 Goshi Matsushima
Goshi Matsushima Yuhki Yanase
Yuhki Yanase Tadashi Nakagawa
Tadashi Nakagawa Mitsuhiro Goda
Mitsuhiro Goda Koichiro Ozawa1,3
Koichiro Ozawa1,3 Toru Hosoi
Toru Hosoi- 1Department of Clinical Pharmacology and Therapeutics, Graduate School of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan
- 2Department of Clinical Pharmacology, Faculty of Pharmaceutical Sciences, Sanyo-Onoda City University, Yamaguchi, Japan
- 3Faculty of Pharmacy, Yasuda Women’s University, Hiroshima, Japan
Endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) have emerged as central regulators of immune cell function and inflammatory processes. The UPR, mediated by three principal ER-resident sensors, IRE1α, PERK and ATF6, maintains cellular homeostasis under stress conditions but also contributes to pathogenesis when dysregulated. Recent studies revealed that the UPR plays critical roles not only in protein folding but also in directing immune cell fate, activation, and cytokine production. Although significant advances have been made, various questions remain regarding the cell-type-specific and context-dependent functions of ER stress responses. Understanding these mechanisms would be crucial for developing targeted therapies. Therefore, in this review, we provide a comprehensive overview of how ER stress and the UPR influence various immune cell types, including monocytes, macrophages, dendritic cells, granulocytes, T cells, B cells, microglia, and astrocytes, within both peripheral and central immune systems.
1 Introduction: ER Stress and UPR
Endoplasmic reticulum (ER) is the largest organelle in the mammalian cell, performing a wide range of essential functions, including the synthesis, transport, and folding of proteins (1–3). It is also the primary site for the synthesis of lipids and steroids, carbohydrate metabolism, and calcium storage (1, 4). A dysfunction in the ER’s protein-folding capacity leads to the accumulation of unfolded or misfolded proteins in the ER, a state known as “ER stress” (5). This stress is triggered by a variety of pathological conditions, including depletion of calcium or redox homeostasis, glucose and energy deprivation, hypoxia, the accumulation of misfolded mutant proteins, and pathogen infection (2, 5–8). To counteract this, cells activate a sophisticated signaling network known as the Unfolded Protein Response (UPR). The UPR is orchestrated by three ER-transmembrane sensors: Inositol-requiring enzyme 1 (IRE1), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor-6 (ATF6) (9) (Figure 1). Both IRE1 and ATF6 exist as two isoforms, α and β, and the α-isoforms are considered the primary mediators of the UPR (10, 11). The primary function of IRE1 is to splice X-box-binding protein 1 (XBP1) mRNA, producing a transcription factor, spliced XBP1 (XBP1s), that alleviates ER stress by enhancing protein-folding and degradation pathways (10, 12). PERK mitigates ER stress by phosphorylating eukaryotic translation initiation factor-2α (eIF2α) to globally attenuate protein synthesis, while paradoxically promoting the translation of the activating transcription factor-4 (ATF4), which can induce the key pro-apoptotic factor C/EBP-homologous protein (CHOP) (10, 12). ATF6 responds to ER stress by trafficking to the Golgi, where cleavage liberates its cytosolic domain, ATF6 fragment (ATF6f), to act as a potent transcription factor that primarily upregulates cytoprotective genes (9, 10, 12). Experimentally, these UPR pathways are often studied by inducing ER stress with chemical agents that promote the accumulation of unfolded proteins, most notably tunicamycin (Tm), which inhibits N-linked glycosylation, and thapsigargin (Tg), which disrupts ER calcium homeostasis (13). In its adaptive phase, the UPR aims to restore homeostasis by attenuating protein translation, upregulating ER chaperones, and enhancing ER-associated degradation (ERAD) of misfolded proteins (14). However, under severe and/or prolonged stress, the UPR switches from a pro-survival to a pro-apoptotic program, triggering cell death (15).
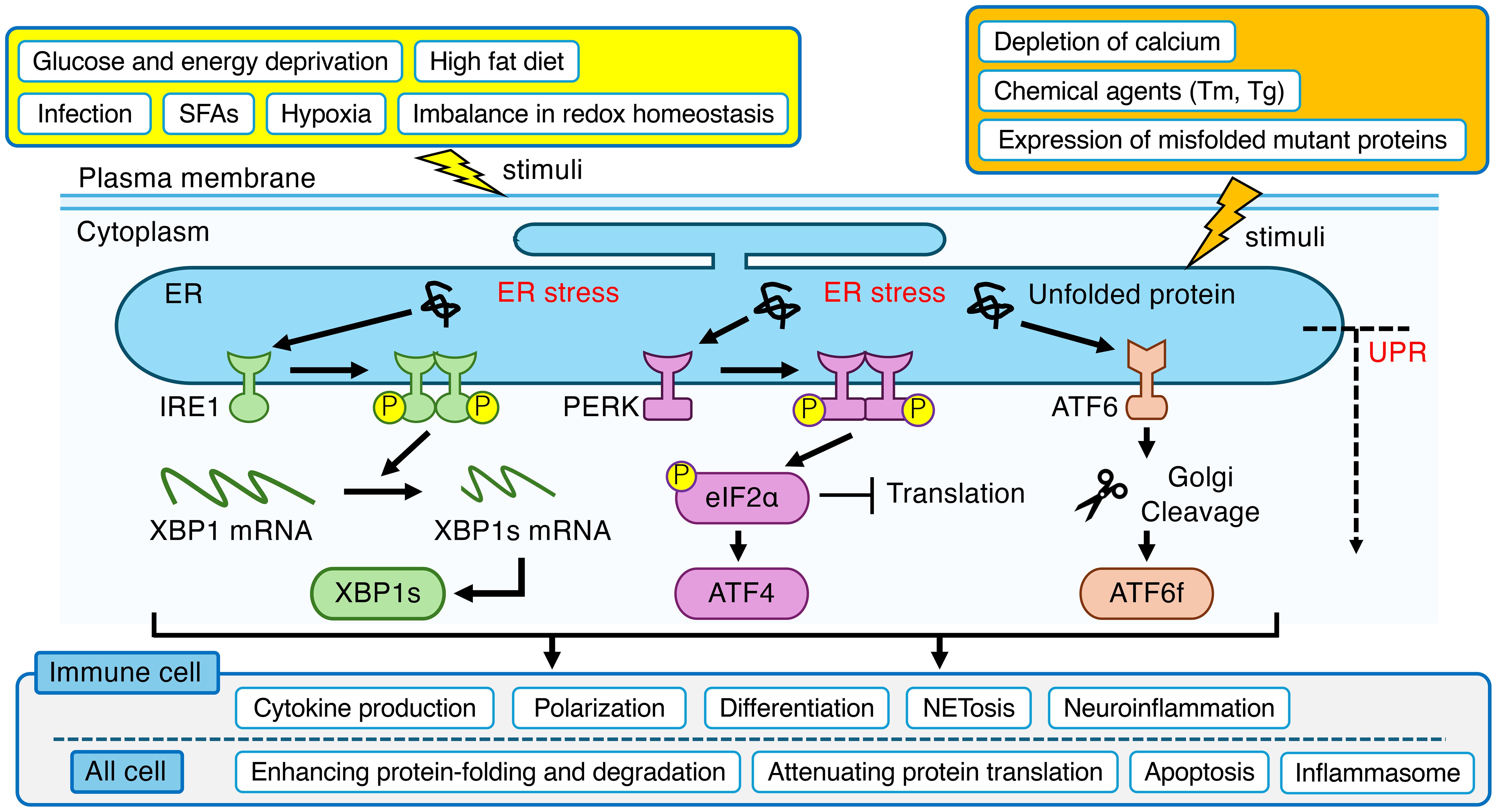
Figure 1. Overview of the ER stress and UPR in mammalian cell. ER stress, triggered by a wide range of physiological and pathological stimuli, leads to the activation of three canonical sensor proteins (IRE1, PERK and ATF6) located on the ER membrane. These pathways orchestrate an adaptive program to restore homeostasis in ER. Beyond this core function, the UPR is intricately linked to the regulation of immune system, modulating critical processes in immune cells such as cytokine production, polarization, differentiation, NETosis, and neuroinflammation. The yellow box indicates indirect stimuli that affect ER function. The orange box indicates direct stimuli that affect ER function. ER, endoplasmic reticulum; UPR, unfolded protein response; IRE1, inositol-requiring enzyme 1; XBP1, X-box-binding protein 1; XBP1s, spliced XBP1; PERK, protein kinase RNA (PKR)-like ER kinase; eIF2α, eukaryotic translation initiation factor-2α; ATF4, activating transcription factor-4; ATF6, activating transcription factor-6; ATF6f, ATF6 fragment; SFAs, saturated fatty acids; Tm, tunicamycin; Tg, thapsigargin; NETs, neutrophil extracellular traps.
Indeed, chronic ER stress and the ensuing dysregulation of the UPR affect the several physical functions of cells, such as secretion of hormones from pancreatic β-cells and adipocytes, resulting in the pathogenesis of numerous human diseases, spanning from metabolic conditions like diabetes and obesity to neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease (16–18). Furthermore, the pathological role of ER stress extends to a range of diseases, including autoimmune conditions like rheumatoid arthritis and systemic lupus erythematosus, as well as cancer (19–23).
A state of chronic and/or low-grade inflammation is now understood to be a key pathological hallmark at the root of a wide range of diseases, including type 2 diabetes, cancer, rheumatoid arthritis and Alzheimer’s disease (24–29). Central to these inflammatory processes are immune cells, whose aberrant activation and dysfunction play key roles in disease progression (30, 31). Within these immune cells, the ER serves as a critical nexus for sensing cellular status and orchestrating adaptive responses. Therefore, ER stress and the UPR may contribute to various diseases by affecting immune cell function and thereby inducing various inflammation.
Currently, the information on the role of ER stress and UPR in immune cells remains insufficient. In this review we focused on and summarized current understanding of the association of ER stress and UPR with immune cell functions.
2 ER stress in immune cells as a driver of inflammation
ER stress has emerged as a critical modulator of the development, activation, and effector functions of both innate and adaptive immune cells. Each of these cell types relies on the ER not only for the synthesis and processing of proteins but also for integrating environmental signals that influence their fate and function. Dysregulated ER stress responses can thus profoundly affect immune homeostasis and contribute to pathological inflammation and tissue damage.
2.1 Peripheral immune cells
2.1.1 Monocytes
Monocytes circulate in the blood, defend against pathogens via phagocytosis, and differentiate into macrophages or dendritic cells (DCs) to support immune responses and tissue repair (32).
Tg-induced ER stress has been shown to induce an inflammatory phenotype in monocytes, leading to the increased mRNA expression of pro-inflammatory cytokines interleukin-6 (IL-6) and IL-8 (33). Furthermore, Tg-induced ER stress in monocytes has been shown to amplify TNF-α production in response to stressors such as lipopolysaccharide (LPS) and palmitic acid (34).
2.1.2 Macrophages
Macrophages are a diverse population of innate immune phagocytes found in all tissues, where they act as sentinels essential for homeostasis, tissue repair, and host defense (35). Conventionally, their activation states are described as a spectrum between two main poles: the pro-inflammatory ‘M1’ phenotype, which is central to host defense against infection, and the anti-inflammatory, tissue-reparative ‘M2’ phenotype (36).
In macrophages, saturated fatty acids (SFAs) engage the IRE1α pathway to promote the activation of the nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome, which in turn leads to the secretion of IL-1β, a pro-inflammatory cytokine closely linked to insulin resistance (37). A key study by Shan et al. demonstrated that myeloid-specific deletion of IRE1α protects mice from diet-induced obesity and insulin resistance by promoting a shift from pro-inflammatory M1 to anti-inflammatory M2 macrophage polarization in adipose tissue (38). Corroborating the therapeutic importance of these genetic findings, pharmacological inhibition of IRE1α has also been shown to be highly effective. In mice with diet-induced obesity, administering a specific inhibitor of IRE1α’s RNase activity (STF-083010) significantly ameliorated insulin resistance and protected against obesity by increasing thermogenesis (39). The primary mechanism involved reducing the accumulation of pro-inflammatory adipose tissue macrophages (ATMs), specifically the ‘M1-like’ CD11c+ and metabolically activated CD9+ subsets, thereby curtailing adipose inflammation (39). While the IRE1α pathway is a clear driver of this process, other branches of the UPR also contribute to the activation of macrophages, including the PERK-ATF4 axis. Indeed, a study demonstrated that deficiency of ATF4, a transcription factor downstream of the PERK pathway, suppresses the SFA-induced expression of the pro-inflammatory cytokine IL-6 in macrophages (40). In addition to these macrophage-intrinsic pathways, a key study demonstrated that in obese mice, high fat diet-induced CHOP expression, particularly within adipocytes, alters the local tissue environment in a way that drives the polarization of ATMs towards the pro-inflammatory M1 phenotype, resulting in the induction of insulin resistance (41).
2.1.3 Dendritic cells
As professional antigen-presenting cells, DCs play critical roles for innate and adaptive immune systems (42). DCs orchestrate the adaptive immune response by presenting captured antigens to naive T cells to provide activation signals, and by producing key instructive cytokines, such as IL-12 and IL-23, for direct T cell differentiation (42–44).
Although dispensable for DCs homeostasis in the steady state, upon activation by both R848 and palmitic acid, ATF6α contributes to the production of critical pro-inflammatory cytokines, including IL-12p70 and IL-6 (45). Beyond the ATF6α pathway, other arms of the UPR also act as potent modulators of cytokine production in activated DCs. It has been demonstrated that inducing ER stress with classical chemical stressors, such as Tm or Tg, in DCs stimulated with pathogen-associated molecular patterns (PAMPs) markedly enhanced the mRNA expression of the pro-inflammatory cytokine IL-23 (46). The underlying mechanism was shown to be dependent on specific UPR branches; the IRE1α pathway was essential for the IL-23 response to the fungal PAMP zymosan, while the PERK pathway was required for the response to the bacterial PAMP LPS. In addition, XBP1s has been shown to be indispensable for the development and survival of DCs (47). Recent reports indicate that tripartite motif containing 29 (TRIM29), known as a member of E3 ubiquitin ligase, promotes PERK-mediated ER stress immune response by inducing SUMOylation and stability of PERK (48). Moreover, TRIM29 is reported to negatively regulate the innate immune response against virus infections by inhibiting production of type I IFNs, such as IFN-α and IFN-β, in DCs and macrophages (49–51). These findings suggest that TRIM29-PERK axis would be a target for ER stress-associated immune disorders.
2.1.4 Granulocytes (neutrophils, eosinophils and basophils)
Granulocytes, which include neutrophils, eosinophils, and basophils, play important roles in inflammation, encompassing both pathogen clearance and immunoregulation (52).
Neutrophils are the most abundant leukocytes and essential first responders in acute inflammation, where they contribute to host defense and tissue repair (53). However, they exacerbate disease through mechanisms including the release of proteases, such as neutrophil elastase, and the formation of Neutrophil Extracellular Traps (NETs), which has established them as a promising therapeutic target for a range of chronic inflammatory conditions (53).
In lupus hyperactivated IRE1α in neutrophils directly drives pathological NETosis, a highly inflammatory process, as demonstrated by the finding that pharmacological inhibition of IRE1α (4μ8C) reduces this process (54).
Eosinophils are now understood to be versatile immunomodulatory cells that bridge innate and adaptive immunity (55). They fulfill this role by modulating the functions of B and T cells and through antigen presentation. Furthermore, they communicate extensively with other innate immune cells, including macrophages and DCs, to regulate the overall inflammatory environment. A study revealed that the IRE1α-XBP1s pathway is selectively and absolutely required for eosinophil differentiation, while being dispensable for the development of other granulocytes like neutrophils (56).
Basophils, rare granulocytes sharing features of both innate and adaptive immunity, contribute to allergic inflammation by expressing the high-affinity IgE receptor (FcϵRI) and releasing mediators, such as histamine, in response to IgE-mediated stimulation (57, 58). The role of ER stress in basophils remains poorly defined, though initial evidence suggests a distinct reliance on the UPR. IgE receptor-mediated stimulation of basophils leads to the activation of the IRE1α pathway (59). However, the physiological significance of this activation is not yet well understood.
2.1.5 T cells
T cells play a pivotal role in directing the adaptive immune response, which ensures the effective and specific clearance of invading pathogens (60). Conversely, dysfunction of T cell development or activity contributes to the pathogenesis of a wide spectrum of human illnesses, such as immunodeficiencies, autoimmune conditions, and allergic disorders. Conventional T cells are broadly divided into two main classes: CD4+ helper T cells that orchestrate the immune response, and CD8+ cytotoxic T cells that eliminate target cells (61).
In tumor-infiltrating T cells, the PERK pathway of the UPR has been identified as a key driver of cellular exhaustion and energy depletion (62). Pharmacological (GSK2606414) or genetic inhibition of the PERK pathway in T cells was shown to preserve their energy reserves and enhance their anti-tumor effector functions, suggesting that targeting ER stress is a promising strategy to bolster T cell-mediated immunity (62). UPR also plays a critical role in shaping the differentiation of specific T helper cell subsets. This is particularly evident in the case of T helper-17 (Th17) cells, a pro-inflammatory subset strongly implicated in the pathogenesis of autoimmune diseases. A key study revealed that ER stress inducers like Tm and Tg, markedly enhance the differentiation of naive T cells into Th17 cells (63). The IRE1α-XBP1s pathway is also crucial for the function of T helper 2 (Th2) cells, a subset involved in allergic responses and anti-helminth immunity. The IRE1α-XBP1s pathway plays a critical role during Th2 cell activation by regulating the expression and secretion of their signature cytokines, as well as their proliferation (64). ER stress also critically influences regulatory T cells (Tregs), which are essential for immune tolerance and inflammation control. ER stress has been shown to mediate the detrimental effects of the stress hormone cortisol on Tregs function (65). Specifically, cortisol exposure in the presence of a specific antigen reduces TGF-β expression from Tregs, an effect that is prevented by the ER stress inhibitor BiP inducer X. Beyond this, ER stress also plays critical roles in the broader dysregulation of Th17/Treg balance (66). For example, the ER stress inhibitor, 4-phenylbutyric acid (4-PBA), has been demonstrated to suppress reactive oxygen species (ROS) production, consequently inhibiting Th17 differentiation and promoting Treg differentiation. In addition, it has been reported that in patients with ulcerative colitis, a synergistic effect between environmental factors and ER stress inhibits the differentiation of Tr1 cells, an IL-10-producing subset of Tregs (67).
2.1.6 B cells and plasma cells
B cells perform a variety of crucial immune functions, including not only their classical roles in antibody production and antigen capture via the B cell receptor (BCR), but also their capacity to act as antigen-presenting cells (68, 69).
The differentiation of B cells into professional antibody-secreting plasma cells is a process critically dependent on the IRE1α-XBP1s pathway (70, 71). These plasma cells can produce a diverse repertoire of antibody classes, each tailored for distinct and specialized effector functions within the immune system (68). While the IRE1α pathway is critical for differentiation, it is also co-opted by malignant plasma cells in multiple myeloma, where its inhibition suppresses the secretion of not only immunoglobulin light chains but also key growth factors and cytokines, such as vascular endothelial growth factor (VEGF), IL-1α, IL-6, and IL-8 (72).
2.2 Neuronal cells
2.2.1 Microglias
Microglia are the resident immune cells of the central nervous system, forming a dynamic and motile network that continuously surveys their local environment (73).
ER stress in microglia has been shown to drive pathological inflammatory responses, while its pharmacological inhibition with 4-PBA is protective (74). Furthermore, a key study utilized mice with a microglia-specific deletion of the ER stress sensor IRE1α. When challenged with a high fat diet, male mice with this deletion were significantly protected from obesity, glucose intolerance, and hypothalamic inflammation compared to their wild-type counterparts (75). In addition to the IRE1α pathway, the PERK branch of the UPR has also been implicated as a key driver of pro-inflammatory microglial polarization. For instance, a study using LPS-stimulated microglia showed that the compound ascorbic acid 6-palmitate significantly inhibited the activation of the PERK/eIF2α pathway (76). This suppression of ER stress, in turn, helped restore the M1/M2 balance by promoting an anti-inflammatory M2 phenotype, evidenced by increased expression of IL-10 and Arginase-1. High glucose, a key feature of diabetes, has also been shown to induce a state of PERK branch activation in microglia. For example, an in vitro study demonstrated that exposing microglial cells to hyperglycemic conditions led to the upregulation of the key ER stress markers CHOP and phosphorylated eIF2α via PERK pathway (77). This activation of the pro-apoptotic ER stress pathway highlighted the vulnerability of microglia to glucotoxicity-induced ER stress.
2.2.2 Astrocytes
Astrocytes are the most abundant glial cells in the central nervous system and are essential for neuronal homeostasis and function (78). Although astrocytes are not classically categorized as immune cells due to their neuroepithelial origin, they are increasingly recognized as key regulators and effectors of neuroinflammatory responses (79, 80).
It has been demonstrated in vitro that inducing ER stress in astrocytes drives the expression of the pro-inflammatory cytokines, TNF-α and IL-6 (81). Crucially, this inflammatory response was significantly suppressed by co-treatment with a specific PERK inhibitor (GSK2606414), providing direct evidence for the PERK pathway’s causal role in driving astrocyte-mediated inflammation. Beyond increasing cytokine production, PERK activation drives astrocytes into a pathological ‘reactivity state,’ causing them to lose neuroprotective functions while gaining neurotoxic properties. Crucially, a landmark study confirmed this causal link, as astrocyte-specific genetic inhibition of the PERK pathway was sufficient to prevent neuronal loss and extend survival in a prion disease model (82). Furthermore, it has been reported that hyperglycemic conditions activate UPR pathways in astrocytes, resulting in an increased secretion of the pro-inflammatory cytokines TNF-α, IL-6 and IL-18 (83).
2.3 ER stress and inflammasome
Inflammasomes function as cytoplasmic platforms that sense PAMPs and/or danger-associated molecular patterns (DAMPs), playing a key role in orchestrating host immune homeostasis in various cells (84–86). Upon activation, they recruit and activate caspase-1, which in turn processes pro-inflammatory cytokines such as IL-1β and IL-18 and induces pyroptosis, a lytic form of programmed cell death (84, 85, 87). Among several types of inflammasomes, the NLRP3 inflammasome is the most extensively studied and appears particularly responsive to cellular stress signals (84). Recent studies have elucidated that ER stress-induced UPR activates NF-κB signaling, leading to promotion of expression of NLRP3 and IL-1β, resulting in activation of inflammasome (88). Moreover, ER stress-induced calcium leakage from the ER into the mitochondria leads to an increase in mitochondrial ROS (mROS) production. These changes act as triggers for various endocrine system diseases. Indeed, in models of fatty liver ischemia/reperfusion, ER stress in macrophages induces mitochondrial calcium overload, which in turn promotes mROS production and activates NLRP3 signaling (89). In the hyperglycemic state of diabetes, elevated ER stress leads to an increase in NLRP3 inflammasome-dependent IL-1β secretion, which causes β-cell dysfunction and promotes obesity and insulin resistance (90). Similar ER stress-inflammasome pathways have been implicated in neurodegenerative disease. In Parkinson’s disease, pathological α-synuclein aggregates have been shown to induce a profound ER stress response in microglia, which subsequently promotes the activation of the NLRP3 inflammasome (91). Thus, ER stress also contributes to the activation of inflammasome which plays central roles in the pathogenesis of chronic inflammation, metabolic diseases, and neurodegenerative conditions.
2.4 Relation of pro-inflammatory cytokines released from immune cells to endocrine cells
The release of pro-inflammatory cytokines from immune cells is a critical driver of widespread endocrine dysfunction. Cytokines, such as TNF-α and IL-6, have been shown to directly impair insulin receptor signaling in metabolic tissues, thereby contributing to the development of insulin resistance (92–94). Moreover, pro-inflammatory cytokines, including IL-1, IL-6, and TNF-α, are implicated in glucocorticoid resistance, thereby compromising the body’s endogenous anti-inflammatory feedback mechanisms (95, 96). In addition to interfering with hormone signaling, pro-inflammatory cytokines induce ER stress in endocrine tissues, resulting in disruptions in protein folding, hormone synthesis, and secretion, and may ultimately promote apoptosis (6). Notably, in pancreatic β-cells, pro-inflammatory cytokines, IL-1β and IFN-γ, amplify ER stress, resulting in the pathogenesis of type 1 diabetes (97). Additionally, IFN-α induces ER stress in thyroid cells, which lead to thyroid cell apoptosis (98).
3 Conclusion
ER stress and the UPR are now recognized as central regulators of immune cell fate and function. Far beyond their canonical roles in protein quality control, the UPR branches, IRE1α, PERK and ATF6, serve as critical signaling hubs that translate environmental and metabolic cues into immune responses. This review highlighted how ER stress shapes inflammation by modulating cytokine production, cell differentiation, and polarization across diverse immune cell types, including monocytes, macrophages, DCs, granulocytes, T cells, B cells, and glial cells such as microglia and astrocytes (Figure 1; Table 1). Notably, dysregulated ER stress skews immune responses toward pathological inflammation, contributing to the progression of metabolic, autoimmune, neurodegenerative, and malignant diseases. However, our understanding remains incomplete. Various questions remain regarding cell-type specificity, temporal dynamics, and the crosstalk between UPR pathways. Future studies should aim to unravel these complexities using conditional genetic models and systems-level approaches. Moreover, targeted pharmacological modulation of specific UPR branches holds therapeutic promise for controlling inflammation without compromising essential ER functions. In conclusion, deciphering the immunological roles of ER stress responses offers a novel and fertile avenue for therapeutic intervention in a wide range of inflammatory diseases.
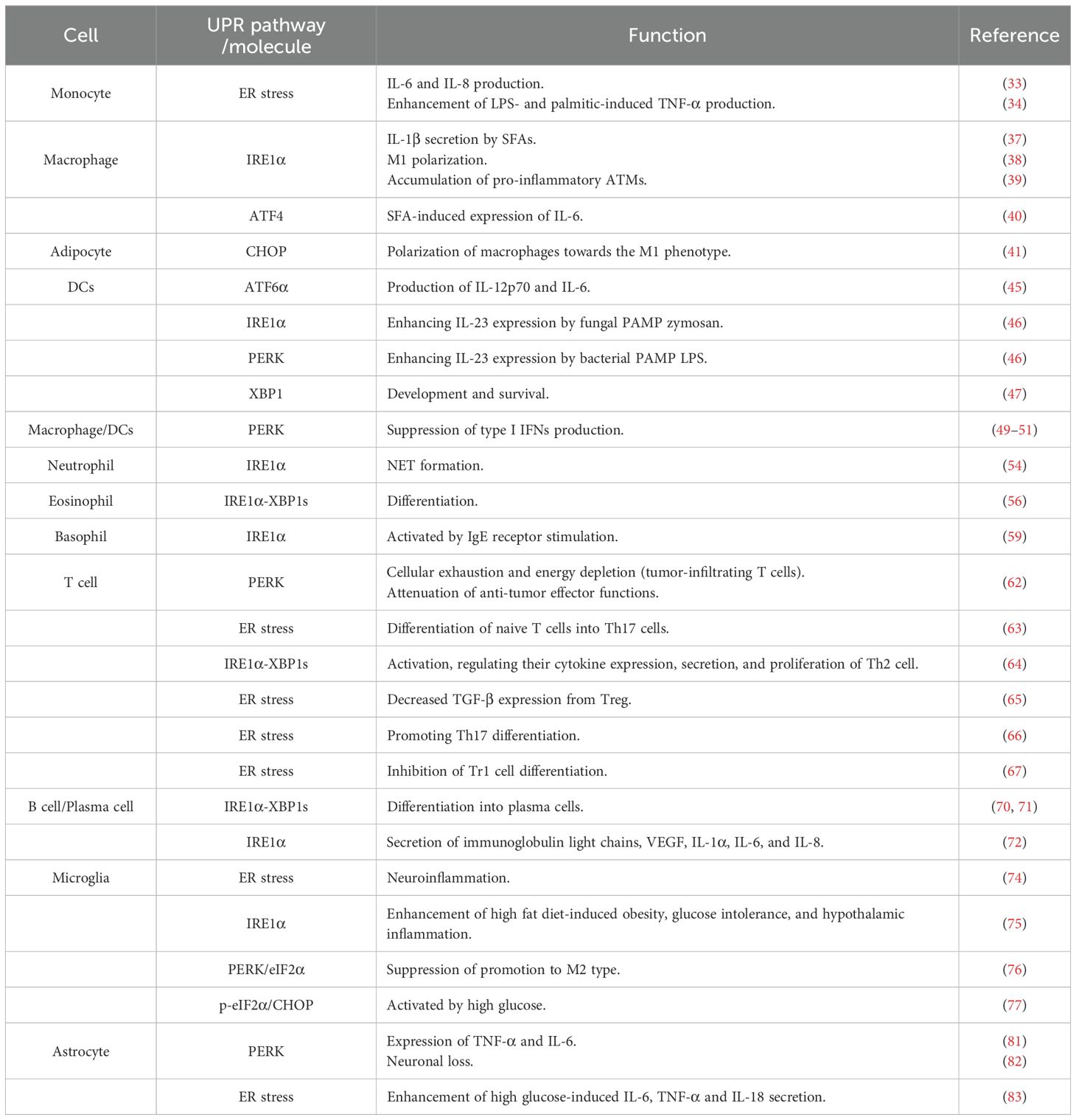
Table 1. Summary of the roles of the UPR in immune cells.
Author contributions
GM: Writing – original draft. YY: Validation, Writing – original draft, Writing – review & editing. TN: Writing – review & editing. MG: Writing – review & editing. KO: Writing – review & editing. TH: Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was partially funded by grants from the Takeda Science Foundation and the Grant-in-Aid for Scientific Research (C) (21K06577) to YY, TH, and KO and the Japan Science and Technology Agency (JST) CREST (JPMJCR2111) to YY. This work was supported by JST SPRING, grant number JPMJSP2132.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schwarz DS and Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. (2016) 73:79–94. doi: 10.1007/s00018-015-2052-6
2. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. (2010) 140:900–17. doi: 10.1016/j.cell.2010.02.034
3. Oakes SA and Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
4. Hwang S, Chang S, Rodriguez PC, and Cubillos-Ruiz JR. Endoplasmic reticulum stress responses in anticancer immunity. Nat Rev Cancer. (2025) 25(9):684–702. doi: 10.1038/s41568-025-00836-5
5. Hetz C and Papa FR. The unfolded protein response and cell fate control. Mol Cell. (2018) 69:169–81. doi: 10.1016/j.molcel.2017.06.017
6. Eizirik DL, Cardozo AK, and Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. (2008) 29:42–61. doi: 10.1210/er.2007-0015
7. Cnop M, Foufelle F, and Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. (2012) 18:59–68. doi: 10.1016/j.molmed.2011.07.010
8. Bettigole SE and Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. (2015) 33:107–38. doi: 10.1146/annurev-immunol-032414-112116
9. Ron D and Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
10. Lin JH, Walter P, and Yen TSB. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. (2008) 3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434
11. Glembotski CC, Rosarda JD, and Wiseman RL. Proteostasis and beyond: ATF6 in ischemic disease. Trends Mol Med. (2019) 25:538–50. doi: 10.1016/j.molmed.2019.03.005
12. Kim P. Understanding the unfolded protein response (UPR) pathway: insights into neuropsychiatric disorders and therapeutic potentials. Biomol Ther (Seoul). (2024) 32:183–91. doi: 10.4062/biomolther.2023.181
13. Oslowski CM and Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. (2011) 490:71–92. doi: 10.1016/B978-0-12-385114-7.00004-0
14. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
15. Tabas I and Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. (2011) 13:184–90. doi: 10.1038/ncb0311-184
16. Yuan S, She D, Jiang S, Deng N, Peng J, and Ma L. Endoplasmic reticulum stress and therapeutic strategies in metabolic, neurodegenerative diseases and cancer. Mol Med. (2024) 30:40. doi: 10.1186/s10020-024-00808-9
17. Scheper W and Hoozemans JJM. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. (2015) 130:315–31. doi: 10.1007/s00401-015-1462-8
18. Hasnain SZ, Lourie R, Das I, Chen ACH, and McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. (2012) 90:260–70. doi: 10.1038/icb.2011.112
19. Chen S, Wang Q, Wang H, and Xia S. Endoplasmic reticulum stress in T cell-mediated diseases. Scand J Immunol. (2023) 98:e13307. doi: 10.1111/sji.13307
20. Barrera M, Aguilera S, Castro I, González S, Carvajal P, Molina C, et al. Endoplasmic reticulum stress in autoimmune diseases: Can altered protein quality control and/or unfolded protein response contribute to autoimmunity? A critical review on Sjögren’s syndrome. Autoimmun Rev. (2018) 17:796–808. doi: 10.1016/j.autrev.2018.02.009
21. Cubillos-Ruiz JR, Mohamed E, and Rodriguez PC. Unfolding anti-tumor immunity: ER stress responses sculpt tolerogenic myeloid cells in cancer. J Immunother Cancer. (2017) 17:5. doi: 10.1186/s40425-016-0203-4
22. Song M and Cubillos-Ruiz JR. Endoplasmic reticulum stress responses in intratumoral immune cells: implications for cancer immunotherapy. Trends Immunol. (2019) 40:128–41. doi: 10.1016/j.it.2018.12.001
23. Zhang W, Shi Y, Oyang L, Cui S, Li S, Li J, et al. Endoplasmic reticulum stress-a key guardian in cancer. Cell Death Discov. (2024) 10:343. doi: 10.1038/s41420-024-02110-3
24. Hunter P. The inflammation theory of disease. EMBO Rep. (2012) 13:968–70. doi: 10.1038/embor.2012.142
25. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
26. Heneka MT, van der Flier WM, Jessen F, Hoozemanns J, Thal DR, Boche D, et al. Neuroinflammation in Alzheimer disease. Nat Rev Immunol. (2025) 25:321–52. doi: 10.1038/s41577-024-01104-7
27. Kiraly M, Foss JF, and Giordano T. Neuroinflammation, its role in Alzheimer’s disease and therapeutic strategie. J Prev Alzheimers Dis. (2023) 10:686–98. doi: 10.14283/jpad.2023.109
28. Leng F and Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. (2021) 17:157–72. doi: 10.1038/s41582-020-00435-y
29. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, and Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y). (2018) 6:575–90. doi: 10.1016/j.trci.2018.06.014
30. Cildir G, Akıncılar SC, and Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med. (2013) 19:487–500. doi: 10.1016/j.molmed.2013.05.001
31. van de Vyver M. Immunology of chronic low-grade inflammation: relationship with metabolic function. J Endocrinol. (2023) 257:e220271. doi: 10.1530/JOE-22-0271
32. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, and Ley K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
33. Carroll TP, Greene CM, O’Connor CA, Nolan AM, O’Neill SJ, and McElvaney NG. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J Immunol. (2010) 184:4538–46. doi: 10.4049/jimmunol.0802864
34. Akhter N, Wilson A, Arefanian H, Thomas R, Kochumon S, Al-Rashed F, et al. Endoplasmic reticulum stress promotes the expression of TNF-α in THP-1 cells by mechanisms involving ROS/CHOP/HIF-1α and MAPK/NF-κB pathways. Int J Mol Sci. (2023) 24:15186. doi: 10.3390/ijms242015186
35. Wynn TA, Chawla A, and Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
36. Liu YC, Zou XB, Chai YF, and Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci. (2014) 10:520–9. doi: 10.7150/ijbs.8879
37. Robblee MM, Kim CC, Abate JP, Valdearcos M, Sandlund KLM, Shenoy MK, et al. Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. (2016) 14:2611–23. doi: 10.1016/j.celrep.2016.02.053
38. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol. (2017) 18:519–29. doi: 10.1038/ni.3709
39. Wu D, Eeda V, Maria Z, Rawal K, Wang A, Herlea-Pana O, et al. Targeting IRE1α improves insulin sensitivity and thermogenesis and suppresses metabolically active adipose tissue macrophages in male obese mice. Elife. (2025) 17:RP100581. doi: 10.7554/eLife.100581
40. Iwasaki Y, Suganami T, Hachiya R, Shirakawa I, Kim-Saijo M, Tanaka M, et al. Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages. Diabetes. (2014) 63:152–61. doi: 10.2337/db13-0757
41. Suzuki T, Gao J, Ishigaki Y, Kondo K, Sawada S, Izumi T, et al. ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Rep. (2017) 18:2045–57. doi: 10.1016/j.celrep.2017.01.076
42. Banchereau J and Teinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
43. Merad M, Sathe P, Helft J, Miller J, and Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
44. McGeachy MJ and Cua DJ. The link between IL-23 and Th17 cell-mediated immune pathologies. Semin Immunol. (2007) 19:372–6. doi: 10.1016/j.smim.2007.10.012
45. Gutiérrez-Ballesteros F, Morales-Reyes J, Fernández D, Geisse A, Arcaya A, Flores-Santibañez F, et al. Normal tissue homeostasis and impairment of selective inflammatory responses in dendritic cells deficient for ATF6α. Front Cell Dev Biol. (2023) 21:1089728. doi: 10.3389/fcell.2023.1089728
46. Márquez S, Fernández JJ, Terán-Cabanillas E, Herrero C, Alonso S, Azogil A, et al. Endoplasmic reticulum stress sensor IRE1α Enhances IL-23 expression by human dendritic cells. Front Immunol. (2017) 19:639. doi: 10.3389/fimmu.2017.00639
47. Iwakoshi NN, Pypaert M, and Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. (2007) 204:2267–75. doi: 10.1084/jem.20070525
48. Wang J, Lu W, Zhang J, Du Y, Fang M, Zhang A, et al. Loss of TRIM29 mitigates viral myocarditis by attenuating PERK-driven ER stress response in male mice. Nat Commun. (2024) 15:3481. doi: 10.1038/s41467-024-44745-x
49. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8:945. doi: 10.1038/s41467-017-00101-w
50. Xing J, Zhang A, Minze LJ, Li XC, and Zhang Z. TRIM29 negatively regulates the type I IFN production in response to RNA virus. J Immunol. (2018) 201:183–92. doi: 10.4049/jimmunol
51. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol. (2016) 17:1373–80. doi: 10.1038/ni.3580
52. Geering B, Stoeckle C, Conus S, and Simon HU. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. (2013) 34:398–409. doi: 10.1016/j.it.2013.04.002
53. Herrero-Cervera A, Soehnlein O, and Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol. (2022) 19:177–91. doi: 10.1038/s41423-021-00832-3
54. Sule G, Abuaita BH, Steffes PA, Fernandes AT, Estes SK, Dobry C, et al. Endoplasmic reticulum stress sensor IRE1α propels neutrophil hyperactivity in lupus. J Clin Invest. (2021) 131:e137866. doi: 10.1172/JCI137866
55. Rosenberg HF, Dyer KD, and Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. (2013) 13:9–22. doi: 10.1038/nri3341
56. Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, Weller PF, et al. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol. (2015) 16:829–37. doi: 10.1038/ni.3225
57. Steiner M, Huber S, Harrer A, and Himly M. The evolution of human basophil biology from neglect towards understanding of their immune functions. BioMed Res Int. (2016) 2016:8232830. doi: 10.1155/2016/8232830
58. Karasuyama H, Obata K, Wada T, Tsujimura Y, and Mukai K. Newly appreciated roles for basophils in allergy and protective immunity. Allergy. (2011) 66:1133–41. doi: 10.1111/j.1398-9995.2011.02613.x
59. Matsushima G, Matsui Y, Okamoto H, Umeda N, Kakimoto M, Mino M, et al. Unique compound with anti-allergic action: inhibition of Lyn kinase activity by KIRA6. Front Pharmacol. (2025) 16:1625798. doi: 10.3389/fphar.2025.1625798
60. Chopp L, Redmond C, O’Shea JJ, and Schwartz DM. From thymus to tissues and tumors: A review of T-cell biology. J Allergy Clin Immunol. (2023) 151:81–97. doi: 10.1016/j.jaci.2022.10.011
61. Ruterbusch M, Pruner KB, Shehata L, and Pepper M. In vivo CD4+ T cell differentiation and function: revisiting the Th1/Th2 paradigm. Annu Rev Immunol. (2020) 38:705–25. doi: 10.1146/annurev-immunol-103019-085803
62. Hurst KE, Lawrence KA, Essman MT, Walton ZJ, Leddy LR, and Jessica E Thaxton JE. Endoplasmic reticulum stress contributes to mitochondrial exhaustion of CD8+ T cells. Cancer Immunol Res. (2019) 7:476–86. doi: 10.1158/2326-6066.CIR-18-0182
63. Brucklacher-Waldert V, Ferreira C, Stebegg M, Fesneau O, Innocentin S, Marie JC, et al. Cellular stress in the context of an inflammatory environment supports TGF-β-independent T helper-17 differentiation. Cell Rep. (2017) 19:2357–70. doi: 10.1016/j.celrep.2017.05.052
64. Pramanik J, Chen X, Kar G, Henriksson J, Gomes T, Park JE, et al. Genome-wide analyses reveal the IRE1a-XBP1 pathway promotes T helper cell differentiation by resolving secretory stress and accelerating proliferation. Genome Med. (2018) 10:76. doi: 10.1186/s13073-018-0589-3
65. Luo J, Zhou C, Wang S, Tao S, Liao Y, Shi Z, et al. Cortisol synergizing with endoplasmic reticulum stress induces regulatory T-cell dysfunction. Immunology. (2023) 170:334–43. doi: 10.1111/imm.13669
66. Chen Y, Chen K, Zhu H, Qin H, Liu J, and Cao X. Methyltransferase Setd2 prevents T cell-mediated autoimmune diseases via phospholipid remodeling. Proc Natl Acad Sci U.S.A. (2024) 121:e2314561121. doi: 10.1073/pnas.2314561121
67. Feng B, Liu H, Yao W, Li Y, Wu G, Yang L, et al. Endoplasmic reticulum stress interferes with the development of type 1 regulating T cells. Inflammation Res. (2024) 73:381–92. doi: 10.1007/s00011-023-01841-w
68. Cyster JG and Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
69. Abebe EC, Dejenie TA, Ayele TM, Baye ND, Teshome AA, and Muche ZT. The role of regulatory B cells in health and diseases: A systemic review. J Inflammation Res. (2021) 14:75–84. doi: 10.2147/JIR.S286426
70. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. (2001) 412:300–7. doi: 10.1038/35085509
71. Hu CCA, Dougan SK, McGehee AM, Love JC, and Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. (2009) 28:1624–36. doi: 10.1038/emboj.2009.117
72. Harnoss JM, Thomas AL, Shemorry A, Marsters SA, Lawrence DA, Lu M, et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc Natl Acad Sci U.S.A. (2019) 116:16420–9. doi: 10.1073/pnas.1906999116
73. Salter MW and Stevens B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
74. Huang W, Liu Y, Li J, Gao Y, Tang J, Yip S, et al. Endoplasmic reticulum stress drives neuroinflammation through lipocalin 2 upregulation in retinal microglia after optic nerve injury. Invest Ophthalmol Vis Sci. (2025) 66:12. doi: 10.1167/iovs.66.5.12
75. Stilgenbauer L, Chen Q, Pungi D, James N, Jayarathne H, Koshko L, et al. Microglial ER stress response via IRE1α regulates diet-induced metabolic imbalance and obesity in mice. Mol Metab. (2025) 95:102128. doi: 10.1016/j.molmet.2025.102128
76. Li Q, Wu Y, Chen XS, Zeng T, Liu LL, Feng ZQ, et al. Ascorbic acid 6-palmitate modulates microglia M1/M2 polarization in lipopolysaccharide-stimulated BV-2 cells via PERK/elF2α mediated endoplasmic reticulum stress. BMC Complement Med Ther. (2022) 22:302. doi: 10.1186/s12906-022-03780-1
77. James AW, Bahader GA, Albassan M, and Shah ZA. The ER chaperone, BIP protects Microglia from ER stress-mediated Apoptosis in Hyperglycemia. Neurochem Int. (2023) 169:105573. doi: 10.1016/j.neuint.2023.105573
78. Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. (2015) 16:249–63. doi: 10.1038/nrn3898
79. Colombo E and Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. (2016) 37:608–20. doi: 10.1016/j.it.2016.06.006
80. Vainchtein ID and Molofsky AV. Astrocytes and microglia: in sickness and in health. Trends Neurosci. (2020) 43:144–54. doi: 10.1016/j.tins.2020.01.003
81. Yang Y, Huang X, Wang W, Sun J, Wang X, Ji C, et al. Endoplasmic reticulum stress of astrocytes in paraventricular nucleus of hypothalamus promotes ventricular electrical instability after acute myocardial infarction in rats. Front Cardiovasc Med. (2025) 12:1574146. doi: 10.3389/fcvm.2025.1574146
82. Smith HL, Freeman OJ, Butcher AJ, Holmqvist S, Humoud I, Schätzl T, et al. Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron. (2020) 105:855–866.e5. doi: 10.1016/j.neuron.2019.12.014
83. Wang G, Cui W, Chen S, Shao Z, Li Y, Wang W, et al. Metformin alleviates high glucose-induced ER stress and inflammation by inhibiting the interaction between caveolin1 and AMPKα in rat astrocytes. Biochem Biophys Res Commun. (2021) 534:908–13. doi: 10.1016/j.bbrc.2020.10.075
84. Chen X, Guo X, Ge Q, Zhao Y, Mu H, and Zhang J. ER stress activates the NLRP3 inflammasome: A novel mechanism of atherosclerosis. Oxid Med Cell Longev. (2019) 7:3462530. doi: 10.1155/2019/3462530
85. Swanson KV, Deng M, and Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
86. Yu HB and Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe. (2008) 4:198–208. doi: 10.1016/j.chom.2008.08.007
87. Ozaki E, Campbell M, and Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflammation Res. (2015) 8:15–27. doi: 10.2147/JIR.S51250
88. Zhou Y, Tong Z, Jiang S, Zheng W, Zhao J, and Zhou X. The roles of endoplasmic reticulum in NLRP3 inflammasome activation. Cells. (2020) 9:1219. doi: 10.3390/cells9051219
89. Li F, Guan Z, Gao Y, Bai Y, Zhan X, Ji X, et al. ER stress promotes mitochondrial calcium overload and activates the ROS/NLRP3 axis to mediate fatty liver ischemic injury. Hepatol Commun. (2024) 8:e0399. doi: 10.1097/HC9.0000000000000399
90. Lv S, Li X, and Wang H. The role of the effects of endoplasmic reticulum stress on NLRP3 inflammasome in diabetes. Front Cell Dev Biol. (2021) 9:663528. doi: 10.3389/fcell.2021.663528
91. Samidurai M, Palanisamy BN, Bargues-Carot A, Hepker M, Kondru N, Manne S, et al. PKC delta activation promotes endoplasmic reticulum stress (ERS) and NLR family pyrin domain-containing 3 (NLRP3) inflammasome activation subsequent to asynuclein-induced microglial activation: involvement of thioredoxin-interacting protein (TXNIP)/thioredoxin (Trx) redoxisome pathway. Front Aging Neurosci. (2021) 13:661505. doi: 10.3389/fnagi.2021.661505
92. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
93. Saltiel AR and Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. (2017) 127:1–4. doi: 10.1172/JCI92035
94. Shoelson SE, Lee J, and Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006) 16:1793–801. doi: 10.1172/JCI29069
95. Pariante CM. Why are depressed patients inflamed? A reflection on 20 years of research on depression, glucocorticoid resistance and inflammation. Eur Neuropsychopharmacol. (2017) 27:554–9. doi: 10.1016/j.euroneuro.2017.04.001
96. Webster JC, Oakley RH, Jewell CM, and Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U.S.A. (2001) 98:6865–70. doi: 10.1073/pnas.121455098
97. Gurzov EN, Ortis F, Cunha DA, Gosset G, Li M, Cardozo AK, et al. Signaling by IL-1beta+IFN-gamma and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ. (2009) 16:1539–50. doi: 10.1038/cdd.2009.99
Keywords: endoplasmic reticulum stress, unfolded protein response, immune cells, PERK (PKR-like endoplasmic reticulum kinase), ATF6 (activating transcription factor 6), IRE1 (inositol-requiring enzyme 1)
Citation: Matsushima G, Yanase Y, Nakagawa T, Goda M, Ozawa K and Hosoi T (2025) Endoplasmic reticulum stress and unfolded protein response in immune cell function. Front. Immunol. 16:1694102. doi: 10.3389/fimmu.2025.1694102
Received: 02 September 2025; Accepted: 13 October 2025;
Published: 24 October 2025.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Tania Angeles Floriano, National Autonomous University of Mexico, MexicoCopyright © 2025 Matsushima, Yanase, Nakagawa, Goda, Ozawa and Hosoi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuhki Yanase, eXlhbmFzZUBoaXJvc2hpbWEtdS5hYy5qcA==; Toru Hosoi, aG9zb2lAcnMuc29jdS5hYy5qcA==