 Dong-Xu Liao
Dong-Xu Liao Xiang An1†
Xiang An1†- 1Department of Hepatobiliary and Pancreatic Surgery, Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
- 2Department of Emergency Surgery, Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
- 3General Practice Center, Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
Hepatocellular carcinoma (HCC) is the predominant type of liver cancer, characterized by high incidence and mortality rates. Despite advancements in surgical and systemic therapies, the prognosis remains poor due to the asymptomatic nature of early-stage HCC. Metabolic reprogramming in HCC cells usually creates an immunosuppressive tumor microenvironment (TME), thereby impeding T cell-mediated antitumor immunity. This review focuses on the metabolic reprogramming patterns in HCC, their impact on T cell function, and the potential of metabolic-immune targeted combination therapies. We emphasize that nutrient competition and the accumulation of inhibitory metabolites are key mechanisms underlying T cell suppression in the TME. This review provides an update on the complex metabolic-immune interactions and helps to identify new therapeutic targets to improve the efficacy of immunotherapy for HCC.
1 Introduction
Hepatocellular carcinoma (HCC), one of the main types of liver cancer, accounts for over 90% of all liver cancer cases. Its incidence and mortality rates rank among the highest globally (1, 2). In 2022, there were approximately 866,136 new cases and 758,725 deaths worldwide. The Asia-Pacific region bore the heaviest burden of HCC, accounting for 71% of global new cases and 42% of deaths in this area (3). In recent years, although significant progress has been made in curative treatments such as surgical resection, liver transplantation, and local ablation, as well as systemic therapies like targeted therapy and immunotherapy, the overall prognosis for HCC remains poor (4–7). The core challenge lies in the fact that HCC is often asymptomatic in its early stages. Most patients present with advanced tumors when clinical symptoms become apparent, thereby missing the opportunity for curative surgery (8). Therefore, there is an urgent need to obtain a deeper understanding of the molecular pathogenesis and identify new therapeutic targets in current HCC fields.
For patients with advanced HCC ineligible for surgery, systemic therapy has been considered as the primary approach to prolong survival and improve life quality (9). Over the past decade, multi-kinase inhibitors such as sorafenib and lenvatinib have become the standard first-line treatments. However, their objective response rate (ORR) typically ranges between 10% and 20%, and patients inevitably develop primary or secondary resistance (10). Nowadays, the emergence of immune checkpoint inhibitors (ICIs), represented by Programmed death receptor-1 (PD-1)/Programmed Death-Ligand 1 (PD-L1) inhibitors, has brought revolutionary breakthroughs in the treatment of advanced HCC (11). ICIs can “release the brakes,” reactivating suppressed T cells and restoring their ability to kill tumor cells by blocking the binding of PD-L1 on the surface of tumor cells to PD-1 on the surface of T cells (12). However, clinical practice has shown that the ORR of ICI monotherapy for HCC is only 15%-20% (13). Even with immune combination therapies like the “atezolizumab + bevacizumab” (T+A regimen), which increase the ORR to approximately 30%, most patients still do not respond (primary resistance) or experience disease progression after initial effectiveness (secondary resistance) (14).
Metabolic reprogramming—the adaptive alteration of cellular metabolic patterns by cancer cells to meet their demands for rapid proliferation and survival—is now recognized as one of the hallmark characteristics of cancer (15, 16). This reprogramming enables them to overcome the limitations of normal metabolism, efficiently acquire and utilize limited nutrients (e.g., glucose and glutamine) to sustain their own survival and support uncontrolled rapid proliferation (17, 18). Given the pervasiveness and central role of these metabolic alterations in tumor biology, cellular metabolic reprogramming and its accompanying changes in energy production and utilization have been unequivocally identified as a hallmark feature of cancer (19). Thus, in-depth investigation into the unique and often aberrant metabolic mechanisms of tumor cells—such as their high dependence on aerobic glycolysis (the Warburg effect) and glutaminolysis—is widely regarded as an extremely promising research field with significant scientific value and clinical application potential (20, 21). Exploring metabolic reprogramming in HCC helps reveal its fundamental biological behavior and opens entirely new avenues for developing novel diagnostic biomarkers, prognostic assessment tools, and therapeutic strategies (22, 23).
The therapeutic bottlenecks of ICIs have profoundly highlighted significant gaps in our understanding of HCC immune evasion mechanisms. Tumors do not simply evade immune surveillance by expressing checkpoint molecules like PD-L1; instead, this evasion is underpinned by a more complex and robust inhibitory network (24–26). At the core of this network lies the “soil” upon which tumors thrive and develop—the tumor microenvironment (TME) (27–29). As an active digestive organ, metabolic changes in HCC profoundly impact the antitumor activity of the immune system within the TME (23, 30, 31). Therefore, it is of great significance to investigate the intrinsic mechanisms leading to ICI treatment resistance, particularly the immunosuppressive mechanisms caused by metabolic alterations in the TME, and identifying new targets and strategies to overcome resistance and sensitize immunotherapy in the field of liver cancer research.
2 Metabolic reprogramming patterns in HCC cells
Metabolic reprogramming has been recognized as one of the ten core hallmarks of cancer (15). This is not merely a passive consequence of malignant proliferation in tumor cells, but a key driver enabling them to actively adapt to the microenvironment and sustain survival, invasion, and metastasis. Compared to normal hepatocytes, HCC cells have undergo significant metabolic alterations to support their biological behaviors of unlimited proliferation, invasion, and metastasis (32). Recent proteomic and metabolomic studies have revealed the remodeling of multiple core metabolic pathways in HCC (33–35).
2.1 Glucose metabolic reprogramming: the Warburg effect and beyond
2.1.1 Glycolysis
It is well-known that enhanced glycolysis is the most classic and extensively studied metabolic feature of HCC cells, known as the “Warburg Effect” (36–38). Cancer cells preferentially convert glucose into lactate even under oxygen-sufficient conditions, rather than fully oxidizing it via mitochondrial tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) (39, 40). This shift is driven by the upregulation of key enzymes and transporters, including glucose transporter 1 (GLUT1) (41), hexokinase 2 (HK2) (42), pyruvate kinase M2 isoform (PKM2) (43), and lactate dehydrogenase A (LDHA) (44). Hexokinase 2 (HK2) enhances glucose uptake by binding to the mitochondrial voltage-dependent anion channel (VDAC) (45, 46). Its inhibitor, Chrysin, can induce cancer cell apoptosis (47). Conversely, high expression of the pyruvate kinase M2 isoform (PKM2) promotes lactate production (48). The natural compound Shikonin effectively blocks glycolysis by inhibiting PKM2 and sensitizes cancer cells to sorafenib (49, 50). Hypoxia-inducible factor HIF-1α plays a central regulatory role in this process. It drives the expression of enzymes such as HK2 and aldolase A (ALDOA) (51, 52). Additionally, metformin can inhibit glycolytic flux by activating the AMPK pathway to inhibit phosphofructokinase 1 (PFK1) (53, 54).
This seemingly inefficient energy production strategy confers the following survival advantages to cancer cells:
1. Rapid energy supply: Accelerated glycolysis generates ATP at a significantly faster rate compared to OXPHOS, meeting the high energy demands of rapidly proliferating cancer cells (55).
2. Biosynthetic precursor provision: Glycolytic intermediates—such as glyceraldehyde-3-phosphate and phosphopentoses—are diverted into the pentose phosphate pathway (PPP) and serine synthesis pathway. These supply precursors for nucleic acids, non-essential amino acids, and reducing equivalents, supporting macromolecular biosynthesis and redox homeostasis (56, 57).
3. Microenvironment acidification: Massive lactate efflux acidifies the TME. This suppresses immune cell activity to promote immune evasion while enhances tumor cell invasiveness and metastatic potential (58–60). Notably, lactate, the end product of glycolysis, is not merely a metabolic waste but can directly regulate gene expression through histone lactylation (such as the H3K18la modification), thereby influencing inflammatory responses within the TME (61).
2.1.2 Pentose phosphate pathway
Pentose Phosphate Pathway (PPP), which is co-activated in HCC, serves not only as an alternative energy source but also as a core strategy for maintaining redox homeostasis and biosynthesis (62). Glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of PPP, drives malignant progression through two key pathways via its overexpression:
Antioxidant Defense: Catalyzes the generation of NADPH to reduce glutathione (GSH), thereby neutralizing reactive oxygen species (ROS) and protecting tumor cells from oxidative damage (63).
Pro-Metastatic Mechanism: Activates STAT3 phosphorylation, inducing the expression of key epithelial-mesenchymal transition (EMT) molecules, promoting invasion and metastasis (64).
Notably, transketolase (TKT) exhibits tissue-specific overexpression in HCC (65). Its abnormal elevation depletes reduced NADP+, leading to ROS accumulation, which subsequently activates pro-survival signals (66, 67). Targeted intervention confirmed: the TKT inhibitor Oxythiamine significantly increases intracellular ROS levels (68). When combined with Sorafenib, it achieved over 60% tumor growth inhibition in a patient-derived xenograft (PDX) model of 292 HCC patients (69). The biological significance of PPP extends beyond antioxidation – the ribose-5-phosphate it generates – provides essential precursors for nucleotide synthesis, directly supplying the DNA/RNA raw materials for rapid tumor cell proliferation (70, 71).
2.1.3 Gluconeogenesis
In sharp contrast, gluconeogenesis is systematically inhibited in HCC (72, 73). Fructose-1,6-bisphosphatase 1 (FBP1) is a key tumor-suppressing enzyme that is inactivated through multi-layered regulation: 1) Epigenetic Silencing: Histone deacetylase-mediated loss of H3K27ac modification suppresses FBP1 promoter activity (ChIP-seq confirmed enrichment of HDAC1/2 at the FBP1 promoter region) (74); and 2) Ubiquitination-Dependent Degradation: The E3 ubiquitin ligase TRIM28 directly binds FBP1, promoting its 26S proteasome-dependent degradation (validated by co-immunoprecipitation assays) (75). FBP1 deficiency releases the inhibition on the glycolytic rate-limiting enzyme PFK1, causing a surge in glycolytic flux (76, 77).
Another key enzyme, phosphoenolpyruvate carboxykinase (PCK1), exhibits a “dual nature”: Under physiological conditions, PCK1 maintains metabolic homeostasis by inhibiting TCA cycle intermediate efflux (cataplerosis) (78).
In HCC, AKT-mediated phosphorylation at Ser90 triggers its aberrant translocation to the endoplasmic reticulum. At the same site, it phosphorylates INSIG1/2 proteins, releases the lipid synthesis transcription factor SREBP1, and unexpectedly drives lipogenesis and tumor progression (79).
Rescue strategies targeting suppressed gluconeogenesis show:
The histone deacetylase inhibitor SAHA upregulates FBP1 expression by 2.5-fold, while lowering he glycolysis rate in HepG2 cells by 40% (74).
The glucocorticoid Dexamethasone restores PCK1/FBP1 expression by activating the transcriptional coactivator PGC-1α, thereby reversing the Warburg effect and reducing tumor volume by 52% in animal models (80, 81).
2.1.4 Tricarboxylic acid cycle
Remodeling of the Tricarboxylic Acid (TCA) Cycle plays a pivotal role in HCC metabolic adaptation (82). The anaplerotic reaction driven by pyruvate carboxylase (PC) replenishes TCA intermediates by generating oxaloacetate (83). Treatment of HepG2 cells with its small molecule inhibitor UK-5099 reduced aspartate synthesis by 70%, causing nucleotide synthesis impairment and cell cycle arrest (84). Conversely, downregulation of isocitrate dehydrogenase 2 (IDH2) promotes extracellular matrix degradation and metastasis by negatively regulating matrix metalloproteinase MMP9 (immunohistochemistry shows a significant negative correlation between IDH2 and MMP9 expression) (85, 86). For the IDH-mutant subtype (mIDH), in a Phase I clinical trial, the oral inhibitor Ivosidenib (AG-120) extended progression-free survival by 3.1 months in advanced HCC patients (NCT02073994) (87, 88).
Furthermore, overexpression of malic enzyme 1 (ME1) drives metastasis through a dual pathway: 1) catalyzing malate decarboxylation to generate NADPH, maintaining a reductive microenvironment (89, 90); and 2) the activation of the ROS-ZEB1 signaling axis induces EMT and suggests its potential as a pan-cancer therapeutic target, as evidenced by an 80% decrease in HCC cell invasiveness following ME1 knockdown with shRNA (91).
2.1.5 Glycolytic metabolites and the epigenetic landscape in hepatocellular carcinoma
Metabolic reprogramming directly modulates the epigenetic landscape of both HCC cells and T cells by altering the abundance of key metabolites. Acetyl-CoA, derived from glucose and lipid metabolism, serves as the primary donor for histone acetylation. Its accumulation in HCC increases histone acetylation levels, thereby activating genes associated with cell proliferation. S-adenosylmethionine (SAM), produced via the methionine cycle, acts as the principal methyl donor for histone methylation. Increased SAM generation promotes histone methylation, leading to the activation of genes involved in tumor progression (92, 93). Alpha-ketoglutarate (α-KG), an intermediate of the TCA cycle, is an essential substrate for histone and DNA demethylation. Its reduction inhibits demethylation processes, thereby promoting tumorigenesis (94, 95).
Furthermore, metabolic reprogramming influences the epigenetic state of T cells. For instance, elevated lactate levels induce histone lactylation (H3K18la) within T cells, which suppresses T-cell activation (96). Future research should focus on elucidating the intricate coupling mechanisms between metabolism and epigenetics. Developing novel therapeutic strategies that target metabolic pathways to restore metabolite balance, reverse epigenetic alterations, inhibit tumor progression, and enhance the efficacy of immunotherapy represents a promising direction.
2.2 Lipid metabolic reprogramming: the de novo synthesized “fuel depot” and signaling hub
Lipid metabolism, including synthesis, storage, and degradation of lipids, is crucial for cellular function and membrane integrity, and its dysregulation is associated with diseases like cancer (97, 98).
2.2.1 Fatty acid uptake
Liver is the central organ of lipid metabolism, and the abnormalities in lipid metabolism in HCC involve multiple aspects (99, 100). In terms of fatty acid uptake and transport, transmembrane transporters such as CD36 and fatty acid transport proteins (FATPs) are upregulated in HCC tissues, promoting fatty acid uptake (101, 102). Meanwhile, the upregulation of CD36 can induce EMT, thereby promoting invasion and metastasis (103, 104). Among fatty acid-binding proteins (FABPs), FABP1 and FABP5 are highly expressed in HCC (105, 106). FABP1 promotes cell migration, angiogenesis, and metastasis through the VEGFR2/SRC and FAK/CDC42 signaling pathways, while FABP5 promotes lipid accumulation and cell proliferation by activating the HIF-1α axis. This is associated with poor prognosis (107, 108). Lipoprotein lipase (LPL) hydrolyzes triglycerides (TAGs) into free fatty acids (FFAs), promoting the uptake of lipoproteins by cells (109–111). Its expression is higher in advanced HCC (stages III/IV), and inhibiting LPL can hinder the proliferation of HCC cells (112).
2.2.2 De novo lipogenesis
Regarding de novo lipogenesis (DNL), which is abnormally activated in HCC and serves as a hallmark feature, provides membrane components and signaling molecules for rapidly proliferating tumor cells (113–115). The expression of key enzymes is significantly increased. For example, ATP citrate lyase (ACLY) cleaves citrate exported from the mitochondria into cytoplasmic acetyl-CoA (a substrate for fatty acid synthesis) (116, 117). Oncogenic drivers such as PI3K/AKT activate ACLY and the Warburg effect through feedback loops (107, 118). Acetyl-CoA carboxylase (ACC) is the rate-limiting enzyme for fatty acid synthesis and catalyzes the formation of malonyl-CoA from acetyl-CoA (119, 120). Its liver-specific inhibitor ND-654 can inhibit hepatic DNL, inflammation, and HCC development (121). Fatty acid synthase (FASN) synthesizes fatty acids such as palmitate from malonyl-CoA and acetyl-CoA (122). Its overexpression indicates a poor prognosis, and inhibiting FASN significantly suppresses the growth and tumorigenesis of HCC cells. Targeting FASN and related de novo lipogenesis is a potential therapeutic strategy (123, 124). Stearoyl-CoA desaturase 1 (SCD1) converts saturated fatty acids into monounsaturated fatty acids (MUFAs) (125). Interfering with SCD1 effectively inhibits HCC progression, while its overexpression is associated with shorter disease-free survival. SCD1 exerts its effects by regulating p53, WNT/β-catenin, EGFR, and autophagy. Inhibiting SCD1 can induce endoplasmic reticulum (ER) stress, which differentiates liver cancer stem cells and thereby sensitizes them to sorafenib (107, 126–128). Acyl-CoA synthase long-chain family member 4 (ACSL4) prefers arachidonic acid, and its overexpression stabilizes c-MYC through the ERK/FBW7/c-MYC axis, promoting tumor formation in vivo and in vitro. It is a potential prognostic marker and therapeutic target (129, 130).
In terms of transcriptional regulation, sterol regulatory element-binding protein 1 (SREBP1) is the core transcriptional activator of de novo lipogenesis (DNL) and is significantly more highly expressed in HCC tumor tissues than in adjacent non-tumor tissues (131–133). Downregulating SREBP1 inhibits the proliferation, migration, and invasion of HCC cells and induces apoptosis and SREBP1 is a prognostic marker for HCC (132). Liver X receptor (LXR), a member of the nuclear receptor superfamily, regulates cholesterol homeostasis and de novo lipogenesis by transactivating SREBP1, FASN, and others (134). LXRα is downregulated in HCC tissues (135–137). LXR agonist T0901317 can upregulate LXRα while downregulate GLUT1 and MMP9, thereby inhibiting HCC progression (136). Nevertheless, long-term use of LXR agonists combined with oxidative stress and a high-fat diet can induce a non-alcoholic steatohepatitis (NASH)-like condition in mice and progress to HCC, demonstrating the complexity of the effects (138).
2.2.3 Fatty acid oxidation
Regarding the dysregulation of fatty acid β-oxidation (FAO), despite the frequent lack of nutrients in tumor centers due to inadequate vascularization, FAO remains an important catabolic pathway for providing energy and anabolic precursors for cell growth (139). Carnitine palmitoyltransferase 1 (CPT1) is the rate-limiting enzyme for FAO and is located on the outer mitochondrial membrane. With abundant glucose, ACC forms a complex with CPT1A to prevent its localization to the mitochondria. During glucose deprivation, the complex dissociates, and free CPT1A localizes to the mitochondrial membrane to enhance FAO (140–142). ACC and CPT1A together protect HCC cells against metabolic stress (143, 144). Acyl-CoA dehydrogenases, including medium-chain (MCAD) and long-chain (LCAD) acyl-CoA dehydrogenases, catalyze the first step of mitochondrial FAO (145–147). They are downregulated by hypoxic stress, inhibiting fatty acid breakdown and promoting HCC cell proliferation. Forced expression of these enzymes can decrease the lipid accumulation caused by hypoxia (148–150). However, knocking down LCAD can promote tumor growth in vivo by inhibiting the Hippo pathway (151). Lipolysis provides the fatty acid substrates for FAO (152). Monoglyceride lipase (MAGL) catalyzes the conversion of monoglycerides into FFAs and glycerol (153, 154). Its high expression in cancer promotes HCC cell proliferation and invasion by generating signaling lipids (including monoglycerides and FFAs), while the NF-κB signaling pathway is involved in MAGL-mediated EMT (155). Adipose triglyceride lipase (ATGL) initiates the hydrolysis of TAGs into diacylglycerols (DAGs) and FFAs (156, 157). High expression of ATGL in HCC tissues leads to elevated levels of DAGs and FFAs, which are associated with poor prognosis. lncRNA-NEAT1 induces abnormal lipolysis in HCC cells by upregulating ATGL, driving cell growth (158).
2.2.4 Cholesterol synthesis
Regarding cholesterol metabolism disorders, dysregulation of cholesterol biosynthesis is a common event in HCC. During the hepatocarcinogenesis, there is new biochemical crosstalk between de novo lipogenesis and the cholesterol biosynthesis pathway (159). HMG-CoA reductase (HMGCR), the rate-limiting enzyme in the mevalonate pathway of cholesterol synthesis, can be inhibited by statins (160–162). For human HCC samples, the upregulation of HMGCR is accompanied by increased mitochondrial cholesterol levels. Inhibiting HMGCR or squalene synthase (the enzyme in the first step of cholesterol biosynthesis) to deplete cholesterol can enhance the sensitivity of HCC cells to chemotherapy (107, 163, 164). Squalene synthase inhibitors (e.g., YM-53601) can synergistically mediate HCC growth arrest and cell death with doxorubicin in vivo (163).
Regarding other related pathways, peroxisome proliferator-activated receptor α (PPARα) regulates the constitutive transcription of genes related to fatty acid transport and TAG homeostasis (165). Long-term administration of PPARα ligands accelerates hepatocyte proliferation, increases ROS production, and leads to HCC in rodents (166–168). The oncogene MYC directly acts as a transcriptional amplifier for specific PPARα target genes (such as KRT23, which promotes hepatocyte proliferation and potential HCC) (169). Cytochrome P450 family 4 (CYP4) is a group of ω-hydroxylases involved in fatty acid transformation. Lowered expression of CYP4 is associated with liver fat accumulation and the pathogenesis of NASH. CYP4Z1 and its pseudogene CYP4Z2P are highly expressed in breast cancer and promote angiogenesis, but their roles in fatty acid metabolism in HCC remain unclear (170, 171).
2.3 Amino acid metabolic reprogramming: glutamine as the central hub
The progression of HCC is accompanied by profound metabolic reprogramming, in which the dysregulation of amino acid metabolism plays a key role in meeting the vigorous biosynthetic demands of tumor cells, maintaining redox homeostasis, and evading immune surveillance (172, 173).
As the central organ of amino acid metabolism, the liver undergoes extensive changes in amino acid uptake, synthesis, degradation, and waste disposal pathways during carcinogenesis, providing an important window into understanding the pathogenesis of HCC and developing targeted therapies (174). Glutamine is an important nitrogen and carbon source for HCC cells, and its abnormal metabolism (glutaminolysis) is a significant characteristic of HCC (175–177). Glutaminase (GLS) is the rate-limiting enzyme for glutaminolysis, with two subtypes playing distinct roles in HCC (178, 179). GLS1 (kidney-type) is universally overexpressed in HCC, promoting cell proliferation and colony formation by activating the AKT/GSK3β/Cyclin D1 axis (180). Its upregulation is associated with advanced clinical pathological features and a stem cell phenotype, regulating cancer stem cell properties through the ROS/WNT/β-catenin pathway (181). Knocking out GLS1 can inhibit tumorigenicity in vivo, suggesting it is an important target for eradicating cancer stem cells (182). GLS2 (liver-type) typically exerts its tumor - suppressing function by promoting ferroptosis, and its regulatory role in glutaminolysis is essential for tumor growth inhibition (183–185). Additionally, GLS2 inhibits EMT through the Dicer-miR-34a-Snail axis, exerting its non-glutamine catabolism function to suppress migration and invasion of HCC cells (186).
To meet the dramatically increased demand for amino acids, HCC cells heavily rely on transmembrane transporters to uptake amino acids from the microenvironment, making key transporters potential therapeutic targets (187, 188). SLC1A5/ASCT2 is the primary Na+-dependent glutamine transporter, whose expression is significantly higher in HCC tumor tissues compared to adjacent non-tumor tissues, and positively correlated with tumor size, making it a potential prognostic indicator (189). SLC7A5/LAT1 forms a heterodimer with 4F2hc (SLC3A2/CD98) to transport large neutral amino acids (such as leucine) (190). LAT1 expression is significantly elevated in HCC lesions and is associated with tumor growth (191, 192). Hippo pathway effectors YAP/TAZ increase amino acid uptake by upregulating LAT1 expression, activating mTORC1, and promoting proliferation (193). Patients with high LAT1 expression have significantly shorter survival rates. Its structural and functional mechanisms are relatively clear, making it a highly promising therapeutic target. The selective inhibitor JPH203 showed partial response or stable disease in a Phase I trial for advanced solid tumors (including cholangiocarcinoma) and is currently in Phase II trials (for cholangiocarcinoma). Another inhibitor, QBS10072S, is undergoing a Phase I trial (including HCC patients) (181, 194).
Glutamic Oxaloacetic Transaminase 1 (GOT1) coordinates glucose and amino acid metabolism to meet nutritional demands (195). Oncogenic KRAS mutations have been found to depend on GOT1 to support long-term cell proliferation (196). lncRNA TMPO-AS1 accelerates HCC progression by targeting the miR-429/GOT1 axis (197). Abnormalities in ornithine cycle (urea cycle) enzymes are common in HCC (197). Carbamoyl Phosphate Synthetase 1 (CPS1), the rate-limiting enzyme in the first step of the urea cycle, is a hypermethylated gene in HCC and is downregulated by aflatoxin B1, thereby inhibiting proliferation and inducing apoptosis (198–201). Argininosuccinate Synthase 1 (ASS1) is another rate-limiting enzyme that is frequently deficient in tumors. ASS1 deficiency in tumors is both a prognostic biomarker and a predictor of sensitivity to arginine-deprivation therapy (202). Due to DNA methylation, ASS1 expression is significantly reduced in HCC patients, and stable silencing of ASS1 promotes migration and invasion (203, 204). ASS1 deficiency increases STAT3 Ser727 phosphorylation and promotes metastasis by upregulating the inhibitor of differentiation 1 (205). ASS1 inhibits HCC metastasis and is a potential diagnostic and therapeutic target (202). Arginase-1 (ARG1) is expressed in the cytoplasm of hepatocytes and is involved in anti-inflammatory, tumor immune, and immunosuppressive functions. Overexpression of ARG1 enhances the viability, migration, and invasion of HCC cells and significantly increases the expression of key EMT factors (vimentin, N-cadherin, β-catenin) (206). Ornithine Transcarbamylase (OTC) is located in the mitochondrial matrix. OTC deficiency disrupts the urea metabolic pathway, leading to elevated blood ammonia levels (potentially life-threatening in severe cases). OTC silencing promotes HCC cell proliferation, and clinical data show that patients with low OTC levels have shorter overall survival (207–210).
Table 1 summarizes the key metabolic pathways, molecules, targeted inhibitors, and therapeutic significance in HCC. It serves as a generalization and supplement to the detailed metabolic reprogramming patterns described earlier. This allows readers to quickly review and compare these key pieces of information, and provides a reference for the subsequent discussion on the impact of metabolic reprogramming on T cells and therapeutic strategies.
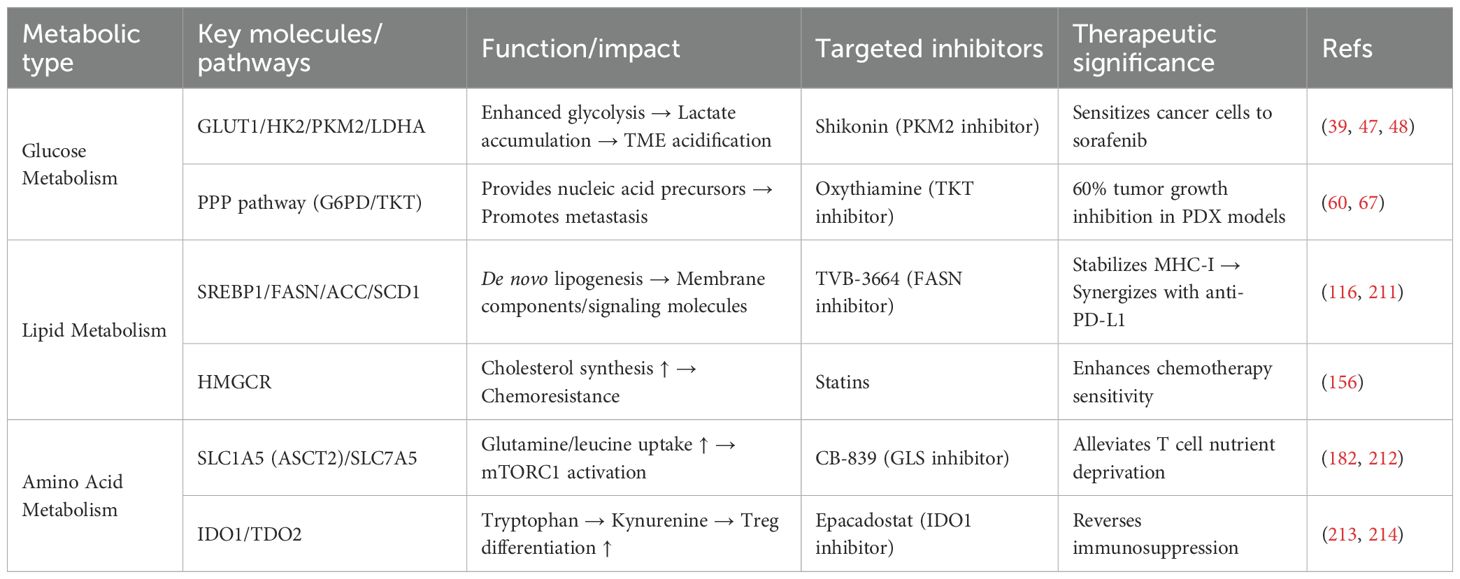
Table 1. Key pathways and targets of metabolic reprogramming in HCC.
3 The alteration of key metabolites in the TME of HCC
TME is a complex ecosystem composed of tumor cells, immune cells, stromal cells, blood vessels, and various cytokines and chemicals (215). Within this ecosystem, tumor cells are not passively present but actively reshape the environment to facilitate their own survival through various means (216). Metabolic reprogramming is one of their most powerful tools for this transformation (217). By altering the uptake, utilization of key nutrients, and secretion of metabolic byproducts, tumor cells can launch a “metabolic war” against the immune system, ultimately establishing an immunosuppressive microenvironment (218).
3.1 Competitive nutrient deprivation
The rapid, uncontrolled proliferation of tumor cells is one of the core biological characteristics (15). This process creates an extremely high dependence on nutrients within the microenvironment. To win this fierce competition for survival, HCC cells have evolved a series of complex metabolic reprogramming strategies. The core of these strategies lies in effectively plundering core resources such as glucose and amino acids from the TME by upregulating the expression of key nutrient transporters on the cell membrane (217, 219). This meets their biosynthetic and energy metabolism demands while causing local nutrient deprivation, thereby suppressing the function of competitors like immune cells (29).
3.1.1 Glucose
“Warburg effect” is one of the most prominent metabolic features of HCC. However, this seemingly inefficient metabolic pattern provides cancer cells with rapid ATP energy and abundant biosynthetic precursors (such as ribose, glycerol, etc.) to support their rapid proliferation (220, 221).
3.1.1.1 Upregulation of glucose transporters
To meet the enormous demand for glucose, HCC cells significantly upregulate the expression of glucose transporters (GLUTs). As the most extensively studied transporter, GLUT1 is highly expressed in the majority of HCC tissues. Its expression level is closely correlated with tumor aggressiveness, glycolytic flux, and poor patient prognosis (222). A study has confirmed that GLUT1 mRNA and protein levels are significantly higher in HCC cells compared to adjacent non-tumor tissues. Knocking down GLUT1 effectively inhibits HCC cell growth and migration (222). GLUT2, also responsible for glucose transport in normal hepatocytes, is upregulated in HCC and associated with poor prognosis (223, 224). GLUT3 has also been shown to be upregulated in HCC, and promote glucose uptake and driving glycolytic reprogramming alongside other GLUTs (225).
The upregulation of GLUTs is regulated by complex signaling networks, which are often abnormally activated in HCC:
PI3K/AKT/mTOR Pathway: This is one of the core signaling pathways in cancer, comprehensively regulating cell growth, proliferation, and metabolism. Its abnormal activation is a major driver of altered glucose metabolism in tumors (226).
HIF-1α: Hypoxia-inducible factor-1α (HIF-1α) is activated under the hypoxic conditions common in the TME. As a key transcription factor, it can directly upregulate the expression of GLUT1, and various glycolytic enzymes, thereby enhancing cell survival capability in hypoxic environments (227, 228).
FOXM1: The transcription factor FOXM1 plays a significant oncogenic role in HCC. FOXM1 can transcriptionally activate GLUT1 expression by directly binding to its promoter region, systematically regulating glycolysis in HCC and promoting tumorigenesis (229).
3.1.2 Glutamine and glutamate
If glucose is the primary fuel for HCC cells, glutamine is their indispensable “secondary fuel” and a key nitrogen source (176). Glutaminolysis provides HCC cells with energy, biosynthetic precursors, and key substances for maintaining redox homeostasis (230).
HCC cells voraciously uptake glutamine and glutamate by upregulating various amino acid transporters, leading to glutamine and glutamate deficiency in the TME (176).
SLC1A5 (ASCT2) is the primary and most extensively studied glutamine transporter. For HCC, SLC1A5 expression is widely upregulated, and its high expression is significantly correlated with tumor growth, metastasis, and poor patient prognosis. Targeted inhibition of SLC1A5 effectively suppresses HCC growth (189, 231).
SLC38A1 (SNAT1) and SLC38A2 (SNAT2): According to research data, these two transporters are significantly upregulated in HCC. Notably, SLC38A2 is even the highest expressed glutamine transporter in certain HCC models. They work synergistically with SLC1A5 to ensure sufficient glutamine supply (232–234).
SLC1A3 (EAAT1/GLAST): As a high-affinity glutamate transporter, SLC1A3 expression in HCC tissues is significantly higher than in normal tissues. Its high expression is closely associated with worse tumor grade, pathological stage, overall survival, and immune evasion phenomena, making it a potential novel prognostic biomarker and therapeutic target (235).
3.1.3 Methionine
Hoffman effect refers to the dependence of cancer cells on methionine. All types of cancer cells exhibit an “addiction” to methionine (236). This phenomenon arises from excessive transmethylation in cancer cells, leading to a greater requirement for methionine compared to normal cells (237). HCC cells commonly exhibit an imbalance in the expression of the methionine adenosyltransferase (MAT) family, characterized by downregulation of the tumor-suppressive MAT1A and upregulation of the oncogenic MAT2A, resulting in abnormalities in the S-adenosylmethionine (SAM) synthesis pathway (238, 239). In diverse human tumor cell lines, ASS1 (argininosuccinate synthase 1) expression is lost, preventing the tumor from regenerating methionine via homocysteine, thereby creating a “methionine auxotrophic” phenotype (240, 241). HCC cells highly express the methionine transporter SLC43A2, and its expression level positively correlates with tumor malignancy (242). This ultimately leads to methionine deficiency in the TME.
3.1.4 Cystine/cysteine
Cystine/cysteine serves as a nutrient supporting HCC growth and participates in the intracellular redox balance of tumor cells (243). Oxidative stress is an inevitable consequence of the rapid metabolism and proliferation of cancer cells, and ferroptosis—an iron-dependent, lipid peroxidation-driven form of programmed cell death—poses a serious threat to cancer cells. HCC cells can establish a robust defense system to resist ferroptosis by reprogramming cysteine metabolism (244).
The core strategy for HCC cells to resist ferroptosis is the upregulation of the cystine/glutamate antiporter, System Xc−. This system consists of two subunits, SLC7A11 and SLC3A2, with SLC7A11 being the core catalytic subunit responsible for its transport function (243). SLC7A11 is abnormally highly expressed in HCC. It transports extracellular cystine (the disulfide-bonded form of two cysteine molecules) into the cell while pumping intracellular glutamate out. This consequently causes cystine deficiency in the TME (245, 246).
3.1.5 Arginine
Arginine plays a complex dual role in tumor biology. It is a precursor for protein synthesis, polyamines, and nitric oxide (NO) generation, and functions as an important signaling molecule (247). The reprogramming of arginine metabolism in HCC cells reflects their high adaptability (23).
A particularly notable phenomenon is the loss or downregulation of expression of argininosuccinate synthase 1 (ASS1), the key rate-limiting enzyme in the arginine synthesis pathway in many HCC cells. This renders these HCC cells “arginine auxotrophs,” dependent on acquiring arginine from the external environment to survive (213, 248). To compensate for this deficiency, HCC cells upregulate cationic amino acid transporters (CATs), such as SLC7A1 (CAT1) and SLC7A2 (CAT2), to enhance arginine uptake, causing arginine deficiency in the TME (249). Simultaneously, tumor cells downregulate enzymes that break down arginine into polyamines (e.g., ARG1) to accumulate high intracellular levels of arginine (206, 250).
3.1.6 Tryptophan
Tumors development requires nutrients and need to evade immune system surveillance and attack. HCC cells exploit tryptophan metabolism to create an immunosuppressive microenvironment through metabolic reprogramming (251, 252).
HCC cells increase tryptophan uptake by upregulating transporters like the L-type amino acid transporter 1 (LAT1/SLC7A5) (193, 253). HCC cells and other cells within their microenvironment (e.g., immune cells) highly express the two key rate-limiting enzymes in the tryptophan catabolic pathway—the kynurenine pathway: indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase 2 (TDO2). IDO1/TDO2 are upregulated in various cancers and associated with poor prognosis (254). In HCC, IDO1 overexpression is closely linked to malignant tumor behavior and an immunosuppressive state (255, 256). Concurrently, the activity of IDO1/TDO2 depletes tryptophan in the TME.
3.2 Oncometabolites
The development and progression of HCC are accompanied by profound cellular metabolic reprogramming. To support their unlimited proliferation, cancer cells preferentially adopt a metabolic pathway that involves glycolysis even in the presence of oxygen, which significantly enhances the efficiency of glucose uptake and utilization. Traditionally, it has been believed that the main consequence of this metabolic pattern is the competition for key nutrients such as glucose with immune cells, leading to “malnutrition” and impaired function of immune cells (257, 258). This is considered a relatively passive form of suppression. However, this perspective overlooks the “output end” of the metabolic processes. In addition to passively consuming nutrients, HCC cells actively secrete metabolites produced during their abnormal metabolic processes, known as “oncometabolites.” These metabolites are “metabolic waste” and biologically active signaling molecules that can act as chemical weapons to directly or indirectly inhibit immune cell functions, reshape the immune microenvironment, and establish a “protective” barrier for tumor growth, invasion, and immune evasion (214, 259). Below, we use the examples of lactate and dog urine acid to illustrate this point.
3.2.1 Lactate
Among all the tumor metabolites, lactate is perhaps the most extensively studied and has the most far-reaching impact. Enhanced aerobic glycolysis metabolism in HCC cells is a key metabolic basis for their proliferation, invasion, and migration, and directly cause the sharp increase in lactate concentration in the TME (260). The large amount of lactate produced within cancer cells needs to be efficiently expelled to maintain a relatively stable intracellular pH and sustain high-throughput glycolysis. This process is primarily mediated by members of the monocarboxylate transporter (MCT) family (261). MCT4 is the main lactate efflux pump, with low affinity for lactate but strong transport capacity, making it particularly suitable for expelling large amounts of lactate from highly glycolytic cells. The expression of MCT4 in HCC tissues is significantly higher than that in adjacent normal liver tissues and is positively correlated with tumor size (262). In HCC, upregulated MCT4 is responsible for pumping lactate produced by cancer cells into the TME, making it a key driver of TME acidification. As a direct product of aerobic glycolysis, lactate concentration in HCC tissues can reach 10–30 mM, far exceeding the 1–2 mM level in normal tissues (263).
3.2.2 Kynurenine
Tryptophan is an essential amino acid in the human body, serving as a key substrate for protein synthesis and as an important signaling molecule regulating immune homeostasis (264). In the TME of HCC, cancer cells systemically activate the tryptophan catabolic pathway by abnormally overexpressing two rate-limiting enzymes, IDO1 and TDO2 (255, 265, 266). These enzymes catalyze the conversion of tryptophan to N-formylkynurenine, triggering a dual pathological effect: local tryptophan depletion and accumulation of the toxic metabolite kynurenine.
3.2.3 Lipid
For HCC, the abnormal and persistent activation of the SREBP pathway leads to a significant increase in de novo lipid synthesis (267). After activation, SREBP triggers the expression of a series of downstream effector enzymes that collectively complete the process of fatty acid synthesis, such as fatty acid synthase (FASN), acetyl-CoA carboxylase (ACC), and stearoyl-CoA desaturase (SCD) (268). Meanwhile, the upregulation of HMGCR in HCC results in increased cholesterol synthesis (269). These lipid metabolic disorders in HCC lead to the accumulation of lipid components such as free fatty acids and cholesterol in the TME.
3.3 Perspectives on spatial metabolomics
The spatial heterogeneity of TME is a critical factor influencing tumor progression and immune responses. Metabolic states vary significantly across different regions of the TME. For example, areas near blood vessels are typically rich in oxygen and nutrients, whereas regions distant from vessels form hypoxic, acidic, and nutrient-deprived “metabolic deserts.” These distinct metabolic niches recruit and shape functionally diverse T-cell subsets (270–272).
Spatial metabolomics, as an emerging technology, enables precise dissection of the spatial distribution of metabolites within tumor tissues, revealing dynamic metabolic changes across different microenvironments. Understanding this spatial distribution is essential for deciphering immunosuppressive mechanisms, as different metabolic niches may influence T-cell functional states through specific metabolites. For instance, lactate accumulation in hypoxic regions may suppress T-cell activity, while nutrient-rich areas may support T-cell proliferation and function (273, 274).
Therefore, the development of region-specific therapeutic strategies must account for this spatial heterogeneity to achieve more precise treatment outcomes. The application of spatial metabolomics will provide new perspectives for understanding TME complexity and offer important insights for developing personalized treatment strategies.
4 Impacts of metabolic reprogramming on T cells in HCC
In HCC, cancer cells undergo a series of profound metabolic shifts to meet their immense demands for energy and biosynthetic precursors required for unlimited proliferation. This selfish metabolic reprogramming creates a unique metabolic niche within the TME of HCC, characterized by extreme scarcity of key nutrients and an accumulation of toxic metabolic byproducts (107). Through its distinct metabolic activities, HCC systematically suppresses the antitumor functions of T cells in two main ways: by competitively consuming essential nutrients required by T cells and by releasing inhibitory metabolites that directly “poison” T cells (31, 275, 276). Figure 1 intuitively illustrates the competition for nutrients between HCC cells and T cells in the tumor microenvironment, as well as the direct damage to T cells caused by the metabolites secreted by tumor cells.
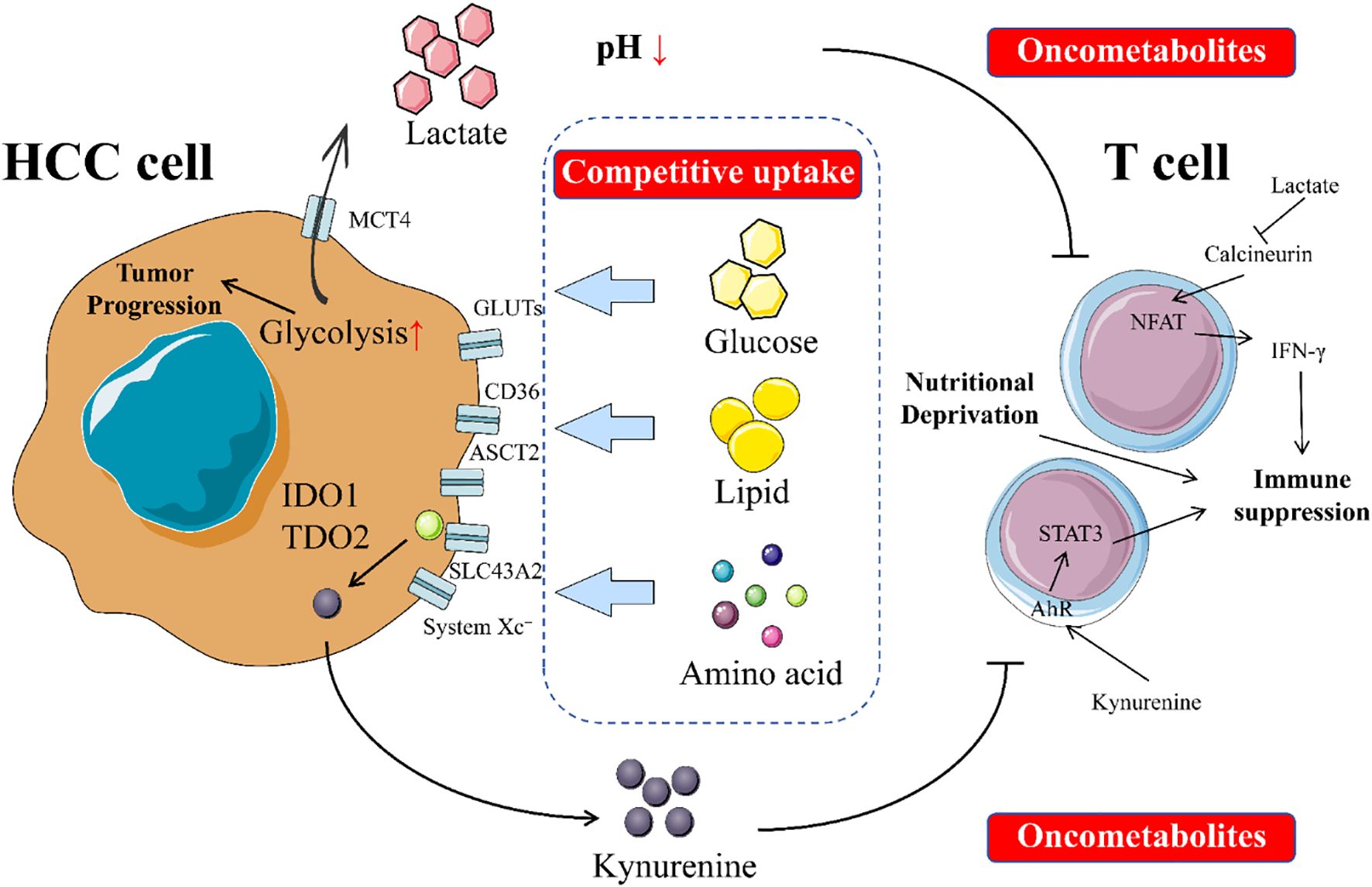
Figure 1. Impacts of metabolic reprogramming on T cells in HCC. In the tumor microenvironment, an intense tug-of-war exists between tumor cells and T cells. Tumor cells compete with T cells for limited nutrients such as glucose, amino acids, and lipids. This competition deprives T cells of the essential nutrients needed for their survival and function, thereby impairing their ability to conduct immune surveillance and kill tumor cells. Additionally, tumor cells secrete oncometabolites that directly harm T cells by disrupting their normal physiological processes, inhibiting their proliferation and activation, and even inducing apoptosis. This interplay not only weakens T cells’ antitumor capabilities but also fosters an environment conducive to tumor growth and spread. The dynamic balance between tumor cells and T cells is crucial for tumor occurrence, development, and immune evasion.
4.1 Nutrient competition and depletion: the “starvation” dilemma of T cells
In the microenvironment, rapidly proliferating tumor cells compete with immune cells responsible for immune surveillance, particularly T cells, for the same limited nutrients. With robust metabolic adaptability and reprogramming capabilities, HCC cells often gain an absolute advantage in the competition for survival essentials such as glucose, amino acids, and fatty acids (277).
4.1.1 Glucose depletion
One of the most well-known metabolic traits of HCC cells is their enhanced glycolytic activity, which leads to excessive glucose consumption. However, glucose is the core energy source essential for effector T cells (Teff) to undergo rapid clonal expansion and exert their cytotoxic functions, such as producing key cytokines like interferon-γ (IFN-γ) (278, 279). When T cells are activated and differentiate into effector T cells, their metabolic mode shifts from relying on oxidative phosphorylation (OXPHOS) to being glycolysis-dominant to meet the energy demands for rapid biosynthesis and functional execution (279). Glucose scarcity in the TME caused by excessive consumption by HCC cells, directly results in a severe energy (ATP) shortage for tumor-infiltrating T cells (TILs) (280).
This energy deficit may trigger a cascade of events: impaired mitochondrial function in T cells, inability to sustain normal physiological activities, weakened proliferation capacity, decreased cytokine secretion, and ultimately functional suppression, leading to a state known as “exhaustion” (281). Thus, this glycolysis-based metabolic competition is widely accepted as a key mechanism driving CD8+T-cell exhaustion and resistance to immunotherapy.
4.1.2 Deprivation of key amino acids
Apart from glucose, amino acids are equally vital for the survival, proliferation, and functional differentiation of T cells. HCC cells exhibit strong predatory behavior in this competition.
4.1.2.1 Glutamine
Many HCC cells display a high dependency on glutamine, often referred to as “glutamine addiction.” This excessive consumption significantly reduces the concentration of glutamine in the TME (282). Glutamine, a non-essential amino acid, is primarily transported into cells via the SLC1A5 transporter. SLC1A5 has been shown to be upregulated during T-cell activation, enhancing glutamine uptake. Deprivation of glutamine blocks the proliferation and cytokine production of mouse T cells in vitro culture systems (283, 284). Lowering tumor cells consumption of glutamine or increasing the levels of glutamine in the TME through exogenous means may potentially improve the efficacy of related cancer immunotherapies.
4.1.2.2 Arginine
Extracellular arginine is transported across membranes via the y+ system of cationic amino acid transporters (including SLC7A1, SLC7A2, and SLC7A3). Knocking out the expression of SLC7A1 using gene-editing technology reduces T-cell arginine uptake, thereby inhibiting T-cell proliferation. Moreover, arginine deprivation leads to T-cell cycle arrest, loss of the Zeta chain in the T-cell antigen receptor (TCR), and decreased T-cell proliferation and cytokine production (285). In mouse models, exogenous arginine supplementation promotes the generation of central memory T cells, thereby enhancing CD8+T-cell-mediated antitumor activity (286). In addition to protein synthesis, arginine is metabolized into various substances, including nitric oxide, proline, ornithine, creatine, guanidinoacetate, and polyamines. The catabolism of arginine into nitric oxide and its derivative peroxynitrite can inhibit antitumor T-cell responses (287).
4.1.2.3 Methionine
Methionine is an essential sulfur-containing amino acid involved in protein synthesis and the production of S-adenosylmethionine (SAM), which is a key methyl donor for DNA, RNA, and protein methylation modifications (288). Methionine uptake relies on various transporters, such as SLC1A5, SLC7A5, SLC7A6, SLC38A2, and SLC43A2. Notably, SLC7A5 is responsible for leucine uptake that is an essential amino acid for T-cell activation (289). As a result, T cells from SLC7A5-deficient mice exhibit impaired antitumor function due to insufficient uptake of leucine and methionine (290). Interestingly, tumor-infiltrating effector CD8+T cells from humans and mice have lower levels of SLC43A2 expression, leading to reduced intracellular levels of methionine and SAM. This further affects the expression of H3K79me2 and STAT5 and weakening T-cell survival and function (291). Additionally, exogenous supplementation of methionine can significantly restore the antitumor activity of effector CD8+T cells (292).
4.1.2.4 Cystine/cysteine
Cystine enters the cell via the glutamate-cystine antiporter system xCT (encoded by SLC7A11) and is rapidly reduced to cysteine. Although SLC7A11-deficient mouse T cells cannot proliferate in vitro, they remain fully activated in vivo. Moreover, in vivo experiments have shown that the deletion of the SLC7A11 gene in T cells has no clear impact on their antitumor response, while the knockout of the SLC7A11 gene in tumor cells can enhance the efficacy of tumor immunotherapy. This phenomenon may be related to the decreased antioxidant capacity of tumor cells and the increased levels of cystine in the microenvironment (293).
4.2 Accumulation of inhibitory metabolites: the “poisonous” environment for T cells
During abnormal and intense metabolic activities, HCC cells actively or passively release large amounts of metabolic byproducts into the TME. These byproducts can severely inhibit T cell function directly or indirectly, collectively creating a “poisonous” environment that leads to T cell dysfunction, exhaustion, or even apoptosis.
4.2.1 Lactate accumulation
Lactate accumulates in large amounts in the TME of HCC. Extremely high concentration of lactate in the TME creates a reverse concentration gradient. This hinders the efflux of lactate produced by T-cell metabolism and drives lactate from the TME into T cells via monocarboxylate transporters such as MCT1, leading to intracellular lactate accumulation and acidification (60, 294). High intracellular lactate concentration competitively inhibits the activity of lactate dehydrogenase (LDH), disrupting the NAD+/NADH redox balance in the cytoplasm and, in turn, negatively feedback-inhibiting key glycolytic rate-limiting enzymes such as phosphofructokinase-1 (PFK-1) (295). Ultimately, this plunges T cells into an “energy crisis,” preventing them from maintaining efficient effector functions. Studies have clearly shown that the acidic environment of the TME (low pH) directly impairs the conformation and enzymatic activity of calcineurin. Calcineurin, a phosphatase whose activity is inhibited, cannot effectively catalyze the dephosphorylation of NFAT proteins (296). This “locks” NFAT in the cytoplasm, preventing it from entering the nucleus to bind to the promoters of target genes, thereby severely weakening or even completely blocking the transcription and secretion of cytokines such as IFN-γ (297). Thus, the acidosis caused by lactate essentially cuts off the “command system” for T-cell-initiated immune responses. Moreover, Tumor-derived lactic acid modulates dendritic cell differentiation, inducing an altered phenotype that reduces IL-12 secretion and contributes to tumor immune escape (212).
4.2.2 Kynurenine
Tryptophan is an amino acid essential for T-cell proliferation and function (298). However, in the TME, tumor cells, stromal cells, or myeloid-derived suppressor cells (MDSCs) highly express indoleamine 2,3-dioxygenase (IDO1) or tryptophan 2,3-dioxygenase (TDO) (299). These enzymes catalyze the catabolism of tryptophan via the kynurenine pathway. This process exerts a dual inhibitory effect: 1) it depletes tryptophan in the TME, causing “nutritional starvation” of T cells and triggering their stagnation and anergy; and 2) its metabolite kynurenine and downstream derivatives, as endogenous ligands of the aryl hydrocarbon receptor (AHR), can be taken up by T cells and activate the AHR signaling pathway within them (211, 300). AHR activation has been shown to inhibit the function of effector T cells and promote the differentiation and proliferation of regulatory T cells (Tregs) with immunosuppressive capabilities, thereby further deepening immune suppression (301).
4.2.3 Lipid overload
Liver is the center of lipid metabolism, and thus HCC is often accompanied by significant lipid metabolic disorders. This finding is not only reflected within tumor cells but also throughout the entire TME, leading to the accumulation of large amounts of lipid components such as free fatty acids (FFAs), cholesterol, and their derivatives (302). This lipid-rich environment is a “double-edged sword” for infiltrating T cells, ultimately leading to functional impairment (303).
Excessive cholesterol uptake and ER stress: Tumor-infiltrating CD8+T cells take up large amounts of lipids from the TME via scavenger receptors (such as CD36) on their surface, attempting to use them as alternative energy sources for fatty acid oxidation (FAO) (304). However, the influx of excessive lipids, particularly free cholesterol, exceeds the cells’ normal metabolic and storage capacities, accumulating within the cells, especially in the endoplasmic reticulum (ER), and causing severe “lipotoxicity”. The accumulation of free cholesterol disrupts the integrity and fluidity of the ER membrane, interfering with proper protein folding and triggering “ER stress” and the broader “unfolded protein response” (UPR) (305). Persistent, unresolved ER stress is a harbinger of cellular dysfunction and apoptosis.
ER stress-driven T-cell exhaustion: ER stress is not a passive damage response. It actively activates a series of downstream signaling pathways that directly push T cells into an “exhaustion” state. Studies have shown that key molecules in ER stress, such as the spliced active form of X-box-binding protein 1 (XBP1s), can act as transcription factors to directly bind to and activate the promoters of genes encoding immune checkpoint molecules related to T-cell exhaustion, such as PD-1, T-cell immunoglobulin and mucin-domain-3 (TIM-3), and lymphocyte-activation gene-3 (LAG-3) (305, 306). The upregulation of these checkpoint molecules makes T cells abnormally sensitive to inhibitory signals from tumor cells, ultimately inducing their dysfunction, proliferative arrest, and apoptosis.
Unlike effector T cells that primarily rely on glycolysis, memory T cells preferentially utilize FAO to sustain their long-term survival and function. This metabolic dichotomy suggests that enhancing FAO through activation of enzymes like PPARα or CPT1A can improve memory T cell function and anti-tumor immunity (307–310). Thus, therapeutic strategies targeting lipid metabolism require precise modulation to effectively suppress tumor metabolism while preserving the critical functions of memory T cells.
Figure 2 comprehensively illustrates the mechanisms by which metabolic reprogramming regulates T cell functions through multiple pathways, including the effects of glucose metabolism, lactate production, and tryptophan metabolism on T cells. Table 2 details the specific effects of changes in various metabolites on T cells and their underlying molecular mechanisms.
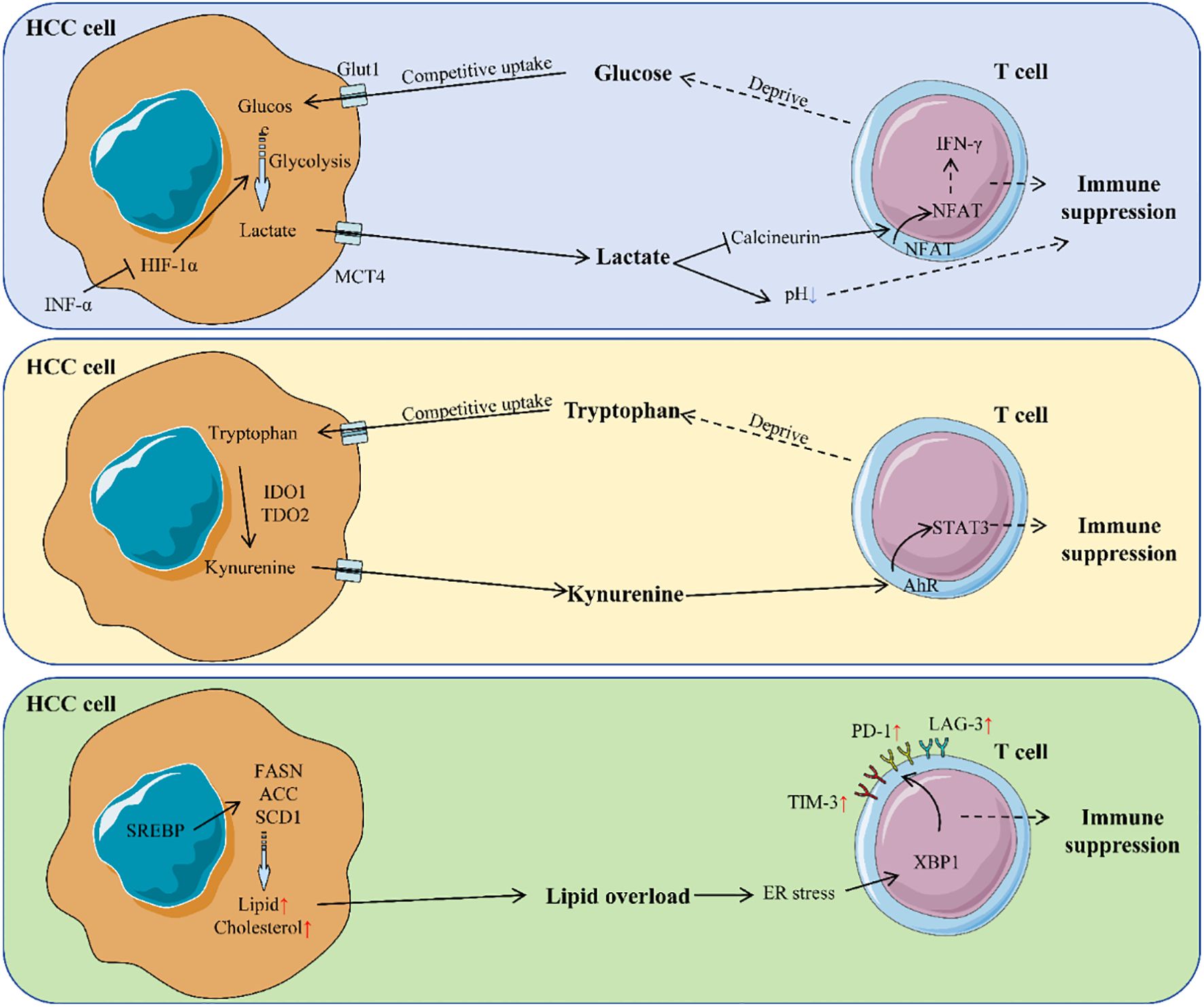
Figure 2. Mechanism of metabolic reprogramming regulating T cells in HCC. HCC cells reprogram metabolism to create an immunosuppressive TME. They consume glucose, produce lactate, and disrupt tryptophan metabolism. Lactate acidifies the TME, impairing T cell activity by inhibiting calcineurin activity in T cells, preventing NFAT from entering the nucleus and thus suppressing the secretion of cytokines like IFN-γ. Tryptophan breakdown generates kynurenine, which activates the AHR pathway in T cells, further suppressing their function. Lipid overload in the TME causes ER stress in T cells, leading to upregulation of immune checkpoint molecules and T cell exhaustion. These metabolic changes collectively inhibit T cell function, promoting immune evasion.
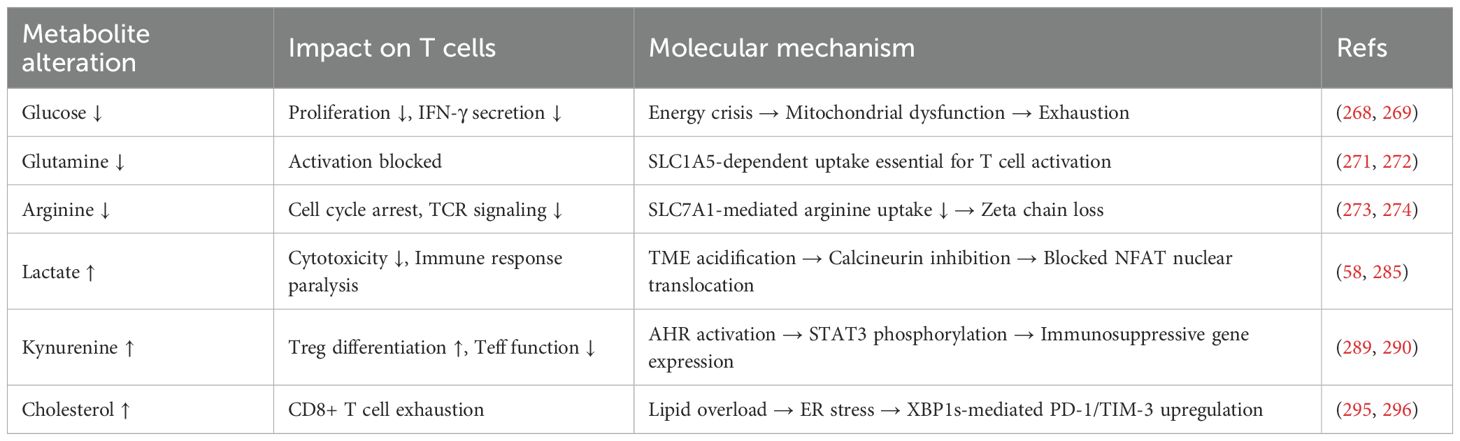
Table 2. Mechanisms of metabolite effects on T cells.
4.3 The impact of gut microbiota on metabolic reprogramming and T-cell responses
The gut microbiota contributes to the pathogenesis of HCC through metabolic regulation and immune modulation. As a complex microbial ecosystem within the human body, its metabolic activities continuously influence host physiology and disease states through the production of bioactive molecules. Recent studies have revealed that gut microbiota and their metabolites play critical roles in HCC initiation and progression, metabolic reprogramming, and shaping the tumor immune microenvironment (311–313). Specific microbial metabolites, such as short-chain fatty acids (SCFAs) and secondary bile acids, directly influence HCC metabolic adaptability and T-cell responses by modulating hepatic metabolic status and immune cell functions (314–316).
4.3.1 Regulation of hepatic metabolism by microbial metabolites
Gut microbial metabolites are transported to the liver via the portal vein, directly regulating the physiological functions of hepatocytes and hepatic immune cells (317). SCFAs, the main products of dietary fiber fermentation by gut bacteria, modulate hepatocyte metabolism through signaling pathways mediated by G protein-coupled receptors (GPR41, GPR43) (318). They alleviate hepatic steatosis by promoting fatty acid oxidation and inhibiting lipid accumulation (319, 320). Under conditions of gut dysbiosis, decreased SCFA levels may exacerbate hepatic metabolic disorders, thereby promoting HCC progression (314). Secondary bile acids, derived from microbial transformation of primary bile acids, regulate hepatic metabolic homeostasis through the farnesoid X receptor (FXR) and Takeda G protein-coupled receptor 5 (TGR5) (321, 322). Studies have demonstrated that abnormal accumulation of secondary bile acids is significantly associated with hepatic steatosis, inflammatory responses, and HCC development (321, 323). Dysbiosis-induced excessive accumulation of secondary bile acids can activate pro-inflammatory signaling pathways, driving hepatic inflammation and fibrosis, thereby creating a favorable environment for HCC initiation (324).
4.3.2 Modulation of T-cell responses by microbial metabolites
Gut microbial metabolites also regulate T-cell function in the liver through immunometabolic reprogramming mechanisms. SCFAs can enhance the differentiation and function of Tregs by influencing immunocyte metabolism, thereby maintaining immune tolerance (325). However, in HCC patients, reduced SCFAs due to dysbiosis may impair Treg function, exacerbate hepatic inflammation, and foster an immunosuppressive tumor microenvironment (326). Secondary bile acids modulate immune cell activity via the TGR5 receptor. Normal activation of TGR5 promotes anti-inflammatory cytokine expression and suppresses excessive inflammatory responses (327, 328). Nevertheless, abnormal accumulation of secondary bile acids caused by dysbiosis may impair TGR5 signaling, weaken anti-inflammatory protective mechanisms, and ultimately promote HCC progression (323, 324).
5 Metabolic-immune targeted combination therapy for HCC
Metabolic reprogramming in HCC can suppress the antitumor activity of T cells. Based on an in-depth understanding of this mechanism, researchers are actively exploring strategies to combine metabolic pathway inhibitors with immune checkpoint inhibitors (ICIs) to achieve a synergistic effect in HCC treatment (31).
5.1 Combination strategies targeting glucose metabolism
It has been reported that IFNα can ameliorate the glucose-deprived state within the TME by reprogramming glucose metabolism in the HCC TME, thereby enhancing antitumor immune activity. Mechanistically, IFNα suppresses HIF1α signaling by inhibiting FBJ osteosarcoma oncogene B (FosB) transcription in HCC cells, leading to reduced glucose consumption capacity in tumor cells and consequently establishing a high-glucose microenvironment. This glucose-enriched milieu promotes transcription of the costimulatory molecule Cd27 in infiltrating CD8+ T cells via the mTOR-FOXM1 signaling pathway, releasing T-cell cytotoxic potential and thereby potentiating the PD-1 blockade-induced immune response (329).
5.2 Combination strategies targeting glutamine metabolism
Glutaminase (GLS) inhibitors, such as CB-839/Telaglenastat, are among the most extensively studied drugs in this field. Multiple preclinical studies have demonstrated that the combination of CB-839 with anti-PD-1/PD-L1 antibodies exhibits significant synergistic antitumor effects in various tumor models, including non-HCC models. The mechanism primarily involves inhibiting glutamine catabolism, thereby alleviating the nutrient deprivation of T cells in the TME and promoting the activation and tumor infiltration of effector T cells (330–332).
5.3 Combination strategies targeting lipid synthesis
The combination of fatty acid synthase (FASN) inhibitors with ICIs has also shown therapeutic potential in HCC models. A study demonstrated that in an orthotopic HCC mouse model, FASN inhibition enhanced the stability of major histocompatibility complex class I (MHC-I) molecules and synergized with anti-PD-L1 blockade to significantly inhibit tumor growth (333). Nevertheless, in another study based on the sgPTEN/c-Met genetically engineered mouse model, no synergistic effect was observed between the FASN inhibitor TVB3664 and the anti-PD-L1 antibody. This suggests that the efficacy of combination therapy may depend on specific tumor genetic backgrounds and microenvironmental characteristics (334). However, the latest detailed preclinical data on the combination of FASN inhibitors with ICIs for HCC treatment, especially studies including systematic immune cell profiling, are also relatively scarce (334, 335).
5.4 Metabolic plasticity, drug resistance, and dynamic metabolic inhibition strategies
Metabolic plasticity is a key adaptive characteristic of cancer cells that enables them to reprogram metabolic pathways to sustain survival and proliferation under metabolic-targeted therapies. In combined therapies for HCC, although metabolic–immune targeting strategies may initially demonstrate considerable anti-tumor efficacy, the metabolic plasticity of cancer cells often leads to acquired resistance. For instance, when GLS inhibitors (e.g., CB-839) are combined with immune checkpoint inhibitors, tumor cells may activate alternative metabolic routes, such as the glutamine synthesis pathway, to circumvent the metabolic stress induced by GLS inhibition (336, 337). Such metabolic reprogramming allows cancer cells to persist under drug pressure, thereby compromising treatment outcomes. This dynamic adaptation of metabolic pathways not only diminishes the efficacy of single-target inhibitors but may also impair immune cell function, as immune activity relies on specific metabolic states.
To overcome the metabolic plasticity of cancer cells, future therapeutic approaches may require the implementation of dynamic or sequential metabolic inhibition strategies. The core concept involves co-targeting multiple metabolic pathways to disrupt the adaptive capacity of cancer cells, thereby enhancing treatment efficacy and reducing the incidence of resistance. For example, initial inhibition of glutamine metabolism using a GLS inhibitor could be followed by combination with a PPP pathway inhibitor (e.g., Oxythiamine) to further restrict metabolic flexibility. Such sequential targeting effectively depletes metabolic resources, impeding cancer cell survival through singular pathway reprogramming. In the context of lipid metabolism, combined administration of FASN inhibitors and HMGCR inhibitors (e.g., statins) may be considered. FASN inhibition reduces lipid synthesis, while statins suppress cholesterol production, thereby dually restricting lipid availability. This combined suppression not only directly affects the metabolic state of cancer cells but may also enhance immune cell function by modulating lipid levels in the tumor microenvironment. Additionally, dynamic metabolic inhibition strategies can be integrated with immunotherapy. For instance, concurrent use of metabolic inhibitors and immune checkpoint blockers may improve immune-mediated recognition and elimination of cancer cells. Such comprehensive therapeutic approaches not only directly suppress tumor metabolism but may also amplify treatment efficacy by remodeling the immune microenvironment.
6 Conclusion
This review provides a comprehensive overview of the metabolic reprogramming in HCC and its profound impact on T cell-mediated antitumor immunity. We have detailed the various metabolic alterations in HCC cells, including enhanced glycolysis (the Warburg effect), lipid metabolism changes, and amino acid metabolism reprogramming, which collectively create an immunosuppressive TME. These metabolic shifts lead to nutrient depletion and the accumulation of inhibitory metabolites, such as lactate and kynurenine, which directly impair T cell function and promote T cell exhaustion.
The innovation of this review lies in its comprehensive analysis of the metabolic-immune interactions in HCC, highlighting the dual role of metabolic reprogramming in fueling tumor growth and suppressing antitumor immunity. By elucidating the mechanisms through which HCC cells outcompete T cells for essential nutrients and produce inhibitory metabolites, this review provides a novel perspective on the therapeutic bottleneck of current immunotherapies. The practicality of this work is evident in its exploration of potential combination therapies targeting metabolic pathways alongside ICIs, offering new strategies to enhance treatment efficacy and overcome resistance in HCC.
This review has several limitations that should be considered. A primary constraint stems from its reliance on published literature, which introduces potential publication bias due to the omission of unpublished or negative results. Moreover, the high degree of heterogeneity in HCC and the dynamic complexity of the TME complicate the extrapolation of findings, as metabolic profiles and their impact on immunity may differ significantly among patients. Although we highlight promising metabolic-immune combination strategies, most supporting evidence comes from preclinical studies. Clinical data remain sparse, with limited sample sizes and follow-up, thereby necessitating further validation of the long-term efficacy and safety of these approaches. Future studies are warranted to address these gaps through expanded clinical trials and mechanistic validation in diverse experimental models.
Future research should focus on several interconnected areas to advance the understanding and treatment of HCC. A deeper exploration of the metabolic adaptations of immune cells within the tumor microenvironment and their dynamic interplay with cancer cell metabolism will help identify novel therapeutic targets. Complementing this, comprehensive and patient-specific metabolic profiling of HCC tumors is essential to fully elucidate the relationship between metabolic reprogramming and immune evasion. The integration of advanced technologies such as spatial metabolomics will be critical to decipher the spatiotemporal heterogeneity of metabolic pathways and provide a foundation for precision medicine. Furthermore, the role of the gut microbiota and its metabolites in the metabolic-immune network of HCC warrants further mechanistic investigation for potential clinical intervention. On the translational front, efforts should prioritize validating the efficacy of metabolism-immunity combination therapies in larger clinical cohorts and developing novel biomarkers to predict patient response, thereby enabling personalized treatment strategies. Finally, exploring additional metabolic pathways and their immunomodulatory effects, coupled with fostering interdisciplinary collaboration to leverage new materials and technologies, will be vital to uncover new therapeutic opportunities and accelerate the translation of basic findings into effective clinical applications for HCC.
Author contributions
D-XL: Conceptualization, Data curation, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. XA: Investigation, Methodology, Writing – original draft. G-XH: Conceptualization, Project administration, Supervision, Validation, Writing – review & editing. T-LY: Conceptualization, Methodology, Project administration, Validation, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
HCC: Hepatocellular carcinoma
TME: Tumor microenvironment
ICI: Immune checkpoint inhibitor
GLUT1: Glucose transporter 1
HK2: Hexokinase 2
PKM2: Pyruvate kinase M2 isoform
LDHA: Lactate dehydrogenase A
HIF-1α: Hypoxia-inducible factor 1α
PFK1: Phosphofructokinase 1
AMPK: AMP-activated protein kinase
PPP: Pentose phosphate pathway
G6PD: Glucose-6-phosphate dehydrogenase
TKT: Transketolase
FBP1: Fructose-1,6-bisphosphatase 1
PCK1: Phosphoenolpyruvate carboxykinase
IDH2: Isocitrate dehydrogenase 2
ME1: Malic enzyme 1
ACLY: ATP citrate lyase
ACC: Acetyl-CoA carboxylase
FASN: Fatty acid synthase
SCD1: Stearoyl-CoA desaturase 1
ACSL4: Acyl-CoA synthase long-chain family member 4
SREBP1: Sterol regulatory element-binding protein 1
LXR: Liver X receptor
FAO: Fatty acid oxidation
CPT1: Carnitine palmitoyltransferase 1
MCAD: Medium-chain acyl-CoA dehydrogenase
LCAD: Long-chain acyl-CoA dehydrogenase
MAGL: Monoglyceride lipase
ATGL: Adipose triglyceride lipase
HMGCR: HMG-CoA reductase
PPARα: Peroxisome proliferator-activated receptor α
CYP4: Cytochrome P450 family 4
GLS: Glutaminase
SLC1A5: Solute carrier family 1 member 5
SLC7A5: Solute carrier family 7 member 5
GOT1: Glutamic oxaloacetic transaminase 1
CPS1: Carbamoyl phosphate synthetase 1
ASS1: Argininosuccinate synthase 1
ARG1: Arginase-1
OTC: Ornithine transcarbamylase
IDO1: Indoleamine 2,3-dioxygenase 1
TDO2: Tryptophan 2,3-dioxygenase 2
MCT: Monocarboxylate transporter
AHR: Aryl hydrocarbon receptor
TIM-3: T-cell immunoglobulin and mucin-domain-3
LAG-3: Lymphocyte-activation gene-3
SCFAs: Short-chain fatty acids
FXR: Farnesoid X receptor
TGR5: Takeda G protein-coupled receptor 5
IFN-γ: Interferon-γ
IL-12: Interleukin-12
PD-1: Programmed death receptor-1
PD-L1: Programmed Death-Ligand 1
References
1. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, and Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. (2019) 16:589–604. doi: 10.1038/s41575-019-0186-y
2. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. (2021) 7:6. doi: 10.1038/s41572-020-00240-3
3. Li Q, Ding C, Cao M, Yang F, Yan X, He S, et al. Global epidemiology of liver cancer 2022: An emphasis on geographic disparities. Chin Med J (Engl). (2024) 137:2334–42. doi: 10.1097/CM9.0000000000003264
4. Vogel A, Meyer T, Sapisochin G, Salem R, and Saborowski A. Hepatocellular carcinoma. Lancet. (2022) 400:1345–62. doi: 10.1016/S0140-6736(22)01200-4
5. Chan Y-T, Zhang C, Wu J, Lu P, Xu L, Yuan H, et al. Biomarkers for diagnosis and therapeutic options in hepatocellular carcinoma. Mol Cancer. (2024) 23:189. doi: 10.1186/s12943-024-02101-z
6. Li M, Bhoori S, Mehta N, and Mazzaferro V. Immunotherapy for hepatocellular carcinoma: The next evolution in expanding access to liver transplantation. J Hepatol. (2024) 81:743–55. doi: 10.1016/j.jhep.2024.05.037
7. Llovet JM, De Baere T, Kulik L, Haber PK, Greten TF, Meyer T, et al. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2021) 18:293–313. doi: 10.1038/s41575-020-00395-0
8. Brown ZJ, Tsilimigras DI, Ruff SM, Mohseni A, Kamel IR, Cloyd JM, et al. Management of hepatocellular carcinoma: A review. JAMA Surg. (2023) 158:410–20. doi: 10.1001/jamasurg.2022.7989
9. Gordan JD, Kennedy EB, Abou-Alfa GK, Beal E, Finn RS, Gade TP, et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline update. J Clin Oncol. (2024) 42:1830–50. doi: 10.1200/JCO.23.02745
10. Zhao Y, Zhang Y-N, Wang K-T, and Chen L. Lenvatinib for hepatocellular carcinoma: From preclinical mechanisms to anti-cancer therapy. Biochim Biophys Acta Rev Cancer. (2020) 1874:188391. doi: 10.1016/j.bbcan.2020.188391
11. Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. (2022) 19:151–72. doi: 10.1038/s41571-021-00573-2
12. Harkus U, Wankell M, Palamuthusingam P, McFarlane C, and Hebbard L. Immune checkpoint inhibitors in HCC: Cellular, molecular and systemic data. Semin Cancer Biol. (2022) 86:799–815. doi: 10.1016/j.semcancer.2022.01.005
13. Shen K-Y, Zhu Y, Xie S-Z, and Qin L-X. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: current status and prospectives. J Hematol Oncol. (2024) 17:25. doi: 10.1186/s13045-024-01549-2
14. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. (2020) 382:1894–905. doi: 10.1056/NEJMoa1915745
15. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
16. Faubert B, Solmonson A, and DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
17. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. (2012) 22:66–79. doi: 10.1016/j.ccr.2012.05.016
18. Park SJ, Yoo HC, Ahn E, Luo E, Kim Y, Sung Y, et al. Enhanced glutaminolysis drives hypoxia-induced chemoresistance in pancreatic cancer. Cancer Res. (2023) 83:735–52. doi: 10.1158/0008-5472.CAN-22-2045
19. Tufail M, Jiang C-H, and Li N. Altered metabolism in cancer: insights into energy pathways and therapeutic targets. Mol Cancer. (2024) 23:203. doi: 10.1186/s12943-024-02119-3
20. You M, Xie Z, Zhang N, Zhang Y, Xiao D, Liu S, et al. Signaling pathways in cancer metabolism: mechanisms and therapeutic targets. Signal Transduct Target Ther. (2023) 8:196. doi: 10.1038/s41392-023-01442-3
21. Park JH, Pyun WY, and Park HW. Cancer metabolism: phenotype, signaling and therapeutic targets. Cells. (2020) 9:2308. doi: 10.3390/cells9102308
22. Dai P, Feng J, Dong Y, Zhang S, Cao J, Cui X, et al. Metabolic reprogramming in hepatocellular carcinoma: an integrated omics study of lipid pathways and their diagnostic potential. J Transl Med. (2025) 23:644. doi: 10.1186/s12967-025-06698-7
23. Park S and Hall MN. Metabolic reprogramming in hepatocellular carcinoma: mechanisms and therapeutic implications. Exp Mol Med. (2025) 57:515–23. doi: 10.1038/s12276-025-01415-2
24. Mergener S and Peña-Llopis S. A new perspective on immune evasion: escaping immune surveillance by inactivating tumor suppressors. Sig Transduct Target Ther. (2022) 7:15. doi: 10.1038/s41392-022-00875-6
25. Galassi C, Chan TA, Vitale I, and Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. (2024) 42:1825–63. doi: 10.1016/j.ccell.2024.09.010
26. Tufail M, Jiang C-H, and Li N. Immune evasion in cancer: mechanisms and cutting-edge therapeutic approaches. Sig Transduct Target Ther. (2025) 10:227. doi: 10.1038/s41392-025-02280-1
27. Tang T, Huang X, Zhang G, Hong Z, Bai X, and Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Target Ther. (2021) 6:72. doi: 10.1038/s41392-020-00449-4
28. Haynes NM, Chadwick TB, and Parker BS. The complexity of immune evasion mechanisms throughout the metastatic cascade. Nat Immunol. (2024) 25:1793–808. doi: 10.1038/s41590-024-01960-4
29. Chen C, Wang Z, Ding Y, and Qin Y. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front Immunol. (2023) 14:1133308. doi: 10.3389/fimmu.2023.1133308
30. Hall Z, Chiarugi D, Charidemou E, Leslie J, Scott E, Pellegrinet L, et al. Lipid remodeling in hepatocyte proliferation and hepatocellular carcinoma. Hepatology. (2021) 73:1028–44. doi: 10.1002/hep.31391
31. Gao B, Lu Y, Lai X, Xu X, Gou S, Yang Z, et al. Metabolic reprogramming in hepatocellular carcinoma: mechanisms of immune evasion and therapeutic implications. Front Immunol. (2025) 16:1592837. doi: 10.3389/fimmu.2025.1592837
32. Tenen DG, Chai L, and Tan JL. Metabolic alterations and vulnerabilities in hepatocellular carcinoma. Gastroenterol Rep (Oxf). (2021) 9:1–13. doi: 10.1093/gastro/goaa066
33. Ng CKY, Dazert E, Boldanova T, Coto-Llerena M, Nuciforo S, Ercan C, et al. Integrative proteogenomic characterization of hepatocellular carcinoma across etiologies and stages. Nat Commun. (2022) 13:2436. doi: 10.1038/s41467-022-29960-8
34. Ruan X, Hu Z, Shen F, Chen Y, Zhong Z, Ou G, et al. Proteomic and metabolomic analysis of platelet related samples reveals energy metabolism disorders in hepatocellular carcinoma. J Transl Med. (2025) 23:654. doi: 10.1186/s12967-025-06694-x
35. Li Z-C, Wang J, Liu H-B, Zheng Y-M, Huang J-H, Cai J-B, et al. Proteomic and metabolomic features in patients with HCC responding to lenvatinib and anti-PD1 therapy. Cell Rep. (2024) 43:113877. doi: 10.1016/j.celrep.2024.113877
36. Iansante V, Choy PM, Fung SW, Liu Y, Chai J-G, Dyson J, et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat Commun. (2015) 6:7882. doi: 10.1038/ncomms8882
37. Zuo Q, He J, Zhang S, Wang H, Jin G, Jin H, et al. PPARγ Coactivator-1α Suppresses metastasis of hepatocellular carcinoma by inhibiting warburg effect by PPARγ-dependent WNT/β-catenin/pyruvate dehydrogenase kinase isozyme 1 axis. Hepatology. (2021) 73:644–60. doi: 10.1002/hep.31280
38. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
39. Kim J and Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. (2006) 66:8927–30. doi: 10.1158/0008-5472.CAN-06-1501
40. Koppenol WH, Bounds PL, and Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. (2011) 11:325–37. doi: 10.1038/nrc3038
41. Huang Q, Li J, Xing J, Li W, Li H, Ke X, et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. (2014) 61:859–66. doi: 10.1016/j.jhep.2014.04.035
42. DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon S-M, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. (2018) 9:446. doi: 10.1038/s41467-017-02733-4
43. Wang F, Hu Y, Wang H, Hu P, Xiong H, Zeng Z, et al. LncRNA FTO-IT1 promotes glycolysis and progression of hepatocellular carcinoma through modulating FTO-mediated N6-methyladenosine modification on GLUT1 and PKM2. J Exp Clin Cancer Res. (2023) 42:267. doi: 10.1186/s13046-023-02847-2
44. Cai J, Song L, Zhang F, Wu S, Zhu G, Zhang P, et al. Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti-PD-1 therapy in hepatocellular carcinoma. Cancer Commun (Lond). (2024) 44:1231–60. doi: 10.1002/cac2.12607
45. Roberts DJ and Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. (2015) 22:248–57. doi: 10.1038/cdd.2014.173
46. Zhang D, Yip YM, and Li L. In silico construction of HK2-VDAC1 complex and investigating the HK2 binding-induced molecular gating mechanism of VDAC1. Mitochondrion. (2016) 30:222–8. doi: 10.1016/j.mito.2016.08.009
47. Xu D, Jin J, Yu H, Zhao Z, Ma D, Zhang C, et al. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J Exp Clin Cancer Res. (2017) 36:44. doi: 10.1186/s13046-017-0514-4
48. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. (2008) 452:230–3. doi: 10.1038/nature06734
49. Liu T, Li S, Wu L, Yu Q, Li J, Feng J, et al. Experimental study of hepatocellular carcinoma treatment by shikonin through regulating PKM2. J Hepatocell Carcinoma. (2020) 7:19–31. doi: 10.2147/JHC.S237614
50. Chen J, Xie J, Jiang Z, Wang B, Wang Y, and Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. (2011) 30:4297–306. doi: 10.1038/onc.2011.137
51. Dinarello A, Betto RM, Diamante L, Tesoriere A, Ghirardo R, Cioccarelli C, et al. STAT3 and HIF1α cooperatively mediate the transcriptional and physiological responses to hypoxia. Cell Death Discov. (2023) 9:226. doi: 10.1038/s41420-023-01507-w
52. Bao X, Zhang J, Huang G, Yan J, Xu C, Dou Z, et al. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. (2021) 12:215. doi: 10.1038/s41419-021-03505-1
53. Hu L, Zeng Z, Xia Q, Liu Z, Feng X, Chen J, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1α/PFKFB3/PFK1 pathway. Life Sci. (2019) 239:116966. doi: 10.1016/j.lfs.2019.116966
54. Ma T, Tian X, Zhang B, Li M, Wang Y, Yang C, et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. (2022) 603:159–65. doi: 10.1038/s41586-022-04431-8
55. Kukurugya MA, Rosset S, and Titov DV. The Warburg Effect is the result of faster ATP production by glycolysis than respiration. Proc Natl Acad Sci U.S.A. (2024) 121:e2409509121. doi: 10.1073/pnas.2409509121
56. Patra KC and Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
57. TeSlaa T, Ralser M, Fan J, and Rabinowitz JD. The pentose phosphate pathway in health and disease. Nat Metab. (2023) 5:1275–89. doi: 10.1038/s42255-023-00863-2
58. Chen S, Xu Y, Zhuo W, and Zhang L. The emerging role of lactate in tumor microenvironment and its clinical relevance. Cancer Lett. (2024) 590:216837. doi: 10.1016/j.canlet.2024.216837
59. Romero-Garcia S, Moreno-Altamirano MMB, Prado-Garcia H, and Sánchez-García FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. (2016) 7:52. doi: 10.3389/fimmu.2016.00052
60. Wang Z-H, Peng W-B, Zhang P, Yang X-P, and Zhou Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine. (2021) 73:103627. doi: 10.1016/j.ebiom.2021.103627
61. Shang X, Cheng B, Zhang C, Zhao C, Wang R, Zhang X, et al. The LDH-H3K18La-nur77 axis potentiates immune escape in small cell lung cancer. Adv Sci (Weinh). (2025):e13608. doi: 10.1002/advs.202413608
62. Kowalik MA, Columbano A, and Perra A. Emerging role of the pentose phosphate pathway in hepatocellular carcinoma. Front Oncol. (2017) 7:87. doi: 10.3389/fonc.2017.00087
63. Jin X, Lv Y, Bie F, Duan J, Ma C, Dai M, et al. METTL3 confers oxaliplatin resistance through the activation of G6PD-enhanced pentose phosphate pathway in hepatocellular carcinoma. Cell Death Differ. (2025) 32:466–79. doi: 10.1038/s41418-024-01406-2
64. Lu M, Lu L, Dong Q, Yu G, Chen J, Qin L, et al. Elevated G6PD expression contributes to migration and invasion of hepatocellular carcinoma cells by inducing epithelial-mesenchymal transition. Acta Biochim Biophys Sin (Shanghai). (2018) 50:370–80. doi: 10.1093/abbs/gmy009
65. Gu X-Y, Zhou Z-J, Yao H, Yang J-L, Gu J, Mu R, et al. The role of transketolase in the immunotherapy and prognosis of hepatocellular carcinoma: a multi-omics approach. Front Immunol. (2025) 16:1529029. doi: 10.3389/fimmu.2025.1529029
66. Zhen X, Zhang M, Hao S, and Sun J. Glucose-6-phosphate dehydrogenase and transketolase: Key factors in breast cancer progression and therapy. BioMed Pharmacother. (2024) 176:116935. doi: 10.1016/j.biopha.2024.116935
67. van der Mijn JC, Panka DJ, Geissler AK, Verheul HM, and Mier JW. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. (2016) 4:14. doi: 10.1186/s40170-016-0154-8
68. Liu C-L, Hsu Y-C, Lee J-J, Chen M-J, Lin C-H, Huang S-Y, et al. Targeting the pentose phosphate pathway increases reactive oxygen species and induces apoptosis in thyroid cancer cells. Mol Cell Endocrinol. (2020) 499:110595. doi: 10.1016/j.mce.2019.110595
69. Xu IM-J, Lai RK-H, Lin S-H, Tse AP-W, Chiu DK-C, Koh H-Y, et al. Transketolase counteracts oxidative stress to drive cancer development. Proc Natl Acad Sci U.S.A. (2016) 113:E725–734. doi: 10.1073/pnas.1508779113
70. Li Y, Yao C-F, Xu F-J, Qu Y-Y, Li J-T, Lin Y, et al. APC/CCDH1 synchronizes ribose-5-phosphate levels and DNA synthesis to cell cycle progression. Nat Commun. (2019) 10:2502. doi: 10.1038/s41467-019-10375-x
71. Kawaguchi T, Nakano D, Koga H, and Torimura T. Effects of a DPP4 inhibitor on progression of NASH-related HCC and the p62/keap1/nrf2-pentose phosphate pathway in a mouse model. Liver Cancer. (2019) 8:359–72. doi: 10.1159/000491763
72. Bian X, Chen H, Yang P, Li Y, Zhang F, Zhang J, et al. Nur77 suppresses hepatocellular carcinoma via switching glucose metabolism toward gluconeogenesis through attenuating phosphoenolpyruvate carboxykinase sumoylation. Nat Commun. (2017) 8:14420. doi: 10.1038/ncomms14420
73. Wang B, Hsu S, Frankel W, Ghoshal K, and Jacob ST. Stat3-mediated activation of miR-23a suppresses gluconeogenesis in hepatocellular carcinoma by downregulating G6PC and PGC-1α. Hepatology. (2012) 56:186–97. doi: 10.1002/hep.25632
74. Yang J, Jin X, Yan Y, Shao Y, Pan Y, Roberts LR, et al. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci Rep. (2017) 7:43864. doi: 10.1038/srep43864
75. Jin X, Pan Y, Wang L, Zhang L, Ravichandran R, Potts PR, et al. MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis. (2017) 6:e312. doi: 10.1038/oncsis.2017.21
76. Son B, Lee S, Kim H, Kang H, Jeon J, Jo S, et al. Decreased FBP1 expression rewires metabolic processes affecting aggressiveness of glioblastoma. Oncogene. (2020) 39:36–49. doi: 10.1038/s41388-019-0974-4
77. Wang M, Huang X, Zhang D, Liu Y, and Liu P. The role of fructose-1,6-bisphosphatase 1 on regulating the cancer progression and drug resistance. Discov Oncol. (2025) 16:346. doi: 10.1007/s12672-025-02112-2
78. Bertinat R, Holyoak T, Gatica R, Jara N, González-Chavarría I, and Westermeier F. The neglected PCK1/glucagon (inter)action in nutrient homeostasis beyond gluconeogenesis: Disease pathogenesis and treatment. Mol Metab. (2025) 94:102112. doi: 10.1016/j.molmet.2025.102112
79. Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. (2020) 580:530–5. doi: 10.1038/s41586-020-2183-2
80. Chen L, Ding L, Wang X, Huang Y, and Gao S-J. Activation of glucocorticoid receptor signaling inhibits KSHV-induced inflammation and tumorigenesis. mBio. (2024) 15:e0301123. doi: 10.1128/mbio.03011-23
81. Puigserver P and Spiegelman BM. Peroxisome proliferator-activated receptor-γ Coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr Rev. (2003) 24:78–90. doi: 10.1210/er.2002-0012
82. Todisco S, Convertini P, Iacobazzi V, and Infantino V. TCA cycle rewiring as emerging metabolic signature of hepatocellular carcinoma. Cancers (Basel). (2019) 12:68. doi: 10.3390/cancers12010068
83. Cappel DA, Deja S, Duarte JAG, Kucejova B, Iñigo M, Fletcher JA, et al. Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metab. (2019) 29:1291–1305.e8. doi: 10.1016/j.cmet.2019.03.014
84. Patterson JN, Cousteils K, Lou JW, Manning Fox JE, MacDonald PE, and Joseph JW. Mitochondrial metabolism of pyruvate is essential for regulating glucose-stimulated insulin secretion. J Biol Chem. (2014) 289:13335–46. doi: 10.1074/jbc.M113.521666
85. Yi W-R, Li Z-H, Qi B-W, Ernest MER, Hu X, and Yu A-X. Downregulation of IDH2 exacerbates the Malignant progression of osteosarcoma cells via increased NF-κB and MMP-9 activation. Oncol Rep. (2016) 35:2277–85. doi: 10.3892/or.2016.4553
86. Tian G-Y, Zang S-F, Wang L, Luo Y, Shi J-P, and Lou G-Q. Isocitrate dehydrogenase 2 suppresses the invasion of hepatocellular carcinoma cells via matrix metalloproteinase 9. Cell Physiol Biochem. (2015) 37:2405–14. doi: 10.1159/000438593
87. Fan B, Mellinghoff IK, Wen PY, Lowery MA, Goyal L, Tap WD, et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Invest New Drugs. (2020) 38:433–44. doi: 10.1007/s10637-019-00771-x
88. Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. (2018) 9:300–5. doi: 10.1021/acsmedchemlett.7b00421
89. Fang X, Zhang J, Li Y, Song Y, Yu Y, Cai Z, et al. Malic enzyme 1 as a novel anti-ferroptotic regulator in hepatic ischemia/reperfusion injury. Adv Sci (Weinh). (2023) 10:e2205436. doi: 10.1002/advs.202205436
90. Lu Y-X, Ju H-Q, Liu Z-X, Chen D-L, Wang Y, Zhao Q, et al. ME1 regulates NADPH homeostasis to promote gastric cancer growth and metastasis. Cancer Res. (2018) 78:1972–85. doi: 10.1158/0008-5472.CAN-17-3155
91. Wen D, Liu D, Tang J, Dong L, Liu Y, Tao Z, et al. Malic enzyme 1 induces epithelial-mesenchymal transition and indicates poor prognosis in hepatocellular carcinoma. Tumour Biol. (2015) 36:6211–21. doi: 10.1007/s13277-015-3306-5
92. Zeng Y, Jiang H, Chen Z, Xu J, Zhang X, Cai W, et al. Histone lactylation promotes multidrug resistance in hepatocellular carcinoma by forming a positive feedback loop with PTEN. Cell Death Dis. (2025) 16:59. doi: 10.1038/s41419-025-07359-9
93. Xia J, Qin X, Zhang L, Liu S, Shi X, and Ren H. Roles and regulation of histone acetylation in hepatocellular carcinoma. Front Genet. (2022) 13:982222. doi: 10.3389/fgene.2022.982222
94. Mi W, Xue Y, Yan H, Zhang Y, Cai X, Zhang S, et al. α-Ketoglutarate dictates AMPK protein synthesis for energy sensing in human cancers. Nat Chem Biol. (2025), 1–13. doi: 10.1038/s41589-025-02013-z
95. Wang Y, Deng P, Liu Y, Wu Y, Chen Y, Guo Y, et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat Commun. (2020) 11:5596. doi: 10.1038/s41467-020-19360-1
96. Raychaudhuri D, Singh P, Chakraborty B, Hennessey M, Tannir AJ, Byregowda S, et al. Histone lactylation drives CD8+ T cell metabolism and function. Nat Immunol. (2024) 25:2140–51. doi: 10.1038/s41590-024-01985-9
97. Bian X, Liu R, Meng Y, Xing D, Xu D, and Lu Z. Lipid metabolism and cancer. J Exp Med. (2021) 218:e20201606. doi: 10.1084/jem.20201606
98. Cheng C, Geng F, Cheng X, and Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). (2018) 38:27. doi: 10.1186/s40880-018-0301-4
99. Jones JG. Hepatic glucose and lipid metabolism. Diabetologia. (2016) 59:1098–103. doi: 10.1007/s00125-016-3940-5
100. Cao L-Q, Xie Y, Fleishman JS, Liu X, and Chen Z-S. Hepatocellular carcinoma and lipid metabolism: Novel targets and therapeutic strategies. Cancer Lett. (2024) 597:217061. doi: 10.1016/j.canlet.2024.217061
101. Hao J-W, Wang J, Guo H, Zhao Y-Y, Sun H-H, Li Y-F, et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat Commun. (2020) 11:4765. doi: 10.1038/s41467-020-18565-8
102. Krammer J, Digel M, Ehehalt F, Stremmel W, Füllekrug J, and Ehehalt R. Overexpression of CD36 and acyl-CoA synthetases FATP2, FATP4 and ACSL1 increases fatty acid uptake in human hepatoma cells. Int J Med Sci. (2011) 8:599–614. doi: 10.7150/ijms.8.599
103. Deng M, Cai X, Long L, Xie L, Ma H, Zhou Y, et al. CD36 promotes the epithelial-mesenchymal transition and metastasis in cervical cancer by interacting with TGF-β. J Transl Med. (2019) 17:352. doi: 10.1186/s12967-019-2098-6
104. Nath A, Li I, Roberts LR, and Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep. (2015) 5:14752. doi: 10.1038/srep14752
105. Seo J, Jeong D-W, Park J-W, Lee K-W, Fukuda J, and Chun Y-S. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun Biol. (2020) 3:638. doi: 10.1038/s42003-020-01367-5
106. Tang W, Sun G, Ji G-W, Feng T, Zhang Q, Cao H, et al. Single-cell RNA-sequencing atlas reveals an FABP1-dependent immunosuppressive environment in hepatocellular carcinoma. J Immunother Cancer. (2023) 11:e007030. doi: 10.1136/jitc-2023-007030
107. Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S, et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. (2022) 12:558–80. doi: 10.1016/j.apsb.2021.09.019
108. Ohata T, Yokoo H, Kamiyama T, Fukai M, Aiyama T, Hatanaka Y, et al. Fatty acid-binding protein 5 function in hepatocellular carcinoma through induction of epithelial-mesenchymal transition. Cancer Med. (2017) 6:1049–61. doi: 10.1002/cam4.1020
109. Nishitsuji K, Hosono T, Uchimura K, and Michikawa M. Lipoprotein lipase is a novel amyloid beta (Abeta)-binding protein that promotes glycosaminoglycan-dependent cellular uptake of Abeta in astrocytes. J Biol Chem. (2011) 286:6393–401. doi: 10.1074/jbc.M110.172106
110. Wang H and Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. (2009) 297:E271–288. doi: 10.1152/ajpendo.90920.2008
111. Goldberg IJ, Eckel RH, and Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. (2009) 50 Suppl:S86–90. doi: 10.1194/jlr.R800085-JLR200
112. Wu Z, Ma H, Wang L, Song X, Zhang J, Liu W, et al. Tumor suppressor ZHX2 inhibits NAFLD-HCC progression via blocking LPL-mediated lipid uptake. Cell Death Differ. (2020) 27:1693–708. doi: 10.1038/s41418-019-0453-z
113. Ning Z, Guo X, Liu X, Lu C, Wang A, Wang X, et al. USP22 regulates lipidome accumulation by stabilizing PPARγ in hepatocellular carcinoma. Nat Commun. (2022) 13:2187. doi: 10.1038/s41467-022-29846-9
114. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. (2011) 140:1071–83. doi: 10.1053/j.gastro.2010.12.006
115. Li S, Liu R, Pan Q, Wang G, Cheng D, Yang J, et al. De novo lipogenesis is elicited dramatically in human hepatocellular carcinoma especially in hepatitis C virus-induced hepatocellular carcinoma. MedComm (2020). (2020) 1:178–87. doi: 10.1002/mco2.15
116. Han Q, Chen C-A, Yang W, Liang D, Lv H-W, Lv G-S, et al. ATP-citrate lyase regulates stemness and metastasis in hepatocellular carcinoma via the Wnt/β-catenin signaling pathway. Hepatobiliary Pancreat Dis Int. (2021) 20:251–61. doi: 10.1016/j.hbpd.2020.05.010
117. Meddeb M, Koleini N, Keykhaei M, Liu T, Rhodehamel M, Mandarano L, et al. ATP-citrate lyase supports cardiac function and NAD+/NADH balance and is depressed in human failing myocardium. JACC Basic Transl Sci. (2025) 10:101301. doi: 10.1016/j.jacbts.2025.04.015
118. Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, and Alifano M. ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett. (2020) 471:125–34. doi: 10.1016/j.canlet.2019.12.010
119. Wang Y, Yu W, Li S, Guo D, He J, and Wang Y. Acetyl-coA carboxylases and diseases. Front Oncol. (2022) 12:836058. doi: 10.3389/fonc.2022.836058
120. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. (2016) 22:1108–19. doi: 10.1038/nm.4181
121. Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of acetyl-coA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. (2019) 29:174–182.e5. doi: 10.1016/j.cmet.2018.08.020
122. Schroeder B, Vander Steen T, Espinoza I, Venkatapoorna CMK, Hu Z, Silva FM, et al. Fatty acid synthase (FASN) regulates the mitochondrial priming of cancer cells. Cell Death Dis. (2021) 12:977. doi: 10.1038/s41419-021-04262-x
123. Che L, Paliogiannis P, Cigliano A, Pilo MG, Chen X, and Calvisi DF. Pathogenetic, prognostic, and therapeutic role of fatty acid synthase in human hepatocellular carcinoma. Front Oncol. (2019) 9:1412. doi: 10.3389/fonc.2019.01412
124. Li Y, Yang W, Zheng Y, Dai W, Ji J, Wu L, et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J Exp Clin Cancer Res. (2023) 42:6. doi: 10.1186/s13046-022-02567-z
125. Sen U, Coleman C, and Sen T. Stearoyl coenzyme A desaturase-1: multitasker in cancer, metabolism, and ferroptosis. Trends Cancer. (2023) 9:480–9. doi: 10.1016/j.trecan.2023.03.003
126. Gutiérrez-Cuevas J, Lucano-Landeros S, López-Cifuentes D, Santos A, and Armendariz-Borunda J. Epidemiologic, genetic, pathogenic, metabolic, epigenetic aspects involved in NASH-HCC: current therapeutic strategies. Cancers (Basel). (2022) 15:23. doi: 10.3390/cancers15010023
127. Sunami Y. NASH, Fibrosis and hepatocellular carcinoma: lipid synthesis and glutamine/acetate signaling. Int J Mol Sci. (2020) 21:6799. doi: 10.3390/ijms21186799
128. Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY, Cheng LKW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol. (2017) 67:979–90. doi: 10.1016/j.jhep.2017.06.015
129. Chen J, Ding C, Chen Y, Hu W, Lu Y, Wu W, et al. ACSL4 promotes hepatocellular carcinoma progression via c-Myc stability mediated by ERK/FBW7/c-Myc axis. Oncogenesis. (2020) 9:42. doi: 10.1038/s41389-020-0226-z
130. Uchihara D, Shimajiri S, Harada Y, Kumamoto K, Oe S, Miyagawa K, et al. Long-chain fatty acyl CoA synthetase 4 expression in pancreatic cancer: a marker for Malignant lesions and prognostic indicator for recurrence. Diagn Pathol. (2025) 20:59. doi: 10.1186/s13000-025-01659-6
131. Su F and Koeberle A. Regulation and targeting of SREBP-1 in hepatocellular carcinoma. Cancer Metastasis Rev. (2024) 43:673–708. doi: 10.1007/s10555-023-10156-5
132. Li C, Yang W, Zhang J, Zheng X, Yao Y, Tu K, et al. SREBP-1 has a prognostic role and contributes to invasion and metastasis in human hepatocellular carcinoma. Int J Mol Sci. (2014) 15:7124–38. doi: 10.3390/ijms15057124
133. Yin F, Feng F, Wang L, Wang X, Li Z, and Cao Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. (2019) 10:672. doi: 10.1038/s41419-019-1884-7
134. Wang B and Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol. (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
135. He J, Yang T, He W, Jiang S, Zhong D, Xu Z, et al. Liver X receptor inhibits the growth of hepatocellular carcinoma cells via regulating HULC/miR-134-5p/FOXM1 axis. Cell Signal. (2020) 74:109720. doi: 10.1016/j.cellsig.2020.109720
136. He X-Y, Zhu M-M, Zheng J, Wang C-Y, Zhao X-K, Zhang B-T, et al. Liver X receptor agonists exert antitumor effects against hepatocellular carcinoma via inducing REPS2 expression. Acta Pharmacol Sin. (2023) 44:635–46. doi: 10.1038/s41401-022-00961-z
137. Long H, Guo X, Qiao S, and Huang Q. Tumor LXR expression is a prognostic marker for patients with hepatocellular carcinoma. Pathol Oncol Res. (2018) 24:339–44. doi: 10.1007/s12253-017-0249-8
138. Shimizu Y, Tamura T, Kemmochi A, Owada Y, Ozawa Y, Hisakura K, et al. Oxidative stress and Liver X Receptor agonist induce hepatocellular carcinoma in Non-alcoholic steatohepatitis model. J Gastroenterol Hepatol. (2021) 36:800–10. doi: 10.1111/jgh.15239
139. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang X-Y, et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
140. Liang K. Mitochondrial CPT1A: Insights into structure, function, and basis for drug development. Front Pharmacol. (2023) 14:1160440. doi: 10.3389/fphar.2023.1160440
141. Serra D, Mera P, Malandrino MI, Mir JF, and Herrero L. Mitochondrial fatty acid oxidation in obesity. Antioxid Redox Signal. (2013) 19:269–84. doi: 10.1089/ars.2012.4875
142. Luo M, Liu Y-Q, Zhang H, Luo C-H, Liu Q, Wang W-Y, et al. Overexpression of carnitine palmitoyltransferase 1A promotes mitochondrial fusion and differentiation of glioblastoma stem cells. Lab Invest. (2022) 102:722–30. doi: 10.1038/s41374-021-00724-0
143. Liu Y, Wang F, Yan G, Tong Y, Guo W, Li S, et al. CPT1A loss disrupts BCAA metabolism to confer therapeutic vulnerability in TP53-mutated liver cancer. Cancer Lett. (2024) 595:217006. doi: 10.1016/j.canlet.2024.217006
144. Ren M, Xu H, Xia H, Tang Q, and Bi F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discov. (2021) 7:125. doi: 10.1038/s41420-021-00504-1
145. Zhang Y, Bharathi SS, Beck ME, and Goetzman ES. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol. (2019) 26:101253. doi: 10.1016/j.redox.2019.101253
146. Bharathi SS, Zhang Y, Gong Z, Muzumdar R, and Goetzman ES. Role of mitochondrial acyl-CoA dehydrogenases in the metabolism of dicarboxylic fatty acids. Biochem Biophys Res Commun. (2020) 527:162–6. doi: 10.1016/j.bbrc.2020.04.105
147. Ikeda Y, Hine DG, Okamura-Ikeda K, and Tanaka K. Mechanism of action of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases. Direct evidence for carbanion formation as an intermediate step using enzyme-catalyzed C-2 proton/deuteron exchange in the absence of C-3 exchange. J Biol Chem. (1985) 260:1326–37. doi: 10.1016/S0021-9258(20)71246-9
148. Cai J, Chen T, Jiang Z, Yan J, Ye Z, Ruan Y, et al. Bulk and single-cell transcriptome profiling reveal extracellular matrix mechanical regulation of lipid metabolism reprograming through YAP/TEAD4/ACADL axis in hepatocellular carcinoma. Int J Biol Sci. (2023) 19:2114–31. doi: 10.7150/ijbs.82177
149. Khare T, Khare S, Angdisen JJ, Zhang Q, Stuckel A, Mooney BP, et al. Defects in long-chain 3-hydroxy acyl-CoA dehydrogenase lead to hepatocellular carcinoma: A novel etiology of hepatocellular carcinoma. Int J Cancer. (2020) 147:1461–73. doi: 10.1002/ijc.32943
150. Ma APY, Yeung CLS, Tey SK, Mao X, Wong SWK, Ng TH, et al. Suppression of ACADM-mediated fatty acid oxidation promotes hepatocellular carcinoma via aberrant CAV1/SREBP1 signaling. Cancer Res. (2021) 81:3679–92. doi: 10.1158/0008-5472.CAN-20-3944
151. Zhao X, Qin W, Jiang Y, Yang Z, Yuan B, Dai R, et al. ACADL plays a tumor-suppressor role by targeting Hippo/YAP signaling in hepatocellular carcinoma. NPJ Precis Oncol. (2020) 4:7. doi: 10.1038/s41698-020-0111-4
152. Grabner GF, Xie H, Schweiger M, and Zechner R. Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nat Metab. (2021) 3:1445–65. doi: 10.1038/s42255-021-00493-6
153. Gil-Ordóñez A, Martín-Fontecha M, Ortega-Gutiérrez S, and López-Rodríguez ML. Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem Pharmacol. (2018) 157:18–32. doi: 10.1016/j.bcp.2018.07.036
154. Galvani F, Scalvini L, Rivara S, Lodola A, and Mor M. Mechanistic modeling of monoglyceride lipase covalent modification elucidates the role of leaving group expulsion and discriminates inhibitors with high and low potency. J Chem Inf Model. (2022) 62:2771–87. doi: 10.1021/acs.jcim.2c00140
155. Zhu W, Zhao Y, Zhou J, Wang X, Pan Q, Zhang N, et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-κB-mediated epithelial-mesenchymal transition. J Hematol Oncol. (2016) 9:127. doi: 10.1186/s13045-016-0361-3
156. Awad D, Cao PHA, Pulliam TL, Spradlin M, Subramani E, Tellman TV, et al. Adipose triglyceride lipase is a therapeutic target in advanced prostate cancer that promotes metabolic plasticity. Cancer Res. (2024) 84:703–24. doi: 10.1158/0008-5472.CAN-23-0555
157. Ruppert PMM and Kersten S. Regulation of adipose tissue metabolism during fasting. Annu Rev Nutr. (2025). doi: 10.1146/annurev-nutr-120524-013857
158. Liu X, Liang Y, Song R, Yang G, Han J, Lan Y, et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol Cancer. (2018) 17:90. doi: 10.1186/s12943-018-0838-5
159. Che L, Chi W, Qiao Y, Zhang J, Song X, Liu Y, et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut. (2020) 69:177–86. doi: 10.1136/gutjnl-2018-317581
160. Zhong S, Li L, Liang N, Zhang L, Xu X, Chen S, et al. Acetaldehyde Dehydrogenase 2 regulates HMG-CoA reductase stability and cholesterol synthesis in the liver. Redox Biol. (2021) 41:101919. doi: 10.1016/j.redox.2021.101919
161. Lu X-Y, Shi X-J, Hu A, Wang J-Q, Ding Y, Jiang W, et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature. (2020) 588:479–84. doi: 10.1038/s41586-020-2928-y
162. Schumacher MM and DeBose-Boyd RA. Posttranslational regulation of HMG coA reductase, the rate-limiting enzyme in synthesis of cholesterol. Annu Rev Biochem. (2021) 90:659–79. doi: 10.1146/annurev-biochem-081820-101010
163. Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G, et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. (2008) 68:5246–56. doi: 10.1158/0008-5472.CAN-07-6161
164. Lu J, Liu X, Fan K, and Lin X. Traditional chinese medicine as a tool for the treatment of hepatocellular carcinoma by targeting pathophysiological mechanism. Cancer Manag Res. (2025) 17:779–92. doi: 10.2147/CMAR.S513729
165. Li Y, Pan Y, Zhao X, Wu S, Li F, Wang Y, et al. Peroxisome proliferator-activated receptors: A key link between lipid metabolism and cancer progression. Clin Nutr. (2024) 43:332–45. doi: 10.1016/j.clnu.2023.12.005
166. Mahmoudvand S, Shokri S, Taherkhani R, and Farshadpour F. Hepatitis C virus core protein modulates several signaling pathways involved in hepatocellular carcinoma. World J Gastroenterol. (2019) 25:42–58. doi: 10.3748/wjg.v25.i1.42
167. Sturm E, Wagner M, and Trauner M. Nuclear receptor ligands in therapy of cholestatic liver disease. Front Biosci (Landmark Ed). (2009) 14:4299–325. doi: 10.2741/3529
168. Tan Y, Wang M, Yang K, Chi T, Liao Z, and Wei P. PPAR-α Modulators as current and potential cancer treatments. Front Oncol. (2021) 11:599995. doi: 10.3389/fonc.2021.599995
169. Kim D, Brocker CN, Takahashi S, Yagai T, Kim T, Xie G, et al. Keratin 23 is a peroxisome proliferator-activated receptor alpha-dependent, MYC-amplified oncogene that promotes hepatocyte proliferation. Hepatology. (2019) 70:154–67. doi: 10.1002/hep.30530
170. Hardwick JP. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem Pharmacol. (2008) 75:2263–75. doi: 10.1016/j.bcp.2008.03.004
171. Leahy C, Osborne N, Shirota L, Rote P, Lee Y-K, Song B-J, et al. The fatty acid omega hydroxylase genes (CYP4 family) in the progression of metabolic dysfunction-associated steatotic liver disease (MASLD): An RNA sequence database analysis and review. Biochem Pharmacol. (2024) 228:116241. doi: 10.1016/j.bcp.2024.116241
172. Vishnoi K, Kumar S, Ke R, Rana A, and Rana B. Dysregulation of immune checkpoint proteins in hepatocellular carcinoma: Impact on metabolic reprogramming. Curr Opin Pharmacol. (2022) 64:102232. doi: 10.1016/j.coph.2022.102232
173. Lu M, Wu Y, Xia M, and Zhang Y. The role of metabolic reprogramming in liver cancer and its clinical perspectives. Front Oncol. (2024) 14:1454161. doi: 10.3389/fonc.2024.1454161
174. Zhao Y, Zhang J, Wang S, Jiang Q, and Xu K. Identification and validation of a nine-gene amino acid metabolism-related risk signature in HCC. Front Cell Dev Biol. (2021) 9:731790. doi: 10.3389/fcell.2021.731790
175. Wu J, Xue R, Jiang R-T, and Meng Q-H. Characterization of metabolic landscape in hepatocellular carcinoma. World J Gastrointest Oncol. (2021) 13:1144–56. doi: 10.4251/wjgo.v13.i9.1144
176. Ye Y, Yu B, Wang H, and Yi F. Glutamine metabolic reprogramming in hepatocellular carcinoma. Front Mol Biosci. (2023) 10:1242059. doi: 10.3389/fmolb.2023.1242059
177. Ziki RA and Colnot S. Glutamine metabolism, a double agent combating or fuelling hepatocellular carcinoma. JHEP Rep. (2024) 6:101077. doi: 10.1016/j.jhepr.2024.101077
178. Katt WP, Lukey MJ, and Cerione RA. A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis. Future Med Chem. (2017) 9:223–43. doi: 10.4155/fmc-2016-0190
179. Matés JM, Campos-Sandoval JA, de Los Santos-Jiménez J, and Márquez J. Glutaminases regulate glutathione and oxidative stress in cancer. Arch Toxicol. (2020) 94:2603–23. doi: 10.1007/s00204-020-02838-8
180. Xi J, Sun Y, Zhang M, Fa Z, Wan Y, Min Z, et al. GLS1 promotes proliferation in hepatocellular carcinoma cells via AKT/GSK3β/CyclinD1 pathway. Exp Cell Res. (2019) 381:1–9. doi: 10.1016/j.yexcr.2019.04.005
181. Lu H, Yin H, Qu L, Ma X, Fu R, and Fan D. Ginsenoside Rk1 regulates glutamine metabolism in hepatocellular carcinoma through inhibition of the ERK/c-Myc pathway. Food Funct. (2022) 13:3793–811. doi: 10.1039/d1fo03728e
182. Li B, Cao Y, Meng G, Qian L, Xu T, Yan C, et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine. (2019) 39:239–54. doi: 10.1016/j.ebiom.2018.11.063
183. Zhang J, Wang C, Chen M, Cao J, Zhong Y, Chen L, et al. Epigenetic silencing of glutaminase 2 in human liver and colon cancers. BMC Cancer. (2013) 13:601. doi: 10.1186/1471-2407-13-601
184. Suzuki S, Venkatesh D, Kanda H, Nakayama A, Hosokawa H, Lee E, et al. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. (2022) 82:3209–22. doi: 10.1158/0008-5472.CAN-21-3914
185. López de la Oliva AR, Campos-Sandoval JA, Gómez-García MC, Cardona C, Martín-Rufián M, Sialana FJ, et al. Nuclear translocation of glutaminase GLS2 in human cancer cells associates with proliferation arrest and differentiation. Sci Rep. (2020) 10:2259. doi: 10.1038/s41598-020-58264-4
186. Kuo T-C, Chen C-K, Hua K-T, Yu P, Lee W-J, Chen M-W, et al. Glutaminase 2 stabilizes Dicer to repress Snail and metastasis in hepatocellular carcinoma cells. Cancer Lett. (2016) 383:282–94. doi: 10.1016/j.canlet.2016.10.012
187. Saito Y and Soga T. Amino acid transporters as emerging therapeutic targets in cancer. Cancer Sci. (2021) 112:2958–65. doi: 10.1111/cas.15006
188. Scalise M, Console L, Rovella F, Galluccio M, Pochini L, and Indiveri C. Membrane transporters for amino acids as players of cancer metabolic rewiring. Cells. (2020) 9:2028. doi: 10.3390/cells9092028
189. Zhang G, Xiao Y, Tan J, Liu H, Fan W, and Li J. Elevated SLC1A5 associated with poor prognosis and therapeutic resistance to transarterial chemoembolization in hepatocellular carcinoma. J Transl Med. (2024) 22:543. doi: 10.1186/s12967-024-05298-1
190. Mastroberardino L, Spindler B, Pfeiffer R, Skelly PJ, Loffing J, Shoemaker CB, et al. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature. (1998) 395:288–91. doi: 10.1038/26246
191. Yi W, Tu M-J, Liu Z, Zhang C, Batra N, Yu A-X, et al. Bioengineered miR-328-3p modulates GLUT1-mediated glucose uptake and metabolism to exert synergistic antiproliferative effects with chemotherapeutics. Acta Pharm Sin B. (2020) 10:159–70. doi: 10.1016/j.apsb.2019.11.001
192. Bothwell PJ, Kron CD, Wittke EF, Czerniak BN, and Bode BP. Targeted suppression and knockout of ASCT2 or LAT1 in epithelial and mesenchymal human liver cancer cells fail to inhibit growth. Int J Mol Sci. (2018) 19:2093. doi: 10.3390/ijms19072093
193. Kim S-Y, Ong Q, Liao Y, Ding Z, Tan AQL, Lim LTR, et al. Genetic ablation of LAT1 inhibits growth of liver cancer cells and downregulates mTORC1 signaling. Int J Mol Sci. (2023) 24:9171. doi: 10.3390/ijms24119171
194. Okano N, Naruge D, Kawai K, Kobayashi T, Nagashima F, Endou H, et al. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Invest New Drugs. (2020) 38:1495–506. doi: 10.1007/s10637-020-00924-3
195. Zhang L, Wu Z, Qiu X, Zhang J, and Cheng S-C. Glutamate oxaloacetate transaminase 1 is dispensable in macrophage differentiation and anti-pathogen response. Commun Biol. (2024) 7:817. doi: 10.1038/s42003-024-06479-w
196. Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, et al. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell. (2018) 33:91–107.e6. doi: 10.1016/j.ccell.2017.12.003
197. Liu X and Shen Z. LncRNA TMPO-AS1 Aggravates the Development of Hepatocellular Carcinoma via miR-429/GOT1 Axis. Am J Med Sci. (2020) 360:711–20. doi: 10.1016/j.amjms.2020.08.010
198. Chen S, Tang Q, Hu M, Song S, Wu X, Zhou Y, et al. Loss of carbamoyl phosphate synthetase 1 potentiates hepatocellular carcinoma metastasis by reducing aspartate level. Adv Sci (Weinh). (2024) 11:e2402703. doi: 10.1002/advs.202402703
199. Zhang S, Hu Y, Wu Z, Zhou X, Wu T, Li P, et al. Deficiency of Carbamoyl Phosphate Synthetase 1 Engenders Radioresistance in Hepatocellular Carcinoma via Deubiquitinating c-Myc. Int J Radiat Oncol Biol Phys. (2023) 115:1244–56. doi: 10.1016/j.ijrobp.2022.11.022
200. Wu T, Luo G, Lian Q, Sui C, Tang J, Zhu Y, et al. Discovery of a carbamoyl phosphate synthetase 1-deficient HCC subtype with therapeutic potential through integrative genomic and experimental analysis. Hepatology. (2021) 74:3249–68. doi: 10.1002/hep.32088
201. Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. (2017) 546:168–72. doi: 10.1038/nature22359
202. Tao X, Zuo Q, Ruan H, Wang H, Jin H, Cheng Z, et al. Argininosuccinate synthase 1 suppresses cancer cell invasion by inhibiting STAT3 pathway in hepatocellular carcinoma. Acta Biochim Biophys Sin (Shanghai). (2019) 51:263–76. doi: 10.1093/abbs/gmz005
203. Caldara M, Dupont G, Leroy F, Goldbeter A, De Vuyst L, and Cunin R. Arginine biosynthesis in Escherichia coli: experimental perturbation and mathematical modeling. J Biol Chem. (2008) 283:6347–58. doi: 10.1074/jbc.M705884200
204. Phillips MM, Sheaff MT, and Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat. (2013) 45:251–62. doi: 10.4143/crt.2013.45.4.251
205. Li T, Zhu Y, Cheng F, Lu C, Jung JU, and Gao S-J. Oncogenic kaposi’s sarcoma-associated herpesvirus upregulates argininosuccinate synthase 1, a rate-limiting enzyme of the citrulline-nitric oxide cycle, to activate the STAT3 pathway and promote growth transformation. J Virol. (2019) 93:e01599–18. doi: 10.1128/JVI.01599-18
206. You J, Chen W, Chen J, Zheng Q, Dong J, and Zhu Y. The oncogenic role of ARG1 in progression and metastasis of hepatocellular carcinoma. BioMed Res Int. (2018) 2018:2109865. doi: 10.1155/2018/2109865
207. Grompe M, Jones SN, and Caskey CT. Molecular detection and correction of ornithine transcarbamylase deficiency. Trends Genet. (1990) 6:335–9. doi: 10.1016/0168-9525(90)90255-5
208. Couchet M, Breuillard C, Corne C, Rendu J, Morio B, Schlattner U, et al. Ornithine transcarbamylase - from structure to metabolism: an update. Front Physiol. (2021) 12:748249. doi: 10.3389/fphys.2021.748249
209. He L, Cai X, Cheng S, Zhou H, Zhang Z, Ren J, et al. Ornithine transcarbamylase downregulation is associated with poor prognosis in hepatocellular carcinoma. Oncol Lett. (2019) 17:5030–8. doi: 10.3892/ol.2019.10174
210. Díaz-Muñoz M and Hernández-Muñoz R. Molecular and biochemical features of the mitochondrial enzyme ornithine transcarbamylase: a possible new role as a signaling factor. Curr Med Chem. (2010) 17:2253–60. doi: 10.2174/092986710791331031
211. Liu Y, Chen S, Wan X, Wang R, Luo H, Chang C, et al. Tryptophan 2,3-dioxygenase-positive matrix fibroblasts fuel breast cancer lung metastasis via kynurenine-mediated ferroptosis resistance of metastatic cells and T cell dysfunction. Cancer Commun (Lond). (2024) 44:1261–86. doi: 10.1002/cac2.12608
212. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. (2006) 107:2013–21. doi: 10.1182/blood-2005-05-1795
213. Zhu Y, Zhou Z, Du X, Lin X, Liang Z-M, Chen S, et al. Cancer cell-derived arginine fuels polyamine biosynthesis in tumor-associated macrophages to promote immune evasion. Cancer Cell. (2025) 43:1045–1060.e7. doi: 10.1016/j.ccell.2025.03.015
214. Liu Y and Yang C. Oncometabolites in cancer: current understanding and challenges. Cancer Res. (2021) 81:2820–3. doi: 10.1158/0008-5472.CAN-20-3730
215. Anderson NM and Simon MC. The tumor microenvironment. Curr Biol. (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
216. El-Tanani M, Rabbani SA, Babiker R, Rangraze I, Kapre S, Palakurthi SS, et al. Unraveling the tumor microenvironment: Insights into cancer metastasis and therapeutic strategies. Cancer Lett. (2024) 591:216894. doi: 10.1016/j.canlet.2024.216894
217. Lin J, Rao D, Zhang M, and Gao Q. Metabolic reprogramming in the tumor microenvironment of liver cancer. J Hematol Oncol. (2024) 17:6. doi: 10.1186/s13045-024-01527-8
218. Bader JE, Voss K, and Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. (2020) 78:1019–33. doi: 10.1016/j.molcel.2020.05.034
219. Cassim S, Raymond V-A, Dehbidi-Assadzadeh L, Lapierre P, and Bilodeau M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle. (2018) 17:903–16. doi: 10.1080/15384101.2018.1460023
220. Zhou Y, Lin F, Wan T, Chen A, Wang H, Jiang B, et al. ZEB1 enhances Warburg effect to facilitate tumorigenesis and metastasis of HCC by transcriptionally activating PFKM. Theranostics. (2021) 11:5926–38. doi: 10.7150/thno.56490
221. Liao M, Yao D, Wu L, Luo C, Wang Z, Zhang J, et al. Targeting the Warburg effect: A revisited perspective from molecular mechanisms to traditional and innovative therapeutic strategies in cancer. Acta Pharm Sin B. (2024) 14:953–1008. doi: 10.1016/j.apsb.2023.12.003
222. Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS, et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol. (2009) 174:1544–52. doi: 10.2353/ajpath.2009.080596
223. Kim YH, Jeong DC, Pak K, Han M-E, Kim J-Y, Liangwen L, et al. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget. (2017) 8:68381–92. doi: 10.18632/oncotarget.20266
224. Heo HJ, Park Y, Lee JH, Kim Y, Kim EK, Kim GH, et al. Clinical big-data-based design of GLUT2-targeted carbon nanodots for accurate diagnosis of hepatocellular carcinoma. Nanoscale. (2022) 14:17053–64. doi: 10.1039/d2nr04238j
225. Gao H, Hao Y, Zhou X, Li H, Liu F, Zhu H, et al. Prognostic value of glucose transporter 3 expression in hepatocellular carcinoma. Oncol Lett. (2020) 19:691–9. doi: 10.3892/ol.2019.11191
226. Hoxhaj G and Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. (2020) 20:74–88. doi: 10.1038/s41568-019-0216-7
227. Chen C, Pore N, Behrooz A, Ismail-Beigi F, and Maity A. Regulation of glut1 mRNA by Hypoxia-inducible Factor-1: INTERACTION BETWEEN H-ras AND HYPOXIA. J Biol Chem. (2001) 276:9519–25. doi: 10.1074/jbc.M010144200
228. Sadlecki P, Bodnar M, Grabiec M, Marszalek A, Walentowicz P, Sokup A, et al. The role of hypoxia-inducible factor-1α, glucose transporter-1, (GLUT-1) and carbon anhydrase IX in endometrial cancer patients. BioMed Res Int. (2014) 2014:616850. doi: 10.1155/2014/616850
229. Shang R, Pu M, Li Y, and Wang D. FOXM1 regulates glycolysis in hepatocellular carcinoma by transactivating glucose transporter 1 expression. Oncol Rep. (2017) 37:2261–9. doi: 10.3892/or.2017.5472
230. Zhou Y, Yu H, Cheng S, Chen Y, He L, Ren J, et al. Glutamate dehydrogenase 1 mediated glutaminolysis sustains HCC cells survival under glucose deprivation. J Cancer. (2022) 13:1061–72. doi: 10.7150/jca.64195
231. Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. (2013) 19:560–70. doi: 10.1158/1078-0432.CCR-12-2334
232. Yang Y, Jia S, Zhu N, Xiao X, Ma Y, Tu K, et al. OTUD5 promotes the growth of hepatocellular carcinoma by deubiquitinating and stabilizing SLC38A1. Biol Direct. (2024) 19:31. doi: 10.1186/s13062-024-00475-0
233. Tambay V, Raymond V-A, Voisin L, Meloche S, and Bilodeau M. Reprogramming of glutamine amino acid transporters expression and prognostic significance in hepatocellular carcinoma. Int J Mol Sci. (2024) 25:7558. doi: 10.3390/ijms25147558
234. Li X, Liang M, Xu Z, Wei Z, and Xu Y. Transcription factor TCF12 activates the transcription level of SLC38A1 to promote the development of hepatocellular carcinoma. Appl Biochem Biotechnol. (2025) 197:5362–77. doi: 10.1007/s12010-025-05263-8
235. Zhi R and Fan F. SLC1A3 is a novel prognostic biomarker associated with immunity and EMT in hepatocellular carcinoma. Discov Oncol. (2024) 15:676. doi: 10.1007/s12672-024-01561-5
236. Kaiser P. Methionine dependence of cancer. Biomolecules. (2020) 10:568. doi: 10.3390/biom10040568
237. Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW, et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med. (2019) 25:825–37. doi: 10.1038/s41591-019-0423-5
238. Li J-T, Yang H, Lei M-Z, Zhu W-P, Su Y, Li K-Y, et al. Dietary folate drives methionine metabolism to promote cancer development by stabilizing MAT IIA. Signal Transduct Target Ther. (2022) 7:192. doi: 10.1038/s41392-022-01017-8
239. Lu SC and Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. (2012) 92:1515–42. doi: 10.1152/physrev.00047.2011
240. Hoffman RM and Erbe RW. High in vivo rates of methionine biosynthesis in transformed human and Malignant rat cells auxotrophic for methionine. Proc Natl Acad Sci U.S.A. (1976) 73:1523–7. doi: 10.1073/pnas.73.5.1523
241. Stern PH, Wallace CD, and Hoffman RM. Altered methionine metabolism occurs in all members of a set of diverse human tumor cell lines. J Cell Physiol. (1984) 119:29–34. doi: 10.1002/jcp.1041190106
242. Lan T, Gao F, Cai Y, Lv Y, Zhu J, Liu H, et al. The protein circPETH-147aa regulates metabolic reprogramming in hepatocellular carcinoma cells to remodel immunosuppressive microenvironment. Nat Commun. (2025) 16:333. doi: 10.1038/s41467-024-55577-0
243. Koppula P, Zhuang L, and Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. (2021) 12:599–620. doi: 10.1007/s13238-020-00789-5
244. Koppula P, Zhang Y, Zhuang L, and Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond). (2018) 38:12. doi: 10.1186/s40880-018-0288-x
245. He F, Zhang P, Liu J, Wang R, Kaufman RJ, Yaden BC, et al. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J Hepatol. (2023) 79:362–77. doi: 10.1016/j.jhep.2023.03.016
246. Chen Q, Zheng W, Guan J, Liu H, Dan Y, Zhu L, et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. (2023) 30:137–51. doi: 10.1038/s41418-022-01051-7
247. Zhu Y, Li H, Hu H, Hu H, and Luo M-L. The arginine metabolism battlefield: how metabolite exchange in the tumor microenvironment shapes an immunosuppressive ecosystem. Cancer Biol Med. (2025). doi: 10.20892/j.issn.2095-3941.2025.0202
248. Kim S, Lee M, Song Y, Lee S-Y, Choi I, Park I-S, et al. Argininosuccinate synthase 1 suppresses tumor progression through activation of PERK/eIF2α/ATF4/CHOP axis in hepatocellular carcinoma. J Exp Clin Cancer Res. (2021) 40:127. doi: 10.1186/s13046-021-01912-y
249. Missiaen R, Anderson NM, Kim LC, Nance B, Burrows M, Skuli N, et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. (2022) 34:1151–1167.e7. doi: 10.1016/j.cmet.2022.06.010
250. Shigematsu Y, Amori G, Kanda H, Takahashi Y, Takazawa Y, Takeuchi K, et al. Decreased ARG1 expression as an adverse prognostic phenotype in non-alcoholic non-virus-related hepatocellular carcinoma. Virchows Arch. (2022) 481:253–63. doi: 10.1007/s00428-022-03318-3
251. Munn DH and Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
252. Krishnamurthy S, Gilot D, Ahn SB, Lam V, Shin J-S, Guillemin GJ, et al. Involvement of kynurenine pathway in hepatocellular carcinoma. Cancers (Basel). (2021) 13:5180. doi: 10.3390/cancers13205180
253. Chai B, Zhang A, Liu Y, Zhang X, Kong P, Zhang Z, et al. KLF7 promotes hepatocellular carcinoma progression through regulating SLC1A5-mediated tryptophan metabolism. J Cell Mol Med. (2024) 28:e70245. doi: 10.1111/jcmm.70245
254. Cheong JE and Sun L. Targeting the IDO1/TDO2-KYN-ahR pathway for cancer immunotherapy - challenges and opportunities. Trends Pharmacol Sci. (2018) 39:307–25. doi: 10.1016/j.tips.2017.11.007
255. Liang X, Gao H, Xiao J, Han S, He J, Yuan R, et al. Abrine, an IDO1 inhibitor, suppresses the immune escape and enhances the immunotherapy of anti-PD-1 antibody in hepatocellular carcinoma. Front Immunol. (2023) 14:1185985. doi: 10.3389/fimmu.2023.1185985
256. Patra T, Cunningham DM, Meyer K, Toth K, Ray RB, Heczey A, et al. Targeting Lin28 axis enhances glypican-3-CAR T cell efficacy against hepatic tumor initiating cell population. Mol Ther. (2023) 31:715–28. doi: 10.1016/j.ymthe.2023.01.002
257. Niu Y, Mayr T, and Muders MH. Competition for nutrients or cell intrinsic programming? - Metabolic mechanisms behind the tumor promoting immune microenvironment in cancer. Signal Transduct Target Ther. (2021) 6:279. doi: 10.1038/s41392-021-00693-2
258. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
259. Dando I, Pozza ED, Ambrosini G, Torrens-Mas M, Butera G, Mullappilly N, et al. Oncometabolites in cancer aggressiveness and tumour repopulation. Biol Rev Camb Philos Soc. (2019) 94:1530–46. doi: 10.1111/brv.12513
260. Niu Z, Yang F, Li H, Wang J, Ni Q, Ma C, et al. MCT4 promotes hepatocellular carcinoma progression by upregulating TRAPPC5 gene. J Hepatocell Carcinoma. (2022) 9:289–300. doi: 10.2147/JHC.S352948
261. Pérez-Tomás R and Pérez-Guillén I. Lactate in the tumor microenvironment: an essential molecule in cancer progression and treatment. Cancers (Basel). (2020) 12:3244. doi: 10.3390/cancers12113244
262. Chen H-L, OuYang H-Y, Le Y, Jiang P, Tang H, Yu Z-S, et al. Aberrant MCT4 and GLUT1 expression is correlated with early recurrence and poor prognosis of hepatocellular carcinoma after hepatectomy. Cancer Med. (2018) 7:5339–50. doi: 10.1002/cam4.1521
263. Lu H, Liu H, Yan R, Ma W, Liu H, Liu R, et al. Carbonic anhydrase 2 facilitates sorafenib resistance by counteracting MCT4-mediated intracellular pH dysregulation in HCC. Cell Rep. (2024) 43:114996. doi: 10.1016/j.celrep.2024.114996
264. Klaessens S, Stroobant V, De Plaen E, and Van den Eynde BJ. Systemic tryptophan homeostasis. Front Mol Biosci. (2022) 9:897929. doi: 10.3389/fmolb.2022.897929
265. Chen H, Sun W, Xie M, Li Z, Chen B, Zhang T, et al. Integrated bioinformatics analysis and experimental validation to understand tryptophan metabolism-related genes in hepatocellular carcinoma. J Cancer. (2024) 15:4879–92. doi: 10.7150/jca.91306
266. Li S, Weng J, Song F, Li L, Xiao C, Yang W, et al. Circular RNA circZNF566 promotes hepatocellular carcinoma progression by sponging miR-4738-3p and regulating TDO2 expression. Cell Death Dis. (2020) 11:452. doi: 10.1038/s41419-020-2616-8
267. Zhang J, Zhang Z, Wu Z, Wang Y, Zhang Z, and Xia L. The switch triggering the invasion process: Lipid metabolism in the metastasis of hepatocellular carcinoma. Chin Med J (Engl). (2024) 137:1271–84. doi: 10.1097/CM9.0000000000003144
268. Bertolio R, Napoletano F, Mano M, Maurer-Stroh S, Fantuz M, Zannini A, et al. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat Commun. (2019) 10:1326. doi: 10.1038/s41467-019-09152-7
269. Zhang Z, Yang J, Liu R, Ma J, Wang K, Wang X, et al. Inhibiting HMGCR represses stemness and metastasis of hepatocellular carcinoma via Hedgehog signaling. Genes Dis. (2024) 11:101285. doi: 10.1016/j.gendis.2024.101285
270. Zhang D, Ni Y, Wang Y, Feng J, Zhuang N, Li J, et al. Spatial heterogeneity of tumor microenvironment influences the prognosis of clear cell renal cell carcinoma. J Transl Med. (2023) 21:489. doi: 10.1186/s12967-023-04336-8
271. Elhanani O, Ben-Uri R, and Keren L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell. (2023) 41:404–20. doi: 10.1016/j.ccell.2023.01.010
272. Bareham B, Dibble M, and Parsons M. Defining and modeling dynamic spatial heterogeneity within tumor microenvironments. Curr Opin Cell Biol. (2024) 90:102422. doi: 10.1016/j.ceb.2024.102422
273. Peralta RM, Xie B, Lontos K, Nieves-Rosado H, Spahr K, Joshi S, et al. Dysfunction of exhausted T cells is enforced by MCT11-mediated lactate metabolism. Nat Immunol. (2024) 25:2297–307. doi: 10.1038/s41590-024-01999-3
274. Gu X-Y, Yang J-L, Lai R, Zhou Z-J, Tang D, Hu L, et al. Impact of lactate on immune cell function in the tumor microenvironment: mechanisms and therapeutic perspectives. Front Immunol. (2025) 16:1563303. doi: 10.3389/fimmu.2025.1563303
275. Nakahara R, Maeda K, Aki S, and Osawa T. Metabolic adaptations of cancer in extreme tumor microenvironments. Cancer Sci. (2023) 114:1200–7. doi: 10.1111/cas.15722
276. Xia J, Wang C, and Li B. Hepatocellular carcinoma cells induce γδ T cells through metabolic reprogramming into tumor-progressive subpopulation. Front Oncol. (2024) 14:1451650. doi: 10.3389/fonc.2024.1451650
277. Lei X, Lei Y, Li J-K, Du W-X, Li R-G, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
278. Fu H, Vuononvirta J, Fanti S, Bonacina F, D’Amati A, Wang G, et al. The glucose transporter 2 regulates CD8+ T cell function via environment sensing. Nat Metab. (2023) 5:1969–85. doi: 10.1038/s42255-023-00913-9
279. Palmer CS, Ostrowski M, Balderson B, Christian N, and Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. (2015) 6:1. doi: 10.3389/fimmu.2015.00001
280. Zhang S, Zhang X, Yang H, Liang T, and Bai X. Hurdle or thruster: Glucose metabolism of T cells in anti-tumour immunity. Biochim Biophys Acta (BBA) - Rev Cancer. (2024) 1879:189022. doi: 10.1016/j.bbcan.2023.189022
281. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. (2013) 123:4479–88. doi: 10.1172/JCI69589
282. Jin J, Byun J-K, Choi Y-K, and Park K-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. (2023) 55:706–15. doi: 10.1038/s12276-023-00971-9
283. Yang Y, Pei T, Liu C, Cao M, Hu X, Yuan J, et al. Glutamine metabolic competition drives immunosuppressive reprogramming of intratumour GPR109A+ myeloid cells to promote liver cancer progression. Gut. (2025) 74:255–69. doi: 10.1136/gutjnl-2024-332429
284. Wu J, Li G, Li L, Li D, Dong Z, and Jiang P. Asparagine enhances LCK signalling to potentiate CD8+ T-cell activation and anti-tumour responses. Nat Cell Biol. (2021) 23:75–86. doi: 10.1038/s41556-020-00615-4
285. Werner A, Amann E, Schnitzius V, Habermeier A, Luckner-Minden C, Leuchtner N, et al. Induced arginine transport via cationic amino acid transporter-1 is necessary for human T-cell proliferation. Eur J Immunol. (2016) 46:92–103. doi: 10.1002/eji.201546047
286. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–842.e13. doi: 10.1016/j.cell.2016.09.031
287. Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, et al. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol. (1999) 162:3356–66. doi: 10.4049/jimmunol.162.6.3356
288. Sanderson SM, Gao X, Dai Z, and Locasale JW. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat Rev Cancer. (2019) 19:625–37. doi: 10.1038/s41568-019-0187-8
289. Huang X, Sun T, Wang J, Hong X, Chen H, Yan T, et al. Metformin reprograms tryptophan metabolism to stimulate CD8+ T-cell function in colorectal cancer. Cancer Res. (2023) 83:2358–71. doi: 10.1158/0008-5472.CAN-22-3042
290. Fernández-Gallego N, Anega B, Luengo-Arias S, Bizkarguenaga M, Gil-Redondo R, Embade N, et al. Restricting SLC7A5-mediated Leucine uptake in T cells prevents acute GVHD and maintains GVT response. EMBO Mol Med. (2025) 17:1631–65. doi: 10.1038/s44321-025-00250-2
291. Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature. (2020) 585:277–82. doi: 10.1038/s41586-020-2682-1
292. Sharma P, Guo A, Poudel S, Boada-Romero E, Verbist KC, Palacios G, et al. Early methionine availability attenuates T cell exhaustion. Nat Immunol. (2025) 26:1384–96. doi: 10.1038/s41590-025-02223-6
293. Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci U.S.A. (2019) 116:9533–42. doi: 10.1073/pnas.1814932116
294. Cappellesso F, Mazzone M, and Virga F. Acid affairs in anti-tumour immunity. Cancer Cell Int. (2024) 24:354. doi: 10.1186/s12935-024-03520-0
295. Liu W, Wang Y, Bozi LHM, Fischer PD, Jedrychowski MP, Xiao H, et al. Lactate regulates cell cycle by remodelling the anaphase promoting complex. Nature. (2023) 616:790–7. doi: 10.1038/s41586-023-05939-3
296. Fumarola C, Petronini PG, and Alfieri R. Impairing energy metabolism in solid tumors through agents targeting oncogenic signaling pathways. Biochem Pharmacol. (2018) 151:114–25. doi: 10.1016/j.bcp.2018.03.006
297. Tillé L, Cropp D, Charmoy M, Reichenbach P, Andreatta M, Wyss T, et al. Activation of the transcription factor NFAT5 in the tumor microenvironment enforces CD8+ T cell exhaustion. Nat Immunol. (2023) 24:1645–53. doi: 10.1038/s41590-023-01614-x
298. Qin R, Zhao C, Wang C-J, Xu W, Zhao J-Y, Lin Y, et al. Tryptophan potentiates CD8+ T cells against cancer cells by TRIP12 tryptophanylation and surface PD-1 downregulation. J Immunother Cancer. (2021) 9:e002840. doi: 10.1136/jitc-2021-002840
299. Tian X, Shen H, Li Z, Wang T, and Wang S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J Hematol Oncol. (2019) 12:84. doi: 10.1186/s13045-019-0772-z
300. Liu X, Yang M, Xu P, Du M, Li S, Shi J, et al. Kynurenine-AhR reduces T-cell infiltration and induces a delayed T-cell immune response by suppressing the STAT1-CXCL9/CXCL10 axis in tuberculosis. Cell Mol Immunol. (2024) 21:1426–40. doi: 10.1038/s41423-024-01230-1
301. Gutiérrez-Vázquez C and Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity. (2018) 48:19–33. doi: 10.1016/j.immuni.2017.12.012
302. Li Z, Guan M, Lin Y, Cui X, Zhang Y, Zhao Z, et al. Aberrant lipid metabolism in hepatocellular carcinoma revealed by liver lipidomics. Int J Mol Sci. (2017) 18:2550. doi: 10.3390/ijms18122550
303. Ke X-Y, Zou M, and Xu C. Lipid metabolism in tumor-infiltrating T cells: mechanisms and applications. Life Metab. (2022) 1:211–23. doi: 10.1093/lifemeta/loac038
304. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity. (2021) 54:1561–1577.e7. doi: 10.1016/j.immuni.2021.05.003
305. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. (2019) 30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002
306. Song M, Sandoval TA, Chae C-S, Chopra S, Tan C, Rutkowski MR, et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. (2018) 562:423–8. doi: 10.1038/s41586-018-0597-x
307. Ma S, Ming Y, Wu J, and Cui G. Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol Immunol. (2024) 21:419–35. doi: 10.1038/s41423-024-01148-8
308. Cao J, Liao S, Zeng F, Liao Q, Luo G, and Zhou Y. Effects of altered glycolysis levels on CD8+ T cell activation and function. Cell Death Dis. (2023) 14:407. doi: 10.1038/s41419-023-05937-3
309. Van den Bossche J and van der Windt GJW. Fatty acid oxidation in macrophages and T cells: time for reassessment? Cell Metab. (2018) 28:538–40. doi: 10.1016/j.cmet.2018.09.018
310. Masuyama S, Mizui M, Morita M, Shigeki T, Kato H, Yamamoto T, et al. Enhanced fatty acid oxidation by selective activation of PPARα alleviates autoimmunity through metabolic transformation in T-cells. Clin Immunol. (2024) 268:110357. doi: 10.1016/j.clim.2024.110357
311. Luo W, Guo S, Zhou Y, Zhao J, Wang M, Sang L, et al. Hepatocellular carcinoma: how the gut microbiota contributes to pathogenesis, diagnosis, and therapy. Front Microbiol. (2022) 13:873160. doi: 10.3389/fmicb.2022.873160
312. Wang Y, Li Y, Lin Y, Cao C, Chen D, Huang X, et al. Roles of the gut microbiota in hepatocellular carcinoma: from the gut dysbiosis to the intratumoral microbiota. Cell Death Discov. (2025) 11:140. doi: 10.1038/s41420-025-02413-z
313. Anwer EKE, Ajagbe M, Sherif M, Musaibah AS, Mahmoud S, ElBanbi A, et al. Gut microbiota secondary metabolites: key roles in GI tract cancers and infectious diseases. Biomedicines. (2025) 13:100. doi: 10.3390/biomedicines13010100
314. Mukhopadhya I and Louis P. Gut microbiota-derived short-chain fatty acids and their role in human health and disease. Nat Rev Microbiol. (2025) 23:635–51. doi: 10.1038/s41579-025-01183-w
315. Wang L-Y, He L-H, Xu L-J, and Li S-B. Short-chain fatty acids: bridges between diet, gut microbiota, and health. J Gastroenterol Hepatol. (2024) 39:1728–36. doi: 10.1111/jgh.16619
316. Mann ER, Lam YK, and Uhlig HH. Short-chain fatty acids: linking diet, the microbiome and immunity. Nat Rev Immunol. (2024) 24:577–95. doi: 10.1038/s41577-024-01014-8
317. Orabi D, Osborn LJ, Fung K, Massey W, Horak AJ, Aucejo F, et al. A surgical method for continuous intraportal infusion of gut microbial metabolites in mice. JCI Insight. (2021) 6:e145607. doi: 10.1172/jci.insight.145607
318. Koh A, De Vadder F, Kovatcheva-Datchary P, and Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. (2016) 165:1332–45. doi: 10.1016/j.cell.2016.05.041
319. Li X, He M, Yi X, Lu X, Zhu M, Xue M, et al. Short-chain fatty acids in nonalcoholic fatty liver disease: New prospects for short-chain fatty acids as therapeutic targets. Heliyon. (2024) 10:e26991. doi: 10.1016/j.heliyon.2024.e26991
320. Münte E and Hartmann P. The role of short-chain fatty acids in metabolic dysfunction-associated steatotic liver disease and other metabolic diseases. Biomolecules. (2025) 15:469. doi: 10.3390/biom15040469
321. Gao Y, Lin J, Ye C, Guo S, and Jiang C. Microbial transformations of bile acids and their receptors in the regulation of metabolic dysfunction-associated steatotic liver disease. Liver Res. (2023) 7:165–76. doi: 10.1016/j.livres.2023.09.002
322. Lee MH, Nuccio S-P, Mohanty I, Hagey LR, Dorrestein PC, Chu H, et al. How bile acids and the microbiota interact to shape host immunity. Nat Rev Immunol. (2024) 24:798–809. doi: 10.1038/s41577-024-01057-x
323. Fleishman JS and Kumar S. Bile acid metabolism and signaling in health and disease: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. (2024) 9:97. doi: 10.1038/s41392-024-01811-6
324. Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS, et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe. (2020) 27:659–670.e5. doi: 10.1016/j.chom.2020.01.021
325. Hu M, Alashkar Alhamwe B, Santner-Nanan B, Miethe S, Harb H, Renz H, et al. Short-chain fatty acids augment differentiation and function of human induced regulatory T cells. Int J Mol Sci. (2022) 23:5740. doi: 10.3390/ijms23105740
326. Yu J, Chen X, Yang X, and Zhang B. Understanding gut dysbiosis for hepatocellular carcinoma diagnosis and treatment. Trends Endocrinol Metab. (2024) 35:1006–20. doi: 10.1016/j.tem.2024.06.003
327. Hu M-M, He W-R, Gao P, Yang Q, He K, Cao L-B, et al. Virus-induced accumulation of intracellular bile acids activates the TGR5-β-arrestin-SRC axis to enable innate antiviral immunity. Cell Res. (2019) 29:193–205. doi: 10.1038/s41422-018-0136-1
328. Sorrentino G, Perino A, Yildiz E, El Alam G, Bou Sleiman M, Gioiello A, et al. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology. (2020) 159:956–968.e8. doi: 10.1053/j.gastro.2020.05.067
329. Hu B, Yu M, Ma X, Sun J, Liu C, Wang C, et al. IFNα Potentiates anti-PD-1 efficacy by remodeling glucose metabolism in the hepatocellular carcinoma microenvironment. Cancer Discov. (2022) 12:1718–41. doi: 10.1158/2159-8290.CD-21-1022
330. Gouda MA, Voss MH, Tawbi H, Gordon M, Tykodi SS, Lam ET, et al. A phase I/II study of the safety and efficacy of telaglenastat (CB-839) in combination with nivolumab in patients with metastatic melanoma, renal cell carcinoma, and non-small-cell lung cancer. ESMO Open. (2025) 10:104536. doi: 10.1016/j.esmoop.2025.104536
331. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) 13:890–901. doi: 10.1158/1535-7163.MCT-13-0870
332. Varghese S, Pramanik S, Williams LJ, Hodges HR, Hudgens CW, Fischer GM, et al. The glutaminase inhibitor CB-839 (Telaglenastat) enhances the antimelanoma activity of T-cell-mediated immunotherapies. Mol Cancer Ther. (2021) 20:500–11. doi: 10.1158/1535-7163.MCT-20-0430
333. Huang J, Tsang WY, Fang X-N, Zhang Y, Luo J, Gong L-Q, et al. FASN inhibition decreases MHC-I degradation and synergizes with PD-L1 checkpoint blockade in hepatocellular carcinoma. Cancer Res. (2024) 84:855–71. doi: 10.1158/0008-5472.CAN-23-0966
334. Wang H, Zhou Y, Xu H, Wang X, Zhang Y, Shang R, et al. Therapeutic efficacy of FASN inhibition in preclinical models of HCC. Hepatology. (2022) 76:951–66. doi: 10.1002/hep.32359
335. Che L, Pilo MG, Cigliano A, Latte G, Simile MM, Ribback S, et al. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle. (2017) 16:499–507. doi: 10.1080/15384101.2017.1282586
336. De Los Santos-Jiménez J, Rosales T, Ko B, Campos-Sandoval JA, Alonso FJ, Márquez J, et al. Metabolic adjustments following glutaminase inhibition by CB-839 in glioblastoma cell lines. Cancers (Basel). (2023) 15:531. doi: 10.3390/cancers15020531
Keywords: hepatocellular carcinoma (HCC), metabolic reprogramming, tumor microenvironment (TME), T cell-mediated antitumor immunity, metabolic-immune targeted combination therapies
Citation: Liao D-X, An X, Huang G-X and Yuan T-L (2025) The impact of metabolic reprogramming in hepatocellular carcinoma on T cell. Front. Immunol. 16:1696113. doi: 10.3389/fimmu.2025.1696113
Received: 31 August 2025; Accepted: 21 October 2025;
Published: 10 November 2025.
Edited by:
Zheng Zhong, University of Michigan, United StatesReviewed by:
Zhennan Wu, Guangzhou University of Chinese Medicine, ChinaChongjian Gao, University of Michigan, United States
Copyright © 2025 Liao, An, Huang and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tong-Ling Yuan, cWluZ3hpbmJpbHVvY2h1bjIzQDE2My5jb20=; Gui-Xiang Huang, aHVhbmdndWl4aWFuZzE5ODlAMTI2LmNvbQ==
†These authors have contributed equally to this work