 Jiexia Wen1,2,3
Jiexia Wen1,2,3 Huagang Liang2,4Min Zhao2,5Bin Xuan2,6
Huagang Liang2,4Min Zhao2,5Bin Xuan2,6 Xiangcai Meng2,6Yang Liu2,6Liwei Wang2,6Li He2,6
Xiangcai Meng2,6Yang Liu2,6Liwei Wang2,6Li He2,6 Tao Zhou2,6Yang Tao1,2
Tao Zhou2,6Yang Tao1,2 Yimin Wang1,2,6*
Yimin Wang1,2,6*- 1Department of Central Laboratory, The First Hospital of Qinhuangdao, Qinhuangdao, Hebei, China
- 2Key Laboratory of Research on Molecular Mechanism of Gastrointestinal Tumors in Qinhuangdao, The First Hospital of Qinhuangdao, Qinhuangdao, Hebei, China
- 3State Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao, China
- 4Department of Thoracic Surgery, The First Hospital of Qinhuangdao, Hebei Medical University, Qinhuangdao, Hebei, China
- 5Department of Pathology, The First Hospital of Qinhuangdao, Hebei Medical University, Qinhuangdao, Hebei, China
- 6Department of General Surgery, The First Hospital of Qinhuangdao, Hebei Medical University, Qinhuangdao, Hebei, China
Ferroptosis is a form of regulated cell death defined in recent years, characterized by iron-dependent accumulation of lipid peroxides. A large body of research has demonstrated that ferroptosis is closely associated with the progression of gastrointestinal tumors (such as gastric cancer, colorectal cancer, and esophageal cancer), and gastrointestinal tumor cells exhibit unique sensitivity to ferroptosis. This indicates that ferroptosis has emerged as a highly promising strategy to combat therapy-resistant colorectal cancer. Although the intrinsic ferroptosis-suppressive and ferroptosis-promoting pathways in gastrointestinal tumors have been fully elucidated, the current understanding of the extrinsic metabolites and pathways that regulate ferroptosis in the pathogenesis of gastrointestinal tumors remains relatively limited. Emerging studies have shown a strong link between gut microbial metabolism and the progression of gastrointestinal tumors. This review summarizes the relevant aspects of gut microbiota metabolism, explores how these gut microbiota-derived metabolites regulate cancer progression through ferroptosis, and proposes that targeting gut microbiota-mediated ferroptosis represents a potential therapeutic approach for gastrointestinal tumors.
1 Introduction
Ferroptosis, a regulated form of cell death distinct from apoptosis, pyroptosis, and necrosis, has attracted considerable attention since its formal characterization in 2012. It is primarily driven by iron-dependent lipid peroxidation, initiated by the accumulation of peroxidized phospholipids in cellular membranes through the action of intracellular iron (1). Excess iron promotes reactive oxygen species (ROS) generation via the Fenton reaction, overwhelming cellular antioxidant defenses, particularly the glutathione (GSH) and glutathione peroxidase 4 (GPX4) system. When ROS production surpasses the cell’s detoxification capacity, they oxidize unsaturated fatty acids within membrane phospholipids (2), leading to lipid peroxide formation that disrupts membrane integrity and causes extensive cellular damage. Morphologically, ferroptosis is marked by mitochondrial abnormalities such as shrinkage, increased membrane density, and loss of cristae (3). Unlike apoptosis, it does not involve cytoplasmic vesicles, chromatin condensation, or nuclear fragmentation; it also lacks the swelling and organelle disintegration typical of necrosis.
Several small molecules modulate ferroptosis, a form of cell death driven by iron-dependent lipid peroxidation. Compounds such as erastin, sulfasalazine, sorafenib, artemisinin derivatives, and buthionine sulfoximine promote ferroptosis by depleting GSH (4). Others, including RSL3, altretamine, DPI17, and FIN56, directly inhibit GPX4 (5–8). Conversely, ferroptosis can be suppressed by iron chelators (e.g., deferoxamine) or specific inhibitors like ferrostatin-1, liproxstatin-1, vitamin E, and others (9). Key protein regulators, including GPX4, p53, SLC7A11, ACSL4, NOX, and NRF2, also critically influence ferroptosis (10). Additionally, the gut microbiota and its metabolites play a significant role: probiotics and metabolites such as short-chain fatty acids (SCFAs), bile acids, and tryptophan derivatives can inhibit ferroptosis and support intestinal health (11). In contrast, bacterial lipopolysaccharide (LPS) promotes ferroptosis by increasing iron accumulation and oxidative stress, exacerbating tissue damage.
Ferroptosis has been increasingly implicated in the pathogenesis of numerous diseases beyond the digestive system, including neurological disorders, ischemia/reperfusion injury, acute kidney injury, cardiovascular diseases, and various cancers (12). Within the gastrointestinal tract, growing evidence specifically links ferroptosis to esophageal, gastric, pancreatic, and colorectal cancers, as well as intestinal I/R injury and inflammatory bowel disease (13). These findings highlight ferroptosis not only as a key disease mechanism but also as a promising therapeutic target. This review summarizes recent advances in understanding the role of ferroptosis in gastrointestinal diseases, with a special focus on the regulatory influence of the gut microbiota and its metabolites. By exploring how microbial components modulate ferroptosis, we further discuss the potential of targeting ferroptosis as a novel treatment strategy for gastrointestinal disorders.
2 Ferroptosis
2.1 Mechanisms of ferroptosis
2.1.1 Lipid peroxidation
Lipid peroxidation is a key executioner in ferroptosis, primarily targeting esterified polyunsaturated fatty acids (PUFAs) in membrane phospholipids, while free PUFAs are largely unaffected. The enzymes ACSL4 and LPCAT3 play crucial roles in synthesizing esterified PUFAs and incorporating arachidonic acid into phospholipids. Knocking down either enzyme reduces the formation of esterified PUFAs and increases cellular resistance to ferroptosis (14), indicating that PUFA availability and esterification are prerequisites for ferroptosis initiation (15). Lipoxygenases (LOXs), particularly ALOX15 (16), are central to enzymatic lipid peroxidation in ferroptosis. ALOX15 catalyzes the oxygenation of arachidonic acid, a process enhanced by the scaffold protein PEBP1 (17). This leads to increased lipid peroxide accumulation, which disrupts membrane integrity and promotes ferroptotic cell death.
2.1.2 Abnormal iron metabolism
Abnormalities in iron metabolism are critical for the initiation of ferroptosis. Elevated intracellular iron levels—often due to uptake of exogenous iron sources such as ferric ammonium citrate, ferric citrate, or ferric chloride hexahydrate—sensitize cells to ferroptosis (18). Excess ferrous iron (Fe2+) participates in Fenton reactions, generating highly reactive hydroxyl radicals (19). Under physiological conditions, iron homeostasis is tightly regulated. Cellular iron uptake is primarily mediated by transferrin receptor 1 (TFR1), which facilitates transferrin-bound iron endocytosis. Knockdown of TFR1 has been shown to inhibit ferroptosis induced by erastin or cystine deprivation. Additionally, intracellular iron levels are modulated by ferritinophagy, an autophagy-like process mediated by nuclear receptor coactivator 4 (NCOA4). NCOA4 targets ferritin for degradation, releasing iron into the labile iron pool (LIP), thereby increasing free iron levels and promoting ferroptosis (20).
2.1.3 GSH depletion
GSH is a key metabolic regulator and antioxidant that neutralizes ROS, protecting lipids, proteins, and DNA from oxidative damage and limiting lipid peroxide accumulation. Ferroptosis can be induced by compounds like erastin and RSL3, which impair the GSH-dependent antioxidant system. Erastin inhibits system Xc−, a cystine/glutamate antiporter, thereby reducing cystine uptake and subsequent GSH synthesis. RSL3 directly inhibits GPX4, which relies on GSH to eliminate lipid peroxides (21). Together, these effects lead to unchecked lipid peroxidation and ferroptosis. Cystine, the oxidized form of cysteine, is essential for GSH synthesis. It is imported into cells via SLC7A11, a key component of system Xc−. Inside the cell, cystine is reduced to cysteine and used for GSH production. By enhancing cystine uptake and GSH synthesis, SLC7A11 helps counteract ferroptosis driven by ROS and iron accumulation. This protective mechanism is often upregulated in tumor cells to promote survival under oxidative stress (22). The cystine/GSH/GPX4 axis is now established as a central pathway regulating ferroptosis, with GPX4 playing a critical role in controlling this cell death process (23).
2.2 Inhibitory pathways of ferroptosis
2.2.1 SLC7A11-GSH-GPX4 pathway
System Xc− is an essential amino acid antiporter composed of a light chain (SLC7A11) and a heavy chain (SLC3A2). It mediates the exchange of extracellular cystine for intracellular glutamate, facilitating cystine uptake, a critical step for GSH biosynthesis. Upon import, cystine is reduced to cysteine, the rate-limiting precursor for GSH. As a key antioxidant, GSH neutralizes lipid peroxides and protects cells from oxidative damage that drives ferroptosis. This detoxification is primarily executed by GPX4, which uses GSH to reduce lipid hydroperoxides into nontoxic lipid alcohols, thereby maintaining membrane integrity (24). Inhibiting GPX4 leads to GSH depletion, accumulation of lipid peroxides, and ultimately induces ferroptosis. GPX4 activity depends not only on GSH availability but is also regulated by epigenetic and post-translational mechanisms that modulate its expression and function (25).
2.2.2 CoQ10-FSP1 pathway
Coenzyme Q10 (CoQ10), also known as ubiquinone, is a lipid-soluble antioxidant that inhibits lipid peroxidation in cell membranes. In the CoQ10–FSP1 pathway, ferroptosis suppressor protein 1 (FSP1) reduces CoQ10 to ubiquinol (CoQ10H2) using NAD(P)H as a cofactor. Ubiquinol then scavenges lipid peroxyl radicals, thereby suppressing lipid peroxidation and ferroptosis (26, 27). This mechanism functions independently of the canonical SLC7A11–GPX4 axis. Additionally, CoQ10 is synthesized via the mevalonate pathway; its inhibition decreases both CoQ10 and GPX4 levels, increasing cellular susceptibility to ferroptosis (28).
2.2.3 GCH1-BH4 pathway
The guanosine 5’-triphosphate (GTP) cyclohydrolase-1 (GCH1)–tetrahydrobiopterin (BH4) pathway represents a key GPX4-independent mechanism that inhibits ferroptosis. BH4 serves as an essential cofactor in nitric oxide production, neurotransmitter synthesis, and aromatic amino acid metabolism. Depletion of BH4 causes nitric oxide synthase uncoupling and elevated ROS levels. Together with dihydrobiopterin (BH2), BH4 forms a redox cycle that scavenges endogenous oxidative free radicals, thereby suppressing ferroptosis (29). This cycle is maintained by dihydrofolate reductase (DHFR) using NADP+/NAD(P)H as a cofactor (29, 30). Notably, supplementation with BH4, but not BH2, confers protection against ferroptosis.
De novo synthesis of BH4 depends on three enzymes: GCH1, 6-pyruvoyltetrahydropterin synthase (PTS), and sepiapterin reductase (SPR). Among these, GCH1 is the rate-limiting enzyme and a central regulator of ferroptosis sensitivity (31). Its deficiency or pharmacological inhibition reduces BH4 levels, leading to oxidative radical accumulation and ferroptosis. Conversely, GCH1 overexpression enhances BH4 biosynthesis, reduces ROS, and promotes CoQ10 production, via synthesis of the precursor 4-hydroxybenzoate, further increasing ferroptosis resistance (30, 32). Thus, the GCH1–BH4 pathway constitutes an important endogenous antioxidant system that inhibits ferroptosis independently of the GPX4-GSH axis.
2.2.4 MBOAT1/2-MUFA pathway
Lipid peroxidation, a key executor of ferroptosis, primarily targets esterified PUFAs within membrane phospholipids. Ferroptosis can be inhibited by monounsaturated fatty acids (MUFAs), which are produced by SCD1 and counteract the detrimental effects of PUFAs (33). Through genome-wide CRISPR activation screens, Liang et al. identified membrane-bound O-acyltransferase 1/2 (MBOAT1/2) as a novel ferroptosis inhibitor. MBOAT1/2 suppresses ferroptosis by remodeling phospholipids via a mechanism independent of GPX4 or FSP1. Specifically, ACSL3 activates MUFAs to form MUFA-CoA, which MBOAT1/2 then uses to synthesize MUFA-containing phospholipids (MUFA-PLs) by transferring the acyl group to lyso-phosphatidylethanolamine (lyso-PE). By replacing oxidizable PUFAs in membranes, MUFA-PLs reduce lipid peroxidation and inhibit ferroptosis (34). The expression of MBOAT2 is regulated by the androgen receptor (AR). AR antagonists lower MBOAT2 levels, and combining them with ferroptosis inducers potently suppresses tumor growth in AR+ prostate cancer. Similarly, MBOAT1 is highly expressed in estrogen receptor (ER)-associated cancers, such as breast, ovary, and endometrium, and is likely regulated by ER. ER antagonists reduce MBOAT1 expression and sensitize cells to ferroptosis inducers (35). Thus, sex hormone receptors play a critical role in regulating ferroptosis.
2.2.5 Mitochondrial DHODH pathway
Dihydroorotate dehydrogenase (DHODH) is a mitochondrial enzyme located on the outer surface of the inner mitochondrial membrane (IMM) and plays a role in important metabolic pathways such as cytochrome P450, purine synthesis, and fatty acid metabolism (36, 37). Studies have shown that the DHODH pathway can protect cells from oxidative stress and mitochondrial damage, including downregulating ROS levels, maintaining mitochondrial membrane potential, and inhibiting apoptosis, which has been proven to be a novel defense system against mitochondrial ferroptosis (38, 39). DHODH regulates mitochondrial lipid peroxidation and ferroptosis by catalyzing ubiquinone (CoQ10)-mediated oxidation of dihydroorotate (DHO) to orotate (OA) and reducing CoQ10 to ubiquinol (CoQ10H2) (38, 40). DHO protects cells from RSL3-induced ferroptosis, and the DHODH inhibitor brequinar (BRQ) is able to promote ferroptosis in GPX4-deficient cancer cells. RSL3-induced ferroptosis in DHODH-deficient cells is not spared by activation of FSP1, suggesting that DHODH acts in an FSP1-independent manner. In addition, studies have shown that DHODH can work in parallel with mitochondrial GPX4 to inhibit ferroptosis (41, 42). Inhibition of DHODH significantly induced mitochondrial lipid peroxidation and ferroptosis in cancer cells with low GPX4 expression, but only made cancer cells with high GPX4 expression sensitive to ferroptosis. In cells with GPX4 knockdown, it was confirmed that mitochondrial GPX4 rather than cytoplasmic GPX4 was able to make cells more resistant to ferroptosis. Therefore, these findings suggest that DHODH and GPX4 act as redundant defense mechanisms in mitochondria to prevent ferroptosis (38) (Figure 1).
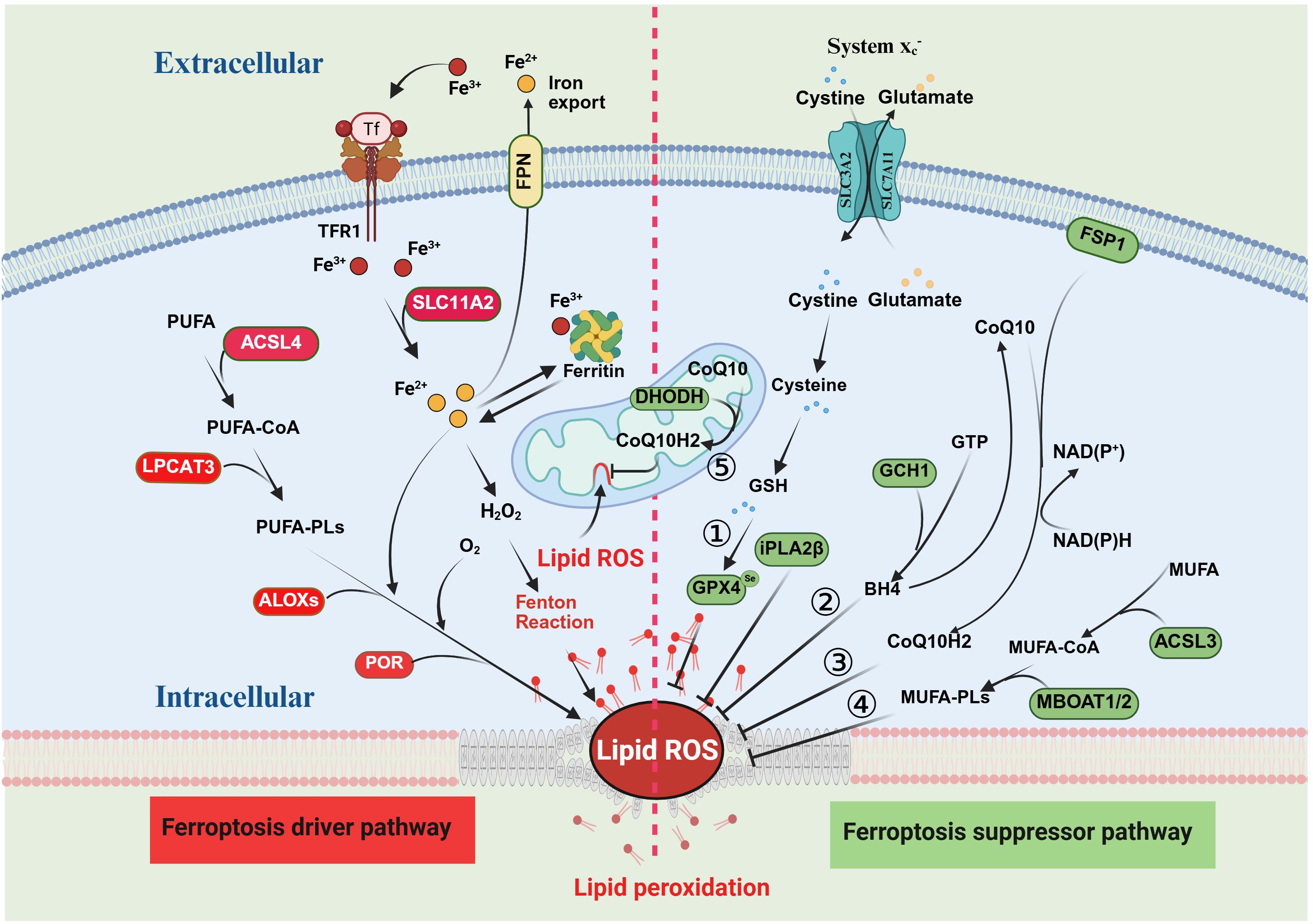
Figure 1. Mechanisms of ferroptosis and suppression. The red section on the left side of the figure illustrates the mechanism of ferroptosis, with proteins in the red boxes representing the key promoters of this process. The green section on the right depicts the major inhibitory pathways of ferroptosis, where proteins in the green boxes denote the main inhibitors. There are five principal inhibitory pathways for ferroptosis: 1) the System Xc−–GPX4 pathway; 2) the CoQ10–FSP1 pathway; 3) the GCH1–BH4 pathway; 4) the MUFA pathway; and 5) the mitochondrial DHODH pathway.
3 Intestinal flora and metabolites in the regulation of ferroptosis
Ferroptosis is characterized by iron overload, excessive ROS production, elevated levels of PUFAs, and extensive lipid peroxidation. Growing evidence highlights a strong relationship between the gut microbiota and ferroptosis. Intestinal probiotics may exert protective effects against ferroptosis by chelating metal ions, scavenging or reducing ROS, and modulating key ferroptosis-related enzymes (43, 44). In addition, microbial metabolites such as SCFAs, bile acids, and tryptophan derivatives are increasingly recognized as critical regulators of this process (45). As summarized in Table 1, these compounds influence ferroptosis through distinct mechanisms.
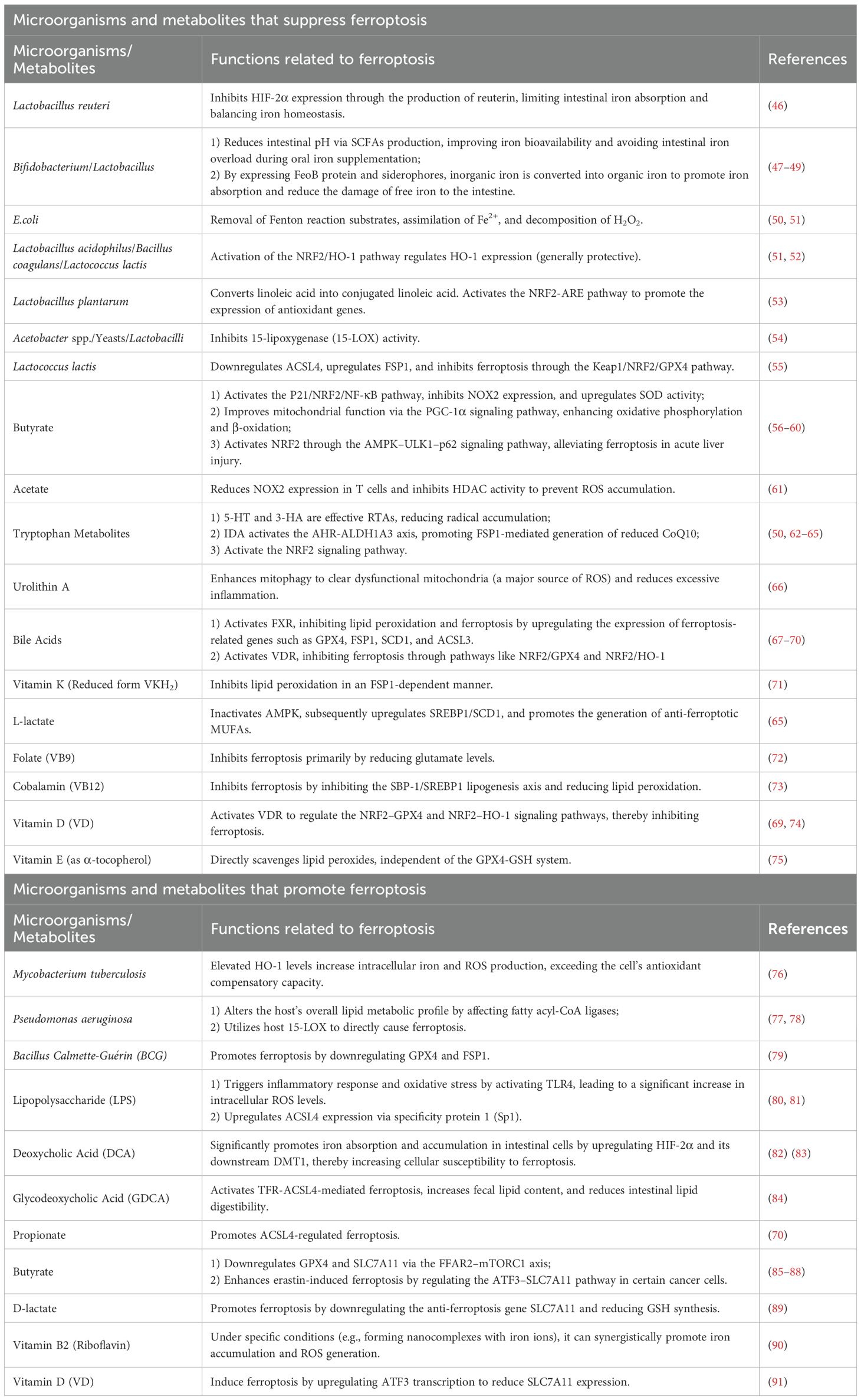
Table 1. Microbial compositions and metabolites associated with ferroptosis.
3.1 Regulation of iron accumulation by intestinal flora and metabolites
Excess free iron, owing to its redox activity, drives ROS generation through the Fenton reaction and serves as a core trigger of ferroptosis. The intestinal microbiota and their metabolites in the regulation of host iron homeostasis via intricate and multilayered networks that complement classical iron metabolic pathways. Certain microbial metabolites directly target key nodes of iron metabolism. For instance, deoxycholic acid (DCA), a secondary bile acid produced by Bacteroidetes such as Clostridium scindens (82), significantly enhances iron absorption and accumulation in intestinal epithelial cells by upregulating hypoxia-inducible factor-2α (HIF-2α) and its downstream effector, divalent metal transporter 1 (DMT1). This mechanism is independent of classical antioxidant systems such as the GPX4-GSH axis but increases cellular susceptibility to ferroptosis by elevating the “iron substrate” necessary for its initiation (83). Conversely, L. reuteri balance this by inhibiting HIF-2α expression to limit intestinal iron absorption through the production of reuterin, demonstrating the fine-tuned balance of iron homeostasis within the microbial community (46).
On the other hand, the gut microbiota and their metabolites can indirectly modulate ferroptosis by regulating systemic iron distribution. They influence plasma ferritin levels (80) and promote the expression of iron-handling molecules such as ferredoxin and hepcidin (92). Together with macrophage-mediated iron recycling, these effects contribute to maintaining iron homeostasis and the stability of systemic iron cycling. In patients with anemia, oral iron supplementation (e.g., FeSO4) has been shown to induce ferroptosis-related intestinal injury (93). Co-administration of probiotics such as Bifidobacterium and Lactobacillus can mitigate this effect by producing SCFAs, which lower intestinal pH and enhance iron bioavailability. This not only corrects anemia more efficiently but also prevents intestinal iron overload and ferroptosis (47). Such probiotic-mediated protection may serve as a compensatory mechanism that alleviates the oxidative stress imposed by iron supplementation on the classical system Xc−–GSH–GPX4 antioxidant axis, primarily by optimizing the local intestinal microenvironment. Furthermore, commensal bacteria can fine-tune iron absorption and utilization by expressing FeoB proteins and siderophores, or by converting inorganic iron into more bioavailable organic forms (48, 49).
Importantly, under conditions of iron overload, the gut microbiota can also confer cellular protection independent of iron metabolism by generating metabolites with intrinsic antioxidant activity. A notable example is the tryptophan-derived metabolite 3-hydroxyanthranilic acid (3-HA), a potent radical-trapping antioxidant. Functionally analogous to FSP1, 3-HA directly scavenges lipid radicals in a manner independent of the GPX4 and CoQ10 systems. Therefore, even when metabolites such as DCA promote intracellular iron accumulation, 3-HA can directly inhibit the execution phase of lipid peroxidation through this parallel antioxidant pathway, thereby preventing the onset of ferroptosis (50, 62). Collectively, these findings illustrate a “multi-level, multi-target” regulatory strategy employed by the gut microbiota to modulate ferroptosis, acting both upstream by controlling iron availability and downstream by directly suppressing lipid peroxidation.
During macrophage-mediated iron recycling, activation of the NRF2/HO-1 pathway activated by probiotics generally exerts protective effects, for example, alleviating renal ischemia-reperfusion injury (51) and mitigating pathology in neurodegenerative disease models (52). The induction of HO-1 represents a key adaptive metabolic node, where bilirubin, a product of HO-1 activity, acts as an endogenous antioxidant that can partially compensate for diminished GSH function. However, this pathway exhibits dual roles. Under specific pathological conditions, such as Mycobacterium tuberculosis (MTB) infection, excessive HO-1-mediated iron release may overwhelm the cell’s antioxidant defenses, leading instead to increased lipid peroxidation and ferroptosis (76) (Figure 2).
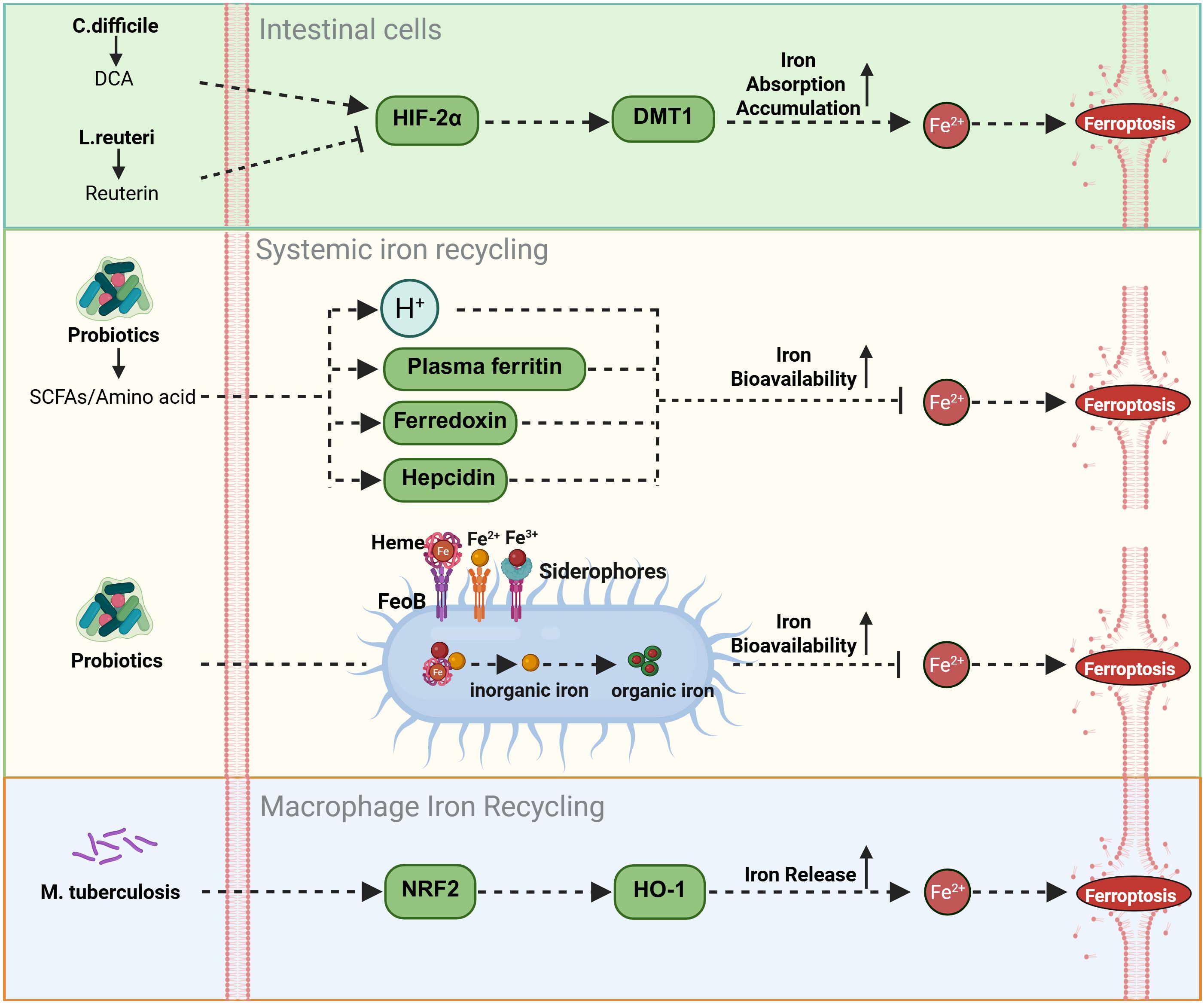
Figure 2. Modulation of iron deposition by gut flora and metabolites. The gut microbiota orchestrates iron metabolism through coordinated mechanisms: at the enterocyte level, Bacteroidetes-derived DCA activates the HIF-2α/DMT1 axis for iron absorption, counterbalanced by L. reuteri-derived reuterin; systemically, probiotics enhance bioavailability via SCFAs/amino acids-induced pH reduction and modulate ferritin/ferredoxin/hepcidin for homeostasis; further precision is achieved via FeoB/siderophore-mediated iron conversion, while macrophage iron recycling is activated through the NRF2/HO-1 pathway.
3.2 Regulation of intestinal flora and metabolites on ROS accumulation
The intestinal flora and their metabolites participate contribute to maintaining intracellular redox homeostasis through complex regulatory networks, thereby playing a critical role in ferroptosis (94). These regulatory mechanisms are multifaceted, involving direct ROS scavenging, modulation of endogenous antioxidant pathways, or regulation of ROS generation sources (Figure 3).
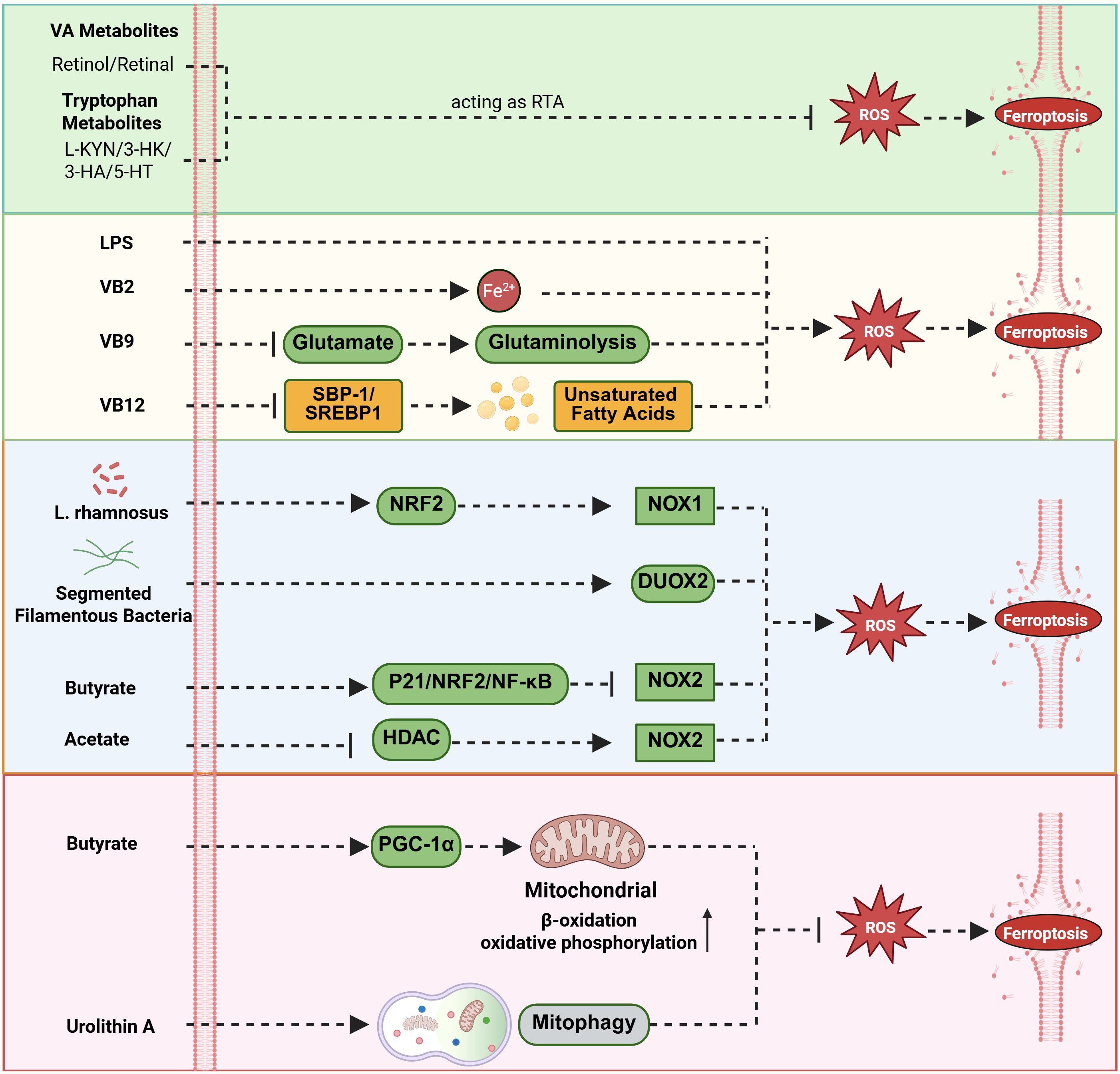
Figure 3. Modulation of ROS accumulation by gut flora and metabolites. The gut microbiota and metabolites regulate intracellular ROS homeostasis and influence the core mechanisms of ferroptosis through a multi-level, interconnected complex network. Vitamin A and tryptophan metabolites act as radical-trapping antioxidants (RTA) to inhibit ferroptosis; LPS significantly increases intracellular ROS levels by activating Toll-like receptor 4 (TLR4) on immune cells such as macrophages; Riboflavin (VB2) promotes iron accumulation and ROS generation, thereby enhancing ferroptosis; Folate (VB9) blocks ferroptosis by reducing glutamate levels; Cobalamin (VB12) inhibits ferroptosis by suppressing the SBP-1/SREBP1 lipogenesis axis; L. rhamnosus upregulates NOX1 expression via the NRF2 pathway; Segmented filamentous bacteria enhance DUOX2 expression levels; conversely, butyrate inhibits NOX2 expression by activating the P21/NRF2/NF-κB pathway, and acetic acid downregulates NOX2 through HDAC inhibition; Regarding the regulation of mitochondrial function, butyrate activates the PGC-1α pathway to enhance mitochondrial function, while urolithin A promotes mitophagy to clear dysfunctional mitochondria and reduce abnormal ROS production.
3.2.1 Metabolites that directly scavenge ROS
A variety of microbial metabolites can function as radical-trapping antioxidants (RTAs), directly neutralizing lipid radicals and thereby inhibiting lipid peroxidation. The actions of these metabolites operate in parallel with classical enzymatic systems such as GPX4 and FSP1, forming a rapid, front-line defense against oxidative damage. Vitamin A derivatives (retinol and all-trans-retinal) and several products of the tryptophan metabolic pathway, including l-kynurenine (L-KYN), 3-hydroxykynurenine (3-HK), 3-HA, and serotonin (5-HT), have all been identified as effective RTAs (50, 75, 95, 96). Functionally analogous to FSP1, these molecules directly quench free radicals within the lipid peroxidation chain reaction, independently of the GPX4–GSH axis, thereby constituting an autonomous and parallel layer of protection against ferroptosis.
3.2.2 Metabolites influencing ROS accumulation
Various microbial metabolites directly or indirectly regulate intracellular ROS levels, thereby profoundly influencing the process of ferroptosis. Among them, LPS, a key component of the cell wall of Gram-negative bacteria, typically triggers a strong inflammatory response and oxidative stress by activating Toll-like receptor 4 (TLR4) on immune cells such as macrophages, leading to a significant increase in intracellular ROS levels. This sharp rise in ROS is an important mechanism for initiating antimicrobial defense in the body, but its excessive accumulation can also cause severe cellular damage (81). Among the vitamin B (VB) metabolites, different members exhibit significant differences in regulating ferroptosis, forming a precise regulatory network. For example, riboflavin (VB2) shows potential value in cancer treatment. Studies have shown that a complex formed by VB2 and ferric chloride encapsulated in nanomaterials can target cancer cells at the tumor site, synergistically promoting the accumulation of iron ions and the generation of ROS within the cancer cells, thereby inducing ferroptosis and effectively inhibiting the growth of breast tumors (90). In contrast, folate (VB9) and cobalamin (VB12) inhibit ferroptosis. Their mechanisms are as follows: folic acid primarily blocks the ferroptosis process by reducing glutamate levels, while cobalamin exerts a protective effect by inhibiting the sterol regulatory element-binding protein (SREBP) binding protein-1 (SBP-1)/SREBP1 lipogenesis axis and reducing lipid peroxidation (72, 73).
3.2.3 Regulation of ROS through NOX enzymes and mitochondrial function
Beyond direct modulation of antioxidant defense systems, the gut microbiota and their metabolites influence ferroptosis susceptibility by regulating the primary sources of ROS generation. With respect to NOX enzyme regulation, Lactobacillus rhamnosus has been shown to upregulate NOX1 expression via the NRF2 signaling pathway (97), while segmented filamentous bacteria enhance Dual Oxidase 2 (DUOX2) expression levels (98); Conversely, certain microbial metabolites, particularly SCFAs, exert inhibitory effects on NOX activity. For instance, butyrate suppresses NOX2 expression and increases superoxide dismutase (SOD) activity through activation of the P21/NRF2/NF-κB pathway (56), whereas acetate downregulates NOX2 in T cells via histone deacetylase (HDAC) inhibition (61). In terms of mitochondrial regulation, butyrate enhances oxidative phosphorylation and β-oxidation and improves mitochondrial efficiency through activation of the PGC-1α signaling pathway, collectively reducing ROS production and inhibiting ferroptosis (57–59). Similarly, urolithin A, a metabolite derived from gut microbial metabolism of ellagic acid, mitigates ferroptosis by promoting mitophagy, the selective clearance of dysfunctional mitochondria, which are major ROS generators (66). This represents a compensatory mechanism that indirectly supports classical antioxidant systems by preserving mitochondrial integrity and maintaining organelle homeostasis.
3.2.4 Metabolites that indirectly regulate ROS through antioxidant pathways
A major group of microbial metabolites contributes to ROS homeostasis indirectly by activating intracellular antioxidant signaling pathways or serving as critical metabolic cofactors. Their principal function is to enhance or regulate the classical ferroptosis defense systems. VB2 strengthens the system Xc−–GPX4 axis through a mechanism independent of IL-17A (99). Similarly, bile acids such as cholic acid and chenodeoxycholic acid (CDCA) activate the Farnesoid X Receptor (FXR), which upregulates the expression of GSH synthase and GPX4, thereby reinforcing this core antioxidant defense network at the transcriptional level (100, 101). Regarding the FSP1-CoQ10 system, the reduced form of vitamin K (VKH2) acts in an FSP1-dependent manner, identifying VKH2 as a key cofactor in this non-canonical antioxidant pathway (71). In addition, the tryptophan-derived metabolite trans-3-indoleacrylic acid (IDA) activates the aryl hydrocarbon receptor (AHR), induces ALDH1A3 expression, and subsequently promotes FSP1-mediated regeneration of reduced CoQ10, thereby strengthening the FSP1–CoQ10 axis (63). Indole-3-acetic acid (IAA), another AHR agonist, enhances antioxidant capacity via two complementary routes. Notably, it promotes SLC7A11-dependent GSH synthesis (64) and provides NADH necessary for CoQ10 biosynthesis (63), effectively bridging the system Xc−–GSH pathway and the CoQ10 regeneration system. Likewise, L-lactate indirectly suppresses ferroptosis by inactivating AMPK, which in turn upregulates SREBP1/SCD1 signaling and promotes the synthesis of anti-ferroptotic MUFAs (65). Through this mechanism, L-lactate supports membrane remodeling to resist lipid peroxidation, complementing the classical ferroptosis defense systems.
In summary, the intestinal microbiota and their metabolites form a multilayered regulatory network that maintains ROS balance. They provide direct antioxidant capacity in parallel with GPX4 and FSP1, modulate classical defense systems via transcriptional and signaling pathways, and control ROS production at its sources through regulation of NOX enzymes and mitochondrial function. Disruption of this intricate network is a critical determinant in the onset of ferroptosis and represents a promising therapeutic target for related diseases.
3.3 Regulation of intestinal flora and metabolites on fatty acid metabolism and lipid peroxidation
The gut microbiota and their metabolites play a pivotal role in lipid homeostasis and ferroptosis by modulating fatty acid metabolism, influencing redox balance, and regulating cell death pathways.
3.3.1 Regulation of fatty acid absorption and metabolism
Gut microbes finally regulate host fatty acid metabolism through the biotransformation of dietary fatty acids and the production of bioactive signaling molecules. Certain bacteria, such as Lactobacillus plantarum, can convert linoleic acid into conjugated linoleic acid and its active intermediates, thereby activating the host NRF2-ARE antioxidant signaling pathway (53). Similarly, commensal bacteria such as Lactobacillus intestinalis metabolize dietary vitamin A into its active derivatives, including retinoic acid (102). These vitamin A metabolites, retinol and all-trans-retinal, not only function as potent RTAs (95) but also generate NADH through the ALDH1A3-catalyzed reaction, providing the reducing power necessary for the FSP1-CoQ10 system (63), In this way, they serve as important metabolic supplements to the canonical FSP1-CoQ10 antioxidant pathway. Other microbial metabolites influence lipid metabolism through distinct mechanisms. Glycodeoxycholic acid (GDCA) promotes ferroptosis by activating the TFR-ACSL4 axis and reducing lipid digestibility (84). LPS upregulates ACSL4 expression via activation of the Sp1 transcription factor in esophageal tissue (80). Additionally, Pseudomonas aeruginosa can alter the host’s lipid metabolic profile by modulating the activity of fatty acyl-CoA ligases (77).
3.3.2 Direct regulation of lipid peroxidation
Microbial metabolites directly modulate lipid peroxidation through multiple mechanisms, forming a complex regulatory network that interfaces with the classical ferroptosis pathways (Figure 4).
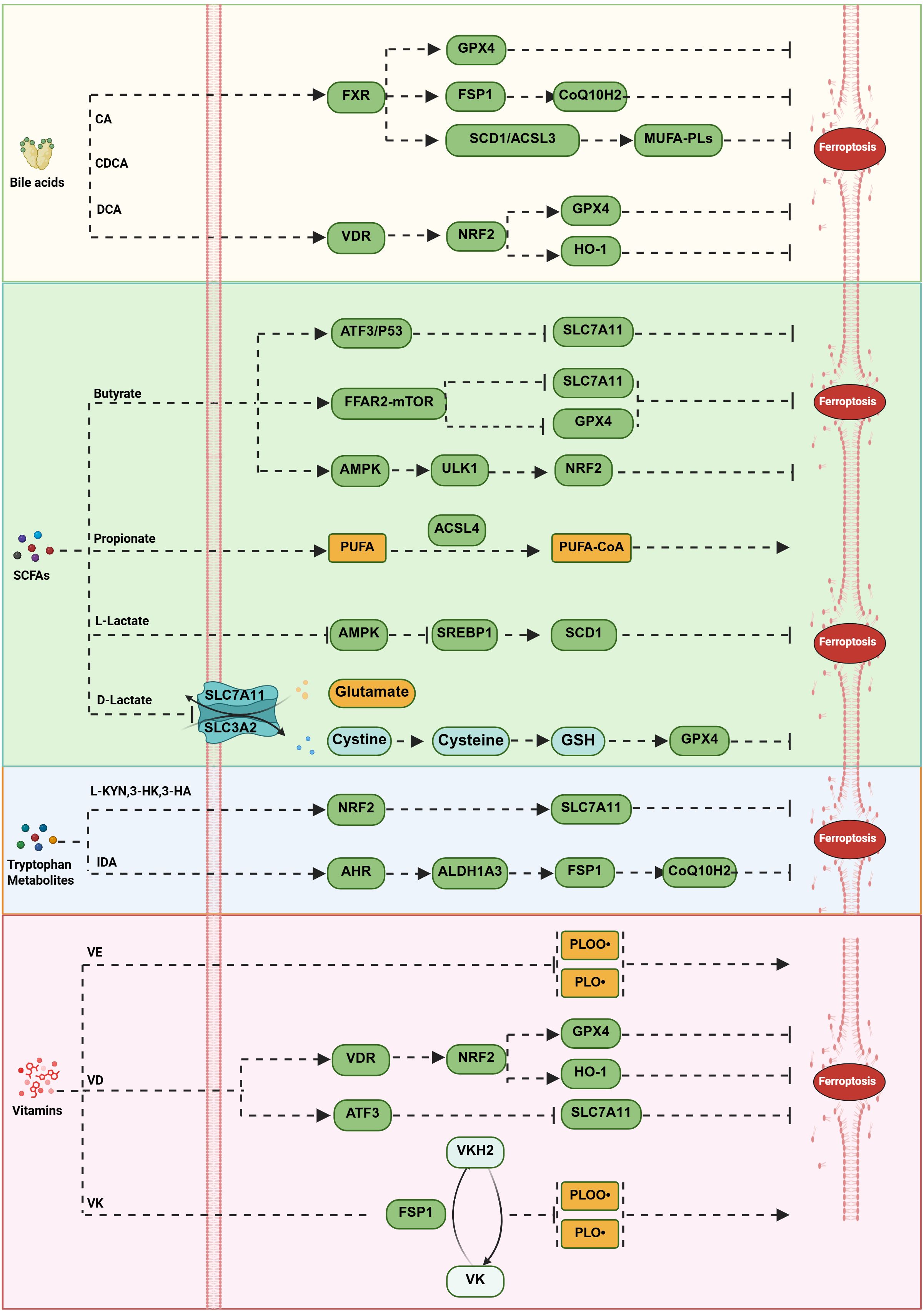
Figure 4. Modulation of lipid peroxidation by gut flora and metabolites. Microbial metabolites regulate lipid peroxidation through multiple mechanisms, forming a complex interaction network with ferroptosis pathways. Bile acids exert dual regulatory effects via nuclear receptors: FXR activation upregulates genes such as GPX4 and FSP1, enhancing the GPX4-GSH and FSP1-CoQ10 systems while inducing SCD1 and ACSL3 to promote the synthesis of monounsaturated fatty acid phospholipids. VDR activation inhibits ferroptosis through pathways like NRF2/GPX4 and NRF2/HO-1. SCFAs exhibit pleiotropic effects: butyrate promotes ferroptosis via the FFAR2–mTORC1 axis by downregulating GPX4 and SLC7A11, mitigates ferroptosis through the AMPK–ULK1–p62 pathway, and also promotes it via ATF3–SLC7A11 and p53–SLC7A11 signaling pathways; propionate promotes ferroptosis by enhancing polyunsaturated fatty acid production and generating more polyunsaturated fatty acid-CoA through ACSL4 action; L-lactate inhibits ferroptosis by suppressing AMPK and upregulating SREBP1 and its downstream target SCD1, whereas D-lactate promotes ferroptosis by downregulating the antiferroptotic gene SLC7A11 and reducing GSH synthesis. Tryptophan metabolites exert protective effects through multiple mechanisms: kynurenine pathway products (L-KYN, 3-HK, and 3-HA) activate NRF2-mediated SLC7A11 expression to prevent ferroptosis; IDA inhibits ferroptosis by enhancing the FSP1-CoQ10 system via the AHR-ALDH1A3 signaling axis. Vitamin metabolites constitute an antioxidant defense system: vitamin E, converted to α-tocopherol, directly scavenges lipid peroxides; VD inhibits ferroptosis through VDR-regulated NRF2–GPX4 and NRF2–HO-1 pathways while inducing it by upregulating ATF3 to reduce SLC7A11 expression; VKH2 suppresses lipid peroxidation in an FSP1-dependent manner.
Bile acids exert dual and context-dependent regulation through nuclear receptor signaling. Activation of the FXR upregulates key antioxidant genes such as GPX4 and FSP1 (67), thereby strengthening both the GPX4-GSH and FSP1-CoQ10 defense systems. Simultaneously, FXR induces SCD1 and ACSL3, promoting the synthesis of anti-ferroptotic MUFA-PLs (68), which provide crucial metabolic support for membrane protection. However, under specific conditions, DCA can sensitize cells to ferroptosis by promoting iron accumulation (83), highlighting its dual and context-dependent role. Activation of the vitamin D receptor (VDR) also confers anti-ferroptotic effects via the NRF2/GPX4 and NRF2/HO-1 signaling axis (69, 70), further broadening the regulatory scope of nuclear receptor-mediated control over classical ferroptosis.
SCFAs participate in a multi-layered regulatory network that can either promote or inhibit ferroptosis. Butyrate can downregulate GPX4 and SLC7A11 via the FFAR2–mTORC1 axis, thereby promoting ferroptosis (85, 86), constituting a direct intervention on the GPX4-GSH system. Other studies indicate that in lung cancer and osteosarcoma cells, butyrate enhances erastin-induced ferroptosis by regulating the ATF3–SLC7A11 pathway (87, 88). Additionally, butyrate can activate NRF2 through the AMPK–ULK1–p62 signaling pathway, subsequently alleviating ferroptosis in acute liver injury (60). Propionate promotes ACSL4-regulated ferroptosis (70), altering lipid substrate composition to increase cellular sensitivity to ferroptosis. Lactate isomers achieve bidirectional regulation via different pathways: L-lactate inhibits ferroptosis by suppressing AMPK and upregulating SREBP1 and its downstream target gene SCD1; whereas D-lactate promotes ferroptosis by downregulating the anti-ferroptosis gene SLC7A11 and reducing GSH synthesis (65, 89). The two isomers achieve fine-tuning of the ferroptosis threshold through distinct signaling pathways.
Tryptophan-derived metabolites contribute additional layers of protection. Metabolites from the kynurenine pathway act as RTAs while also activating the NRF2 pathway (50, 62), providing both direct antioxidant defense and indirect enhancement of the GPX4-GSH system. 5-HT directly reduces peroxidized phospholipids (62, 96), functioning independently of known classical antioxidant mechanisms. Moreover, IDA reinforces the FSP1-CoQ10 pathway via the AHR-ALDH1A3 signaling axis (63), exemplifying a novel mode through which microbial metabolites modulate ferroptosis defenses.
Vitamin metabolites constitute a crucial antioxidant defense system: Vitamin E, converted to α-tocopherol, can directly scavenge lipid peroxides (75). This action is independent of the GPX4-GSH system, providing an independent antioxidant safeguard. Vitamin D (VD), through VDR, regulates the NRF2–GPX4 and NRF2–HO-1 signaling pathways (69, 74), thereby inhibiting ferroptosis; VD can also induce ferroptosis by upregulating ATF3 transcription to reduce SLC7A11 expression (91). The reduced form of VKH2 inhibits lipid peroxidation in an FSP1-dependent manner (103), forming a regulatory branch parallel and independent to the classical FSP1-CoQ10 system.
3.3.3 Regulation via enzyme systems and pathogen interactions
Microorganisms regulate lipid peroxidation through enzymatic systems and direct host-pathogen interactions, forming complex intersections with the classical ferroptosis pathways. LOXs are core executors of lipid peroxidation, and microbial modulation of LOX activity significantly influences ferroptosis susceptibility. For instance, gut dysbiosis can promote neuroinflammation through activation of 5-LOX, whereas beneficial microbes such as Acetobacter, yeast, and Lactobacillus exert protective effects by inhibiting 15-LOX activity (54). In contrast, pathogenic bacteria like Pseudomonas aeruginosa exploit host 15-LOX to induce ferroptosis directly (78), a mechanism independent of, yet potentially synergistic with, GPX4 dysfunction. Similarly, Mycobacterium bovis BCG infection promotes ferroptosis by downregulating GPX4 and FSP1 (79), directly impairing two major antioxidant defense systems. Conversely, engineered probiotics such as Lactococcus lactis MG1363-pMG36e-GLP-1 have been shown to inhibit ferroptosis via activation of the Keap1/NRF2/GPX4 signaling pathway (55), demonstrating the therapeutic potential of beneficial microbes in reinforcing host ferroptosis resistance.
Collectively, these findings indicate that microbial metabolites and interactions do not simply replicate the actions of classical ferroptosis pathways. Instead, they form a highly integrated defense network that interconnects with canonical systems through independent regulatory modules, metabolic support, and compensatory mechanisms. This multi-layered regulation provides new insight into the physiological and pathological roles of ferroptosis and identifies promising microbial and metabolic targets for therapeutic intervention in ferroptosis-related diseases.
4 Ferroptosis and digestive tumors
Ferroptosis plays crucial roles in the proliferation, invasion, and metastasis of gastrointestinal tumors, as well as in the resistance of these tumors to radiotherapy and chemotherapy (Table 2).
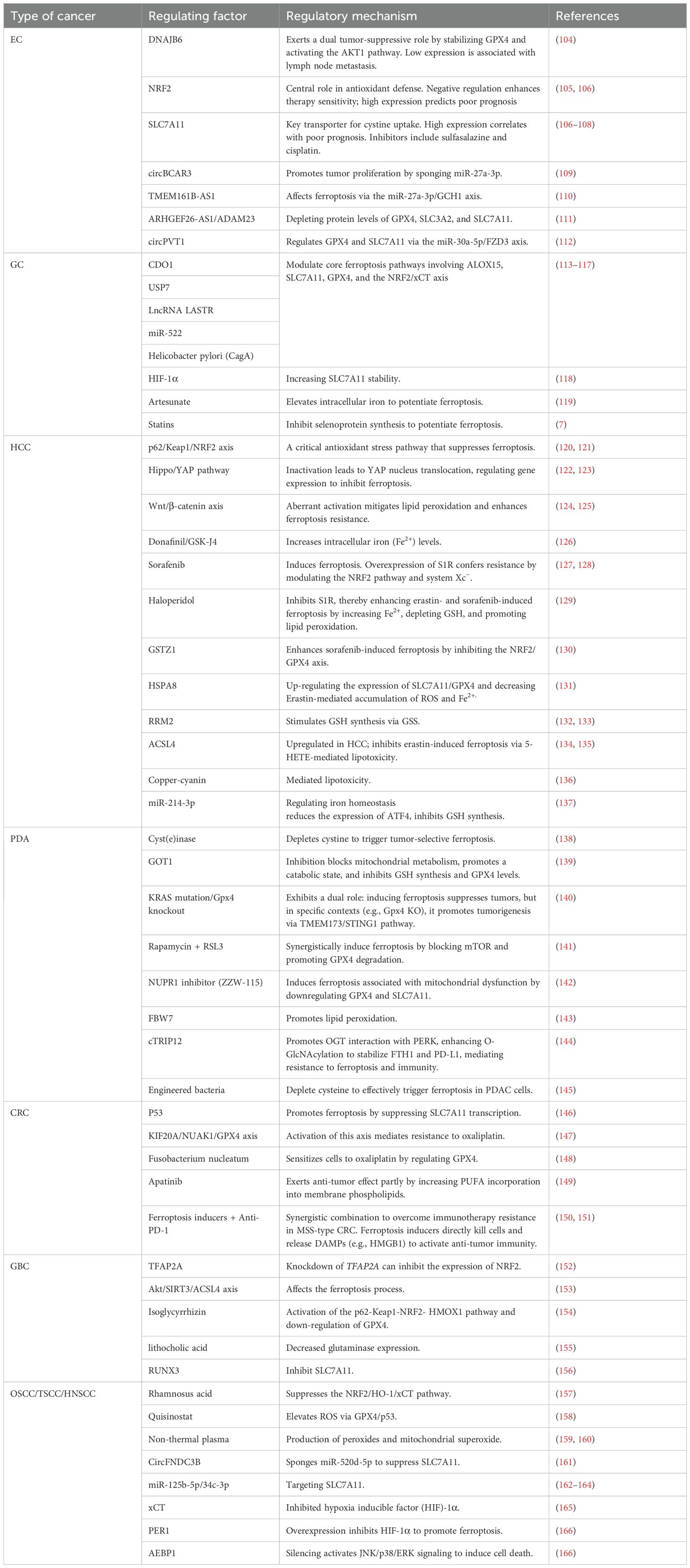
Table 2. Role of ferroptosis in digestive tract tumor.
4.1 Ferroptosis and esophageal cancer
Esophageal carcinoma (EC) remains one of the most prevalent and lethal malignancies worldwide. Treatment of advanced or unresectable EC continues to face the major challenge of resistance to radiotherapy and chemotherapy. In recent years, ferroptosis, an iron-dependent form of regulated cell death characterized by lipid peroxidation, has emerged as a promising mechanism to overcome therapeutic resistance. Evidence suggests that conventional radiotherapy and chemotherapy can induce ferroptosis to a certain degree, and precise modulation of this process may offer an effective strategy to reverse treatment resistance (167).
At the molecular level, ferroptosis is governed by a complex multi-layered regulatory network. GPX4, the core negative regulator of ferroptosis, can be directly inhibited by small molecules such as RSL3 (168), and indirectly modulated by chaperone proteins including HSP27 (169) and DNAJB6. Notably, reduced expression of DNAJB6 is closely associated with lymph node metastasis in EC (104). DNAJB6 exerts a dual tumor-suppressive role by stabilizing GPX4 and activating the signaling AKT1 pathway (170). Concurrently, the transcription factor NRF2 plays a central role in cellular antioxidant defense, and its expression level is a critical determinant of therapeutic response. Downregulation of NRF2 enhances sensitivity to radiotherapy and chemotherapy (105), whereas its overexpression correlates with poor clinical prognosis (106). Natural compounds such as liensinine (171) and polygalacin D (172) have been shown to promote ferroptosis by modulating NRF2 expression. The NRF2-ARF interaction also represents a key mechanism underlying p53-independent ferroptosis (173). SLC7A11, the cystine/glutamate antiporter, functions as another pivotal regulatory hub in ferroptosis. Pharmacological inhibitors of SLC7A11, such as sulfasalazine (107) and cisplatin (108), act through distinct mechanisms to disrupt redox homeostasis. Clinical studies further confirm that elevated SLC7A11 expression is significantly associated with poor prognosis in EC patients (106).
Recent discoveries involving competing endogenous RNA (ceRNA) networks have added a layer of post-transcriptional regulation to ferroptosis in EC. For example, circBCAR3 promotes tumor proliferation by sponging miR-27a-3p (109); TMEM161B-AS1 modulates ferroptosis via the miR-27a-3p/GCH1 axis (110); ARHGEF26-AS1 promotes ferroptosis through the miR-372-3p/ADAM23 pathway (111); and circPVT1 regulates GPX4 and SLC7A11 expression via the miR-30a-5p/FZD3 axis (112). Collectively, these findings reveal a complex post-transcriptional regulatory network that links non-coding RNAs to ferroptosis control, offering novel molecular entry points for the development of targeted therapeutic strategies in esophageal cancer.
In the realm of radio- and chemosensitization, accumulating evidence indicates that ionizing radiation can enhance the susceptibility of EC cells to ferroptosis by upregulating the ferroptosis marker gene PTGS2 and promoting lipid peroxidation (174). Targeting SCD1 (175) or epigenetically silencing METTL3 (176) which affects the m6A modification of SLC7A11 and GPX4, can trigger ferroptosis and improve radiosensitivity. Significant progress has also been achieved in chemotherapy-related ferroptosis research. Multiple conventional anticancer drugs have been shown to promote ferroptosis through diverse mechanisms, including inhibition of system Xc−, depletion of intracellular GSH, or modulation of GPX4 activity. For example, PLK1 knockdown increases lipid peroxidation and enhances sensitivity to paclitaxel and cisplatin (177). Similarly, depletion of HMGA1 (178) or ALDH5A1 (179) disrupts intracellular redox homeostasis, while PARP inhibitors (180) or silencing of GLRX5 (181) can potentiate cisplatin sensitivity via ferroptosis, particularly in specific genedeficient models.
Therapeutic intervention strategies are becoming increasingly diverse. Small-molecule inhibitors such as erastin and RSL3 play important roles by targeting system Xc− and GPX4, respectively. Inhibition of SLC7A11 effectively reverses chemoresistance in p53-deficient EC cells (182), while NRF2 inhibitors such as Brusatol (183) enhance radiosensitivity by promoting lipid peroxidation. Natural compounds also display unique advantages. Ferulic acid (184) and oridonin (185) induce ferroptosis by directly inhibiting system Xc− activity, whereas berbamine facilitates GPX4 degradation (186). Alantolactone (187) and licochalcone A (188) indirectly promote ferroptosis by modulating signaling pathways, including p53 and ROS. In addition, realgar (189) and brusatol (190) enhance chemosensitivity by suppressing NRF2 activity, highlighting the therapeutic potential of natural compounds in ferroptosis regulation.
The integration of photodynamic therapy (PDT) and immunotherapy has opened new therapeutic frontiers in EC. Photosensitizers such as talaporfin (191) and 5-aminolevulinic acid (5-ALA) effectively induce ferroptosis by generating ROS and inhibiting the system Xc−/GPX4 axis. Moreover, ALA-PDT modulates serum high-mobility group box 1 (HMGB1) levels and macrophage morphology, thereby contributing to enhanced antitumor immunity (192). In immunotherapy, inhibition of NQO1 has been shown to simultaneously induce ferroptosis and increase responsiveness to immune checkpoint blockade (193). Furthermore, advanced nanomaterials offer promising synergistic strategies. For instance, Fe-MOF/CP (194) and 2DG@FS-Nb (195) nanoparticles can deplete intracellular GSH and regulate macrophage polarization states, significantly enhancing anti-tumor immune responses when combined with PD-1 inhibitors.
The advent of nanotechnology has further propelled the development of ferroptosis-based therapies. Iron-based nanozymes effectively induce ferroptosis by catalyzing the Fe³+/Fe2+ redox cycle and depleting intracellular GSH (196). In addition, various nano-drug delivery systems not only improve drug targeting and bioavailability (197) but also exhibit synergistic therapeutic effects when combined with radiotherapy (198) or chemotherapy (199), achieving precise induction of ferroptosis and amplifying immunogenic cell death in a coordinated manner (200).
In summary, ferroptosis-targeted therapy in EC has progressed from single-target approaches to a new stage of multi-target, synergistic regulation. Close cross-regulation exists among several key molecular pathways. For instance, p53 can induce ferroptosis by inhibiting SLC7A11 (201), while its mutation status influences miR-27a-3p expression (109). Similarly, the Hippo-YAP pathway modulates ferroptosis sensitivity by regulating TFRC and ACSL4 (202) and exhibits reciprocal interactions with GPX4 and SLC7A11. These findings suggest that simultaneously targeting multiple regulatory nodes may yield superior therapeutic outcomes. With the deepening understanding of ferroptosis mechanisms and their integration with conventional modalities such as radiotherapy, chemotherapy, and immunotherapy, ferroptosis-targeted strategies are poised to offer new avenues for improving the prognosis of EC patients.
4.2 Ferroptosis and gastric cancer
Ferroptosis, an iron-dependent, lipid peroxidation-driven form of regulated cell death, has emerged as a critical mechanism in gastric cancer (GC), profoundly influencing tumorigenesis, therapeutic responsiveness, and tumor microenvironment (TME) dynamics. The therapeutic induction of ferroptosis holds dual strategic value. First, it directly suppresses tumor proliferation and metastasis. Targeting key negative regulators such as GPX4, or applying natural ferroptosis inducers like DHPO, can effectively trigger ferroptotic cell death and synergize with emerging modalities such as CAR-T therapy (113, 203). Second, ferroptosis represents a promising approach to overcoming chemotherapy resistance. Classical inducers such as erastin and RSL3 sensitize GC cells to standard chemotherapeutic agents, including cisplatin (204, 205). Moreover, ferroptosis exerts potent immunomodulatory effects by remodeling the immunosuppressive TME, characterized by elevated ROS levels and reduced expression of immune checkpoints such as PD-L1 (206, 207). The combination of erastin with PD-1 inhibitors demonstrates synergistic tumor clearance, highlighting the dual mechanism of ferroptosis inducers: direct cytotoxicity against cancer cells and enhancement of antitumor immunity.
The execution of ferroptosis in GC is governed by a highly intricate, multi-layered regulatory network. At the molecular level, a diverse array of factors, including non-coding RNAs (e.g., miR-522, LncRNA LASTR), regulatory proteins (e.g., USP7, ATF3, CDO1), and Helicobacter pylori infection (through virulence factors such as CagA), precisely modulate core ferroptosis pathways involving ALOX15, SLC7A11, GPX4, and the NRF2/xCT axis (113–117). Within the TME, cellular interactions further shape ferroptotic susceptibility. The elevated iron demand of cancer cells sustains their rapid proliferation, whereas immune infiltrates exert contrasting effects: CD8+ T cells can promote ferroptosis by secreting IFN-γ, which downregulates SLC3A2/SLC7A11 (208), while M2-polarized macrophages often suppress ferroptosis and facilitate tumor progression. Moreover, ferroptosis-associated danger signals such as HMGB1 can reinforce this pro-tumor state by promoting M2 macrophage polarization, establishing a self-perpetuating feedback loop (209). Pathophysiological conditions, including hypoxia, hypoxia-induced factor 1α (HIF-1α)/PMAN signaling has been shown to inhibit ferroptosis, thereby conferring survival advantages to tumor cells under oxygen-deprived conditions (118).
The successful clinical translation of ferroptosis-based therapies depends on the identification of reliable biomarkers and the development of innovative therapeutic modalities. Biomarkers such as GPX4 expression levels (210) accumulation of the lipid peroxidation product 4-HNE, and the transport activity of ABCC2 have emerged as valuable indicators for patient stratification, prognosis prediction, and therapeutic monitoring (211, 212). Therapeutically, beyond the classical system Xc− inhibitors (e.g., erastin) and GPX4 inhibitors (e.g., RSL3), multiple agents have been explored to enhance ferroptosis through diverse mechanisms: artesunate increases intracellular iron levels; statins inhibit seleno protein synthesis; and microenvironment-responsive nanocarriers enable targeted delivery of ferroptosis inducers (7, 119, 213, 214). In addition, emerging metabolic targets such as MAT2A exhibit synergistic effects when combined with established ferroptosis inducers, particularly in tumors exhibiting methionine dependency (215).
In summary, targeting ferroptosis represents a promising and multifaceted therapeutic strategy in GC. Nonetheless, several obstacles hinder its clinical implementation, including tumor plasticity that promotes adaptive resistance, limitations in the precision and efficiency of drug delivery systems, and the ferroptosis-suppressive nature of the TME. Future breakthroughs will require a deeper mechanistic understanding of ferroptosis regulation within the TME, alongside the integration of nanotechnology, immunotherapy, and clinical oncology. Such interdisciplinary approaches hold the potential to transform ferroptosis from a mechanistic concept into a clinically controllable and precise anticancer strategy.
4.3 Ferroptosis and liver cancer
In hepatocellular carcinoma (HCC), ferroptosis, an iron-dependent form of regulated cell death, has emerged as a significant focus in cancer research. As the central organ responsible for iron metabolism, the liver is particularly vulnerable to disturbances in iron homeostasis, with iron overload acting as a direct trigger of ferroptosis (216). Although the induction of ferroptosis can effectively suppress HCC progression, tumor cells frequently develop adaptive resistance by activating key signaling pathways or altering the expression of ferroptosis-related genes, thereby making ferroptosis both a therapeutic challenge and an attractive target (217).
The core regulatory mechanism of ferroptosis revolves around the cellular antioxidant defense network, with the GPX4-dependent pathway playing a fundamental role (218). The proper function of this system relies on adequate intracellular GSH levels and the enzymatic activity of GPX4. Specifically, the system Xc− transporter, particularly the SLC7A11 subunit, facilitates the uptake of extracellular cystine, a rate-limiting precursor for GSH synthesis (219). GSH serves as a crucial cofactor for GPX4, reducing pro-ferroptotic lipid peroxides into non-toxic lipid alcohols, thereby preventing ferroptotic cell death (218). Consequently, any factor that disrupts system Xc− activity (220), depletes GSH, or inhibits GPX4 function (221) promotes the accumulation of lipid peroxides and ultimately triggers ferroptosis.
Beyond the canonical GPX4 axis, several signaling pathways intricately fine-tune ferroptosis regulation in HCC. The p62/Keap1/NRF2 pathway constitutes a major antioxidant defense mechanism: p62 competitively binds Keap1, leading to NRF2 stabilization and activation. Activated NRF2 upregulates a series of antioxidant and iron metabolism-related genes, collectively suppressing ferroptosis (120, 121). Similarly, inactivation of the Hippo pathway allows its downstream effector YAP to translocate into the nucleus, where it modulates gene expression to inhibit ferroptosis and promote tumor growth (122, 123). Moreover, aberrant activation of the Wnt/β-catenin signaling cascade synergistically enhances ferroptosis resistance in HCC cells, in part by attenuating lipid peroxidation (124, 125). Collectively, these interconnected signaling networks form a complex defense system that enables HCC cells to evade ferroptotic death.
Ferroptosis regulators can be broadly classified into inducers and inhibitors based on their functional roles. Ferroptosis inducers are further categorized by their mechanisms of action into system Xc− inhibitors, GPX4 inhibitors, and GSH-depleting agents (222). Notably, some compounds, such as FINO2, do not directly inhibit GPX4 or system Xc− but instead inactivate GPX4 enzymatic activity indirectly through iron oxidation (223). Conversely, β-mercaptoethanol can bypass system Xc− inhibition by directly facilitating cystine uptake into cells (224, 225). Ferroptosis inhibitors, on the other hand, including endogenous antioxidants, synthetic RTAs, members of the lysyl oxidase family, and iron chelators, can effectively prevent or mitigate ferroptotic cell death. In recent years, non-coding RNAs, including lncRNAs, miRNAs, and circRNAs, have emerged as novel and versatile regulators of ferroptosis. By modulating genes and proteins involved in iron metabolism, lipid metabolism, and antioxidant defense, non-coding RNAs play crucial roles in HCC development, therapeutic resistance, and treatment response (226, 227).
Growing evidence highlights the therapeutic potential of targeting ferroptosis in HCC. For example, the combination of donafenib and GSK-J4 induces ferroptosis by upregulating heme oxygenase-1 (HMOX1), thereby elevating intracellular Fe2+ concentrations (126). Conversely, in sorafenib-treated HCC cells, overexpression of the sigma-1 receptor (S1R) reduces oxidative stress via modulation of the NRF2 pathway and system Xc−, conferring ferroptosis resistance (127, 128). Conversely, haloperidol-mediated inhibition of S1R enhances erastin- and sorafenib-induced ferroptosis by increasing Fe2+ accumulation, depleting GSH, and promoting lipid peroxidation (129). Similarly, the recombinant protein GSTZ1 enhances sorafenib-induced ferroptosis by suppressing the NRF2/GPX4 axis (130). During HBV-associated hepatocarcinogenesis, HSPA8 inhibits ferroptosis by upregulating SLC7A11 and GPX4, thereby reducing erastin-induced ROS and Fe2+ accumulation. Inhibition of HSPA8 not only suppresses tumor growth but also sensitizes HBV-positive HCC cells to ferroptosis, highlighting its therapeutic relevance (131). Likewise, the ribonucleotide reductase regulatory subunit M2 (RRM2), frequently overexpressed in HCC tissues, promotes GSH biosynthesis via glutathione synthetase (GSS), thus protecting cells from ferroptosis and contributing to sorafenib resistance (132, 133). Additionally, long-chain ACSL4 is upregulated in HCC and inhibits erastin-induced ferroptosis through 5-hydroxyeicosatetraenoic acid (5-HETE)-mediated lipotoxicity (134, 135). In contrast, inhibition of copper cyanide, a ferroptosis suppressor, enhances Fe2+ and ROS accumulation, thereby promoting erastin- and RSL3-induced ferroptosis (136). Non-coding RNAs also play key regulatory roles in HCC ferroptosis. For instance, miR-214-3p promotes ferroptosis by downregulating ATF4 and inhibiting GSH synthesis (137). Such findings highlight non-coding RNAs as promising molecular targets for modulating ferroptotic sensitivity in HCC.
In summary, ferroptosis serves as a central mechanism in the pathogenesis, progression, and treatment responsiveness of HCC. The regulation of this process involves a highly interconnected network centered on the system Xc−/GSH/GPX4 axis, along with multiple signaling pathways, including p62/Keap1/NRF2, Hippo/YAP, and Wnt/β-catenin. Therapeutic strategies that target these pathways, particularly in combination with ncRNA-based interventions or small-molecule modulators, represent a powerful approach for precision treatment of HCC, offering broad prospects for translational research and clinical application.
4.4 Ferroptosis and pancreatic cancer
Pancreatic ductal adenocarcinoma (PDAC), a highly aggressive malignancy, develops through a well-characterized progression from normal ductal epithelium to precancerous lesions and ultimately to invasive carcinoma. Although this multistep process is influenced by diverse genetic and environmental factors, the persistent activation of oncogenic KRAS within the inflammatory TME is recognized as the core mechanism (228). Intriguingly, ferroptosis, an iron-dependent, regulated form of cell death characterized by the accumulation of lipid peroxides, was originally identified as a Ras mutation-dependent process upon its discovery (229). This origin-related association suggests the particular therapeutic potential of targeting ferroptosis in KRAS-mutant pancreatic cancer. However, accumulating evidence has revealed a paradoxical duality in the role of ferroptosis within PDAC: under certain conditions, it acts as a potent tumor-suppressive mechanism, whereas in others, it paradoxically promotes tumor initiation and progression.
This dual nature has been clearly demonstrated in multiple genetically engineered mouse models (GEMMs). In the KPFSR model, tamoxifen-induced systemic deletion of Slc7a11 triggered tumor-selective ferroptosis and significantly inhibited tumor growth. Similarly, cystine depletion using cyst(e)inase reproduced significant tumor suppression in the KPC model (138). Mechanistically, beyond directly disrupting cystine uptake, targeting downstream metabolic nodes can also effectively induce ferroptosis. For instance, inhibition of cytosolic aspartate aminotransferase disrupts cellular metabolic balance and enhances autophagic flux, increases the labile iron pool, and ultimately promotes ferroptotic cell death (139). Paradoxically, in pancreas-specific Gpx4 knockout KC models or under high-iron diet conditions, ferroptosis instead facilitated tumorigenesis (140). Mechanistic investigations revealed that ferroptosis-induced oxidative DNA damage results in the release of 8-hydroxyguanosine (8-OHG), which activates the TMEM173/STING1-dependent DNA sensing pathway, promoting macrophage recruitment and activation during the early stages of tumor formation. Another study demonstrated that autophagy-dependent ferroptosis enables tumor cells to release mutant KRASG12D protein via exosomes, driving macrophage polarization toward the pro-tumorigenic M2 phenotype (140). This seemingly contradictory behavior of ferroptosis in PDAC likely depends on contextual factors, including differences in genetic background (such as TP53 mutation status) and the pleiotropic functions of key regulators like GPX4 and SLC7A11, which participate in both ferroptosis-dependent and ferroptosis-independent cellular pathways.
The influence of ferroptosis in pancreatic cancer extends well beyond tumor cell death; it profoundly reshapes the tumor immune microenvironment through the release of diverse signaling molecules. On one hand, ferroptosis can promote M2-type tumor-associated macrophage (TAM) polarization via the release of KRASG12D, thereby promoting an immunosuppressive milieu (140). On the other hand, ferroptosis can also activate anti-tumor immune responses by stimulating the production of pro-inflammatory cytokines in an NF-κB-dependent manner. This occurs through the release of decorin, which binds to the AGER receptor on macrophages, driving immune activation (230). Similarly, HMGB1, released during ferroptosis, functions as a damage-associated molecular pattern (DAMP) molecule recognized by antigen-presenting cells, triggering innate and adaptive immune responses (209). Collectively, these findings indicate that the net immunological outcome of ferroptosis in pancreatic cancer depends on the immune microenvironment depends on multiple contextual factors, including the specific signaling molecules released, their spatiotemporal dynamics, and the phenotypes of the immune cells that receive these signals. Although bioinformatics analyses have preliminarily constructed association networks linking ferroptosis regulators to patterns of immune cell infiltration (231), these correlations remain predictive and require rigorous experimental validation.
Given the broad resistance of pancreatic cancer to conventional chemotherapy, therapeutically inducing ferroptosis has emerged as a promising approach to overcome drug resistance. Targeting key regulatory nodes has shown encouraging results: the combination of rapamycin and RSL3 efficiently induces ferroptosis by synergistically inhibiting mTOR signaling and promoting GPX4 degradation (141). Drug repositioning also holds potential: zalcitabine, for instance, induces mitochondrial DNA stress and activates the STING1 pathway, leading to autophagy-dependent ferroptosis (232). Similarly, the NUPR1 inhibitor ZZW-115 triggers ferroptosis associated with mitochondrial dysfunction by downregulating GPX4 and SLC7A11 expression (142). Among natural products, a triple combination of piperlongumine, cortinin A, and sulfasalazine demonstrates synergistic efficacy in inducing ferroptosis (233). Moreover, artesunate and its active metabolite dihydroartemisinin exhibit selective cytotoxicity against RAS-mutant pancreatic cancer cells, primarily through mechanisms involving iron metabolism dysregulation (234). In terms of combination therapies, ferroptosis inducers such as erastin and RSL3 can enhance the cytotoxicity of gemcitabine (143), while the co-administration of dihydroartemisinin and cisplatin exerts synergistic anti-tumor effects by simultaneously inducing ferroptosis and exacerbating DNA damage (235).
Chemotherapy resistance remains a major obstacle in the clinical management of pancreatic cancer, and the ferroptosis pathway plays a complex and context-dependent role in this process. Tumor cells can evade ferroptotic death by activating adaptive stress responses. For instance, the gemcitabine-induced ATF4-HSPA5 axis stabilizes GPX4, thereby suppressing ferroptosis and promoting drug resistance (236). To overcome these resistance mechanisms, several sensitization strategies have been developed. The tumor suppressor FBW7 inhibits SCD1 expression by promoting NR4A1 degradation, which alters membrane lipid composition and enhances susceptibility to lipid peroxidation (143). TRIM21 counteracts ferroptosis by ubiquitinating and degrading the arachidonic acid metabolism enzyme EPHX1, a process that can be pharmacologically targeted by bezafibrate (237). In KRAS-mutant tumors, the KRAS/ERK1 axis promotes ALOX15B degradation via ABHD17C, while methylprotodioscin disrupts this interaction to restore ALOX15B’s pro-ferroptotic activity (238). A particularly noteworthy finding involves the circular RNA cTRIP12, which facilitates the interaction between OGT and PERK, thereby enhancing O-GlcNAcylation and stabilizing ferritin heavy chain and PD-L1. This dual stabilization mediates resistance to both ferroptosis and immune checkpoint therapy, making cTRIP12 a promising target for combination treatment (144). Additional resistance mechanisms have also been elucidated. KRAS mutations can upregulate TMOD3, promoting F-actin polymerization, which in turn enhances autophagosome–lysosome fusion and accelerates ACSL4 degradation. This process inhibits ferroptosis and contributes to resistance against PD-1 antibodies (239). In parallel, targeting cysteine metabolism, a critical component of ferroptosis defense, has shown promise. Engineered bacteria designed to deplete cysteine effectively induce ferroptosis in PDAC cells by precisely disrupting cysteine utilization (145).
In summary, ferroptosis exerts a context-dependent dual role in pancreatic cancer: it can act as a potent tumor-suppressive mechanism, directly eliminating malignant cells, yet under certain conditions, it can also release pro-tumorigenic signaling molecules that reshape the tumor microenvironment. Preclinical studies have demonstrated the potential of ferroptosis-targeted therapies to overcome both apoptosis and immune resistance. Future research should focus on identifying the molecular switches that dictate the pro- or anti-tumor fate of ferroptosis, elucidating its immunogenic mechanisms, and developing low-toxicity, clinically translatable ferroptosis inducers for optimized combination therapies.
4.5 Ferroptosis and colorectal cancer
Ferroptosis, an iron-dependent, lipid peroxidation-driven form of regulated cell death, plays a central role in the initiation, progression, and therapeutic resistance of colorectal cancer (CRC). Its core regulatory mechanism revolves around the system Xc–GPX4 antioxidant axis, the disruption of which directly leads to the accumulation of lipid peroxides and subsequent cell death. In CRC, a complex molecular network finely regulates this process. For instance, p53 promotes ferroptosis by suppressing SLC7A11 transcription (146), whereas activation of the NRF2 signaling pathway enables cancer cells to evade ferroptosis through SLC7A11 upregulation (240). Furthermore, the KIF20A/NUAK1/GPX4 axis has been identified as a critical mechanism conferring resistance to oxaliplatin-based chemotherapy (147). A profound bidirectional relationship exists between ferroptosis and therapeutic resistance: while certain targeted drugs may inadvertently inhibit ferroptosis and promote drug resistance, the deliberate induction of ferroptosis represents a promising approach to overcoming such resistance. Beyond its direct cytotoxic effects on tumor cells, ferroptosis also influences the TME by releasing diverse signaling molecules that remodel immune responses and correlate closely with patient prognosis.
Within the unique setting of the colorectum, the gut microbiota serves as a crucial “external regulator,” adding a layer of complexity to ferroptosis modulation. Microbially derived metabolites, including vitamins, bile acids, SCFAs, and tryptophan metabolites, exert highly context-dependent, bidirectional effects on ferroptosis. Most vitamins and certain bile acids inhibit ferroptosis by upregulating GPX4 or activating antioxidant pathways such as NRF2, thereby potentially enhancing tumor survival. In contrast, SCFAs (such as butyrate) tend to promote ferroptosis and suppress tumor growth by downregulating SLC7A11 and GPX4 expression. Interestingly, some metabolites, such as VB2 and specific bile acids, display dual functions, either promoting or suppressing ferroptosis depending on the microenvironmental context (11). Collectively, this intricate microbiota-mediated regulation of ferroptosis enriches our understanding of host–microbe interactions in colorectal cancer and highlights promising opportunities to modulate ferroptosis through dietary, probiotic, or microbial-targeted interventions.
Building on these mechanistic insights, targeting the key metabolic nodes of ferroptosis, namely, iron, amino acid, and lipid metabolism, has emerged as a highly promising therapeutic strategy. At the iron metabolism level, modulating the activity of proteins such as the transferrin receptor or ferritin to expand the intracellular LIP effectively induces ferroptosis, a mechanism exploited by agents like vitamin C (241). Within amino acid metabolism, the system Xc–GPX4 axis represents the central therapeutic target. Compounds such as erastin, which inhibits SLC7A11, or RSL3, which directly inactivates GPX4, can robustly trigger ferroptotic cell death. Notably, the gut bacterium Fusobacterium nucleatum has been shown to enhance oxaliplatin sensitivity by modulating GPX4 expression (148). Regarding lipid metabolism, the promotion of PUFA incorporation into membrane phospholipids, a process catalyzed by ACSL4, provides the essential biochemical “fuel” for ferroptosis (242). Indeed, drugs such as apatinib exert part of their anti-tumor efficacy through this pathway (149).
Perhaps even more compelling is the combination of ferroptosis inducers with immune checkpoint inhibitors, which has opened a new frontier for overcoming immunotherapy resistance in CRC, particularly in microsatellite stable (MSS) patients (150). This combined strategy operates through multiple synergistic mechanisms: ferroptosis inducers (e.g., RSL3, sorafenib) directly promote tumor cell death (243, 244), while the release of DAMPs, such as HMGB1, from ferroptotic cells activates dendritic cells and recruits CD8+ T lymphocytes, transforming an immunosuppressive “cold” tumor microenvironment into an immunostimulatory “hot” one (151). Cutting-edge research is now focused on identifying natural ferroptosis-inducing compounds from traditional medicines, such as artemisinin and ginsenoside Rh3, that act through multiple regulatory pathways (245), as well as developing intelligent nanodelivery systems capable of tumor-targeted co-delivery of ferroptosis inducers and immunotherapeutic agents (246). These advances aim to precisely modulate tumor metabolism while simultaneously triggering robust anti-tumor immunity, heralding a new era of integrated metabolic intervention and immunotherapy in CRC treatment.
4.6 Ferroptosis in other digestive cancers
Compared with the major digestive system malignancies discussed above, research on ferroptosis in gallbladder cancer (GBC) and oral cavity cancer, particularly oral squamous cell carcinoma (OSCC), remains in its infancy. Current evidence is largely derived from preliminary in vitro studies, and the field is still primarily descriptive, focusing on the identification of potential ferroptosis regulators rather than the construction of comprehensive mechanistic networks or translational frameworks.
In GBC, several molecular factors have been implicated in ferroptosis regulation. Induction of ferroptosis appears to be promoted by TFAP2A knockdown (which suppresses NRF2) (152), activation of the Akt/SIRT3/ACSL4 axis (153), and treatment with compounds such as isoglycyrrhizin, which acts through the p62-Keap1-NRF2-HMOX1 pathway and GPX4 downregulation (154), or lithocholic acid, which inhibits glutaminase and depletes GSH (155). Conversely, RUNX3 may inhibit ferroptosis via a p53-dependent mechanism that activates GADD45A and subsequently suppresses SLC7A11 expression (156).
In OSCC, several regulators have also been identified that modulate sensitivity to treatments such as cisplatin and PDT. Ferroptosis inducers include rhamnazin (which inhibits the NRF2/HO-1/xCT pathway) (157), Quisinostat (which elevates ROS via the GPX4/p53 axis) (158), and non-thermal plasma (159, 160). Resistance mechanisms, on the other hand, involve molecules such as circFNDC3B and specific miRNAs (e.g., miR-125b-5p, miR-34c-3p) that target and suppress SLC7A11/xCT (161–164), as well as activation of the IL-6/JAK2/STAT3 signaling pathway (165). Furthermore, PER1 overexpression promotes ferroptosis by inhibiting HIF-1α, while AEBP1 silencing activates JNK/p38/ERK signaling to induce cell death (166). Collectively, these findings provide preliminary evidence that ferroptosis plays a functional role in GBC and OSCC; however, further in vivo validation and mechanistic dissection are essential to clarify its biological significance and assess the therapeutic potential of ferroptosis-targeted interventions in these cancers.
5 Ferroptosis in tumor therapy
The high uptake of iron by tumors and the bidirectional effect of iron metabolism on tumor microenvironment provide more means for clinical anti-tumor cells. Ferroptosis has become a key new target for tumor therapy.
5.1 Chemotherapy and ferroptosis
Chemotherapy remains a primary treatment for malignant tumors, but its efficacy is often limited by drug resistance. Recent studies have indicated that ferroptosis is closely associated with chemotherapy resistance. Combining ferroptosis inducers with chemotherapeutic agents may help overcome clinical challenges and improve therapeutic outcomes. In gastric cancer, STAT3 acts as a negative regulator of ferroptosis. The STAT3 inhibitor W1131 has demonstrated anti-tumor effects by inducing ferroptosis and may synergize with chemotherapy to further suppress tumor growth (247). Similarly, kaurane-type diterpenoids promote both ferroptosis and apoptosis by inhibiting peroxidase activity of Prdx I/II, and exhibit synergistic anti-tumor effects when combined with cisplatin (248). Sorafenib, a first-line treatment for advanced hepatocellular carcinoma, often faces resistance issues. Metallothionein IG (MTIG), involved in oxidative stress response, inhibits SRF-induced ferroptosis. NRF2 activation is crucial for MTIG expression, and targeting this pathway may help attenuate SRF resistance (249, 250). However, chemotherapy-induced ferroptosis may also damage normal tissues (251). Thus, balancing cancer cell elimination with protection of healthy cells is essential in treatment design. Table 3 summarizes ferroptosis-targeting agents currently used clinically or with strong translational potential.
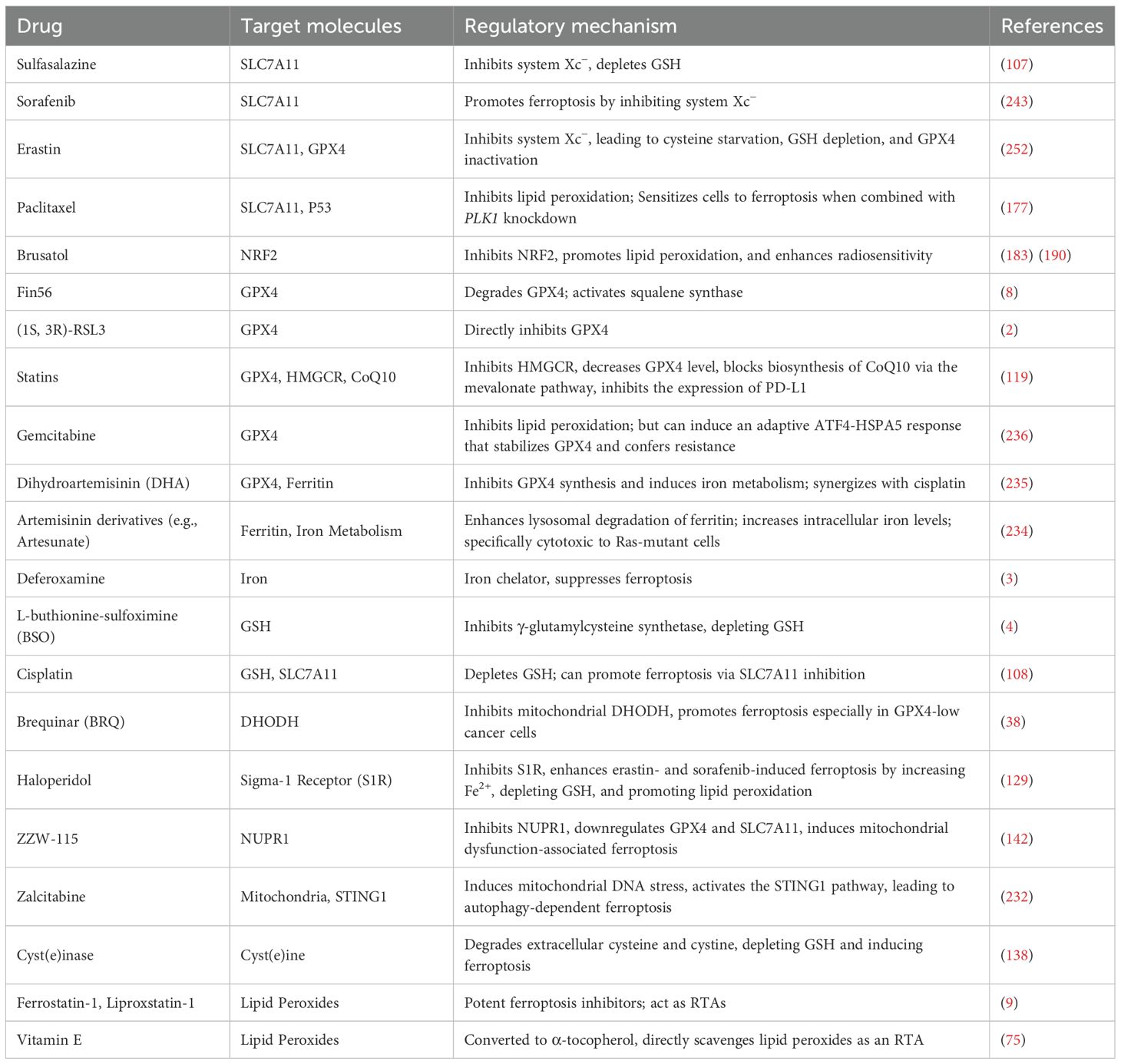
Table 3. Application of ferroptosis-related drugs in tumor therapy.
5.2 Immunotherapy and ferroptosis
Immunotherapy achieves anti-tumor effects by activating the immune system and enhancing one’s ability to fight cancer. Ferroptosis not only has direct anti-tumor effects, but also enhances the immunogenicity of cancer cells and the sensitivity of immunotherapy, bringing new opportunities for tumor treatment, especially in addressing the problem of drug resistance to immunotherapy drugs (252). Immunocheckpoint inhibitors (ICIs) mainly promote lipid peroxidation-dependent ferroptosis of tumor cells by activating CD8+ T cells, releasing IFN-γ, down-regulating the expression of SLC7A11 and SLC3A2 (208). Clinically, ACSL4 has improved the survival rate of cancer patients treated with ICIs (253). Similarly, in melanoma patients, a decrease in SLC3A2 expression is strongly associated with an increase in the efficacy of ICIs (254). Damage-associated molecules released by ferroptosis cells promote dendritic cell maturation and induce CD8+ T cell activation. Activated CD8+ T cells inhibit ferroptosis by binding to TYRO3 and promote the development of a pro-tumor microenvironment by decreasing the M1/M2 macrophage ratio, leading to resistance to anti-PD-1/PD-L1 therapy (255). Studies have found that anti-PD-1 drugs and TGFβ inhibitors can synergistically enhance the immune response in TME, resulting in an increase in the content of H2O2 in M1 macrophages, promoting Fenton reaction, and inducing ferroptosis of tumor cells (256). It is hypothesized that the long-term effects of ferroptosis on tumor immunotherapy depend on the interaction between tumor cells and various immune cell subsets.
5.3 Radiotherapy and ferroptosis
Radiotherapy produces ROS and free radicals through ionizing radiation to destroy chemical bonds, leading to DNA damage and exerting anti-tumor effects. Radiotherapy significantly induced the expression of ACSL4, which in turn promoted ferroptosis of tumor cells, increased the sensitivity of tumors to radiotherapy, and effectively inhibited tumor growth (174). ACSL4 deletion or treatment with liproxstatin-1, an inhibitor of ferroptosis, significantly reduced 4-hydroxynonenal (4-HNE) levels, while the level of 4-HNE in esophageal cancer tissues after radiotherapy was significantly associated with better clinical prognosis of cancer patients (257). Radiotherapy can induce the expression of SLC7A11 and GPX4, and promote radioresistance by inhibiting ferroptosis. Inhibition of SLC7A11 or GPX4 with ferroptosis inducers can increase the sensitivity of radioresistant tumor cells and xenografts to radiotherapy (174). Hypoxia microenvironment is an important feature of solid malignant tumors. Radioresistance caused by hypoxia is the main reason for its poor radiotherapy effect (258, 259). In addition, the up-regulation of HIF-1α caused by hypoxia is also an important factor in radioresistance (260, 261). HIF-1α-mediated ferroptosis inhibition is closely related to poor prognosis and treatment tolerance of hypoxic tumors. Therefore, regulating ferroptosis to reduce HIF-1α-induced radioresistance may be an effective strategy (164). Whole lactoferrin can down-regulate the expression of HIF-1α, improve the hypoxic microenvironment of breast cancer, and promote radiation-induced DNA damage, improving the radiotherapy effect of breast cancer (262). In addition, studies have found that IFN-γ released by immunotherapy-activated CD8+ T cells and radiotherapy-activated macrophages can independently and synergistically inhibit SLC7A11.This leads to a decrease in cystine uptake, which in turn increases lipid peroxidation and ferroptosis, making tumor cells sensitive to radiotherapy (263). Therefore, in clinical practice, radiotherapy is usually combined with chemotherapy, targeted therapy or immunotherapy to enhance the clearance of tumor cells.
5.4 Nanomedicine and ferroptosis
Nanotechnology has achieved remarkable success in cancer treatment. The ferroptosis mediated by Fe3O4-SAS @ PLT assembled by sulfasalazine (SAS) -loaded magnetic nanoparticles (Fe3O4) and platelet (PLT) membrane can not only induce tumor-specific immune response, but also effectively repolarize immunosuppressive M2 into anti-tumor M1, regulating the anti-tumor interaction between tumor cells and macrophages (264). In the nanoparticle SRF@Hb-Ce6, which constructed by connecting hemoglobin (Hb) with photosensitizer chlorin e6 (Ce6) and loading sorafenib, Hb utilizes its own oxygen and iron to provide oxygen for PDT while providing iron for ferroptosis. PDT enhances ferroptosis by inducing IFN-γ secretion by immune cells (265). In addition, the nanoparticles SRF @ MPDA-SPIO which constructed based on superparamagnetic iron oxide (SPIO) also have the functions of inducing ferroptosis and PDT (266). Polyethylene glycol-modified ultra-small silicon nanoparticles (C′ dots) for tumor imaging can bind and enrich iron ions, which can be endocytosed into cells and lead to an increase in intracellular iron content, causing iron metabolism disorders in tumor cells (267, 268). Ultrasmall single-crystal Fe nanoparticles (bcc-USINP) were synthesized by one-step high-temperature pyrolysis, providing a simple, safe and efficient tumor-responsive zero-valent iron (Fe) delivery system, which can selectively release a large amount of iron ions in weakly acidic TIME and promote Fenton reaction. Bcc-USINP can cause ferroptosis of tumor cells at lower concentrations (269). Nanomaterials have the advantages of promoting drug dissolution, improving absorption and precise targeting, and show great promise for combining them with ferroptosis in cancer treatment.
6 Conclusion
During the development and therapeutic response of digestive tract cancers, ferroptosis—an iron-dependent, programmed form of cell death intricately regulated by the gut microbiota and their metabolites—plays a critical role. In recent years, mechanistic studies of ferroptosis in gastrointestinal tumors have made substantial progress, gradually uncovering the key molecules and signaling pathways involved. However, the interplay between ferroptosis and other regulated cell death pathways, such as autophagy and apoptosis, remains incompletely understood. Moreover, clinical detection and application of ferroptosis-related biomarkers or therapies are still at an early stage, and the development of ferroptosis-targeted anti-cancer drugs remains limited. This persistent gap between basic research and clinical translation continues to impede the full recognition and therapeutic utilization of ferroptosis in GI cancer management.
Based on the evidence presented, we propose a coherent integrative model in which gut microbial metabolites serve as central conductors, fine-tuning the ferroptosis orchestra within the tumor microenvironment. This model posits that the balance between ferroptosis-suppressive and ferroptosis-promoting signals from the gut microbiota can decisively influence cancer cell fate.
The model highlights two primary, opposing forces. On one hand, beneficial microbes and their metabolites primarily act as ferroptosis inhibitors, which can paradoxically support tumor cell survival. Key mechanisms include: SCFAs like butyrate, which enhance mitochondrial function and activate the cytoprotective NRF2 pathway; bile acids that activate nuclear receptors (FXR/VDR) to upregulate a ferroptosis-defense network; tryptophan derivatives that activate the AHR receptor to bolster cellular antioxidant systems; and vitamin metabolites that provide crucial redox regulation - VK forms a defense branch independent of GPX4 through an FSP1-dependent pathway, while vitamin A and E derivatives act as direct radical-trapping antioxidants providing chemical protection; and probiotics that help maintain systemic iron homeostasis and activate protective pathways.
In contrast, gut dysbiosis and pathogenic bacteria can drive ferroptosis, a process that can be harnessed for therapy or contribute to tissue damage. This occurs through several mechanisms: Pathogen-associated molecules like LPS and specific bacteria can upregulate pro-ferroptotic enzymes such as ACSL4 and ALOX15; altered bile acid profiles can promote iron uptake and lipid peroxidation; the depletion of protective metabolites like SCFAs indirectly lifts the brakes on ferroptosis; and notably, VB2 (VB2) demonstrates context-dependent duality - under specific microenvironmental conditions (such as when forming nanocomplexes with iron ions), VB2 can transform from a “protector” into an “attacker,” synergistically promoting iron accumulation and ROS generation, thereby exhibiting pro-ferroptotic activity.
This dual perspective positions the gut microbiota at the core of ferroptosis regulation, offering novel strategies for cancer treatment. Interventions such as probiotics, prebiotics, fecal microbiota transplantation, direct administration of pro-ferroptotic metabolites, or elimination of ferroptosis-suppressive microbes can modulate the balance of ferroptosis—suppressing it in normal tissues to reduce damage, or selectively inducing it in tumors to overcome drug resistance.
While the potential of harnessing the gut microbiome to modulate ferroptosis is a compelling strategy for cancer therapy, it is crucial to acknowledge the significant limitations of the current research landscape. The evidence summarized herein is predominantly derived from in vitro cell cultures and animal models. These pre-clinical systems cannot fully replicate the complexity of the human tumor microenvironment or the dynamic ecosystem of the human gut, creating a substantial translational gap before these findings can be applied clinically.
A pivotal, yet often unaddressed, question is the physiological relevance of the microbial metabolites involved. Many studies utilize supraphysiological concentrations to elicit clear ferroptotic responses in vitro. It remains unclear whether the local or systemic levels of metabolites like SCFAs, bile acids, or tryptophan derivatives in humans are sufficient to significantly influence ferroptosis pathways in tumors. Furthermore, human data are largely correlative, making it difficult to establish causality between microbial shifts and ferroptosis modulation in cancer patients.
To advance this field, future research must bridge these translational gaps. Priority directions include conducting human studies that integrate microbiome and metabolomic profiling with ferroptosis biomarkers in patient biopsies; determining the pharmacologically and physiologically achievable concentrations of key microbial metabolites, and launching early-phase clinical trials that combine ferroptosis inducers with microbiome-targeted interventions. Addressing these limitations will be essential to building a realistic and mechanistically grounded framework for translating ferroptosis-based strategies into clinical oncology.
Overall, ferroptosis plays a fundamental role in the proliferation, invasion, metastasis, and resistance to radiotherapy and chemotherapy in gastrointestinal cancers, highlighting its immense potential as a therapeutic target. As research continues to unravel the intricate regulatory networks governing ferroptosis and its interplay with the tumor microenvironment, this form of cell death is poised to become a central target for improving clinical outcomes in patients with gastrointestinal malignancies. Harnessing the microbiota-metabolism-ferroptosis axis for metabolic intervention may serve as a crucial bridge between basic discovery and clinical translation, paving the way for next-generation, precision cancer therapies.
Author contributions
JW: Writing – original draft, Writing – review & editing. HL: Writing – original draft, Writing – review & editing. MZ: Writing – original draft, Writing – review & editing. BX: Writing – original draft, Writing – review & editing. XM: Writing – original draft, Writing – review & editing. YL: Writing – original draft, Writing – review & editing. LW: Writing – original draft, Writing – review & editing. LH: Writing – original draft, Writing – review & editing. TZ: Writing – original draft, Writing – review & editing. YT: Writing – original draft, Writing – review & editing. YW: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Hebei Province Foreign Talent Introduction Program (Study on the Interventional Effect of Magnetic-Targeted Stem Cells in Liver Injury, J-STD-SL (2025) No. 3), Hebei Natural Science Foundation (Grant No. C2025107013), the National Natural Science Foundation of China (Grant No. 81802372), the Natural Science Foundation of Hebei Province (Grant No. H2020107005 & H2020107002) and the Scientific and Technological Project of the Hebei Province of China (Grant No. 14397702D).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zheng J and Conrad M. Ferroptosis: when metabolism meets cell death. Physiol Rev. (2025) 105:651–706. doi: 10.1152/physrev.00031.2024
2. Lee J, Seo Y, and Roh JL. Emerging therapeutic strategies targeting GPX4-mediated ferroptosis in head and neck cancer. Int J Mol Sci. (2025) 26. doi: 10.3390/ijms26136452
3. Žalytė E. Ferroptosis, metabolic rewiring, and endometrial cancer. Int J Mol Sci. (2023) 25. doi: 10.3390/ijms25010075
4. Wang K, Zhang Z, Tsai HI, Liu Y, Gao J, Wang M, et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. (2021) 28:1222–36. doi: 10.1038/s41418-020-00644-4
5. Fu HJ, Zhou XY, Qin DL, Qiao Q, Wang QZ, Li SY, et al. Inhibition of ferroptosis delays aging and extends healthspan across multiple species. Adv Sci (Weinh). (2025) 12:e2416559. doi: 10.1002/advs.202416559
6. Wang L, Huang H, Li X, Ouyang L, Wei X, Xie J, et al. A review on the research progress of traditional Chinese medicine with anti-cancer effect targeting ferroptosis. Chin Med. (2023) 18:132. doi: 10.1186/s13020-023-00838-1
7. Xia X, Fan X, Zhao M, and Zhu P. The relationship between ferroptosis and tumors: A novel landscape for therapeutic approach. Curr Gene Ther. (2019) 19:117–24. doi: 10.2174/1566523219666190628152137
8. Shu L, Luo P, Ren C, Lai Z, Zhang X, Wu C, et al. Inhalable fibroin hybrid liposomes for the targeted treatment of lung adenocarcinoma via autophagy-enhanced ferroptosis. ACS Nano. (2025) 19:31656–76. doi: 10.1021/acsnano.5c09493
9. Zhao X, Zhang M, He J, Li X, and Zhuang X. Emerging insights into ferroptosis in cholangiocarcinoma (Review). Oncol Lett. (2024) 28:606. doi: 10.3892/ol.2024.14739
10. Zhao Y, Linkermann A, Takahashi M, Li Q, and Zhou X. Ferroptosis in cardiovascular disease: regulatory mechanisms and therapeutic implications. Eur Heart J. (2025) 46:3247–60. doi: 10.1093/eurheartj/ehaf374
11. Cui W, Hao M, Yang X, Yin C, and Chu B. Gut microbial metabolism in ferroptosis and colorectal cancer. Trends Cell Biol. (2025) 35:341–51. doi: 10.1016/j.tcb.2024.08.006
12. Zou P, He Q, Xia H, and Zhong W. Ferroptosis and its impact on common diseases. PeerJ. (2024) 12. doi: 10.7717/peerj.18708
13. Zhang R, Kang R, and Tang D. Ferroptosis in gastrointestinal cancer: from mechanisms to implications. Cancer Lett. (2023) 561:216147. doi: 10.1016/j.canlet.2023.216147
14. Jiang C, Yan Y, Long T, Xu J, Chang C, Kang M, et al. Ferroptosis: a potential therapeutic target in cardio-cerebrovascular diseases. Mol Cell Biochem. (2025) 480:4379–99. doi: 10.1007/s11010-025-05262-7
15. Kagan VE, Tyurina YY, Sun WY, Vlasova II, Dar H, Tyurin VA, et al. Redox phospholipidomics of enzymatically generated oxygenated phospholipids as specific signals of programmed cell death. Free Radic Biol Med. (2020) 147:231–41. doi: 10.1016/j.freeradbiomed.2019.12.028
16. Deng S, Cao H, Li T, Wang X, Meng J, Zeng T, et al. Lachnospiraceae-bacterium alleviates ischemia-reperfusion injury in steatotic donor liver by inhibiting ferroptosis via the Foxo3-Alox15 signaling pathway. Gut Microbes. (2025) 17:2460543. doi: 10.1080/19490976.2025.2460543
17. Lin XM, Pan MH, Sun J, Wang M, Huang ZH, Wang G, et al. Membrane phospholipid peroxidation promotes loss of dopaminergic neurons in psychological stress-induced Parkinson’s disease susceptibility. Aging Cell. (2023) 22:e13970. doi: 10.1111/acel.13970
18. Ru Q, Li Y, Chen L, Wu Y, Min J, and Wang F. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct Target Ther. (2024) 9:271. doi: 10.1038/s41392-024-01969-z
19. Lee J and Roh JL. Ferroptosis: iron release mechanisms in the bioenergetic process. Cancer metastasis Rev. (2025) 44:36. doi: 10.1007/s10555-025-10252-8
20. Zhao H, Wang Z, and Wang H. The role of NCOA4-mediated ferritinophagy in the ferroptosis of hepatocytes: A mechanistic viewpoint. Pathology Res Pract. (2025) 270. doi: 10.1016/j.prp.2025.155996
21. Ruan B, Dong J, Wei F, Huang Z, Yang B, Zhang L, et al. DNMT aberration-incurred GPX4 suppression prompts osteoblast ferroptosis and osteoporosis. Bone Res. (2024) 12:68. doi: 10.1038/s41413-024-00365-1
22. Zhang HL, Guo YQ, Liu S, Ye ZP, Li LC, Hu BX, et al. Galectin-13 reduces membrane localization of SLC7A11 for ferroptosis propagation. Nat Chem Biol. (2025) 21:1531–43. doi: 10.1038/s41589-025-01888-2
23. Zheng J and Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. (2020) 32:920–37. doi: 10.1016/j.cmet.2020.10.011
24. Koppula P, Zhuang L, and Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. (2021) 12:599–620. doi: 10.1007/s13238-020-00789-5
25. Cui C, Yang F, and Li Q. Post-translational modification of GPX4 is a promising target for treating ferroptosis-related diseases. Front Mol Biosci. (2022) 9:901565. doi: 10.3389/fmolb.2022.901565
26. Deng H, Zhao L, Ge H, Gao Y, Fu Y, Lin Y, et al. Ubiquinol-mediated suppression of mitochondria-associated ferroptosis is a targetable function of lactate dehydrogenase B in cancer. Nat Commun. (2025) 16:2597. doi: 10.1038/s41467-025-57906-3
27. Chen X, Yu X, Zhong S, Sha P, Li R, Xu X, et al. ALDH2/eIF3E interaction modulates protein translation critical for cardiomyocyte ferroptosis in acute myocardial ischemia injury. Circulation. (2025). doi: 10.1161/circulationaha.125.075220
28. Sui Y, Geng X, Wang Z, Zhang J, Yang Y, and Meng Z. Targeting the regulation of iron homeostasis as a potential therapeutic strategy for nonalcoholic fatty liver disease. Metabolism: Clin Exp. (2024) 157:155953. doi: 10.1016/j.metabol.2024.155953
29. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. (2020) 16:1351–60. doi: 10.1038/s41589-020-0613-y
30. Hu Q, Wei W, Wu D, Huang F, Li M, Li W, et al. Blockade of GCH1/BH4 axis activates ferritinophagy to mitigate the resistance of colorectal cancer to erastin-induced ferroptosis. Front Cell Dev Biol. (2022) 10:810327. doi: 10.3389/fcell.2022.810327
31. Cronin SJF, Rao S, Tejada MA, Turnes BL, Licht-Mayer S, Omura T, et al. Phenotypic drug screen uncovers the metabolic GCH1/BH4 pathway as key regulator of EGFR/KRAS-mediated neuropathic pain and lung cancer. Sci Transl Med. (2022) 14:eabj1531. doi: 10.1126/scitranslmed.abj1531
32. Du J, Zhu X, Zhang Y, Huang X, Wang X, Yang F, et al. CTRP13 attenuates atherosclerosis by inhibiting endothelial cell ferroptosis via activating GCH1. Int Immunopharmacol. (2024) 143:113617. doi: 10.1016/j.intimp.2024.113617
33. Kazmirczak F, Vogel NT, Prisco SZ, Patterson MT, Annis J, Moon RT, et al. Ferroptosis-mediated inflammation promotes pulmonary hypertension. Circ Res. (2024) 135:1067–83. doi: 10.1161/circresaha.123.324138
34. Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. (2023) 186:2748–64 e22. doi: 10.1016/j.cell.2023.05.003
35. Belavgeni A, Tonnus W, and Linkermann A. Cancer cells evade ferroptosis: sex hormone-driven membrane-bound O-acyltransferase domain-containing 1 and 2 (MBOAT1/2) expression. Signal Transduct Target Ther. (2023) 8:336. doi: 10.1038/s41392-023-01593-3
36. Teng D, Swanson KD, Wang R, Zhuang A, Wu H, Niu Z, et al. DHODH modulates immune evasion of cancer cells via CDP-Choline dependent regulation of phospholipid metabolism and ferroptosis. Nat Commun. (2025) 16:3867. doi: 10.1038/s41467-025-59307-y
37. Yang C, Zhao Y, Wang L, Guo Z, Ma L, Yang R, et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat Cell Biol. (2023) 25:836–47. doi: 10.1038/s41556-023-01146-4
38. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
39. Cao J, Chen X, Chen L, Lu Y, Wu Y, Deng A, et al. DHODH-mediated mitochondrial redox homeostasis: a novel ferroptosis regulator and promising therapeutic target. Redox Biol. (2025) 85:103788. doi: 10.1016/j.redox.2025.103788
40. Boukalova S, Hubackova S, Milosevic M, Ezrova Z, Neuzil J, and Rohlena J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165759. doi: 10.1016/j.bbadis.2020.165759
41. Wong KH, Wang Y, Wang X, Yin Y, Feng K, and Chen M. Unsaturated fatty acid-doped liposomes deliver piperine to deactivate defensive mechanism for ferroptosis in cancer therapy. J Controlled release: Off J Controlled Release Soc. (2025) 382:113656. doi: 10.1016/j.jconrel.2025.113656
42. Deng R, Fu L, Liang H, Ai X, Liu F, Li N, et al. Inhibition of mitochondrial complex I induces mitochondrial ferroptosis by regulating CoQH2 levels in cancer. Cell Death Dis. (2025) 16:254. doi: 10.1038/s41419-025-07510-6
43. Zha X, Liu X, Wei M, Huang H, Cao J, Liu S, et al. Microbiota-derived lysophosphatidylcholine alleviates Alzheimer’s disease pathology via suppressing ferroptosis. Cell Metab. (2025) 37:169–86.e9. doi: 10.1016/j.cmet.2024.10.006
44. Jin Z, Yang Y, Cao Y, Wen Q, Xi Y, Cheng J, et al. The gut metabolite 3-hydroxyphenylacetic acid rejuvenates spermatogenic dysfunction in aged mice through GPX4-mediated ferroptosis. Microbiome. (2023) 11:212. doi: 10.1186/s40168-023-01659-y
45. Asnicar F, Berry SE, Valdes AM, Nguyen LH, Piccinno G, Drew DA, et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat Med. (2021) 27:321–32. doi: 10.1038/s41591-020-01183-8
46. Das NK, Schwartz AJ, Barthel G, Inohara N, Liu Q, Sankar A, et al. Microbial metabolite signaling is required for systemic iron homeostasis. Cell Metab. (2020) 31:115–30 e6. doi: 10.1016/j.cmet.2019.10.005
47. Fikri B, Ridha NR, Putri SH, Salekede SB, Juliaty A, Tanjung C, et al. Effects of probiotics on immunity and iron homeostasis: A mini-review. Clin Nutr ESPEN. (2022) 49:24–7. doi: 10.1016/j.clnesp.2022.03.031
48. Saha M, Sarkar S, Sarkar B, Sharma BK, Bhattacharjee S, and Tribedi P. Microbial siderophores and their potential applications: a review. Environ Sci pollut Res Int. (2016) 23:3984–99. doi: 10.1007/s11356-015-4294-0
49. Ren Z, Zhao Z, Wang Y, and Huang K. Preparation of selenium/zinc-enriched probiotics and their effect on blood selenium and zinc concentrations, antioxidant capacities, and intestinal microflora in canine. Biol Trace Elem Res. (2011) 141:170–83. doi: 10.1007/s12011-010-8734-x
50. Fiore A, Zeitler L, Russier M, Gross A, Hiller MK, Parker JL, et al. Kynurenine importation by SLC7A11 propagates anti-ferroptotic signaling. Mol Cell. (2022) 82:920–32 e7. doi: 10.1016/j.molcel.2022.02.007
51. Zhang P, Han X, Zhang X, and Zhu X. Lactobacillus acidophilus ATCC 4356 alleviates renal ischemia-reperfusion injury through antioxidant stress and anti-inflammatory responses and improves intestinal microbial distribution. Front Nutr. (2021) 8:667695. doi: 10.3389/fnut.2021.667695
52. Song X, Zhao Z, Zhao Y, Jin Q, and Li S. Protective Effects of Bacillus coagulans JA845 against D-Galactose/AlCl(3)-Induced Cognitive Decline, Oxidative Stress and Neuroinflammation. J Microbiol Biotechnol. (2022) 32:212–9. doi: 10.4014/jmb.2111.11031
53. Miyamoto J, Igarashi M, Watanabe K, Karaki SI, Mukouyama H, Kishino S, et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat Commun. (2019) 10:4007. doi: 10.1038/s41467-019-11978-0
54. Villarreal-Soto SA, Bouajila J, Pace M, Leech J, Cotter PD, Souchard JP, et al. Metabolome-microbiome signatures in the fermented beverage, Kombucha. Int J Food Microbiol. (2020) 333:108778. doi: 10.1016/j.ijfoodmicro.2020.108778
55. Yue M, Wei J, Chen W, Hong D, Chen T, and Fang X. Neurotrophic Role of the Next-Generation Probiotic Strain L. lactis MG1363-pMG36e-GLP-1 on Parkinson’s Disease via Inhibiting Ferroptosis. Nutrients. (2022) 14. doi: 10.3390/nu14224886
56. Kim SY, Chae CW, Lee HJ, Jung YH, Choi GE, Kim JS, et al. Sodium butyrate inhibits high cholesterol-induced neuronal amyloidogenesis by modulating NRF2 stabilization-mediated ROS levels: involvement of NOX2 and SOD1. Cell Death Dis. (2020) 11:469. doi: 10.1038/s41419-020-2663-1
57. Rose S, Bennuri SC, Davis JE, Wynne R, Slattery JC, Tippett M, et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl Psychiatry. (2018) 8:42. doi: 10.1038/s41398-017-0089-z
58. Li X, Dong L, Li A, Yi J, Brotto M, and Zhou J. Butyrate ameliorates mitochondrial respiratory capacity of the motor-neuron-like cell line NSC34-G93A, a cellular model for ALS. Biomolecules. (2022) 12. doi: 10.3390/biom12020333
59. Chapkin RS, Navarro SL, Hullar MAJ, and Lampe JW. Diet and gut microbes act coordinately to enhance programmed cell death and reduce colorectal cancer risk. Dig Dis Sci. (2020) 65:840–51. doi: 10.1007/s10620-020-06106-8
60. Yang C-J, Chang H-C, Sung P-C, Ge M-C, Tang H-Y, Cheng M-L, et al. Oral fecal transplantation enriches Lachnospiraceae and butyrate to mitigate acute liver injury. Cell Rep. (2024) 43:113591. doi: 10.1016/j.celrep.2023.113591
61. Al-Harbi NO, Nadeem A, Ahmad SF, Alotaibi MR, AlAsmari AF, Alanazi WA, et al. Short chain fatty acid, acetate ameliorates sepsis-induced acute kidney injury by inhibition of NADPH oxidase signaling in T cells. Int Immunopharmacol. (2018) 58:24–31. doi: 10.1016/j.intimp.2018.02.023
62. Liu D, Liang CH, Huang B, Zhuang X, Cui W, Yang L, et al. Tryptophan metabolism acts as a new anti-ferroptotic pathway to mediate tumor growth. Adv Sci (Weinh). (2023) 10:e2204006. doi: 10.1002/advs.202204006
63. Cui W, Guo M, Liu D, Xiao P, Yang C, Huang H, et al. Gut microbial metabolite facilitates colorectal cancer development via ferroptosis inhibition. Nat Cell Biol. (2024) 26:124–37. doi: 10.1038/s41556-023-01314-6
64. Peng Y, Ouyang L, Zhou Y, Lai W, Chen Y, Wang Z, et al. AhR Promotes the Development of Non-small cell lung cancer by Inducing SLC7A11-dependent Antioxidant Function. J Cancer. (2023) 14:821–34. doi: 10.7150/jca.82066
65. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z, et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. (2020) 33:108487. doi: 10.1016/j.celrep.2020.108487
66. D’Amico D, Andreux PA, Valdes P, Singh A, Rinsch C, and Auwerx J. Impact of the natural compound urolithin A on health, disease, and aging. Trends Mol Med. (2021) 27:687–99. doi: 10.1016/j.molmed.2021.04.009
67. Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev. (2020) 34:526–43. doi: 10.1101/gad.334219.119
68. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. (2019) 26:420–32 e9. doi: 10.1016/j.chembiol.2018.11.016
69. Xu P, Lin B, Deng X, Huang K, Zhang Y, and Wang N. VDR activation attenuates osteoblastic ferroptosis and senescence by stimulating the Nrf2/GPX4 pathway in age-related osteoporosis. Free Radic Biol Med. (2022) 193:720–35. doi: 10.1016/j.freeradbiomed.2022.11.013
70. Wei Y, Liu W, Wang R, Chen Y, Liu J, Guo X, et al. Propionate promotes ferroptosis and apoptosis through mitophagy and ACSL4-mediated ferroptosis elicits anti-leukemia immunity. Free Radic Biol Med. (2024) 213:36–51. doi: 10.1016/j.freeradbiomed.2024.01.005
71. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. (2022) 608:778–83. doi: 10.1038/s41586-022-05022-3
72. Wang P, Huang Y, Sun B, Chen H, Ma Y, Liu Y, et al. Folic acid blocks ferroptosis induced by cerebral ischemia and reperfusion through regulating folate hydrolase transcriptional adaptive program. J Nutr Biochem. (2024) 124:109528. doi: 10.1016/j.jnutbio.2023.109528
73. Qin S, Wang Y, Li L, Liu J, Xiao C, Duan D, et al. Early-life vitamin B12 orchestrates lipid peroxidation to ensure reproductive success via SBP-1/SREBP1 in Caenorhabditis elegans. Cell Rep. (2022) 40:111381. doi: 10.1016/j.celrep.2022.111381
74. Cai Y, Li X, Tan X, Wang P, Zhao X, Zhang H, et al. Vitamin D suppresses ferroptosis and protects against neonatal hypoxic-ischemic encephalopathy by activating the Nrf2/HO-1 pathway. Transl Pediatr. (2022) 11:1633–44. doi: 10.21037/tp-22-397
75. Conrad M and Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. (2019) 15:1137–47. doi: 10.1038/s41589-019-0408-1
76. Yang S, Ouyang J, Lu Y, Harypursat V, and Chen Y. A dual role of heme oxygenase-1 in tuberculosis. Front Immunol. (2022) 13:842858. doi: 10.3389/fimmu.2022.842858
77. Ning K, Lu K, Chen Q, Guo Z, Du X, Riaz F, et al. Epigallocatechin gallate protects mice against methionine-choline-deficient-diet-induced nonalcoholic steatohepatitis by improving gut microbiota to attenuate hepatic injury and regulate metabolism. ACS Omega. (2020) 5:20800–9. doi: 10.1021/acsomega.0c01689
78. Dar HH, Anthonymuthu TS, Ponomareva LA, Souryavong AB, Shurin GV, Kapralov AO, et al. A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO(*) sabotage of theft-ferroptosis. Redox Biol. (2021) 45:102045. doi: 10.1016/j.redox.2021.102045
79. Ma C, Wu X, Zhang X, Liu X, and Deng G. Heme oxygenase-1 modulates ferroptosis by fine-tuning levels of intracellular iron and reactive oxygen species of macrophages in response to Bacillus Calmette-Guerin infection. Front Cell Infect Microbiol. (2022) 12:1004148. doi: 10.3389/fcimb.2022.1004148
80. Liu S, Tang Y, Liu L, Yang L, Li P, Liu X, et al. Proteomic analysis reveals that ACSL4 activation during reflux esophagitis contributes to ferroptosis-mediated esophageal mucosal damage. Eur J Pharmacol. (2022) 931:175175. doi: 10.1016/j.ejphar.2022.175175
81. Geng J, Shi Y, Zhang J, Yang B, Wang P, Yuan W, et al. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat Commun. (2021) 12:3519. doi: 10.1038/s41467-021-23683-y
82. Funabashi M, Grove TL, Wang M, Varma Y, McFadden ME, Brown LC, et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature. (2020) 582:566–70. doi: 10.1038/s41586-020-2396-4
83. Wang C, Chu Q, Dong W, Wang X, Zhao W, Dai X, et al. Microbial metabolite deoxycholic acid-mediated ferroptosis exacerbates high-fat diet-induced colonic inflammation. Mol Metab. (2024) 84:101944. doi: 10.1016/j.molmet.2024.101944
84. Liu S, Gao Z, He W, Wu Y, Liu J, Zhang S, et al. The gut microbiota metabolite glycochenodeoxycholate activates TFR-ACSL4-mediated ferroptosis to promote the development of environmental toxin-linked MAFLD. Free Radic Biol Med. (2022) 193:213–26. doi: 10.1016/j.freeradbiomed.2022.10.270
85. Wang G, Qin S, Chen L, Geng H, Zheng Y, Xia C, et al. Butyrate dictates ferroptosis sensitivity through FFAR2-mTOR signaling. Cell Death Dis. (2023) 14:292. doi: 10.1038/s41419-023-05778-0
86. He Y, Ling Y, Zhang Z, Mertens RT, Cao Q, Xu X, et al. Butyrate reverses ferroptosis resistance in colorectal cancer by inducing c-Fos-dependent xCT suppression. Redox Biol. (2023) 65. doi: 10.1016/j.redox.2023.102822
87. Bi R, Hu R, Jiang L, Wen B, Jiang Z, Liu H, et al. Butyrate enhances erastin-induced ferroptosis of lung cancer cells via modulating the ATF3/SLC7A11 pathway. Environ Toxicol. (2024) 39:529–38. doi: 10.1002/tox.23857
88. Nie J, Ling Y, Jin M, Chen Z, Liu W, Shen W, et al. Butyrate enhances erastin-induced ferroptosis of osteosarcoma cells via regulating ATF3/SLC7A11 pathway. Eur J Pharmacol. (2023) 957:176009. doi: 10.1016/j.ejphar.2023.176009
89. Lv M, Gong Y, Liu X, Wang Y, Wu Q, Chen J, et al. CDK7-YAP-LDHD axis promotes D-lactate elimination and ferroptosis defense to support cancer stem cell-like properties. Signal Transduct Target Ther. (2023) 8:302. doi: 10.1038/s41392-023-01555-9
90. Zhou M, Yuan M, Jin Y, Zhou Q, Yu Y, Li J, et al. Vitamin B2-based ferroptosis promoter for sono-enhanced nanocatalytic therapy of triple-negative breast cancer. Advanced Funct Materials. (2023) 33. doi: 10.1002/adfm.202303899
91. Guo S, Zhao W, Zhang W, Li S, Teng G, and Liu L. Vitamin D promotes ferroptosis in colorectal cancer stem cells via SLC7A11 downregulation. Oxid Med Cell Longevity. (2023) 2023. doi: 10.1155/2023/4772134
92. Bessman NJ, Mathieu JRR, Renassia C, Zhou L, Fung TC, Fernandez KC, et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science. (2020) 368:186–9. doi: 10.1126/science.aau6481
93. Zhao N, Liu JM, Yang FE, Ji XM, Li CY, Lv SW, et al. A novel mediation strategy of DSS-induced colitis in mice based on an iron-enriched probiotic and in vivo bioluminescence tracing. J Agric Food Chem. (2020) 68:12028–38. doi: 10.1021/acs.jafc.0c05260
94. Qi X, Zhang Y, Guo H, Hai Y, Luo Y, and Yue T. Mechanism and intervention measures of iron side effects on the intestine. Crit Rev Food Sci Nutr. (2020) 60:2113–25. doi: 10.1080/10408398.2019.1630599
95. Jakaria M, Belaidi AA, Bush AI, and Ayton S. Vitamin A metabolites inhibit ferroptosis. BioMed Pharmacother. (2023) 164:114930. doi: 10.1016/j.biopha.2023.114930
96. Chidley C, Darnell AM, Gaudio BL, Lien EC, Barbeau AM, Vander Heiden MG, et al. A CRISPRi/a screening platform to study cellular nutrient transport in diverse microenvironments. Nat Cell Biol. (2024) 26:825–38. doi: 10.1038/s41556-024-01402-1
97. Darby TM, Naudin CR, Luo L, and Jones RM. Lactobacillus rhamnosus GG-induced expression of leptin in the intestine orchestrates epithelial cell proliferation. Cell Mol Gastroenterol Hepatol. (2020) 9:627–39. doi: 10.1016/j.jcmgh.2019.12.004
98. Grasberger H, Gao J, Nagao-Kitamoto H, Kitamoto S, Zhang M, Kamada N, et al. Increased expression of DUOX2 is an epithelial response to mucosal dysbiosis required for immune homeostasis in mouse intestine. Gastroenterology. (2015) 149:1849–59. doi: 10.1053/j.gastro.2015.07.062
99. Li X, Yang J, Shi E, Lu Y, Song X, Luo H, et al. Riboflavin alleviates fluoride-induced ferroptosis by IL-17A-independent system Xc(-)/GPX4 pathway and iron metabolism in testicular Leydig cells. Environ pollut. (2024) 344:123332. doi: 10.1016/j.envpol.2024.123332
100. Tschuck J, Theilacker L, Rothenaigner I, Weiss SAI, Akdogan B, Lam VT, et al. Farnesoid X receptor activation by bile acids suppresses lipid peroxidation and ferroptosis. Nat Commun. (2023) 14:6908. doi: 10.1038/s41467-023-42702-8
101. Liu CX, Gao Y, Xu XF, Jin X, Zhang Y, Xu Q, et al. Bile acids inhibit ferroptosis sensitivity through activating farnesoid X receptor in gastric cancer cells. World J Gastroenterol. (2024) 30:485–98. doi: 10.3748/wjg.v30.i5.485
102. Bonakdar M, Czuba LC, Han G, Zhong G, Luong H, Isoherranen N, et al. Gut commensals expand vitamin A metabolic capacity of the mammalian host. Cell Host Microbe. (2022) 30:1084–92 e5. doi: 10.1016/j.chom.2022.06.011
103. Xie Y, Li S, Wu D, Wang Y, Chen J, Duan L, et al. Vitamin K: infection, inflammation, and auto-immunity. J Inflammation Res. (2024) 17:1147–60. doi: 10.2147/JIR.S445806
104. Jiang B, Zhao Y, Shi M, Song L, Wang Q, Qin Q, et al. DNAJB6 promotes ferroptosis in esophageal squamous cell carcinoma. Dig Dis Sci. (2020) 65:1999–2008. doi: 10.1007/s10620-019-05929-4
105. Kawasaki Y, Okumura H, Uchikado Y, Kita Y, Sasaki K, Owaki T, et al. Nrf2 is useful for predicting the effect of chemoradiation therapy on esophageal squamous cell carcinoma. Ann Surg Oncol. (2014) 21:2347–52. doi: 10.1245/s10434-014-3600-2
106. Feng L, Zhao K, Sun L, Yin X, Zhang J, Liu C, et al. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J Transl Med. (2021) 19:367. doi: 10.1186/s12967-021-03042-7
107. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, and Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. (2015) 212:555–68. doi: 10.1084/jem.20140857
108. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res Treat. (2018) 50:445–60. doi: 10.4143/crt.2016.572
109. Xi Y, Shen Y, Wu D, Zhang J, Lin C, Wang L, et al. CircBCAR3 accelerates esophageal cancer tumorigenesis and metastasis via sponging miR-27a-3p. Mol Cancer. (2022) 21:145. doi: 10.1186/s12943-022-01615-8
110. Lu M, Li J, Fan X, Xie F, Fan J, and Xiong Y. Novel immune-related ferroptosis signature in esophageal cancer: an informatics exploration of biological processes related to the TMEM161B-AS1/hsa-miR-27a-3p/GCH1 regulatory network. Front Genet. (2022) 13:829384. doi: 10.3389/fgene.2022.829384
111. Chen C, Zhao J, Liu JN, and Sun C. Mechanism and role of the neuropeptide LGI1 receptor ADAM23 in regulating biomarkers of ferroptosis and progression of esophageal cancer. Dis Markers. (2021) 2021:9227897. doi: 10.1155/2021/9227897
112. Yao W, Wang J, Meng F, Zhu Z, Jia X, Xu L, et al. Circular RNA circPVT1 inhibits 5-fluorouracil chemosensitivity by regulating ferroptosis through miR-30a-5p/FZD3 axis in esophageal cancer cells. Front Oncol. (2021) 11:780938. doi: 10.3389/fonc.2021.780938
113. Guan X, Wang Y, Yu W, Wei Y, Lu Y, Dai E, et al. Blocking ubiquitin-specific protease 7 induces ferroptosis in gastric cancer via targeting stearoyl-coA desaturase. Adv Sci (Weinh). (2024) 11:e2307899. doi: 10.1002/advs.202307899
114. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. (2020) 19:43. doi: 10.1186/s12943-020-01168-8
115. Crowe SE. Helicobacter pylori infection. N Engl J Med. (2019) 380:1158–65. doi: 10.1056/NEJMcp1710945
116. Ni H, Qin H, Sun C, Liu Y, Ruan G, Guo Q, et al. MiR-375 reduces the stemness of gastric cancer cells through triggering ferroptosis. Stem Cell Res Ther. (2021) 12:325. doi: 10.1186/s13287-021-02394-7
117. Wei F, Meng Y, Zhu DX, and Wu J. Mechanism and role of ferroptosis in the development of gastric cancer. Clin Exp Med. (2025) 25:182. doi: 10.1007/s10238-025-01722-y
118. Lin Z, Song J, Gao Y, Huang S, Dou R, Zhong P, et al. Hypoxia-induced HIF-1alpha/lncRNA-PMAN inhibits ferroptosis by promoting the cytoplasmic translocation of ELAVL1 in peritoneal dissemination from gastric cancer. Redox Biol. (2022) 52:102312. doi: 10.1016/j.redox.2022.102312
119. Stockwell BR and Jiang X. The chemistry and biology of ferroptosis. Cell Chem Biol. (2020) 27:365–75. doi: 10.1016/j.chembiol.2020.03.013
120. Xiaofang S, Zhanhui O, Ruochan C, Xiaohua N, De C, Rui K, et al. Erratum: Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2025) 82:E38–E9. doi: 10.1097/HEP.0000000000001384
121. Li Y, Xu B, Ren X, Wang L, Xu Y, Zhao Y, et al. Correction: Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. Cell Mol Biol Lett. (2023) 28:69. doi: 10.1186/s11658-023-00478-1
122. Ren X, Wang X, Yan Y, Chen X, Cai Y, Liang Q, et al. Integrative bioinformatics and experimental analysis revealed TEAD as novel prognostic target for hepatocellular carcinoma and its roles in ferroptosis regulation. Aging (Albany NY). (2022) 14:961–74. doi: 10.18632/aging.203853
123. Wesener MC, Weiler SME, Bissinger M, Klessinger TF, Rose F, Merker S, et al. CRKL enhances YAP signaling through binding and JNK/JUN pathway activation in liver cancer. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25158549
124. Shree Harini K, Ezhilarasan D, and Mani U. Molecular insights on intracellular Wnt/beta-catenin signaling in alcoholic liver disease. Cell Biochem Funct. (2024) 42:e3916. doi: 10.1002/cbf.3916
125. Fu H, Li W, Liu J, Tang Q, Weng Z, Zhu L, et al. Ellagic acid inhibits dihydrotestosterone-induced ferroptosis and promotes hair regeneration by activating the wnt/beta-catenin signaling pathway. J Ethnopharmacol. (2024) 330:118227. doi: 10.1016/j.jep.2024.118227
126. Zheng C, Zhang B, Li Y, Liu K, Wei W, Liang S, et al. Donafenib and GSK-J4 synergistically induce ferroptosis in liver cancer by upregulating HMOX1 expression. Adv Sci (Weinh). (2023) 10:e2206798. doi: 10.1002/advs.202206798
127. Wang J, Shanmugam A, Markand S, Zorrilla E, Ganapathy V, and Smith SB. Sigma 1 receptor regulates the oxidative stress response in primary retinal Muller glial cells via NRF2 signaling and system xc(-), the Na(+)-independent glutamate-cystine exchanger. Free Radic Biol Med. (2015) 86:25–36. doi: 10.1016/j.freeradbiomed.2015.04.009
128. Bai T, Lei P, Zhou H, Liang R, Zhu R, Wang W, et al. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med. (2019) 23:7349–59. doi: 10.1111/jcmm.14594
129. Bai T, Wang S, Zhao Y, Zhu R, Wang W, and Sun Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun. (2017) 491:919–25. doi: 10.1016/j.bbrc.2017.07.136
130. Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. (2021) 12:426. doi: 10.1038/s41419-021-03718-4
131. Wang Y, Zhao M, Zhao L, Geng Y, Li G, Chen L, et al. HBx-induced HSPA8 stimulates HBV replication and suppresses ferroptosis to support liver cancer progression. Cancer Res. (2023) 83:1048–61. doi: 10.1158/0008-5472.CAN-22-3169
132. Yang Y, Lin J, Guo S, Xue X, Wang Y, Qiu S, et al. RRM2 protects against ferroptosis and is a tumor biomarker for liver cancer. Cancer Cell Int. (2020) 20:587. doi: 10.1186/s12935-020-01689-8
133. Yang PM, Lin LS, and Liu TP. Sorafenib inhibits ribonucleotide reductase regulatory subunit M2 (RRM2) in hepatocellular carcinoma cells. Biomolecules. (2020) 10. doi: 10.3390/biom10010117
134. Ndiaye H, Liu JY, Hall A, Minogue S, Morgan MY, and Waugh MG. Immunohistochemical staining reveals differential expression of ACSL3 and ACSL4 in hepatocellular carcinoma and hepatic gastrointestinal metastases. Biosci Rep. (2020) 40. doi: 10.1042/BSR20200219
135. Seibt TM, Proneth B, and Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. (2019) 133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014
136. Shang Y, Luo M, Yao F, Wang S, Yuan Z, and Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. (2020) 72:109633. doi: 10.1016/j.cellsig.2020.109633
137. Bai T, Liang R, Zhu R, Wang W, Zhou L, and Sun Y. MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J Cell Physiol. (2020) 235:5637–48. doi: 10.1002/jcp.29496
138. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. (2020) 368:85–9. doi: 10.1126/science.aaw9872
139. Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. (2021) 12:4860. doi: 10.1038/s41467-021-24859-2
140. Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. (2020) 11:6339. doi: 10.1038/s41467-020-20154-8
141. Liu Y, Wang Y, Liu J, Kang R, and Tang D. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. (2021) 28:55–63. doi: 10.1038/s41417-020-0182-y
142. Liu J, Song X, Kuang F, Zhang Q, Xie Y, Kang R, et al. NUPR1 is a critical repressor of ferroptosis. Nat Commun. (2021) 12:647. doi: 10.1038/s41467-021-20904-2
143. Ye Z, Zhuo Q, Hu Q, Xu X, Mengqi L, Zhang Z, et al. FBW7-NRA41-SCD1 axis synchronously regulates apoptosis and ferroptosis in pancreatic cancer cells. Redox Biol. (2021) 38:101807. doi: 10.1016/j.redox.2020.101807
144. Lin H, Zhu S, Chen Y, Lu J, Xie C, Liao C, et al. Targeting cTRIP12 counteracts ferroptosis resistance and augments sensitivity to immunotherapy in pancreatic cancer. Drug Resist Update. (2025) 81:101240. doi: 10.1016/j.drup.2025.101240
145. Qiao C, Wang L, Huang C, Jia Q, Bao W, Guo P, et al. Engineered bacteria manipulate cysteine metabolism to boost ferroptosis-based pancreatic ductal adenocarcinoma therapy. Adv Mater. (2025) 37:e2412982. doi: 10.1002/adma.202412982
146. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. (2017) 20:1692–704. doi: 10.1016/j.celrep.2017.07.055
147. Yang C, Zhang Y, Lin S, Liu Y, and Li W. Suppressing the KIF20A/NUAK1/Nrf2/GPX4 signaling pathway induces ferroptosis and enhances the sensitivity of colorectal cancer to oxaliplatin. Aging (Albany NY). (2021) 13:13515–34. doi: 10.18632/aging.202774
148. Zhang Y and Xie J. Targeting ferroptosis regulators by natural products in colorectal cancer. Front Pharmacol. (2024) 15:1374722. doi: 10.3389/fphar.2024.1374722
149. Tian X, Li S, and Ge G. Apatinib promotes ferroptosis in colorectal cancer cells by targeting ELOVL6/ACSL4 signaling. Cancer Manag Res. (2021) 13:1333–42. doi: 10.2147/CMAR.S274631
150. Yan H, Talty R, Aladelokun O, Bosenberg M, and Johnson CH. Ferroptosis in colorectal cancer: a future target? Br J Cancer. (2023) 128:1439–51. doi: 10.1038/s41416-023-02149-6
151. Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C, et al. Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med. (2023) 15:eadg3049. doi: 10.1126/scitranslmed.adg3049
152. Huang HX, Yang G, Yang Y, Yan J, Tang XY, and Pan Q. TFAP2A is a novel regulator that modulates ferroptosis in gallbladder carcinoma cells via the Nrf2 signalling axis. Eur Rev Med Pharmacol Sci. (2020) 24:4745–55. doi: 10.26355/eurrev_202005_21163
153. Wang Z, Li W, Wang X, Zhu Q, Liu L, Qiu S, et al. Isoliquiritigenin induces HMOX1 and GPX4-mediated ferroptosis in gallbladder cancer cells. Chin Med J (Engl). (2023) 136:2210–20. doi: 10.1097/CM9.0000000000002675
154. Li W, Wang Z, Lin R, Huang S, Miao H, Zou L, et al. Lithocholic acid inhibits gallbladder cancer proliferation through interfering glutaminase-mediated glutamine metabolism. Biochem Pharmacol. (2022) 205:115253. doi: 10.1016/j.bcp.2022.115253
155. Cai C, Zhu Y, Mu J, Liu S, Yang Z, Wu Z, et al. DNA methylation of RUNX3 promotes the progression of gallbladder cancer through repressing SLC7A11-mediated ferroptosis. Cell Signal. (2023) 108:110710. doi: 10.1016/j.cellsig.2023.110710
156. Balachander K and Paramasivam A. Ferroptosis: An emerging therapeutic target for oral cancer. Oral Oncol. (2022) 131:105970. doi: 10.1016/j.oraloncology.2022.105970
157. Han L, Li L, and Wu G. Induction of ferroptosis by carnosic acid-mediated inactivation of Nrf2/HO-1 potentiates cisplatin responsiveness in OSCC cells. Mol Cell Probes. (2022) 64:101821. doi: 10.1016/j.mcp.2022.101821
158. Wang X, Liu K, Gong H, Li D, Chu W, Zhao D, et al. Death by histone deacetylase inhibitor quisinostat in tongue squamous cell carcinoma via apoptosis, pyroptosis, and ferroptosis. Toxicol Appl Pharmacol. (2021) 410:115363. doi: 10.1016/j.taap.2020.115363
159. Sato K, Shi L, Ito F, Ohara Y, Motooka Y, Tanaka H, et al. Non-thermal plasma specifically kills oral squamous cell carcinoma cells in a catalytic Fe(II)-dependent manner. J Clin Biochem Nutr. (2019) 65:8–15. doi: 10.3164/jcbn.18-91
160. Zhu T, Shi L, Yu C, Dong Y, Qiu F, Shen L, et al. Ferroptosis promotes photodynamic therapy: supramolecular photosensitizer-inducer nanodrug for enhanced cancer treatment. Theranostics. (2019) 9:3293–307. doi: 10.7150/thno.32867
161. Yang J, Cao XH, Luan KF, and Huang YD. Circular RNA FNDC3B protects oral squamous cell carcinoma cells from ferroptosis and contributes to the Malignant progression by regulating miR-520d-5p/SLC7A11 axis. Front Oncol. (2021) 11:672724. doi: 10.3389/fonc.2021.672724
162. Yu Y, MohamedAl-Sharani H, and Zhang B. EZH2-mediated SLC7A11 upregulation via miR-125b-5p represses ferroptosis of TSCC. Oral Dis. (2023) 29:880–91. doi: 10.1111/odi.14040
163. Sun K, Ren W, Li S, Zheng J, Huang Y, Zhi K, et al. MiR-34c-3p upregulates erastin-induced ferroptosis to inhibit proliferation in oral squamous cell carcinomas by targeting SLC7A11. Pathol Res Pract. (2022) 231:153778. doi: 10.1016/j.prp.2022.153778
164. Yang Y, Tang H, Zheng J, and Yang K. The PER1/HIF-1alpha negative feedback loop promotes ferroptosis and inhibits tumor progression in oral squamous cell carcinoma. Transl Oncol. (2022) 18:101360. doi: 10.1016/j.tranon.2022.101360
165. Li M, Jin S, Zhang Z, Ma H, and Yang X. Interleukin-6 facilitates tumor progression by inducing ferroptosis resistance in head and neck squamous cell carcinoma. Cancer Lett. (2022) 527:28–40. doi: 10.1016/j.canlet.2021.12.011
166. Zhou Q, Wang X, Zhang Y, Wang L, and Chen Z. Inhibition of AEBP1 predisposes cisplatin-resistant oral cancer cells to ferroptosis. BMC Oral Health. (2022) 22:478. doi: 10.1186/s12903-022-02503-9
167. Feng H and Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PloS Biol. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
168. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. (2017) 551:247–50. doi: 10.1038/nature24297
169. Liu CC, Li HH, Lin JH, Chiang MC, Hsu TW, Li AF, et al. Esophageal cancer stem-like cells resist ferroptosis-induced cell death by active hsp27-GPX4 pathway. Biomolecules. (2021) 12. doi: 10.3390/biom12010048
170. Yu VZ, Wong VC, Dai W, Ko JM, Lam AK, Chan KW, et al. Nuclear localization of DNAJB6 is associated with survival of patients with esophageal cancer and reduces AKT signaling and proliferation of cancer cells. Gastroenterology. (2015) 149:1825–36 e5. doi: 10.1053/j.gastro.2015.08.025
171. Zhang X, Yuan S, Fan H, Zhang W, and Zhang H. Liensinine alleviates sepsis-induced acute liver injury by inhibiting the NF-kappaB and MAPK pathways in an Nrf2-dependent manner. Chem Biol Interact. (2024) 396:111030. doi: 10.1016/j.cbi.2024.111030
172. Xiao S, Liu N, Yang X, Ji G, and Li M. Polygalacin D suppresses esophageal squamous cell carcinoma growth and metastasis through regulating miR-142-5p/Nrf2 axis. Free Radic Biol Med. (2021) 164:58–75. doi: 10.1016/j.freeradbiomed.2020.11.029
173. Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. (2017) 68:224–32 e4. doi: 10.1016/j.molcel.2017.09.009
174. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. (2020) 30:146–62. doi: 10.1038/s41422-019-0263-3
175. Luo H, Wang X, Song S, Wang Y, Dan Q, and Ge H. Targeting stearoyl-coa desaturase enhances radiation induced ferroptosis and immunogenic cell death in esophageal squamous cell carcinoma. Oncoimmunology. (2022) 11:2101769. doi: 10.1080/2162402X.2022.2101769
176. Li N, Yi X, He Y, Huo B, Chen Y, Zhang Z, et al. Targeting ferroptosis as a novel approach to alleviate aortic dissection. Int J Biol Sci. (2022) 18:4118–34. doi: 10.7150/ijbs.72528
177. Zhao M, Lu T, Bi G, Hu Z, Liang J, Bian Y, et al. PLK1 regulating chemoradiotherapy sensitivity of esophageal squamous cell carcinoma through pentose phosphate pathway/ferroptosis. BioMed Pharmacother. (2023) 168:115711. doi: 10.1016/j.biopha.2023.115711
178. Yang JY, Lei XY, He KY, Guo JR, Liu MJ, Li JQ, et al. HMGA1 drives chemoresistance in esophageal squamous cell carcinoma by suppressing ferroptosis. Cell Death Dis. (2024) 15:158. doi: 10.1038/s41419-024-06467-2
179. Song K, Ma C, Maswikiti EP, Gu B, Wang B, Wang N, et al. Downregulation of ALDH5A1 suppresses cisplatin resistance in esophageal squamous cell carcinoma by regulating ferroptosis signaling pathways. Mol Carcinog. (2024) 63:1892–906. doi: 10.1002/mc.23778
180. Prosz A, Sahgal P, Huffman BM, Sztupinszki Z, Morris CX, Chen D, et al. Mutational signature-based identification of DNA repair deficient gastroesophageal adenocarcinomas for therapeutic targeting. NPJ Precis Oncol. (2024) 8:87. doi: 10.1038/s41698-024-00561-6
181. Lee J, You JH, Shin D, and Roh JL. Inhibition of glutaredoxin 5 predisposes cisplatin-resistant head and neck cancer cells to ferroptosis. Theranostics. (2020) 10:7775–86. doi: 10.7150/thno.46903
182. Lei G, Zhang Y, Hong T, Zhang X, Liu X, Mao C, et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene. (2021) 40:3533–47. doi: 10.1038/s41388-021-01790-w
183. Ballout F, Lu H, Chen Z, Hu T, Chen L, Washington MK, et al. Targeting NRF2 sensitizes esophageal adenocarcinoma cells to cisplatin through induction of ferroptosis and apoptosis. Antioxidants (Basel). (2022) 11. doi: 10.3390/antiox11101859
184. Cao Y, Zhang H, Tang J, and Wang R. Ferulic acid mitigates growth and invasion of esophageal squamous cell carcinoma through inducing ferroptotic cell death. Dis Markers. (2022) 2022:4607966. doi: 10.1155/2022/4607966
185. Zhang J, Wang N, Zhou Y, Wang K, Sun Y, Yan H, et al. Oridonin induces ferroptosis by inhibiting gamma-glutamyl cycle in TE1 cells. Phytother Res. (2021) 35:494–503. doi: 10.1002/ptr.6829
186. Peng H, He Y, Hu Y, Sheng S, Maitiyasen M, Li J, et al. Berbamine promotes ferroptosis of esophageal squamous cell carcinoma by facilitating USP51-mediated GPX4 ubiquitination and degradation. BioMed Pharmacother. (2024) 179:117309. doi: 10.1016/j.biopha.2024.117309
187. Lu Z, Zhang G, Zhang Y, Hua P, Fang M, Wu M, et al. Isoalantolactone induces apoptosis through reactive oxygen species-dependent upregulation of death receptor 5 in human esophageal cancer cells. Toxicol Appl Pharmacol. (2018) 352:46–58. doi: 10.1016/j.taap.2018.05.026
188. Liu J, Deng L, Wang L, Qian D, He C, Ren Q, et al. Licochalcone A induces G2/M phase arrest and apoptosis via regulating p53 pathways in esophageal cancer: In-vitro and in-vivo study. Eur J Pharmacol. (2023) 958:176080. doi: 10.1016/j.ejphar.2023.176080
189. Zhang X, Yang R, Wang H, Cao C, Zhao W, Duan L, et al. Inhibition of p62-keap1-nrf2 pathway activation by realgar promotes the inhibition of esophageal cancer cell proliferation, migration, and ferroptosis. Curr Drug Delivery. (2024) 21:236–48. doi: 10.2174/1567201820666221226105655
190. Zhu X, Huang N, Ji Y, Sheng X, Huo J, Zhu Y, et al. Brusatol induces ferroptosis in oesophageal squamous cell carcinoma by repressing GSH synthesis and increasing the labile iron pool via inhibition of the NRF2 pathway. BioMed Pharmacother. (2023) 167:115567. doi: 10.1016/j.biopha.2023.115567
191. Kojima Y, Tanaka M, Sasaki M, Ozeki K, Shimura T, Kubota E, et al. Induction of ferroptosis by photodynamic therapy and enhancement of antitumor effect with ferroptosis inducers. J Gastroenterol. (2024) 59:81–94. doi: 10.1007/s00535-023-02054-y
192. Cunderlikova B, Kluckova K, Babal P, Mlkvy P, and Teplicky T. Modifications of DAMPs levels in extracellular environment induced by aminolevulinic acid-based photodynamic therapy of esophageal cancer cells. Int J Radiat Biol. (2024) 100:802–16. doi: 10.1080/09553002.2024.2310002
193. Yuan Z, Wang X, Qin B, Hu R, Miao R, Zhou Y, et al. Targeting NQO1 induces ferroptosis and triggers anti-tumor immunity in immunotherapy-resistant KEAP1-deficient cancers. Drug Resist Update. (2024) 77:101160. doi: 10.1016/j.drup.2024.101160
194. Bai T, Xue P, Shao S, Yan S, and Zeng X. Cholesterol depletion-enhanced ferroptosis and immunotherapy via engineered nanozyme. Adv Sci (Weinh). (2024) 11:e2405826. doi: 10.1002/advs.202405826
195. Dai Y, Guo Z, Leng D, Jiao G, Chen K, Fu M, et al. Metal-coordinated NIR-II nanoadjuvants with nanobody conjugation for potentiating immunotherapy by tumor metabolism reprogramming. Adv Sci (Weinh). (2024) 11:e2404886. doi: 10.1002/advs.202404886
196. Yang J, Ren B, Cai H, Xiong W, Feng J, Fan Q, et al. Cyclic catalysis of intratumor Fe(3+/2+) initiated by a hollow mesoporous iron sesquioxide nanoparticle for ferroptosis therapy of large tumors. Biomaterials. (2025) 313:122793. doi: 10.1016/j.biomaterials.2024.122793
197. Han S, Zou J, Xiao F, Xian J, Liu Z, Li M, et al. Nanobiotechnology boosts ferroptosis: opportunities and challenges. J Nanobiotechnology. (2024) 22:606. doi: 10.1186/s12951-024-02842-5
198. Zhou LL, Guan Q, Zhou W, Kan JL, Teng K, Hu M, et al. A multifunctional covalent organic framework nanozyme for promoting ferroptotic radiotherapy against esophageal cancer. ACS Nano. (2023) 17:20445–61. doi: 10.1021/acsnano.3c06967
199. Shi J, Ma W, Deng J, Zheng S, Xia F, Liu X, et al. Self-assembled hyaluronic acid nanomicelle for enhanced cascade cancer chemotherapy via self-sensitized ferroptosis. Carbohydr Polym. (2024) 343. doi: 10.1016/j.carbpol.2024.122489
200. Shen J, Feng K, Yu J, Zhao Y, Chen R, Xiong H, et al. Responsive and traceless assembly of iron nanoparticles and (131)I labeled radiopharmaceuticals for ferroptosis enhanced radio-immunotherapy. Biomaterials. (2025) 313:122795. doi: 10.1016/j.biomaterials.2024.122795
201. Li X, Xiong W, Wang Y, Li Y, Cheng X, and Liu W. p53 activates the lipoxygenase activity of ALOX15B via inhibiting SLC7A11 to induce ferroptosis in bladder cancer cells. Lab investigation; J Tech Methods Pathol. (2023) 103:100058. doi: 10.1016/j.labinv.2022.100058
202. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. (2019) 572:402–6. doi: 10.1038/s41586-019-1426-6
203. Cheng X, Dai E, Wu J, Flores NM, Chu Y, Wang R, et al. Atlas of metastatic gastric cancer links ferroptosis to disease progression and immunotherapy response. Gastroenterology. (2024) 167:1345–57. doi: 10.1053/j.gastro.2024.07.038
204. Ma W, Hu N, Xu W, Zhao L, Tian C, and Kamei KI. Ferroptosis inducers: A new frontier in cancer therapy. Bioorg Chem. (2024) 146:107331. doi: 10.1016/j.bioorg.2024.107331
205. Jin S, Wang H, Zhang Z, and Yan M. Targeting ferroptosis: small-molecule inducers as novel anticancer agents. Anticancer Agents Med Chem. (2025) 25:517–32. doi: 10.2174/0118715206342278241008081126
206. Zhao H, Ao L, Sorina, Wei Y, Yin HZ, Zhang N, et al. Ferroptosis and gastric cancer: from molecular mechanisms to clinical implications. Front Immunol. (2025) 16:1581928. doi: 10.3389/fimmu.2025.1581928
207. Hou J, Wang B, Li J, and Liu W. Ferroptosis and its role in gastric and colorectal cancers. Korean J Physiol Pharmacol. (2024) 28:183–96. doi: 10.4196/kjpp.2024.28.3.183
208. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
209. Wen Q, Liu J, Kang R, Zhou B, and Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
210. Renner N, Schob F, Pape R, Suciu I, Spreng AS, Uckert AK, et al. Modeling ferroptosis in human dopaminergic neurons: Pitfalls and opportunities for neurodegeneration research. Redox Biol. (2024) 73:103165. doi: 10.1016/j.redox.2024.103165
211. Sun Z, Zhang T, Ahmad MU, Zhou Z, Qiu L, Zhou K, et al. Comprehensive assessment of immune context and immunotherapy response via noninvasive imaging in gastric cancer. J Clin Invest. (2024) 134. doi: 10.1172/JCI175834
212. Wang Y, Gan X, Cheng X, Jia Y, Wang G, Tang X, et al. ABCC2 induces metabolic vulnerability and cellular ferroptosis via enhanced glutathione efflux in gastric cancer. Clin Transl Med. (2024) 14:e1754. doi: 10.1002/ctm2.1754
213. Kiani BH, Kayani WK, Khayam AU, Dilshad E, Ismail H, and Mirza B. Artemisinin and its derivatives: a promising cancer therapy. Mol Biol Rep. (2020) 47:6321–36. doi: 10.1007/s11033-020-05669-z
214. Li R, Ning Y, Yuan Y, and Yang X. Molecular mechanisms of ferroptosis and its effects on bladder cancer. Zhong nan da xue xue bao Yi xue ban = J Cent South Univ Med Sci. (2024) 49:286–95. doi: 10.11817/j.issn.1672-7347.2024.230352
215. Ma M, Kong P, Huang Y, Wang J, Liu X, Hu Y, et al. Activation of MAT2A-ACSL3 pathway protects cells from ferroptosis in gastric cancer. Free Radic Biol Med. (2022) 181:288–99. doi: 10.1016/j.freeradbiomed.2022.02.015
216. Wu L, Ding Q, Wang X, Li P, Fan N, Zhou Y, et al. Visualization of dynamic changes in labile iron(II) pools in endoplasmic reticulum stress-mediated drug-induced liver injury. Anal Chem. (2020) 92:1245–51. doi: 10.1021/acs.analchem.9b04411
217. Pan R, Zhao Z, Xu D, Li C, and Xia Q. GPX4 transcriptionally promotes liver cancer metastasis via GRHL3/PTEN/PI3K/AKT axis. Transl Res. (2024) 271:79–92. doi: 10.1016/j.trsl.2024.05.007
218. Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, and Vergely C. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. (2022) 24. doi: 10.3390/ijms24010449
219. Jyotsana N, Ta KT, and DelGiorno KE. The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front Oncol. (2022) 12:858462. doi: 10.3389/fonc.2022.858462
220. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via slc7a11-mediated ferroptosis. Circ Res. (2020) 127:486–501. doi: 10.1161/CIRCRESAHA.120.316509
221. Huang W, Guo Y, Qian Y, Liu X, Li G, Wang J, et al. Ferroptosis-inducing compounds synergize with docetaxel to overcome chemoresistance in docetaxel-resistant non-small cell lung cancer cells. Eur J Med Chem. (2024) 276:116670. doi: 10.1016/j.ejmech.2024.116670
222. Hu X, Zhang P, Li S, Zhang J, Wang D, Wang Z, et al. Mitochondrial GCN5L1 acts as a novel regulator for iron homeostasis to promote sorafenib sensitivity in hepatocellular carcinoma. J Transl Med. (2024) 22:593. doi: 10.1186/s12967-024-05404-3
223. Wang LL, Mai YZ, Zheng MH, Yan GH, and Jin JY. A single fluorescent probe to examine the dynamics of mitochondria-lysosome interplay and extracellular vesicle role in ferroptosis. Dev Cell. (2024) 59:517–28 e3. doi: 10.1016/j.devcel.2024.01.003
224. Chen X, Wang J, Yang P, Liu HY, Zhong S, Lu C, et al. SENP3 sensitizes macrophages to ferroptosis via de-SUMOylation of FSP1. Redox Biol. (2024) 75:103267. doi: 10.1016/j.redox.2024.103267
225. Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. (2021) 33:174–89 e7. doi: 10.1016/j.cmet.2020.12.007
226. Wang H, Fleishman JS, Cheng S, Wang W, Wu F, Wang Y, et al. Epigenetic modification of ferroptosis by non-coding RNAs in cancer drug resistance. Mol Cancer. (2024) 23:177. doi: 10.1186/s12943-024-02088-7
227. Sun L, Cao H, Wang Y, and Wang H. Regulating ferroptosis by non-coding RNAs in hepatocellular carcinoma. Biol Direct. (2024) 19:80. doi: 10.1186/s13062-024-00530-w
228. Waters AM and Der CJ. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb Perspect Med. (2018) 8. doi: 10.1101/cshperspect.a031435
229. Costa I, Barbosa DJ, Benfeito S, Silva V, Chavarria D, Borges F, et al. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol Ther. (2023) 244:108373. doi: 10.1016/j.pharmthera.2023.108373
230. Liu J, Zhu S, Zeng L, Li J, Klionsky DJ, Kroemer G, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy. (2022) 18:2036–49. doi: 10.1080/15548627.2021.2008692
231. Shi Y, Wang Y, Dong H, Niu K, Zhang W, Feng K, et al. Crosstalk of ferroptosis regulators and tumor immunity in pancreatic adenocarcinoma: novel perspective to mRNA vaccines and personalized immunotherapy. Apoptosis. (2023) 28:1423–35. doi: 10.1007/s10495-023-01868-8
232. Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, and Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. (2021) 17:948–60. doi: 10.1080/15548627.2020.1739447
233. Yamaguchi Y, Kasukabe T, and Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. (2018) 52:1011–22. doi: 10.3892/ijo.2018.4259
234. Chen G, Guo G, Zhou X, and Chen H. Potential mechanism of ferroptosis in pancreatic cancer. Oncol Lett. (2020) 19:579–87. doi: 10.3892/ol.2019.11159
235. Du J, Wang X, Li Y, Ren X, Zhou Y, Hu W, et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. (2021) 12:705. doi: 10.1038/s41419-021-03996-y
236. Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. (2017) 77:2064–77. doi: 10.1158/0008-5472.CAN-16-1979
237. Fan X, Dai Y, Mo C, Li H, Luan X, Wang B, et al. TRIM21 promotes tumor growth and gemcitabine resistance in pancreatic cancer by inhibiting EPHX1-mediated arachidonic acid metabolism. Adv Sci (Weinh). (2025) 12:e2413674. doi: 10.1002/advs.202413674
238. Li M, Yu X, Liu Y, Ouyang S, Wu L, Chen X, et al. KRAS/ABHD17C/ALOX15B axis promotes pancreatic cancer progression via ferroptosis evasion. Adv Sci (Weinh). (2025) 12:e04470. doi: 10.1002/advs.202504470
239. He Z, Zheng D, Li F, Chen L, Wu C, Zeng Z, et al. TMOD3 accelerated resistance to immunotherapy in KRAS-mutated pancreatic cancer through promoting autophagy-dependent degradation of ASCL4. Drug Resist Update. (2025) 78:101171. doi: 10.1016/j.drup.2024.101171
240. Sun R, Lin Z, Wang X, Liu L, Huo M, Zhang R, et al. AADAC protects colorectal cancer liver colonization from ferroptosis through SLC7A11-dependent inhibition of lipid peroxidation. J Exp Clin Cancer Res. (2022) 41:284. doi: 10.1186/s13046-022-02493-0
241. Lorenzato A, Magri A, Matafora V, Audrito V, Arcella P, Lazzari L, et al. Vitamin C restricts the emergence of acquired resistance to EGFR-targeted therapies in colorectal cancer. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12030685
242. Chen C, Yang Y, Guo Y, He J, Chen Z, Qiu S, et al. CYP1B1 inhibits ferroptosis and induces anti-PD-1 resistance by degrading ACSL4 in colorectal cancer. Cell Death Dis. (2023) 14:271. doi: 10.1038/s41419-023-05803-2
243. Tang HH, Hsu CP, Su PY, Tsai SP, Doan LH, Chen CY, et al. Targeting GPX4 to overcome sorafenib resistance of human hepatocellular carcinoma by inducing ferroptosis. J Cell Physiol. (2025) 240:e70078. doi: 10.1002/jcp.70078
244. Li J, Mao S, Yang S, Lou Y, Zhao X, Chen J, et al. Targeting JMJD6/PPARγ/GPX4 axis overcomes ferroptosis resistance and enhances therapeutic efficacy in hepatocellular carcinoma. Oncogene. (2025) 44:4377–90. doi: 10.1038/s41388-025-03581-z
245. Chen JF, Wu SW, Shi ZM, and Hu B. Traditional Chinese medicine for colorectal cancer treatment: potential targets and mechanisms of action. Chin Med. (2023) 18:14. doi: 10.1186/s13020-023-00719-7
246. Sun D, Zhang J, Wang L, Yu Z, O’Driscoll CM, and Guo J. Nanodelivery of immunogenic cell death-inducers for cancer immunotherapy. Drug Discov Today. (2021) 26:651–62. doi: 10.1016/j.drudis.2020.11.029
247. Ouyang S, Li H, Lou L, Huang Q, Zhang Z, Mo J, et al. Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol. (2022) 52:102317. doi: 10.1016/j.redox.2022.102317
248. Sun Y, Qiao Y, Liu Y, Zhou J, Wang X, Zheng H, et al. ent-Kaurane diterpenoids induce apoptosis and ferroptosis through targeting redox resetting to overcome cisplatin resistance. Redox Biol. (2021) 43:101977. doi: 10.1016/j.redox.2021.101977
249. Wang Y, Wei Z, Pan K, Li J, and Chen Q. The function and mechanism of ferroptosis in cancer. Apoptosis. (2020) 25:786–98. doi: 10.1007/s10495-020-01638-w
250. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. (2016) 64:488–500. doi: 10.1002/hep.28574
251. Zhang S, Liu Q, Chang M, Pan Y, Yahaya BH, Liu Y, et al. Chemotherapy impairs ovarian function through excessive ROS-induced ferroptosis. Cell Death Dis. (2023) 14:340. doi: 10.1038/s41419-023-05859-0
252. Gong D, Chen M, Wang Y, Shi J, and Hou Y. Role of ferroptosis on tumor progression and immunotherapy. Cell Death Discov. (2022) 8:427. doi: 10.1038/s41420-022-01218-8
253. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. (2022) 40:365–78 e6. doi: 10.1016/j.ccell.2022.02.003
254. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. (2020) 585:113–8. doi: 10.1038/s41586-020-2623-z
255. Jiang Z, Lim SO, Yan M, Hsu JL, Yao J, Wei Y, et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J Clin Invest. (2021) 131. doi: 10.1172/JCI139434
256. Zhang F, Li F, Lu GH, Nie W, Zhang L, Lv Y, et al. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano. (2019) 13:5662–73. doi: 10.1021/acsnano.9b00892
257. Ji Q, Fu S, Zuo H, Huang Y, Chu L, Zhu Y, et al. ACSL4 is essential for radiation-induced intestinal injury by initiating ferroptosis. Cell Death Discov. (2022) 8:332. doi: 10.1038/s41420-022-01127-w
258. Vaupel P and Semenza GL. Master role of hypoxia in cancer progression: major insights during ISOTT’s half-century. Adv Exp Med Biol. (2024) 1463:15–20. doi: 10.1007/978-3-031-67458-7_3
259. Suvac A, Ashton J, and Bristow RG. Tumour hypoxia in driving genomic instability and tumour evolution. Nat Rev Cancer. (2025) 25:167–88. doi: 10.1038/s41568-024-00781-9
260. Dong Y, Ge X, Guo Q, Ou X, Liu C, Wang Y, et al. Mycobacterium bovis frd operon phase variation hijacks succinate signaling to drive immunometabolic rewiring and pathogenicity. Nat Commun. (2025) 16:6538. doi: 10.1038/s41467-025-61824-9
261. Wang G, Mao W, Zhang Y, Yang H, Zhu M, Li Y, et al. Multiomics and systematic analyses reveal the roles of hemoglobin and the HIF-1 pathway in polycystic ovary syndrome. Adv Sci (Weinh). (2025) 12:e2411679. doi: 10.1002/advs.202411679
262. Zhang Z, Lu M, Chen C, Tong X, Li Y, Yang K, et al. Holo-lactoferrin: the link between ferroptosis and radiotherapy in triple-negative breast cancer. Theranostics. (2021) 11:3167–82. doi: 10.7150/thno.52028
263. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. (2019) 9:1673–85. doi: 10.1158/2159-8290.CD-19-0338
264. Jiang Q, Wang K, Zhang X, Ouyang B, Liu H, Pang Z, et al. Platelet membrane-camouflaged magnetic nanoparticles for ferroptosis-enhanced cancer immunotherapy. Small. (2020) 16:e2001704. doi: 10.1002/smll.202001704
265. Xu T, Ma Y, Yuan Q, Hu H, Hu X, Qian Z, et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS Nano. (2020) 14:3414–25. doi: 10.1021/acsnano.9b09426
266. Guan Q, Guo R, Huang S, Zhang F, Liu J, Wang Z, et al. Mesoporous polydopamine carrying sorafenib and SPIO nanoparticles for MRI-guided ferroptosis cancer therapy. J Control Release. (2020) 320:392–403. doi: 10.1016/j.jconrel.2020.01.048
267. Wang Y, Sun T, and Jiang C. Nanodrug delivery systems for ferroptosis-based cancer therapy. J Controlled release: Off J Controlled Release Soc. (2022) 344:289–301. doi: 10.1016/j.jconrel.2022.01.034
268. Cheng K, Guo Q, Shen Z, Yang W, Zhou Y, Sun Z, et al. Frontiers of ferroptosis research: An analysis from the top 100 most influential articles in the field. Front Oncol. (2022) 12:948389. doi: 10.3389/fonc.2022.948389
Keywords: ferroptosis, digestive tract tumors, gut microbiota metabolites, tumor microenvironment, cancer therapy
Citation: Wen J, Liang H, Zhao M, Xuan B, Meng X, Liu Y, Wang L, He L, Zhou T, Tao Y and Wang Y (2025) Harnessing the gut microbiome to modulate ferroptosis: a metabolic strategy for the treatment of digestive tract cancers. Front. Immunol. 16:1703964. doi: 10.3389/fimmu.2025.1703964
Received: 12 September 2025; Accepted: 27 October 2025;
Published: 10 November 2025.
Edited by:
Marina De Bernard, University of Padua, ItalyReviewed by:
Attinder Chadha, University of California, San Francisco, United StatesRuoxi Zhang, University of Texas Southwestern Medical Center, United States
Copyright © 2025 Wen, Liang, Zhao, Xuan, Meng, Liu, Wang, He, Zhou, Tao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yimin Wang, ZHJ3YW5neWltaW5AaGVibXUuZWR1LmNu