autophagy pathways. Cell Prolif. (2024) 57:e13560. doi: 10.1111/cpr.13560
197. Lindemann C, Marschall V, Weigert A, Klingebiel T, and Fulda S. Smac mimetic-induced upregulation of CCL2/MCP-1 triggers migration and invasion of glioblastoma cells and influences the tumor microenvironment in a paracrine manner. Neoplasia. (2015) 17:481–9. doi: 10.1016/j.neo.2015.05.002
198. Tang CH and Tsai CC. CCL2 increases MMP-9 expression and cell motility in human chondrosarcoma cells via the Ras/Raf/MEK/ERK/NF-κB signaling pathway. Biochem Pharmacol. (2012) 83:335–44. doi: 10.1016/j.bcp.2011.11.013
199. Chang YH, Huang YL, Tsai HC, Chang AC, Ko CY, Fong YC, et al. Chemokine ligand 2 promotes migration in osteosarcoma by regulating the miR-3659/MMP-3 axis. Biomedicines. (2023) 11. doi: 10.3390/biomedicines11102768
200. Liu Q, Zhang H, Jiang X, Qian C, Liu Z, and Luo D. Factors involved in cancer metastasis: a better understanding to "seed and soil" hypothesis. Mol Cancer. (2017) 16:176. doi: 10.1186/s12943-017-0742-4
201. Groblewska M, Litman-Zawadzka A, and Mroczko B. The role of selected chemokines and their receptors in the development of gliomas. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21103704
202. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
203. Zhang Y, Wang X, Gu Y, Liu T, Zhao X, Cheng S, et al. Complement C3 of tumor-derived extracellular vesicles promotes metastasis of RCC via recruitment of immunosuppressive myeloid cells. Proc Natl Acad Sci. (2025) 122:e2420005122. doi: 10.1073/pnas.2420005122
204. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. (2015) 12:244–57. doi: 10.1016/j.celrep.2015.06.024
205. Rosenbaum SR, Hughes CJ, Fields KM, Purdy SC, Gustafson A, Wolin A, et al. An EYA3/NF-κB/CCL2 signaling axis suppresses cytotoxic NK cells in the pre-metastatic niche to promote triple negative breast cancer metastasis. bioRxiv. (2024). doi: 10.1101/2024.07.31.606072
206. Lee SH, Lee D, Choi J, Oh HJ, Ham I-H, Ryu D, et al. Spatial dissection of tumour microenvironments in gastric cancers reveals the immunosuppressive crosstalk between <em<CCL2+</em< fibroblasts and <em<STAT3</em<-activated macrophages. Gut. (2024) 74:714–27. doi: 10.1136/gutjnl-2024-332901
207. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. (2020) 19:41. doi: 10.1186/s12943-020-01165-x
208. Wei X, Wang H, Liu H, Wang J, Zhou P, Li X, et al. Disruption of tumor-intrinsic PGAM5 increases anti-PD-1 efficacy through the CCL2 signaling pathway. J Immunother Cancer. (2025) 13. doi: 10.1136/jitc-2024-009993
209. Choi J, Lee HJ, Yoon S, Ryu HM, Lee E, Jo Y, et al. Blockade of CCL2 expression overcomes intrinsic PD-1/PD-L1 inhibitor-resistance in transglutaminase 2-induced PD-L1 positive triple negative breast cancer. Am J Cancer Res. (2020) 10:2878–94.
210. Eferl R. CCL2 at the crossroad of cancer metastasis. Jakstat. (2013) 2:e23816. doi: 10.4161/jkst.23816
211. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
212. Piao C, Cai L, Qiu S, Jia L, Song W, and Du J. Complement 5a enhances hepatic metastases of colon cancer via monocyte chemoattractant protein-1-mediated inflammatory cell infiltration. J Biol Chem. (2015) 290:10667–76. doi: 10.1074/jbc.M114.612622
213. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. (2015) 212:1043–59. doi: 10.1084/jem.20141836
214. Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell. (2012) 22:91–105. doi: 10.1016/j.ccr.2012.05.023
215. Roblek M, Protsyuk D, Becker PF, Stefanescu C, Gorzelanny C, Glaus Garzon JF, et al. CCL2 is a vascular permeability factor inducing CCR2-dependent endothelial retraction during lung metastasis. Mol Cancer Res. (2019) 17:783–93. doi: 10.1158/1541-7786.MCR-18-0530
216. Iwamoto H, Izumi K, and Mizokami A. Is the C-C motif ligand 2-C-C chemokine receptor 2 axis a promising target for cancer therapy and diagnosis? Int J Mol Sci. (2020) 21:9328. doi: 10.3390/ijms21239328
217. Scott AM, Wolchok JD, and Old LJ. Antibody therapy of cancer. Nat Rev Cancer. (2012) 12:278–87. doi: 10.1038/nrc3236
218. S0916, A Phase II, Window Trial of the Anti-CCR2 Antibody MLN1202 in Patients With Bone Metastases (2009). Available online at: https://clinicaltrials.gov/study/NCT01015560 (Accessed October 15, 2025).
219. An Open-Label, Phase 1b, Multi-Arm Study to Evaluate the Safety, Tolerability, and Pharmacodynamics of Investigational Treatments in Combination With Standard of Care Immune Checkpoint Inhibitors in Patients With Advanced Melanoma (2016). Available online at: https://clinicaltrials.gov/study/NCT02723006 (Accessed October 10, 2025).
220. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
221. Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. (2013) 71:1041–50. doi: 10.1007/s00280-013-2099-8
222. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. (2015) 10:111–23. doi: 10.1007/s11523-014-0320-2
223. An Open-Label, Multicenter, Phase 2 Study of Single-Agent CNTO 888 (an Anti-CCL2 Monoclonal Antibody) for the Treatment of Subjects With Metastatic Castrate-Resistant Prostate Cancer (2009). Available online at: https://clinicaltrials.gov/study/NCT00992186 (Accessed October 13, 2025).
224. Fetterly GJ, Aras U, Meholick PD, Takimoto C, Seetharam S, McIntosh T, et al. Utilizing pharmacokinetics/pharmacodynamics modeling to simultaneously examine free CCL2, total CCL2 and carlumab (CNTO 888) concentration time data. J Clin Pharmacol. (2013) 53:1020–7. doi: 10.1002/jcph.140
225. Bonapace L, Coissieux M-M, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. (2014) 515:130–3. doi: 10.1038/nature13862
226. Finlay J, Roberts CM, Dong J, Zink JI, Tamanoi F, and Glackin CA. Mesoporous silica nanoparticle delivery of chemically modified siRNA against TWIST1 leads to reduced tumor burden. Nanomedicine. (2015) 11:1657–66. doi: 10.1016/j.nano.2015.05.011
227. Shen S, Zhang Y, Chen KG, Luo YL, and Wang J. Cationic polymeric nanoparticle delivering CCR2 siRNA to inflammatory monocytes for tumor microenvironment modification and cancer therapy. Mol Pharm. (2018) 15:3642–53. doi: 10.1021/acs.molpharmaceut.7b00997
228. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. (2018) 67:1112–23. doi: 10.1136/gutjnl-2017-313738
229. Yang F, Yuan C, Chen F, Qin ZS, Schmitt NC, Lesinski GB, et al. Combined IL6 and CCR2 blockade potentiates antitumor activity of NK cells in HPV-negative head and neck cancer. J Exp Clin Cancer Res. (2024) 43:76. doi: 10.1186/s13046-024-03002-1
230. Fang W, Kozai Y, Acevedo DS, Brodine R, Gorrepati HS, Arviso N, et al. Cooperative CCL2/CCR2 and HGF/MET signaling enhances breast cancer growth and invasion associated with metabolic reprogramming. Cancer Biol Ther. (2025) 26:2535824. doi: 10.1080/15384047.2025.2535824
231. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. (2013) 31:760–8. doi: 10.1007/s10637-012-9869-8
Keywords: chemokine ligand 2, tumor microenvironment, immune oncology, immune cells, tumor metastasis, targeted therapy
Citation: Jin Y, Wang Y and Yang R (2025) Chemokine ligand 2: beyond chemotaxis—a multifaceted role in tumor progression. Front. Immunol. 16:1685474. doi: 10.3389/fimmu.2025.1685474
Received: 14 August 2025; Accepted: 28 October 2025;
Published: 18 November 2025.
Edited by:
Francesca Coperchini, University of Pavia, ItalyReviewed by:
Poupak Fallahi, University of Pisa, ItalyEric Ubil, University of Kentucky, United States
Copyright © 2025 Jin, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Yang, eXJzaHRjbUAxNjMuY29t