 Liping Chen
Liping Chen Qianping Huang
Qianping Huang Peipei Zhou
Peipei Zhou- 1The First Affiliated Hospital of Guangzhou Medical University, State Key Laboratory of Respiratory Diseases, Guangzhou, Guangdong, China
- 2Guangzhou National Laboratory, Bio-Island, Guangzhou, Guangdong, China
Effective anti-tumor immunity critically depends on functional CD8+ T cells, yet in almost all solid tumors, these cells become dysfunctional, exhausted, or spatially excluded. This breakdown of immune surveillance arises not only from cell-intrinsic T cell exhaustion but also from multimodal communication among tumor, stromal, and immune cells within the tumor microenvironment (TME). This communication is mediated not only through direct receptor-ligand interactions but also through a suite of indirect mechanisms, such as metabolic competition, secretion of immunosuppressive metabolites and cytokines, extracellular vesicle exchange, and even mitochondrial transfer via tunneling nanotubes or membrane transfer through T cell trogocytosis. Together, these suppressive interactions impair CD8+ T cell metabolism, effector function, and persistence, thereby enabling tumor immune evasion. In this review, we summarize current understanding of how multimodal cell-cell communication, including immune checkpoints, metabolic reprogramming, and stromal crosstalk, cooperatively drive CD8+ T cell dysfunction. We also highlight emerging therapeutic strategies aimed at rewiring these suppressive networks, with emphasis on translational potential. A deeper understanding of the spatial, molecular, and metabolic context of CD8+ T cell suppression offers new avenues to enhance the efficacy of cancer immunotherapies.
1 Introduction
CD8+ cytotoxic T lymphocytes (CTLs) are central mediators of anti-tumor immunity, capable of directly eliminating malignant cells through perforin-granzyme release and Fas-FasL signaling (1, 2). Their activation requires tumor antigens presentation by dendritic cells (DCs), co-stimulation signals (e.g., CD28-B7), and pro-inflammation cytokines [e.g., interleukin (IL)-12, interferon-gamma (IFN-γ)], leading to clonal expansion and cytotoxic effector functions acquisition (3). Upon antigen-specific activation, CTLs proliferate and differentiate into two major subsets: effector CD8+ T cells, characterized by high expression of granzyme, perforin, and IFN-γ, which eliminate target tumor cells; and memory CD8+ T cells that possess self-renewal and multilineage differentiation capacities, providing a cellular reservoir for long-term immune surveillance (4, 5).
Under chronic antigen exposure, however, CTLs gradually lose effector function and upregulate inhibitory receptors such as programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4), this dysfunctional state is termed T cell exhaustion (3). This trajectory of CD8+ T cell differentiation and dysfunction proceeds through successive stages: naïve T cells → activated T cells → stem-like progenitor of exhausted T cells (Tpex) → effector-like or intermediate exhausted T cells → terminal exhausted T cells (6, 7). TME provides spatial niches that critically shape this progression (8). Tertiary lymphoid structures (TLS) and perivascular regions, enriched with DCs, maintain TCF1+ Tpex cells, which preserve responsiveness to immune checkpoint blockade (ICB) (9–12). In contrast, tumor margins are enriched with CD103+ tissue-resident memory T cells (Trm) associated with favorable patient prognosis, while the immunosuppressive and hypoxic tumor core drives T cells towards terminal exhaustion, reinforced by persistent antigen exposure (13–15).
This dysfunctional state is further exacerbated by immunosuppressive factors in the TME, including tumor‐associated macrophages (TAMs, e.g., IRF8+) (16) and inhibitory cytokine networks (17), ultimately impairing antitumor immunity. Preclinical and clinical studies consistently demonstrate that in solid tumors, CD8+ T cells become functionally exhausted and metabolically impaired due to persistent antigen exposure and immunosuppressive mechanisms within the TME (18, 19). These mechanisms include direct inhibition by tumor and stromal cells, as well as indirect suppression via metabolic competition and soluble mediators, collectively impairing CD8+ T cell function and antitumor immunity (8, 20, 21).
A critical axis of immune evasion involves direct cell-to-cell interactions that drive CD8+ T cell dysfunction. Tumor cells exploit a repertoire of inhibitory ligands [e.g., PD-L1, B7 homolog 3 (B7-H3), and human leukocyte antigen (HLA)-E)] to engage checkpoint receptors [PD-1, Lymphocyte-activation gene 3 (LAG-3), Natural killer group 2 member A (NKG2A)] on T cells, thereby blunting TCR signaling and cytotoxicity activity. Immune cells such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) further suppress CTLs through mechanisms including CTLA-4-mediated blockade of co-stimulation and PD-L1 expression. Cancer-associated fibroblasts (CAFs) reinforce this suppression both by expressing ligands such as PD-L1 and carcinoembryonic antigen-related cell adhesion molecule 1 (Ceacam-1), and by physically restricting CD8+ T cells infiltration into tumor niches. Together, these interactions highlight the complexity of contact-dependent immunosuppression and underscore the limitations of current checkpoint blockade therapies.
Beyond direct contact, the TME imposes indirect suppression through metabolic hijacking, stromal crosstalk, and biochemical perturbations. Tumor cells aggressively outcompete T cells for essential nutrients including glucose and arginine, while releasing immunosuppressive metabolites such as lactate, adenosine and kynurenine. Extracellular vesicles, tunneling nanotubes and T cell trogocytosis further exacerbate suppression by transferring inhibitory cargos, such as dysfunctional mitochondria, inhibitory miRNAs, or even membrane fragments, to T cells. Meanwhile, cytokines (e.g., TGF-β) and ions (e.g., Mg²+, ammonia) disrupt T cell metabolism, signaling and epigenetic programming. Stromal components such as CAFs and MDSCs amplify these effects by remodeling the extracellular matrix, secreting suppressive cytokines, and inducing hypoxia. Collectively, these processes create a hostile metabolic and structural niche that sustains T cell dysfunction.
These multimodal pathways act synergistically to impair CD8+ T cell cytotoxicity and persistence, and spatial access into tumors, ultimately enabling immune evasion. Overcoming this coordinated suppression remains a major challenge in current cancer immunotherapy. In this review, we summarize recent advances in understanding the mechanisms of multimodal cell-cell communication, including immune checkpoint signaling, metabolic interference, and stromal crosstalk, that collectively drives CD8+ T cell dysfunction (Figure 1). We further discuss emerging therapeutic strategies designed to disrupt these suppressive networks and restore anti-tumor immunity, with particular attention to combinatorial approaches with translational potential. A precise understanding of the spatial and molecular dynamics of CD8+ T cell suppression will be pivotal for overcoming resistance to current immunotherapies.

Figure 1. Multifaceted regulation of CD8+ T cell function within the TME. (A) Differentiation and exhaustion of CD8+ T cells under chronic antigen stimulation. Effector-like or intermediate exhausted T cells (Teff); Exhausted T cells (Tex); Tissue-resident memory T cells (Trm). (B) The TME exerts dual effects on CD8+ T cells: it can promote T cell activation and effector functions, while simultaneously driving exhaustion and dysfunction. Left panel (Activation): Dendritic cells prime CD8+ T cells through integrated signals, which collectively enhance T cell proliferation, migration, differentiation, cytokine production, and cytotoxic capacity. Right panel (Suppression): Tumor cells suppress CD8+ T cell function through multiple mechanisms: (1) immunosuppressive ligand-receptor interactions [programmed cell death ligand 1 (PD-L1)-PD-1, transforming growth factor β (TGF-β)-TGF β receptor (TGF-βR)]; (2) nutrient competition (glucose, lipids, and amino acids); (3) tumor-derived exosomes; (4) intercellular material transfer via nanotubes and trogocytosis; (5) cytokines; and (6) release of immunosuppressive cytokines or metabolites (Mg2+, lithium, and ammonia). These inhibitor cues collectively drive upregulation of checkpoint receptors, diminished proliferation, and self-renewal capacity, reduced cytokine production, and impaired cytotoxicity, ultimately driving CD8+ T cells toward exhaustion. Image created with bioRender.com, with permission. Created in BioRender. Zhou, P. (2025) https://BioRender.com/e66x2mi.
2 Direct cell-to-cell interactions suppressing CD8+ T cell function
The direct interaction between CD8+ T cells and other cells in TME, including tumor cells, other immune cells and CAFs, is crucial for shaping anti-tumor immune responses. Direct contact through receptor ligand engagement and immunological synapses regulates CD8+ T cell activation, effector function, and exhaustion. While stimulatory signals enhance cytotoxicity, some interaction induced inhibitory pathways blunt TCR signaling, cytokine production, and proliferation. This section reviews how tumor cells, immune cells, and CAFs suppress CD8+ T cells function through surface expressed inhibitory molecules and checkpoint receptor-ligand interactions (Figure 2).
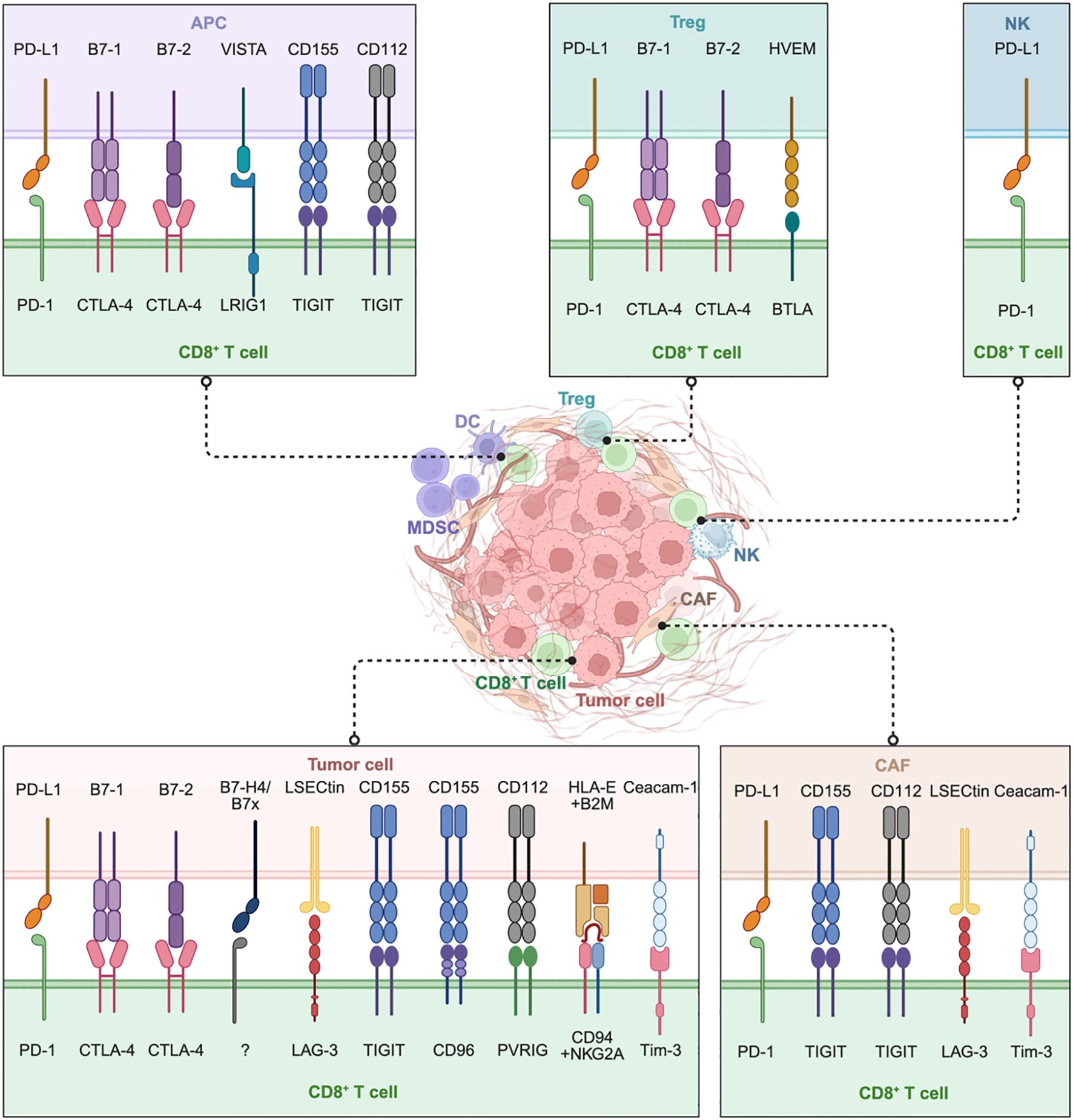
Figure 2. Direct cell-to-cell contact plays a critical role in the suppression of CD8+ T cells within the TME. Tumor cells inhibit CTLs by engaging inhibitory ligands with corresponding receptors, while multiple immune cells, including antigen-presenting cells (APCs), regulatory T cells (Tregs), NK cells (NKs), and specialized CD8+ Tregs, further suppress CD8+ T cells through checkpoint molecules like PD-1, CTLA-4, and VISTA. Cancer-associated fibroblasts (CAFs) uniquely contribute by engaging in direct inhibitory signaling (such as PD-L1-PD-1, Ceacam-1-Tim-3) and by imposing physical barriers that restrict T cell infiltration. Collectively, this intricate intercellular communication network drives CD8+ T cell dysfunction and exhaustion. Targeting these specific interactions, particularly beyond PD-1 and CTLA-4 (e.g., LAG-3, TIGIT, VISTA, PVRIG, CD96, NKG2A) and disrupting CAF-mediated suppression represent promising approaches to great reinvigorate CD8+ T cell anti-tumor responses. Image created with bioRender.com, with permission. Created in BioRender. Zhou, P. (2025) https://BioRender.com/kjhqxg8.
2.1 Tumor cell-to-CD8+ T cell interactions
Tumor cells directly inhibit infiltrating CD8+ T cells by engaging multiple inhibitory ligands. The PD-1-PD-L1 axis remains a dominant pathway: IFN-γ produced by activated T cells induces PD-L1 expression on tumor cells (22), which in turn binds PD-1 on CD8+ T cells, delivering potent inhibitory signals that attenuate TCR signaling (e.g., reduced ZAP70 phosphorylation), cytokine secretion (e.g., IFN-γ), and cytotoxic activity, ultimately driving CD8+ T cells into a dysfunctional state. Similarly, B7 ligands B7-1 (CD80) and B7-2 (CD86) on tumor cells engage CTLA-4 on activated CD8+ T cells, outcompeting CD28 and thereby blocking co-stimulatory signals required for T cell activation, leading to CD8+ T cell anergy.
Beyond these classical checkpoints, emerging ligand-receptor pathways are increasingly recognized. B7x-B7-H4, a member of the B7 family broadly expressed across tumors, which binds unidentified inhibitory receptor on activated, but not resting CD8+ T cells (23–25). B7-H4 inhibits CD8+ T cell responses at an early stage primarily by arresting cell cycle progression, suppressing TCR signaling, and reducing IL-2 production (26). Liver and lymph node sinusoidal endothelial cell C-type lectin (LSECtin), expressed in the liver and on multiple tumor types (e.g., melanoma), suppresses anti-tumor immunity by binding LAG-3 on CD8+ T cells, where its KIEELE motif has been identified as structurally and functionally essential for LAG-3’s inhibitory capacity. LAG-3 signaling inhibits effector T cell function by associating with CD3, where co-engagement suppresses proliferation, IFN-γ secretion, and calcium mobilization (27, 28).
T cell immunoglobulin and ITIM domain (TIGIT), an Ig superfamily member specifically expressed in immune cells, binds CD155 on tumor cells, directly inhibiting effector CD8+ T cell function (27). CD96, which also binds CD155, antagonizes the activating receptor CD226. Although CD96-mediated intracellular signaling remains incompletely characterized, its cytoplasmic ITIM domain suggests inhibitory potential (29). Notably, CD155hi lung adenocarcinoma (LUAD) cells dramatically reduce IFN-γ production in CD8+ T cells, thereby suppressing antitumor immunity (30). Poliovirus receptor-related protein 2 (PVRL2), also known as CD112, expressed by tumor cells and tumor-associated myeloid cells, binds the late-induced inhibitory receptor PVRIG (CD112R) on activated CD8+ T cells. The PVRL2-PVRIG axis, mediated by PVRIG’s ITIM domain, diminishes IL-12 receptor expression, suppresses cytotoxicity, and promotes CD8+ T cell exhaustion (31, 32).
Additional interactions further reinforce this suppressive network. HLA-E-Qa-1b complexes, presenting specific peptides processed by endoplasmic reticulum aminopeptidase 1-2 (ERAP1-2), engage the inhibitory natural killer cell group 2 member A (NKG2A)-CD94 heterodimer on a subset of CD8+ tumor-infiltrating lymphocytes (TILs), leading to suppression of TCR signaling and consequent impairment of cytotoxic effector function (33, 34). Ceacam-1-Tim-3 interactions have also been implicated, although current support comes primarily from clinical evidence rather than experimental validation (35).
Collectively, these inhibitory dyads converge to restrain CD8+ T cell cytotoxicity and persistence, underscoring the importance of multi-targeted checkpoint blockade.
2.2 Immune cell-to-CD8+ T cell interactions
Multiple immune cell populations within the TME suppress CD8+ T cell function through direct contact. Antigen-presenting cells (APCs), including DCs and macrophages, inhibit CD8+ T cells through classic immune evasion pathways, like PD-L1-PD-1 axis (36–39). APCs also express VISTA (V-domain immunoglobulin suppressor of T cell activation), functioning as a ligand for immunoglobulin-like domains 1 (LRIG1) on CD8+ T cells in a “trans” configuration, contributing to T cell inhibition and quiescence (40–42). Furthermore, constitutive expression of CD80-CD86 on APCs allows binding of CTLA-4 on activated CD8+ T cells (43, 44). CTLA-4 not only transmits intrinsic inhibitory signals but also, on Tregs, mediates the trans-endocytosis and degradation of CD80-CD86 from the APC surface, thereby limiting co-stimulation for other T cells (43, 45). Follicular dendritic cells (FDCs) express CD112 and CD155, which engage TIGIT on TILs, promoting a dysfunctional state characterized by high co-expression of PD-1, and diminished production of IFN-γ, tumor necrosis factor-alpha (TNF-α), and IL-2 (46). Natural Killer (NK) cells upregulate PD-L1 upon tumor recognition and IL-18 stimulation, generating PD-L1hi NK cells that directly suppress CD8+ T cell proliferation in PD-L1-PD-1-dependent manner (47). CD45RA− CCR7− (C-C motif chemokine receptor 7) Tregs exhibit upregulated CD80/CD86 expression alongside reduced HLA-DR, enabling potent suppression of CD8+ T cell function through dual mechanisms: IL-10 secretion and cell-contact-dependent inhibition mediated by CD80/CD86-CTLA-4 interaction, as evidenced by diminished IFN-γ, granzyme B production, and proliferation (48). Herpes virus entry mediator (HVEM, also TNFRSF14), a member of the TNF receptor superfamily expressed by both immune and non-immune cells that is frequently upregulated in malignancies, engages B and T lymphocyte attenuator (BTLA) on T cells to trigger co-inhibitory signaling, thereby suppressing TCR-mediated activation and impairing cytotoxic effector function (49, 50). Intriguingly, CD8+ T cells themselves may acquire suppression function. For example, a subset of CD8+ T cells, identified in humans as CD8+HLA-DR+ T cells, can adopt regulatory functions, further constraining effector responses (51). LRIG1, expressed on CD8+ T cells, interact with VISTA in cis or trans to suppresses anti-tumor immunity by inducing quiescence in CD8+ T cells and limiting the development of effector T cells from progenitor and memory-like cells (40). In summary, the effectiveness of CD8+ T cells in controlling tumors are significantly limited by an inhibitory interaction established immunosuppressive network in the TME.
2.3 CAFs-to-CD8+ T cell interactions
CAFs suppress CD8+ T cell function through both checkpoint signaling and structural modulation of the TME. CAFs frequently express PD-L1 (52), reciprocally upregulated through crosstalk with tumor cells via contact or soluble factors, which directly binds PD-1 on CD8+ T cells and correlates with poor prognosis in cancers like esophageal carcinoma. Like tumor cells and FDCs, CAFs also express CD155 and CD112, engaging TIGIT on TILs. TIGIT+ PD-1+ T cells exhibit reduced IFN-γ, TNF-α, and IL-2 production and impaired cytotoxicity, marking dysfunctional CD8+ T effector memory cells (TEM) cells. Dual blockade of TIGIT and PD-1 reverses this exhaustion, restoring antitumor responses (46). In hepatic tissues, LSECtin on hepatic CAFs engages LAG-3 on CD8+ T cells via the KIEELE motif, recruiting inhibitory signals through CD3 to suppress proliferation, IFN-γ, and calcium flux, dampening antitumor immunity (27). Moreover, CAFs express other immunosuppressive ligands: Ceacam-1 binds TIM-3 on CD8+ T cells, reinforcing exhaustion (27, 53). Beyond checkpoint ligands, CAFs remodel the extracellular matrix, restrict CD8+ T cells infiltration, and secret cytokines and exosomes that further impair function.
Through these diverse roles, CAFs act as key regulators of immune exclusion and resistance to immunotherapy. Targeting CAFs-CD8+ T cells interactions represents a promising strategy for successful cancer immunotherapies combination with checkpoint blockade.
2.4 Therapeutic strategies targeting direct cell-cell interactions
Immune checkpoints such as PD-1 and CTLA-4 are critical regulators of immune tolerance, preventing excessive immune activation. Tumors exploit this mechanism through ligand overexpression (e.g., PD-L1) to suppress T-cell function and facilitate immune escape. ICB therapies targeting PD-1-PD-L1, CTLA-4, and LAG-3 have significantly improved survival in multiple cancers (54, 55). However, complete response rates remain limited (56), largely due to tumor heterogeneity and the complexity of the immunosuppressive in TME, underscoring the need for stratified and context-specific immunotherapy approaches (15).
The functional state of CD8+ T cells, which serve as the core effector cells in antitumor immunity, is not shaped by a single signal but instead by integrated crosstalk with diverse cell populations in the TME (57, 58). Accordingly, immunotherapy strategies are shifting from a T cell-centric focus toward approaches that modulate the cellular interactions within the TME to promote effective antitumor immunity. The central therapeutic goal is to enhance T cell recognition and effector function while simultaneously blocking tumor immune evasion pathways.
Immune checkpoint inhibitors (ICIs) represent the most direct strategy (49). Anti–PD-1-PD-L1 antibodies restore effector functions of CD8+ T cells (such as cytokine secretion and cytotoxicity) by disrupting PD-L1-PD-1 inhibitory axis (49). Beyond classical ICIs, novel checkpoints such as TIGIT have been identified (59, 60). While anti-TIGIT monotherapy or combination therapy with anti–PD-1 has shown potential in some clinical trials, these approached remain insufficient to fully reinvigorate CD8+ T cells, particularly in patients with advanced or high tumor burden (61, 62). To enhance TIGIT-targeted immunotherapy, combination regimens are being developed, including anti–CTLA-4 or anti-vascular endothelial growth factor (VEGF) agents in triple blockade (e.g., TIGIT + PD-1-PD-L1 + CTLA-4 or + VEGF), or combinations with chemotherapy (59). In addition, multiple bispecific and trispecific antibodies have also entered clinical development, showing preliminary potential in overcoming resistance.
Beyond checkpoint inhibition, targeting interactions between CD8+ T cells and other immune cells offers additional therapeutic avenues (57). For example, anti-CTLA-4 antibodies (e.g., ipilimumab) function in part by depleting intertumoral Tregs, thereby relieving suppression on CD8+ T cells (63). Additionally, combination therapy with doxorubicin and IL-12 has been shown to shift receptor signaling in tumor infiltrating CD8+ T cells toward immunostimulatory pathways while reducing Treg infiltration, thus enhancing local effector activity (64).
CAFs present another major challenge to restrict CD8+ T cell infiltration and function by constructing both physical and biochemical barriers (65). Overcoming CAF-mediated immunosuppression is thus critical for restoring CD8+ T cell-mediated antitumor activity (66). In triple-negative breast cancer (TNBC), CAFs are particularly important therapeutic targets. Huo et al. engineered a CAF-targeted nanosystem co-loaded with a TGF-β inhibitor (LY3200882) and PD-L1 siRNA. Upon matrix metalloproteinase-2 (MMP2)-responsive release, LY3200882 preferentially modulates CAF activity, reducing extracellular matrix deposition and enhancing T-cell infiltration. Simultaneously, PD-L1 siRNA downregulates PD-L1 expression in both tumor cells and CAFs. This dual-action strategy effectively reverses CAF-driven immunosuppression, remodels the TME, and suppresses TNBC progression (67).
3 Indirect suppression via TME
The TME exerts profound indirect suppression on CD8+ T cell responses, orchestrating a complex network of metabolic, biochemical and structural barriers that shape anti-tumor immunity. Mounting evidence indicates that tumors co-opt multifaceted pathways, including metabolic reprogramming, cytokine induction, receptor modulation, and immune checkpoint activation, to systemically impair CD8+ T cell effector function, thereby fostering tumor progression. These immunosuppressive circuits are increasingly recognized as critical drivers of tumor immune evasion, positioning them as attractive therapeutic targets for restoring anti-tumor immunity. This section focuses on indirect TME-driven suppression, delineating how tumor cells and stromal elements orchestrate CD8+ T cell suppression through metabolic competition (e.g., nutrient deprivation), intercellular communication (eg. exosomes, tunneling nanotubes, or trogocytosis), and microenvironmental perturbations (eg. cytokine networks, ionic imbalances, or ammonia accumulation) (Table 1). Collectively, these mechanisms establish an immunosuppressive niche that subverts CD8+ T cell surveillance and therapeutic efficacy.
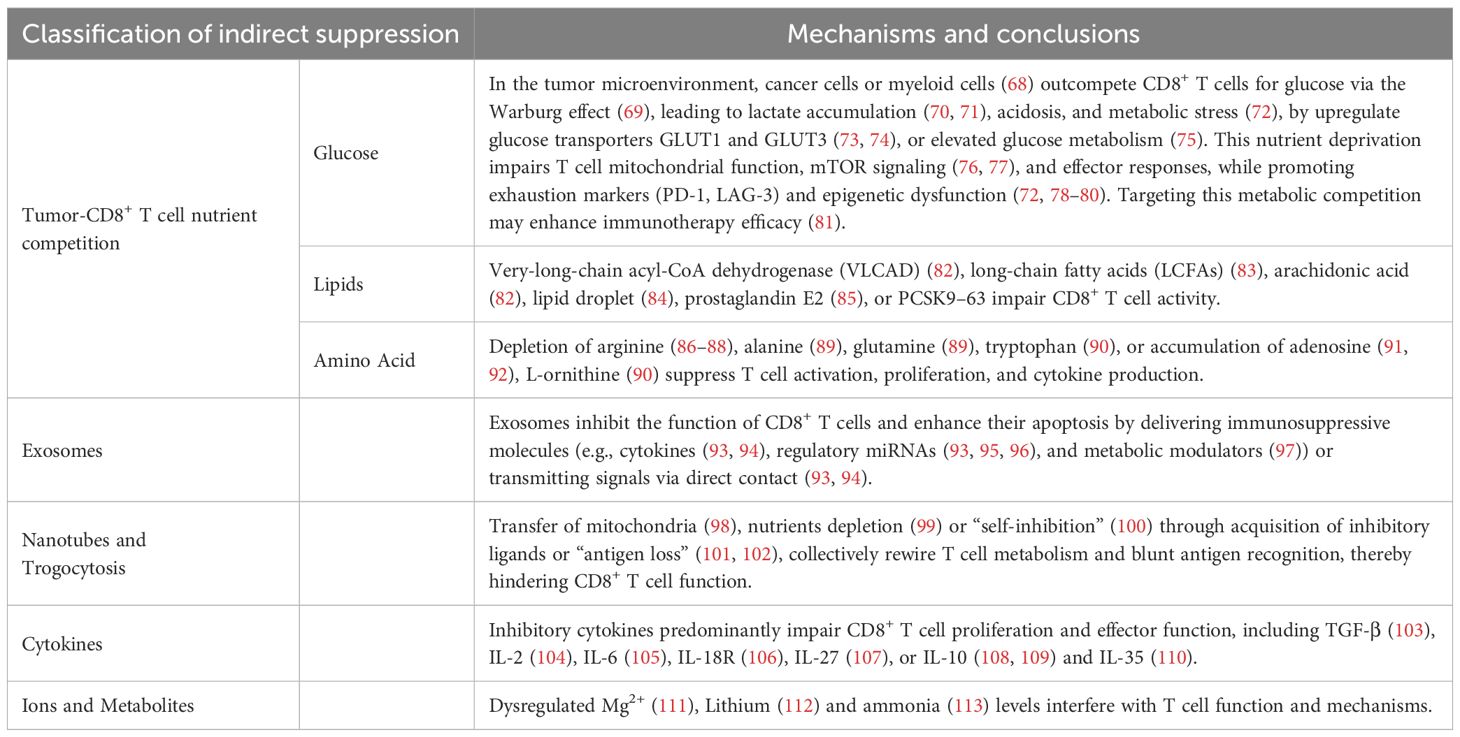
Table 1. Indirect regulation of CD8+ T cell dysfunction and exhaustion by the TME.
3.1 Tumor-CD8+ T cell nutrient competition
The availability of nutrients within the TME has emerged as a pivotal determinant of CD8+ T cell function. Compelling evidence indicates that enhanced nutrient uptake, glycolytic flux, and oxidative metabolism collectively potentiate CD8+ T cell proliferation and effector differentiation within tumors. This metabolic adaptation is essential for sustaining anti-tumor responses. Nevertheless, the TME frequently imposes profound metabolic constraints, including nutrient deprivation and lipid accumulation, that directly impair CD8+ T cell effector responses and immune surveillance. Strategies to overcome these barriers show therapeutic promise.
3.1.1 Glucose
Glucose metabolism plays a pivotal role in the TME, impacting both tumor progression and the functional capabilities of TILs (Figure 3). Tumor cells exploit the Warburg effect, consuming glucose and releasing lactate, which drives extracellular acidosis, hypoxia, disordered vasculature, and dense extracellular matrix within the TME (69, 73, 79, 114, 115). This nutrient competition restricts glucose availability to TILs, resulting in mitochondrial dysfunction and altered lipid metabolism, ultimately hindering T cell effector function and persistence. To sustain growth, tumor cells upregulate glucose transporters such as GLUT1 and GLUT3, and avidly consuming glucose and glutamine to promote T cell exhaustion and immune evasion (73, 74). In renal cell carcinoma, elevated tumor glycolysis corelates with reduced effector CD8+ T cells (75). Nutrient deprivation triggers AMP-activated protein kinase (AMPK) activation, while suppressing mTOR thereby disrupting T cell differentiation (116). Moreover, dysregulation of glucose metabolism through pathways such as PI3K/AKT/mTOR signaling further impacts T cell activation, Ca²+ signaling, and O-GlcNAcylation, all of which are essential for T cell effector function (76, 77, 117).

Figure 3. Metabolic Reprogramming in the TME Driving CD8+ T Cell Dysfunction. Glucose, lipid, and amino acid metabolism in the TME collectively impair CD8+ T cell function through nutrient competition, metabolite accumulation, and inhibitory signaling (1). Tumor cells and myeloid cells mediated glucose uptake and lactate accumulation suppress glycoses and mTOR activity in T cells. (2) Accumulation of long-chain fatty acids (LCFAs) and lipid abnormalities within T cells causes lipotoxicity and mitochondrial dysfunction. (3) Amino acid depletion by enzymes such as arginase 1 (ARG1) and IDO disrupts TCR signaling and generates immunosuppressive metabolites such as adenosine. These metabolic pathways collectively drive T cell dysfunction and represent potential therapeutic targets. Image created with bioRender.com, with permission. Created in BioRender. Zhou, P. (2025) https://BioRender.com/8h0fjul.
Emerging evidence challenges the notion that immune dysfunction arises solely from tumor-driven nutrient deprivation. Reinfeld et al. demonstrated that myeloid cells, rather than T cells or tumor cells, exhibit the highest glucose uptake, while tumor cells preferentially rely on glutamine metabolism (68). These distinct metabolic programs are governed by intrinsic cellular programming mechanisms including differential mTORC1 activity and metabolic gene expression, rather than extracellular nutrient competition (68). Moreover, inhibiting glutamine metabolism was further shown to enhance glucose uptake across multiple cell types, suggesting a feedback mechanism between glucose and glutamine utilization. These findings emphasizes that immune metabolic dysfunction in the TME is shaped not only by nutrient deprivation but also by cell type-specific cellular metabolic programming, providing novel directions for metabolism-based therapeutic strategies.
Beyond nutrient depletion, additional metabolic barriers, including lactate accumulation, acidic pH, hypoxia, and elevated ROS, further contribute to T cell dysfunction by reprogramming metabolism and upregulating immune checkpoint expression (72). Notably, PD-L1 blockade has been shown to enhance T cell infiltration and metabolic fitness in glycolysis-low tumors (78). Conversely, inhibition of lactate dehydrogenase (LDHA) impairs CD8+ T cell migration, proliferation, and effector functions (70), while blockade of OGR1 in melanoma restores CD8+ T cell cytotoxic activity (71).
Together, glucose dysregulation in the TME not only hinders T cell effector functions but also increases the immune checkpoint expression and exhaustion, constituting a key mechanism of tumor immune evasion. These insights underscore the therapeutic potential of reprograming glucose metabolism by enhancing T cell glycolytic capacity, restraining tumor glycolysis, or targeting glutamine-glucose metabolic crosstalk, to overcome metabolic barriers and enhance immunotherapeutic efficacy (81).
3.1.2 Lipids
The interplay between lipids and CD8+ T cell dysfunction within the TME has attracted growing interest, revealing complex mechanisms by which lipid accumulation and metabolism shape anti-tumor immunity. Lipid metabolism dichotomizes into opposing immunomodulatory pathways within the TME: one suppresses CD8+ T cell effector function (118–120), while the other sustains or enhances CD8+ T cell activation (121). This section highlights the specific immunosuppressive lipids present in the TME and delineate the mechanisms by which they impair CD8+ T cell activity (Figure 3). For example, intrapancreatic CD8+ T cells exhibit downregulation of very-long-chain acyl-CoA dehydrogenase (VLCAD), exacerbating the accumulation of lipotoxic long-chain fatty acids (LCFAs) and VLCFAs (82). Metabolic reprogramming through enforced VLCAD expression enhanced intratumorally T cell survival and persistence in a pancreatic ductal adenocarcinoma (PDA) mouse model, overcoming a major immunotherapy hurdle (82). LCFAs such as palmitate impede CD8+ T cell proliferation and effector cytokine production (83). Among unsaturated fatty acids, oleic acid and linoleic acid exert divergent effects on tumor progression: linoleic acid reprograms tumor-infiltrating CD8+ T cells from an exhausted phenotype towards a memory-like state, potentiating their effector function (122). Arachidonic acid induces ferroptosis in tumor cells but may concurrently trigger ferroptosis in tumor-infiltrating CD8+ T cells (82). The TME induces lipid droplet accumulation in dysfunctional CD8+ TILs through acetyl-CoA carboxylase-mediated metabolic reprogramming (84). Prostaglandin E2 impairs IL-2 sensing in human CD8+ T cells, promoting oxidative stress and ferroptosis (85). Cholesterol and its derivatives critically modulate CD8+ T cell function in context-dependent manner: cholesterol enhances TCR signaling, yet tumor cells derived PCSK9 dysregulates CD8+ T cell cholesterol metabolism, thereby suppressing TCR signaling (123), while the oxysterol 27-hydroxycholesterol facilitates metastasis, an effect potently suppressed by CYP27A1 inhibition (124). Notably, in pancreatic tumors, CD8+ T cell accumulation of LCFAs impairs mitochondrial function and fatty acid catabolism, recapitulating the proliferative and cytokine defects observed upon in vitro palmitate treatment (82). Rather than serving as an energy source, these accumulated lipids impair mitochondrial function and induce transcriptional reprogramming of lipid metabolism pathways, ultimately hampering CD8+ T cell metabolic fitness and anti-tumor activity (82).
3.1.3 Amino acid
The TME orchestrates a complex metabolic interplay where amino acid availability profoundly impacts the functionality of CD8+ T cells through diverse mechanisms (Figure 3). Amino acids serve as critical substrates for various cellular processes such as protein synthesis, epigenetic modifications (e.g., SAM-dependent methylation), and energy metabolism, making them highly contested resources between tumor cells and T cells. For example, in activated T cells, extracellular alanine is preferentially utilized for protein synthesis rather than catabolism. Arginine catabolism by arginase 1 (ARG1) and inducible nitric oxide synthase (iNOS) impairs TCR function by downregulating the CD3ξ chain expression (86). Moreover, ARG1-containing extracellular vesicles can traffic to draining lymph nodes, where their uptake by dendritic cells suppresses antigen-specific T-cell proliferation, as demonstrated in ovarian carcinoma models (87, 88). Adenosine further compromises T cell function and metabolic fitness through the A2AR/PKA/mTORC1 pathway, dampening both peripheral and tumor-infiltrating CD8+ T cells (91, 92). Alanine deprivation delays the activation of naive and memory T cells (125), although it has limited effects on T cell effector function. In contrast, glutamine deprivation restricts metabolic flexibility, while SLC7A11, a multi-pass transmembrane protein, driven cysteine depletion promotes oxidative stress (89). L-ornithine has been shown to suppress T cell functionality, as observed in murine models of chronic viral infection where altered expression of hepatic urea cycle enzymes results in L-ornithine accumulation, leading to the inhibition of virus-specific CD8+ T cell responses (126). Similarly, tryptophan depletion triggers GCN2-mediated stress responses that suppress mTOR signaling, further restricting T cell activity (90).
Collectively, these metabolic perturbations disrupt T cell activation, proliferation, and the production of effector molecules, thereby contributing to immunotherapy resistance. Targeting this metabolic axis offers novel therapeutic strategies, such as inhibiting ARG1 or GLS in combination with immune checkpoint blockade, may restore amino acid homeostasis and reinvigorate antitumor immunity. Such strategies highlight a promising frontier that integrates metabolic and immunological intervention to overcome treatment resistance.
3.2 Exosomes
In various cancers, exosomes derived from tumor cells or stromal cells carry molecular cargo that induces dysfunction or exhaustion of CD8+ T cells, thereby facilitating tumor progression and resistance to immunotherapy. Exosomes suppress CD8+ T cell function and promote their apoptosis through two primary mechanisms: (1) delivery of immunosuppressive molecules and (2) ligand-receptor interactions that trigger contact-dependent signaling.
In the first route, exosomes transport inhibitory factors including cytokines [e.g., TGF-β (93), IL-8 (94)], regulatory miRNAs [e.g., microRNAs (93, 95) and circRNA (96)], and metabolic modulators (97) (e.g., lactate dehydrogenase LDHA and lactate). These cargos collectively impair T cell activation, disrupt inflammatory signaling pathways (eg. STAT1-IFN-γ) and compromise glycolytic metabolism. In the second route, exosome surface ligands, including PD-L1 (127) and FasL, engage corresponding receptors on CD8+ T cells, driving exhaustion or apoptosis. Together, these coordinated immunosuppressive actions establish exosomes as critical mediators of T cell dysfunction in cancer, while also presenting potential therapeutic targets for enhancing immunotherapies. Recent studies demonstrate the breadth of this regulation. For example, Fan Xu et al. showed that IL-8 in exosomes derived from prostate cancer cells hyperactivates peroxisome proliferators-activated receptors (PPARα) in recipient CD8+ T cells, which downregulates GLUT1 and hexokinase 2 to reduce glucose utilization while upregulating Carnitine O-palmitoyltransferase 1 and peroxisomal acyl-coenzyme A oxidase 1 to enhance fatty acid catabolism, ultimately exacerbating CD8+ T cell starvation and promoting cellular exhaustion (94). Non-small cell lung cancer (NSCLC) cells release circUSP7 via exosome secretion, which upregulates SHP2 expression by sponging miR-934, thereby inhibiting CD8+ T cell secretion of IFN-γ, TNF-α, granzyme B, and perforin and ultimately suppressing CD8+ T cell function (128). Another example is the exosome circCCAR1, which is taken up by CD8+ T cells and induces CD8+ T cell dysfunction by stabilizing PD-1 protein (96). Collectively, these studies delineate a complex network whereby tumor and stromal cell-derived exosomes carry diverse molecular cargos, including circRNAs, cytokines and proteins, that induce CD8+ T cell dysfunction, in addition offering novel opportunities for therapeutic targets.
3.3 Nanotubes and trogocytosis
The contribution of nanotubes and trogocytosis in regulating CD8+ T cell function within the TME has become an emerging area, particularly regarding intercellular mitochondrial transfer and its consequences on T cell efficacy. Mitochondrial dysfunction in CD8+ T cells represents a fundamental driver of T cell exhaustion in tumor contexts, making these intercellular communication mechanisms highly relevant to tumor immune evasion.
Current evidence reveals that nanotube-mediated mitochondrial transfer exhibits dual functionality. On one hand, nanotubes can restore T cell metabolic activity by delivering functional mitochondria; on the other hand, tumor cells often exploit this progress to transfer dysfunctional mitochondria containing mutations or oxidative damage, thereby promoting T cell failure. The principal inhibitory mechanisms of nanotubes toward CD8+ T cells encompass metabolic subversion through mitochondrial hijacking (98) and nutrient deprivation (99). Using multimodal imaging and metabolic profiling, Tanmoy Saha et al. demonstrated that cancer cells hijack mitochondria from immune cells via tunneling nanotubes, simultaneously depleting immune cell function while metabolically empowering tumor cells (129). In contrast, Jeremy G. Baldwin et al. showed that bone marrow stromal cells transfer healthy mitochondria to CD8+ T cells through intercellular nanotubes, thereby restoring CD8+ T cell function and promoting anti-tumor responses (98). Together, these findings highlight the complex, context-dependent role of nanotubes in immune regulation and underscore their potential as therapeutic targets in cancer immunotherapy.
Trogocytosis, the direct transfer of membrane fragments and regulatory molecules during cell-cell contact, also play a crucial role on T cell function. In the TME, CD8+ T cells that acquire inhibitory molecules from APCs or tumor cells can undergo suppression of cytokine production and proliferation through reverse signaling (45). Mechanistically, trogocytosis in CD8+ T cells, where they acquire inhibitory ligands or pMHC complexes, can promote immune evasion, leading to T cell exhaustion mainly through “self-inhibition” (100) and “antigen loss” (101). For example, Lu et al. demonstrated that activation of trogocytosis in intratumoral CTL through the ATF3-CH25H axis dampened the anti-tumor immune response (100). Notably, CD8+ T cells engage in cell-to-cell material exchange by obtaining pMHC from APCs or tumor cells in a TCR-dependent manner, may themselves become targets for killing by neighboring CD8+ T cells (101, 102). While trogocytosis may prolong antigen receptor engagement and transiently enhance activation, sustained or excessive trogocytosis promote exhaustion (130). From a translational perspective, engineering CAR-T cells to resistant trogocytosis or to avoid the acquisition of inhibitory signals could improve their persistence and therapeutic efficacy in tumors (100, 131).
3.4 Cytokines
Multiple studies have elucidated the pivotal roles of cytokine signaling and the inhibitory receptor upregulation in driving CD8+ T cell dysfunction within the TME. Cytokines impair CD8+ T cell proliferation, cytotoxicity (e.g., granzyme B and perforin expression), and effector functions by inducing exhaustion, metabolic inhibition, and apoptosis. For example, TGF-β and IL-2 suppress CD8+ T cell proliferation and cytotoxic activity (103, 104). Mechanistically, TGF-β reduces CXCR3 expression by binding to the CXCR3 promoter through Smad2, thereby diminishing CD8+ T cell responsiveness to CXCL10. Ablation of the TGF-β receptor I (ALK5) restores CXCR3 expression, enhances T cell infiltration and cytotoxicity, and promotes tumor regression, these effects are partially reversed by CXCR3 blockade. Furthermore, chronic TGF-β1 signaling orchestrates terminal dysfunction of CD8+ T cells through stable epigenetic reprogramming (17). Rebalancing TGF-β1-BMP signaling, for instance with BMP4 agonist SB4, preserves effector-memory programs, reduces exhaustion marker expression, enhances anti-tumor responses, and synergizes with ICB by restoring T cells responsive state.
IL-2 plays pivotal roles in regulating CD8+ T cell proliferation, effector function, exhaustion, memory formation, and metabolic adaptability (132). Recent findings underscore the context-dependent effects of IL-2: while elevated IL-2 transiently enhance the proliferation and effector functions of CD25hi CD8+ T cells, they also accelerate exhaustion (133–135). In chronic stimulatory settings such as tumor microenvironments, sustained IL-2 signaling drives CD8+ T cell exhaustion through STAT5-mediated tryptophan hydroxylase 1 upregulation, generating 5-hydroxytryptophan that promotes inhibitory receptor expression and suppress effector function, revealing a conserved metabolic-epigenetic axis of T cell dysfunction in both mouse and human systems (104). Clinically, high-dose interleukin-2 (HD IL-2) has been employed for the treatment of advanced melanoma and renal cell carcinoma (136, 137), whereas low-dose recombinant human IL-2 selectively modulates the abundance of regulatory T (Treg) cells, follicular helper T (TFH) cells and IL-17-producing helper T (TH17) cells (138). Through these effects, IL-2 promotes the development and survival of Treg cells while inhibiting the differentiation of TFH and TH17 subsets, thereby reshaping the immune milieu. Currently, multiple IL-2-based products are under clinical and pre-clinical investigation, requiring evaluation of their effects to reprogram dysfunctional state of anti-tumor CD8+ T cells. Modulation of CD8+ T cell exhaustion programs by IL-2 to promote the generation of effector cells with stem-like properties provides the immunological rationale for the combination therapy of IL-2 with PD-1 blockade (136, 139). Furthermore, engineered IL-2 partial agonists have been shown to preserve the stem-like properties and mitochondrial fitness of CD8+ T cells, thereby enhancing anti-tumor immunity (140). In parallel, IL-6-STAT3 signaling, activated by STK31, also promotes CD8+ T cell exhaustion in tumors (105), while IL-18 released in the TME through inflammasome activation drives T-cell exhaustion via IL2-STAT5 and AKT-mTOR signaling downstream of IL-18R (106).
Cytokine pathways also intersect with inhibitory receptor regulation. IL-27 upregulates PD-1 expression via STAT1 signaling yet paradoxically sustains CD8+ T cell activity and synergizes with PD-1- PD-L1 blockade (107). Although IL-10 is classically categorized as immunosuppressive through its ability to induce inhibition, recent work suggests that IL-10 alleviates T cell exhaustion by promoting oxidative phosphorylation (OXPHOS) in PD-1+ TIM-3+ CD8+ T cells. An IL-10-Fc fusion protein acts through IL-10 receptors on T cells to specifically enhance OXPHOS, proliferation and cytotoxicity in this subset, thereby reversing exhaustion and enhancing anti-tumor response (108, 109). Conversely, Treg-derived IL-10 and IL-35 cooperatively upregulate the expression of multiple inhibitory receptors and drive BLIMP1-dependent exhaustion of tumor infiltration CD8+ T cells, further impeding antitumor immunity (110).
Collectively, these findings underscore the central role of cytokine-mediated signaling networks and inhibitory receptor upregulation in orchestrating CD8+ T cell dysfunction within TME, emphasizing the therapeutic potential of targeting these pathways to reinvigorate anti-tumor immunity.
3.5 Ions and metabolites (Mg2+, Lithium and Ammonia)
The immune function of CD8+ T cells is profoundly affected by various ions and metabolites that modulate signaling and metabolic fitness. Magnesium (Mg²+) functions as a critical second messenger that regulates CD8+ T cell activity through metabolic circuits that sustain effector functions. Deficiency of intracellular free Mg²+ impairs NKG2D receptor expression on both NK cells and CD8+ T cells, thereby compromising cytotoxic responses against pathogens such as Epstein-Barr virus (111). Lithium, widely used in psychiatric treatment, also exerts immunomodulatory effects on CD8+ T cells. Mechanistically, cytoplasmic lactate promotes lysosomal proton influx, meanwhile lithium prevents lysosomal acidification by inhibiting vacuolar ATPase, thereby restoring diacylglycerol-PKCθ signaling to recruit monocarboxylate transporter 1 to mitochondria. This enabled lactic acid transport into mitochondria for CD8+ T-cell energy production (112). Ammonia functions as a potent immunosuppressive metabolite within the TME. Elevated ammonia levels reprogram T cell metabolism, leading to exhaustion and proliferation arrest (113). Mechanistically, ammonia accumulation increases lysosomal pH, impairs lysosomal ammonia trapping capacity. This causes ammonia reflux into mitochondria, triggering mitochondrial damage and subsequent cell death (141). Collectively, these findings highlight distinct roles for ions and metabolites in shaping CD8+ T anti-tumor immunity.
Indirect suppression in the TME operates through tightly interconnected metabolic, vesicular, structural, and cytokine mediated pathways. These circuits converge CD8+ T cells to impair metabolism, signaling, and effector function, driving exhaustion and immune escape. Understanding and therapeutically targeting these mechanisms will be essential for restoring durable anti-tumor immunity.
3.6 Integrative strategies to restore T cell function
The progress of immunotherapy has been driven by advances in immune checkpoint research, leading to the clinical approval of adoptive T cell therapy (142, 143). However, CAR-T cell therapies show limited efficacy in many solid tumors and are often linked to immune-related adverse events (144–146). Studies have shown that impaired mitochondrial quality in TILs reduces cytokine secretion and increases the expression of co-inhibitory receptors, while tertiary lymphoid structures in several cancers characterized by chronic inflammatory signaling (147). Moreover, the TME frequently lacks the pro-inflammatory cues or innate immune activation required for optimal T cell priming and expansion, thereby constraining therapeutic efficacy.
To overcome these barriers, emerging strategies aim to synergize innate immune activation with pro-inflammatory stimuli, extending therapeutic benefit beyond checkpoint inhibition, including nutritional interventions (148), oncolytic viruses (149), cGAS-STING agonists (150, 151), cytokine therapy (152), mitochondrial function modulation (153), and vaccine development (149, 154). Addressing metabolic dysregulation, such as lactic acid accumulation in TME (155–157), is particularly critical for maintaining T-cell stemness, emphasizing the importance of mitochondrial fitness in adoptive transfer approaches. Although IL-2 monotherapy showed early promise in metastatic renal cell carcinoma (RCC) and melanoma, its clinical utility was limited by toxicity and Treg activation, prompting a shift toward combination regimens (158, 159). Similarly, pharmacological activation of K+ channels, such as with riluzole, a non-specific activator of the KCa3.1 channel, enhances cisplatin uptake in colorectal cancer patients with cisplatin resistance (160).
Improving the efficacy of ICIs requires addressing secondary inhibitory barriers in the TME, including immune-suppressive metabolite accumulation (113), nutrient competition (68), ion imbalances (e.g., high potassium environment), hypoxia, and acidosis-related metabolic hindrances (161, 162). Overcoming these multifactorial constraints is essential for fully unleashing the cytotoxic potential of T cells. Preclinical studies demonstrate that multi-targeted approaches can enhance antitumor efficacy, such as M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β (163). In addition, innovative platforms such as nanotube- and exosome-based drug delivery systems (164) and CRISPR-Cas9-based genetic engineering (165) are expanding therapeutic possibilities in personalized gene therapy.
4 Discussion
Effective antitumor immunity critically depends on functional CD8+ T cells, whose suppression within the TME constitutes a major immune escape mechanism. This suppression occurs through two major routes: (1) Direct cell-to-cell interactions, including tumor cell-CD8+ T cell contact (e.g., PD-L1-PD-1), inhibitory signals from CAFs, and immune cell crosstalk (e.g., DC-macrophage-T cell interactions); and (2) Indirect TME-driven mechanisms, such as metabolic competition (nutrient deprivation), intercellular communication (exosomes, tunneling nanotubes, T cell trogocytosis), and microenvironmental perturbations involving immunosuppressive cytokine networks (TGF-β), ionic imbalances (e.g., Mg²+ deficiency), and metabolite accumulation (e.g., ammonia).
Within this suppressive networks, CD8+ T cell function is progressively impaired by diverse suppressive cues. Recent studies highlight that tumors directly suppress CD8+ T cells via inhibitory ligand-receptor interactions, most prominently through the PD-1-PD-L1 axis and the CTLA-4-B7-1 (CD80)-B7-2 (CD86) pathway (56, 86, 166, 167). Additionally, APCs and CAFs suppress CD8+ T cell function by engaging CTLA-4 on activated CD8+ T cells, thereby constraining the availability of co-stimulatory signals. ICB therapies targeting PD-1-PD-L1, CTLA-4, and LAG-3 have improved survival in multiple cancers (54, 55). However, complete and durable responses remain limited, largely due to tumor heterogeneity, compensatory pathways, and the multifaceted suppressive networks in the TME (56, 168, 169). These limitations underscore the need for complementary or combinatorial strategies that extend beyond classical checkpoint inhibition. A2AR antagonists counteract adenosine-mediated immunosuppression in the TME, thereby restoring T cell-mediated tumor killing (170, 171). Currently, several A2AR antagonists (e.g., AZD4635, CPI-444, AB928) have advanced into Phase II clinical development for indications including prostate cancer and NSCLC (172). Notably, although these candidates vary in developmental stage and tumor type, they demonstrate synergistic effects when combined with PD-1/PD-L1 inhibitors, exhibiting superior antitumor activity compared to either agent alone (171). These findings highlight the potential of targeting metabolic pathways and nutrient competition presents promising avenue to enhance effector responses (169, 173, 174).
Despite these advances, our understanding of how direct and indirect communication networks suppress CD8+ T cells in TME remain incomplete. A key challenge lies in decoding these interactions at sufficient resolution, cutting-edge platforms such as spatial resolved transcriptomics, single-cell CRISPR screening (175), nanotherapeutics (176, 177) are now being leveraged to dissect TME-T cell interaction at cellular and molecular levels. Likewise, clinical strategies like CAR-T cell therapy (178, 179) and bispecific antibodies (180) provided translational opportunities for targeting these networks. In particularly, extracellular vesicle-mediated signaling (e.g., exosomes and tunneling nanotubes) represents an underexplored mechanism of tumor-driven immune evasion and a potential strategy of novel therapeutic targets.
Importantly, the functional state of CD8+ T cells is tightly dictated by their local microenvironment niche, which is defined by spatial position and communicative interactions with neighboring cells. Ligand-receptor pairs are emerging as critical determinants of these intercellular communication (181, 182). Advances in single-cell and spatial multi-omics allow the dissection of these networks at both cellular and molecularly levels (183). In the parallel, advanced computational framworks enable the systematic analysis of immune infiltration, inference of cell phenotypes, spatial mapping of cellular interactions, and discovery of novel cell-cell communication events, with tools such as CellTalker, PyMINEr, CCCExplorer, SoptSC, NicheNet, CellPhoneDB, CellChat, and CSOmap (184–186).
Another major clinical challenge is the early prediction of immunotherapy efficacy (187). Platforms such as the gel-liquid interface co-culture model have recapitulated human immunity and tumor microenvironment interactions and identified circulating tumor-reactive T cells as biomarkers of treatment response in lung cancers (188). Integration of such ex vivo systems with omics and computational pipelines may accelerate biomarkers discovery.
Therapeutic strategies is increasingly focused on multi-target synergistic interventions (54). Dual-blockade strategies, such as combined PD-1-PD-L1 and TIGIT blockade (189), and tri-blockade regimes, integrating epigenetic modulators (e.g., HDAC inhibitors) with anti-angiogenic agents and PD-1 antibodies, have shown promise in refractory solid tumors by simultaneously remodeling the TME and restoring T cell function (190). Beyond blockade, and emerging therapeutic approach aims to sustain long-term T-cell function by preventing over-activation. An Fc-attenuated LAG-3-TCR bispecific antibody has been engineered to suppress T cell activity independently of MHC-II, demonstrating therapeutic potential in autoimmune models and offering a new avenue for sustaining T-cell function in cancer immunotherapy (191).
Collectively, the intrinsic cellular composition of the TME, coupled with pervasive immune evasion and multifaceted crosstalk, highlights the need for integrative therapeutic strategies that simultaneously target direct inhibitory interactions, metabolic competition, and intercellular communication.
Author contributions
LC: Writing – original draft, Writing – review & editing. QH: Writing – original draft, Writing – review & editing. PZ: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the National Natural Science Foundation of China (NSFC82572098) (to P.Z.), the Young Talent Program of China (HJJH 2025-2027) (to P.Z.), and the Guangzhou National Laboratory Start-up Funding (GZNL2025C01038) (to P.Z.).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
TME: Tumor microenvironment
CTLs: Cytotoxic T lymphocytes
DCs: Dendritic cells
IL-12: interleukin-12
IFN-γ: Interferon-gamma
PD-1: Programmed cell death protein 1
CTLA-4: Cytotoxic T-lymphocyte associated protein 4
Tmp: Memory precursor T cells
Tpex: Progenitor of exhausted T cells
TLS: Tertiary lymphoid structures
TCF1: T-cell factor 1
ICB: Immune checkpoint blockade
Trm: Tissue-resident memory T cells
TAMs: Tumor‐associated macrophages
TGF-βR: Transforming growth factor β receptor
B7-H3: B7 homolog 3
HLA: Human leukocyte antigen
LAG-3: Lymphocyte-activation gene 3
NKG2A: Natural killer group 2 member A
Tregs: Regulatory T cells
MDSCs: Myeloid-derived suppressor cells
CAFs: Cancer-associated fibroblasts
Ceacam-1: Carcinoembryonic antigen-related cell adhesion molecule 1
ZAP70: Zeta-chain-associated protein kinase 70
LSECtin: Liver and lymph node sinusoidal endothelial cell C-type lectin
LUAD: Lung adenocarcinoma
PVRL2: Poliovirus receptor-related protein 2
ERAP1-2: Endoplasmic reticulum aminopeptidase 1-2
NKG2A: Natural killer cell group 2 member A
TILs: Tumor-infiltrating lymphocytes
APCs: Antigen-presenting cells
VISTA: V-domain immunoglobulin suppressor of T cell activation
LRIG1: Ligand for immunoglobulin-like domains 1
FDCs: Follicular dendritic cells
TNF-α: Tumor necrosis factor-alpha
NK: Natural Killer
CCR7: C-C motif chemokine receptor 7
HVEM: Herpes virus entry mediator
BTLA: B and T lymphocyte attenuator
ICIs: Immune checkpoint inhibitors
VEGF: Vascular endothelial growth factor
TNBC: Triple-negative breast cancer
MMP2: Matrix metalloproteinase-2
GLUT1: Glucose transporters glucose transporter 1
AMPK: AMP-activated protein kinase
LDHA: Lactate dehydrogenase
VLCAD: Very-long-chain acyl-CoA dehydrogenase
LCFAs: Long-chain fatty acids
PDA: Pancreatic ductal adenocarcinoma
ARG1: Arginine catabolism by arginase 1
PPARα: peroxisome proliferators-activated receptors
NSCLC: Non-small cell lung cancer
iNOS: Inducible nitric oxide synthase
STAT3: Signal Transducer and Activator of Transcription 3
Mg²⁺: Magnesium
OXPHOS: Oxidative phosphorylation
scTCR-seq: Single-cell T cell receptor sequencing.
References
1. Reina-Campos M, Scharping NE, and Goldrath AW. CD8(+) T cell metabolism in infection and cancer. Nat Rev Immunol. (2021) 21:718–38. doi: 10.1038/s41577-021-00537-8
2. Golstein P and Griffiths GM. An early history of T cell-mediated cytotoxicity. Nat Rev Immunol. (2018) 18:527–35. doi: 10.1038/s41577-018-0009-3
3. Baessler A and Vignali DAA. T cell exhaustion. Annu Rev Immunol. (2024) 42:179–206. doi: 10.1146/annurev-immunol-090222-110914
4. Gebhardt T, Park SL, and Parish IA. Stem-like exhausted and memory CD8(+) T cells in cancer. Nat Rev Cancer. (2023) 23:780–98. doi: 10.1038/s41568-023-00615-0
5. Chung HK, McDonald B, and Kaech SM. The architectural design of CD8+ T cell responses in acute and chronic infection: Parallel structures with divergent fates. J Exp Med. (2021) 218:e20201730. doi: 10.1084/jem.20201730
6. Sun L, Su Y, Jiao A, Wang X, and Zhang B. T cells in health and disease. Signal Transduct Target Ther. (2023) 8:235. doi: 10.1038/s41392-023-01471-y
7. Hu Y, Zhao Q, Qin Y, Mei S, Wang B, Zhou H, et al. CARD11 signaling regulates CD8(+) T cell tumoricidal function. Nat Immunol. (2025) 26:1113–26. doi: 10.1038/s41590-025-02192-w
8. Philip M and Schietinger A. CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol. (2022) 22:209–23. doi: 10.1038/s41577-021-00574-3
9. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
10. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature. (2017) 548:537–42. doi: 10.1038/nature23477
11. Petitprez F, de Reyniès A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. (2020) 577:556–60. doi: 10.1038/s41586-019-1906-8
12. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. (2020) 577:561–5. doi: 10.1038/s41586-019-1914-8
13. Parra ER. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Front Mol Biosci. (2021) 8:668340. doi: 10.3389/fmolb.2021.668340
14. Feng Y, Ma W, Zang Y, Guo Y, Li Y, Zhang Y, et al. Spatially organized tumor-stroma boundary determines the efficacy of immunotherapy in colorectal cancer patients. Nat Commun. (2024) 15:10259. doi: 10.1038/s41467-024-54710-3
15. Wang XQ, Danenberg E, Huang CS, Egle D, Callari M, Bermejo B, et al. Spatial predictors of immunotherapy response in triple-negative breast cancer. Nature. (2023) 621:868–76. doi: 10.1038/s41586-023-06498-3
16. Nixon BG, Kuo F, Ji L, Liu M, Capistrano K, Do M, et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity. (2022) 55:2044–58.e5. doi: 10.1016/j.immuni.2022.10.002
17. Saadey AA, Yousif A, Osborne N, Shahinfar R, Chen YL, Laster B, et al. Rebalancing TGFbeta1/BMP signals in exhausted T cells unlocks responsiveness to immune checkpoint blockade therapy. Nat Immunol. (2023) 24:280–94. doi: 10.1038/s41590-022-01384-y
18. Scirgolea C, Sottile R, De Luca M, Susana A, Carnevale S, Puccio S, et al. NaCl enhances CD8(+) T cell effector functions in cancer immunotherapy. Nat Immunol. (2024) 25:1845–57. doi: 10.1038/s41590-024-01923-9
19. Yang MQ, Zhang SL, Sun L, Huang LT, Yu J, Zhang JH, et al. Targeting mitochondria: restoring the antitumor efficacy of exhausted T cells. Mol Cancer. (2024) 23:260. doi: 10.1186/s12943-024-02175-9
20. Chi H, Deng S, Xu K, Zhang Y, Song T, Yu J, et al. SEMA3G-NRP1 signaling functions as an immune checkpoint that enables tumor immune evasion by impairing T-cell cytotoxicity. Cancer Res. (2025) 85:912–24. doi: 10.1158/0008-5472.CAN-24-2223
21. Zheng Y, Yao Y, Ge T, Ge S, Jia R, Song X, et al. Amino acid metabolism reprogramming: shedding new light on T cell anti-tumor immunity. J Exp Clin Cancer Res. (2023) 42:291. doi: 10.1186/s13046-023-02845-4
22. Jenkins RW, Thummalapalli R, Carter J, Cañadas I, and Barbie DA. Molecular and genomic determinants of response to immune checkpoint inhibition in cancer. Annu Rev Med. (2018) 69:333–47. doi: 10.1146/annurev-med-060116-022926
23. Zang X, Loke P, Kim J, Murphy K, Waitz R, and Allison JP. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci U S A. (2003) 100:10388–92. doi: 10.1073/pnas.1434299100
24. John P, Pulanco MC, Galbo PM Jr., Wei Y, Ohaegbulam KC, Zheng D, et al. The immune checkpoint B7x expands tumor-infiltrating Tregs and promotes resistance to anti-CTLA-4 therapy. Nat Commun. (2022) 13:2506. doi: 10.1038/s41467-022-30143-8
25. Li Y, Liu Y, Zhao N, Yang X, Li Y, Zhai F, et al. Checkpoint regulator B7x is epigenetically regulated by HDAC3 and mediates resistance to HDAC inhibitors by reprogramming the tumor immune environment in colorectal cancer. Cell Death Dis. (2020) 11:753. doi: 10.1038/s41419-020-02968-y
26. MacGregor HL and Ohashi PS. Molecular pathways: evaluating the potential for B7-H4 as an immunoregulatory target. Clin Cancer Res. (2017) 23:2934–41. doi: 10.1158/1078-0432.CCR-15-2440
27. Anderson AC, Joller N, and Kuchroo VK. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
28. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. (2014) 74:3418–28. doi: 10.1158/0008-5472.CAN-13-2690
29. Feng M, Wu Z, Zhou Y, Wei Z, Tian E, Mei S, et al. BCL9 regulates CD226 and CD96 checkpoints in CD8(+) T cells to improve PD-1 response in cancer. Signal Transduct Target Ther. (2021) 6:313. doi: 10.1038/s41392-021-00730-0
30. Zhang H, Liu Q, Lei Y, Zhou J, Jiang W, Cui Y, et al. Direct interaction between CD155 and CD96 promotes immunosuppression in lung adenocarcinoma. Cell Mol Immunol. (2021) 18:1575–7. doi: 10.1038/s41423-020-00538-y
31. Zeng T, Cao Y, Jin T, Tian Y, Dai C, and Xu F. The CD112R/CD112 axis: a breakthrough in cancer immunotherapy. J Exp Clin Cancer Res. (2021) 40:285. doi: 10.1186/s13046-021-02053-y
32. Murter B, Pan X, Ophir E, Alteber Z, Azulay M, Sen R, et al. Mouse PVRIG has CD8(+) T cell-specific coinhibitory functions and dampens antitumor immunity. Cancer Immunol Res. (2019) 7:244–56. doi: 10.1158/2326-6066.CIR-18-0460
33. Tsao HW, Anderson S, Finn KJ, Perera JJ, Pass LF, Schneider EM, et al. Targeting the aminopeptidase ERAP enhances antitumor immunity by disrupting the NKG2A-HLA-E inhibitory checkpoint. Immunity. (2024) 57:2863–78.e12. doi: 10.1016/j.immuni.2024.10.013
34. Eugène J, Jouand N, Ducoin K, Dansette D, Oger R, Deleine C, et al. The inhibitory receptor CD94/NKG2A on CD8(+) tumor-infiltrating lymphocytes in colorectal cancer: a promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod Pathol. (2020) 33:468–82. doi: 10.1038/s41379-019-0322-9
35. Yang F, Zeng Z, Li J, Ren X, and Wei F. TIM-3 and CEACAM1 are prognostic factors in head and neck squamous cell carcinoma. Front Mol Biosci. (2021) 8:619765. doi: 10.3389/fmolb.2021.619765
36. Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. (2021) 53:1334–47. doi: 10.1038/s41588-021-00911-1
37. Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. (2020) 580:257–62. doi: 10.1038/s41586-020-2134-y
38. Ding S, Qiao N, Zhu Q, Tong Y, Wang S, Chen X, et al. Single-cell atlas reveals a distinct immune profile fostered by T cell-B cell crosstalk in triple negative breast cancer. Cancer Commun (Lond). (2023) 43:661–84. doi: 10.1002/cac2.12429
39. Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature. (2015) 521:94–8. doi: 10.1038/nature14395
40. Ta HM, Roy D, Zhang K, Alban T, Juric I, Dong J, et al. LRIG1 engages ligand VISTA and impairs tumor-specific CD8(+) T cell responses. Sci Immunol. (2024) 9:eadi7418. doi: 10.1126/sciimmunol.adi7418
41. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. (2011) 208:577–92. doi: 10.1084/jem.20100619
42. Martin AS, Molloy M, Ugolkov A, von Roemeling RW, Noelle RJ, Lewis LD, et al. VISTA expression and patient selection for immune-based anticancer therapy. Front Immunol. (2023) 14:1086102. doi: 10.3389/fimmu.2023.1086102
43. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
44. Walker LS and Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. (2011) 11:852–63. doi: 10.1038/nri3108
45. Tekguc M, Wing JB, Osaki M, Long J, and Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U S A. (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
46. Josefsson SE, Beiske K, Blaker YN, Førsund MS, Holte H, Østenstad B, et al. TIGIT and PD-1 mark intratumoral T cells with reduced effector function in B-cell non-hodgkin lymphoma. Cancer Immunol Res. (2019) 7:355–62. doi: 10.1158/2326-6066.CIR-18-0351
47. Sierra JM, Secchiari F, Nuñez SY, Iraolagoitia XLR, Ziblat A, Friedrich AD, et al. Tumor-experienced human NK cells express high levels of PD-L1 and inhibit CD8(+) T cell proliferation. Front Immunol. (2021) 12:745939. doi: 10.3389/fimmu.2021.745939
48. Mao FY, Kong H, Zhao YL, Peng LS, Chen W, Zhang JY, et al. Increased tumor-infiltrating CD45RA(-)CCR7(-) regulatory T-cell subset with immunosuppressive properties foster gastric cancer progress. Cell Death Dis. (2017) 8:e3002. doi: 10.1038/cddis.2017.388
49. Zappasodi R, Merghoub T, and Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. (2018) 33:581–98. doi: 10.1016/j.ccell.2018.03.005
50. Guruprasad P, Carturan A, Zhang Y, Cho JH, Kumashie KG, Patel RP, et al. The BTLA-HVEM axis restricts CAR T cell efficacy in cancer. Nat Immunol. (2024) 25:1020–32. doi: 10.1038/s41590-024-01847-4
51. Machicote A, Belén S, Baz P, Billordo LA, and Fainboim L. Human CD8(+)HLA-DR(+) regulatory T cells, similarly to classical CD4(+)Foxp3(+) cells, suppress immune responses via PD-1/PD-L1 axis. Front Immunol. (2018) 9:2788. doi: 10.3389/fimmu.2018.02788
52. Kawasaki K, Noma K, Kato T, Ohara T, Tanabe S, Takeda Y, et al. PD-L1-expressing cancer-associated fibroblasts induce tumor immunosuppression and contribute to poor clinical outcome in esophageal cancer. Cancer Immunol Immunother. (2023) 72:3787–802. doi: 10.1007/s00262-023-03531-2
53. Du Y, Shi J, Wang J, Xun Z, Yu Z, Sun H, et al. Integration of pan-cancer single-cell and spatial transcriptomics reveals stromal cell features and therapeutic targets in tumor microenvironment. Cancer Res. (2024) 84:192–210. doi: 10.1158/0008-5472.CAN-23-1418
54. Borgeaud M, Sandoval J, Obeid M, Banna G, Michielin O, Addeo A, et al. Novel targets for immune-checkpoint inhibition in cancer. Cancer Treat Rev. (2023) 120:102614. doi: 10.1016/j.ctrv.2023.102614
55. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. (2021) 18:345–62. doi: 10.1038/s41571-021-00473-5
56. Carlino MS, Larkin J, and Long GV. Immune checkpoint inhibitors in melanoma. Lancet. (2021) 398:1002–14. doi: 10.1016/S0140-6736(21)01206-X
57. Farhood B, Najafi M, and Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. (2019) 234:8509–21. doi: 10.1002/jcp.27782
58. Luri-Rey C, Teijeira Á, Wculek SK, de Andrea C, Herrero C, Lopez-Janeiro A, et al. Cross-priming in cancer immunology and immunotherapy. Nat Rev Cancer. (2025) 25:249–73. doi: 10.1038/s41568-024-00785-5
59. Zhang P, Liu X, Gu Z, Jiang Z, Zhao S, Song Y, et al. Targeting TIGIT for cancer immunotherapy: recent advances and future directions. biomark Res. (2024) 12:7. doi: 10.1186/s40364-023-00543-z
60. Noel S, Lee K, Gharaie S, Kurzhagen JT, Pierorazio PM, Arend LJ, et al. Immune checkpoint molecule TIGIT regulates kidney T cell functions and contributes to AKI. J Am Soc Nephrol. (2023) 34:755–71. doi: 10.1681/ASN.0000000000000063
61. Wienke J, Visser LL, Kholosy WM, Keller KM, Barisa M, Poon E, et al. Integrative analysis of neuroblastoma by single-cell RNA sequencing identifies the NECTIN2-TIGIT axis as a target for immunotherapy. Cancer Cell. (2024) 42:283–300.e8. doi: 10.1016/j.ccell.2023.12.008
62. Rousseau A, Parisi C, and Barlesi F. Anti-TIGIT therapies for solid tumors: a systematic review. ESMO Open. (2023) 8:101184. doi: 10.1016/j.esmoop.2023.101184
63. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. (2018) 33:649–63.e4. doi: 10.1016/j.ccell.2018.02.010
64. Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G, et al. T-cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors. Clin Cancer Res. (2018) 24:2920–34. doi: 10.1158/1078-0432.CCR-17-1365
65. Biffi G and Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev. (2021) 101:147–76. doi: 10.1152/physrev.00048.2019
66. Zhang Z, Yu Y, Zhang Z, Li D, Liang Z, Wang L, et al. Cancer-associated fibroblasts-derived CXCL12 enhances immune escape of bladder cancer through inhibiting P62-mediated autophagic degradation of PDL1. J Exp Clin Cancer Res. (2023) 42:316. doi: 10.1186/s13046-023-02900-0
67. Zhang P, Qin C, Liu N, Zhou X, Chu X, Lv F, et al. The programmed site-specific delivery of LY3200882 and PD-L1 siRNA boosts immunotherapy for triple-negative breast cancer by remodeling tumor microenvironment. Biomaterials. (2022) 284:121518. doi: 10.1016/j.biomaterials.2022.121518
68. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. (2021) 593:282–8. doi: 10.1038/s41586-021-03442-1
69. Vander Heiden MG, Cantley LC, and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
70. Wang K, Zhang Y, and Chen ZN. Metabolic interaction: tumor-derived lactate inhibiting CD8(+) T cell cytotoxicity in a novel route. Signal Transduct Target Ther. (2023) 8:52. doi: 10.1038/s41392-023-01320-y
71. Cao L, Li W, Yang X, Zhang W, Li M, Zhang H, et al. Inhibition of host Ogr1 enhances effector CD8(+) T-cell function by modulating acidic microenvironment. Cancer Gene Ther. (2021) 28:1213–24. doi: 10.1038/s41417-021-00354-0
72. Song BS, Moon JS, Tian J, Lee HY, Sim BC, Kim SH, et al. Mitoribosomal defects aggravate liver cancer via aberrant glycolytic flux and T cell exhaustion. J Immunother Cancer. (2022) 10:e004337. doi: 10.1136/jitc-2021-004337
73. Wu L, Jin Y, Zhao X, Tang K, Zhao Y, Tong L, et al. Tumor aerobic glycolysis confers immune evasion through modulating sensitivity to T cell-mediated bystander killing via TNF-alpha. Cell Metab. (2023) 35:1580–96.e9. doi: 10.1016/j.cmet.2023.07.001
74. Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. (2013) 16:1373–82. doi: 10.1038/nn.3510
75. Yang G, Cheng J, Xu J, Shen C, Lu X, He C, et al. Metabolic heterogeneity in clear cell renal cell carcinoma revealed by single-cell RNA sequencing and spatial transcriptomics. J Transl Med. (2024) 22:210. doi: 10.1186/s12967-024-04848-x
76. Chen Y, Xu Z, Sun H, Ouyang X, Han Y, Yu H, et al. Regulation of CD8(+) T memory and exhaustion by the mTOR signals. Cell Mol Immunol. (2023) 20:1023–39. doi: 10.1038/s41423-023-01064-3
77. Huang M, Yu X, Wang Q, Jiang Z, Li X, Chen W, et al. The immune checkpoint TIGIT/CD155 promotes the exhaustion of CD8 + T cells in TNBC through glucose metabolic reprogramming mediated by PI3K/AKT/mTOR signaling. Cell Commun Signal. (2024) 22:35. doi: 10.1186/s12964-023-01455-z
78. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, et al. CTLA-4 blockade drives loss of T(reg) stability in glycolysis-low tumours. Nature. (2021) 591:652–8. doi: 10.1038/s41586-021-03326-4
79. Cheng H, Qiu Y, Xu Y, Chen L, Ma K, Tao M, et al. Extracellular acidosis restricts one-carbon metabolism and preserves T cell stemness. Nat Metab. (2023) 5:314–30. doi: 10.1038/s42255-022-00730-6
80. Franco F, Jaccard A, Romero P, Yu YR, and Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab. (2020) 2:1001–12. doi: 10.1038/s42255-020-00280-9
81. Guerrero JA, Klysz DD, Chen Y, Malipatlolla M, Lone J, Fowler C, et al. GLUT1 overexpression in CAR-T cells induces metabolic reprogramming and enhances potency. Nat Commun. (2024) 15:8658. doi: 10.1038/s41467-024-52666-y
82. Manzo T, Prentice BM, Anderson KG, Raman A, Schalck A, Codreanu GS, et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med. (2020) 217:e20191920. doi: 10.1084/jem.20191920
83. Lim SA, Su W, Chapman NM, and Chi H. Lipid metabolism in T cell signaling and function. Nat Chem Biol. (2022) 18:470–81. doi: 10.1038/s41589-022-01017-3
84. Hunt EG, Hurst KE, Riesenberg BP, Kennedy AS, Gandy EJ, Andrews AM, et al. Acetyl-CoA carboxylase obstructs CD8(+) T cell lipid utilization in the tumor microenvironment. Cell Metab. (2024) 36:969–83.e10. doi: 10.1016/j.cmet.2024.02.009
85. Morotti M, Grimm AJ, Hope HC, Arnaud M, Desbuisson M, Rayroux N, et al. PGE(2) inhibits TIL expansion by disrupting IL-2 signalling and mitochondrial function. Nature. (2024) 629:426–34. doi: 10.1038/s41586-024-07352-w
86. Cane S, Barouni RM, Fabbi M, Cuozzo J, Fracasso G, Adamo A, et al. Neutralization of NET-associated human ARG1 enhances cancer immunotherapy. Sci Transl Med. (2023) 15:eabq6221. doi: 10.1126/scitranslmed.abq6221
87. Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun. (2019) 10:3000. doi: 10.1038/s41467-019-10979-3
88. Xiao J, Wang S, Chen L, Ding X, Dang Y, Han M, et al. 25-Hydroxycholesterol regulates lysosome AMP kinase activation and metabolic reprogramming to educate immunosuppressive macrophages. Immunity. (2024) 57:1087–104.e7. doi: 10.1016/j.immuni.2024.03.021
89. Yang L, Chu Z, Liu M, Zou Q, Li J, Liu Q, et al. Amino acid metabolism in immune cells: essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J Hematol Oncol. (2023) 16:59. doi: 10.1186/s13045-023-01453-1
90. St Paul M, Saibil SD, Kates M, Han S, Lien SC, Laister RC, et al. Ex vivo activation of the GCN2 pathway metabolically reprograms T cells, leading to enhanced adoptive cell therapy. Cell Rep Med. (2024) 5:101465. doi: 10.1016/j.xcrm.2024.101465
91. Mastelic-Gavillet B, Navarro Rodrigo B, Decombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J Immunother Cancer. (2019) 7:257. doi: 10.1186/s40425-019-0719-5
92. Chen S, Akdemir I, Fan J, Linden J, Zhang B, and Cekic C. The expression of adenosine A2B receptor on antigen-presenting cells suppresses CD8(+) T-cell responses and promotes tumor growth. Cancer Immunol Res. (2020) 8:1064–74. doi: 10.1158/2326-6066.CIR-19-0833
93. Shang A, Gu C, Wang W, Wang X, Sun J, Zeng B, et al. Exosomal circPACRGL promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-beta1 axis. Mol Cancer. (2020) 19:117. doi: 10.1186/s12943-020-01235-0
94. Xu F, Wang X, Huang Y, Zhang X, Sun W, Du Y, et al. Prostate cancer cell-derived exosomal IL-8 fosters immune evasion by disturbing glucolipid metabolism of CD8(+) T cell. Cell Rep. (2023) 42:113424. doi: 10.1016/j.celrep.2023.113424
95. Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, et al. Endoplasmic reticulum stress causes liver cancer cells to release exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology. (2019) 70:241–58. doi: 10.1002/hep.30607
96. Yang C, Wu S, Mou Z, Zhou Q, Dai X, Ou Y, et al. Exosome-derived circTRPS1 promotes Malignant phenotype and CD8+ T cell exhaustion in bladder cancer microenvironments. Mol Ther. (2022) 30:1054–70. doi: 10.1016/j.ymthe.2022.01.022
97. Zheng J, Yan X, Lu T, Song W, Li Y, Liang J, et al. CircFOXK2 promotes hepatocellular carcinoma progression and leads to a poor clinical prognosis via regulating the Warburg effect. J Exp Clin Cancer Res. (2023) 42:63. doi: 10.1186/s13046-023-02624-1
98. Baldwin JG, Heuser-Loy C, Saha T, Schelker RC, Slavkovic-Lukic D, Strieder N, et al. Intercellular nanotube-mediated mitochondrial transfer enhances T cell metabolic fitness and antitumor efficacy. Cell. (2024) 187:6614–30 e21. doi: 10.1016/j.cell.2024.08.029
99. Ikeda H, Kawase K, Nishi T, Watanabe T, Takenaga K, Inozume T, et al. Publisher Correction: Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature. (2025) 639:E5. doi: 10.1038/s41586-025-08764-y
100. Lu Z, McBrearty N, Chen J, Tomar VS, Zhang H, De Rosa G, et al. ATF3 and CH25H regulate effector trogocytosis and anti-tumor activities of endogenous and immunotherapeutic cytotoxic T lymphocytes. Cell Metab. (2022) 34:1342–58 e7. doi: 10.1016/j.cmet.2022.08.007
101. Huang JF, Yang Y, Sepulveda H, Shi W, Hwang I, Peterson PA, et al. TCR-Mediated internalization of peptide-MHC complexes acquired by T cells. Science. (1999) 286:952–4. doi: 10.1126/science.286.5441.952
102. Pagliano O, Morrison RM, Chauvin JM, Banerjee H, Davar D, Ding Q, et al. Tim-3 mediates T cell trogocytosis to limit antitumor immunity. J Clin Invest. (2022) 132:e152864. doi: 10.1172/JCI152864
103. Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A, et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat Commun. (2020) 11:1749. doi: 10.1038/s41467-020-15404-8
104. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. (2021) 22:358–69. doi: 10.1038/s41590-020-00850-9
105. Li S, Lin J, Huang L, Hu S, Wang M, Sun W, et al. STK31 drives tumor immune evasion through STAT3-IL-6 mediated CD8(+) T cell exhaustion. Oncogene. (2025) 44:1452–62. doi: 10.1038/s41388-024-03271-2
106. Lutz V, Hellmund VM, Picard FSR, Raifer H, Ruckenbrod T, Klein M, et al. IL18 receptor signaling regulates tumor-reactive CD8+ T-cell exhaustion via activation of the IL2/STAT5/mTOR pathway in a pancreatic cancer model. Cancer Immunol Res. (2023) 11:421–34. doi: 10.1158/2326-6066.CIR-22-0398
107. Breart B, Williams K, Krimm S, Wong T, Kayser BD, Wang L, et al. IL-27 elicits a cytotoxic CD8(+) T cell program to enforce tumour control. Nature. (2025) 639:746–53. doi: 10.1038/s41586-024-08510-w
108. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell. (2011) 20:781–96. doi: 10.1016/j.ccr.2011.11.003
109. Guo Y, Xie YQ, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat Immunol. (2021) 22:746–56. doi: 10.1038/s41590-021-00940-2
110. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
111. Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. (2013) 341:186–91. doi: 10.1126/science.1240094
112. Ma J, Tang L, Tan Y, Xiao J, Wei K, Zhang X, et al. Lithium carbonate revitalizes tumor-reactive CD8(+) T cells by shunting lactic acid into mitochondria. Nat Immunol. (2024) 25:552–61. doi: 10.1038/s41590-023-01738-0
113. Bell HN, Huber AK, Singhal R, Korimerla N, Rebernick RJ, Kumar R, et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab. (2023) 35:134–49.e6. doi: 10.1016/j.cmet.2022.11.013
114. Cheng J, Yan J, Liu Y, Shi J, Wang H, Zhou H, et al. Cancer-cell-derived fumarate suppresses the anti-tumor capacity of CD8(+) T cells in the tumor microenvironment. Cell Metab. (2023) 35:961–78.e10. doi: 10.1016/j.cmet.2023.04.017
115. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. (2021) 22:205–15. doi: 10.1038/s41590-020-00834-9
116. Ma S, Ming Y, Wu J, and Cui G. Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol Immunol. (2024) 21:419–35. doi: 10.1038/s41423-024-01148-8
117. Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, et al. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and Malignancy. Nat Immunol. (2016) 17:712–20. doi: 10.1038/ni.3439
118. Turner JA, Fredrickson MA, D’Antonio M, Katsnelson E, MacBeth M, Van Gulick R, et al. Lysophosphatidic acid modulates CD8 T cell immunosurveillance and metabolism to impair anti-tumor immunity. Nat Commun. (2023) 14:3214. doi: 10.1038/s41467-023-38933-4
119. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. (2021) 33:1001–12.e5. doi: 10.1016/j.cmet.2021.02.015
120. Yang X, Deng B, Zhao W, Guo Y, Wan Y, Wu Z, et al. FABP5(+) lipid-loaded macrophages process tumour-derived unsaturated fatty acid signal to suppress T-cell antitumour immunity. J Hepatol. (2025) 82:676–89. doi: 10.1016/j.jhep.2024.09.029
121. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. (2022) 40:365–78.e6. doi: 10.1016/j.ccell.2022.02.003
122. Nava Lauson CB, Tiberti S, Corsetto PA, Conte F, Tyagi P, Machwirth M, et al. Linoleic acid potentiates CD8(+) T cell metabolic fitness and antitumor immunity. Cell Metab. (2023) 35:633–50.e9. doi: 10.1016/j.cmet.2023.02.013
123. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. (2019) 30:143–56.e5. doi: 10.1016/j.cmet.2019.04.002
124. Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun. (2017) 8:864. doi: 10.1038/s41467-017-00910-z
125. Ron-Harel N, Ghergurovich JM, Notarangelo G, LaFleur MW, Tsubosaka Y, Sharpe AH, et al. T cell activation depends on extracellular alanine. Cell Rep. (2019) 28:3011–21.e4. doi: 10.1016/j.celrep.2019.08.034
126. Lercher A, Bhattacharya A, Popa AM, Caldera M, Schlapansky MF, Baazim H, et al. Type I interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity. (2019) 51:1074–87.e9. doi: 10.1016/j.immuni.2019.10.014
127. Guan L, Wu B, Li T, Beer LA, Sharma G, Li M, et al. HRS phosphorylation drives immunosuppressive exosome secretion and restricts CD8(+) T-cell infiltration into tumors. Nat Commun. (2022) 13:4078. doi: 10.1038/s41467-022-31713-6
128. Chen SW, Zhu SQ, Pei X, Qiu BQ, Xiong D, Long X, et al. Cancer cell-derived exosomal circUSP7 induces CD8(+) T cell dysfunction and anti-PD1 resistance by regulating the miR-934/SHP2 axis in NSCLC. Mol Cancer. (2021) 20:144. doi: 10.1186/s12943-021-01448-x
129. Saha T, Dash C, Jayabalan R, Khiste S, Kulkarni A, Kurmi K, et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol. (2022) 17:98–106. doi: 10.1038/s41565-021-01000-4
130. Manfredi F, Stasi L, Buonanno S, Marzuttini F, Noviello M, Mastaglio S, et al. Harnessing T cell exhaustion and trogocytosis to isolate patient-derived tumor-specific TCR. Sci Adv. (2023) 9:eadg8014. doi: 10.1126/sciadv.adg8014
131. Zhai Y, Du Y, Li G, Yu M, Hu H, Pan C, et al. Trogocytosis of CAR molecule regulates CAR-T cell dysfunction and tumor antigen escape. Signal Transduct Target Ther. (2023) 8:457. doi: 10.1038/s41392-023-01708-w
132. Zhou P. Emerging mechanisms and applications of low-dose IL-2 therapy in autoimmunity. Cytokine Growth Factor Rev. (2022) 67:80–8. doi: 10.1016/j.cytogfr.2022.06.003
133. Rosen DB, Kvarnhammar AM, Laufer B, Knappe T, Karlsson JJ, Hong E, et al. TransCon IL-2 beta/gamma: a novel long-acting prodrug with sustained release of an IL-2Rbeta/gamma-selective IL-2 variant with improved pharmacokinetics and potent activation of cytotoxic immune cells for the treatment of cancer. J Immunother Cancer. (2022) 10:e004991. doi: 10.1136/jitc-2022-004991
134. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, and Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. (2010) 32:79–90. doi: 10.1016/j.immuni.2009.11.012
135. Boyman O, Kovar M, Rubinstein MP, Surh CD, and Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. (2006) 311:1924–7. doi: 10.1126/science.1122927
136. Buchbinder EI, Dutcher JP, Daniels GA, Curti BD, Patel SP, Holtan SG, et al. Therapy with high-dose Interleukin-2 (HD IL-2) in metastatic melanoma and renal cell carcinoma following PD1 or PDL1 inhibition. J Immunother Cancer. (2019) 7:49. doi: 10.1186/s40425-019-0522-3
137. Davar D, Ding F, Saul M, Sander C, Tarhini AA, Kirkwood JM, et al. High-dose interleukin-2 (HD IL-2) for advanced melanoma: a single center experience from the University of Pittsburgh Cancer Institute. J Immunother Cancer. (2017) 5:74. doi: 10.1186/s40425-017-0279-5
138. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight. (2021) 6:e147474. doi: 10.1172/jci.insight.147474
139. Hashimoto M, Ramalingam SS, and Ahmed R. Harnessing CD8 T cell responses using PD-1-IL-2 combination therapy. Trends Cancer. (2024) 10:332–46. doi: 10.1016/j.trecan.2023.11.008
140. Mo F, Yu Z, Li P, Oh J, Spolski R, Zhao L, et al. An engineered IL-2 partial agonist promotes CD8(+) T cell stemness. Nature. (2021) 597:544–8. doi: 10.1038/s41586-021-03861-0
141. Zhang H, Liu J, Yuan W, Zhang Q, Luo X, Li Y, et al. Ammonia-induced lysosomal and mitochondrial damage causes cell death of effector CD8(+) T cells. Nat Cell Biol. (2024) 26:1892–902. doi: 10.1038/s41556-024-01503-x
142. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. (2014) 124:1070–80. doi: 10.1182/blood-2013-10-535245
143. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, and Gottschalk S. Inducible activation of myD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. (2017) 7:1306–19. doi: 10.1158/2159-8290.CD-17-0263
144. Verdun N and Marks P. Secondary cancers after chimeric antigen receptor T-cell therapy. N Engl J Med. (2024) 390:584–6. doi: 10.1056/NEJMp2400209
145. Steffin DHM, Muhsen IN, Hill LC, Ramos CA, Ahmed N, Hegde M, et al. Long-term follow-up for the development of subsequent Malignancies in patients treated with genetically modified IECs. Blood. (2022) 140:16–24. doi: 10.1182/blood.2022015728
146. Bishop DC, Clancy LE, Simms R, Burgess J, Mathew G, Moezzi L, et al. Development of CAR T-cell lymphoma in 2 of 10 patients effectively treated with piggyBac-modified CD19 CAR T cells. Blood. (2021) 138:1504–9. doi: 10.1182/blood.2021010813
147. Schumacher TN and Thommen DS. Tertiary lymphoid structures in cancer. Science. (2022) 375:eabf9419. doi: 10.1126/science.abf9419
148. Khodabakhshi A, Akbari ME, Mirzaei HR, Seyfried TN, Kalamian M, and Davoodi SH. Effects of Ketogenic metabolic therapy on patients with breast cancer: A randomized controlled clinical trial. Clin Nutr. (2021) 40:751–8. doi: 10.1016/j.clnu.2020.06.028
149. Das K, Belnoue E, Rossi M, Hofer T, Danklmaier S, Nolden T, et al. A modular self-adjuvanting cancer vaccine combined with an oncolytic vaccine induces potent antitumor immunity. Nat Commun. (2021) 12:5195. doi: 10.1038/s41467-021-25506-6
150. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. (2020) 30:966–79. doi: 10.1038/s41422-020-00395-4
151. Bukhalid RA, Duvall JR, Lancaster K, Catcott KC, Malli Cetinbas N, Monnell T, et al. XMT-2056, a HER2-directed STING agonist antibody-drug conjugate, induces innate antitumor immune responses by acting on cancer cells and tumor-resident immune cells. Clin Cancer Res. (2025) 31:1766–82. doi: 10.1158/1078-0432.CCR-24-2449
152. Bhagwat AS, Torres L, Shestova O, Shestov M, Mellors PW, Fisher HR, et al. Cytokine-mediated CAR T therapy resistance in AML. Nat Med. (2024) 30:3697–708. doi: 10.1038/s41591-024-03271-5
153. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. (2007) 11:37–51. doi: 10.1016/j.ccr.2006.10.020
154. Li H, Hu Y, Li J, He J, Yu G, Wang J, et al. Intranasal prime-boost RNA vaccination elicits potent T cell response for lung cancer therapy. Signal Transduct Target Ther. (2025) 10:101. doi: 10.1038/s41392-025-02191-1
155. Melendez AV, Velasco Cardenas RM, Lagies S, Strietz J, Siukstaite L, Thomas OS, et al. Novel lectin-based chimeric antigen receptors target Gb3-positive tumour cells. Cell Mol Life Sci. (2022) 79:513. doi: 10.1007/s00018-022-04524-7
156. Guan X, Rodriguez-Cruz V, and Morris ME. Cellular uptake of MCT1 inhibitors AR-C155858 and AZD3965 and their effects on MCT-mediated transport of L-lactate in murine 4T1 breast tumor cancer cells. AAPS J. (2019) 21:13. doi: 10.1208/s12248-018-0279-5
157. Zhuang L, Scolyer RA, Murali R, McCarthy SW, Zhang XD, Thompson JF, et al. Lactate dehydrogenase 5 expression in melanoma increases with disease progression and is associated with expression of Bcl-XL and Mcl-1, but not Bcl-2 proteins. Mod Pathol. (2010) 23:45–53. doi: 10.1038/modpathol.2009.129
158. Raeber ME, Sahin D, and Boyman O. Interleukin-2-based therapies in cancer. Sci Transl Med. (2022) 14:eabo5409. doi: 10.1126/scitranslmed.abo5409
159. Su EW, Moore CJ, Suriano S, Johnson CB, Songalia N, Patterson A, et al. IL-2Ralpha mediates temporal regulation of IL-2 signaling and enhances immunotherapy. Sci Transl Med. (2015) 7:311ra170. doi: 10.1126/scitranslmed.aac8155
160. Pillozzi S, D’Amico M, Bartoli G, Gasparoli L, Petroni G, Crociani O, et al. The combined activation of K(Ca)3.1 and inhibition of K(v)11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br J Cancer. (2018) 118:200–12. doi: 10.1038/bjc.2017.392
161. Verma NK, Wong BHS, Poh ZS, Udayakumar A, Verma R, Goh RKJ, et al. Obstacles for T-lymphocytes in the tumour microenvironment: Therapeutic challenges, advances and opportunities beyond immune checkpoint. EBioMedicine. (2022) 83:104216. doi: 10.1016/j.ebiom.2022.104216
162. Arner EN and Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. (2023) 41:421–33. doi: 10.1016/j.ccell.2023.01.009
163. Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci Transl Med. (2018) 10:eaan5488. doi: 10.1126/scitranslmed.aan5488
164. Ansari MA, Thiruvengadam M, Venkidasamy B, Alomary MN, Salawi A, Chung IM, et al. Exosome-based nanomedicine for cancer treatment by targeting inflammatory pathways: Current status and future perspectives. Semin Cancer Biol. (2022) 86:678–96. doi: 10.1016/j.semcancer.2022.04.005
165. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
166. Xue G, Li X, Kalim M, Fang J, Jiang Z, Zheng N, et al. Clinical drug screening reveals clofazimine potentiates the efficacy while reducing the toxicity of anti-PD-1 and CTLA-4 immunotherapy. Cancer Cell. (2024) 42:780–96.e6. doi: 10.1016/j.ccell.2024.03.001
167. Lim DW, Kao HF, Suteja L, Li CH, Quah HS, Tan DS, et al. Clinical efficacy and biomarker analysis of dual PD-1/CTLA-4 blockade in recurrent/metastatic EBV-associated nasopharyngeal carcinoma. Nat Commun. (2023) 14:2781. doi: 10.1038/s41467-023-38407-7
168. Arrieta VA, Dmello C, McGrail DJ, Brat DJ, Lee-Chang C, Heimberger AB, et al. Immune checkpoint blockade in glioblastoma: from tumor heterogeneity to personalized treatment. J Clin Invest. (2023) 133:e163447. doi: 10.1172/JCI163447
169. Zemek RM, Anagnostou V, Pires da Silva I, Long GV, and Lesterhuis WJ. Exploiting temporal aspects of cancer immunotherapy. Nat Rev Cancer. (2024) 24:480–97. doi: 10.1038/s41568-024-00699-2
170. Allard B, Beavis PA, Darcy PK, and Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. (2016) 29:7–16. doi: 10.1016/j.coph.2016.04.001
171. Thompson EA and Powell JD. Inhibition of the adenosine pathway to potentiate cancer immunotherapy: potential for combinatorial approaches. Annu Rev Med. (2021) 72:331–48. doi: 10.1146/annurev-med-060619-023155
172. Xia C, Yin S, To KKW, and Fu L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol Cancer. (2023) 22:44. doi: 10.1186/s12943-023-01733-x
173. Guo D, Tong Y, Jiang X, Meng Y, Jiang H, Du L, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IkappaBalpha. Cell Metab. (2022) 34:1312–24.e6. doi: 10.1016/j.cmet.2022.08.002
174. Wang J, Jia W, Zhou X, Ma Z, Liu J, and Lan P. CBX4 suppresses CD8(+) T cell antitumor immunity by reprogramming glycolytic metabolism. Theranostics. (2024) 14:3793–809. doi: 10.7150/thno.95748
175. Zhou P, Shi H, Huang H, Sun X, Yuan S, Chapman NM, et al. Single-cell CRISPR screens in vivo map T cell fate regulomes in cancer. Nature. (2023) 624:154–63. doi: 10.1038/s41586-023-06733-x
176. Xiao Q, Tan M, Yan G, and Peng L. Revolutionizing lung cancer treatment: harnessing exosomes as early diagnostic biomarkers, therapeutics and nano-delivery platforms. J Nanobiotechnology. (2025) 23:232. doi: 10.1186/s12951-025-03306-0
177. Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, and Whiteside TL. Clinical significance of PD-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res. (2018) 24:896–905. doi: 10.1158/1078-0432.CCR-17-2664
178. Chen AXY, Yap KM, Kim JS, Sek K, Huang YK, Dunbar PA, et al. Rewiring endogenous genes in CAR T cells for tumour-restricted payload delivery. Nature. (2025) 644:241–51. doi: 10.1038/s41586-025-09212-7
179. Liu Z, Shi M, Ren Y, Xu H, Weng S, Ning W, et al. Recent advances and applications of CRISPR-Cas9 in cancer immunotherapy. Mol Cancer. (2023) 22:35. doi: 10.1186/s12943-023-01738-6
180. Wang SJ, Dougan SK, and Dougan M. Immune mechanisms of toxicity from checkpoint inhibitors. Trends Cancer. (2023) 9:543–53. doi: 10.1016/j.trecan.2023.04.002
181. Bischoff P, Trinks A, Obermayer B, Pett JP, Wiederspahn J, Uhlitz F, et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene. (2021) 40:6748–58. doi: 10.1038/s41388-021-02054-3
182. Anderson NM and Simon MC. The tumor microenvironment. Curr Biol. (2020) 30:R921–R5. doi: 10.1016/j.cub.2020.06.081
183. Janesick A, Shelansky R, Gottscho AD, Wagner F, Williams SR, Rouault M, et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat Commun. (2023) 14:8353. doi: 10.1038/s41467-023-43458-x
184. Armingol E, Officer A, Harismendy O, and Lewis NE. Deciphering cell-cell interactions and communication from gene expression. Nat Rev Genet. (2021) 22:71–88. doi: 10.1038/s41576-020-00292-x
185. Su J, Song Y, Zhu Z, Huang X, Fan J, Qiao J, et al. Cell-cell communication: new insights and clinical implications. Signal Transduct Target Ther. (2024) 9:196. doi: 10.1038/s41392-024-01888-z
186. Ren X, Zhong G, Zhang Q, Zhang L, Sun Y, and Zhang Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. (2020) 30:763–78. doi: 10.1038/s41422-020-0353-2
187. Saad MB, Hong L, Aminu M, Vokes NI, Chen P, Salehjahromi M, et al. Predicting benefit from immune checkpoint inhibitors in patients with non-small-cell lung cancer by CT-based ensemble deep learning: a retrospective study. Lancet Digit Health. (2023) 5:e404–e20. doi: 10.1016/S2589-7500(23)00082-1
188. Li K, Liu C, Sui X, Li C, Zhang T, Zhao T, et al. An organoid co-culture model for probing systemic anti-tumor immunity in lung cancer. Cell Stem Cell. (2025) 32:1218–34.e7. doi: 10.1016/j.stem.2025.05.011
189. Shi G, Huang X, Ma L, Li H, Zhong J, Wang J, et al. First-line tislelizumab and ociperlimab combined with gemcitabine and cisplatin in advanced biliary tract cancer (ZSAB-TOP): a multicenter, single-arm, phase 2 study. Signal Transduct Target Ther. (2025) 10:260. doi: 10.1038/s41392-025-02356-y
190. Wang M, Chen Y, Tian L, Wu C, Chen J, Hu J, et al. Vascular normalization augments the anti-tumor efficacy of combined HDAC inhibitor with immunotherapy in solid tumors. Cancer Discov. (2025) 15:1883–904. doi: 10.1158/2159-8290.c.8016675
Keywords: tumor microenvironment, CD8+ T cell, multimodal cell-cell communication, suppression, dysfunction
Citation: Chen L, Huang Q and Zhou P (2025) Multimodal cell-cell communication driving CD8+ T cell dysfunction and immune evasion. Front. Immunol. 16:1691746. doi: 10.3389/fimmu.2025.1691746
Received: 24 August 2025; Accepted: 21 October 2025;
Published: 05 November 2025.
Edited by:
Tapas Patra, Sri Shankara Cancer Hospital and Reseaech Center, IndiaReviewed by:
Suresh Satpati, Department of Genomic Medicine, United StatesWendy Mao, BioNTech (USA), United States
Copyright © 2025 Chen, Huang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peipei Zhou, emhvdV9wZWlwZWlAZ3psYWIuYWMuY24=
†These authors have contributed equally to this work