 Ana Belén Moreno-Castaño1,2,3*
Ana Belén Moreno-Castaño1,2,3* Gontzal Iraola3,4
Gontzal Iraola3,4 Núria Martínez-Cibrián2,3,5Nil Albiol2,3,5
Núria Martínez-Cibrián2,3,5Nil Albiol2,3,5 Daniel-Nicolás Marco Prats3,4Julia Martinez-Sanchez1,2
Daniel-Nicolás Marco Prats3,4Julia Martinez-Sanchez1,2 Pedro Castro2,3,4†
Pedro Castro2,3,4† Maribel Diaz-Ricart1,2,3†
Maribel Diaz-Ricart1,2,3†- 1Hemostasis and Erythropathology Laboratory, Hematopathology, Pathology Department, Centre Diagnòstic Biomèdic (CDB), Hospital Clínic Barcelona, Barcelona, Spain
- 2Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 3Facultat de Medicina i Ciències de la Salut, Universitat de Barcelona, Barcelona, Spain
- 4Medical Intensive Care Unit, Hospital Clínic Barcelona, Barcelona, Spain
- 5Hematology Department, Institut del Càncer i Malalties de la Sang (ICAMS), IDIBAPS, Hospital Clínic Barcelona, Barcelona, Spain
Chimeric antigen receptor (CAR) T-cell therapy has revolutionized the treatment of relapsed or refractory hematologic malignancies. While its clinical efficacy is well established, CAR T-cell therapy is frequently associated with severe immune-mediated toxicities, including cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), coagulopathy, and hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS). Increasing evidence suggests that endothelial dysfunction, hemostatic imbalance, and complement activation are key contributors to the pathogenesis of these complications. Substantial research efforts have focused on identifying circulating biomarkers capable of predicting toxicity onset and severity, as well as stratifying patients at risk for early non-relapse mortality. In this review, we summarize the current understanding of the pathophysiological mechanisms underlying early CAR T cell–related toxicities, with particular emphasis on biomarkers of endotheliopathy and related pathways involved in their development. We focus on highlighting translational biomarkers with potential diagnostic, prognostic, and monitoring value that could be implemented in clinical practice to improve patient risk stratification, differential diagnosis, and therapeutic follow-up.
1 Introduction
Chimeric antigen receptor (CAR) T-cell therapy now constitutes a mainstay in the treatment of hematologic malignancies, offering remarkable efficacy and durable responses in various disease settings, often with manageable toxicity profiles (1, 2). However, CAR T-cell therapy is associated with a distinct spectrum of particular toxicities driven by immune system hyperactivation, triggered by downstream effects of CAR T-cell-target interactions, monocyte and macrophage stimulation, endothelial dysfunction, and release of proinflammatory cytokines. The associated toxicities include cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and immune effector cell associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS) (3–5).
Despite advances in preventive and therapeutic strategies to reduce the risk of immune-related complications, such as the use of bridging therapies to shrink tumor burden before lymphodepletion, and early intervention upon symptom onset, non-relapse mortality (NRM) remains a concern. A recent meta-analysis reported unexpectedly high NRM rates, which vary depending on the specific CAR T-cell construct used (6).
Endothelial dysfunction is increasingly recognized as a key factor in the development and persistence of early CAR T cell–related toxicities. While the importance of endothelial health in immune-mediated complications is well established in allogeneic hematopoietic stem cell transplantation (allo-HSCT), its contribution to CAR T cell–associated toxicities is only beginning to be elucidated. Early HCT-related complications—such as sinusoidal obstruction syndrome (SOS), engraftment syndrome (ES), capillary leak syndrome (CLS), transplant-associated thrombotic microangiopathy (TA-TMA), acute graft-versus-host disease (aGVHD), and vascular idiopathic pneumonia syndrome (vascular-IPS)— all feature endothelial injury as a central pathophysiological component (7). Given the therapeutic similarities between HCT and CAR T cell therapy, together with the overlapping clinical manifestations of their toxicities, endothelial dysfunction emerges as a plausible shared pathogenic substrate in both settings.
This review aims to examine the role of endothelial dysfunction in CAR T-cell toxicities. We will first provide an overview of CAR T-cell structure, clinical use, and outcomes; then analyze the underlying pathophysiological mechanisms associated with endothelial injury, evaluate emerging biomarkers and scoring systems; and discuss potential therapeutic strategies targeting the endothelium-immune cell axis.
2 CAR T-cell structure, clinical use and outcomes
CAR T-cells are autologous T lymphocytes that are genetically engineered to express a chimeric antigen receptor targeting tumor-specific antigens. While new designs are being explored, second-generation CARs are still the most used. These usually include a single-chain variable fragment (scFv) derived from an antibody recognizing a particular antigen, linked to intracellular signaling domains such as CD3ζ and one costimulatory molecule (CD28 or 4-1BB), connected through a hinge and transmembrane region (1). Third-generation CARs combine both CD28 and 4-1BB domains to improve T-cell activation. In contrast, fourth-generation CARs, also called “TRUCKS” or “armored CARs,” add extra features like cytokine secretion and immune checkpoint regulation to enhance antitumor effects (3).
The clinical workflow for CAR T-cell therapy generally includes: 1) eligibility screening to confirm adequate organ function and performance status; 2) interruption or modification of disease-directed therapy to control tumor burden before apheresis (if necessary); 3) apheresis to collect mononuclear cells, which are sent for CAR T-cell manufacturing; 4) bridging therapy, when indicated, to prevent disease progression during the manufacturing period; 5) lymphodepleting chemotherapy, typically fludarabine plus cyclophosphamide administered over three consecutive days, to promote CAR T-cell expansion; and 6) CAR T-cell infusion (8, 9). Lymphodepletion is critical for in vivo CAR T expansion (10–12), and both the use of bridging therapy and achieving disease control before CAR T-cell infusion have been associated with improved outcomes (8, 12–14).
Currently, seven CAR T-cell products have received FDA approval. Two are directed against B-cell maturation antigen (BCMA) (idecabtagene vicleucel and ciltacabtagene autoleucel), for the treatment of multiple myeloma. Five target CD19: tisagenlecleucel, axicabtagene ciloleucel, brexucabtagene autoleucel, lisocabtagene maraleucel, and obecabtagene autoleucel. Additionally, numerous academic CAR T-cell products are in use in specific countries or centers (e.g., varnimcabtagene autoleucel and cesnicabtagene autoleucel in Spain (15), relmacabtagene autoleucel, equecabtagene autoleucel, and inaticabtagene autoleucel in China (16). CD19-directed CAR T-cell therapies are approved for a variety of B-cell malignancies, including B-cell acute lymphoblastic leukemia (B-ALL: tisa-cel, brexu-cel, obe-cel), diffuse large B-cell lymphoma (DLBCL: tisa-cel, axi-cel, liso-cel), mantle cell lymphoma (MCL: brexu-cel, liso-cel), follicular lymphoma (FL: tisa-cel, axi-cel, liso-cel), and chronic lymphocytic leukemia (CLL: liso-cel only).
CAR T-cell kinetics, clinical outcomes, B-cell aplasia duration, and persistence vary significantly across indications and products (2). In CD19-directed CAR T-cell therapy, B-cell aplasia is strongly correlated with CAR T-cell persistence (17). Generally, CD28-based constructs exhibit rapid expansion and shorter persistence, while 4-1BB-based products display slower expansion but longer in vivo survival. Other factors influencing persistence include T-cell subset composition (e.g., naive vs. memory T-cells), disease type, and the characteristics of the tumor microenvironment (TME) (2). Clinical outcomes vary across diseases. Reported overall response rates (ORR), complete response (CR) rates, median progression-free survival (mPFS), and overall survival (mOS) are summarized in Table 1.
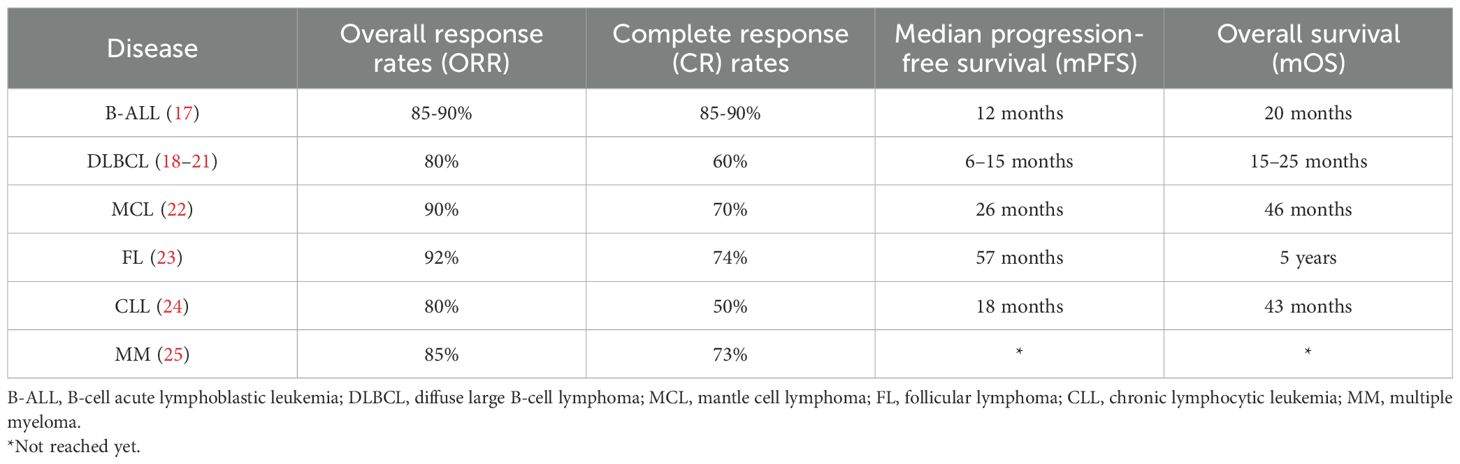
Table 1. CAR T-cell clinical outcomes according to different diseases.
Although CAR T-cells are primary used for the treatment of malignancies, autoimmune diseases and even infectious disorders can also be targeted as well, with promising results emerging for autoimmune indications and several ongoing trials ongoing, mostly involving CD19-directed products (26, 27).
Several factors are associated with long-term responses to CAR T-cell therapy. Although these also vary by disease, lower tumor burden at the time of infusion is consistently associated with deeper responses and prolonged survival (2). Conversely, the presence of dense TME impairs CAR T-cell function and is linked to poorer outcomes (28–31).
3 CAR T-cell-associated toxicities
Cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS) are well-recognized immune-mediated toxicities following CAR T cell infusion. These syndromes share a common initial pathophysiology, involving immune system hyperactivation, driven by the interplay between infused CAR T-cells and host immune cells, leading to a surge in proinflammatory cytokines and, in some cases, an associated coagulopathy. Other immune-related toxicities, such as immune effector cell-associated hematotoxicity (ICAHT), tumor inflammation-associated neurotoxicity (TIAN), and movement and neurocognitive treatment-emergent adverse events (MNTs), have also been described but fall outside the scope of this review (32–34).
3.1 Cytokine release syndrome
CRS is a systemic inflammatory syndrome characterized by elevated levels of circulating inflammatory cytokines, such as interleukin-6 (IL-6), interferon γ (IFN-γ) and IL-1, alongside widespread immune cell activation. CRS can occur in multiple clinical contexts, including infections, autoimmune conditions, malignancies, and other immunotherapies (35). It was first described following CAR T-cell therapy in 2010, in a patient with metastatic colon cancer treated with an ERBB2-targeted CAR T-cell product. After infusion, the patient developed respiratory distress, bilateral pulmonary infiltrates, and multiorgan failure, ultimately dying five days after despite intensive medical intervention. Serum cytokine analysis revealed high levels of IFNγ, granulocyte macrophage-colony stimulating factor (GM-CSF), tumor necrosis factor-α (TNF-α), IL-6, and IL-10 (36).
CAR T-cells become activated upon recognition of tumor-associated antigens, releasing large quantities of perforin, granzymes, and proinflammatory cytokines that induce pyroptosis (inflammatory cell death) in tumor cells (37–39). This process triggers the release of damage-associated molecular patterns (DAMPs), which activate monocytes and macrophages via pattern recognition receptors (PRRs), further amplifying the inflammatory response. Interleukin-1β (IL-1β), generated through inflammasome activation during macrophage pyroptosis, plays a critical role in sustaining and amplifying this cytokine cascade (38, 40–42).
Among the released cytokines, interleukin-6 (IL-6) is central to the systemic manifestations of CRS. IL-6, in combination with its soluble receptor, activates endothelial cells via the GP130 signaling pathway, increasing capillary permeability and driving the development of capillary leak syndrome, hypotension and coagulopathy (40). IL-6 receptor (IL-6R) blockade with tocilizumab -a monoclonal antibody targeting IL-6R- is a cornerstone of CRS management (43).
CRS is the most common early toxicity following CAR-T cell infusion (44). It usually occurs within the first 14 days post-infusion, with a median onset of 2–7 days. Fever is typically the first manifestation, followed by chills, sinus tachycardia, hypotension, hypoxemia, and dyspnea (32). Reported incidence ranges from 42% to 92%, with severe cases occurring in 0-24% depending on the CAR T-cell product (45–48). CRS-related mortality has been linked to respiratory failure, cardiac arrest, or multiorgan dysfunction (6).
Risk factors for CRS include both patient- and product-related variables. Patient-related risks encompass disease type, high tumor burden, active infection, low platelet count, elevated systemic inflammatory markers, and biomarkers of endothelial activation such as angiopoietin-2 and von Willebrand factor. Product-related risks include the use of CD28 costimulatory domains, high CAR T-cell dose or expansion kinetics, and fludarabine-based lymphodepletion regimens (3, 49).
CRS should be graded according to the 2019 American Society for Transplantation and Cellular Therapy (ASTCT) consensus criteria, based on fever, blood pressure, and respiratory status (4).
First-line treatment consists of supportive care and administration of tocilizumab, while corticosteroids are typically reserved for cases that are refractory to initial therapy. In severe or steroid-resistant presentations, additional agents such as anakinra (IL-1 receptor antagonist), siltuximab (anti-IL-6), ruxolitinib (JAK2 inhibitor), emapalumab (anti-INF-γ), antithymocyte globuline, and/or cyclophosphamide may be considered (50–54) (Figure 1).
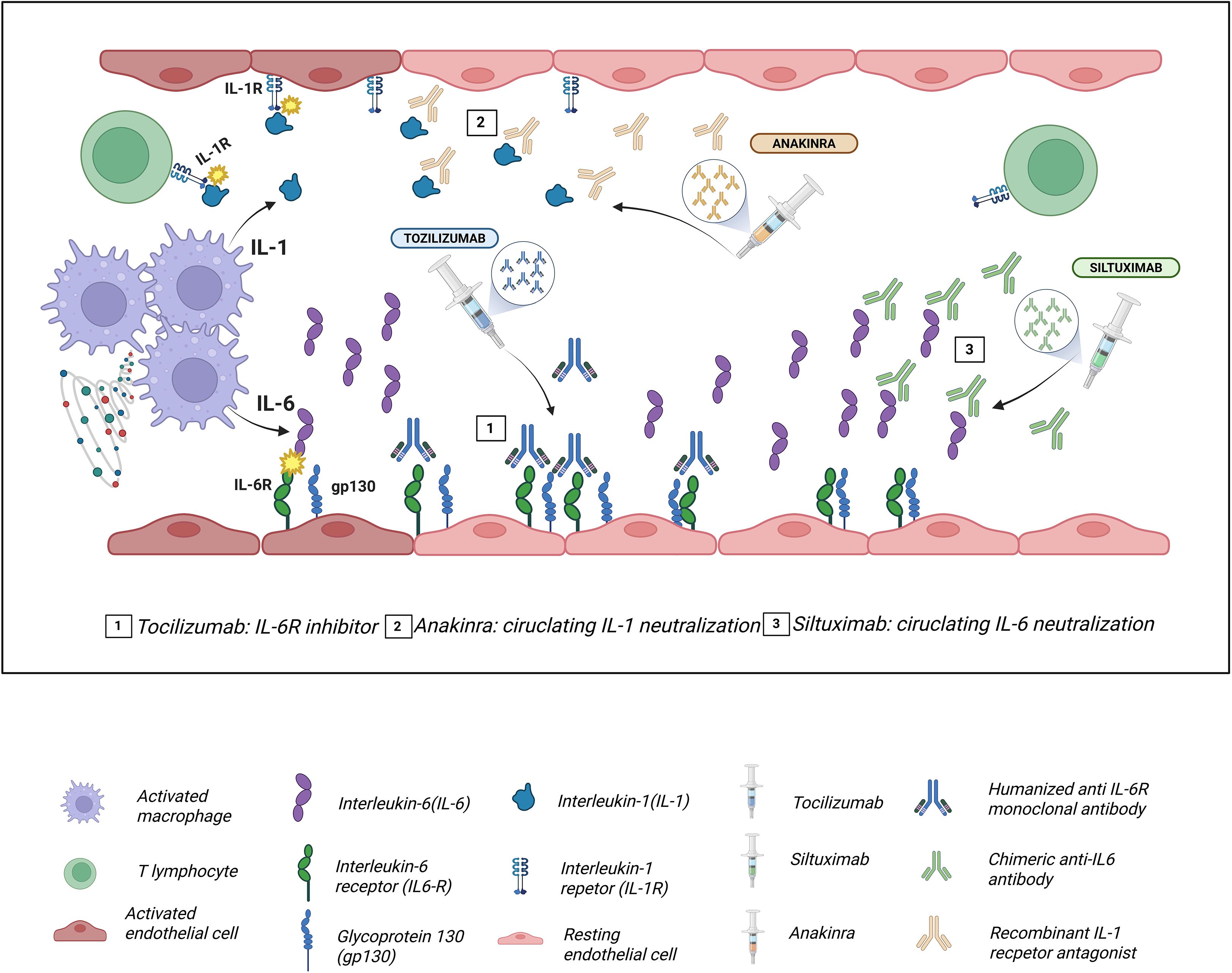
Figure 1. Molecular targets of the main drugs used in the management of cytokine release syndrome (CRS). Tocilizumab, a humanized monoclonal antibody against IL-6R, blocks both classical and trans interleukin-6 (IL-6) signaling, preventing Glycoprotein (gp130)/JAK-STAT3 activation and attenuating endothelial dysfunction, capillary leak, and hypotension characteristic of CRS. Siltuximab binds directly to circulating IL-6, neutralizing it and preventing its interaction with interleukin-6 receptor (IL-6R), thereby reducing systemic inflammatory amplification. Anakinra, a recombinant interleukin-1 receptor antagonist (IL-1R1a), inhibits IL-1α/IL-1β signaling via MyD88 and NF-κB, decreasing macrophage-mediated inflammation and contributing to the control of refractory CRS.
3.2 Immune effector cell-associated neurotoxicity syndrome
ICANS comprises a spectrum of neurological manifestations, typically manifesting as encephalopathy characterized by inattention, disorientation, expressive aphasia, tremor, impaired fine motor skills, and decreased level of consciousness. Additional findings may include focal or generalized weakness, myoclonus, seizures, and, in severe cases, cerebral edema leading to increased intracranial pressure and brain herniation (40, 55).
While systemic inflammation contributes to ICANS, its pathophysiology is largely driven by blood brain barrier (BBB) disruption, increased vascular permeability, and glial cell injury (3). The BBB is a highly selective interface composed of specialized cerebrovascular endothelial cells, pericytes, and astrocytes that maintain central nervous system (CNS) homeostasis (56). Elevated circulating cytokines, more prominently IL-1, IL-6, TNF-α, interact with their specific receptors and activate proinflammatory signaling pathways such as NF-κB, MAPK and JAK-STAT (57, 58). This activation within the vascular endothelium triggers the release of Weibel-Palade bodies containing von Willebrand factor (VWF) and angiopoietin 2 (Ang-2) (59, 60). VWF promotes microthrombus formation and cerebral thrombotic microangiopathy (61), while Ang-2 inhibits the Tie2 signaling pathway, thereby destabilizing the endothelium and increasing BBB permeability, which facilitates cytokine entry into the CNS (62, 63).
Pericytes, which are essential for maintaining microvascular integrity, become stressed by cytokines and secrete IL-6 and VEGF-A (both permeability-inducing mediators), thereby amplifying endothelial injury (64). Moreover, Parker et al. identified CD19 and its chaperone CD81 in human brain pericytes, with sustained CD19 expression from development though adulthood. This variable expression may lead to off-target interactions between anti-CD19 CAR T-cells and pericytes, contributing to an enhanced BBB breakdown. Interestingly, the same study demonstrated that CD19 expression in mouse brain cells is markedly lower than in human brain cells, which may help explain the lower incidence of ICANS and highlights the translational limitations of these models (65).
Monocyte infiltration into the CNS and activation of resident microglia lead to the release of IL-1β, IL-6, and other pro-inflammatory mediators within the brain parenchyma (43). Astrocyte activation upon exposure to inflammatory cytokines further contributes to the development of cerebral edema, and IL-1β-induced VEGF-A disrupts endothelial tight junctions, exacerbating BBB leakiness (56). The accumulation of leukocytes and cytokines in the CNS promotes neuroinflammation, and in situ activation of microglia and macrophages induces the release of neurotoxic metabolites, such as glutamate and quinolinic acid, leading to neuronal excitotoxicity via NMDA receptor stimulation (40, 66).
ICANS incidence varies between 0-63%, with severe cases in up to 31% of patients (47, 48, 67–69), widely depending on the CAR T-cell product and disease context. Risk factors mirror those for CRS and include early-onset and severe CRS (70).
ICANS usually develops within the first 10 days after infusion, most often after CRS, though concurrent or independent onset has been reported (4, 58), as well as delayed presentations (71).
ICANS grading follows the ASTCT consensus, which includes four domains: the 10-point immune effector cell-associated encephalopathy (ICE) score (which should be routinely monitored after infusion), level of consciousness, presence and type of seizures, motor deficits, and signs of cerebral edema (4).
First-line treatment consists of supportive care and corticosteroids. In refractory cases, anakinra, siltuximab, and intrathecal or systemic chemotherapy may be considered (50–52, 54, 72, 73).
3.3 Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome
IEC-HS is a rare, life-threatening hyperinflammatory syndrome, phenotypically resembling secondary hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome, and is directly related to immune effector cell therapy. It is characterized by uncontrolled immune activation and multi-organ failure.
Pathophysiologically, IEC-HS results from sustained CAR T-cell and CD8+ T-cell activity, compounded by profound and persistent NK cell lymphopenia, which impairs immune regulation (74). This leads to high systemic levels of IL-1β, IFN-γ, and IL-18, in addition to IL-6, perpetuating pathological immune activation, including massive macrophage stimulation reflected in extreme hyperferritinemia, cytopenias, and multi-organ dysfunction.
CAR T-cell expansion in this setting may occur independently of antigen presence and it is associated with uncontrolled T cell proliferation. The distinct cytokine profile, combined with persistent NK lymphopenia, suggests IEC-HS represents a late-stage variant of CRS with a different immunopathogenesis. requiring therapeutic strategies beyond IL-6 blockade (74, 75).
Clinically, IEC-HS shares several features with CRS, such as fever and multiorgan dysfunction. However, it is particularly characterized by progressive or new-onset cytopenias, marked hyperferritinemia, coagulopathy (including hypofibrinogenemia) and/or elevated liver enzymes (74). IEC-HS often emerges during or after the resolution of CRS and should not be misclassified as severe CRS alone. The absence of standardized diagnostic criteria prior to recent consensus definitions likely contributed to its underrecognition (5). Efforts have been made to identify biomarkers capable of distinguishing IEC-HS from CRS (76); however, as these markers fall outside the scope of this review and are not primarily related to endothelial injury, they are not discussed in detail herein.
The syndrome also shares risk factors with CRS, including high tumor burden and elevated CAR T-cell doses, and is more frequently reported with CD22- or BCMA-targeted products than with CD19-directed constructs (74, 77).
Treatment includes corticosteroids and anakinra as first-line therapy. In refractory cases, escalation may include the addition of agents such as ruxolitinib, emapalumab, or low-dose etoposide (5).
3.4 Coagulopathy
Coagulopathy may develop in the setting of CRS, ICANS, or IEC-HS, as a result of widespread immune activation and endothelial injury. This culminates in a prothrombotic state characterized by Weibel–Palade body exocytosis, release of VWF and factor VIII, and exposure of tissue factor, initiating the coagulation cascade (78, 79). Enhanced thrombin generation leads to consumption of fibrinogen, clotting factors, and platelets, while secondary fibrinolysis elevates D‐dimer levels (63). Simultaneously, depletion of natural anticoagulants such as antithrombin and proteins C and S disrupts regulatory mechanisms, perpetuating microthrombosis and endothelial damage, exacerbating capillary leak syndrome and contributing to organ dysfunction (80, 81). In severe cases, the coagulopathy may fulfill diagnostic criteria for disseminated intravascular coagulation (DIC) (82).
Although endothelial dysfunction and coagulopathy are closely linked, overt consumption coagulopathy with clinical bleeding or thrombosis usually occurs only in patients with severe CRS or ICANS (56, 83). The highest risk of clinical coagulopathy is within the first month after infusion (84) and, while not frequent, its mortality rates are concerning (85).
4 The endothelium in CAR T-cell toxicities
Endothelial dysfunction has been identified as a cornerstone in the development and perpetuation of CAR T-cell-associated toxicities.
CAR T-cell recipients often show signs of endothelial dysfunction even prior to infusion. Most patients have undergone multiple lines of therapy, including cytotoxic agents known to cause endothelial injury. Additionally, a substantial proportion may have received prior allogeneic or autologous stem cell transplantation (SCT), which has been independently associated with endothelial damage (86, 87).
Just before CAR T-cell infusion, lymphodepleting chemotherapy, typically a combination of fludarabine and cyclophosphamide, has also been associated to further endothelial damage (87–89). Finally, the cytokine storm that ensues after infusion exerts a direct and severe insult on the vascular endothelium (90). As described previously in the pathophysiology of CRS, ICANS, and IEC-HS, endothelial activation and damage play a central role in mediating clinical toxicity. The interconnected pathways linking endothelial dysfunction with CRS and ICANS are illustrated in Figure 2.
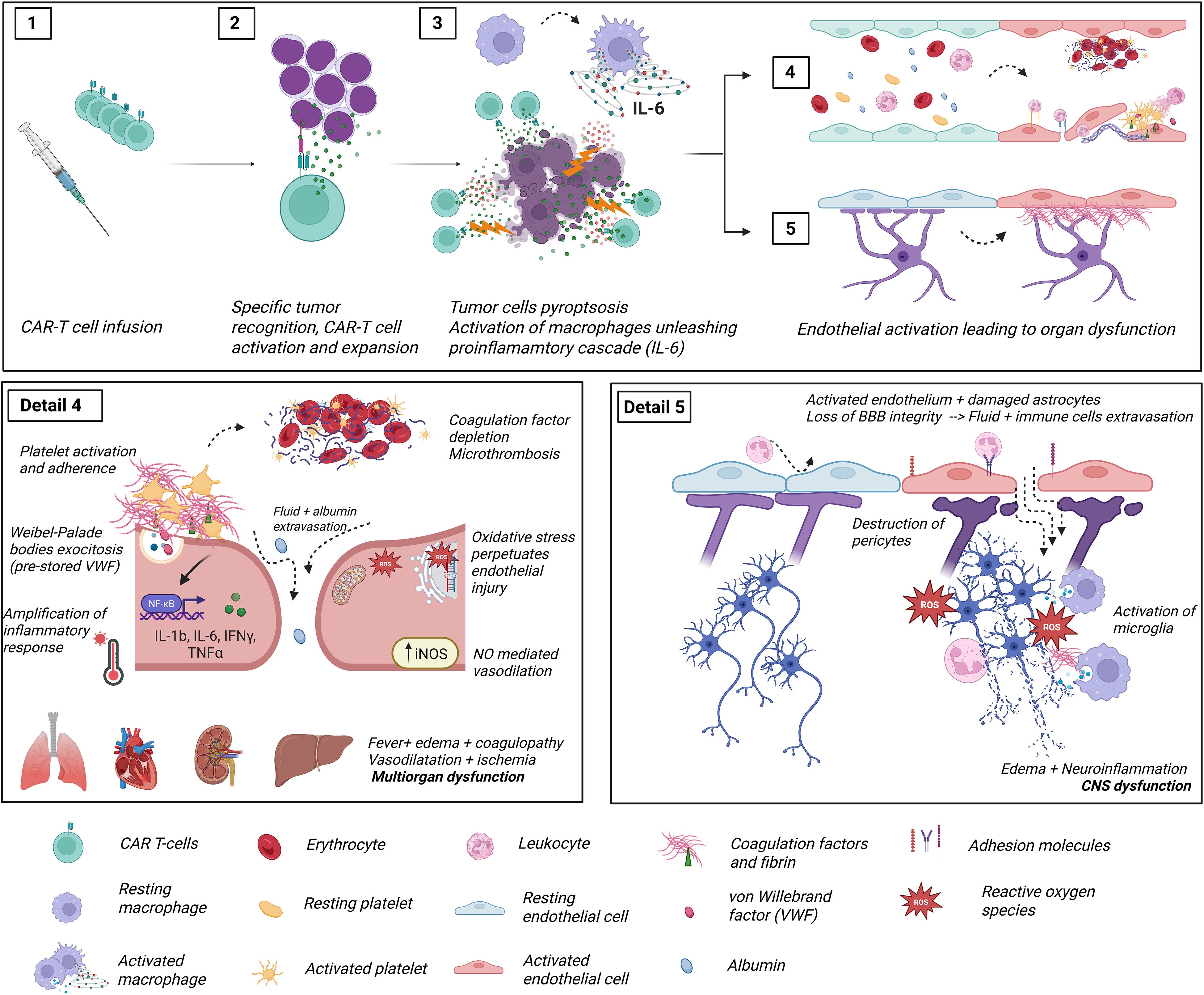
Figure 2. Diagram representing the pathophysiology of CAR-T cell treatment toxicities and its relationship with endothelial cells. Detail 1: CAR T-cells are infused into the patient. Detail 2: CAR T-cells recognize tumor antigens through their chimeric receptor, leading to activation and clonal expansion of the CAR T-cell population. Detail 3: Activated CAR T-cells release induce pyroptosis (inflammatory cell death) of tumor cells, triggering the release of high levels of damage-associated molecular patterns (DAMPs), which are recognized by pattern recognition receptors (PRRs) on host macrophages and monocytes to secrete additional cytokines (IL-6, TNFα…). These pro-inflammatory cytokines have an activating effect on ubiquitous endothelial layers. Detail 4: Longitudinal section of a blood vessel showing the normal disposition of an endothelial layer (blue cells). The red cells depict activated endothelial cells and their consequences (Panel 4 detail): endothelial cells lose their tight junctions, and fluid extravasation occurs. Additionally, dysfunctional endothelial cells switch their secretory phenotype of anticoagulant and fibrinolytic factors towards a hypercoagulative and hypofibrinolytic state due to increased release of PAI-1 and preformed TF and VWF. Intravascular microthrombi and liberation of neutrophil extracellular traps result in turbulent blood flow and agglutination of circulating cells, ultimately leading to tissue hypoperfusion. In addition, detached endothelial cells pass to circulation as Circulating Endothelial Cells (CECs), exposing the extracellular matrix, highly reactive to circulating platelets. The activation of iNOS increases NO availability, with both a vasodilatory effect in smooth muscle and oxidative cell damage. Ultimately, dysfunctional endothelial cells further amplify the cytokine release syndrome via upregulation of nuclear factor kappa B, which is linked to the inflammatory and oxidative stress responses. Detail 5: Longitudinal section of blood-brain barrier (BBB) showing analogous activation of endothelial cells responsible for ICANS. The pericytes, a mainstay of the BBB regulation, become inured following endothelial activation, leading to fluid and cell leakage into the cerebral parenchyma. This penetration of the central nervous system is followed by activation of microglia and neuroinflammation, responsible for the typical clinical picture of ICANS. This figure was created with BioRender.com (Diaz-Ricart, M. (2025) https://BioRender.com). CECs: circulant endothelial cells; CRS, Cytokine release syndrome; ICANS, Immune effector cell associated neurotoxicity syndrome; IFNγ, interferon gamma; IL, interleukin; iNOS, inducible nitric oxide synthase; NO, nitric oxide; NETs, neutrophil extracellular traps; PAI-1, plasminogen activator inhibitor 1; ROS, reactive oxygen species; TF, tissue factor; TNFα, tumor necrosis factor α; VWF, Von Willebrand factor.
Endothelial cells exposed to inflammatory stimuli undergo a phenotypic shift. They upregulate adhesion molecules for leukocytes and platelets, such as vascular cell adhesion molecule-1 (VCAM-1) and VWF, respectively, while also increasing the expression of pro-angiogenic mediators, such as vascular endothelial growth factor (VEGF) and angiopoetin-2 (Ang-2). Simultaneously, the intercellular junction protein VE-Cadherin is downregulated, and the synthesis of key vasodilators and antiplatelet agents, such as prostacyclin and nitric oxide, is impaired. These molecular changes result in an endothelium that is procoagulant, proinflammatory, hyperpermeable, vasoplegic, and prone to excessive proliferation (91, 92).
The clinical manifestations of endothelial dysfunction vary depending on the severity of the insult and the anatomical site of endothelial injury (87).
There are mechanistic similarities between the endothelial damage observed in CAR T-related toxicities and that seen after allo-SCT, including the release of common inflammatory cytokines (e.g., IL-1, IL-6, TNFα), followed by enhanced leukocyte adhesion and migration due to increased expression of endothelial adhesion molecules (93).
5 Circulating biomarkers and surrogate indices of endothelial dysfunction, hemostatic imbalance, and innate immune activation in CAR T-cell toxicities
Multiple biomarkers reflecting endothelial dysfunction and associated pathways, including coagulation and complement activation, have been studied to elucidate their role in the pathogenesis of CAR T-cell toxicities and to assess their potential predictive value for toxicity severity and treatment outcomes. In this section, we review key laboratory parameters and classify them into the following categories: (1) biomarkers of endothelial dysfunction, (2) surrogate indices of endothelial stress, (3) biomarkers of coagulopathy (basic coagulation parameters and activators, fibrinolysis parameters and fibrin degradation products, and primary hemostasis parameters), and (4) markers of innate immunity activation. This framework aims to facilitate understanding of the molecular networks that contribute to CAR T-cell–related complications and their potential clinical utility.
5.1 Biomarkers of endothelial dysfunction
5.1.1 Angiopoietin-Tie2 axis and angiogenic factors
The angiopoietin axis, comprising angiopoietin-1 (Ang-1), angiopoietin-2 (Ang-2), and their ratio (Ang-2:Ang-1) has been implicated in the development of CAR T-cell toxicities. Both ligands bind to the Tie2 receptor tyrosine kinase on endothelial cells, with opposing effects: Ang-1 promotes endothelial stability, while Ang-2, released in response to endothelial injury, destabilizes cell-cell junctions, enhances vascular permeability, and induces proliferative signaling (94). Elevated Ang-2 levels have been associated with severe CRS and were already detectable at the time of lymphodepletion in patients who subsequently developed severe toxicity (49). Increased post-infusion Ang-2 levels also predicted the need for ICU admission (95), and the Ang-2:Ang-1 ratio correlated with organ dysfunction in severe CRS cases (96). Growth differentiation factor-15 (GDF-15) is a stress-responsive cytokine produced by cardiomyocytes, endothelial cells, and other cell types in response to metabolic or oxidative stress, inflammation or cellular senescence (97). GDF-15 has been found elevated in CAR T-cell recipients compared to healthy controls and in this setting is considered a marker of endothelial injury (98).
5.1.2 TNF receptor I
TNFRI, a receptor for TNF-α, is upregulated in endothelial cells in response to inflammation. Its engagement leads to NF-κB and AP-1 (two families of transcription factors) activation, resulting in increased expression of leukocyte adhesion molecules (99). Although TNFRI is not endothelial- specific, elevated circulating levels have been observed at the onset of CAR T-cell toxicities compared to post-infusion values (95).
5.1.3 Adhesion molecules and endothelial junction proteins
Activated endothelial cells upregulate proteins involved in leukocyte adhesion and transmigration, including VCAM-1 and intercellular adhesion molecule-1 (ICAM-1). Soluble forms of these molecules have been found elevated in patients with severe CRS (96) and coagulopathy (81, 100). In contrast, platelet endothelial cell adhesion molecule (PECAM-1), which maintains intercellular junction integrity, is typically downregulated during endothelial stress, contributing to increased vascular permeability.
5.1.4 Von Willebrand Factor
VWF is a multimeric protein with multiple functions, most prominently mediating platelet adhesion to the exposed subendothelium, promoting platelet-platelet interactions, and stabilizing coagulation factor VIII. It is released from Weibel-Palade bodies in response to endothelial injury (101). Several studies have demonstrated that VWF levels increase during CRS and correlate with its severity (80, 96). Notably, elevated VWF levels at the time of lymphodepletion have been associated with subsequent CRS development (49). More recently, VWF has been proposed as part of a biomarker panel to differentiate CRS from sepsis, another common and serious early complication following CAR T-cell infusion (95).
5.1.5 Soluble Suppression of tumorgenicity 2 protein
ST2 is a member of the interleukin-1 (IL-1) transmembrane receptor superfamily and is expressed by various cell types, including endothelial cells (102). Its soluble form (sST2) acts as a decoy receptor for IL-33, impairing its anti-inflammatory and antifibrotic signaling. sST2 is a recognized marker of cardiovascular stress and inflammation (103). In CAR T-cell patients, sST2 levels increased significantly at the onset of toxicities and showed predictive potential for treatment-related complications (95).
5.2 Surrogate indices of endothelial dysfunction: EASIX and related scores
Direct measurement of endothelial biomarkers is not routinely available in clinical practice, as these assays may be time-consuming or require specialized platforms. Therefore, surrogate indices derived from standard laboratory parameters have been proposed to approximate endothelial dysfunction in the context of CAR T- cell therapy.
5.2.1 EASIX
The Endothelial Activation Stress Index (EASIX) is calculated using the formula: (LDH [U/L] x Creatinine [mg/dL])/Platelet count [109/L]. Although composed of nonspecific variables that may be altered by multiple causes in complex hematologic patients, EASIX has emerged as a valuable surrogate marker of endotheliopathy. Originally developed in the setting of allo-HSCT, EASIX has consistently shown predictive value for complications and poor outcomes (104–106). More recently, its application has been extended to CAR T-cell therapy. Several studies have demonstrated that elevated pre-infusion EASIX scores correlate with the development and severity of CAR T-cell-related toxicities (107–109).
5.2.2 Modified and simplified EASIX (m-EASIX, s-EASIX)
In an effort to improve performance or simplify calculation, various adaptations of the EASIX formula have been evaluated. The modified EASIX (m-EASIX) replaces creatinine with C-reactive protein. It has shown good predictive accuracy for severe CRS, ICANS (110–112) and consumptive coagulopathy (80). The simplified EASIX (s-EASIX) omits creatinine from the formula. Despite its simplicity, it has demonstrated utility in predicting severe CRS (94, 107, 113) and ICANS (114), although its validation has often been limited to specific patient subsets and CAR T-cell constructs (107, 115).
5.2.3 EASIX-based cytokine augmented models
Building upon the original index, some authors have incorporated inflammatory biomarkers, such as IL-6, IL-10, or ferritin, into EASIX-based models. These scores considering cytokine levels have also demonstrated predictive value for early identification of patients at risk for severe CRS, ICANS, and coagulopathy (116–118).
5.3 Biomarkers of coagulopathy
These biomarkers can be grouped into three main categories: basic coagulation parameters and activators, fibrinolysis-related markers and fibrin degradation products, and markers of primary hemostasis.
5.3.1 Basic coagulation parameters and activators
Prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT) have been observed in patients with CRS, correlating with its severity (80, 83, 100). These alterations reflect the consumption of coagulation factors during systemic inflammation. Additionally, fibrinogen levels may decrease significantly in patients with severe CRS (83, 84) or IEC-HS (119, 120), where hypofibrinogenemia constitutes a diagnostic criterion (121). Of note, low fibrinogen concentrations have also been reported following tocilizumab administration (122).
Other studies have demonstrated that patients experiencing severe CRS or ICANS exhibit decreased levels of antithrombin (80), elevated tissular factor expression (81), and increased thrombin generation, as reflected by higher plasma levels of prothrombin fragment 1 + 2 (100). These findings confirm the presence of dysregulated coagulation and its mechanistic link to endotheliopathy in promoting a prothrombotic state.
5.3.2 Fibrinolysis markers and fibrin degradation products
Activation of the coagulation cascade triggers a compensatory response by the fibrinolytic system, where plasmin degrades fibrin clots. The balance of fibrinolytic activity is critical in inflammatory conditions such as CRS and IEC-HS.
The soluble urokinase-type plasminogen activator receptor (suPAR) is the soluble form of uPAR, a glycoprotein expressed in the membrane of immune cells. It is a marker of immune activation and fibrinolysis, generated upon the binding of urokinase plasminogen activator (uPA) to its membrane receptor (uPAR), leading to suPAR release into plasma. Active uPA cleaves plasminogen into plasmin but also activates the complement system (123). Although suPAR levels may vary across clinical settings (124, 125), in CAR T-cell recipients they have been shown to be elevated compared to controls, and to correlate with m-EASIX scores pre-infusion, and EASIX scores at two weeks post-infusion (98).
Plasminogen activator inhibitor-1 (PAI-1) is a serine protease inhibitor produced by endothelial cells and platelets. By inhibiting uPA and tissular plasminogen activator (t-PA), it exerts a strong antifibrinolytic effect. Elevated PAI-1 levels have been proposed as a potential biomarker to distinguish CRS from sepsis, being significantly higher in the latter (95).
As a result of increased fibrinolysis, fibrin degradation products (FDPs) such as D-dimer are commonly elevated in high-grade CRS (80, 83, 84, 100), reflecting the downstream effects of uncontrolled thrombin and fibrin generation.
5.3.3 Primary hemostasis markers
The relationship between endothelial damage and activation of primary hemostasis is particularly evident following the enhanced release of VWF into the plasma and extracellular space. This promotes platelet adhesion and aggregation, ultimately leading to platelet consumption. Thrombocytopenia has been observed in patients with severe CRS, particularly in those with coexisting laboratory evidence of coagulopathy (80).
ADAMTS-13 (A Disintegrin and Metalloproteinase with thrombospondin type 1 motif, member 13) is an enzyme that cleaves ultra-large VWF multimers and regulates platelet adhesion. Lower levels of ADAMTS-13 have been observed in patients with severe ICANS compared to those with milder forms (56), as well as at post-infusion time compared to baseline values in CAR T- cell recipients. These findings suggest a role for microangiopathic features in the pathogenesis of neurotoxicity and other thrombotic complications.
5.4 Biomarkers of innate immune activation
CAR T-cell-associated toxicities involve not only endothelial dysfunction and coagulation disturbances, but also profound activation of the innate immune system. Inflammatory cytokines, complement activation and neutrophil-derived mediators all contribute to the immunopathogenesis of toxicities such as CRS, ICANS, and IEC-HS.
5.4.1 Complement activation
The alternative complement pathway can be initiated by cytokine-induced neutrophil activation and subsequent C3b release (126), as well as by coagulation-derived proteases such as thrombin and plasmin, which can cleave complement components to generate the anaphylatoxins C3a and C5a. Terminal complement activation results in the formation of the membrane attack complex (MAC, C5b9). Its soluble form, sC5b9, has been shown to be elevated in CAR T-cell patients compared to healthy controls and correlates with both EASIX and m-EASIX scores at different time points (98). Moreover, biomarker panels including sC5b9 have demonstrated strong performance in predicting severe CAR T-cell-related toxicities and in differentiating CRS from sepsis (95). Recently, increased sC5b9 have also been associated with grade ≥ 2 ICANS and higher m-EASIX scores (127).
5.4.2 Neutrophil extracellular traps
NETs are web-like structures composed of DNA, histones, and granule proteins released by neutrophils in response to inflammatory stimuli. Originally described as part of the antimicrobial response, NETs also play a pathological role in cancer and immunotherapy-related syndromes. In the tumor microenvironment, NETs promote immune suppression and tumor progression. In the context of CAR T-cell therapy, they may physically impede CAR-T function (128, 129).
NETs have been observed to increase after CAR T-cell infusion and have emerged as promising biomarkers for severe toxicity prediction. They have also shown utility in distinguishing CRS from sepsis, particularly when included as part of multiparameter biomarker panels (95).
6 Potential clinical applications of biomarkers
The potential clinical applications of the biomarkers discussed above are summarized in Table 2. Biomarkers have been categorized according to their underlying biological process (endothelial dysfunction, hemostatic imbalance, and innate immune activation including complement pathways), and their proposed clinical utility. These include prediction of mortality, prediction of toxicity onset and severity, and differential diagnosis, particularly to distinguish CAR T-toxicities from clinically overlapping syndromes such as sepsis).

Table 2. Circulating biomarkers and surrogate scores of endothelial dysfunction, hemostasis misbalance, and other linked pathways, and their potential clinical uses in each moment during CAR T-cell immunotherapy.
Moreover, the utility of these biomarkers has been mapped across distinct time points in the CAR T-cell therapy timeline: pre-lymphodepletion, between lymphodepletion and CAR T-cell infusion, early post-infusion period, and toxicity onset.
7 Future perspectives and conclusions
As observed in complications that appear early after other forms of cellular therapy (7), the endotheliopathy and its related pathways are critical in the pathophysiology of CAR T-cell-associated toxicities. The key points of the present review are as follows:
● There is compelling evidence supporting the utility of circulating biomarkers for diagnostic confirmation, early prediction, severity stratification, and even non-relapse mortality prognosis.
● Most of the biomarkers reviewed remain confined to research settings. Their translation into clinical practice will require rigorous analytical validation, inter-laboratory standardization, and the development of evidence-based clinical guidelines delineating their diagnostic and therapeutic relevance.
● Growing recognition of the endothelium as a therapeutic target has prompted the evaluation of pharmacological agents with endothelial-protective properties for the management of CAR T-cell toxicities (133).
● To date, most interventional strategies involving endothelial modulation have focused on enhancing vascular permeability to facilitate CAR T-cell infiltration into solid tumors (134–136).
● The therapeutic strategies, aimed at stabilizing the endothelium, may offer a promising avenue to mitigate immune-mediated toxicity. These strategies may involve modulation of: proinflammatory cytokines, reactive oxygen species (ROS), adhesion molecules, and complement components (137).
● None of the mentioned interventions has reached advanced clinical development. However, several are currently under evaluation in investigator-initiated trials (138, 139).
o Defibrotide, despite exhibiting a favorable safety profile, showed limited efficacy in preventing ICANS in patients treated with axicabtagene ciloleucel (139).
o Statins, when initiated before immunotherapy and maintained during treatment, and combined with intrathecal dexamethasone, were associated with a reduction in the incidence and severity of ICANS (140).
o Complement inhibitors have shown potential for the treatment of immunotherapy-related thrombotic microangiopathy (141).
o In vitro, the combined blockade of TNFα and IL1β using adalimumab and an anti-IL-1β antibody, respectively, demonstrated a synergistic effect in preventing endothelial activation, warranting further clinical investigation (138).
o Fourth-generation CAR T-cell constructs may carry an increased risk of immune-related toxicities. However, certain designs aim to reduce these effects by incorporating negative-feedback loops: a tocilizumab-secreting CAR T-cell design was shown to lower toxicity while unexpectedly increasing efficacy (142). Another promising approach involves spatially confined CAR T-cell activation and cytokine release, which could be advantageous in selected tumor types or anatomical locations. In a recent study, anti-CD19 CAR T-cells engineered to secrete IL-12 specifically within hypoxic environments achieved high efficacy rates and no detectable toxicity in DLBCL mouse models (143). To date, however, none of these strategies have been clinically validated as safer alternatives to second-generation CAR T-cells.
● The incorporation of biomarkers into clinical decision-making, including patient selection, preemptive toxicity management, and monitoring of endothelial-targeted therapies, remains an unmet need and a promising area of future research and personalized immunotherapy.
● In parallel, integrating multi-omic approaches with artificial intelligence–based predictive models holds great potential to refine risk stratification and develop more robust, individualized predictors of CAR T-cell related toxicities (144).
Author contributions
AM-C: Conceptualization, Project administration, Writing – original draft.. GI: Writing – original draft. NM-C: Writing – original draft. NA: Writing – original draft. DP: Writing – original draft. JM-S: Writing – original draft. PC: Writing – review & editing. MD-R: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was partially supported by ‘Instituto de Salud Carlos III’ (PI22/00684, PI22/00240 and PI22/00367) from the Spanish Ministry of Health and the ‘Departament de Recerca i Universitats de la Generalitat de Catalunya’ (2021-SGR-01118).
Acknowledgments
We would like to thank the laboratory technicians Marc Pino, Patricia Molina, Lidia Martín, Estefanía García, Pilar Gómez, Alejandro Román and Mireia Garre for their technical support at different stages of the research.
Conflict of interest
MD-R has received honoraria from Jazz Pharmaceuticals and Sanofi for lectures, and from Novartis, Zacros and Sysmex to the institution. PC has received honoraria from Gilead, Pfizer and Alexion.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, and He B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
2. Cappell KM and Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
3. Morris EC, Neelapu SS, Giavridis T, and Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
4. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood marrow Transplant. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
5. Hines MR, Knight TE, McNerney KO, Leick MB, Jain T, Ahmed S, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome. Transplant Cell Ther. (2023) 29:438.e1–.e16. doi: 10.1016/j.jtct.2023.03.006
6. Cordas Dos Santos DM, Tix T, Shouval R, Gafter-Gvili A, Alberge JB, Cliff ERS, et al. A systematic review and meta-analysis of nonrelapse mortality after CAR T cell therapy. Nat Med. (2024) 30:2667–78. doi: 10.1038/s41591-024-03084-6
7. Moreno-Castaño AB, Salas MQ, Palomo M, Martinez-Sanchez J, Rovira M, Fernández-Avilés F, et al. Early vascular endothelial complications after hematopoietic cell transplantation: Role of the endotheliopathy in biomarkers and target therapies development. Front Immunol. (2022) 13:1050994. doi: 10.3389/fimmu.2022.1050994
8. Amini L, Silbert SK, Maude SL, Nastoupil LJ, Ramos CA, Brentjens RJ, et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat Rev Clin Oncol. (2022) 19:342–55. doi: 10.1038/s41571-022-00607-3
9. Kröger N, Gribben J, Chabannon C, Yakoub-Agha I, and Einsele H. The EBMT/EHA CAR-T cell handbook. Kröger N, Gribben J, Chabannon C, Yakoub-Agha I, and Einsele H, editors. Cham (CH: Springer (2022).
10. Shah NN, Lee DW, Yates B, Yuan CM, Shalabi H, Martin S, et al. Long-term follow-up of CD19-CAR T-cell therapy in children and young adults with B-ALL. J Clin Oncol. (2021) 39:1650–9. doi: 10.1200/JCO.20.02262
11. Gauthier J, Bezerra ED, Hirayama AV, Fiorenza S, Sheih A, Chou CK, et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell Malignancies. Blood. (2021) 137:323–35. doi: 10.1182/blood.2020006770
12. Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood. (2019) 133:1876–87. doi: 10.1182/blood-2018-11-887067
13. Bhaskar ST, Dholaria BR, Sengsayadeth SM, Savani BN, and Oluwole OO. Role of bridging therapy during chimeric antigen receptor T cell therapy. EJHaem. (2022) 3:39–45. doi: 10.1002/jha2.335
14. Roddie C, Neill L, Osborne W, Iyengar S, Tholouli E, Irvine D, et al. Effective bridging therapy can improve CD19 CAR-T outcomes while maintaining safety in patients with large B-cell lymphoma. Blood advances. (2023) 7:2872–83. doi: 10.1182/bloodadvances.2022009019
15. Ortíz-Maldonado V, Rives S, Castellà M, Alonso-Saladrigues A, Benítez-Ribas D, Caballero-Baños M, et al. CART19-BE-01: A multicenter trial of ARI-0001 cell therapy in patients with CD19(+) relapsed/refractory Malignancies. Mol Ther. (2021) 29(2):636–44. doi: 10.1016/j.ymthe.2020.09.027
16. Wang Y, Lv L, Song Y, Wei X, Zhou H, Liu Q, et al. Inaticabtagene autoleucel in adult relapsed or refractory B-cell acute lymphoblastic leukemia. Blood advances. (2025) 9:836–43. doi: 10.1182/bloodadvances.2024014182
17. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
18. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
19. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
20. Neelapu SS, Jacobson CA, Ghobadi A, Miklos DB, Lekakis LJ, Oluwole OO, et al. Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. (2023) 141(19):2307–15. doi: 10.1182/blood.2022018893
21. Westin JR, Oluwole OO, Kersten MJ, Miklos DB, Perales MA, Ghobadi A, et al. Survival with axicabtagene ciloleucel in large B-cell lymphoma. New Engl J Med. (2023) 389:148–57. doi: 10.1056/NEJMoa2301665
22. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. Three-year follow-up of KTE-X19 in patients with relapsed/refractory mantle cell lymphoma, including high-risk subgroups, in the ZUMA-2 study. J Clin Oncol. (2023) 41:555–67. doi: 10.1200/JCO.21.02370
23. Neelapu SS, Chavez JC, Sehgal AR, Epperla N, Ulrickson M, Bachy E, et al. Three-year follow-up analysis of axicabtagene ciloleucel in relapsed/refractory indolent non-Hodgkin lymphoma (ZUMA-5). Blood. (2024) 143:496–506. doi: 10.1182/blood.2023021243
24. Siddiqi T, Maloney DG, Kenderian SS, Brander DM, Dorritie K, Soumerai J, et al. Lisocabtagene maraleucel in chronic lymphocytic leukaemia and small lymphocytic lymphoma (TRANSCEND CLL 004): a multicentre, open-label, single-arm, phase 1–2 study. Lancet (London England). (2023) 402:641–54. doi: 10.1016/S0140-6736(23)01052-8
25. San-Miguel J, Dhakal B, Yong K, Spencer A, Anguille S, Mateos MV, et al. Cilta-cel or standard care in lenalidomide-refractory multiple myeloma. New Engl J Med. (2023) 389:335–47. doi: 10.1056/NEJMoa2303379
26. Morte-Romea E, Pesini C, Pellejero-Sagastizábal G, Letona-Giménez S, Martínez-Lostao L, Aranda SL, et al. CAR Immunotherapy for the treatment of infectious diseases: a systematic review. Front Immunol. (2024) 15:1289303. doi: 10.3389/fimmu.2024.1289303
27. Müller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Völkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - A case series with follow-up. New Engl J Med. (2024) 390(8):687–700. doi: 10.1056/NEJMoa2308917
28. Holland EM, Yates B, Ling A, Yuan CM, Wang HW, Stetler-Stevenson M, et al. Characterization of extramedullary disease in B-ALL and response to CAR T-cell therapy. Blood advances. (2022) 6:2167–82. doi: 10.1182/bloodadvances.2021006035
29. Ortiz-Maldonado V, Alonso-Saladrigues A, Español-Rego M, Martínez-Cibrián N, Faura A, Magnano L, et al. Results of ARI-0001 CART19 cell therapy in patients with relapsed/refractory CD19-positive acute lymphoblastic leukemia with isolated extramedullary disease. Am J Hematol. (2022) 97(6):731–9. doi: 10.1002/ajh.26519
30. Zugasti I, Tormo-Ratera M, Oliver-Caldés A, Soler-Perromat JC, González-Calle V, Moreno DF, et al. Clinical impact of [18F]FDG-PET/CT in ARI0002h treatment, a CAR-T against BCMA for relapsed/refractory multiple myeloma. Blood Adv. (2025) 9:571–82. doi: 10.1182/bloodadvances.2024014360
31. Dima D, Abdallah AO, Davis JA, Awada H, Goel U, Rashid A, et al. Impact of extraosseous extramedullary disease on outcomes of patients with relapsed-refractory multiple myeloma receiving standard-of-care chimeric antigen receptor T-cell therapy. Blood Cancer J. (2024) 14:90. doi: 10.1038/s41408-024-01068-w
32. Brudno JN and Kochenderfer JN. Current understanding and management of CAR T cell-associated toxicities. Nat Rev Clin Oncol. (2024) 21:501–21. doi: 10.1038/s41571-024-00903-0
33. Karschnia P and Dietrich J. Neurological complications of CAR T cell therapy for cancers. Nat Rev Neurol. (2025) 21(8):422–31. doi: 10.1038/s41582-025-01112-8
34. Strongyli E, Evangelidis P, Sakellari I, Gavriilaki M, and Gavriilaki E. Change in neurocognitive function in patients who receive CAR-T cell therapies: A steep hill to climb. Pharm (Basel Switzerland). (2024) 17(5):591. doi: 10.3390/ph17050591
35. Fajgenbaum DC and June CH. Cytokine storm. New Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
36. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
37. Zhang Y, Qin D, Shou AC, Liu Y, Wang Y, and Zhou L. Exploring CAR-T cell therapy side effects: mechanisms and management strategies. J Clin Med. (2023) 12(19):6124. doi: 10.3390/jcm12196124
38. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
39. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Sci (New York NY). (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
40. Xiao X, Huang S, Chen S, Wang Y, Sun Q, Xu X, et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J Exp Clin Cancer research: CR. (2021) 40:367. doi: 10.1186/s13046-021-02148-6
41. Gong T, Liu L, Jiang W, and Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
42. Liu D, Xu X, Dai Y, Zhao X, Bao S, Ma W, et al. Blockade of AIM2 inflammasome or α1-AR ameliorates IL-1β release and macrophage-mediated immunosuppression induced by CAR-T treatment. J Immunother Cancer. (2021) 9(1):e001466. doi: 10.1136/jitc-2020-001466
43. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
44. Frey N and Porter D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biol Blood marrow Transplant. (2019) 25:e123–e7. doi: 10.1016/j.bbmt.2018.12.756
45. Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. New Engl J Med. (2022) 386:640–54. doi: 10.1056/NEJMoa2116133
46. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet (London England). (2021) 398:491–502. doi: 10.1016/S0140-6736(21)01222-8
47. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. New Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
48. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. (2019) 20:31–42. doi: 10.1016/S1470-2045(18)30864-7
49. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. (2017) 130:2295–306. doi: 10.1182/blood-2017-06-793141
50. Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol. (2022) 33:259–75. doi: 10.1016/j.annonc.2021.12.003
51. Gazeau N, Liang EC, Wu QV, Voutsinas JM, Barba P, Iacoboni G, et al. Anakinra for refractory cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome after chimeric antigen receptor T cell therapy. Transplant Cell Ther. (2023) 29:430–7. doi: 10.1016/j.jtct.2023.04.001
52. Bajwa A, Zhao Q, Geer M, Lin C, Westholder J, Maakaron J, et al. Siltuximab for chimeric antigen receptor T-cell therapy-related CRS and ICANS: a multicenter retrospective analysis. Blood advances. (2025) 9:170–5. doi: 10.1182/bloodadvances.2024013688
53. Schuelke MR, Bassiri H, Behrens EM, Canna S, Croy C, DiNofia A, et al. Emapalumab for the treatment of refractory cytokine release syndrome in pediatric patients. Blood advances. (2023) 7:5603–7. doi: 10.1182/bloodadvances.2023010712
54. Jain MD, Smith M, and Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141(20):2430–42. doi: 10.1182/blood.2022017414
55. Karschnia P, Jordan JT, Forst DA, Arrillaga-Romany IC, Batchelor TT, Baehring JM, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. (2019) 133:2212–21. doi: 10.1182/blood-2018-12-893396
56. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
57. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204–18. doi: 10.18632/oncotarget.23208
58. Kang S and Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. (2021) 53:1116–23. doi: 10.1038/s12276-021-00649-0
59. Bernardo A, Ball C, Nolasco L, Moake JF, and Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. (2004) 104:100–6. doi: 10.1182/blood-2004-01-0107
60. Giddings JC and Shall L. Enhanced release of von Willebrand factor by human endothelial cells in culture in the presence of phorbol myristate acetate and interleukin 1. Thromb Res. (1987) 47:259–67. doi: 10.1016/0049-3848(87)90139-3
61. Gust J, Ponce R, Liles WC, Garden GA, and Turtle CJ. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
62. Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. (2006) 12:235–9. doi: 10.1038/nm1351
63. Schwameis M, Schörgenhofer C, Assinger A, Steiner MM, and Jilma B. VWF excess and ADAMTS13 deficiency: a unifying pathomechanism linking inflammation to thrombosis in DIC, malaria, and TTP. Thromb haemostasis. (2015) 113:708–18. doi: 10.1160/TH14-09-0731
64. Pinto SN and Krenciute G. The mechanisms of altered blood-brain barrier permeability in CD19 CAR T-cell recipients. Int J Mol Sci. (2024) 25(1):644. doi: 10.3390/ijms25010644
65. Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell. (2020) 183:126–42.e17. doi: 10.1016/j.cell.2020.08.022
66. Heyes MP, Saito K, Major EO, Milstien S, Markey SP, and Vickers JH. A mechanism of quinolinic acid formation by brain in inflammatory neurological disease. Attenuation of synthesis from L-tryptophan by 6-chlorotryptophan and 4-chloro-3-hydroxyanthranilate. Brain. (1993) 116:1425–50. doi: 10.1093/brain/116.6.1425
67. Shah BD, Bishop MR, Oluwole OO, Logan AC, Baer MR, Donnellan WB, et al. KTE-X19 anti-CD19 CAR T-cell therapy in adult relapsed/refractory acute lymphoblastic leukemia: ZUMA-3 phase 1 results. Blood. (2021) 138:11–22. doi: 10.1182/blood.2020009098
68. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet (London England). (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
69. Oliver-Caldés A, González-Calle V, Cabañas V, Español-Rego M, Rodríguez-Otero P, Reguera JL, et al. Fractionated initial infusion and booster dose of ARI0002h, a humanised, BCMA-directed CAR T-cell therapy, for patients with relapsed or refractory multiple myeloma (CARTBCMA-HCB-01): a single-arm, multicentre, academic pilot study. Lancet Oncol. (2023) 24:913–24. doi: 10.1016/S1470-2045(23)00222-X
70. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
71. Nie EH, Su YJ, Baird JH, Agarwal N, Bharadwaj S, Weng WK, et al. Clinical features of neurotoxicity after CD19 CAR T-cell therapy in mantle cell lymphoma. Blood advances. (2024) 8:1474–86. doi: 10.1182/bloodadvances.2023011896
72. Shah NN, Johnson BD, Fenske TS, Raj RV, and Hari P. Intrathecal chemotherapy for management of steroid-refractory CAR T-cell-associated neurotoxicity syndrome. Blood advances. (2020) 4:2119–22. doi: 10.1182/bloodadvances.2020001626
73. Zurko JC, Johnson BD, Aschenbrenner E, Fenske TS, Hamadani M, Hari P, et al. Use of early intrathecal therapy to manage high-grade immune effector cell-associated neurotoxicity syndrome. JAMA Oncol. (2022) 8:773–5. doi: 10.1001/jamaoncol.2022.0070
74. Lichtenstein DA, Schischlik F, Shao L, Steinberg SM, Yates B, Wang HW, et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood. (2021) 138:2469–84. doi: 10.1182/blood.2021011898
75. Hines MR, Keenan C, Maron Alfaro G, Cheng C, Zhou Y, Sharma A, et al. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br J haematology. (2021) 194:701–7. doi: 10.1111/bjh.17662
76. Campodonico E, Angelillo P, Doglio M, Ugolini A, Lupo Stanghellini MT, Noviello M, et al. Biomarkers for an early diagnosis of immune effector cell associated-hemophagocytic syndrome. Front Immunol. (2025) 16:1635062. doi: 10.3389/fimmu.2025.1635062
77. Kennedy VE, Wong C, Huang CY, Thiruvengadam SK, Wolf J, Martin TG, et al. Macrophage activation syndrome-like (MAS-L) manifestations following BCMA-directed CAR T cells in multiple myeloma. Blood advances. (2021) 5:5344–8. doi: 10.1182/bloodadvances.2021005020
78. Matsumoto H, Annen S, Mukai N, Ohshita M, Ogawa S, Okita M, et al. Association of endotheliopathy with coagulofibrinolytic reactions and disseminated intravascular coagulation after trauma: a retrospective observational study. Sci Rep. (2024) 14:29630. doi: 10.1038/s41598-024-81123-5
79. Dusse LM, Alpoim PN, Lwaleed BA, de Sousa LP, Carvalho M, and Gomes KB. Is there a link between endothelial dysfunction, coagulation activation and nitric oxide synthesis in preeclampsia? Clinica Chim Acta. (2013) 415:226–9. doi: 10.1016/j.cca.2012.10.006
80. Galli E, Sorà F, Hohaus S, Fresa A, Pansini I, Autore F, et al. Endothelial activation predicts disseminated intravascular coagulopathy, cytokine release syndrome and prognosis in patients treated with anti-CD19 CAR-T cells. Br J haematology. (2023) 201:86–94. doi: 10.1111/bjh.18596
81. Jiang H, Liu L, Guo T, Wu Y, Ai L, Deng J, et al. Improving the safety of CAR-T cell therapy by controlling CRS-related coagulopathy. Ann Hematol. (2019) 98:1721–32. doi: 10.1007/s00277-019-03685-z
82. Buechner J, Grupp SA, Hiramatsu H, Teachey DT, Rives S, Laetsch TW, et al. Practical guidelines for monitoring and management of coagulopathy following tisagenlecleucel CAR T-cell therapy. Blood advances. (2021) 5:593–601. doi: 10.1182/bloodadvances.2020002757
83. Wang Y, Qi K, Cheng H, Cao J, Shi M, Qiao J, et al. Coagulation disorders after chimeric antigen receptor T cell therapy: analysis of 100 patients with relapsed and refractory hematologic Malignancies. Biol Blood marrow Transplant. (2020) 26:865–75. doi: 10.1016/j.bbmt.2019.11.027
84. Bindal P, Patell R, Chiasakul T, Lauw MN, Ko A, Wang TF, et al. A meta-analysis to assess the risk of bleeding and thrombosis following chimeric antigen receptor T-cell therapy: Communication from the ISTH SSC Subcommittee on Hemostasis and Malignancy. J Thromb haemostasis: JTH. (2024) 22:2071–80. doi: 10.1016/j.jtha.2024.03.021
85. Song Z, Tu D, Tang G, Liu N, Tai Z, Yang J, et al. Hemophagocytic lymphohistiocytosis and disseminated intravascular coagulation are underestimated, but fatal adverse events in chimeric antigen receptor T-cell therapy. Haematologica. (2023) 108:2067–79. doi: 10.3324/haematol.2022.281455
86. Lickefett B, Chu L, Ortiz-Maldonado V, Warmuth L, Barba P, Doglio M, et al. Lymphodepletion - an essential but undervalued part of the chimeric antigen receptor T-cell therapy cycle. Front Immunol. (2023) 14:1303935. doi: 10.3389/fimmu.2023.1303935
87. Gavriilaki E, Sakellari I, and Gavriilaki M. Anagnostopoulos A. A new era in endothelial injury syndromes: toxicity of CAR-T cells and the role of immunity. Int J Mol Sci. (2020) 21(11):3886. doi: 10.3390/ijms21113886
88. Martinez-Sanchez J, Pascual-Diaz R, Palomo M, Moreno-Castaño AB, Ventosa H, Salas MQ, et al. Mafosfamide, a cyclophosphamide analog, causes a proinflammatory response and increased permeability on endothelial cells in vitro. Bone marrow Transplant. (2023) 58:407–13. doi: 10.1038/s41409-023-01912-w
89. Eissner G, Multhoff G, Gerbitz A, Kirchner S, Bauer S, Haffner S, et al. Fludarabine induces apoptosis, activation, and allogenicity in human endothelial and epithelial cells: protective effect of defibrotide. Blood. (2002) 100:334–40. doi: 10.1182/blood.V100.1.334
90. Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. (2008) 103:398–406. doi: 10.1007/s00395-008-0733-0
91. Carreras E and Diaz-Ricart M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplant (2011) 46(12):1495–502. doi: 10.1038/bmt.2011.65
92. Cooke KR, Jannin A, and Ho V. The contribution of endothelial activation and injury to end-organ toxicity following allogeneic hematopoietic stem cell transplantation. Biol Blood marrow Transplant. (2008) 14:23–32. doi: 10.1016/j.bbmt.2007.10.008
93. Palomo M, Moreno-Castaño AB, Salas MQ, Escribano-Serrat S, Rovira M, Guillen-Olmos E, et al. Endothelial activation and damage as a common pathological substrate in different pathologies and cell therapy complications. Front Med. (2023) 10:1285898. doi: 10.3389/fmed.2023.1285898
94. García-Román J and Zentella-Dehesa A. Vascular permeability changes involved in tumor metastasis. Cancer letters. (2013) 335:259–69. doi: 10.1016/j.canlet.2013.03.005
95. Moreno-Castaño AB, Fernández S, Ventosa H, Palomo M, Martinez-Sanchez J, Ramos A, et al. Characterization of the endotheliopathy, innate-immune activation and hemostatic imbalance underlying CAR-T cell toxicities: laboratory tools for an early and differential diagnosis. J Immunother Cancer. (2023) 11(4):e006365. doi: 10.1136/jitc-2022-006365
96. Hong F, Shi M, Cao J, Wang Y, Gong Y, Gao H, et al. Predictive role of endothelial cell activation in cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukaemia. J Cell Mol Med. (2021) 25:11063–74. doi: 10.1111/jcmm.17029
97. di Candia AM, de Avila DX, Moreira GR, Villacorta H, and Maisel AS. Growth differentiation factor-15, a novel systemic biomarker of oxidative stress, inflammation, and cellular aging: Potential role in cardiovascular diseases. Am Heart J plus: Cardiol Res practice. (2021) 9:100046. doi: 10.1016/j.ahjo.2021.100046
98. Gavriilaki E, Demosthenous C, Evangelidis P, Bousiou Z, Batsis I, Vardi A, et al. Soluble urokinase-type plasminogen activator receptor (suPAR), growth differentiation factor-15 (GDF-15), and soluble C5b-9 (sC5b-9) levels are significantly associated with endothelial injury indices in CAR-T cell recipients. Int J Mol Sci. (2024) 25(20):11028. doi: 10.3390/ijms252011028
99. Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res. (2002) 4 Suppl 3:S109–16. doi: 10.1186/ar576
100. Jess J, Yates B, Dulau-Florea A, Parker K, Inglefield J, Lichtenstein D, et al. CD22 CAR T-cell associated hematologic toxicities, endothelial activation and relationship to neurotoxicity. J Immunother Cancer. (2023) 11(6):e005898. doi: 10.1136/jitc-2022-005898
101. Atiq F and O’Donnell JS. Novel functions for von Willebrand factor. Blood. (2024) 144:1247–56. doi: 10.1182/blood.2023021915
102. Zhang J, Chen Z, Ma M, and He Y. Soluble ST2 in coronary artery disease: Clinical biomarkers and treatment guidance. Front Cardiovasc Med. (2022) 9:924461. doi: 10.3389/fcvm.2022.924461
103. Netala VR, Hou T, Wang Y, Zhang Z, and Teertam SK. Cardiovascular biomarkers: tools for precision diagnosis and prognosis. Int J Mol Sci. (2025) 26(7):3218. doi: 10.3390/ijms26073218
104. Muratore E, Gambuti G, Leardini D, Baccelli F, Venturelli F, Larcinese L, et al. The EASIX score as a predictor of sinusoidal obstruction syndrome and nonrelapse mortality in paediatric patients receiving allogeneic haematopoietic stem cell transplantation. Bone Marrow Transplant. (2025) 60:346–52. doi: 10.1038/s41409-024-02489-8
105. Nawas MT, Sanchez-Escamilla M, Devlin SM, Maloy MA, Ruiz JD, Sauter CS, et al. Dynamic EASIX scores closely predict nonrelapse mortality after allogeneic hematopoietic cell transplantation. Blood advances. (2022) 6:5898–907. doi: 10.1182/bloodadvances.2022007381
106. Peña M, Salas MQ, Mussetti A, Moreno-Gonzalez G, Bosch A, Patiño B, et al. Pretransplantation EASIX predicts intensive care unit admission in allogeneic hematopoietic cell transplantation. Blood advances. (2021) 5:3418–26. doi: 10.1182/bloodadvances.2021004812
107. Tomasik J, Avni B, Grisariu S, Elias S, Zimran E, Stepensky P, et al. Endothelial activation and stress index score as a prognostic factor of cytokine release syndrome in CAR-T patients - A retrospective analysis of multiple myeloma and large B-cell lymphoma cohorts. Archivum immunologiae therapiae experimentalis. (2024) 72(1). doi: 10.2478/aite-2024-0018
108. Korell F, Penack O, Mattie M, Schreck N, Benner A, Krzykalla J, et al. EASIX and severe endothelial complications after CD19-directed CAR-T cell therapy-A cohort study. Front Immunol. (2022) 13:877477. doi: 10.3389/fimmu.2022.877477
109. Frenking JH, Zhou X, Wagner V, Hielscher T, Kauer J, Mai EK, et al. EASIX-guided risk stratification for complications and outcome after CAR T-cell therapy with ide-cel in relapsed/refractory multiple myeloma. J Immunother Cancer. (2024) 12(10):e009220. doi: 10.1136/jitc-2024-009220
110. Zhao Y, Zhang X, Zhang M, Guo R, Zhang Y, Pu Y, et al. Modified EASIX scores predict severe CRS/ICANS in patients with acute myeloid leukemia following CLL1 CAR-T cell therapy. Ann Hematol. (2024) 103:969–80. doi: 10.1007/s00277-024-05617-y
111. Pennisi M, Sanchez-Escamilla M, Flynn JR, Shouval R, Alarcon Tomas A, Silverberg ML, et al. Modified EASIX predicts severe cytokine release syndrome and neurotoxicity after chimeric antigen receptor T cells. Blood advances. (2021) 5:3397–406. doi: 10.1182/bloodadvances.2020003885
112. Zandaki D, Selukar S, Bi Y, Li Y, Zinsky M, Bonifant CL, et al. EASIX and m-EASIX predict CRS and ICANS in pediatric and AYA patients after CD19-CAR T-cell therapy. Blood advances. (2025) 9:270–9. doi: 10.1182/bloodadvances.2024014027
113. Gavriilaki E, Tzannou I, Batsis I, Tsonis I, Liga M, Gkirkas K, et al. EASIX and m-EASIX predict severe cytokine release syndrome and overall survival after CAR T-cell therapy. Blood vessels Thromb hemostasis. (2024) 1:100025. doi: 10.1016/j.bvth.2024.100025
114. Goel U, Dima D, Davis JA, Rashid A, Wesson W, Williams L, et al. Development of an Endothelial Activation and Stress Index (EASIX)-based predictive model for cytokine release syndrome and neurotoxicity after B-cell maturation antigen directed chimeric antigen receptor T-cell therapy for relapsed/refractory multiple myeloma. Bone Marrow Transplant. (2025). doi: 10.1016/j.jtct.2025.01.625
115. de Boer JW, Keijzer K, Pennings ERA, van Doesum JA, Spanjaart AM, Jak M, et al. Population-based external validation of the EASIX scores to predict CAR T-cell-related toxicities. Cancers. (2023) 15(22):5443. doi: 10.3390/cancers15225443
116. Barker K, Marco T, Husnain M, and Katsanis E. Addition of phosphorous and IL6 to m-EASIX score improves detection of ICANS and CRS, as well as CRS progression. Cancers. (2025) 17(6):918. doi: 10.3390/cancers17060918
117. Wang X, Li C, Luo W, Zhang Y, Huang Z, Xu J, et al. IL-10 plus the EASIX score predict bleeding events after anti-CD19 CAR T-cell therapy. Ann Hematol. (2023) 102:3575–85. doi: 10.1007/s00277-023-05477-y
118. Greenbaum U, Strati P, Saliba RM, Torres J, Rondon G, Nieto Y, et al. CRP and ferritin in addition to the EASIX score predict CAR-T-related toxicity. Blood advances. (2021) 5:2799–806. doi: 10.1182/bloodadvances.2021004575
119. Sandler RD, Tattersall RS, Schoemans H, Greco R, Badoglio M, Labopin M, et al. Diagnosis and management of secondary HLH/MAS following HSCT and CAR-T cell therapy in adults; A review of the literature and a survey of practice within EBMT centres on behalf of the autoimmune diseases working party (ADWP) and transplant complications working party (TCWP). Front Immunol. (2020) 11:524. doi: 10.3389/fimmu.2020.00524
120. Zu C, Wu S, Zhang M, Wei G, Xu H, Cui J, et al. A distinct cytokine network distinguishes chimeric antigen receptor T cell (CAR-T)-associated hemophagocytic lymphohistiocytosis-like toxicity (carHLH) from severe cytokine release syndrome following CAR-T therapy. Cytotherapy. (2023) 25:1167–75. doi: 10.1016/j.jcyt.2023.06.008
121. Ortí G, Dachy G, Graham CE, Peric Z, Alarcon A, del Bufalo F, et al. Less frequent complications following CAR T-cell therapies: hemophagocytic lymphohistiocytosis, graft-versus-host disease, thrombotic microangiopathy, coagulation disorders and secondary Malignancies: best practice recommendations from the EBMT Practice Harmonization and Guidelines Committee. Bone Marrow Transplantation. (2025) 60:751–8. doi: 10.1038/s41409-025-02567-5
122. Perl M, Herfeld K, Harrer DC, Höpting M, Schweiger M, Sterz U, et al. Tocilizumab administration in cytokine release syndrome is associated with hypofibrinogenemia after chimeric antigen receptor T-cell therapy for hematologic Malignancies. Haematologica. (2024) 109:2969–77. doi: 10.3324/haematol.2023.284564
123. Manfredi M, Van Hoovels L, Benucci M, De Luca R, Coccia C, Bernardini P, et al. Soluble urokinase plasminogen activator receptor (suPAR) in autoimmune rheumatic and non rheumatic diseases. J Pers Med. (2023) 13(4):688. doi: 10.3390/jpm13040688
124. Lin JC, Chen XD, Xu ZR, Zheng LW, and Chen ZH. Association of the circulating supar levels with inflammation, fibrinolysis, and outcome in severe burn patients. Shock (Augusta Ga). (2021) 56:948–55. doi: 10.1097/SHK.0000000000001806
125. Rasmussen LJH, Petersen JEV, and Eugen-Olsen J. Soluble urokinase plasminogen activator receptor (suPAR) as a biomarker of systemic chronic inflammation. Front Immunol. (2021) 12:780641. doi: 10.3389/fimmu.2021.780641
126. Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. (2011) 117:1340–9. doi: 10.1182/blood-2010-05-283564
127. Bezerra E, Durisek G III, Zhao Q, Denlinger N, Voorhees T, Sanchez-Petitto G, et al. Association between post-CART terminal complement complex levels and immune effector cell-associated neurotoxicity syndrome (ICANS). Blood. (2024) 144:3446. doi: 10.1182/blood-2024-198872
128. Stepanov A, Wang W, Hou Y, Ivan Chernikov IC, Bissonnette R, Tetz G, et al. Abstract B048: Neutrophil extracellular traps-targeting DNase I enhances the therapeutic efficacy of CAR-T cell adoptive immunotherapy in a syngeneic murine metastasis model. Cancer Res. (2024) 84:B048–B. doi: 10.1158/1538-7445.TUMBODY-B048
129. Volkov DV, Tetz GV, Rubtsov YP, Stepanov AV, and Gabibov AG. Neutrophil extracellular traps (NETs): opportunities for targeted therapy. Acta naturae. (2021) 13:15–23. doi: 10.32607/actanaturae.11503
130. Gavriilaki E, Tzannou I, Vardi A, Tsonis I, Liga M, Girkas K, et al. Easix indices predict CRS and overall survival in adult CAR-T cell recipients. Blood. (2023) 142:6905. doi: 10.1182/blood-2023-179506
131. Acosta-Medina AA, Johnson IM, Bansal R, Hathcock M, Kenderian SJ, Durani U, et al. Pre-lymphodepletion & infusion endothelial activation and stress index as predictors of clinical outcomes in CAR-T therapy for B-cell lymphoma. Blood Cancer J. (2023) 13:7. doi: 10.1038/s41408-022-00777-4
132. Holtzman NG, Xie H, Bentzen S, Kesari V, Bukhari A, El Chaer F, et al. Immune effector cell-associated neurotoxicity syndrome after chimeric antigen receptor T-cell therapy for lymphoma: predictive biomarkers and clinical outcomes. Neuro-oncology. (2021) 23:112–21. doi: 10.1093/neuonc/noaa183
133. Demosthenous C, Evangelidis P, Gatsis A, Mitroulis I, Vakalopoulou S, Vardi A, et al. Endothelial injury following CAR-T cell immunotherapy for hematological Malignancies. Cancers. (2025) 17(17):2876. doi: 10.3390/cancers17172876
134. Wachholz GE, Akbari P, Huijbers EJM, Jalan P, van Beijnum JR, and Griffioen AW. Targeting endothelial cell anergy to improve CAR T cell therapy for solid tumors. Biochim Biophys Acta Rev cancer. (2024) 1879:189155. doi: 10.1016/j.bbcan.2024.189155
135. Zhuang X, Maione F, Robinson J, Bentley M, Kaul B, Whitworth K, et al. CAR T cells targeting tumor endothelial marker CLEC14A inhibit tumor growth. JCI Insight. (2020) 5(19):e138808. doi: 10.1172/jci.insight.138808
136. Akbari P, Katsarou A, Daghighian R, van Mil L, Huijbers EJM, Griffioen AW, et al. Directing CAR T cells towards the tumor vasculature for the treatment of solid tumors. Biochim Biophys Acta Rev cancer. (2022) 1877:188701. doi: 10.1016/j.bbcan.2022.188701
137. Zhang Q, Zhu X, and Xiao Y. The critical role of endothelial cell in the toxicity associated with chimeric antigen receptor T cell therapy and intervention strategies. Ann hematology. (2024) 103:2197–206. doi: 10.1007/s00277-024-05640-z
138. Chen Y, Li R, Shang S, Yang X, Li L, Wang W, et al. Therapeutic potential of TNFα and IL1β Blockade for CRS/ICANS in CAR-T therapy via ameliorating endothelial activation. Front Immunol. (2021) 12:623610. doi: 10.3389/fimmu.2021.623610
139. Jacobson CA, Rosenthal AC, Arnason J, Agarwal S, Zhang P, Wu W, et al. A phase 2 trial of defibrotide for the prevention of chimeric antigen receptor T-cell-associated neurotoxicity syndrome. Blood advances. (2023) 7:6790–9. doi: 10.1182/bloodadvances.2023009961
140. Sumransub N, Seichter C, Kent K, Shanley R, Hu M, O’Leary D, et al. Simvastatin with intrathecal dexamethasone reduces neurotoxicity in adults receiving chimeric antigen receptor (CAR) T-cells treatment. Blood. (2023) 142:3493. doi: 10.1182/blood-2023-187070
141. Wheeler M, Skiles JL, and Rahrig AL. Thrombotic microangiopathy following CAR-T cell therapy successfully treated with eculizumab in a pediatric patient. Transplant Cell Ther. (2024) 30:S129. doi: 10.1016/j.jtct.2024.01.019
142. Lin MY, Nam E, Shih RM, Shafer A, Bouren A, Ayala Ceja M, et al. Self-regulating CAR-T cells modulate cytokine release syndrome in adoptive T-cell therapy. J Exp Med. (2024) 221(6):e20221988. doi: 10.1084/jem.20221988
143. Zhou W, Miao J, Cheng Z, Wang Z, Wang J, Guo H, et al. Hypoxia-regulated secretion of IL-12 enhances antitumor activity and safety of CD19 CAR-T cells in the treatment of DLBCL. Mol Ther oncolytics. (2023) 30:216–26. doi: 10.1016/j.omto.2023.08.009
Keywords: biomarkers, biomarker panels, CAR T toxicities, coagulopathy, CRS, ICANS, IEC-HS, prediction
Citation: Moreno-Castaño AB, Iraola G, Martínez-Cibrián N, Albiol N, Prats D-NM, Martinez-Sanchez J, Castro P and Diaz-Ricart M (2025) Endothelial dysfunction and hemostatic imbalance in CAR T-cell-associated toxicities: pathophysiological insights and the role of circulating biomarkers. Front. Immunol. 16:1699894. doi: 10.3389/fimmu.2025.1699894
Received: 05 September 2025; Accepted: 20 October 2025;
Published: 03 November 2025.
Edited by:
Eleni Gavriilaki, Aristotle University of Thessaloniki, GreeceReviewed by:
Magdiel Pérez-Cruz, PEREZ-DAVIN Biomedical Research Institute, United StatesPaschalis Evangelidis, Aristotle University of Thessaloniki, Greece
Copyright © 2025 Moreno-Castaño, Iraola, Martínez-Cibrián, Albiol, Prats, Martinez-Sanchez, Castro and Diaz-Ricart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Belén Moreno-Castaño, YW1vcmVuY2E1OEBkb2N0LnViLmVkdQ==
†These authors have contributed equally to this work and share senior authorship