 Wei-Jia Yang1*†
Wei-Jia Yang1*† Qing-Ping Kang1†Li-Ming Liang2†Qian Zhou3Xiao-Min Gong1Min Dou1Cui-Juan Huang1Ying Lin1
Qing-Ping Kang1†Li-Ming Liang2†Qian Zhou3Xiao-Min Gong1Min Dou1Cui-Juan Huang1Ying Lin1- 1Department of Eugenics and Genetics, Guilin People’s Hospital, Guilin, Guangxi, China
- 2Genetic Metabolism Laboratory, Guilin Women and Children’s Hospital, Guilin, Guangxi, China
- 3Genetic and Precision Medicine Lab, The First Affiliated Hospital of Guilin Medical University, Guilin, Guangxi, China
Objective: The aim of this study is to examine the relationship between hematological parameters, hemoglobin electrophoresis findings, and phenotypic characteristics in individuals carrying rare thalassemia gene variants in Northern Guangxi, China.
Methods: Peripheral blood samples were collected from 3,890 individuals (including 834 couples) who tested positive for thalassemia at the Prenatal Diagnosis Center of Guilin People’s Hospital between March 2019 and March 2025. Standard thalassemia genotyping was performed using Gap-PCR and PCR-reverse dot blot (PCR-RDB) assays to detect common α- and β-thalassemia mutations prevalent in southern China. Participants with negative results genotype-phenotype discordance underwent extended molecular testing to detect rare thalassemia variants. In cases where both partners were identified as carriers, amniotic fluid samples were collected from pregnant women for prenatal diagnosis.
Results: Thalassemia major was diagnosed in 13 fetuses, with elective termination of two affected pregnancies. The detection rate for common thalassemia mutations was 44.27% (1,722/3,890), while rare variants were identified in 1.72% (67/3,890). Among participants with negative results from conventional genotyping, the detection rate of rare mutations increased to 26.38% (67/254). A total of 42 rare thalassemia variants were found, including 25 α-thalassemia, 14 β-thalassemia, and 3 δ-thalassemia mutations. A novel 4.3 kb deletion (chr16:176935–181274DEL), encompassing the α1 gene and a recombined non-functional gene-X-Y-Z segment, was reported for the first time. The -α4.3/–SEA genotype was associated with HbH disease.
Conclusion: A substantial frequency of rare thalassemia gene mutations was identified in the Northern Guangxi population, contributing to the regional mutational landscape. These rare genotypes were associated with distinctive hematological and hemoglobin electrophoretic features. Characteristic phenotypic patterns, combined with specific laboratory parameters, facilitated preliminary inference of genotypes and supported the application of targeted diagnostic approaches. This strategy may improve diagnostic accuracy, reduce missed or incorrect diagnoses, and enhance prenatal and postnatal management strategies.
1 Introduction
Thalassemia is the most prevalent and clinically significant monogenic disorder worldwide. It is characterized by hemolytic anemia resulting from deletions or mutations in genes involved in globin chain synthesis, which disrupt the balance of α- and β-globin production and subsequently shorten erythrocyte lifespan. In China, approximately 30 million individuals are carriers of thalassemia-related genetic variants, with an estimated 300,000 individuals affected by thalassemia major (TM) or thalassemia intermedia (TI), both of which require clinical intervention (1). Carriers generally exhibit normal life expectancy and development; however, offspring of two carriers have a 25% risk of inheriting TM or TI.
Management of TM typically necessitates long-term blood transfusion and iron chelation therapy, contributing to significant financial, temporal, and physical burdens. Although hematopoietic stem cell transplantation represents a potentially curative option, its application is limited by high cost and restricted availability. Consequently, preventive strategies remain central to disease control. Since 2010, the government-initiated “Guangxi Thalassemia Prevention and Control Plan” in Guangxi, China, has adopted an integrated approach comprising free premarital screening, pre-pregnancy assessment, prenatal screening, and prenatal diagnosis. Accurate thalassemia genotyping serves as a cornerstone of this program.
Conventional genetic testing methods for thalassemia, such as reverse dot blot hybridization, have been routinely used in clinical practice for over two decades. These methods facilitate the detection of 3 common α-thalassemia deletions, 3 α-globin point mutations, and 18 β-globin point mutations. However, a proportion of thalassemia cases remain undiagnosed using these standard approaches. Currently, over 130 α-thalassemia and more than 300 β-thalassemia mutation types have been identified (2). Tang et al. examined 72 participants suspected of carrying rare thalassemia mutations and identified uncommon α- or β-globin gene variants in 49 cases through a combination of next-generation sequencing (NGS), third-generation sequencing (TGS), and chromosome microarray analysis/copy number variation (CNV) sequencing (3).
Yin et al. reported 5 rare thalassemia cases among 20 samples using TGS (4). Zhuang et al. conducted genetic screening in a cohort of 6,174 participants, identifying 2,390 carriers (38.71%) of α- or β-globin gene mutations, including 40 individuals with rare or novel variants (5). Peng et al. developed a TGS-based method known as Comprehensive Analysis of Thalassemia Alleles, which successfully identified 10 clinically relevant variants including 3 structural variants and 7 single nucleotide variants among 100 participants who presented with abnormal hematologic parameters or hemoglobin electrophoresis results but had negative findings on standard genetic tests (6). These studies used methods such as GAP-polymerase chain reaction (PCR), NGS, TGS, and other molecular techniques to improve the detection of rare thalassemia mutations.
Guilin, located in northeastern Guangxi, is recognized as a region with a high prevalence of thalassemia. Guilin People’s Hospital has been conducting thalassemia genetic and prenatal diagnostic testing for the past six years. The subsequent section outlines the findings of the institution from its screening for rare thalassemia variants.
2 Participants and methods
2.1 Study participants
Group A included 3,890 participants who provided peripheral blood samples at the Prenatal Diagnosis Center of Guilin People’s Hospital between March 2019 and March 2025. Participants ranged in age from 2 to 48 years and comprised of 1,826 males and 2,064 females. Among these, 834 samples were from couples undergoing joint screening.
Group B included 13 participants (9 males and 4 females) aged 3 to 44 years, who were identified through retrospective data review conducted during the same period. These participants presented with clinical features indicative of thalassemia but exhibited discordant findings on conventional genotyping.
Group C consisted of 834 couples who underwent routine thalassemia genotyping at the same center within the same timeframe. For couples in which both partners were identified as thalassemia carriers, prenatal diagnostic testing was recommended. In such cases, amniotic fluid samples were collected from the pregnant women for fetal thalassemia gene analysis.
2.1.1 Inclusion criteria
(1) Positive thalassemia screening with negative findings on conventional genotyping was defined by the presence of at least one of the following six criteria: 1) mean corpuscular volume (MCV) < 82 fL; 2) mean corpuscular hemoglobin (MCH) < 27 pg; 3) HbA2 < 2.4% on hemoglobin (Hb) electrophoresis; 4) HbA2 ≥ 3.5%; 5) elevated fetal hemoglobin (HbF) levels (typically ≥ 2%); or 6) abnormal hemoglobin profiles. Cases meeting any of these parameters were classified as screen-positive.
(2) Cases with a prior clinical diagnosis of TM or TI, presenting with mild anemia but negative results on conventional genotyping, were re-evaluated. Participants who continued to test negative upon retesting were subsequently included for further analysis of rare thalassemia gene variants.
2.1.2 Exclusion criteria
Cases involving iron deficiency anemia, immune-mediated hemolysis, or other hematologic disorders were excluded from the analysis.
2.2 Informed consent selection
The study protocol was approved by the Ethics Committee of Guilin People’s Hospital (approval numbers 2022-072KY, 2023-120KY). Written informed consent was obtained from all participants. For participants under 18 years of age, consent was provided by a legal guardian.
2.3 Laboratory methods
2.3.1 Analysis of blood routine parameters
Whole blood cell analysis was conducted using the Sysmex XE-5000 fully automated hematology analyzer.
2.3.2 Hb electrophoresis detection and analysis
HB component quantification was conducted using the Sebia Capillarys 2 Flex Piercing system, a fully automated capillary electrophoresis platform.
2.3.3 Serum ferritin detection
SF levels were measured using the Roche cobas e801 automated chemiluminescence immunoassay system.
2.3.4 Thalassemia gene detection
2.3.4.1 Conventional thalassemia gene detection
Genomic DNA was extracted from peripheral blood and amniotic fluid samples using a nucleic acid extraction kit provided by Shenzhen Yilifang Biotechnology Co., Ltd. A total of 25 common thalassemia genotypes were assessed. GAP-PCR was used to find four α-thalassemia deletions: αα/-α3.7, αα/-α4.2, αα/–SEA, and αα/–Thai. RDB-PCR was applied for the identification of three α-globin point mutations: αα/ααCS, αα/ααWS, and αα/ααQS.
Eighteen β-thalassemia mutations were also analyzed, including CD41–42, CD43, IVS-II-654, IVS-II-28, IVS-II-29, IVS-II-30, IVS-II-32, CD71–72, βE, CD17, CD31, CD37, CD14–15, CD27–28, IVS-I-1, IVS-I-5, CAP + 1, and IntM.
Samples suspected of carrying rare thalassemia variants were referred to third-party laboratories for further analysis, including Yaneng Bioscience Co., Ltd., Shenzhen Yilifang Biotechnology Co., Ltd., BGI Genomics, and Beijing Berry Genomics Biotechnology Co., Ltd.
2.3.4.2 Rare thalassemia detection technologies
The following methods were used for the detection of thalassemia gene variants:
(1) Nested PCR;
(2) Multiplex ligation-dependent probe amplification (MLPA);
(3) Sanger sequencing (first-generation sequencing);
(4) NGS;
(5) TGS;
(6) Long-read index PCR based on the novel CycloneSEQ nanopore sequencing platform (Shenzhen BGI Genomics). Long-read sequencing was conducted using the CycloneSEQ WT-02 single-molecule nanopore sequencer, developed by BGI Genomics. This protocol, approved by the BGI Genomics Institutional Review Board (IRB24094), enables comprehensive detection of genetic variants in the HBA, HBB, HBD, and HBG loci, including single nucleotide variations (SNVs), deletions, and structural variations.
3 Results
3.1 Results from Group C – prenatal diagnosis in carrier couples
Among the 834 couples screened, 93 were identified as carrying genetically identical thalassemia genotypes. Prenatal diagnosis was performed in 48 couples, each involving a pregnant woman carrying a fetus at risk, while data from the remaining carrier couples who underwent testing at external institutions were excluded from analysis. No rare genotypes were identified among the 48 couples. TM was diagnosed in 13 fetuses, with the corresponding genotypes and pregnancy outcomes presented in Table 1.
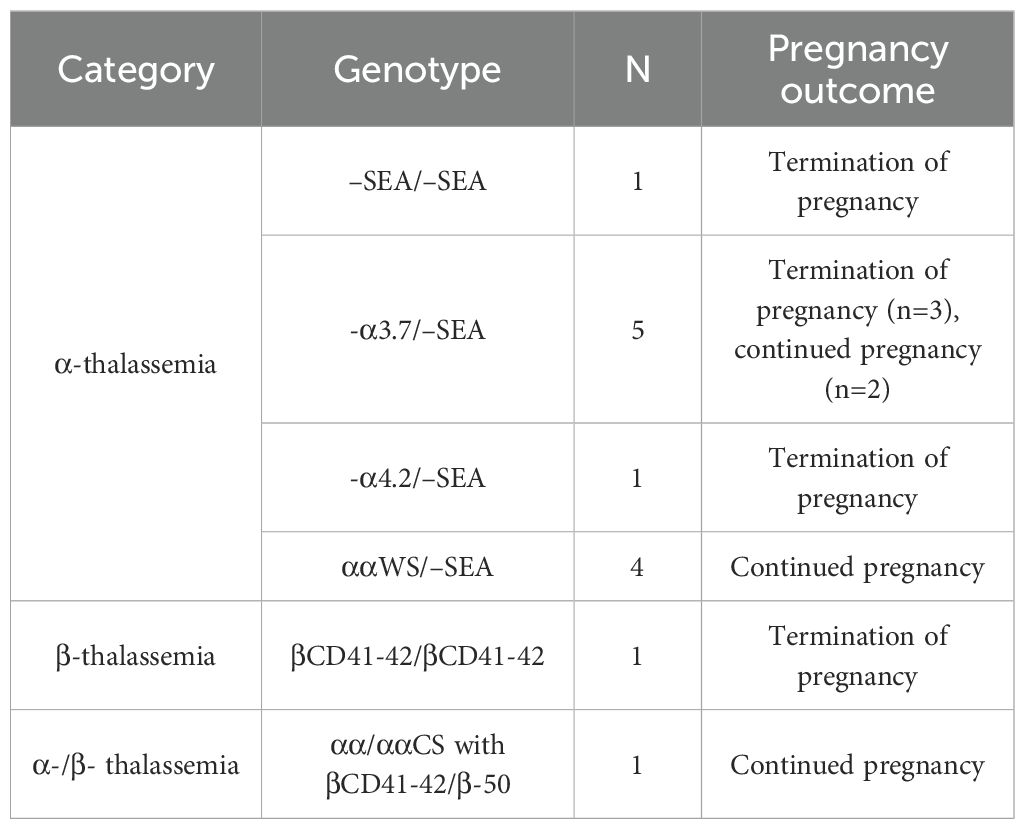
Table 1. Prenatal genotype diagnosis of 13 fetuses with thalassemia major.
3.2 Results from Group A – routine genotyping and identification of discordant cases
Among the 3,890 participants in Group A, routine thalassemia genotyping of peripheral blood samples identified 1,081 cases of α-thalassemia, 551 cases of β-thalassemia, and 90 cases with combined α- and β-thalassemia mutations, resulting in an overall detection rate of 44.27% (1,722/3,890). The remaining 2,168 participants tested negative for common thalassemia genotypes. Following the exclusion of 1,375 cases associated with hematological conditions such as infections and iron deficiency anemia, 763 cases remained with negative results on routine genotyping but positive outcomes on thalassemia screening, representing 19.61% (763/3,890).
3.3 Rare thalassemia genotyping in discordant and referred cases
Of the 763 cases with negative results on routine genotyping but positive thalassemia screening, all individuals were contacted by telephone and invited to participate in rare thalassemia gene testing. A total of 241 participants provided informed consent. Including 13 additional cases from Group B, a total of 254 participants underwent rare thalassemia genotyping, resulting in the identification of 67 cases (refer to Supplementary Tables 1-7). The detection rate for rare thalassemia mutations in this cohort from northern Guangxi was 1.72% (67/3,890), with a detection rate of 26.38% (67/254) among those who tested negative for common genotypes. A total of 42 rare genotypes were identified, consisting of 25 α-thalassemia, 14 β-thalassemia, and 3 δ-thalassemia variants.
3.3.1 Copy number variations
Five rare CNV types involving the α- and β-globin gene clusters were identified, encompassing eight cases (excluding Thailand-type deletions due to coverage by the detection kit). Among these, the α-globin cluster included the α2.4 deletion, the 4.3 deletion (Figure 1A), the family-α90_93(-8bp)(AGCTTCGG) variant, and the family-HS-40 deletion, as well as one duplication event. Within the β-globin gene cluster, two rare CNVs were found: one of Taiwan-type heterozygosity (Figure 1B) and one case of Southeast Asian (SEA) deletion with heterozygosity involving the SEA type (Figure 1C).
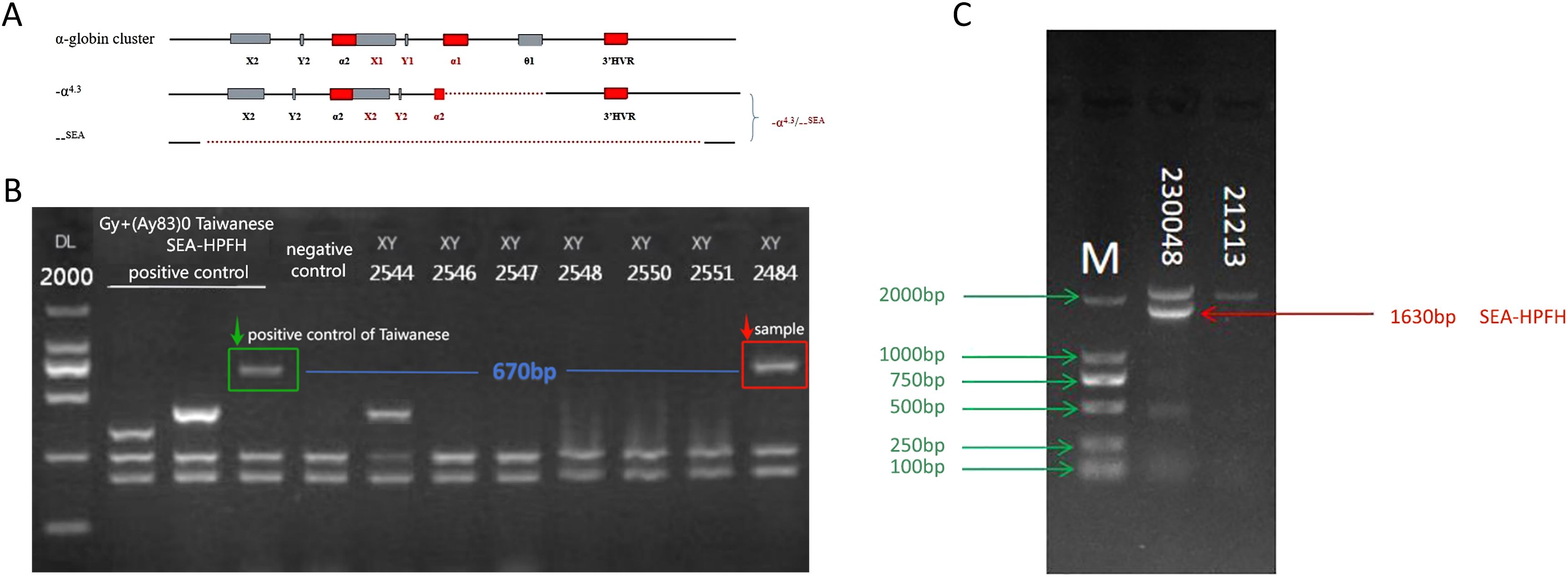
Figure 1. Rare CNVs in the α- and β-globin gene clusters. (A) −α4.3/−−SEA deletion (rare 4.3& kb deletion: chr16:176935–181274DEL) involving the α1 gene and a non-functional X–Y–Z segment. (B) Sample 2484: GAP-PCR agarose electrophoresis showing a 670& bp band consistent with the positive control for the Taiwan-type deletion. (C) Sample 230048: GAP-PCR agarose electrophoresis revealing a 1630& bp band indicative of SEA-HPFH deletion.
3.3.2 Rare SNVs in the α-globin gene cluster
Seven rare SNVs were identified within the α-globin gene cluster. These included ααCD30 (HBA2: c.91_93delGAG) with SEA heterozygosity, HBA2: c.168dup (Figure 2A), and three intronic variants: IVS-I-117 (G > A), IVS-II-34 (G > A), and IVS-II-55 (T > G). Among these, IVS-II-55 (T > G) was observed in one case coexisting with both the α-thalassemia Constant Spring (CS) mutation and δ-thalassemia (Figures 2B–E). Additionally, one case involved a PolyA signal mutation (AATAAA > AATAAG) co-occurring with β-thalassemia (Figures 2F–G).
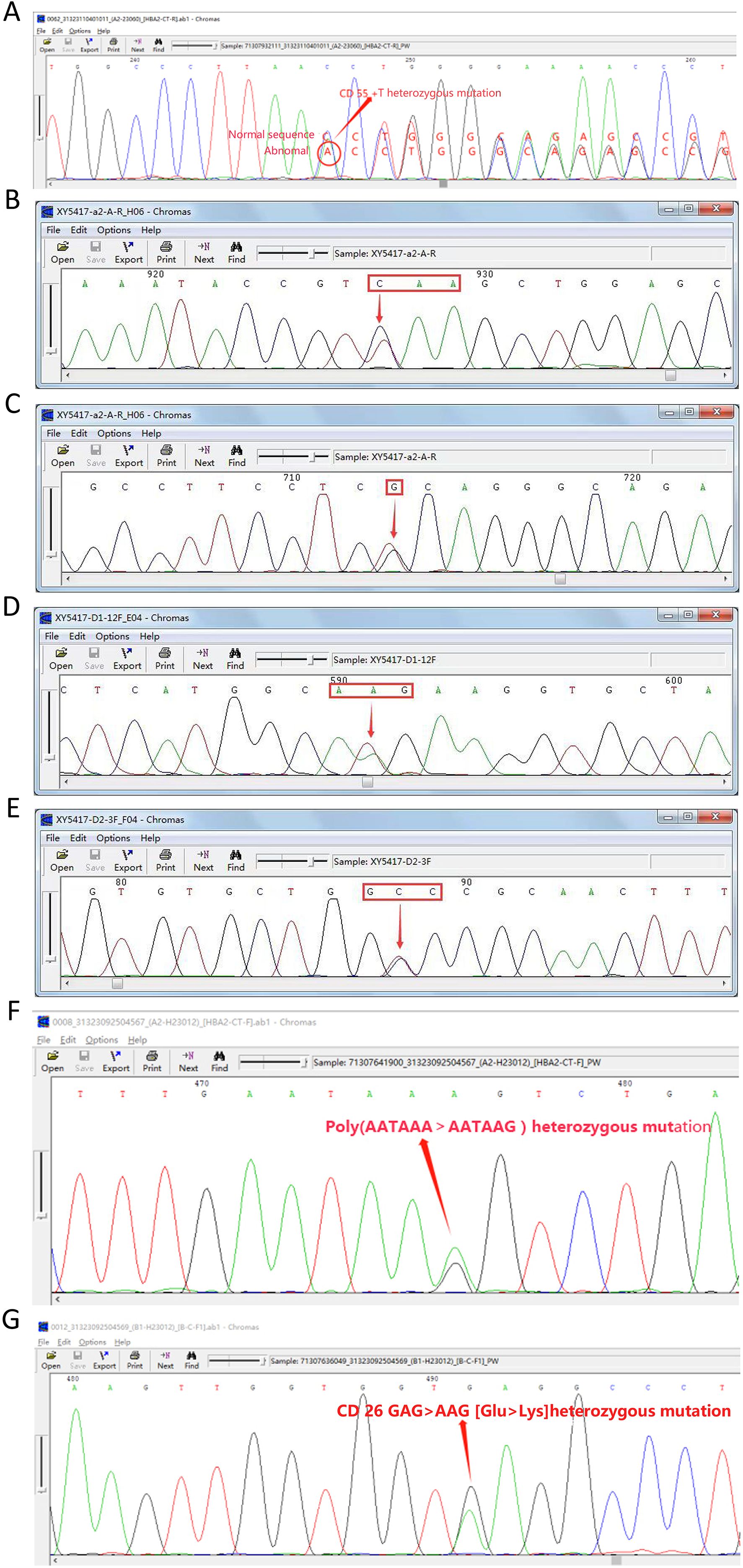
Figure 2. Seven rare SNVs in the α-globin gene cluster. (A) Codon 55 insertion: HBA2:c.168dup (αα/ααHBA2:c.168dup). (B) Nonsense mutation: CS (TAA > CAA). (C) IVS-II-55 (T > G). (D) CD65 (AAG > ATG). (E) CD115 (GCC > GTC). (F) PolyA signal mutation: *HBA2:c.94A > G (AATAAA > AATAAG). (G) β-globin mutation: βCD26 sequencing diagram.
3.3.3 Rare SNVs in the β-globin gene cluster
Five rare SNVs were detected within the β-globin gene cluster. These included three cases of βn/βCD30 (A > G), one case of βn/β−31 (A > C), and three intronic variants: IVS-II-5 (G > C), IVS-II-81C, and IVS-II-672 (A > C).
3.3.4 Hong Kong-type and triplicated α-globin genotypes
Hong Kong-type and triplicated α-globin genotypes were identified and included three cases of HKαα/αα, one case of HKαα/ααCS, three cases of HKαα/ααWS, and one case of HKαα/−−SEA. Two triplicated α-globin arrangements were detected: two cases of β41–42/αααaaa3.7 and two cases of β17/αααaaa4.2.
3.3.5 Structural hemoglobin variants in the α-globin cluster
Rare Hb structural variant genotypes within the α-globin gene cluster included two cases of −α4.2-Hb Q-Thailand/αα (Figures 3A–B), one case of −α4.2-Hb Q-Thailand/−α3.7, and one case of −α4.2-Hb Q-Thailand/−−SEA (Figures 3C–D). Additionally, one case of αα/ααHb G-Honolulu (HbA2: c.91 G > C) was identified (Figures 3E–F), along with one case of αα/ααHb Hekinan (HbA1: c.84 G > T) coexisting with Gγ-158C > T and Aγ-158C > T variants (Figures 3G–I).
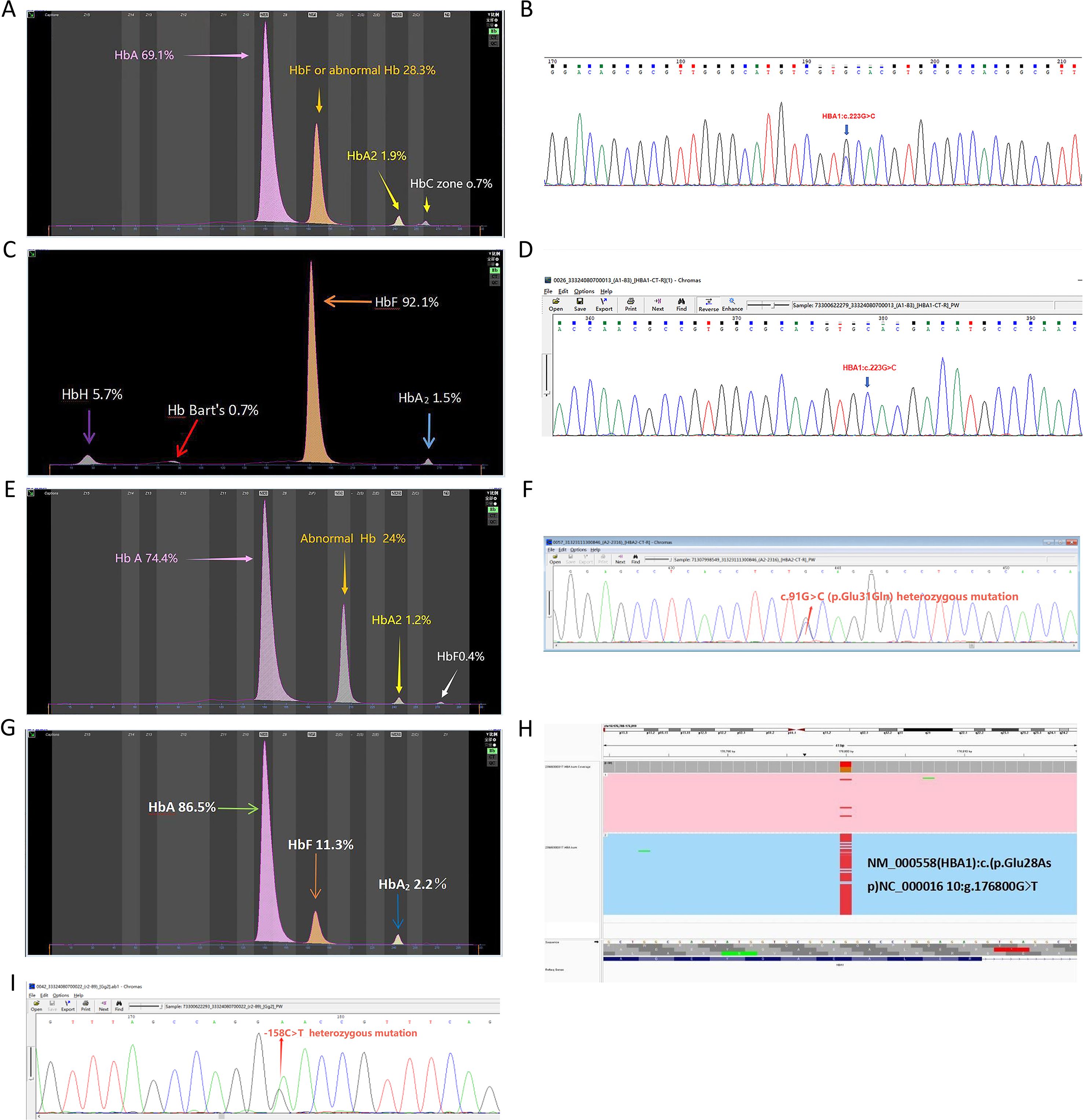
Figure 3. Rare hemoglobin structural variant genotypes in the α-globin gene cluster. (A) Electrophoresis pattern of αα/−α4.2Hb Q-Thailand (HBA1:c.223G > C), presenting abnormal Hb in zones F and (C, B) Sequencing diagram of αα/−α4.2Hb Q-Thailand. (C) Electrophoresis pattern of −−SEA/−α4.2Hb Q-Thailand, with abnormal Hb bands in zones F, Z15 (HbH), and Z12 (Hb Bart’s). (D) Sequencing diagram of −−SEA/−α4.2 Hb Q-Thailand. (E) Electrophoresis pattern of Hb G-Honolulu (HBA2:c.91G > C) with abnormal Hb in zone (D) (F) Sequencing diagram of Hb G-Honolulu. (G) Electrophoresis pattern of Hb Hekinan (HBA1:c.84G > T). (H) TGS sequencing diagram of Hb Hekinan. (I) Sequencing diagram of Gγ-158C > T and Aγ-158C > T variants.
3.3.6 Structural hemoglobin variants in the β-globin cluster
Within the β-globin gene cluster, Hb New York (HBB: c.341T > A) was the most frequently detected variant, identified in 13 cases. Additional variants included four cases of Hb J-Bangkok (HBB: c.170G > A), and single cases of Hb G-Taipei (HBB: c.68A > G) (Figures 4A–B), Hb O-Arab (HBB: c.364G > A) (Figures 4C–D), and Hb Barcelona (HBB: c.283G > C) (Figures 4E–F).

Figure 4. Rare hemoglobin structural variants in the β-globin gene cluster.(A) Electrophoresis pattern of Hb G-Taipei (HBB:c.68A > G), presenting abnormal Hb in zone D. (B) Sequencing diagram of Hb G-Taipei. (C) Electrophoresis pattern of HbO-Arab (HBB:c.364G > A), showing abnormal Hb overlapping with HbA2 and HbF. (D) Sequencing diagram of HbO-Arab. (E) Electrophoresis pattern of Hb Barcelona (HBB:c.283G > C). (F) Sequencing diagram of Hb Barcelona.
3.3.7 δ-thalassemia variants
Two cases of δ-thalassemia CD10 (−G) (HBD: c.31delG) were identified. An additional case exhibited compound mutations involving δ-thalassemia CD65 (A > T) and CD115 (C > T), along with α-thalassemia ααCS/ααIVS-II-55 genotype.
3.4 Group B – genotype–phenotype discordance
Retrospective analysis of clinical records in Group B identified 13 cases exhibiting genotype–phenotype discordance. Subsequent rare thalassemia gene analysis confirmed the presence of variants in 9 of these cases.
4 Discussion
4.1 Rare variants and analysis of α-globin gene cluster CNVs
4.1.1 2.4 deletion
A case with the -α2.4/αα genotype was identified. Previous reports by researchers in China characterized this deletion as involving a minimal genomic fragment, commonly associated with microcytic hypochromia and reduced HbA2 levels, but typically without apparent clinical manifestations (7, 8). However, when co-inherited with the αα/–SEA, the resulting -α2.4/–SEA combination may lead to a deletional form of HbH disease, which can manifest as mild to moderate anemia. Clinical management in such cases necessitates individualized genetic counseling and risk assessment during pregnancy.
4.1.2 -α90-93(-8bp) (AGCTTCGG) mutation
Two cases of the rare SEA/−α90-93 (−8& bp)(AGCTTCGG) mutation were identified. The first reported instance of the αα/−α90-93(−8& bp)(AGCTTCGG) genotype in Guangxi was documented by Li et al. (9) DNA sequencing localized the 8& bp deletion to the α2 gene, spanning nucleotides 34162 to 34171, and the mutation has been predicted to result in α+-thalassemia. When present in compound heterozygosity with the SEA deletion, this genotype leads to HbH disease, typically characterized by the presence of HbH and HbBart’s bands on hemoglobin electrophoresis. The clinical phenotype is similar to that observed in non-deletional HbH disease. Participants with the ααCS−−SEA genotype generally do not require regular transfusion support, though prenatal diagnostic evaluation is recommended. NGS is an appropriate method for detecting such mutations.
4.1.3 HS-40 deletion
Two cases of the rare –HS-40/-α3.7 genotype were identified. Heterozygous deletion of the HS-40 regulatory region is typically associated with mild anemia. When co-inherited with the −α3.7 deletion, the resulting genotype may lead to HbH disease. Along with the SEA deletion, this genotype has been linked to Bart’s hydrops fetalis (10–12).
4.1.4 First report of 4.3 deletion
A 44-year-old male, initially diagnosed with the conventional thalassemia genotype αα/–SEA, was re-evaluated using long-read sequencing conducted on the CycloneSEQ WT-02 single-molecule nanopore sequencer developed by BGI Genomics. The sequencing results identified a −-α4.3/–SEA genotype. This specific deletion has not been previously reported in the literature.
4.1.5 α duplication
A 30-year-old female was identified as carrying an α-globin gene duplication, confirmed through testing at Berry TGS. The duplication spanned chr16:173490–175917, encompassing a 2.427& kb segment between the HBA2 and HBA1 genes. No previous reports have documented duplications within this specific region.
Rare CNVs within the α-globin gene cluster are typically associated with decreased HbA2 levels and are often accompanied by microcytic hypochromic features. The presence of HbH and/or HbBart’s bands on hemoglobin electrophoresis is indicative of HbH disease.
4.2 Rare variants and analysis of CNVs in the β-globin gene cluster
4.2.1 Taiwan type
The three most common deletion-associated β-thalassemia variants in China, caused by large-scale deletions within the β-globin gene cluster, include Chinese type Gγ+(Aγδβ)0 thalassemia, the SEA-Hereditary Persistence of Fetal Hemoglobin (HPFH), and the Taiwan type. These variants are typically characterized by microcytic hypochromic anemia accompanied by significantly elevated HbF levels. In this study, one adult male heterozygote with the βTaiwanese/βN genotype (Taiwan type) was identified using GAP-PCR, consistent with previous findings reported by several members of the study team (13–15). Wang et al. documented five cases of the Taiwan type in Huadu, Guangdong Province, noting that mean HbA2 levels were higher than those observed in participants with Chinese type or SEA, whereas HbF levels were comparatively lower (15). Clinically, the presence of microcytic hypochromic anemia, elevated HbA2, and increased HbF levels along with negative results from conventional genetic testing should prompt the use of GAP-PCR to differentiate among the Chinese type, SEA, and Taiwan type.
4.2.2 Southeast Asian deletion SEA-HPFH
An adult male in this study was diagnosed with β17/βSEA-HPFH via Gap-PCR. Phenotypic descriptions of β-thalassemia associated with the SEA-HPFH deletion predominantly indicate mild to moderate anemia, with long-term survival generally not requiring regular blood transfusion support (16, 17).
Conventional genetic techniques are limited in detecting CNVs within the β-globin gene cluster. Accordingly, the integration of GAP-PCR with other diagnostic approaches and hematological parameters is recommended to achieve a comprehensive genetic and phenotypic assessment.
4.3 Rare variants and analysis of α-globin gene cluster SNVs
4.3.1 ααCD30 heterozygote
The ααCD30 (HBA2: c.91_93delGAG) heterozygous variant is a rare mutation located within exon 1 of the HBA2 gene, affecting protein translation. The resulting clinical phenotype, α+→α0, has been documented by multiple members of the study team across southeastern, southwestern, and southern regions of China (5, 6, 8, 18). Lin et al. reported 12 cases exhibiting microcytic hypochromic anemia and reduced HbA2 levels (8). In this study, an adult male was identified with moderate anemia, significantly low HbA2, a prominent presence of HbH bands, and a small number of HbBart’s bands. The rare genotype ααCD30/–SEA was found, consistent with findings reported by Ren et al. and Feng et al., and was classified as HbH disease (19, 20).
4.3.2 HBA2: c.168dup heterozygous variation
One case of the relatively rare HBA2 SNV, HBA2: c.168dup, was identified in this study. This variant, also referred to as codon 55/56 (+T), was previously reported in the heterozygous state by Peng et al. (6)
4.3.3 Intron variation
A case of ααIVS-I-117(G > A)/αα was identified in this study, presenting without anemia or microcytic hypochromic features, consistent with a previously reported case by Lin et al. in Guangxi (8). The IVS-II-55(T > G) heterozygous variant has been documented in multiple studies, typically in the absence of anemia or microcytic hypochromia, and is often associated with slightly reduced HbA2 levels (8, 21). This variant is frequently observed in combination with IVS-II-119(−G) and the SEA heterozygote in individuals lacking a clinical phenotype (22). In this study, one case of IVS-II-55(T > G) was detected in conjunction with a CS point mutation and δ-thalassemia. The IVS-II-34 (HBA2: c.300 + 34G > A) heterozygote is rarely reported in the literature; however, a case was identified in this study, with hematological parameters comparable to those described by Lin and Peng (8, 23).
4.3.4 PolyA (AATAAA > AATAAG) (HBA2:c. *94A > G)
PolyA mutations arise from various base substitutions or deletions within the HBA2 globin gene (24). Baysal et al. analyzed 84 chromosomes carrying α-thalassemia in the United Arab Emirates and reported that 47.4% harbored PolyA1 mutations (25). This mutation is considered extremely rare in China. Recently, Zhuang et al. described an individual with α+-thalassemia carrying the αPolyA1 mutation, who exhibited normal MCV (84& fL) and hemoglobin levels (113×10¹²g/L), with a mildly reduced MCH (26.5& pg) (26). In this study, one patient with this mutation along with the βCD26 (GAG > AAG) heterozygous variant, resulted in an imbalance of α/β globin chain synthesis. Notably, this patient did not exhibit anemia or microcytic hypochromic features. Hemoglobin electrophoresis data were not available for this case.
4.4 Rare variants and analysis of β-globin gene cluster SNVs
4.4.1 βN/βCD30 (A > G) (HBB c.91A > G)
In this study, two adult women and one 7-year-old girl were identified as heterozygous for the c.91A > G mutation. All three participants presented with mild anemia, microcytic hypochromic features, and elevated HbA2 levels. Similar findings were reported by Luo (7). The c.91A > G mutation affects the sequence upstream of the 5′ splice junction of the first intron, disrupting normal mRNA splicing and resulting in a β0 thalassemia.
4.4.2 βN/βIVS II-5 (G > C) (HBB: c.315 + 5G > C)
The IVS-II-5 (G > C) mutation, located at the fifth nucleotide of intron 2 in the β-globin gene, is classified as β+ thalassemia. In this study, two adult cases were identified with microcytic hypochromic features in the absence of anemia. Comparable cases have been documented by Yang and Ouyang (27, 28).
4.4.3 βN/β IVS-II-81C
Multiple reports in the literature indicated that participants heterozygous for this variant typically do not exhibit anemia or microcytic hypochromia and often present with normal or elevated HbA2 levels (14, 28). In this study, one case was identified in which this variant coexisted with the SEA heterozygote, resulting in a mitigation of the α/β imbalance and the absence of a clinical phenotype.
4.4.4 βN/βIVS-II-672 (HBB c.316-179)
No anemia, microcytic hypochromia, or abnormalities in HbA2 levels were observed. Similar findings have been documented by Huang et al. and Zhuang et al. (14, 26)
4.4.5 βN/β-31 (A > C) (HBB c.-81A > C)
This heterozygous variant was identified in an adult female who exhibited no anemia or microcytic hypochromia and presented with an elevated HbA2 level, consistent with findings reported by Lin and Peng (18, 23). The c.-81A > C mutation is located within the 5′ untranslated region of the HBB gene, a non-coding region implicated in the regulation of mRNA stability, pre-mRNA splicing, and translation initiation. Disruptions in this region may contribute to the pathogenesis of thalassemia.
The microcytic hypochromic features and reduced HbA2 levels commonly observed in SNVs of the α-globin gene cluster are consistent with those seen in CNVs. Additionally, elevated HbA2 levels associated with SNVs in the β-globin gene cluster are frequently used as an initial screening indicator for α-thalassemia prior to confirmatory genetic testing.
4.5 Hong Kong type and triad
Hong Kong type and triad represent distinct forms of α-thalassemia. Hematological parameters in carriers of the HKαα/αα and anti-HKαα/αα genotypes typically fall within normal reference ranges. Laboratory identification is primarily based on GAP-PCR followed by agarose gel electrophoresis, where the presence of two bands (a normal 3.7& kb band and a weaker 3.7& kb band) suggests the Hong Kong type genotype (e.g., HKαα/αα, HKαα/−α3.7, or αα/−α3.7), while the presence of three bands (normal, 3.7, and SEA) indicates a likely combination of the Hong Kong type with the SEA heterozygote (29–31).
In complex cases involving the Hong Kong type, triad, 3.7& kb, and 4.2& kb deletions, confirmation through nested PCR combined with MLPA or TGS is essential. In this study, hematological parameters for individuals with the HKαα/αα genotype were within the normal range, consistent with findings reported by Huang et al. (14) One case of HKαα/ααCS presented with slightly reduced MCH and normal HbA2 levels. Similarly, Lin reported two cases with mildly reduced MCH and HbA2 levels as low as 1.5% (11). Hematological profiles of the HKαα/−−SEA genotype were nearly indistinguishable from those observed in αα/−−SEA carriers, aligning with the reports of Lin (8), Huang (14), and Liang (31).
From these findings, when one partner carries the αα/−−SEA genotype and the other carries HKαα/α, prenatal diagnostic testing is not considered necessary. The phenotype of the HKαα/−−SEA genotype differs significantly from those of αα3.7/−−SEA and αα4.2/−−SEA, both of which are associated with mild to moderate anemia. In cases where the fetus is diagnosed with either the αα3.7/−−SEA or αα4.2/−−SEA genotype, further counseling and discussion regarding pregnancy outcomes should be conducted with the pregnant individual and their family.
The α-globin gene cluster contains three homologous sequence elements like X-, Y-, and Z-box fragments. During meiosis, misalignment and unequal homologous recombination among these regions can result in triplication of the α-globin gene. Recombination between the homologous Z2 and Z1 boxes generates the −α3.7 single-gene deletion allele and the corresponding αααanti-3.7 triplication allele. Similarly, recombination between the X2 and X1 boxes produces the −α4.2 deletion allele and the αααanti-4.2 triplication allele. Conventional PCR-based detection techniques may misclassify αααanti-4.2 and αααanti-3.7 alleles as αα, as these alleles retain intact α2 and α1 genes. When co-inherited with β-thalassemia, the resulting imbalance in the β/α globin chain ratio can exacerbate the clinical severity, potentially leading to intermediate β-thalassemia (12).
In this group, no participants with αααanti-3.7/αα or αααanti-4.2/αα genotypes were identified. However, four cases were detected with complex triplication-associated β-thalassemia, including two individuals with β41–42/αααaaa3.7 and two with β17/αααaaa4.2 genotypes. All four cases presented with clinical TI and required blood transfusion. These findings are consistent with those reported by Ren et al. (32)
4.6 Rare Hb variants
4.6.1 Rare α Hb variants
Hb Q-Thailand (HBA1: c.223G > C) arises from a GAC→CAC substitution at codon 74 of the HBA1 gene, resulting in the replacement of aspartic acid with histidine at the N-terminal region of the α1-globin chain. This variant is frequently associated with the −α4.2 deletion type of thalassemia. Clinically, Hb Q-Thailand is often asymptomatic and may be misclassified as a simple −α4.2 deletion, potentially leading to missed diagnoses. Previous studies have identified Hb Q-Thailand as the most prevalent hemoglobin variant in Shaoguan, Guangdong (0.17%, 17/10,285), particularly among the Hakka population in Meizhou, Guangdong Province, and it is also widely distributed across Southeast Asia (33).
In this group, two cases of −α4.2-Hb Q-Thailand/αα were detected, without clinical anemia, microcytic hypochromia, or elevated HbA2 levels. Abnormal hemoglobin bands were observed in the F and C zones on capillary electrophoresis, consistent with prior reports (5, 8, 20, 27). However, in complex genotypes involving large-fragment deletions such as co-inheritance with the SEA deletion (−α4.2-Hb Q-Thailand/−−SEA) HbH disease may develop. In such cases, capillary electrophoresis indicates the absence of the HbA band, with abnormal bands appearing in the F, Z15 (HbH), and Z12 (Hb Bart’s) zones. According to the research, anemia associated with the HbH-4.2Q Thailand genotype may be more severe than that observed in HbH-WS, and comparable to deletional HbH disease (e.g., −α4.2/−−SEA). In this study, a pregnant participant with the −α4.2-Hb Q-Thailand/−−SEA genotype had a hemoglobin level of 79& g/L, excluding iron deficiency anemia associated with pregnancy (11).
Hb G-Honolulu (HbA2: c.91G > C) involves a glutamine-to-glutamic acid substitution at codon 31 of the α-globin chain, resulting in altered retention time (RT) of hemoglobin subcomponents during specific cation-exchange high-performance liquid chromatography (HPLC) assays. Previous studies have indicated that this variant may interfere with HbA1c measurement, occasionally producing an “E window” rather than a numerical value (34). In such cases, the E window is smaller than that typically observed with the Hb E variant, suggesting the presence of an alternative hemoglobin variant. Subsequent genetic testing confirmed the presence of Hb G-Honolulu. In this study, the abnormal hemoglobin band corresponding to this variant was located in the D zone. The absence of clinical anemia and microcytic hypochromia was consistent with findings reported by Luo and Yang (11, 27).
Hb Hekinan (HbA1: c.84G > T) was first described in Japan in 1988 and results from a point mutation at codon 27 of the HBA1 gene, replacing glutamate with aspartate. Due to the absence of a net charge change, this variant is detectable by HPLC but may not be identified by capillary or cellulose acetate electrophoresis, increasing the risk of missed diagnosis. In this study, the variant was detected by TGS and was associated with elevated HbF levels resulting from γ-thalassemia, despite the absence of conventional thalassemia gene mutations. Chen reported a family with the Hb Hekinan variant in combination with the αα/−−SEA genotype, without significant hematological abnormalities or clinical anemia (35). It was hypothesized that the mutation in the HBA1 gene characterized by reduced functional output triggered compensatory upregulation of the HBA2 gene, partially offsetting α1 chain deficiency and resulting in a mild clinical phenotype. In this study, Hb Hekinan was associated with significantly elevated HbF levels due to coexisting Gγ-158C > T and Aγ-158C > T mutations, without anemia or microcytic hypochromia, though HbA2 levels were reduced. No previous reports have documented this specific complex phenotype.
4.6.2 Rare β-Hb variants
Hb New York (HBB: c.341T > A) is a hemoglobin variant characterized by the substitution of valine with glutamic acid at position 113 of the β-globin chain [β113 (G15) Val→Glu, GTG > GAG]. This replacement of a hydrophobic residue with an acidic one reduces the overall hydrophobicity of the β-chain and results in a structurally less stable hemoglobin molecule compared to Hb A, thereby increasing its susceptibility to degradation. Despite its relative frequency, Hb New York is typically not associated with significant hematological abnormalities or clinical anemia. On capillary hemoglobin electrophoresis, this variant presents as an abnormal band in the Z11 zone. Among the heterozygous individuals identified in this study, the mean concentration of abnormal hemoglobin was 43.05& ±& 3.96%, consistent with findings from multiple prior studies (20, 27, 28, 33, 36).
Hb J-Bangkok (HBB: c.170G > A) results from a point mutation at codon 56 of the β-globin gene (GGC > GAC), leading to the substitution of glycine with aspartic acid. This amino acid replacement induces a conformational change in the β-globin chain. Hb J-Bangkok is the second most common hemoglobin variant following HbE and Hb New York among heterozygotes. Clinically, its presentation is similar to that of Hb New York. On capillary electrophoresis, Hb J-Bangkok is observed as an abnormal hemoglobin band overlapping with the HbA2 band in the Z12 zone. In this study, four heterozygous cases of Hb J-Bangkok were identified, with an abnormal hemoglobin concentration of 43.05& ±& 3.96%, consistent with data reported in earlier studies (5, 11, 20, 27, 33).
Hb G-Taipei (HBB: c.68A > G) is a point mutation at codon 22 of the β-globin gene, resulting in the substitution of glutamic acid with glycine (Glu→Gly). HPLC analysis of HbA1c in participants carrying this variant has presented a RT longer than that of HbA0, attributed to the increased positive charge introduced by the mutation, which leads to an apparent elevation in HbA1c content (37). In this study, capillary electrophoresis of an Hb G-Taipei heterozygote demonstrated that 42.7% of the abnormal hemoglobin bands were located in the D zone. No clinical signs of anemia or hematological abnormalities were observed. These findings are consistent with reports from previous studies (27, 33).
HbO-Arab (HBB: c.364G > A) is a rare β-globin variant resulting from a substitution of glutamic acid with lysine at codon 121 (β121 Glu→Lys), effectively replacing normal HbA. Heterozygous carriers of HbO-Arab typically present with normal hemoglobin levels (38). In this study, one case of HbO-Arab was identified, with abnormal hemoglobin bands overlapping both HbA2 and HbF regions on capillary electrophoresis. Miniar Kalai et al. reported that individuals homozygous for HbO-Arab exhibited mild to moderate anemia (38). Van Gammeren et al. described a newborn carrying both HbO-Arab (HBB: c.364G > A) and Hb D-Los Angeles (HBB: c.664G > C), who developed mild microcytic anemia within one year (39). Elbashir et al. reported 13 African American individuals aged 2.7 to 62.5 years with Hb S/O, all of whom presented with hemolytic anemia (40). These findings indicate that the clinical phenotype associated with HbO-Arab is highly variable and may become more severe when co-inherited with other hemoglobin variants or β-thalassemia, often exceeding the clinical impact of the three more common Hb variants.
Hb Barcelona (HBB: c.283G > C) is a rare hemoglobin variant caused by the substitution of aspartic acid with histidine at position 94 of the β-globin chain. This mutation was first described by Aguilar et al. in a Spanish family presenting with mild polycythemia (41). Functional studies by Phillips et al. demonstrated that Hb Barcelona exhibits approximately a two-fold increase in oxygen affinity compared to normal HbA, likely accounting for the polycythemia observed in carriers without associated anemia (42). In this study, 34.8% of abnormal hemoglobin was detected in a case of Hb Barcelona, closely matching the 37% reported by Phillips et al. (42)
A common characteristic of abnormal hemoglobin variants, if involving α- or β-globin chains, is the appearance of abnormal bands on hemoglobin electrophoresis. These bands may vary in detectability; while some are readily identified, others are more subtle and may require confirmation through NGS or TGS. Unlike typical thalassemia syndromes, abnormal hemoglobinopathies do not consistently present with microcytic hypochromic features, which helps in their differential diagnosis. Furthermore, it should be noted that Hb Q-Thailand is frequently associated with the −α4.2 deletion. When co-inherited with the 4.2 deletion interlocking, the resulting genotype may resemble HbH disease, increasing the risk of missed diagnosis if not appropriately investigated.
4.7 δ-thalassemia
δ-thalassemia is primarily caused by point mutations. Both δ0- and δ+-thalassemia, if in the homozygous or heterozygous state, generally do not produce clinical symptoms. The principal hematologic feature is a reduced HbA2 level observed during hemoglobin analysis. To date, 19 distinct mutation types have been reported, including −77 T > C, −65 A > C, and CD30 G > C (43). Zhang et al. reported a δ-thalassemia prevalence of 0.49% in the Yunnan population, which is slightly higher than that observed in other southern Chinese populations (0.4%) (44).
In this study, δ-globin gene mutations were identified in 87 of 195 samples (44.62%). The most frequently observed mutation was −77 T > C, accounting for 88.51% of cases (77/87), followed by −30 T > C (3.45%, 3/87) and a start codon mutation (Met > Ile) (2.30%, 2/87). Two heterozygous cases were detected with the HBD:c.31delG mutation, both presenting with an HbA2 level of 1.4%. Additionally, one case of α-thalassemia with the genotype ααCS/ααIVS-II-55 was found to co-occur with δ-thalassemia mutations (CD65 A > T and CD115 C > T), exhibiting an even lower HbA2 level of 0.7% and no evidence of clinical anemia.
5 Conclusion
A total of 42 rare thalassemia gene mutations were identified in the Northern Guangxi population, including the first reported case of −α4.3/−−SEA, a rare 4.3& kb deletion (chr16:176935–181274DEL) encompassing the α1 gene and involving regional recombination with a non-functional −X–Y–Z segment. This finding contributes to the expanding mutational spectrum of thalassemia in the region. Each rare thalassemia variant was associated with distinct hematological characteristics and hemoglobin electrophoresis profiles. Clinically, rare genotypes may be inferred through integration of phenotypic features and corresponding hematological parameter patterns. The selection of appropriate diagnostic methods or a combination of multiple methods can improve diagnostic accuracy, reduce the likelihood of missed or incorrect diagnoses, and support effective prenatal and postnatal management strategies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Guilin People’s Hospital (Approval number: 2023-120KY). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
W-JY: Formal Analysis, Funding acquisition, Methodology, Resources, Software, Visualization, Writing – original draft, Writing – review & editing. Q-PK: Investigation, Project administration, Software, Supervision, Validation, Visualization, Writing – review & editing. L-ML: Data curation, Investigation, Methodology, Writing – review & editing. QZ: Methodology, Project administration, Validation, Writing – review & editing. X-MG: Formal Analysis, Investigation, Methodology, Resources, Writing – review & editing. MD: Investigation, Project administration, Supervision, Validation, Writing – review & editing. C-JH: Methodology, Resources, Visualization, Writing – review & editing. YL: Methodology, Project administration, Supervision, Visualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. (1) Funding Source: Open Project of NHC Key Laboratory of Thalassemia Medicine. Project Title: Precise Diagnosis and Intervention Strategies for Thalassemia with Genotype-Phenotype Inconsistency. Grant Number: GJWJWDP202303. (2) Funding Source: Self-funded Project of Western Medicine Category, Health Commission of Guangxi Zhuang Autonomous Region. Project Title: Study on Genotyping of Silent and Rare Types of Thalassemia in Pregnant Couples, Grant Number: Z-C20221640.
Acknowledgments
We are particularly grateful to all the people who have given us help on our article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1695120/full#supplementary-material
Abbreviations
CATSA, Comprehensive Analysis of Thalassemia Alleles; TM, Thalassemia major; TI, Thalassemia intermedia; HSCT, hematopoietic stem cell transplantation; TGS, Third Generation Sequencing; NGS, Next Generation Sequencing; CMA, Chromosome Microoarray Analysis; GAP-PCR, GAP Polymerase Chain Reaction; Hb, hemoglobin, MCV, mean corpuscular volume; MCH, meancorpuscular hemoglobin; SF, Serum ferritin; MLPA, Multiplex ligation-dependent probe amplification; SNV, Single nucleotide variation; SV, structural variation; CNV, copy number variation; HPFH, Hereditary Persistence of Fetal Hemoglobin; HbF: fetal hemoglobin; HPLC, High Performance Liquid Chromatography
References
1. Wang WD, Hu F, Zhou DH, Gale RP, Lai YR, Yao HX, et al. Thalassaemia in China. Blood Rev. (2023) 60:101074. doi: 10.1016/j.blre.2023.101074
2. Xian JJ, Wang YC, He JC, Li SY, He WZ, Mang XY, et al. Molecular epidemiology and hematologic characterization of thalassemia in guangdong province, southern China. Clin Appl Thromb Hemost. (2022) 28:10760296221119807. doi: 10.1177/10760296221119807
3. Tang HS, Xiong Y, Tang JQ, Wang XH, Wang Y, Huang LP, et al. Screening and diagnosis of rare thalassemia variants: is third-generation sequencing enough? Arch Pathol Lab Med. (2025) 149:e1–e10. doi: 10.5858/arpa.2023-0382-OA
4. Yin ZZ, Yao J, Wei FX, Chen CY, Yan HM, and Zhang M. Targeted next-generation sequencing reveals a large novel β-thalassemia deletion that removes the entire HBB gene. Hemoglobin. (2022) 46:290–5. doi: 10.1080/03630269.2022.2145964
5. Zhuang J, Wang J, Huang N, Zheng Y, and Xu L. Application of third-generation sequencing technology for identifying rare α- and β-globin gene variants in a Southeast Chinese region. BMC Med Genomics. (2024) 17:241. doi: 10.1186/s12920-024-02014-2
6. Peng CT, Zhang HX, Ren J, Chen H, DU Z, Zhou T, et al. Analysis of rare thalassemia genetic variants based on third-generation sequencing. Sci Rep. (2022) 12:9907. doi: 10.1038/s41598-022-14038-8
7. Luo S, Chen X, Zeng D, Tang N, Yuan DJ, Zhong QY, et al. The value of single-molecule real-time technology in the diagnosis of rare thalassemia variants and analysis of phenotype-genotype correlation. J Hum Genet. (2022) 67:183–95. doi: 10.1038/s10038-021-00983-1
8. Lin L, Zuo YJ, Chen BY, Zhou CF, Jiang YY, Wang L, et al. Phenotypic analysis of hemoglobinopathies caused by rare variations in the alpha-globin gene in Guangxi Zhuang Autonomous Region. Chin J Endemiol. (2024) 43:285–94. doi: 10.3760/cma.j.cn231583-20230104-00002
9. Li Y, Liang L, Tian M, Qin T, and Wu X. Detection of Hb H disease caused by a novel mutation and –SEA deletion using capillary electrophoresis. J Clin Lab Anal. (2019) 33:e22949. doi: 10.1002/jcla.22949
10. Wu MY, He Y, Yan JM, and Li DZ. A novel selective deletion of the major α-globin regulatory element (MCS-R2) causing α-thalassaemia. Br J Haematol. (2017) 176:984–6. doi: 10.1111/bjh.14005
11. Luo S, Chen XY, Zhong QY, Wang QH, Xu ZH, Qin LQ, et al. Analysis of rare thalassemia caused by HS-40 regulatory site deletion. Hematology. (2020) 25:286–91. doi: 10.1080/16078454.2020.1799587
12. Mota NO, Kimura EM, Ferreira RD, Pedroso GA, Albuquerque , Ribeiro DM DM, et al. Rare α0-thalassemia deletions detected by MLPA in five unrelated Brazilian patients. Genet Mol Biol. (2017) 40:768–73. doi: 10.1590/1678-4685-GMB-2016-0330
13. Li YM, Zhang SL, Kan LJ, Zhang B, Tang HM, Li R, and Zhang XM. Molecular characteristics and phenotypes of thalassemia gene variations. J Lab Med. (2019) 34:613–6. doi: 10.3969/j.issn.1673-8640.2019.07.009
14. Huang YY, Huang J, Ye LH, and Shen XL. Analysis of the detection of rare genotypes of thalassemia in the population of Laibin area, Guangxi. J Public Health Prev Med. (2023) 34:131–5. doi: 10.3969/j.issn.1006-2483.2023.01.031
15. Wang JC, Yao CZ, Huang YL, Liu L, Yuan TL, Qin DQ, et al. Differential diagnosis of the three most common deletion types of beta thalassemia in Chinese. Chin J Exp Hematol. (2021) 29:1247–50. doi: 10.19746/j.cnki.issn.1009-2137.2021.04.035
16. Ouyang H, Hu XN, Xu WH, Lan XE, Zhu SF, Liu DX, et al. A rare case of deletion type beta thalassemia and its family analysis. Lab Med Clin. (2020) 17:2447–8. doi: 10.3989/j.issn.1672-9455.2020.17.006
17. Ju AP, Fu XT, Liu YX, Lin K, Li N, and Li XC. Genotypes and hematological phenotypes of two common deletion types of beta thalassemia. Lab Med Clin. (2023) 20:177–85. doi: 10. 3969/j. issn.1672-9455.2023.02.008
18. Lin F, Yang LY, Lin M, Zheng XB, Lu M, Qiu ML, et al. Rare gene mutations of thalassemia in southern Chinese populations. Chin J Med Genet. (2017) 34:792–6. doi: 10.3760/cma.j.issn.1003-9406.2017.06.002
19. Ren ZM, Li WJ, Xing ZH, Fu XY, Zhang JY, Chen YS, et al. Detecting rare thalassemia in children with anemia using third-generation sequencing. Hematology. (2023) 28:2241226. doi: 10.1080/16078454.2023.2241226
20. Feng J, Cui D, Li C, Yang YS, Li QL, Li XM, et al. The comprehensive analysis of thalassemia alleles (CATSA) based on single-molecule real-time technology (SMRT) is a more powerful strategy in the diagnosis of thalassemia caused by rare variants. Clin Chim Acta. (2023) 551:117619. doi: 10.1016/j.cca.2023.117619
21. Chen F and Chen MF. Analysis of detection results of 121 cases of rare thalassemia gene mutations. Int Med Health Herald. (2024) 30)13:2190–7. doi: 10.3760/cma.j.issn.1007-1245.2024.13.015
22. Liang WD, Zeng JW, Xue JQ, Lin Y, and Chen XL. Identification of a rare alpha thalassemia gene mutation in a family and its genetic analysis. J Mol Diagn Ther. (2021) 13:76–8. doi: 10.19930/j.cnki.jmdt.2021.01.019
23. Peng C, He J, Zhou SH, Zeng G, Li HY, Wang WH, et al. Analysis of 21,618 cases of thalassemia gene detection in Changsha area. Chin J Prenat Diagn (Electronic Edition). (2022) 2)3:21–6. doi: 2.13470/j.cnoi.cjpd.2022.03.004
24. Kalle Kwaifa I, Lai MI, and Md Noor S. Non-deletional alpha thalassaemia: a review. Orphanet J Rare Dis. (2020) 15:166. doi: 10.1186/s13023-020-01429-1
25. Baysal E. α-thalassemia syndromes in the United Arab Emirates. Hemoglobin. (2011) 35:574–80. doi: 10.3109/03630269.2011.634698
26. Zhuang JL, Chen CN, Fu WY, Wang YB, Zhuang QM, Lu YL, et al. Third-generation sequencing as a new comprehensive technology for identifying rare α- and β-globin gene variants in thalassemia alleles in the chinese population. Arch Pathol Lab Med. (2023) 147:208–14. doi: 10.5858/arpa.2021-0510-OA
27. Yang FD, Gao WH, Liang LS, Xie YN, Xia LH, Huang GQ, et al. Analysis of thalassemia gene detection results in 9236 pregnant women in Foshan. J Mol Diagn Ther. (2020) 12:1147–51. doi: 10.3969/j.issn.1674-6929.2020.09.004
28. Ouyang H, Hu XN, Huang YY, Luo JX, Liu DX, Xu WH, et al. Analysis of 41 cases of rare beta thalassemia gene mutations. Chin J Birth Health Genet. (2021) 29:824–7. doi: 10.13404/j.cnki.cjbhh.20211014.001
29. Gu YY, Wu BY, Lin L, Cai G, and Gu MM. Genotype study of abnormal -alpha 3.7 deletion band in thalassemia. J Shanghai Jiaotong Univ (Med Sci). (2020) 40:1081–5. doi: 10.3969/J.ISSN.1674-8115.2020.08.013
30. Chen J, Ban SW, and Chen W. Genetic diagnosis and clinical phenotype analysis of Hb H disease with alpha thalassemia in Wuzhou area. Lab Med Clin. (2019) 16:2049–51. doi: 10.3969/j.issn.1672-9455.2019.14.028
31. Liang L, Zhao L, Tian M, Qin T, Wu X, Lao ZC, et al. Screening and identification of Hong Kong type combined with Southeast Asian type deletion thalassemia. Chin J Clin Med. (2020) 13:982–5. doi: 10.3969/j.issn.1674-3806.2020.10.07
32. Ren ZM, Huang LL, Liu SX, Li CG, and Chen YS. Clinical analysis of 2 cases of beta thalassemia combined with alpha globin gene triplet. J Clin Pediatr. (2021) 39:338–40. doi: 10.3969/j.issn.1000-3606.2021.05.004
33. Ma Z, Fan S, Liu J, Liu Y, Guo Y, and Huang W. Molecular characterization of hemoglobinopathies and thalassemias in Northern Guangdong Province, China. Med (Baltimore). (2021) 100:e27713. doi: 10.1097/MD.0000000000027713
34. Cheng WL, Neo SF, Chew S, Sethi SK, and Loh TP. HbG-Honolulu interferes with some cation-exchange HPLC HbA1c assays. Clin Chem Lab Med. (2016) 54:e77–9. doi: 10.1515/cclm-2015-0640
35. Chen XY and Lin FQ. Analysis of a family with rare Hb Hekinan combined with Southeast Asian type alpha thalassemia. Chin J Birth Health Genet. (2022) 30:686–9. doi: 10.13404/j.cnki.cjbhh.2022.04.037
36. Lan JF. Analysis of rare thalassemia gene mutations and prenatal diagnosis strategies in Fangchenggang area of Guangxi. Physic Online. (2023) 13:13–5.
37. Zhang XM, Wen DM, Xu SN, Suo MH, and Chen YQ. Effects of hemoglobin variants HbJ Bangkok, HbE, HbG Taipei, and HbH on analysis of glycated hemoglobin via ion-exchange high-performance liquid chromatography. J Clin Lab Anal. (2018) 32:e22214. doi: 10.1002/jcla.22214
38. Kalai M, Moumni I, Ouragini H, Chaouechi D, Boudriga I, and Menif S. Coinheritance of hbO arab/β0-thalassemia with severe manifestation in newborn. Am J Perinatol. (2024) 41:594–7. doi: 10.1055/s-0042-1743185
39. van Gammeren AJ, Pelkmans L, Endschot CCWV, Roelofsen-de Beer RJAC, and Harteveld CL. An Unusual Compound Heterozygosity for Hb O-Arab (HBB: c.364G>A) and Hb D-Los Angeles (HBB: c.364G>C). Hemoglobin. (2020) 44:61–3. doi: 10.1080/03630269.2019.1710530
40. Elbashir I and Elsayed Yousif TY. Molecular detection of hemoglobin O-arab in the Sudanese population. Int J Gen Med. (2023) 16:3323–30. doi: 10.2147/IJGM.S421140
41. Aguilar i Bascompte JL, Wajcman H, Poyart C, and Labie D. Hemoglobin Barcelona beta 94 (FG1) Asp leads to His: a new hemoglobin Variant with increased oxygen affinity. Nouv Rev Fr Hematol (1978). (1981) 23:267–73.
42. Phillips SE, Perutz MF, Poyart C, and Wajcman H. Structure and function of haemoglobin Barcelona Asp FG1(94) beta leads to His. J Mol Biol. (1983) 164:477–80. doi: 10.1016/0022-2836(83)90062-1
43. Zhang JW and Long JF. Hemoglobin and hemoglobinopathies. Guangxi: Guangxi Science and Technology Publishing House (2003).
Keywords: hemoglobin electrophoresis, phenotype, rare globin variation, genotype thalassemia, molecular diagnosis
Citation: Yang W-J, Kang Q-P, Liang L-M, Zhou Q, Gong X-M, Dou M, Huang C-J and Lin Y (2025) Genotypic and phenotypic characterization of rare globin variants in Northern Guangxi, China. Front. Immunol. 16:1695120. doi: 10.3389/fimmu.2025.1695120
Received: 29 August 2025; Accepted: 13 October 2025;
Published: 28 October 2025.
Edited by:
Qiao Liu, Children’s Hospital of Chongqing Medical University, ChinaReviewed by:
Lucia Carmela Cosenza, University of Ferrara, ItalyCatherine Lynn T. Silao, University of the Philippines Manila, Philippines
Copyright © 2025 Yang, Kang, Liang, Zhou, Gong, Dou, Huang and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Jia Yang, d2VpamlheWFuZ3l3akAxMjYuY29t
†These authors have contributed equally to this work