 Manuel Ruiz-Pablos
Manuel Ruiz-Pablos Bruno Paiva
Bruno Paiva Aintzane Zabaleta
Aintzane Zabaleta- 1Faculty of Biological Sciences, Universidad Complutense de Madrid, Madrid, Spain
- 2Clinica Universidad de Navarra, Centro de Investigación Médica Aplicada (CIMA), IdiSNA, Instituto de Investigación Sanitaria de Navarra, Pamplona, Spain
The prevalence of autoimmune diseases in contemporary human populations poses a challenge for both medicine and evolutionary biology. This review explores how the ancestral human leukocyte antigen class II (HLA-II) haplotypes DR2-DQ6, DR4-DQ8 and DR3-DQ2 could play a central role in susceptibility to these diseases. We propose that these haplotypes, selected in historical contexts of high infectious pressure, may have been maintained because of their ability to elicit strong T-cell responses against pathogens; however, that antigenic promiscuity may be associated with an increased tendency toward immune hyperreactivity in modern environments. This hyperreactivity, involving proinflammatory cytokines including interferon-gamma (IFN-γ), could contribute to the breakdown of tolerance and the emergence of autoimmunity and related clinical phenomena (e.g., Long COVID, myalgic encephalomyelitis/chronic fatigue syndrome and post-vaccination syndromes), although the evidence for the latter remains limited. Finally, we discuss how chronic infections, immunotherapies, vaccination, obesity and chronic physical stressors may exacerbate this susceptibility and consider the therapeutic implications of integrating HLA-II profiling into clinical practice.
1 Introduction
Autoimmune diseases, such as multiple sclerosis, rheumatoid arthritis, celiac disease, systemic lupus erythematosus, type 1 diabetes and Sjögren’s syndrome, represent an evolutionary paradox: they are harmful to the organism, yet they are linked to genes that have persisted throughout human evolution (1–3). The principal system involved in this paradox is HLA-II, which plays a central role in antigen presentation and CD4+ T-cell activation (2).
Autoreactive lymphocytes can arise stochastically through T-cell receptor (TCR) and B-cell receptor (BCR) gene rearrangements (4, 5), as well as by somatic hypermutation in B cells (6, 7). These processes occur physiologically during thymic selection and within germinal centers (7–9), and may be further amplified in ectopic lymphoid structures under conditions of chronic inflammation (10, 11). In this context, both self-antigens and neoantigens, peptides generated by somatic mutations or post-translational modifications, can be presented by HLA-II molecules, potentially triggering the activation of autoreactive clones in genetically susceptible individuals (12–14). Whether such clones remain quiescent or progress to pathogenicity depends largely on the peptide-binding repertoire of HLA class II molecules (12, 13, 15, 16).
In this context, the ancestral HLA-II haplotypes DR2-DQ6, DR4-DQ8 and DR3-DQ2 (corresponding, respectively, to HLA-DRB1*15:01-DQA1*01:02-DQB1*06:02, HLA-DRB1*04:01-DQA1*03:01-DQB1*03:02 and HLA-DRB1*03:01-DQA1*05:01-DQB1*02:01) stand out for their evolutionary capacity to confront lethal pathogens, but also for their association with autoimmune diseases (1, 2, 17–20).
This review examines how these ancestral haplotypes have contributed to human survival against infections and how their antigenic promiscuity translates into an increased risk of autoimmune disease. We also explore modern triggers, such as chronic infections, obesity, physical trauma, immunotherapies and vaccines, that exacerbate this risk in individuals carrying these haplotypes.
We propose an integrative model linking HLA class II haplotypes, antigen persistence, and the breakdown of immune tolerance (Figure 1).
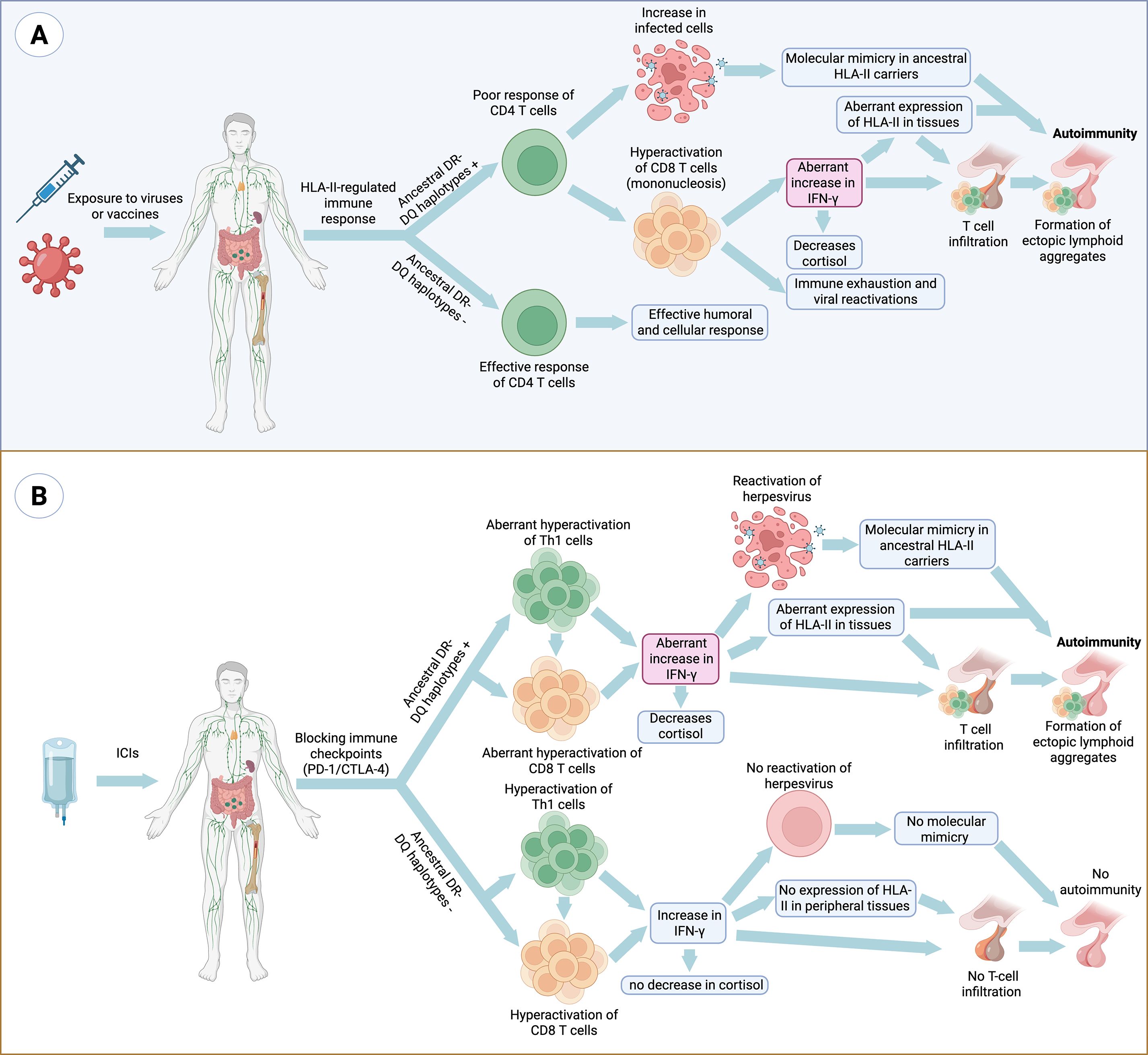
Figure 1. Mechanisms of autoimmunity mediated by ancestral HLA class II haplotypes in response to viral infections, vaccination and immune checkpoint inhibition. (A) Certain vaccine-derived antigens or pathogens with antigen persistence and immune-evasion strategies, such as Epstein–Barr virus, may display in carriers of ancestral HLA class II haplotypes (DR2–DQ6, DR3–DQ2 and DR4–DQ8) a limited CD4⁺ T-cell recognition of specific epitopes. This limitation favors antigen persistence and the compensatory expansion of CD8⁺ T cells. Chronic stimulation and sustained interferon-γ production by CD8⁺ T cells and NK cells promote ectopic HLA class II expression, molecular mimicry, the formation of lymphoid aggregates and an increase in antigen presentation to CD4⁺ T cells. Moreover, ancestral HLA class II haplotypes exhibit greater peptide-binding promiscuity, including binding of self-peptides through molecular mimicry, and generate stronger Th1 responses (IFN-γ) compared with other haplotypes, which is associated with reduced cortisol levels and the establishment of a sustained pro-inflammatory microenvironment. The combination of chronic immune hyperreactivity and persistent antigenic stimulation progressively leads to immune exhaustion and loss of tolerance, thereby promoting autoimmunity. (B) A parallel mechanism occurs following immune checkpoint blockade (ICIs, anti-PD-1/anti-CTLA-4), where T-cell disinhibition exacerbates hyperactivation in carriers of ancestral HLA haplotypes. This favors viral reactivation, molecular mimicry and aberrant HLA-II expression in peripheral tissues, thereby amplifying T-cell infiltration, ectopic lymphoid aggregate formation and the risk of autoimmunity.
2 Search strategy and selection criteria
This manuscript is a narrative review based on structured literature searches performed principally in PubMed, supplemented by searches in Scopus, Google Scholar, and preprint servers (medRxiv/bioRxiv) up to 30 September 2025. Search strategies combined controlled vocabulary (MeSH) and free-text terms using Boolean operators. Key terms and their combinations included concepts covering HLA class II haplotypes (e.g., “HLA class II”, “HLA-DRB1”, “DQB1”, “DR2-DQ6”, “DR3-DQ2”, “DR4-DQ8”), autoimmunity (e.g., “autoimmune disease”, “multiple sclerosis”, “type 1 diabetes”, “rheumatoid arthritis”, “systemic lupus erythematosus”, “celiac disease”), infectious triggers and chronic infections (e.g., “EBV”, “CMV”, “Mycobacterium tuberculosis”, “chronic infection”), immunotherapies and vaccines (e.g., “checkpoint inhibitor”, “immune-related adverse events”, “vaccine”, “adjuvant”), and related mechanistic topics (e.g., “immune exhaustion”, “IFN-gamma”, “HLA phylogeny”, “antigenic promiscuity”, “molecular mimicry”), among others.
Inclusion criteria comprised original research articles, mechanistic animal studies (including humanized HLA transgenic models), systematic reviews and meta-analyses, and relevant clinical reports that addressed the immunogenetic or evolutionary role of HLA-II haplotypes in infection or autoimmunity. A single preprint from medRxiv (21) met the inclusion criteria and is identified as a preprint in the reference list. Exclusion criteria included articles not available in English and publications without primary immunogenetic or mechanistic relevance to the topics addressed.
Titles and abstracts were screened by the authors, and full texts of potentially relevant articles were reviewed. Additional references were identified by manual reference-list screening of key publications. The aim was not to conduct a systematic review or meta-analysis but to produce an integrative, up-to-date narrative synthesis of available evidence linking ancestral HLA-II haplotypes to immune hyperreactivity and autoimmunity.
3 Evolutionary context of HLA haplotypes
Human evolution has been shaped by multiple environmental, pathogenic and social factors that favored the selection of genetic variants that maximize survival against diverse threats, particularly infectious diseases (1, 20, 22). Among these variants, HLA haplotypes occupy a central position because they are involved in antigen presentation to T cells and thereby mediate the immune response to pathogens (1, 20). The haplotypes DR2-DQ6, DR4-DQ8 and DR3-DQ2 are among the most prevalent in Caucasian human populations (19, 23–25) and have a complex evolutionary history, linked both to resistance to lethal infections in the past and to susceptibility to autoimmune diseases in the present (1, 20, 22). This relationship poses an evolutionary paradox: haplotypes that in a primitive environment conferred a survival advantage against acute extracellular infections may, in the modern context of increased life expectancy, persistent latent infections, widespread vaccination, immunotherapies, and proinflammatory conditions such as obesity, instead predispose to harmful autoimmune responses.
These three ancestral haplotypes account for approximately 90% of autoimmune diseases (20), including type 1 diabetes, as recently confirmed by a multi-ancestry genome-wide association study (GWAS) (26). Given that thousands of HLA-II haplotypes exist in the human population, it is intriguing to understand why these three haplotypes are particularly associated with autoimmunity (1, 20). If these genes were broadly deleterious, natural selection would likely have eliminated them over time (20). Instead, their high allele frequencies in autoimmunity suggest that they were advantageous in historical contexts, being essential for species survival under those conditions (20). It is hypothesized that, over millennia, these genes accumulated variants enabling them to present a wider diversity of pathogen-derived peptides, thereby activating larger numbers of CD4+ T cells to defend the host against infection (20). However, we propose that the alternative hypothesis advanced by Mangalam et al. is more compelling: because these haplotypes are ancestral, they may have been inherently more promiscuous in antigen recognition from their origins, which allowed them to persist through evolutionary bottlenecks (1). This promiscuous antigen recognition is illustrated in Mangalam et al., which show that epitopes presented by certain HLA-DR alleles (for example, HLA-DRB1*15:01) have a broader binding capacity not only to autoantigenic peptides but also to infectious agents, such as hepatitis viruses (1, 20). Similar findings have been reported for the alleles HLA-DRB1*03:01 and HLA-DRB1*04:01 (1, 20). Such promiscuity in the recognition of pathogen-derived peptides would have conferred a crucial adaptive advantage for species protection throughout history.
4 HLA-DRB1 lineages and their phylogenetic history
In humans there are 13 allelic lineages of HLA-DRB1 (22). According to the phylogenetic relationships among DRB genes in primates (hominoids, New World monkeys and Old-World monkeys) described by Bontrop et al., the HLA-DRB1*04, *03 and *02 lineages are the oldest, with HLA-DRB1*04 being the most ancestral lineage (27). Yasukochi and Satta identified two main groups of HLA-DRB1 alleles, designated Group A and Group B, based on a phylogenetic analysis (22). The alleles HLA-DRB1*04 and HLA-DRB1*15 belong to Group B, which is characterized by trans-species polymorphism (22). These lineages, shared with chimpanzees and other primates (for example Patr-DRB1*02 and Patr-DRB1*07), have a very ancient origin, predating the human–chimpanzee divergence approximately 6–7 million years ago (22). By contrast, HLA-DRB1*03 is associated with Group A, which has a more recent origin (22).
Phylogenetic data indicate that Group B is older, with an estimated time to the most recent common ancestor (TMRCA) of about 41 million years, whereas Group A has a TMRCA of approximately 29 million years (22). This pattern suggests that alleles such as HLA-DRB1*04 and HLA-DRB1*15 have been maintained by balancing selection because of their functional importance in immune responses (22). Although HLA-DRB1*03 emerged more recently, it has also been conserved due to its functional relevance (22). Moreover, the HLA-DRB1*03 lineage has been identified in humans, chimpanzees, bonobos, gorillas and orangutans, reinforcing its antiquity (28). A putative “proto-HLA-DRB1*03” ancestral lineage appears to have diverged in the last ~5 million years, giving rise to human-specific lineages such as HLA-DRB1*08, *11, *13 and *14 (28).
Taken together, these phylogenetic trees are consistent with the antiquity of these alleles and with the possibility that their long-term conservation reflects functional importance for immune responses. The HLA-DRB1*04 (DR4) and HLA-DRB1*15 (DR2) lineages are among the most ancient, followed by HLA-DRB1*03 (DR3), which is somewhat more recent. The persistence of these lineages over millions of years underscores their effectiveness against acute infections and their capacity to recognize a broader repertoire of pathogen antigens.
4.1 Consolidation of ancestral HLA haplotypes: genetic bottlenecks and positive selection
The study of the evolution of HLA haplotypes in Homo sapiens and their relationship with adaptation to pathogens over time is essential to understand how natural selection has shaped human genetic diversity (1, 20, 22). Diversity among HLA molecules has been maintained through long-term host–pathogen coevolution, with HLA loci being the most polymorphic in the human genome (1, 20, 22). This diversity is reflected in the persistence of allelic lineages and the sharing of trans-species polymorphisms between closely related species, such as chimpanzees and humans (22). Modern humans, after their exodus from Africa, encountered novel pathogen repertoires outside their continent of origin, which may have favored local positive selection of certain HLA alleles, as observed in patterns of ancestral haplotype frequencies across different populations (1, 20, 22).
During the evolution of Homo sapiens, ancestral HLA haplotypes became established primarily due to their ability to present key pathogen-derived antigens that affected early human populations (1, 20, 22). This consolidation, influenced by genetic bottlenecks such as migrations and glaciations, resulted in the selection of genetic variants that improved survival against lethal infections (1, 20). However, it also contributed to a reduction in genetic diversity that has persisted to the present.
Migration out of Africa (~50,000–55,000 years ago):
As humans migrated into Asia, Europe, and other continents, they encountered new pathogens and environmental conditions (1, 29). In this context, positive selection favored HLA alleles that maximized immune responses against these local pathogens (1).
Historical pandemics:
At various times in human history, pandemics have exerted profound impacts on the selection of genetic variants (30). The Black Death (1346–1353) imposed strong selective pressure on the human immune system, altering the frequency of several class II HLA alleles, some of which increased in frequency such as HLA-DRB1*13, while others decreased following the pandemic (31). However, the HLA-DRB1*15:01 and HLA-DRB1*03:01 haplotypes not only remained stable in frequency before and after the outbreak, but were also among the most frequent alleles in the population at both times, which could indicate that they already provided a sufficient level of protection against Yersinia pestis, thereby avoiding additional selective pressure on them (31). This stability in their prevalence over time supports the hypothesis that these ancestral haplotypes have been effective in immune responses against multiple pathogens.
Similarly, influenza virus has been a key factor in the evolution of the HLA repertoire (32). The allele HLA-DRB1*04:01 has been observed to confer greater resistance to influenza (H1N1) by eliciting robust immune responses through more effective T cell activation and increased interferon-gamma (IFN-γ) production (32). In animal model studies, individuals carrying HLA-DRB1*04:01 showed faster recovery and greater cross-protection against different influenza strains compared with the allele HLA-DRB1*04:02 (32). This immunological advantage could have favored its selection over time, despite its association with autoimmune diseases such as rheumatoid arthritis (33). Similarly, during the 1918 Spanish influenza pandemic, ancestral haplotypes were observed to influence the severity of respiratory infections (34, 35). In this context, HLA-DRB1*04:01 and HLA-DRB1*03:01, by conferring greater cross-immunity with different influenza strains, could have provided an additional immunological advantage against the Spanish influenza virus, favoring their selection in subsequent generations (35). A more recent study reinforces this hypothesis by demonstrating that immunization with conserved CD4+ T-cell epitopes between the 2009 pandemic H1N1 and prior seasonal strains reduced viral load in HLA-DR3 transgenic mouse models (35). This suggests that cell-mediated immunity driven by these alleles can confer partial protection against infection and modulate disease severity, even in the absence of cross-reactive antibodies.
However, not all historical cases show a protective effect for these alleles. In the case of medieval leprosy, an ancient DNA study of skeletal remains from infected individuals at the St. Jørgen leprosarium (Odense, Denmark) showed that the allele HLA-DRB1*15:01, rather than conferring protection, was significantly associated with increased susceptibility to lepromatous leprosy, similar to observations in modern populations in India, China, and Brazil (36). Nonetheless, this allele persisted in the population, suggesting that it may have conferred immunological advantages in other infectious contexts.
4.2 Genetic variability and adaptation of the HLA class II system
The HLA class II system, located on chromosome 6, is notable for its high genetic variability, which reflects positive selection exerted by pathogen exposure throughout human evolution (1, 20, 22). In African populations, where Homo sapiens originated, there is greater diversity of HLA class II alleles (1, 22). As humans migrated out of Africa, they faced novel pathogens and population bottlenecks that favored the selection of specific haplotypes such as DR2-DQ6, DR4-DQ8, and DR3-DQ2, which were particularly effective at presenting pathogen antigens and eliciting robust immune responses (1, 2, 17–20, 22). These haplotypes facilitated efficient activation of CD4+ T lymphocytes and the secretion of proinflammatory cytokines such as IFN-γ and IL-17, which are essential for survival during viral and bacterial epidemics (1, 20, 32, 37, 38). Moreover, coexpression of HLA class II molecules allows individuals who are homozygous or heterozygous for ancestral haplotypes to experience a synergistic effect, increasing T cell activation and the secretion of inflammatory cytokines (1).
However, this capacity to recognize a broad diversity of antigens, although advantageous from an evolutionary standpoint, carries a dual impact. In the modern environment, characterized by reduced mortality from acute infections due to medical advances, but increased exposure to chronic immunological stimuli (persistent infections, vaccinations, immunotherapies), this capacity may predispose carriers of these haplotypes to an exaggerated inflammatory response and the development of autoimmune diseases (20). Indeed, these HLA class II variants could contribute to dysregulated immune activation in the presence of a chronic triggering stimulus (e.g., persistent infections), creating an immunological imbalance that favors autoimmunity in genetically susceptible individuals. This persistence is consistent with host–pathogen co-evolution: many viruses have selected immune-evasion and persistence strategies (latency or long-term infections) that favor coexistence with the host rather than lethality, producing continuous antigenic exposure that can be deleterious when it coincides with promiscuous HLA-class-II haplotypes (12, 39–41) rather than lethality, producing continuous antigenic exposure that can be deleterious when it coincides with promiscuous HLA-class-II haplotypes (12, 39–41).
4.3 The survival paradox: from protection to autoimmunity
The survival paradox lies in the fact that the same haplotypes that conferred an evolutionary advantage against lethal infections in the past can now generate harmful autoimmune responses (1, 19, 20, 22). Haplotypes DR2-DQ6, DR4-DQ8 and DR3-DQ2 were favored for their ability to elicit effective immune responses against deadly infections, but in a modern environment, where lethal infections are less common and chronic stimuli such as latent infections or systemic inflammation predominate, these haplotypes may contribute to increased risk of autoimmunity (1, 19, 20, 22). Crucially, the emergence and persistence of pathogenic autoreactive clones depends not only on HLA-II–restricted peptide presentation but also on the host T- and B-cell receptor repertoire: although TCR and BCR sequences are generated stochastically by V(D)J recombination (and by somatic hypermutation in B cells), HLA-dependent selection processes in the thymus and in germinal centers strongly shape which clones are deleted, tolerated or expanded, such that HLA alleles indirectly influence which autoreactive clones survive and potentially become pathogenic (42–46). In other words, the persistence of these haplotypes suggests an adaptive balance in which the immune response that provides protection against acute infections can become hyperreactive and increase autoimmune risk in the presence of chronic infections (Figure 2).
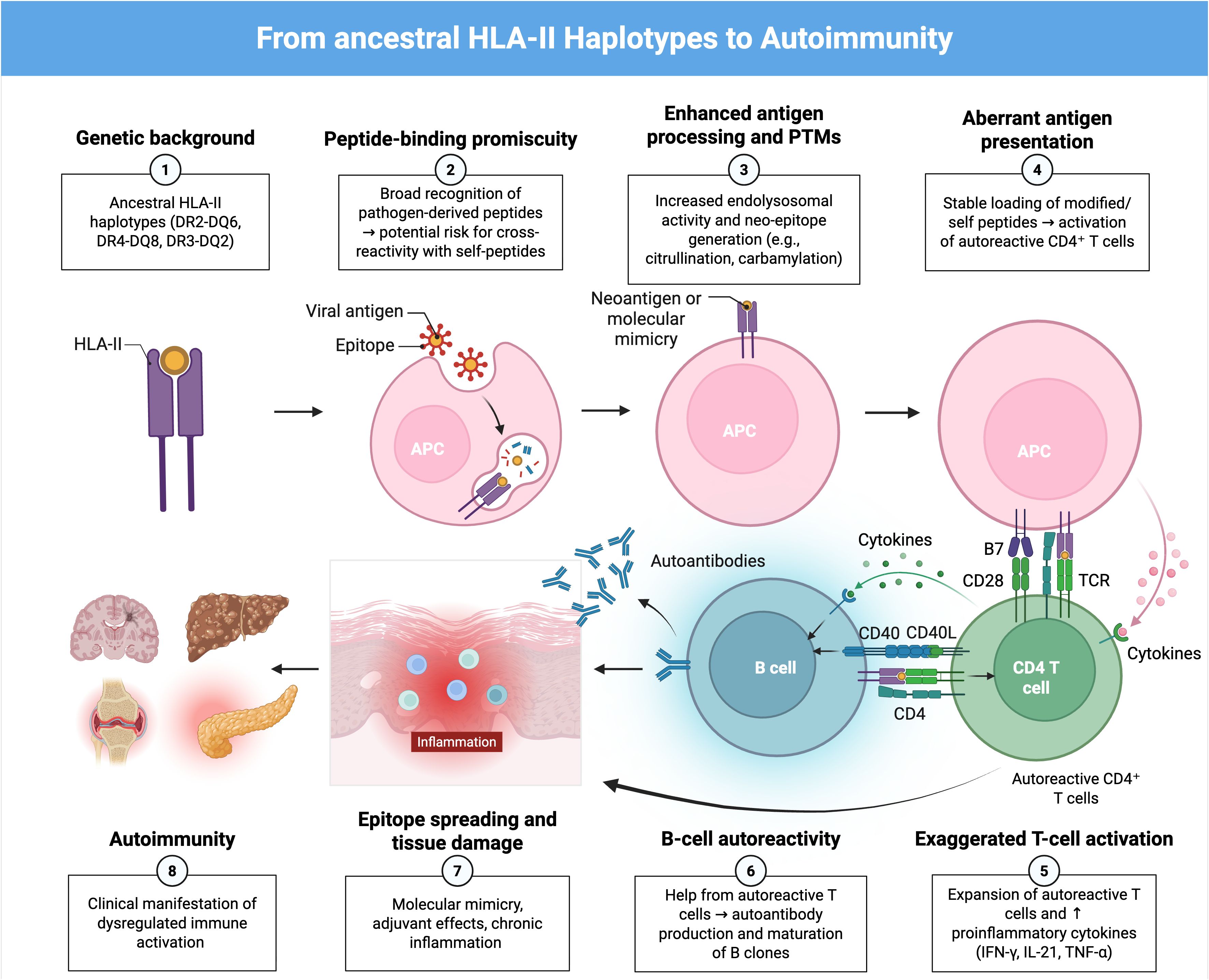
Figure 2. Proposed pathway of autoimmunity driven by ancestral HLA-II haplotypes.Ancestral HLA-II haplotypes (such as DR2-DQ6, DR4-DQ8, and DR3-DQ2) exhibit high peptide-binding promiscuity, facilitating the presentation of a wide variety of pathogen-derived peptides. Inflammation induced by these infections, particularly through IFN-γ, together with the generation of neoantigens via post-translational modifications, stimulates MHC-II expression even in non-professional antigen-presenting cells and increases the presentation of self-peptides, which can lead to the activation of autoreactive CD4- T cells via TCR-MHC-II recognition and B7-CD28 co-stimulation. Activated T cells proliferate and secrete proinflammatory cytokines (IFN-γ, IL-21, TNF-α), amplifying inflammation and promoting B-cell autoreactivity. These B cells present the same antigen on MHC-II and receive help from T cells through CD40-CD40L interactions, driving their differentiation into memory and plasma cells that produce autoantibodies. The combination of epitope spreading, molecular mimicry, and adjuvant-induced activation leads to chronic inflammation, tissue damage, and the clinical manifestation of autoimmune diseases.
Molecular-level mechanisms can reconcile how an evolutionarily advantageous HLA-II repertoire becomes pathogenic in modern contexts (13). Allele-specific peptide-binding preferences (determined by the composition of pockets within the HLA-DR/DQ binding groove) result in selective presentation of particular self-peptides or post-translationally modified peptides (e.g. citrullinated or deamidated peptides) that otherwise escape thymic deletion (47). For example, HLA-DQ2/DQ8 preferentially present deamidated gliadin peptides in coeliac disease (48), HLA-DRB1 shared-epitope alleles bind citrullinated vimentin/filaggrin peptides in rheumatoid arthritis (49), and HLA-DQB1*03:02 (DQ8) presents insulin B-chain peptides implicated in type 1 diabetes (50). Concurrently, inflammatory cytokines such as IFN-γ activate the JAK–STAT1–CIITA pathway, driving ectopic upregulation of HLA-II on non-professional antigen presenting cells and facilitating local presentation of these auto-epitopes to autoreactive CD4+ T cells (51). Together, allele-dependent peptide selection and inflammation-driven HLA-II expression provide a molecular route from pathogen-focused immunity to loss of self-tolerance (13).
5 Ancestral HLA class II haplotypes and resistance to acute infections
Alleles HLA-DRB1*15, HLA-DRB1*04 and HLA-DRB1*03 have been extensively studied for their capacity to induce intense immune responses (19, 20, 52, 53). These alleles have shown efficacy in clearing rapidly replicating pathogens due to their ability to induce a strong T helper 1 (Th1)-type response mediated by IFN-γ and TNF-α (19, 20, 52).
In dengue virus infection, the ancestral alleles HLA-DRB1*04:01 and HLA-DRB1*15:01 have shown greater resistance to dengue virus infection, recognizing a larger number of epitopes and generating a more efficient IFN-γ response compared with other alleles and closely related allelic variants, such as *04:03 and *15:02 (38). Similarly, infants who inherit HLA-DRB1*15:01 have been observed to have a higher probability of reverting to HIV (human immunodeficiency virus)–seronegative status despite being born with maternal anti-HIV antibodies (54). This phenomenon could be explained by a more effective immune response provided by this allele, possibly via increased IFN-γ production, which would contribute to improved immune control during early exposure. Likewise, in visceral leishmaniasis, the presence of HLA-DRB1*15:01 favors higher IFN-γ/IL-10 ratios, promoting a robust protective Th1 response (37, 55).
Furthermore, the DR2-DQ6 haplotype produces high levels of IFN-γ and is associated with an efficient response against intracellular pathogens such as Mycobacterium tuberculosis, a feature particularly critical in regions where tuberculosis was endemic (1). A meta-analysis of HLA class II variability and hepatitis B infection indicated that the HLA-DR4 allele is significantly associated with the ability to clear hepatitis B virus (HBV) (56). Similarly, alleles HLA-DRB1*04:01 and HLA-DRB1*15:01 have been linked to greater efficacy in clearing hepatitis C virus (57).
The study by Rodríguez et al. (2008) demonstrated that transgenic mice expressing HLA-DR2 and HLA-DR3 controlled viral encephalomyelitis induced by Theiler’s murine encephalomyelitis virus (TMEV) through increased production of IFN-γ and IL-2 by CD4+ T cells, allowing effective viral clearance without the need for antigen presentation by HLA class I (52). This mechanism is consistent with a scenario in which these haplotypes could have been favored by selection for rapid responses to lethal infections, potentially providing a survival advantage under high pathogen pressure (e.g., historical epidemics and pandemics). Moreover, multiple model systems have shown that IFN-γ is essential for control of this virus in the central nervous system (CNS) (58–60), which supports the hypothesis that haplotypes associated with greater IFN-γ production during acute infection may be more effective at eliminating intracellular pathogens.
These examples support the hypothesis that ancestral HLA class II haplotypes may have contributed to resistance against highly lethal infections, potentially favoring survival in populations exposed to such pathogens (61). However, this same intense activation becomes detrimental in the context of chronic intracellular infections, where pathogens (e.g., herpesviruses such as EBV or CMV, and certain intracellular bacteria) have co-evolved with the host to develop sophisticated immune-evasion strategies, including inhibition of antigen presentation, establishment of latency or persistent reservoirs, and induction of IL-10-dominated immunosuppressive milieus (62–64). Such evasion prevents complete clearance and leads to sustained antigenic stimulation and chronic inflammation (65), contributing to the development of autoimmune diseases such as multiple sclerosis (66). This phenomenon explains why ancestral haplotypes were positively selected in epidemic and pandemic contexts, yet today their hyperreactivity may predispose to autoimmune disorders and poorer regulation of chronic infections.
6 Ancestral HLA class II haplotypes and immune hyperreactivity in chronic infections
Ancestral HLA class II haplotypes, such as DR2-DQ6, DR3-DQ2 and DR4-DQ8, have evolved under infectious pressure, which has favored a hyperactive immune response (1, 19, 20, 22). Although this reactivity provides an advantage in detecting intracellular pathogens, its persistence does not always guarantee elimination of the microorganism and, in many cases, leads to chronic inflammation, tissue damage and an increased risk of autoimmune disease (20). The inability of the immune system to eradicate certain pathogens is due to microbial evasion strategies that neutralize or modulate the immune response, allowing their survival within the host despite constant T cell activation (19, 20, 53).
The relationship between HLA haplotypes and susceptibility or resistance to chronic infections, such as hepatitis B and tuberculosis, illustrates this paradox. The DR2-DQ6 haplotype has been observed to exert a protective effect against initial acute hepatitis B infection (67–71). In addition, HLA-DRB1*15:01 is associated with an enhanced humoral response to the hepatitis B surface antigen (HBsAg), promoting more efficient anti-HBsAg antibody production via activation of CD4+ T cells (72). However, this same haplotype has also been associated with an increased risk of chronic infection (73–75). This duality suggests that initial resistance to infection may be linked to an exaggerated immune response that, over time, leads to immune exhaustion and progression to chronic infection.
The hypothesis explaining this paradox is based on the cytokine hyperresponse induced by these ancestral haplotypes during the acute phase of infection. The strong initial immune response facilitates partial viral clearance but fails to eradicate the virus completely because of viral evasion mechanisms. This same hyperimmunity can be exploited by pathogens that have developed evasion strategies, resulting in chronic inflammation and tissue damage. HBV employs strategies such as inhibition of antigen presentation and induction of T cell anergy, which allow it to persist despite a robust initial response (76, 77). This leads to functional exhaustion of T cells and chronic inflammation that favor progression to chronic hepatitis (76).
Similarly, in tuberculosis, certain HLA haplotypes have been linked to protection against initial infection but also to an increased risk of progressive latent tuberculosis (1, 78, 79), certain HLA haplotypes have been linked to protection against initial infection but also to an increased risk of progressive latent tuberculosis (1, 78, 79). This reinforces the idea that the same immunological advantage that protects against initial infection can, under conditions of pathogen persistence due to evasion mechanisms, become a disadvantage by promoting a chronic inflammatory environment that favors the microorganism’s persistence in the host.
These findings underscore the complexity of the interaction between host genetics and the evolution of infectious diseases. The persistence of these haplotypes in the population could be explained by their advantage in protecting against lethal acute infections, despite the cost of increased predisposition to chronicity. This initial evolutionary advantage would have increased the fitness of carrier individuals, allowing them to reach reproductive age and transmit these alleles to their offspring. The fact that these haplotypes have been maintained in the human gene pool suggests that the benefit of effective protection against acute infections outweighed the cost of greater susceptibility to chronic infection and long-term immune exhaustion.
M. tuberculosis evades the immune response by blocking phagosome–lysosome fusion within macrophages, thereby permitting intracellular survival (80). Furthermore, within the granuloma it induces production of IL-10 and TGF-β, creating a localized immunosuppressive environment that reduces macrophage and T cell activation and hinders pathogen clearance (80). In this context, the DR2-DQ6 haplotype has been associated with greater susceptibility to pulmonary tuberculosis and a less effective response for controlling the infection (78, 79). The mutation from DQ6.1 to DQ6.2 of the DR2-DQ6 haplotype in European populations increased the production of IFN-γ and IL-17, improving the response to intracellular pathogens (1). However, it also intensified inflammation, which increased susceptibility to autoimmune diseases such as multiple sclerosis (1).
Both M. tuberculosis and Epstein-Barr virus (EBV) have been implicated in the induction of autoimmune diseases in individuals carrying the HLA-DRB1*15:01-DQB1*06:02 haplotype (81–86). In the case of EBV, hyperreactivity of HLA class II haplotypes such as HLA-DRB1*15 (12, 13, 15, 16).
In this context, the ancestral HLA-II haplotypes DR2-DQ6,:01-DQB1*06:02 induces intense activation of CD4+ and CD8+ T cells (53, 84). However, EBV evades the immune system by downregulating antigen presentation in infected B cells and promoting an IL-10–mediated immunosuppressive milieu (87). Moreover, this haplotype contributes to poor recognition of EBV-infected cells, further hindering viral clearance (Figure 1) (53). As a result, the virus persists in the organism, provoking continuous immune hyperactivation with chronic inflammation that can contribute to the development of autoimmune diseases such as multiple sclerosis (20, 53, 84). In addition, despite CD8+ T cell expansion, their antiviral function is deficient due to inadequate help from HLA- DRB1*15:01-restricted CD4+ T cells, which prevents effective elimination of the virus (53, 84). It should be noted that susceptibility to multiple sclerosis is also observed in other ancestral haplotypes such as DR4-DQ8 and DR3-DQ2 (17).
Similarly, M. tuberculosis, by inducing a persistent inflammatory response with elevated IFN-γ and IL-17 production in individuals with the DR2-DQ6 haplotype, can break immune tolerance and contribute to a chronic proinflammatory state that facilitates CNS autoimmunity via molecular mimicry (80, 85, 86, 88). This phenomenon is demonstrated in the experimental autoimmune encephalomyelitis (EAE) model, where M. tuberculosis antigens in complete Freund’s adjuvant are used to induce autoimmunity against myelin in susceptible animals, suggesting that prolonged exposure to this pathogen could be an additional environmental factor in the pathogenesis of multiple sclerosis in genetically susceptible individuals (81, 85, 89, 90). This process is related to the cross-restriction by HLA class II and cross-reactivity of CD4+ T cells to hyperstimulatory antigens in infections such as herpes simplex virus type 2, influenza A virus and M. tuberculosis, which could favor dysregulated immune responses and contribute to the development of autoimmunity (85, 86, 91).
Cytomegalovirus (CMV) is another pathogen that exploits immune hyperreactivity without being eradicated (92–94). HLA-DRB1*15:01 has been associated with a reduced CD4+ T cell response to the CMV pp65 antigen and with increased susceptibility to CMV infections following hematopoietic stem cell transplantation (95). This suggests that individuals carrying HLA-DRB1*15:01 may have impaired control of CMV, leading to chronic inflammation, accelerated immunosenescence, immune exhaustion and long-term impairment of immune responses (95, 96). The virus elicits a strong immune response, but this hyperreactivity paradoxically leads to immune exhaustion and dysfunction over time (92–94, 97). CMV employs several evasion mechanisms, such as interfering with T cell activation and altering dendritic cell function, which hinder presentation of viral antigens (96, 98, 99). In addition, CMV induces production of IL-10 and TGF-β, creating an immunosuppressive milieu that limits effective T cell activity at sites of infection (96, 99). This chronic activation of the immune system contributes to CD8+ T cell exhaustion, compromises immune surveillance, favors periodic viral reactivation and may promote the development of autoimmunity (92, 96, 97, 100).
In the case of Mycobacterium leprae, the pathogen responsible for lepromatous leprosy, its association with HLA-DRB1*15:01 promotes a Th2-type immune response characterized by excessive IL-10 and TGF-β production at foci of infection (36, 101). This blocks macrophage activation and intracellular killing of the pathogen, permitting its proliferation within Schwann cells and establishing a localized state of immunosuppression (101, 102). Despite strong systemic immune hyperreactivity, the infection is perpetuated and results in CD8+ T cell exhaustion and chronic tissue damage, while sustained inflammation favors autoimmunity in susceptible individuals (101).
The alleles HLA-DRB1*15, HLA-DRB1*04 and HLA-DRB1*03 have been associated with increased baseline immune hyperactivity, resulting in constant activation of CD4+ and CD8+ T cells (19, 20, 53). However, pathogen evasion mechanisms, often evolved specifically to blunt host immunity, can impair effective responses regardless of the individual’s HLA-II haplotype, meaning that even hyperreactive haplotypes may fail to clear certain intracellular infections (53, 103). Thus, the common mechanism among these pathogens is the interaction between high HLA-II reactivity and microbial evasion strategies. The capacity of these haplotypes to present a larger number of epitopes favors expansion of CD4- T cells with both increased clonal breadth and heightened effector activity (e.g. IFN-γ); however, this also includes an increased pool of autoreactive T cells that escape thymic deletion (20). This may create a vicious cycle in individuals with hyperreactive HLA-II haplotypes: the immune system detects the pathogen, but, unable to eliminate it completely, immune activation persists. Consequently, chronic activation of CD8+ T cells occurs which, under persistent stimulation, become exhausted and lose functionality (76, 92, 96, 97, 101, 104). In an attempt to compensate for this exhaustion, a shift toward a Th2 response is observed, with increased production of IL-4 and IL-10 and greater B cell activation, which amplifies the humoral response (101, 105). While humoral responses can contribute to pathogen control, effective clearance of many intracellular microbes typically requires Th1-mediated cellular immunity; therefore, a shift toward Th2 may reduce tissue-damaging Th1 activity but can also impair pathogen eradication, favoring persistence (106, 107). This shift not only perpetuates the infection by weakening cellular immunity, but may also promote autoimmunity, since autoantibody production and chronic inflammation maintain dysregulated immune activation. Autoimmunity can arise via failures at multiple checkpoints: defective central tolerance in the thymus, dysregulated germinal-center reactions driving autoreactive B-cell expansion and somatic hypermutation, or ectopic/tertiary lymphoid structures in inflamed tissues that sustain local autoreactive responses (10, 44, 108, 109). Moreover, chronic inflammation associated with T cell exhaustion is also linked to a Th2 cytokine profile within the tumor microenvironment, which could influence cancer progression by promoting an immunosuppressive milieu (101).
Thus, the evolutionary pressure that selected highly reactive and promiscuous haplotypes for defense against acute infections now acts as a predisposing factor for chronic inflammatory and autoimmune diseases in contemporary populations.
In contrast, HLA-II haplotypes with lower functional reactivity may have been favored in certain populations as a strategy of controlled symbiosis, permitting coexistence with chronic pathogens without triggering uncontrolled inflammation. This balance between protection and harm exemplifies how immune system evolution has shaped susceptibility to infectious and autoimmune diseases today.
While evolutionary models provide a coherent framework, quantifying the net fitness trade-offs empirically remains challenging because autoimmune phenotypes typically present post-reproductively and because modern environmental changes (longer lifespan, altered pathogen exposure, demographic shifts) complicate direct measurements of historical selection.
7 Persistence of ancestral haplotypes in the present day
The prevalence of these ancestral haplotypes in modern populations reflects not only positive selection exerted by historical infectious pressure but also the persistence of conserved genetic mechanisms (1, 19, 20, 22, 36). Although in a modern context these variants may contribute to autoimmune disease risk, their retention over millennia attests to their importance in protection against lethal pathogens in past environments (1, 19, 20, 22).
For example, DR2-DQ6 has been shown to induce more robust responses to infections such as dengue virus or visceral leishmaniasis, although its prevalence has also been associated with multiple sclerosis (37, 38, 53). DR4-DQ8, while associated with greater resistance to infections such as HBV, has also been linked to autoimmune diseases such as rheumatoid arthritis and type 1 diabetes (33, 56, 110, 111). Similarly, DR3-DQ2 can optimize the immune response to viral infections but is also implicated in autoimmune disorders such as celiac disease and systemic lupus erythematosus (1, 112, 113).
Natural selection therefore favored strong immune responses to lethal infections without accounting for the long-term effects of immune hyperactivation. In other words, natural selection promoted robust immune defenses against acute infections, enabling individuals to reach reproductive age. Autoimmunity, which frequently manifests after reproductive age, likely had a limited impact on evolutionary fitness, facilitating persistence of these alleles. In the modern world, however, factors such as prolonged exposure to latent infections, increased life expectancy and an inflammatory environment exacerbated by pollution and other environmental factors alter this balance, converting ancestral advantages into susceptibilities. From an evolutionary perspective, this phenomenon can be explained as an adaptive trade-off: the immune mechanisms selected ensured immediate survival in a high-infectious-pressure environment, although they resulted in increased autoimmunity later in life, after reproduction had typically occurred.
8 Triggering factors of autoimmunity
8.1 Chronic infections
This duality raises a key question: how can haplotypes that were evolutionarily selected for their effectiveness in the immune response to acute infections simultaneously cause severe dysfunctions when confronted with pathogens that employ advanced immune-evasion mechanisms, such as latent pathogens, and thereby predispose to the development of autoimmune diseases?
8.1.1 Failure to control latent infections or pathogens with extensive immune-evasion mechanisms
Ancestral HLA haplotypes, such as DR2-DQ6, DR4-DQ8 and DR3-DQ2, represent evolutionary adaptations to acute infections, but in the context of latent infections or pathogens with advanced immune-evasion strategies, such as EBV, their immune responsiveness can be allele-specific attenuated for certain viral epitopes (1, 19, 20, 53). For example, EBV establishes latency in B lymphocytes; in this setting, HLA-DRB1*15:01 (a component of DR2-DQ6) has been associated with attenuated CD4+ T-cell–mediated control of EBV-infected B cells in humanized-mouse models, resulting in higher levels of infected cells and impaired CD4 help to CD8+ responses, which can favor persistence and episodic reactivation of the virus (53). The combination of allele-specific limitations in antiviral CD4 help and sustained antigenic stimulation provides a mechanistic route whereby viral persistence may drive chronic immune activation and increase the risk of autoreactivity.
This dysfunction of the CD4+ T cell response facilitates expansion of virus-transformed cells (latency) and perpetuates low-grade chronic inflammation (53, 84, 100). In carriers of DR2-DQ6 haplotypes, the inability to mount an effective response could create an environment in which the virus alternates more frequently between latency and lytic replication, rather than bei. Unlike other HLA haplotypes that might limit latent viral load and reduce reactivation frequency, DR2-DQ6 favors viral persistence and continuous immune stimulation (53, 114, 115). This phenomenon is not limited to EBV but exemplifies a broader pattern: in carriers of highly reactive haplotypes such as DR2-DQ6, DR4-DQ8 and DR3-DQ2, the mere presence of a latent pathogen may be sufficient to sustain persistent immune hyperactivation due to ongoing antigenic persistence (1, 20, 100, 116, 117).
8.1.2 Compensatory hyperactivation of CD8+ T cells and natural killer cells
In response to deficient CD4+ T cell functionality, the immune system engages alternative pathways to control latent or chronic infection (100, 114, 118, 119). Hyperactivation of CD8+ T cells becomes a critical compensatory response attempting to control the infection (114, 118, 119). CD8+ T cells are responsible for immune surveillance of infected or transformed cells (114). In individuals with HLA haplotypes such as DR2-DQ6, DR4-DQ8, or DR3-DQ2, who have a genetic predisposition to suboptimal CD4+ responses against certain latent pathogens, this dysfunction may force CD8+ T cells to assume a more active role to compensate for the lack of CD4+ control (53, 114). This hypothesis is supported by converging data indicating reduced CD4+ helper capacity in carriers of these haplotypes, higher latent viral loads (as seen in EBV infections), and sustained hyperactivity of the CD8+ compartment under chronic antigenic stimulation (20, 53, 84, 101, 114, 119). This process results in increased production of IFN-γ, a key effector that, while effective against infection, also amplifies inflammation (118).
Concurrently, natural killer (NK) cells, crucial in the early response to viral infections, are activated by cellular stress signals and suboptimal viral antigen presentation (120). IFN-γ production by NK cells further amplifies the inflammatory signal, creating an environment that favors tissue dysfunction (120–122). At this stage, IFN-γ production is therefore dominated by CD8+ T cells and NK cells, while the contribution of CD4+ T cells remains limited, reflecting their secondary role in an imbalanced immune response (123).
8.1.3 Increased compensatory humoral response
As the immune system attempts to compensate for deficient functional CD4+ T cells, particularly Th1 subsets, a shift toward greater activation of CD4+ Th2 cells is observed (101). This Th2 polarization promotes B cell activation and increased antibody production, constituting a compensatory humoral response to the poor control of infection mediated by the deficient cellular response (105). However, sustained activation of the B-cell compartment can also lead to autoantibody generation, even in the absence of active infection, thereby increasing the risk of autoimmunity and tissue damage (105, 124). Studies in autoimmune diseases and chronic viral infections such as EBV and HIV have documented how loss of Th1 control can induce humoral dysregulation with pathological consequences (125, 126).
Consequently, although this humoral response seeks to assist in infection control, excessive Th2 and B cell activation is not only ineffective against intracellular pathogens but may also induce immune responses directed against self-tissues, exacerbating immune dysfunction (101).
8.1.4 Increased aberrant HLA class II expression and autoimmunity
As inflammation persists, the high levels of IFN-γ produced associated with these ancestral haplotypes and the chronic inflammatory response induce aberrant HLA class II expression on nonprofessional antigen-presenting cells in peripheral tissues (i.e., in the cells of any tissue affected by inflammation), compared with less-reactive haplotypes (127–129). This process may increase the likelihood of presentation of autoantigens and neoantigens and the subsequent activation of autoreactive CD4+ T lymphocytes, which in turn initiate immune responses against host tissues (129, 130). At this point, CD4+ T cells, previously limited in their contribution to the immune response, begin to play a central role in perpetuating the inflammatory cycle (130). Autoreactive CD4+ T cells are activated and produce additional IFN-γ, further reinforcing inflammation and establishing a vicious cycle of tissue damage (130, 131).
Moreover, autoreactive B cells gain prominence because, by presenting autoantigens to autoreactive CD4+ T cells, they promote T cell activation and expansion, leading to pathogenic T–B cooperation (132). Once activated, CD4+ T cells provide co-stimulatory signals (such as CD40L) and cytokines that drive differentiation of B cells into autoantibody-producing plasma cells, amplifying the humoral response and chronic inflammation (133). The inflammatory cycle involves not only T and B cells but is also amplified by continuous production of proinflammatory cytokines (134). This process further intensifies chronic inflammation and contributes to disease progression in autoimmunity (135). Thus, activation of autoreactive CD4+ T and B lymphocytes generates an environment conducive to the emergence of autoimmune diseases.
Ancestral HLA-II haplotypes are associated with amplified pro-inflammatory cytokine responses, particularly IFN-γ, which can sustain a chronic inflammatory milieu and increase the risk of autoimmunity (1, 19, 20, 52). An example of this phenomenon was observed in a study using transgenic murine models expressing human HLA alleles (32). Specifically, two strains expressing HLA-DRB1*04:01 and HLA-DRB1*04:02, respectively, were compared, and although both developed immunity to influenza infection, only the *0401 mice generated a more intense cross-protective immunity (32). This response was associated with more pronounced activation of CD4+ T cells, including memory autoreactive Th17 cells against self-epitopes, suggesting an increased propensity for autoreactivity under conditions of intense immune activation (32).
8.1.5 Immune exhaustion
As hyperactivity of CD8+ T cells persists, the phenomenon of immune exhaustion begins to develop (104). In this process, CD8+ T cells exposed to prolonged activation and stress undergo loss of function, limiting their ability to control infection effectively and perpetuating chronic inflammation (100, 101, 104). This exhaustion is characterized by expression of inhibitory markers such as programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte–associated protein 4 (CTLA-4), lymphocyte activation gene-3 (LAG-3) and T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), indicators of a functionally compromised state of CD8+ T cells (76, 104). In addition, NK cells responding to chronic inflammation may indirectly promote CD8- T-cell exhaustion, thereby fostering viral persistence and immunological dysfunction (122).
The PD-1 receptor, in particular, is associated with inhibition of T cell activation, reducing their capacity to produce IFN-γ and other inflammatory cytokines (97, 104). This self-regulatory mechanism, although essential to prevent damage from excessive inflammation, also contributes to failure to eliminate pathogens that are latent or possess advanced immune-evasion mechanisms (97, 104, 123). With immune exhaustion, CD8- T cells are unable to sustain effective responses, favoring infection persistence; the resulting prolonged antigen exposure may, in turn, increase susceptibility to autoimmunity (65, 76, 104, 136, 137).
8.2 Immunotherapies
Immune checkpoint inhibitors (ICIs), such as monoclonal antibodies directed against PD-1, programmed death-ligand 1 (PD-L1) and CTLA-4, have revolutionized cancer treatment by enhancing T cell activity against tumor cells (138). However, in individuals with ancestral HLA haplotypes (DR2-DQ6, DR4-DQ8 and DR3-DQ2), this therapeutic strategy can exacerbate underlying inflammatory mechanisms and increase the risk of autoimmunity, an adverse effect known as ICI-mediated immune toxicity (138–142).
In carriers of HLA haplotypes such as DR2-DQ6, DR4-DQ8 and DR3-DQ2, which evolved to mount robust proinflammatory responses to acute infections (1), blockade of PD-1 and CTLA-4 interferes with physiological immune-regulatory mechanisms that limit excessive inflammation (138–143).
By releasing the immunological “brake” represented by these molecules, ICIs induce hyperactivation of CD8+ and CD4+ T lymphocytes, resulting in an exacerbated cellular response with increased production of inflammatory cytokines such as IFN-γ, similar to the hyperresponse observed in carriers of ancestral haplotypes during acute infections, in comparison with other haplotypes (1, 144–146).
This increase in IFN-γ promotes aberrant HLA class II expression on nonprofessional antigen-presenting cells in peripheral tissues, facilitating presentation of autoantigens and activation of autoreactive CD4+ T lymphocytes (128, 129, 147). This process constitutes a critical point in the development of T cell–mediated autoimmunity (129). While ICIs can indeed reactivate pre-existing autoreactive lymphocytes, the higher incidence and severity of ICI-related autoimmunity in carriers of certain ancestral HLA-II haplotypes suggests that HLA genotype also modulates the generation, persistence, and effector phenotype of such clones, thereby increasing the likelihood that immune disinhibition induced by ICIs will precipitate clinically overt autoimmunity (138–142, 148–150). Recent clinical and translational studies indicate that specific HLA genotypes modulate the risk and phenotype of immune-related adverse events during checkpoint inhibitor therapy, suggesting a potential role for HLA profiling in toxicity risk stratification (151). Therefore, in individuals with these ancestral haplotypes, this inflammatory milieu can amplify chronic inflammation and lead to autoimmune diseases, partially recapitulating the mechanism observed in chronic infections with latent or highly immune-evasive pathogens (129, 138–143).
8.3 Vaccines
Vaccines are fundamental tools for prevention of infectious diseases (152). However, in individuals with ancestral HLA haplotypes such as DR2-DQ6, DR4-DQ8 and DR3-DQ2 they can trigger exacerbated inflammatory responses (20, 152–156). This phenomenon would occur especially in the context of vaccines that induce high levels of proinflammatory cytokine production, such as IFN-γ, generating an immunological environment that mimics the mechanisms that predispose to autoimmunity after infections, particularly in genetically predisposed individuals (1, 20, 152, 157).
Whereas DR2-DQ6 has solid clinical evidence, particularly in the case of post-Pandemrix® narcolepsy following the H1N1 campaign, where carriers of the HLA-DQB1*06:02 allele showed a markedly increased risk associated with exacerbated IFN-γ responses and autoimmune/inflammatory syndrome induced by adjuvants (ASIA)-type phenomena, direct human vaccination studies are still lacking for DR3-DQ2 and DR4-DQ8 (152, 154, 155). Moreover, although not prophylactic vaccines against pathogens, transgenic murine models have shown that immunization with proinsulin peptides in the context of DR3-DQ2 induces autoimmune diabetes, and immunization with type II collagen in DR4-DQ8 carriers induces experimental arthritis (158, 159). These findings demonstrate how an “external” antigen can trigger autoimmunity in the presence of these haplotypes, a mechanism that could be emulated after vaccination with pathogen antigens that present similar amino-acid sequences (molecular mimicry) in susceptible individuals (152).
Carriers of ancestral HLA-II haplotypes tend to mount strong antigen-specific cellular responses, with marked IFN-γ production (1, 20). This evolutionary profile, optimized for combating acute infections, can amplify the risk of chronic inflammation and activation of autoreactive T lymphocytes in situations where a prolonged or exacerbated immune stimulus is generated, as might occur with certain vaccines (1, 20, 156). In these individuals, the interaction between genetics and vaccine design could predispose to development of T cell–mediated autoimmune diseases exacerbated by systemic inflammation (152, 156, 160).
mRNA-based and viral-vector vaccines differ significantly in design and the immunological impact they generate (161). mRNA vaccines, such as those developed for SARS-CoV-2, deliver a fragment of messenger RNA encapsulated in lipid nanoparticles that encodes a viral antigen (161). This type of vaccine has an approximate half-life of around 30 days in the organism, since the mRNA is degraded, limiting the duration of antigen expression (162). Although these vaccines elicit robust CD8+ T cell responses and substantial IFN-γ production, the transient nature of the stimulus minimizes the risk of sustained chronic inflammation (163). This is particularly relevant for individuals with ancestral HLA haplotypes, since the limited duration of the stimulus could reduce the probability of developing a persistent autoimmune response. Nevertheless, isolated cases of autoimmunity following mRNA vaccination have been reported, indicating that despite their more transient nature they are not entirely free of this risk (164, 165).
An example of this interaction was observed in individuals vaccinated against SARS-CoV-2: recent studies have shown that specific alleles such as HLA-DRB1*15:01 are associated with an increased T cell response to the spike antigen after the first and second vaccine doses, demonstrating a more intense immune response in carriers of this allele (166, 167). However, this association with enhanced immunogenicity does not establish a causal link to autoimmunity, which would require dedicated studies assessing post-vaccination autoimmune outcomes in carriers of these alleles.
On the other hand, viral-vector vaccines use attenuated or nonreplicating viruses, such as adenoviruses, to deliver genes encoding the viral antigen (168). Unlike mRNA vaccines, viral vectors can persist in the organism for a longer period, resulting in sustained immune stimulation (169, 170). This persistence may support antigen expression for months, so subsequent exposures can trigger potent T cell responses directed against antigen-expressing cells, which may translate into chronic tissue damage (171). Consequently, this prolonged stimulus could amplify IFN-γ production in individuals with ancestral HLA haplotypes, creating a chronic inflammatory environment that favors activation of autoreactive T lymphocytes and aberrant HLA-II expression in peripheral tissues (157, 167, 172–174). The latter phenomenon facilitates autoantigen presentation and perpetuates the inflammatory cycle, thereby increasing the risk of developing autoimmune diseases (157, 164, 173, 174).
It is important to clarify that not all vaccines with potent immunostimulatory capacity belong to the viral-vector category (175). A relevant example is Pandemrix®, the vaccine used during the H (176). This formulation can also generate a hyperimmune response, amplifying IFN-γ production in carriers of ancestral HLA haplotypes, which has been linked to autoimmune adverse events in genetically susceptible populations (154, 177, 178).
This concern materialized in a paradigmatic case during mass vaccination with Pandemrix® against H1N1 (177). In Finland and Sweden, a marked increase in narcolepsy cases was reported among adolescents and young adults, particularly in carriers of the HLA-DQB1*06:02 allele (154, 177). Presence of this allele was strongly associated with increased risk of narcolepsy in vaccine-exposed cohorts, even in heterozygotes, consistent with an interaction between genetic profile and vaccine-associated risk (154). Studies also indicated that anti-A/H1N1 antibody levels were significantly higher in individuals younger than 13 years, and that both patients and HLA-DQB1*06:02-positive controls showed an increased serological response (154, 177). Epidemiologic evidence for vaccine-associated autoimmunity is strongest in specific, well-characterized episodes (e.g., Pandemrix-associated narcolepsy), whereas most other vaccine–autoimmunity links remain limited to case reports or small series. Hence, generalization should be cautious: large, HLA-stratified vaccine safety cohorts are necessary to quantify absolute and relative risks across haplotypes.
In addition to intrinsic vaccine design, adjuvants included in many vaccine formulations constitute a critical factor, especially in predisposed individuals (157, 179). These compounds act via the “adjuvant effect”: coadministration of an antigen with microbial-derived components activates Toll-like receptors (TLRs) on antigen-presenting cells (APCs), which not only induces release of proinflammatory cytokines but also increases expression of antigen-presentation molecules such as HLA-II and of co-stimulatory molecules such as B7-1/2 (CD80/CD86), even on nonprofessional cells (for example, fibroblasts or endothelial cells) (157, 180). In the case of classical adjuvants such as aluminum salts, designed to produce localized inflammation at the injection site, this activation enhances IFN-γ and other proinflammatory cytokine production (181, 182). In carriers of ancestral HLA haplotypes (DR2-DQ6, DR3-DQ2, DR4-DQ8), whose baseline inflammatory response is already more intense, such increases in HLA-II and costimulatory signaling could trigger aberrant IFN-γ production and expansion of autoreactive T cells, emulating an autoimmune process similar to that induced by chronic infections (20, 65, 153, 183, 184). Likewise, more advanced adjuvants, such as oil-in-water emulsions like MF59 or TLR4 agonists, may further exacerbate this disproportionate immune activation, increasing the risk of autoimmunity in these individuals (157, 180, 185, 186).
The prolonged stimulus generated by viral-vector vaccines and certain adjuvants can reproduce features of chronic infection in terms of sustained inflammation (65, 157, 171, 187). In individuals with ancestral HLA haplotypes, this prolonged inflammation could induce aberrant HLA-II expression in nonimmune cells and activate autoreactive T cells, creating an inflammatory milieu that perpetuates tissue damage (20, 153, 157). This process contributes to the development of autoimmune conditions such as inflammatory arthritis, myocarditis, autoimmune encephalitis and autoimmune hepatitis (157, 179, 188, 189).
Although vaccines are essential for control of infectious diseases, they can trigger dysregulated immune responses in individuals with ancestral HLA haplotypes because of these individuals’ tendency to generate intense inflammation. Therefore, vaccine design, adjuvant selection and post-vaccination monitoring of genetically susceptible subjects are crucial to minimize the risk of vaccine-induced autoimmunity. While mRNA vaccines may appear safer due to their transient antigenic stimulus, clinical cases of autoimmunity following their use demonstrate that genetic predisposition is the primary factor, with antigen-exposure duration acting as a secondary modulator of an immune process preconditioned by the host genome.
8.4 Allergies
Chronic exposure to allergens can induce a persistent inflammatory milieu, especially in genetically predisposed individuals, which may favor development of autoimmune diseases (20, 190, 191). Sensitization to allergens responsible for allergic rhinitis, asthma or dermatitis involves an exaggerated immune response mediated by CD4+ T cells and B cells (192, 193). This activation generates a Th2 response characterized by production of cytokines such as IL-4, IL-5 and IL-13, promoting chronic inflammation in the airways, skin or other affected tissues (192, 194). However, in individuals with ancestral HLA-II haplotypes such as DR2-DQ6, DR4-DQ8 or DR3-DQ2, the immune response can diverge toward a more intense Th1 profile, increasing production of IFN-γ and TNF-α (20, 153, 191). Immunogenetic studies have shown that alleles such as HLA-DRB1*15, HLA-DRB1*03 and HLA-DRB1*04 are significantly associated with allergen-specific IgE responses to diverse allergens, supporting their direct involvement in allergic processes (195–197). This suggests that, in carriers of these haplotypes, chronic allergen exposure could induce more intense and sustained inflammation that may create a milieu favorable to autoimmunity (20, 191).
The study by Germundson et al. (2022) supports the influence of HLA-II alleles on allergic responses, showing differences in antibody production and inflammation according to the expressed allele (198). It was observed that both male and female HLA-DRB1*03:01 mice, as well as female HLA-DRB1*15:01 mice, exhibited significantly increased β-lactoglobulin (BLG)-specific IgE production, indicating a more intense allergic response (198). Conversely, sensitized HLA-DQ8 mice (HLA-DQA1*03:01-DQB1*03:02) of both sexes, and to a lesser extent male HLA-DRB1*15:01 mice, showed robust increases in BLG-specific IgG1, suggesting a different but equally relevant immune response in allergic sensitization (198). Additionally, sensitized HLA-DRB1*15:01 mice displayed sex-specific behavioral changes, with males showing altered mobility and anxiety-like behavior, while females exhibited impaired spatial memory. A significant shortening of villi was also detected in sensitized males carrying HLA-DRB1*03:01 or HLA-DRB1*15:01 alleles (198). Even the HLA-DRB1*15:01-DQB1*06:02 haplotype was associated with cow’s-milk allergy, particularly with higher humoral responses to BLG (199). This suggests that, in carriers of these haplotypes, chronic allergen exposure could induce more intense and sustained inflammation that may promote an environment favorable to autoimmunity (20, 191, 198).
Persistent inflammation can erode immunological tolerance, promoting the activation of autoreactive T cells and the production of autoantibodies by B cells (200, 201). Proinflammatory mediators, notably IFN-γ, can upregulate HLA-II on non-professional antigen-presenting cells, thereby facilitating local presentation of self-antigens, including neoantigens, and focal immune activation (200, 202, 203). In combination with tissue dysfunction and co-factors (e.g., chronic infections, metabolic stress, or physical trauma), this milieu favors loss of tolerance and the emergence of autoimmune pathology (201, 204–206).
8.5 Obesity
Obesity, as a state of low-grade chronic inflammation, can mimic the inflammatory responses observed in persistent infections and increase expression of HLA class II on nonprofessional antigen-presenting cells (207–211).
Hyperlipidemia, a feature of obesity, activates Toll-like receptors (TLRs), particularly TLR4, in macrophages and dendritic cells, stimulating NF-κB signaling and the production of cytokines such as TNF-α, IL-6 and IFN-γ (208). Similarly, hyperglycemia induces oxidative stress and endoplasmic reticulum stress, which activate the unfolded protein response (UPR) and perpetuate inflammatory signaling (208). The combination of these pathways amplifies systemic inflammation, which in individuals carrying ancestral HLA class II haplotypes could enhance HLA-II expression in peripheral tissues and facilitate activation of autoreactive T lymphocytes (153).
Moreover, increased intestinal permeability in obesity permits translocation of lipopolysaccharide (LPS) into the systemic circulation, triggering metabolic endotoxemia (208). LPS activate TLR4, exacerbating NF-κB–mediated inflammation (208). In carriers of ancestral haplotypes, this persistent inflammation could drive dysregulated activation of T and B cells, perpetuating an inflammatory cycle that favors development of autoimmune diseases (153).
The chronic inflammatory state derived from obesity sustains IFN-γ production (211), which in proinflammatory ancestral haplotypes prone to intense immune responses and may further amplify HLA class II expression on non-immune cells (153). This phenomenon, documented in HLA-transgenic models and genetic studies, could facilitate autoantigen presentation and promote activation of autoreactive T cells (17). This inflammatory cycle is particularly detrimental in genetically predisposed individuals, where metabolic stress and systemic inflammation potentiate progression toward autoimmune disease (17, 153).
Therefore, in the context of obesity, individuals with proinflammatory HLA class II haplotypes may experience amplification of systemic inflammation, aberrant HLA-II expression and an increased likelihood of developing autoimmune diseases (153, 207, 211). Moreover, epidemiological data show that excess adiposity can synergize with HLA risk alleles to raise autoimmune risk, most notably adolescent obesity interacting with HLA-DRB1*15 to increase multiple sclerosis susceptibility, and overweight/obesity interacting with high-risk HLA genotypes to promote latent autoimmune diabetes in adults (212, 213).
8.6 Chronic physical stress
Chronic physical stress, such as recurrent physical trauma, hypoxia or persistent injury, generates a sustained inflammatory state that activates NF-κB signaling (214, 215). This activation, mediated by proinflammatory cytokines such as TNF-α and IL-1, leads to increased production of IFN-γ, primarily mediated by T lymphocytes and natural killer (NK) cells (216, 217). IFN-γ in turn activates the JAK–STAT pathway, especially STAT1, which regulates transcription of inflammatory genes and HLA-II expression, thereby amplifying the immune response (218, 219). NF-κB and STAT1 signaling pathways thus act in concert to potentiate chronic inflammation (220, 221). In carriers of ancestral HLA class II haplotypes such as DR2-DQ6, DR4-DQ8 and DR3-DQ2, this overproduction of IFN-γ and concurrent activation of both pathways could intensify inflammatory responses due to greater genetic sensitivity to these mediators (1, 20, 52).
Although physical stress and tissue trauma have been linked to the onset or exacerbation of autoimmune diseases (e.g., psoriasis, psoriatic arthritis, rheumatoid arthritis) (205, 222), direct evidence of interaction between specific HLA-II haplotypes and physical stress is limited. Notably, a large Swedish study reported a synergistic effect between head trauma and HLA-DRB1*15:01 on multiple sclerosis risk, supporting the concept that HLA genotype can modulate the autoimmune consequences of tissue injury (223).
8.7 Transplantation
In the transplantation setting, the presence of proinflammatory HLA class II haplotypes, such as DR4-DQ8, DR2-DQ6 and DR3-DQ2, has been associated with increased production of proinflammatory cytokines after the procedure, promoting a persistent systemic inflammatory milieu (19, 224). This exacerbated immunological profile can intensify activation of T and B cells, increase the risk of acute and chronic graft rejection, and contribute to post-transplant tissue injury (224–226).
Several studies have reported that carriers of these haplotypes exhibit higher levels of mediators such as IFN-γ, TNF-α and IL-6, together with sustained activation of NF-κB and STAT1 pathways, thereby potentiating the inflammatory response (19, 20, 52, 153, 227). This hyperreactive immune state not only compromises graft function but has also been associated with higher post-transplant morbidity and mortality, underscoring the prognostic importance of HLA genetic profile in clinical outcomes (19, 224). For example, pancreas transplants in patients carrying HLA-DR3 and/or HLA-DR4 have shown higher risk of recurrence of type 1 diabetes (226).
9 Discussion
The relationship between HLA haplotypes and the immune response represents a central framework for understanding how evolution has shaped both defense against lethal infections and current susceptibility to inflammatory and autoimmune diseases (1, 20, 22). Ancestral HLA class II haplotypes (DR2-DQ6, DR4-DQ8 and DR3-DQ2) have been selected for their ability to induce rapid and intense immune responses to acute infections, mediated primarily by production of proinflammatory Th1 cytokines such as IFN-γ and TNF-α (1, 19, 20, 52). This adaptive advantage, which in historical contexts may have meant the difference between survival and death during high-mortality epidemics, can today translate into an increased propensity for exaggerated immune responses, systemic inflammation and autoimmunity (1, 2, 17–20, 22, 228, 229). The conceptual model depicted in Figure 1 integrates these evolutionary pressures with the mechanistic pathways that may link ancestral HLA-II haplotypes to immune hyperreactivity and autoimmunity.
This hyperreactivity may be related to the high promiscuity of antigen presentation by the DR2-DQ6, DR4-DQ8 and DR3-DQ2 haplotypes, which permits recognition of a broad repertoire of both pathogenic and self-peptides; this enhances their efficacy in clearing acute infections but raises the risk of autoimmunity in contexts of persistent antigenic exposure, such as chronic infections (1, 20).
In the modern environment, characterized by lower mortality from acute infections but high persistence of pathogens with sophisticated immune-evasion strategies, such as Mycobacterium tuberculosis, Epstein–Barr virus or citomegalovirus, immunological hyperreactivity can become a double-edged sword (81–86, 92, 95). The intense inflammatory response, although initially protective, can be maintained chronically without achieving pathogen eradication, generating a persistent microenvironment that promotes loss of immune tolerance and autoimmunity (1, 19, 20). Mechanisms such as molecular mimicry, bystander activation and antigenic persistence can convert an initially protective response into a pathogenic pathway that promotes autoimmunity in genetically susceptible individuals (230). Sustained T cell activation and continuous production of proinflammatory cytokines, such as IFN-γ, sustain a systemic inflammatory state that causes tissue damage (1). In this context, the immune response not only fails to eliminate the infection but may also contribute to pathogen persistence and to development of autoimmune diseases, as observed in multiple sclerosis, active tuberculosis or visceral leishmaniasis (1, 20, 37, 53, 85).
This same mechanism could extend beyond classical autoimmune diseases (such as multiple sclerosis, rheumatoid arthritis, lupus, celiac disease or type 1 diabetes) and might explain increased susceptibility to conditions characterized by systemic inflammation and immune dysregulation (Table 1), such as long COVID, myalgic encephalomyelitis/chronic fatigue syndrome and certain post-vaccination syndromes (179, 231–239). In all of these conditions, immunological profiles have been described that include sustained activation of T and B lymphocytes, increased IFN-γ, autoantibody production and ectopic, sustained HLA-II expression (which could be more pronounced compared with less-reactive haplotypes), mechanisms that are congruent with the pathophysiology observed in carriers of these haplotypes (234, 235, 238, 240–247). Several studies have even linked the development of post-vaccination syndromes and ME/CFS with alleles belonging to the DR2-DQ6, DR4-DQ8 and DR3-DQ2 haplotypes (184, 239, 248–252). In the case of long COVID, for example, although some genetic analyses suggest involvement of the HLA class II region, robust and replicated studies conclusively linking the DR2-DQ6, DR4-DQ8 or DR3-DQ2 haplotypes to long COVID risk are still lacking (21, 253). However, haplotypes such as HLA-DRB1*15:01-DQA1*01:02-DQB1*06:02 (DR2-DQ6) have been shown to bind numerous peptides from the SARS-CoV-2 spike protein with high affinity, which favors stronger immune responses (166, 254–257). This hyperreactivity, although beneficial against extracellular pathogens, has also been associated with higher autoimmune risk (153), suggesting a possible indirect role of these haplotypes in susceptibility to long COVID. Indeed, HLA-DRB1*15:01 has also been associated with increased risk of severe COVID-19 (258).
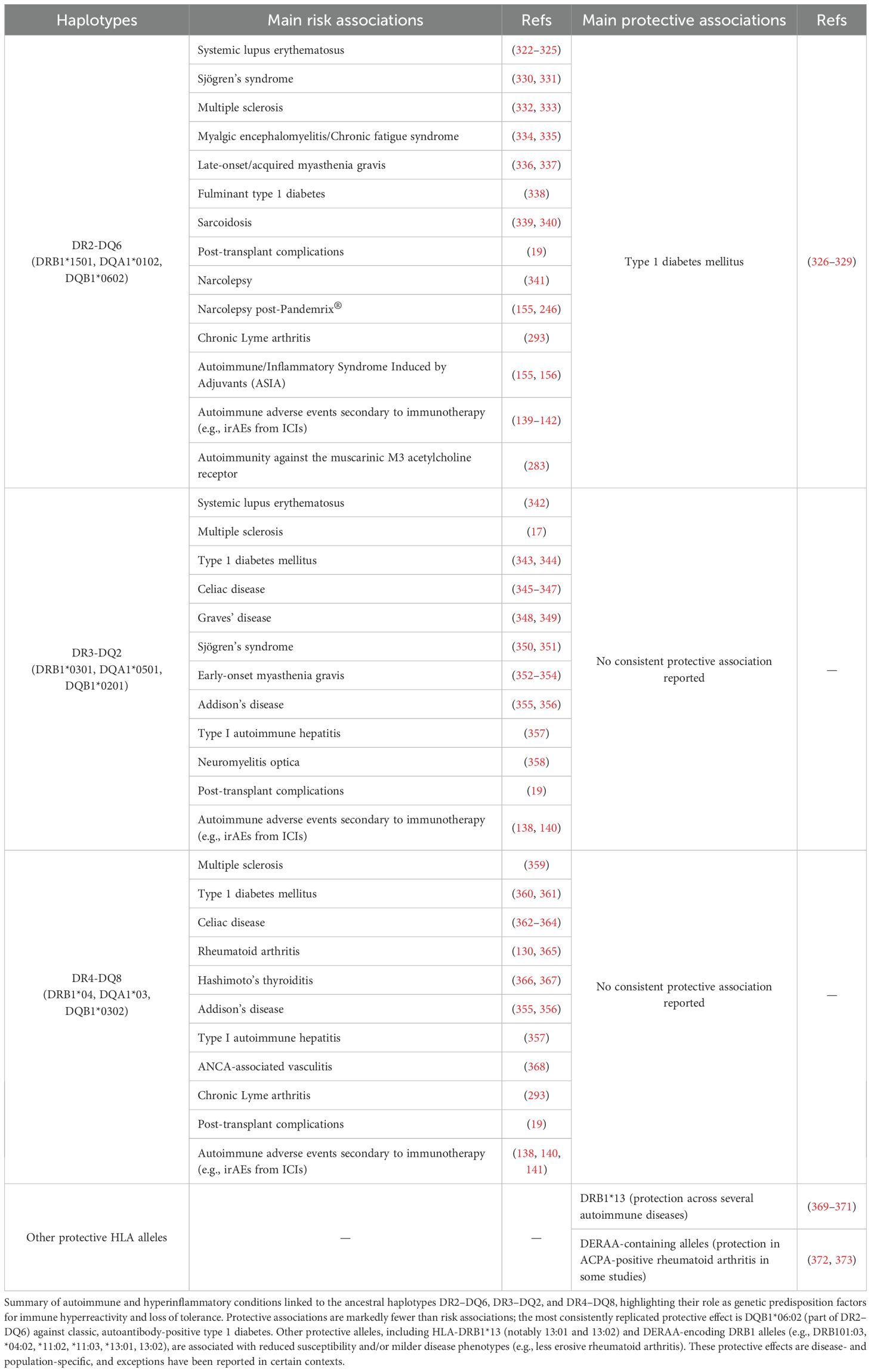
Table 1. Autoimmune and inflammatory diseases associated with ancestral HLA-II haplotypes.
Beyond these associations, the pathophysiology of post-viral syndromes such as long COVID and ME/CFS may require integration of neuroendocrine and autonomic mechanisms to explain the constellation of profound fatigue, cognitive dysfunction, exertional intolerance and dysautonomia present in many patients (259–263). In this regard, persistent antigenic stimulation in genetically susceptible hosts (HLA-II) could favor the development of autoimmunity directed against endocrine and autonomic targets (21, 230, 248, 253, 264, 265); this is especially relevant for understanding the clinical presentation of long COVID and ME/CFS (261, 266, 267). Frequently, and not mutually exclusively but rather complementarily, two pathogenic pathways coexist and converge on the same clinical phenotype:
i. Autoimmunity directed at the hypothalamic–pituitary–adrenal (HPA) axis and hypocortisolism (261). Cortisol, produced by the adrenal glands under control of the HPA axis, is the principal endogenous anti-inflammatory regulator (268). The HPA axis may be affected by direct viral injury, by neuroinflammation, or by autoimmunity against the pituitary (hypophysitis) following infection, leading to central or functional hypocortisolism (269–274). Relative and sustained hypocortisolism diminishes the capacity to suppress inflammatory responses, promotes neuroinflammation, and reduces tolerance to exertion and stress, thereby perpetuating the chronic inflammation observed in long COVID and ME/CFS (260–262).
ii. Autoimmunity against cholinergic receptors and loss of parasympathetic tone (266, 267, 275–277). Acetylcholine released by the vagus nerve constitutes a potent anti-inflammatory pathway (278, 279); the emergence of autoantibodies or antigen-specific T-cell responses directed against muscarinic (e.g., M3) or nicotinic receptors can block cholinergic signaling and reduce the “vagal brake” (280–284). This leads to dysautonomia (POTS, orthostatic intolerance), relatively increased sympathetic activity and loss of inflammatory control, conditions that facilitate chronification of the immune response (267, 277, 285). Recent data show associations between M3-reactive responses and certain HLA-DR genotypes, suggesting possible relevance of haplotypes such as HLA-DRB1*15:01–DQA1*01:02–DQB1*06:02 (DR2-DQ6) in this context (283). Moreover, the clinical benefit observed in some studies with cholinergic modulators (e.g., pyridostigmine) supports the pathophysiological plausibility of this pathway (286–289).
These mechanisms are not mutually exclusive and frequently potentiate one another (279). Loss of vagal tone favors sympathetic predominance which, in settings of sustained activation, can ultimately uncouple or attenuate the response of the HPA axis (279, 290, 291). Conversely, a sustained state of hypocortisolism reduces hormonal control of inflammation and may diminish cholinergic modulation, closing a pro-inflammatory feedback loop that facilitates chronification and immune exhaustion (261, 279, 292). Therefore, HLA-II susceptibility could facilitate viral persistence or reactivation (e.g., EBV, SARS-CoV-2, other herpesviruses), fueling continuous antigenic stimulation or neoantigen exposure that promotes generation of autoantibodies or autoreactive T cell responses directed against the pituitary, cholinergic receptors or other neuronal targets, thereby closing the pathogenic circuit. The same pattern can be observed in other clinical models: in chronic Lyme arthritis (Borrelia burgdorferi), HLA-DR4 alleles (and to a lesser extent DR2) are overrepresented in chronic, refractory forms, suggesting that these lineages favor both immune-mediated persistence of infection and development of joint autoimmunity (293, 294). Similarly, in multiple sclerosis the risk haplotype HLA-DRB1*15:01 (DR2) is associated with impaired immune control of EBV, hindering recognition and elimination of EBV-infected cells (53) and, together with EBV immune-evasion factors (vIL-10, BNLF2a) that reduce antigen presentation (62), promotes viral persistence and chronic antigenic stimulation that can trigger reactivity against myelin (295, 296). Taken together, these models illustrate how an HLA-II predisposition that facilitates microbial persistence can, through persistent antigenic or neoantigen presentation, secondarily trigger autoimmunity (arthritis in Lyme disease; demyelination in multiple sclerosis) and suggest that similar mechanisms may be operating in long COVID, myalgic encephalomyelitis/CFS and certain post-vaccination syndromes. However, although mechanistic models (viral immune evasion plus hyperinflammatory host responses) are plausible and supported by animal and in vitro work, human data remain largely associative; prospective studies linking allele-specific antigen presentation, pathogen load/persistence and downstream autoimmunity are still needed to move from correlation to causation.
Far from representing an inherent “weakness” of ancestral haplotypes, this phenomenon suggests an evolutionary trade-off in which the same immune response that conferred an advantage against lethal infections under high infectious-mortality conditions may be counterproductive in the modern environment (1, 20, 22, 228). Rather than an intrinsic deficiency in defense against chronic infections, what occurs is a mismatch between pathogen characteristics and the immune response induced by these haplotypes (61, 228, 229, 297). Intracellular pathogens that possess advanced immune-evasion mechanisms require more nuanced, less hyperreactive control strategies, which may account for the relative inefficacy of highly reactive HLA haplotypes in chronic infection contexts (1, 20, 123, 298).
It is important to note that the persistence of these haplotypes in the human population reflects an adaptive equilibrium (1, 20). While DR2-DQ6, DR4-DQ8 and DR3-DQ2 confer an advantage against acute infections by generating more effective immune responses, this same mechanism renders carriers more prone to exaggerated immune responses in chronic infections, increasing autoimmune disease risk (Table 2) (1, 20, 22, 61, 228). This balance between protection from acute infections and risk of autoimmunity following chronic infection may have been crucial for survival in earlier environments where lethal infections were far more common than chronic infections (61, 299–302). These observations provide indirect support for an evolutionary trade-off between infection resistance and autoimmune susceptibility; however, genomic signals can be influenced by demographic factors and association-study biases (e.g., overrepresentation of European populations), so functional validation is required for each allele and context.
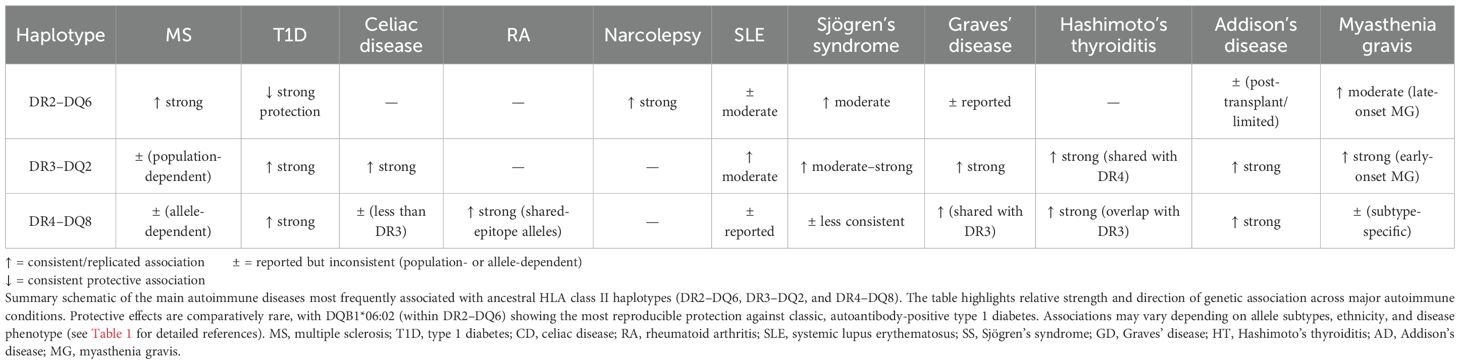
Table 2. Summary schematic of major HLA class II haplotype associations with autoimmune diseases.
Therefore, the evolution of these HLA haplotypes must be understood in terms of historical evolutionary fitness. The advantage conferred by rapid clearance of acute infections likely outweighed the costs associated with autoimmune diseases, which were less prevalent in populations historically exposed to high infectious mortality (1). In this sense, the persistence of these haplotypes reflects an adaptation that favored short-term survival against immediate threats posed by virulent, lethal pathogens, despite adverse long-term health consequences.
Hence, the persistence of these haplotypes in modern populations, with particularly high frequencies in northern Europe and Scandinavia (23–25, 303), reflects this evolutionary balance where the benefit against acute infections outweighed the cost of autoimmunity, which often manifested after reproductive age (1, 61, 302, 304, 305). Today, however, shifts in pathogen ecology, increased life expectancy and exposure to novel chronic inflammatory stimuli, such as latent infections, vaccinations, obesity or chronic stress, have converted these adaptive advantages into pathophysiological vulnerabilities (Figure 3) (306–310).
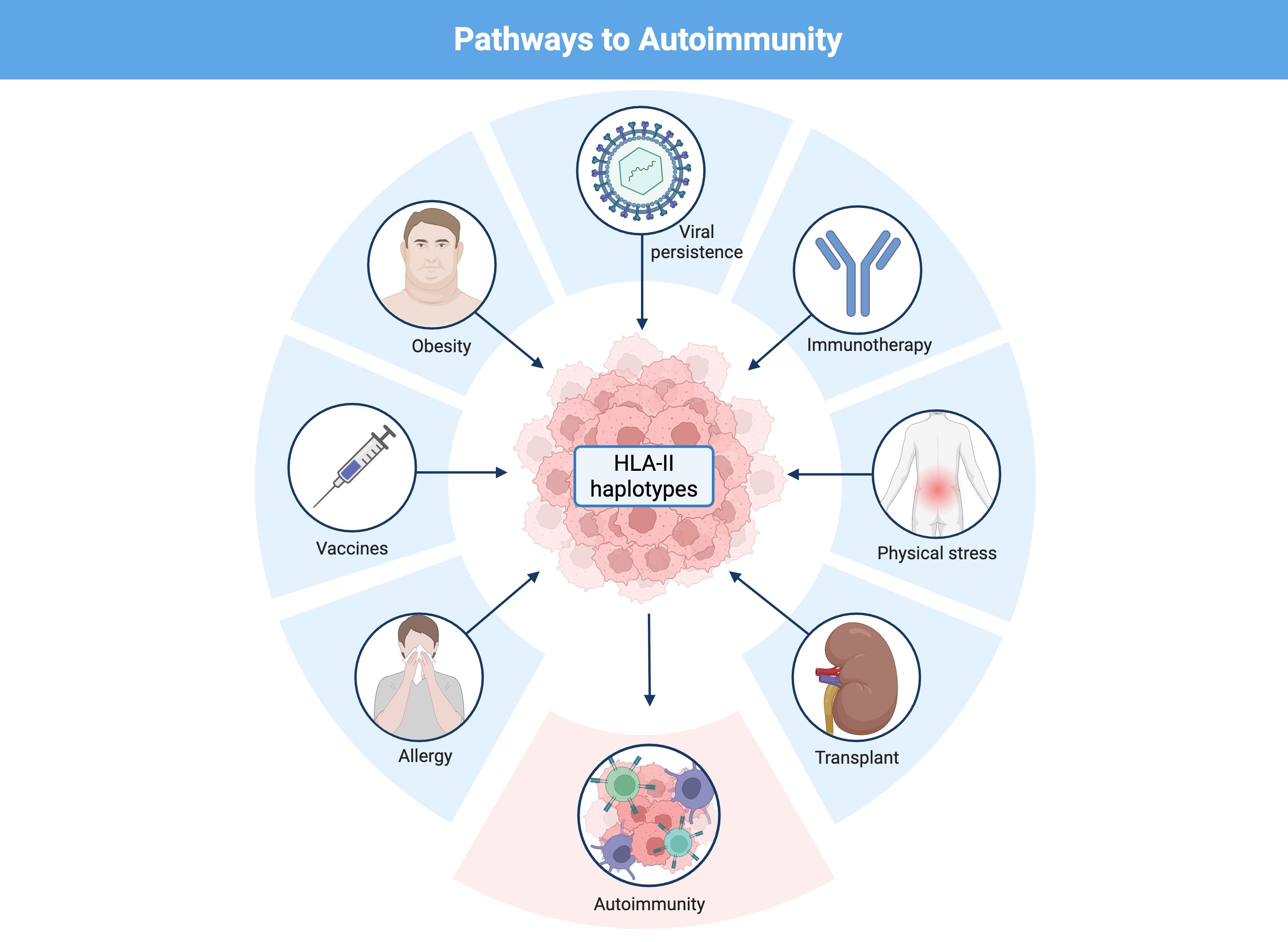
Figure 3. Pathways linking ancestral HLA-II haplotypes to autoimmunity. Schematic representation of the interplay between genetic predisposition and chronic immune activation in the development of autoimmunity. Ancestral HLA-II haplotypes (DR2-DQ6, DR3-DQ2, DR4-DQ8) confer evolutionary advantages against acute infections by promoting strong T cell responses, but in the modern context they predispose to immune hyperreactivity. Persistent antigenic stimulation, often derived from intracellular pathogens such as viral infections (e.g., Epstein–Barr virus), but also triggered by vaccines, immunotherapy, obesity, chronic physical stress, allergy, or transplantation, can sustain proinflammatory Th1/Th17 responses, continuous IFN-γ production, and aberrant HLA-II expression in peripheral tissues. In genetically susceptible individuals, this persistent inflammatory microenvironment promotes loss of tolerance, activation of autoreactive T and B cells, and the development of autoimmune disease.
This phenomenon highlights the complexity of immune system evolution, where the same immune response that is protective in an acute infection context can be detrimental in the setting of chronic infections (1, 61). Modern interventions in the form of immunomodulatory therapies and treatments for autoimmune diseases may help mitigate the negative effects of this immune hyperreactivity, but they also underscore the need for a more holistic approach that considers the evolutionary history of the human immune system (2, 311).
10 Concluding remarks and future directions
The paradoxical “weakness” of ancestral HLA haplotypes with respect to chronic infections should not be interpreted as an immunological deficiency but rather as an evolutionary trade-off whereby the same immunological trait that ensured survival against lethal pathogens in the past can now increase the risk of autoimmune disease in the present (1, 19, 20, 61). This balance between protection and risk reflects how evolution has shaped the human immune system to confront the infectious threats of its time, and raises questions about how changes in pathogen environments affect human health today.
Understanding this duality allows not only a reinterpretation of autoimmunity as the consequence of an evolutionary trade-off but also the proposal of more personalized preventive and therapeutic strategies. In the future, integrating HLA-II profiling into risk assessment could help identify individuals susceptible not only to classical autoimmune diseases but also to post-infectious or post-vaccinal inflammatory syndromes, allowing adjustment of immunological stimulus intensity and development of safer, more effective interventions.
Progress will require a multidimensional, prioritized approach that moves from association to mechanism and enables safe, HLA-informed translation into the clinic (26, 312). First, well-powered, multi-ancestry prospective cohorts with high-resolution HLA sequencing and harmonized phenotyping are essential to define population- and ancestry-specific effects and to reduce Eurocentric bias in current datasets (26). Second, allele-resolved, paired immunopeptidomics on affected and matched control tissues (or validated surrogate tissues) should be used to determine whether implicated haplotypes present broader or disease-relevant peptide repertoires in vivo; recent methodological advances make these experiments increasingly feasible on limited clinical samples (15, 313). Third, integrate improved in-silico HLA-II presentation models with empirical peptidomics to prioritize candidate peptides for downstream testing, contemporary predictors have substantially improved performance across HLA-DR/DQ/DP loci and can reduce the candidate search space for functional assays (314, 315). Fourth, deploy high-throughput antigen/TCR discovery platforms and pooled functional screens (e.g., TCR-mapping and barcoded peptide/TCR assays) to validate T-cell reactivity and establish whether peptides presented by specific alleles elicit pathogenic autoreactive T-cell responses (316, 317). Finally, complement molecular work with translational studies that include HLA stratification in vaccine surveillance and immunotherapy cohorts, paired single-cell/spatial transcriptomics and standardized longitudinal phenotyping; integrate data into open, interoperable resources so findings are reproducible and can inform HLA-guided preventive or therapeutic strategies (318–321).
This work has important limitations: much of the evidence is correlational (genomic signals and population associations) and may be affected by demographic factors, population structure and biases in association studies (e.g., overrepresentation of European populations). In addition, HLA allele imputation (statistical estimation of variants not directly genotyped) and heterogeneity in clinical definitions of post-infectious phenotypes (e.g., long COVID) limit comparability between studies; therefore, the evolutionary and mechanistic hypotheses discussed here require allele- and context-specific functional validation.
Author contributions
MR-P: Writing – original draft, Writing – review & editing. BP: Writing – original draft, Writing – review & editing. AZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by The Solve ME/CFS Initiative under the RAMSAY GRANT PROGRAM 2019 and through donations for chronic fatigue syndrome research through the Helpify crowdfunding platform.
Acknowledgments
All Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mangalam AK, Taneja V, and David CS. HLA class II molecules influence susceptibility vs protection in inflammatory diseases by determining the cytokine profile. J Immunol. (2013) 190:513. doi: 10.4049/JIMMUNOL.1201891
2. Song Y, Li J, and Wu Y. Evolving understanding of autoimmune mechanisms and new therapeutic strategies of autoimmune disorders. Signal Transduction Targeted Ther. (2024) 9:1–40. doi: 10.1038/s41392-024-01952-8
3. Yamamoto Y, Shirai Y, Edahiro R, Kumanogoh A, and Okada Y. Large-scale cross-trait genetic analysis highlights shared genetic backgrounds of autoimmune diseases. Immunol Med. (2025) 48:1–10. doi: 10.1080/25785826.2024.2394258
4. Griesemer AD, Sorenson EC, and Hardy MA. The role of the thymus in tolerance. Transplantation. (2010) 90:465–74. doi: 10.1097/TP.0B013E3181E7E54F
5. Lo WL and Huseby ES. The partitioning of TCR repertoires by thymic selection. J Exp Med. (2024) 221:e20230897. doi: 10.1084/JEM.20230897
6. Guo W, Smith D, Aviszus K, Detanico T, Heiser RA, and Wysocki LJ. Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J Exp Med. (2010) 207:2225–37. doi: 10.1084/JEM.20092712
7. Victora GD and Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2022) 40:413–42. doi: 10.1146/ANNUREV-IMMUNOL-120419-022408/CITE/REFWORKS
8. Adamopoulou E, Tenzer S, Hillen N, Klug P, Rota IA, Tietz S, et al. Exploring the MHC-peptide matrix of central tolerance in the human thymus. Nat Commun. (2013) 4:1–9. doi: 10.1038/NCOMMS3039;SUBJMETA
9. Young C and Brink R. Germinal centers and autoantibodies. Immunol Cell Biol. (2020) 98:480–9. doi: 10.1111/IMCB.12321
10. Corsiero E, Nerviani A, Bombardieri M, and Pitzalis C. Ectopic lymphoid structures: Powerhouse of autoimmunity. Front Immunol. (2016) 7:430/BIBTEX. doi: 10.3389/FIMMU.2016.00430/BIBTEX
11. Lucchesi D and Bombardieri M. The role of viruses in autoreactive B cell activation within tertiary lymphoid structures in autoimmune diseases. J Leukoc Biol. (2013) 94:1191–9. doi: 10.1189/JLB.0413240
12. Matzaraki V, Kumar V, Wijmenga C, and Zhernakova A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. (2017) 18:1–21. doi: 10.1186/S13059-017-1207-1/FIGURES/2
13. Arshad S, Cameron B, and Joglekar AV. Immunopeptidomics for autoimmunity: unlocking the chamber of immune secrets. NPJ Syst Biol Appl. (2025) 11:1–11. doi: 10.1038/S41540-024-00482-X;SUBJMETA
14. Symonds P, Marcu A, Cook KW, Metheringham RL, Durrant LG, and Brentville VA. Citrullinated epitopes identified on tumour MHC class II by peptide elution stimulate both regulatory and th1 responses and require careful selection for optimal anti-tumour responses. Front Immunol. (2021) 12:764462/BIBTEX. doi: 10.3389/FIMMU.2021.764462/BIBTEX
15. Stražar M, Park J, JG A, HB T, TK P, DR P, et al. HLA-II immunopeptidome profiling and deep learning reveal features of antigenicity to inform antigen discovery. Immunity. (2023) 56:1681–1698.e13. doi: 10.1016/j.immuni.2023.05.009
16. Ishigaki K, Lagattuta KA, Luo Y, James EA, Buckner JH, and Raychaudhuri S. HLA autoimmune risk alleles restrict the hypervariable region of T cell receptors. Nat Genet. (2022) 54:393–402. doi: 10.1038/S41588-022-01032-Z
17. Luckey D, Bastakoty D, and Mangalam AK. Role of HLA class II genes in susceptibility and resistance to multiple sclerosis: Studies using HLA transgenic mice. J Autoimmun. (2011) 37:122–8. doi: 10.1016/j.jaut.2011.05.001
18. Klein J and Sato A. The HLA system. New Engl J Med. (2000) 343:782–6. doi: 10.1056/NEJM200009143431106
19. Lund KP, Eriksson F, Hauge AW, Mora-Jensen HI, Sørensen SS, and Bruunsgaard H. The HLA-DR4-DQ8 phenotype of the recipient is associated with increased mortality after kidney transplantation. Clin Immunol. (2021) 226:108711. doi: 10.1016/J.CLIM.2021.108711
20. Mangalam AK, Rajagopalan G, Taneja V, and David CS. HLA class II transgenic mice mimic human inflammatory diseases. Adv Immunol. (2008) 97:65–147. doi: 10.1016/S0065-2776(08)00002-3
21. Chaudhary NS, Weldon CH, Nandakumar P, Team 23andMe Research, Holmes MV, and Aslibekyan S. Multi-ancestry GWAS of Long COVID identifies immune-related loci and etiological links to chronic fatigue syndrome, fibromyalgia and depression. MedRxiv. (2024) 2024:10.07.24315052. doi: 10.1101/2024.10.07.24315052
22. Yasukochi Y and Satta Y. A human-specific allelic group of the MHC DRB1 gene in primates. J Physiol Anthropol. (2014) 33:14. doi: 10.1186/1880-6805-33-14
23. Lande A, Andersen I, Egeland T, Lie BA, and Viken MK. -C, -B, -DRB1, -DQB1 and -DPB1 allele and haplotype frequencies in 4514 healthy Norwegians. Hum Immunol. (2018) 79:527–9. doi: 10.1016/J.HUMIMM.2018.04.012
24. Hauge AW, Ekstrøm CT, Mehrabi HR, and Bruunsgaard H. Healthy danes from eastern Denmark. HLA. (2021) 97:95–7. doi: 10.1111/TAN.14101;PAGE:STRING:ARTICLE/CHAPTER
25. Allele frequency net database . Available online at: http://www.allelefrequencies.net/hla6003x.asp (Accessed August 21, 2025).
26. Michalek DA, Tern C, Zhou W, Robertson CC, Farber E, Campolieto P, et al. A multi-ancestry genome-wide association study in type 1 diabetes. Hum Mol Genet. (2024) 33:958–68. doi: 10.1093/HMG/DDAE024
27. Bontrop RE, Otting N, De Groot NG, and Doxiadis GGM. Major histocompatibility complex class II polymorphisms in primates. Immunol Rev. (1999) 167:339–50. doi: 10.1111/j.1600-065X.1999.tb01403.x
28. Von Salomé J, Gyllensten U, and Bergström TF. Full-length sequence analysis of the HLA-DRB1 locus suggests a recent origin of alleles. Immunogenetics. (2007) 59:261–71. doi: 10.1007/S00251-007-0196-8/METRICS
29. O’Connell JF, Allen J, Williams MAJ, Williams AN, Turney CSM, Spooner NA, et al. When did homo sapiens first reach Southeast Asia and Sahul? Proc Natl Acad Sci U.S.A. (2018) 115:8482–90. doi: 10.1073/PNAS.1808385115/-/DCSUPPLEMENTAL
30. Pittman KJ, Glover LC, Wang L, and Ko DC. The legacy of past pandemics: common human mutations that protect against infectious disease. PloS Pathog. (2016) 12:e1005680. doi: 10.1371/JOURNAL.PPAT.1005680
31. Immel A, Key FM, Szolek A, Barquera R, Robinson MK, Harrison GF, et al. Analysis of genomic DNA from medieval plague victims suggests long-term effect of yersinia pestis on human immunity genes. Mol Biol Evol. (2021) 38:4059–76. doi: 10.1093/MOLBEV/MSAB147
32. Luckey D, Weaver EA, Osborne DG, Billadeau DD, and Taneja V. Immunity to Influenza is dependent on MHC II polymorphism: study with 2 HLA transgenic strains. Sci Rep. (2019) 9:1–10. doi: 10.1038/s41598-019-55503-1
33. Wysocki T, Olesińska M, and Paradowska-Gorycka A. Current understanding of an emerging role of HLA-DRB1 gene in rheumatoid arthritis–from research to clinical practice. Cells. (2020) 9:1127. doi: 10.3390/CELLS9051127
34. McMichael AJ. Legacy of the influenza pandemic 1918: The host T cell response. BioMed J. (2018) 41:242. doi: 10.1016/J.BJ.2018.08.003
35. Moise L, Tassone R, Latimer H, Terry F, Levitz L, Haran JP, et al. Immunization with cross-conserved H1N1 influenza CD4+T-cell epitopes lowers viral burden in HLA DR3 transgenic mice. Hum Vaccin Immunother. (2013) 9:2060–8. doi: 10.4161/HV.26511
36. Krause-Kyora B, Nutsua M, Boehme L, Pierini F, Pedersen DD, Kornell SC, et al. Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval Europeans. Nat Commun. (2018) 9:1569. doi: 10.1038/s41467-018-03857-x
37. Singh T, Fakiola M, Oommen J, Singh AP, Singh AK, Smith N, et al. Epitope binding characteristics for risk versus protective DRB1 alleles for visceral leishmaniasis. J Immunol. (2018) 200:2727. doi: 10.4049/JIMMUNOL.1701764
38. Weiskopf D, Angelo MA, Grifoni A, O’Rourke PH, Sidney J, Paul S, et al. HLA-DRB1 alleles are associated with different magnitudes of dengue virus–specific CD4+ T-cell responses. J Infect Dis. (2016) 214:1117. doi: 10.1093/INFDIS/JIW309
39. King AA, Shrestha S, Harvill ET, and Bjørnstad ON. Evolution of acute infections and the invasion-persistence trade-off. Am Nat. (2009) 173:446–55. doi: 10.1086/597217
40. Cressler CE, McLeod DV, Rozins C, Van Den Hoogen J, and Day T. The adaptive evolution of virulence: a review of theoretical predictions and empirical tests. Parasitology. (2016) 143:915–30. doi: 10.1017/S003118201500092X
41. Draborg AH, Duus K, and Houen G. Epstein-barr virus in systemic autoimmune diseases. Clin Dev Immunol. (2013) 2013:535738. doi: 10.1155/2013/535738
42. Schatz DG and Swanson PC. V(D)J recombination: Mechanisms of initiation. Annu Rev Genet. (2011) 45:167–202. doi: 10.1146/ANNUREV-GENET-110410-132552/CITE/REFWORKS
43. Di Noia JM and Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. (2007) 76:1–22. doi: 10.1146/ANNUREV.BIOCHEM.76.061705.090740/CITE/REFWORKS
44. Klein L, Kyewski B, Allen PM, and Hogquist KA. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat Rev Immunol. (2014) 14:377–91. doi: 10.1038/NRI3667;SUBJMETA
45. Košmrlj A, Jha AK, Huseby ES, Kardar M, and Chakraborty AK. How the thymus designs antigen-specific and self-tolerant T cell receptor sequences. Proc Natl Acad Sci. (2008) 105:16671–6. doi: 10.1073/PNAS.0808081105
46. Logunova NN, Kriukova VV, Shelyakin PV, Egorov ES, Pereverzeva A, Bozhanova NG, et al. MHC-II alleles shape the CDR3 repertoires of conventional and regulatory naïve CD4+ T cells. Proc Natl Acad Sci. (2020) 117:13659–69. doi: 10.1073/PNAS.2003170117
47. James EA, Moustakas AK, Bui J, Nouv R, Papadopoulos GK, and Kwok WW. The binding of antigenic peptides to HLA-DR is influenced by interactions between pocket 6 and pocket 9. J Immunol. (2009) 183:3249–58. doi: 10.4049/JIMMUNOL.0802228
48. Tollefsen S, Arentz-Hansen H, Fleckenstein B, Molberg Ø, Ráki M, Kwok WW, et al. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J Clin Invest. (2006) 116:2226–36. doi: 10.1172/JCI27620
49. Holoshitz J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr Opin Rheumatol. (2010) 22:293–8. doi: 10.1097/BOR.0B013E328336BA63
50. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest. (2017) 127:2881–91. doi: 10.1172/JCI94549
51. Zhao M, Flynt FL, Hong M, Chen H, Gilbert CA, Briley NT, et al. MHC class II transactivator (CIITA) expression is upregulated in multiple myeloma cells by IFN-γ. Mol Immunol. (2007) 44:2923–32. doi: 10.1016/j.molimm.2007.01.009
52. Rodriguez M, Zoecklein L, Kerkvliet JG, Pavelko KD, Papke L, Howe CL, et al. Human HLA-DR transgenes protect mice from fatal virus-induced encephalomyelitis and chronic demyelination. J Virol. (2008) 82:3369–80. doi: 10.1128/JVI.02243-07/ASSET/CC18D86C-AD19-430B-8F11-ADE7931609B7/ASSETS/GRAPHIC/ZJV0070804160008.JPEG
53. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated immune control of Epstein–Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol. (2021) 51:64–75. doi: 10.1002/EJI.202048655
54. Winchester R, Chen Y, Rose S, Selby J, and Borkowsky W. Major histocompatibility complex class II DR alleles DRB1*1501 and those encoding HLA-DR13 are preferentially associated with a diminution in maternally transmitted human immunodeficiency virus 1 infection in different ethnic groups: Determination by an automated sequence-based typing method. Proc Natl Acad Sci U.S.A. (1995) 92:12374–8. doi: 10.1073/PNAS.92.26.12374
55. Alves C, Souza T, Meyer I, Toralles MBP, and Brites C. Immunogenetics and infectious diseases: special reference to the mayor histocompatibility complex. Braz J Infect Dis. (2006) 10:122–31. doi: 10.1590/S1413-86702006000200010
56. Yan ZH, Fan Y, Wang XH, Mao Q, Deng GH, and Wang YM. Relationship between HLA-DR gene polymorphisms and outcomes of hepatitis B viral infections: A meta-analysis. World J Gastroenterology : WJG. (2012) 18:3119. doi: 10.3748/WJG.V18.I24.3119
58. Murray PD, McGavern DB, Pease LR, and Rodriguez M. Cellular sources and targets of IFN-γ-mediated protection against viral demyelination and neurological deficits. Eur J Immunol. (2002) 32:606–15. doi: 10.1002/1521-4141(200203)32:3<606::AID-IMMU606>3.0.CO;2-D
59. Rodriguez M, Zoecklein LJ, Howe CL, Pavelko KD, Gamez JD, Nakane S, et al. Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial theiler’s murine encephalomyelitis virus infection. J Virol. (2003) 77:12252–65. doi: 10.1128/JVI.77.22.12252-12265.2003
60. Rodriguez M, Pavelko K, and Coffman RL. Gamma interferon is critical for resistance to Theiler’s virus-induced demyelination. J Virol. (1995) 69:7286–90. doi: 10.1128/JVI.69.11.7286-7290.1995
61. Meyer D, Vitor VR, Bitarello BD, Débora DY, and Nunes K. A genomic perspective on HLA evolution. Immunogenetics. (2018) 70:5–27. doi: 10.1007/s00251-017-1017-3
62. Jochum S, Moosmann A, Lang S, Hammerschmidt W, and Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PloS Pathog. (2012) 8:e1002704. doi: 10.1371/JOURNAL.PPAT.1002704
63. Gabor F, Jahn G, Sedmak DD, and Sinzger C. In vivo Downregulation of MHC Class I Molecules by HCMV Occurs During All Phases of Viral Replication but Is Not Always Complete. Front Cell Infect Microbiol. (2020) 10:283/BIBTEX. doi: 10.3389/FCIMB.2020.00283/BIBTEX
64. Bejarano MT and Masucci MG. Interleukin-10 abrogates the inhibition of epstein-barr virus–induced B-cell transformation by memory T-cell responses. Blood. (1998) 92:4256–62. doi: 10.1182/BLOOD.V92.11.4256
65. Yin R, Melton S, Huseby ES, Kardar M, and Chakraborty AK. How persistent infection overcomes peripheral tolerance mechanisms to cause T cell–mediated autoimmune disease. Proc Natl Acad Sci U.S.A. (2024) 121:e2318599121. doi: 10.1073/PNAS.2318599121/SUPPL_FILE/PNAS.2318599121.SAPP.PDF
66. Vietzen H, Berger SM, Kühner LM, Furlano PL, Bsteh G, Berger T, et al. Ineffective control of Epstein-Barr-virus-induced autoimmunity increases the risk for multiple sclerosis. Cell. (2023) 186:5705–5718.e13. doi: 10.1016/j.cell.2023.11.015
67. Tălăngescu A, Calenic B, Mihăilescu DF, Tizu M, Marunelu I, Constantinescu AE, et al. Molecular analysis of HLA genes in Romanian patients with chronic hepatitis B virus infection. Curr Issues Mol Biol. (2024) 46:1064. doi: 10.3390/CIMB46020067
68. Luo J, Jin AN, Wu XD, and Yu RB. Meta-analysis on the relationship between HLA-DRB1 gene polymorphism and chronic hepatitis B in Chinese population. World J Gastoentrol. (2006) 14(31):3050–4. doi: 10.11569/wcjd.v14.i31.3050
69. Yang G, Liu J, Han S, Xie H, Du R, Yan Y, et al. Association between hepatitis B virus infection and HLA-DRB1 genotyping in Shaanxi Han patients in northwestern China. Tissue Antigens. (2007) 69:170–5. doi: 10.1111/J.1399-0039.2006.00744.
70. Almarri A and Batchelor JR. HLA and hepatitis B infection. Lancet. (1994) 344:1194–5. doi: 10.1016/S0140-6736(94)90510-X
71. Wang L, Zou ZQ, and Wang K. Clinical relevance of HLA gene variants in HBV infection. J Immunol Res. (2016) 2016:9069375. doi: 10.1155/2016/9069375
72. Li X, Zhou Q, Lu Z, Huang R, Lin D, Xu J, et al. Association of HLA-DRB1 alleles with status of antibodies to hepatitis B surface and e antigen. J Med Virol. (2024) 96:e29867. doi: 10.1002/JMV.29867
73. Kankonkar S and Shankarkumar U. HLA DRB alleles in chronic hepatitis B infected patients. Int J Hum Genet. (2008) 8:331–4. doi: 10.1080/09723757.2008.11886048
74. Akhter R, Shirin A, Tabassum S, and Hossen M. Association of HLA DRB1*15 gene among acute and chronic hepatitis B infected Bangladeshi patients. Bangladesh J Infect Dis. (2018) 5:3–9. doi: 10.3329/BJID.V5I1.37709
75. Patel N. HLA class II genotyping in chronic hepatitis B infection. J Assoc Physicians India (2003) 51:779–81.
76. Fisicaro P, Barili V, Rossi M, Montali I, Vecchi A, Acerbi G, et al. Pathogenetic mechanisms of T cell dysfunction in chronic HBV infection and related therapeutic approaches. Front Immunol. (2020) 11:849/XML. doi: 10.3389/FIMMU.2020.00849/XML
77. Jan RH, Lin YL, Chen LK, Huang MT, Wang LC, and Chiang BL. Hepatitis B virus surface antigen can activate dendritic cells and modulate T helper type immune response. Microbiol Immunol. (2011) 55:51–9. doi: 10.1111/J.1348-0421.2010.00284.X
78. Harishankar M, Selvaraj P, and Bethunaickan R. Influence of genetic polymorphism towards pulmonary tuberculosis susceptibility. Front Med (Lausanne). (2018) 5:213/XML. doi: 10.3389/FMED.2018.00213/XML
79. Singh AK, Abhimanyu, Yadav AB, Sharma S, Garg R, Bose M, et al. HLA-DRB1*1501 and VDR polymorphisms and survival of mycobacterium tuberculosis in human macrophages exposed to inhalable microparticles. Pharmacogenomics. (2013) 14:531–40. doi: 10.2217/PGS.13.12
80. Chandra P, Grigsby SJ, and Philips JA. Immune evasion and provocation by Mycobacterium tuberculosis. Nat Rev Microbiol. (2022) 20:750–66. doi: 10.1038/s41579-022-00763-4
81. Kaushansky N, Altmann DM, David CS, Lassmann H, and Ben-Nun A. DQB1*0602 rather than DRB1*1501 confers susceptibility to multiple sclerosis-like disease induced by proteolipid protein (PLP). J Neuroinflamm. (2012) 9:1–15. doi: 10.1186/1742-2094-9-29/FIGURES/6
82. Kaushansky N, Altmann DM, Ascough S, David CS, Lassmann H, and Ben-Nun A. HLA-DQB1*0602 determines disease susceptibility in a new “Humanized” Multiple sclerosis model in HLA-DR15 (DRB1*1501;DQB1*0602) transgenic mice. J Immunol. (2009) 183:3531–41. doi: 10.4049/JIMMUNOL.0900784
83. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated immune control of Epstein-Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol. (2021) 51:64–75. doi: 10.1002/EJI.202048655
84. Olsson T. Epstein Barr virus infection and immune defense related to HLA-DR15: consequences for multiple sclerosis. Eur J Immunol. (2021) 51:56–9. doi: 10.1002/EJI.202049030
85. Babu Chodisetti S, Rai PK, Gowthaman U, Pahari S, and Agrewala JN. Potential T cell epitopes of Mycobacterium tuberculosis that can instigate molecular mimicry against host: implications in autoimmune pathogenesis. BMC Immunol. (2012) 13:1–10. doi: 10.1186/1471-2172-13-13/TABLES/1
86. Birnbaum G, Kotilinek L, and Albrecht L. Spinal fluid lymphocytes from a subgroup of multiple sclerosis patients respond to mycobacterial antigens. Ann Neurol. (1993) 34:18–24. doi: 10.1002/ANA.410340106
87. Ressing ME, Van Leeuwen D, Verreck FAW, Gomez R, Heemskerk B, Toebes M, et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc Natl Acad Sci U.S.A. (2003) 100:11583–8. doi: 10.1073/pnas.2034960100
88. Churilov LP, Normatov MG, Ling H, Zhuang M, Kudlay D, and Starshinova A. Autoimmune diseases and molecular mimicry in tuberculosis. Biol 2024 Vol 13 Page. (1083) 2024:13:1083. doi: 10.3390/BIOLOGY13121083
89. Ballerini C. Experimental autoimmune encephalomyelitis. Methods Mol Biol. (2021) 2285:375–84. doi: 10.1007/978-1-0716-1311-5_27
90. Feliciano-Sanchez A, García-Gil R, Cubas-Nuñez L, Castillo-Villalba J, Fuentes-Maestre J, Fil M, et al. Development and optimisation of an animal model for the study of ganglion cells in degenerative diseases of the retina and optic nerve. Archivos la Sociedad Española Oftalmología (English Edition). (2019) 94:263–72. doi: 10.1016/J.OFTALE.2019.01.011
91. Yousef S, Planas R, Chakroun K, Hoffmeister-Ullerich S, Binder TMC, Eiermann TH, et al. TCR bias and HLA cross-restriction are strategies of human brain-infiltrating JC virus-specific CD4+ T cells during viral infection. J Immunol. (2012) 189:3618–30. doi: 10.4049/JIMMUNOL.1201612
92. Tavenier J, Margolick JB, and Leng SX. T-cell immunity against cytomegalovirus in HIV infection and aging: relationships with inflammation, immune activation, and frailty. Med Microbiol Immunol. (2019) 208:289–94. doi: 10.1007/S00430-019-00591-Z/METRICS
93. Mueller MC, Kern WV, Usadel S, Pauly MC, Cathomen T, and Salzer U. Assessing the differential impact of chronic CMV and treated HIV infection on CD8+ T-cell differentiation in a matched cohort study: is CMV the key? AIDS Res Ther. (2021) 18:1–9. doi: 10.1186/S12981-021-00361-Z/FIGURES/3
94. Poloni C, Szyf M, Cheishvili D, and Tsoukas CM. Are the healthy vulnerable? Cytomegalovirus seropositivity in healthy adults is associated with accelerated epigenetic age and immune dysregulation. J Infect Dis. (2022) 225:443–52. doi: 10.1093/INFDIS/JIAB365
95. Hyun YS, Jo HA, Lee YH, Kim SM, Baek IC, Sohn HJ, et al. Comprehensive analysis of CD4+ T cell responses to CMV pp65 antigen restricted by single HLA-DR, -DQ, and -DP allotype within an individual. Front Immunol. (2021) 11:602014/BIBTEX. doi: 10.3389/FIMMU.2020.602014/BIBTEX
96. Varani S, Frascaroli G, Landini MP, and Söderberg-Nauclér C. Human cytomegalovirus targets different subsets of antigen-presenting cells with pathological consequences for host immunity: implications for immunosuppression, chronic inflammation and autoimmunity. Rev Med Virol. (2009) 19:131–45. doi: 10.1002/RMV.609
97. Sester U, Presser D, Dirks J, Gärtner BC, Köhler H, and Sester M. PD-1 expression and IL-2 loss of cytomegalovirus- specific t cells correlates with viremia and reversible functional anergy. Am J Transplant. (2008) 8:1486–97. doi: 10.1111/j.1600-6143.2008.02279.x
98. Redeker A, Remmerswaal EBM, van der Gracht ETI, Welten SPM, Höllt T, Koning F, et al. The contribution of cytomegalovirus infection to immune senescence is set by the infectious dose. Front Immunol. (2018) 8:1953/BIBTEX. doi: 10.3389/FIMMU.2017.01953/BIBTEX
99. Rölle A and Olweus J. Dendritic cells in cytomegalovirus infection: Viral evasion and host countermeasures. APMIS. (2009) 117:413–26. doi: 10.1111/J.1600-0463.2009.02449.X
100. Brooks DG, Teyton L, Oldstone MBA, and McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol. (2005) 79:10514–27. doi: 10.1128/JVI.79.16.10514-10527.2005/ASSET/A4044A75-6A47-4943-9657-C3FE36E6BD5E/ASSETS/GRAPHIC/ZJV0160566930007.JPEG
101. Park AJ, Rendini T, Martiniuk F, and Levis WR. Leprosy as a model to understand cancer immunosurveillance and T cell anergy. J Leukoc Biol. (2016) 100:47–54. doi: 10.1189/JLB.5RU1215-537RR
102. Silva MJA, Silva CS, Brasil TP, Alves AK, dos Santos EC, Frota CC, et al. An update on leprosy immunopathogenesis: systematic review. Front Immunol. (2024) 15:1416177/XML. doi: 10.3389/FIMMU.2024.1416177/XML
103. Finlay BB and McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. (2006) 124:767–82. doi: 10.1016/J.CELL.2006.01.034
104. Saeidi A, Zandi K, Cheok YY, Saeidi H, Wong WF, Lee CYQ, et al. T-cell exhaustion in chronic infections: Reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front Immunol. (2018) 9:2569/XML. doi: 10.3389/FIMMU.2018.02569/XML
105. Chen JS and Eisenbarth SC. A ten-acious cytokine for a tenacious infection. Sci Immunol. (2019) 4:eaaw7720. doi: 10.1126/SCIIMMUNOL.AAW7720
106. Mosmann TR and Coffman RL. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. (1989) 7:145–73. doi: 10.1146/ANNUREV.IY.07.040189.001045/CITE/REFWORKS
107. Flynn JAL, Chan J, Triebold KJ, Dalton DK, Stewart TA, and Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. (1993) 178:2249–54. doi: 10.1084/JEM.178.6.2249
108. Pitzalis C, Jones GW, Bombardieri M, and Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. (2014) 14:447–62. doi: 10.1038/nri3700
109. Vinuesa CG, Sanz Ĩ, and Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. (2009) 9:845–57. doi: 10.1038/NRI2637
110. Nguyen C, Varney MD, Harrison LC, and Morahan G. Definition of high-risk type 1 diabetes HLA-DR and HLA-DQ types using only three single nucleotide polymorphisms. Diabetes. (2013) 62:2135–40. doi: 10.2337/DB12-1398
111. Odeh R, Alassaf A, Alhawari H, Jafar H, Awidi A, Bani Hani F, et al. Immunogenetic profiling of type 1 diabetes in Jordan: a case-control study on HLA-associated risk and protection. . J Pediatr Endocrinol Metab. (2025) 38(11):1144–53. doi: 10.1515/JPEM-2025-0402
112. Chowdhary VR, Dai C, Tilahun AY, Hanson JA, Smart MK, Grande JP, et al. A central role for HLA-DR3 in anti-smith antibody responses and glomerulonephritis in a transgenic mouse model of spontaneous lupus. J Immunol. (2015) 195:4660–7. doi: 10.4049/JIMMUNOL.1501073
113. Renaudineau Y, Charras A, Natoli V, Congy-Jolivet N, Haldenby S, Liu X, et al. Across ancestries, HLA-B∗08:01∼DRB1∗03:01 (DR3) and HLA-DQA∗01:02 (DR2) increase the risk to develop juvenile-onset systemic lupus erythematosus through low complement C4 levels. J Transl Autoimmun. (2025) 10:100268. doi: 10.1016/j.jtauto.2025.100268
114. Olsson T. Epstein Barr virus infection and immune defense related to HLA-DR15: consequences for multiple sclerosis. Eur J Immunol. (2021) 51:56. doi: 10.1002/EJI.202049030
115. Agostini S, Mancuso R, Guerini FR, D’Alfonso S, Agliardi C, Hernis A, et al. HLA alleles modulate EBV viral load in multiple sclerosis. J Transl Med. (2018) 16:1–9. doi: 10.1186/S12967-018-1450-6/FIGURES/6
116. Godot V, Harraga S, Beurton I, Tiberghien P, Sarciron E, Gottstein B, et al. Resistance/susceptibility to Echinococcus multilocularis infection and cytokine profile in humans. II. Influence HLA B8 DR3 DQ2 haplotype. Clin Exp Immunol. (2000) 121:491. doi: 10.1046/J.1365-2249.2000.01309.X
117. Lenzi M, Frisoni M, Mantovani V, Ricci P, Muratori L, Francesconi R, et al. Haplotype HLA-B8-DR3 confers susceptibility to hepatitis C virus-related mixed cryoglobulinemia. Blood. (1998) 91:2062–6. doi: 10.1182/BLOOD.V91.6.2062
118. Kolls JK, Habetz S, Shean MK, Vazquez C, Brown JA, Lei D, et al. IFN-gamma and CD8+ T cells restore host defenses against Pneumocystis carinii in mice depleted of CD4+ T cells. J Immunol. (1999) 162:2890–4. doi: 10.4049/jimmunol.162.5.2890
119. Christensen JP, Bartholdy C, Wodarz D, and Thomsen AR. Depletion of CD4+ T cells precipitates immunopathology in immunodeficient mice infected with a noncytocidal virus. J Immunol. (2001) 166:3384–91. doi: 10.4049/JIMMUNOL.166.5.3384
120. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44. doi: 10.1126/SCIENCE.1198687
121. Cordeiro PAS, Assone T, Prates G, Tedeschi MRM, Fonseca LAM, and Casseb J. The role of IFN-γ production during retroviral infections: an important cytokine involved in chronic inflammation and pathogenesis. Rev Inst Med Trop Sao Paulo. (2022) 64:e64. doi: 10.1590/S1678-9946202264064
122. Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8 + T-cell immunity. Proc Natl Acad Sci U.S.A. (2012) 109:1210–5. doi: 10.1073/PNAS.1118834109/SUPPL_FILE/PNAS.201118834SI.PDF
123. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486. doi: 10.1038/NRI3862
124. Fillatreau S, Manfroi B, and Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. (2021) 17:98–108. doi: 10.1038/S41584-020-00544-4;SUBJMETA=1313,249,250,631;KWRD=AUTOIMMUNE+DISEASES,IMMUNOLOGY
125. Sun K, Wu C, Kong Q, Hu J, Shi L, Pi Y, et al. Lymphocytes in patients with chronic active epstein-barr virus disease exhibited elevated PD-1/PD-L1 expression and a prevailing th2 immune response. Mediterr J Hematol Infect Dis. (2024) 16:e2024037. doi: 10.4084/MJHID.2024.037
126. Clerici M and Shearer GM. A TH1→TH2 switch is a critical step in the etiology of HIV infection. Immunol Today. (1993) 14:107–11. doi: 10.1016/0167-5699(93)90208-3
127. Scott NA, Zhao Y, Krishnamurthy B, Mannering SI, Kay TWH, and Thomas HE. IFNγ-Induced MHC Class II expression on islet endothelial cells is an early marker of insulitis but is not required for diabetogenic CD4+T cell migration. Front Immunol. (2018) 9:2800/BIBTEX. doi: 10.3389/FIMMU.2018.02800/BIBTEX
128. Muczynski KA, Anderson SK, and Pious D. Discoordinate surface expression of IFN-γ-induced HLA class II proteins in nonprofessional antigen-presenting cells with absence of DM and class II colocalization. J Immunol. (1998) 160:3207–16. doi: 10.4049/JIMMUNOL.160.7.3207
129. Ishina IA, Zakharova MY, Kurbatskaia IN, Mamedov AE, Belogurov AA, and Gabibov AG. MHC class II presentation in autoimmunity. Cells 2023 Vol 12 Page 314. (2023) 12:314. doi: 10.3390/CELLS12020314
130. Chemin K, Gerstner C, and Malmström V. Effector functions of CD4+ T cells at the site of local autoimmune inflammation-lessons from rheumatoid arthritis. Front Immunol. (2019) 10:353. doi: 10.3389/fimmu.2019.00353
131. Dumitriu IE. The life (and death) of CD4+CD28null T cells in inflammatory diseases. Immunology. (2015) 146:185–93. doi: 10.1111/IMM.12506
132. Choi SC and Morel L. B cell contribution of the CD4+ T cell inflammatory phenotypes in systemic lupus erythematosus. Autoimmunity. (2017) 50:37–41. doi: 10.1080/08916934.2017.1280028
133. Vinuesa CG, Linterman MA, Yu D, and Maclennan ICM. Follicular helper T cells. Annu Rev Immunol. (2016) 34:335–68. doi: 10.1146/ANNUREV-IMMUNOL-041015-055605/CITE/REFWORKS
134. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduction Targeted Ther. (2021) 6:1 2021. doi: 10.1038/s41392-021-00658-5
135. Sun L, Su Y, Jiao A, Wang X, and Zhang B. T cells in health and disease. Signal Transduction Targeted Ther. (2023) 8:1–50. doi: 10.1038/s41392-023-01471-y
136. Collier JL, Weiss SA, Pauken KE, Sen DR, and Sharpe AH. Not-so-opposite ends of the spectrum: CD8+ T cell dysfunction across chronic infection, cancer and autoimmunity. Nat Immunol. (2021) 22:7 2021. doi: 10.1038/s41590-021-00949-7
137. Kahan SM, Wherry EJ, and Zajac AJ. T cell exhaustion during persistent viral infections. Virology. (2015) 479–480:180–93. doi: 10.1016/J.VIROL.2014.12.033
138. Akturk HK, Kahramangil D, Sarwal A, Hoffecker L, Murad MH, and Michels AW. Immune checkpoint inhibitor-induced Type 1 diabetes: a systematic review and meta-analysis. Diabetic Med. (2019) 36:1075–81. doi: 10.1111/DME.14050
139. Kobayashi T, Iwama S, Sugiyama D, Yasuda Y, Okuji T, Ito M, et al. Original research: Anti-pituitary antibodies and susceptible human leukocyte antigen alleles as predictive biomarkers for pituitary dysfunction induced by immune checkpoint inhibitors. J Immunother Cancer. (2021) 9:2493. doi: 10.1136/JITC-2021-002493
140. Jiang N, Yu Y, Zhang M, Tang Y, Wu D, Wang S, et al. Association between germ-line HLA and immune-related adverse events. Front Immunol. (2022) 13:952099/BIBTEX. doi: 10.3389/FIMMU.2022.952099/BIBTEX
141. Berner F and Flatz L. Autoimmunity in immune checkpoint inhibitor-induced immune-related adverse events: A focus on autoimmune skin toxicity and pneumonitis. Immunol Rev. (2023) 318:37–50. doi: 10.1111/IMR.13258
142. Yano S, Ashida K, Sakamoto R, Sakaguchi C, Ogata M, Maruyama K, et al. Human leucocyte antigen DR15, a possible predictive marker for immune checkpoint inhibitor–induced secondary adrenal insufficiency. Eur J Cancer. (2020) 130:198–203. doi: 10.1016/j.ejca.2020.02.049
143. De Filette JMK, Pen JJ, Decoster L, Vissers T, Bravenboer B, van der Auwera BJ, et al. Immune checkpoint inhibitors and type 1 diabetes mellitus: a case report and systematic review. Eur J Endocrinol. (2019) 181:363–74. doi: 10.1530/EJE-19-0291
144. Dougan M and Pietropaolo M. Time to dissect the autoimmune etiology of cancer antibody immunotherapy. J Clin Invest. (2020) 130:51–61. doi: 10.1172/JCI131194
145. Wu L, Tsang VHM, Sasson SC, Menzies AM, Carlino MS, Brown DA, et al. Unravelling checkpoint inhibitor associated autoimmune diabetes: from bench to bedside. Front Endocrinol (Lausanne). (2021) 12:764138/XML. doi: 10.3389/FENDO.2021.764138/XML
146. Jorgovanovic D, Song M, Wang L, and Zhang Y. Roles of IFN-γ in tumor progression and regression: a review. biomark Res. (2020) 8:1 2020. doi: 10.1186/S40364-020-00228-X
147. Wijdeven RH, van Luijn MM, Wierenga-Wolf AF, Akkermans JJ, van den Elsen PJ, Hintzen RQ, et al. Chemical and genetic control of IFN γ-induced MHCII expression. EMBO Rep. (2018) 19:45553. doi: 10.15252/EMBR.201745553/SUPPL_FILE/EMBR201745553-SUP-0005-SDATAEV.PDF
148. Postow MA, Sidlow R, and Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. New Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMRA1703481/SUPPL_FILE/NEJMRA1703481_DISCLOSURES.PDF
149. Lin JS, Wang DY, Mamlouk O, Glass WF, Abdelrahim M, Yee C, et al. Immune checkpoint inhibitor associated reactivation of primary membranous nephropathy responsive to rituximab. J Immunother Cancer. (2020) 8:e001287. doi: 10.1136/JITC-2020-001287
150. Yin Q, Wu L, Han L, Zheng X, Tong R, Li L, et al. Immune-related adverse events of immune checkpoint inhibitors: a review. Front Immunol. (2023) 14:1167975/XML. doi: 10.3389/FIMMU.2023.1167975/XML
151. Gandarillas S, Newland ES, Toppmeyer D, Stephenson R, Denzin L, and Dasgeb B. HLA inherence as a potential parameter in checkpoint inhibitor-associated autoimmune adverse event assessment. Front Med (Lausanne). (2023) 10:1288844/BIBTEX. doi: 10.3389/FMED.2023.1288844/BIBTEX
152. Segal Y and Shoenfeld Y. Vaccine-induced autoimmunity: the role of molecular mimicry and immune crossreaction. Cell Mol Immunol. (2018) 15:586–94. doi: 10.1038/CMI.2017.151;KWRD=BIOMEDICINE
153. Mangalam AK, Taneja V, and David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. J Immunol. (2013) 190:513–9. doi: 10.4049/JIMMUNOL.1201891
154. Lernmark Å. Environmental factors in the etiology of type 1 diabetes, celiac disease, and narcolepsy. Pediatr Diabetes. (2016) 17:65–72. doi: 10.1111/PEDI.12390
155. Hallberg P, Smedje H, Eriksson N, Kohnke H, Daniilidou M, Öhman I, et al. Pandemrix-induced narcolepsy is associated with genes related to immunity and neuronal survival. EBioMedicine. (2019) 40:595–604. doi: 10.1016/j.ebiom.2019.01.041
156. Seida I, Alrais M, Seida R, Alwani A, Kiyak Z, Elsalti A, et al. Autoimmune/inflammatory syndrome induced by adjuvants (ASIA): past, present, and future implications. Clin Exp Immunol. (2023) 213:87–101. doi: 10.1093/CEI/UXAD033
157. Guimarães LE, Baker B, Perricone C, and Shoenfeld Y. Vaccines, adjuvants and autoimmunity. Pharmacol Res. (2015) 100:190–209. doi: 10.1016/J.PHRS.2015.08.003
158. Verhagen J, Yusuf N, Smith EL, Whettlock EM, Naran K, Arif S, et al. Proinsulin peptide promotes autoimmune diabetes in a novel HLA-DR3-DQ2-transgenic murine model of spontaneous disease. Diabetologia. (2019) 62:2252–61. doi: 10.1007/S00125-019-04994-8/FIGURES/5
159. Nabozny GH, Baisch JM, Cheng S, Cosgrove D, Griffiths MM, Luthra HS, et al. HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: a novel model for human polyarthritis. J Exp Med. (1996) 183:27–37. doi: 10.1084/JEM.183.1.27
160. Mustelin T and Andrade F. Autoimmunity: the neoantigen hypothesis. Front Immunol. (2024) 15:1432985. doi: 10.3389/FIMMU.2024.1432985
161. Rijkers GT, Weterings N, Obregon-Henao A, Lepolder M, Dutt TS, van Overveld FJ, et al. Antigen presentation of mRNA-based and virus-vectored SARS-coV-2 vaccines. Vaccines. (2021) 9:848. doi: 10.3390/VACCINES9080848
162. Boros LG, Kyriakopoulos AM, Brogna C, Piscopo M, McCullough PA, and Seneff S. Long-lasting, biochemically modified mRNA, and its frameshifted recombinant spike proteins in human tissues and circulation after COVID-19 vaccination. Pharmacol Res Perspect. (2024) 12:e1218. doi: 10.1002/PRP2.1218
163. Samaan P, Korosec CS, Budylowski P, Chau SLL, Pasculescu A, Qi F, et al. mRNA vaccine-induced SARS-CoV-2 spike-specific IFN-γ and IL-2 T-cell responses are predictive of serological neutralization and are transiently enhanced by pre-existing cross-reactive immunity. J Virol. (2025) 99:e0168524. doi: 10.1128/JVI.01685-24/SUPPL_FILE/JVI.01685-24-S0001.DOCX
164. Rodríguez Y, Rojas M, Beltrán S, Polo F, Camacho-Domínguez L, Morales SD, et al. Autoimmune and autoinflammatory conditions after COVID-19 vaccination. New Case Rep updated literature review. J Autoimmun. (2022) 132:102898. doi: 10.1016/J.JAUT.2022.102898
165. Camacho-Domínguez L, Rodríguez Y, Polo F, Restrepo Gutierrez JC, Zapata E, Rojas M, et al. COVID-19 vaccine and autoimmunity. A new case of autoimmune hepatitis and review of the literature. J Transl Autoimmun. (2022) 5:100140. doi: 10.1016/J.JTAUTO.2022.100140
166. Astbury S, Reynolds CJ, Butler DK, Muñoz-Sandoval DC, Lin KM, Pieper FP, et al. HLA-DR polymorphism in SARS-CoV-2 infection and susceptibility to symptomatic COVID-19. Immunology. (2022) 166:68–77. doi: 10.1111/IMM.13450
167. Xie J, Mothe B, Alcalde Herraiz M, Li C, Xu Y, Jödicke AM, et al. Relationship between HLA genetic variations, COVID-19 vaccine antibody response, and risk of breakthrough outcomes. Nat Commun. (2024) 15:1–11. doi: 10.1038/s41467-024-48339-5
168. Balar PC and Chavda VP. Nonreplicating viral vectors for vaccine delivery. In: Viral Vectors for Vaccine Delivery. (Balar PC, Chavda VP) (2025), 125–47. doi: 10.1002/9781394271566.CH5
169. Provine NM and Klenerman P. Adenovirus vector and mRNA vaccines: Mechanisms regulating their immunogenicity. Eur J Immunol. (2023) 53:2250022. doi: 10.1002/EJI.202250022
170. Tatsis N, Fitzgerald JC, Reyes-Sandoval A, Harris-McCoy KC, Hensley SE, Zhou D, et al. Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines. Blood. (2007) 110:1916–23. doi: 10.1182/BLOOD-2007-02-062117
171. Byrnes AP, MacLaren RE, and Chariton HM. Immunological instability of persistent adenovirus vectors in the brain: peripheral exposure to vector leads to renewed inflammation, reduced gene expression, and demyelination. J Neurosci. (1996) 16:3045–55. doi: 10.1523/JNEUROSCI.16-09-03045.1996
172. Netea MG and Joosten LAB. Beyond adaptive immunity: induction of trained immunity by COVID-19 adenoviral vaccines. J Clin Invest. (2023) 133:e166467. doi: 10.1172/JCI166467
173. Singhal V and Gupta S. Post-COVID-19 vaccination and thyrotoxicosis (ASIA syndrome): single-centre experience from India with review of literature. Indian J Endocrinol Metab. (2023) 27:524–9. doi: 10.4103/IJEM.IJEM_202_23
174. Jara LJ, Vera-Lastra O, Mahroum N, Pineda C, and Shoenfeld Y. Autoimmune post-COVID vaccine syndromes: does the spectrum of autoimmune/inflammatory syndrome expand? Clin Rheumatol. (2022) 41:1603. doi: 10.1007/S10067-022-06149-4
175. Ghattas M, Dwivedi G, Lavertu M, and Alameh MG. Vaccine technologies and platforms for infectious diseases: current progress, challenges, and opportunities. Vaccines. (2021) 9:1490. doi: 10.3390/VACCINES9121490
176. Walker WT and Faust SN. Monovalent inactivated split-virion AS03-adjuvanted pandemic influenza A (H1N1) vaccine. Expert Rev Vaccines. (2010) 9:1385–98. doi: 10.1586/ERV.10.141
177. Buonocore SM and van der Most RG. Narcolepsy and H1N1 influenza immunology a decade later: What have we learned? Front Immunol. (2022) 13:902840/ENDNOTE. doi: 10.3389/FIMMU.2022.902840/ENDNOTE
178. Giarola-Silva S, Coelho-dos-Reis JGA, Mourão MM, Campi-Azevedo AC, Nakagaki Silva EE, Luiza-Silva M, et al. Distinct patterns of cellular immune response elicited by influenza non-adjuvanted and AS03-adjuvanted monovalent H1N1(pdm09) vaccine. Antiviral Res. (2017) 144:70–82. doi: 10.1016/J.ANTIVIRAL.2017.05.009
179. Shoenfeld Y and Agmon-Levin N. ‘ASIA’ – Autoimmune/inflammatory syndrome induced by adjuvants. J Autoimmun. (2011) 36:4–8. doi: 10.1016/J.JAUT.2010.07.003
180. Pulendran B S, Arunachalam P, and O’Hagan DT. Emerging concepts in the science of vaccine adjuvants. Nat Rev Drug Discov. (2021) 20:454–75. doi: 10.1038/S41573-021-00163-Y;SUBJMETA=250,590,631;KWRD=IMMUNOLOGY,VACCINES
181. Su Z, Boucetta H, Shao J, Huang J, Wang R, Shen A, et al. Next-generation aluminum adjuvants: Immunomodulatory layered double hydroxide NanoAlum reengineered from first-line drugs. Acta Pharm Sin B. (2024) 14:4665–82. doi: 10.1016/J.APSB.2024.09.012
182. Mohr E, Cunningham AF, Toellner KM, Bobat S, Coughlan RE, Bird RA, et al. IFN-γ produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine. Proc Natl Acad Sci U.S.A. (2010) 107:17292–7. doi: 10.1073/PNAS.1004879107/SUPPL_FILE/PNAS.201004879SI.PDF
183. Cohen Tervaert JW, Martinez-Lavin M, Jara LJ, Halpert G, Watad A, Amital H, et al. Autoimmune/inflammatory syndrome induced by adjuvants (ASIA) in 2023. Autoimmun Rev. (2023) 22:103287. doi: 10.1016/J.AUTREV.2023.103287
184. Borba V, Malkova A, Basantsova N, Halpert G, Andreoli L, Tincani A, et al. Classical examples of the concept of the ASIA syndrome. Biomolecules. (2020) 10:1–17. doi: 10.3390/BIOM10101436
185. Kuroda Y, Nacionales DC, Akaogi J, Reeves WH, and Satoh M. Autoimmunity induced by adjuvant hydrocarbon oil components of vaccine. Biomedicine Pharmacotherapy. (2004) 58:325–37. doi: 10.1016/J.BIOPHA.2004.04.009
186. Farrugia M and Baron B. The role of toll-like receptors in autoimmune diseases through failure of the self-recognition mechanism. Int J Inflam. (2017) 2017:8391230. doi: 10.1155/2017/8391230
187. Thomas CE, Schiedner G, Kochanek S, Castro MG, and Löwenstein PR. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: Toward realistic long-term neurological gene therapy for chronic diseases. Proc Natl Acad Sci U.S.A. (2000) 97:7482–7. doi: 10.1073/PNAS.120474397/SUPPL_FILE/4743TABLE3.HTML
188. Mansour K, Chadli Z, Ghachem I, Ben FN, Ben RH, Ben FN, et al. Seronegative acute encephalitis following COVID-19 vaccines: a case series of an overlooked diagnosis with literature review. Eur J Clin Pharmacol. (2023) 79:975–87. doi: 10.1007/S00228-023-03510-7/TABLES/2
189. Trontzas IP, Kyriakoulis KG, Vathiotis IA, Syrigos A, Kounadis G, Siasiakou S, et al. Vaccine-related autoimmune hepatitis: emerging association with SARS-coV-2 vaccination or coincidence? Vaccines (Basel). (2022) 10:2073. doi: 10.3390/VACCINES10122073/S1
190. Krishna MT, Subramanian A, Adderley NJ, Zemedikun DT, Gkoutos GV, and Nirantharakumar K. Allergic diseases and long-term risk of autoimmune disorders: longitudinal cohort study and cluster analysis. Eur Respir J. (2019) 54:1900476. doi: 10.1183/13993003.00476-2019
191. Bartůňková J, Kayserová J, and Shoenfeld Y. Allergy and autoimmunity: Parallels and dissimilarity: The Yin and Yang of Immunopathology. Autoimmun Rev. (2009) 8:302–8. doi: 10.1016/J.AUTREV.2008.09.004
192. Aldakheel FM. Allergic diseases: A comprehensive review on risk factors, immunological mechanisms, link with COVID-19, potential treatments, and role of allergen bioinformatics. Int J Environ Res Public Health. (2021) 18:12105. doi: 10.3390/IJERPH182212105
193. Utsugi M. Antigen presenting cells in allergy. Nippon Rinsho Japanese J Clin Med. (2009) 67:2076–81. doi: 10.1067/MAI.2001.117457/ASSET/EDDFD34E-4E01-4F7F-9472-9624CD9B0DE0/MAIN.ASSETS/GR5.JPG
194. Wang J, Zhou Y, Zhang H, Hu L, Liu J, Wang L, et al. Pathogenesis of allergic diseases and implications for therapeutic interventions. Signal Transduction Targeted Ther. (2023) 8:1–30. doi: 10.1038/s41392-023-01344-4
195. Howell WM and Holgate ST. HLA genetics and allergic disease. Thorax. (1995) 50:815–8. doi: 10.1136/THX.50.8.815
196. Chauhan B, Hutcheson PS, Slavin RG, and Bellone CJ. MHC restriction in allergic bronchopulmonary aspergillosis. Front Bioscience. (2003) 8:140–8. doi: 10.2741/971/PDF
197. Vercelli D. Remembrance of things past: HLA genes come back on the allergy stage. J Allergy Clin Immunol. (2012) 129:846–7. doi: 10.1016/j.jaci.2011.12.998
198. Dolence J, Smeekens J, Khodoun M, Germundson DL, Nookala S, Smith NA, et al. HLA-II alleles influence physical and behavioral responses to a whey allergen in a transgenic mouse model of cow’s milk allergy. Front Allergy. (2022) 3:870513. doi: 10.3389/FALGY.2022.870513
199. Savilahti EM, Ilonen J, Kiviniemi M, Saarinen KM, Vaarala O, and Savilahti E. Human leukocyte antigen (DR1)-DQB1*0501 and (DR15)-DQB1*0602 haplotypes are associated with humoral responses to early food allergens in children. Int Arch Allergy Immunol. (2010) 152:169–77. doi: 10.1159/000265538
200. Horst AK, Kumashie KG, Neumann K, Diehl L, and Tiegs G. Antigen presentation, autoantibody production, and therapeutic targets in autoimmune liver disease. Cell Mol Immunol. (2021) 18:92–111. doi: 10.1038/S41423-020-00568-6;SUBJMETA=249,250,38,631;KWRD=AUTOIMMUNITY,IMMUNOLOGICAL+DISORDERS
201. Chavda VP, Feehan J, and Apostolopoulos V. Inflammation: the cause of all diseases. Cells 2024 Vol 13 Page. (1906) 2024:13:1906. doi: 10.3390/CELLS13221906
202. Lipski DA, Dewispelaere R, Foucart V, Caspers LE, DeFrance M, Bruyns C, et al. MHC class II expression and potential antigen-presenting cells in the retina during experimental autoimmune uveitis. J Neuroinflamm. (2017) 14:1–22. doi: 10.1186/S12974-017-0915-5/TABLES/4
203. Wang MY, Qiao Y, Wei SJ, Su ZL, and Lu HY. MHC class II of different non-professional antigen-presenting cells mediate multiple effects of crosstalk with CD4+T cells in lung diseases. Front Med (Lausanne). (2025) 12:1388814/BIBTEX. doi: 10.3389/FMED.2025.1388814/BIBTEX
204. Sundaresan B, Shirafkan F, Ripperger K, and Rattay K. The role of viral infections in the onset of autoimmune diseases. Viruses. (2023) 15:782. doi: 10.3390/V15030782
205. Muhsen A, Hertz A, and Amital H. The association between physical trauma and autoimmune articular and dermatological disorders. Autoimmun Rev. (2025) 24:103711. doi: 10.1016/J.AUTREV.2024.103711
206. Schett G and Neurath MF. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun. (2018) 9:1–8. doi: 10.1038/S41467-018-05800-6;SUBJMETA=127,250,256,4020,4023,631,692;KWRD=CYTOKINES,GASTROENTEROLOGY,INFLAMMATION,RHEUMATOLOGY
207. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
208. Catrysse L and van Loo G. Inflammation and the metabolic syndrome: the tissue-specific functions of NF-κB. Trends Cell Biol. (2017) 27:417–29. doi: 10.1016/J.TCB.2017.01.006/ASSET/59470FC9-5AF4-4A4A-B4B5-12E1CB58A24E/MAIN.ASSETS/GR1.SML
209. Lumeng CN, Bodzin JL, and Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
210. Morris DL, Cho KW, DelProposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G, et al. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes. (2013) 62:2762–72. doi: 10.2337/DB12-1404
211. Deng T, Lyon CJ, Minze LJ, Lin J, Zou J, Liu JZ, et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab. (2013) 17:411–22. doi: 10.1016/J.CMET.2013.02.009/ATTACHMENT/8D322151-F4F1-4D3C-9F8C-91B905B6D1D1/MMC1.PDF
212. Hedström AK, Bomfim IL, Barcellos L, GianFrancesco M, Schaefer C, Kockum I, et al. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology. (2014) 82:865–72. doi: 10.1212/WNL.0000000000000203
213. Hjort R, Löfvenborg JE, Ahlqvist E, Alfredsson L, Andersson T, Grill V, et al. Interaction between overweight and genotypes of HLA, TCF7L2, and FTO in relation to the risk of latent autoimmune diabetes in adults and type 2 diabetes. J Clin Endocrinol Metab. (2019) 104:4815–26. doi: 10.1210/JC.2019-00183
214. Oliver KM, Taylor CT, and Cummins EP. Hypoxia. Regulation of NFκB signalling during inflammation: The role of hydroxylases. . Arthritis Res Ther. (2009) 11:1–8. doi: 10.1186/AR2575/METRICS
215. Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, and Yezierski RP. Traumatic spinal cord injury induces nuclear factor-κB activation. J Neurosci. (1998) 18:3251–60. doi: 10.1523/JNEUROSCI.18-09-03251.1998
216. Fischer H, Hedlund G, Kalland T, Sjögren HO, and Dohlsten M. Independent regulation of IFN-gamma and tumor necrosis factor by IL-1 in human T helper cells. J Immunol. (1990) 145:3767–72. doi: 10.4049/jimmunol.145.11.3767
217. Almishri W, Santodomingo-Garzon T, Le T, Stack D, Mody CH, and Swain MG. TNFα Augments cytokine-induced NK cell IFNγ Production through TNFR2. J Innate Immun. (2016) 8:617–29. doi: 10.1159/000448077
218. Lee YJ and Benveniste EN. Stat1 alpha expression is involved in IFN-gamma induction of the class II transactivator and class II MHC genes. J Immunol. (1996) 157:1559–68. doi: 10.4049/jimmunol.157.4.1559
219. Hu X and Ivashkiv LB. Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity. (2009) 31:539–50. doi: 10.1016/j.immuni.2009.09.002
220. Fu Y, Xiang Y, Wang Y, Liu Z, Yang D, Zha J, et al. The STAT1/HMGB1/NF-κB pathway in chronic inflammation and kidney injury after cisplatin exposure. Theranostics. (2023) 13:2757–73. doi: 10.7150/THNO.81406
221. Publishing Asia B and Yoshimura A. Signal transduction of inflammatory cytokines and tumor development. Cancer Sci. (2006) 97:439–47. doi: 10.1111/J.1349-7006.2006.00197.X
222. Anam K, Amare M, Naik S, Szabo KA, and Davis TA. Severe tissue trauma triggers the autoimmune state systemic lupus erythematosus in the MRL/++ lupus-prone mouse. Lupus. (2009) 18:318–31. doi: 10.1177/0961203308097479
223. Johansson E, Alfredsson L, Strid P, Kockum I, Olsson T, and Hedström AK. Head trauma results in manyfold increased risk of multiple sclerosis in genetically susceptible individuals. J Neurol Neurosurg Psychiatry. (2024) 95:554. doi: 10.1136/JNNP-2023-332643
224. Evans PC, Smith S, Hirschfield G, Rigopoulou E, Wreghitt TG, Wight DGD, et al. Recipient HLA-DR3, tumour necrosis factor-α promoter allele-2 (tumour necrosis factor-2) and cytomegalovirus infection are interrelated risk factors for chronic rejection of liver grafts. J Hepatol. (2001) 34:711–5. doi: 10.1016/S0168-8278(00)00101-X
225. Alelign T, Ahmed MM, Bobosha K, Tadesse Y, Howe R, and Petros B. Kidney transplantation: the challenge of human leukocyte antigen and its therapeutic strategies. J Immunol Res. (2018) 2018:5986740. doi: 10.1155/2018/5986740
226. Ballou C, Barton F, Payne EH, Berney T, Villard J, Meier RPH, et al. Matching for HLA-DR excluding diabetogenic HLA-DR3 and HLA-DR4 predicts insulin independence after pancreatic islet transplantation. Front Immunol. (2023) 14:1110544/BIBTEX. doi: 10.3389/FIMMU.2023.1110544/BIBTEX
227. Durinovic-Belló I, Jelinek E, Schlosser M, Eiermann T, Boehm BO, Karges W, et al. Class III alleles at the insulin VNTR polymorphism are associated with regulatory T-cell responses to proinsulin epitopes in HLA-DR4, DQ8 individuals. Diabetes. (2005) 54:S18–24. doi: 10.2337/DIABETES.54.SUPPL_2.S18
228. Corona E, Dudley JT, and Butte AJ. Extreme evolutionary disparities seen in positive selection across seven complex diseases. PloS One. (2010) 5:e12236. doi: 10.1371/JOURNAL.PONE.0012236
229. Sams A and Hawks J. Celiac disease as a model for the evolution of multifactorial disease in humans. Hum Biol. (2014) 86:19–36. doi: 10.3378/027.086.0102
230. Fujinami RS, Von Herrath MG, Christen U, and Whitton JL. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin Microbiol Rev. (2006) 19:80–94. doi: 10.1128/CMR.19.1.80-94.2006/ASSET/A5E8C342-5DE8-4E39-A77B-80040660F270/ASSETS/GRAPHIC/ZCM0010621610003.JPEG
231. Komaroff AL and Lipkin WI. ME/CFS and Long COVID share similar symptoms and biological abnormalities: road map to the literature. Front Med (Lausanne). (2023) 10:1187163. doi: 10.3389/FMED.2023.1187163
232. Rasa-Dzelzkaleja S, Krumina A, Capenko S, Nora-Krukle Z, Gravelsina S, Vilmane A, et al. The persistent viral infections in the development and severity of myalgic encephalomyelitis/chronic fatigue syndrome. J Transl Med. (2023) 21:1–12. doi: 10.1186/S12967-023-03887-0/FIGURES/8
233. Rasa S, Nora-Krukle Z, Henning N, Eliassen E, Shikova E, Harrer T, et al. Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J Transl Med. (2018) 16:268. doi: 10.1186/S12967-018-1644-Y
234. Yin K, Peluso MJ, Luo X, Thomas R, Shin MG, Neidleman J, et al. Long COVID manifests with T cell dysregulation, inflammation and an uncoordinated adaptive immune response to SARS-CoV-2. Nat Immunol. (2024) 25:218–25. doi: 10.1038/S41590-023-01724-6;SUBJMETA=250,2514,255,326,4130,596,631;KWRD=SARS-COV-2,VIRAL+INFECTION
235. Blomberg J, Gottfries C-G, Elfaitouri A, Rizwan M, and Rosén A. Infection elicited autoimmunity and myalgic encephalomyelitis/chronic fatigue syndrome: an explanatory model. Front Immunol. (2018) 9:229. doi: 10.3389/FIMMU.2018.00229
236. Arron HE, Marsh BD, Kell DB, Khan MA, Jaeger BR, and Pretorius E. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: the biology of a neglected disease. Front Immunol. (2024) 15:1386607/XML. doi: 10.3389/FIMMU.2024.1386607/XML
237. Ollila HM, Sharon E, Lin L, Sinnott-Armstrong N, Ambati A, Yogeshwar SM, et al. Narcolepsy risk loci outline role of T cell autoimmunity and infectious triggers in narcolepsy. Nat Commun. (2023) 14:1–13. doi: 10.1038/S41467-023-36120-Z;TECHMETA=43,45;SUBJMETA=205,208,2138,4017,631,692;KWRD=GENETICS,GENOME-WIDE+ASSOCIATION+STUDIES,MOLECULAR+MEDICINE
238. Seida I, Seida R, Elsalti A, and Mahroum N. Vaccines and autoimmunity—From side effects to ASIA syndrome. Med (B Aires). (2023) 59:364. doi: 10.3390/MEDICINA59020364
239. Soriano A, Nesher G, and Shoenfeld Y. Predicting post-vaccination autoimmunity: Who might be at risk? Pharmacol Res. (2015) 92:18–22. doi: 10.1016/J.PHRS.2014.08.002
240. Krishna BA, Lim EY, Metaxaki M, Jackson S, Mactavous L, Lyons PA, et al. Spontaneous, persistent, T cell–dependent IFN-γ release in patients who progress to Long Covid. Sci Adv. (2024) 10:9379. doi: 10.1126/SCIADV.ADI9379/SUPPL_FILE/SCIADV.ADI9379_SM.PDF
241. Santopaolo M, Gregorova M, Hamilton F, Arnold D, Long A, Lacey A, et al. Prolonged T-cell activation and long COVID symptoms independently associate with severe COVID-19 at 3 months. Elife. (2023) 12:e85009. doi: 10.7554/ELIFE.85009
242. Yang T, Yang Y, Wang D, Li C, Qu Y, Guo J, et al. The clinical value of cytokines in chronic fatigue syndrome. J Transl Med. (2019) 17:213. doi: 10.1186/S12967-019-1948-6
243. Maes M, Bosmans E, and Kubera M. Increased expression of activation antigens on CD8+ T lymphocytes in Myalgic Encephalomyelitis/chronic fatigue syndrome: inverse associations with lowered CD19+ expression and CD4+/CD8+ ratio, but no associations with (auto)immune, leaky gut, oxidative and nitrosative stress biomarkers. Neuro Endocrinol Lett. (2015) 36:439–46.
244. Eaton-Fitch N, Rudd P, Er T, Hool L, Herrero L, and Marshall-Gradisnik S. Immune exhaustion in ME/CFS and long COVID. JCI Insight. (2024) 9(20):e183810. doi: 10.1172/JCI.INSIGHT.183810
245. Loebel M, Grabowski P, Heidecke H, Bauer S, Hanitsch LG, Wittke K, et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav Immun. (2016) 52:32–9. doi: 10.1016/J.BBI.2015.09.013
246. Vuorela A, Freitag TL, Leskinen K, Pessa H, Härkönen T, Stracenski I, et al. Enhanced influenza A H1N1 T cell epitope recognition and cross-reactivity to protein-O-mannosyltransferase 1 in Pandemrix-associated narcolepsy type 1. Nat Commun. (2021) 12:17. doi: 10.1038/s41467-021-22637-8
247. Halma M and Varon J. Breaking the silence: Recognizing post-vaccination syndrome. Heliyon. (2025) 11:e43478. doi: 10.1016/J.HELIYON.2025.E43478
248. Smith J, Fritz EL, Kerr JR, Cleare AJ, Wessely S, and Mattey DL. Association of chronic fatigue syndrome with human leucocyte antigen class II alleles. J Clin Pathol. (2005) 58:860–3. doi: 10.1136/jcp.2004.022681
249. Keller RH, Lane JL, Klimas N, Reiter WM, Fletcher MA, van Riel F, et al. Association between HLA class II antigens and the chronic fatigue immune dysfunction syndrome. Clin Infect Dis. (1994) 18 Suppl 1:S154–6. doi: 10.1093/clinids/18.supplement_1.s154
250. Wang T, Yin J, Miller AH, and Xiao C. A systematic review of the association between fatigue and genetic polymorphisms. Brain Behav Immun. (2017) 62:230. doi: 10.1016/J.BBI.2017.01.007
251. Arango MT, Perricone C, Kivity S, Cipriano E, Ceccarelli F, Valesini G, et al. HLA-DRB1 the notorious gene in the mosaic of autoimmunity. Immunol Res. (2017) 65:82–98. doi: 10.1007/S12026-016-8817-7
252. Mitchell LA, Tingle AJ, MacWilliam L, Home C, Keown P, Gaur LK, et al. HLA-DR class II associations with rubella vaccine-induced joint manifestations. J Infect Dis. (1998) 177:5–12. doi: 10.1086/513807
253. Georgopoulos A, James L, and Peterson P. Human leukocyte antigen (HLA) at the root of persistent antigens and long COVID. J Immunol Sci. (2025) 9:1–3. doi: 10.29245/2578-3009/2025/1.1257
254. Gutiérrez-Bautista JF, Sampedro A, Gómez-Vicente E, Rodríguez-Granger J, Reguera JA, Cobo F, et al. HLA class II polymorphism and humoral immunity induced by the SARS-coV-2 mRNA-1273 vaccine. Vaccines (Basel). (2022) 10:402. doi: 10.3390/VACCINES10030402/S1
255. Mentzer AJ, O’Connor D, Bibi S, Chelysheva I, Clutterbuck EA, Demissie T, et al. Human leukocyte antigen alleles associate with COVID-19 vaccine immunogenicity and risk of breakthrough infection. Nat Med. (2023) 29:1 2022. doi: 10.1038/s41591-022-02078-6
256. Ragone C, Meola S, Fiorillo PC, Penta R, Auriemma L, Tornesello ML, et al. HLA does not impact on short-medium-term antibody response to preventive anti-SARS-cov-2 vaccine. Front Immunol. (2021) 12:734689. doi: 10.3389/FIMMU.2021.734689
257. Bertinetto FE, Magistroni P, Mazzola GA, Costa C, Elena G, Alizzi S, et al. The humoral and cellular response to mRNA SARS-CoV-2 vaccine is influenced by HLA polymorphisms. HLA. (2023) 102:301–15. doi: 10.1111/TAN.15049
258. Hajeer A, Jawdat D, Massadeh S, Aljawini N, Abedalthagafi MS, Arabi YM, et al. Association between human leukocyte antigen alleles and COVID-19 disease severity. J Infect Public Health. (2024) 17:102498. doi: 10.1016/J.JIPH.2024.102498
259. Dani M, Dirksen A, Taraborrelli P, Torocastro M, Panagopoulos D, Sutton R, et al. Autonomic dysfunction in ‘long COVID’: rationale, physiology and management strategies. Clin Med. (2021) 21:e63–7. doi: 10.7861/CLINMED.2020-0896
260. Zhang YD and Wang LN. Research progress in the treatment of chronic fatigue syndrome through interventions targeting the hypothalamus-pituitary-adrenal axis. Front Endocrinol (Lausanne). (2024) 15:1373748. doi: 10.3389/FENDO.2024.1373748
261. Ruiz-Pablos M, Paiva B, and Zabaleta A. Hypocortisolemic ASIA: a vaccine- and chronic infection-induced syndrome behind the origin of long COVID and myalgic encephalomyelitis. Front Immunol. (2024) 15:1422940/PDF. doi: 10.3389/FIMMU.2024.1422940/PDF
262. Kanczkowski W, Beuschlein F, and Bornstein SR. Is there a role for the adrenal glands in long COVID? Nat Rev Endocrinol. (2022) 18:8 2022. doi: 10.1038/s41574-022-00700-8
263. Yavropoulou MP, Tsokos GC, Chrousos GP, and Sfikakis PP. Protracted stress-induced hypocortisolemia may account for the clinical and immune manifestations of Long COVID. Clin Immunol. (2022) 245:109133. doi: 10.1016/J.CLIM.2022.109133
264. Proal AD, Aleman S, Bomsel M, Brodin P, Buggert M, Cherry S, et al. Targeting the SARS-CoV-2 reservoir in long COVID. Lancet Infect Dis. (2025) 25:e294–306. doi: 10.1016/S1473-3099(24)00769-2
265. Underhill JA, Mahalingam M, Peakman M, and Wessely S. Lack of association between HLA genotype and chronic fatigue syndrome. Eur J Immunogenetics. (2001) 28:425–8. doi: 10.1046/j.1365-2370.2001.00235.x
266. Achleitner M, Steenblock C, Dänhardt J, Jarzebska N, Kardashi R, Kanczkowski W, et al. Clinical improvement of Long-COVID is associated with reduction in autoantibodies, lipids, and inflammation following therapeutic apheresis. Mol Psychiatry. (2023) 28:2872–7. doi: 10.1038/S41380-023-02084-1;TECHMETA
267. Bynke A, Julin P, Gottfries CG, Heidecke H, Scheibenbogen C, and Bergquist J. Autoantibodies to beta-adrenergic and muscarinic cholinergic receptors in Myalgic Encephalomyelitis (ME) patients – A validation study in plasma and cerebrospinal fluid from two Swedish cohorts. Brain Behav Immun Health. (2020) 7:100107. doi: 10.1016/J.BBIH.2020.100107
268. Liberman AC, Budziñski ML, Sokn C, Gobbini RP, Steininger A, and Arzt E. Regulatory and mechanistic actions of glucocorticoids on T and inflammatory cells. Front Endocrinol (Lausanne). (2018) 9:235/FULL. doi: 10.3389/FENDO.2018.00235/FULL
269. Menotti S, di Filippo L, Terenzi U, Chiloiro S, and De Marinis L. Hypophysitis in COVID-19: a systematic review. Pituitary. (2024) 27:874–88. doi: 10.1007/S11102-024-01462-4/METRICS
270. Taieb A, Nassim BHS, Asma G, Jabeur M, Ghada S, and Asma BA. The growing understanding of the pituitary implication in the pathogenesis of long COVID-19 syndrome: A narrative review. Adv Respir Med. (2024) 92:96–109. doi: 10.3390/ARM92010013
271. Alzahrani AS, Mukhtar N, Aljomaiah A, Aljamei H, Bakhsh A, AlSudani N, et al. The impact of COVID-19 viral infection on the hypothalamic-pituitary-adrenal axis. Endocrine Pract. (2021) 27:83–9. doi: 10.1016/j.eprac.2020.10.014
272. Tate W, Walker M, Sweetman E, Helliwell A, Peppercorn K, Edgar C, et al. Molecular mechanisms of neuroinflammation in ME/CFS and long COVID to sustain disease and promote relapses. Front Neurol. (2022) 13:877772/BIBTEX. doi: 10.3389/FNEUR.2022.877772/BIBTEX
273. Tomas C, Newton J, and Watson S. A review of hypothalamic-pituitary-adrenal axis function in chronic fatigue syndrome. ISRN Neurosci. (2013) 2013:1–8. doi: 10.1155/2013/784520
274. Menotti S, Giampietro A, Raia S, Veleno M, Angelini F, Tartaglione T, et al. Unveiling the etiopathogenic spectrum of hypophysitis: A narrative review. J Pers Med. (2023) 13:1210. doi: 10.3390/JPM13081210
275. Yamamoto S, Ouchi Y, Nakatsuka D, Tahara T, Mizuno K, Tajima S, et al. Reduction of [11C](+)3-MPB binding in brain of chronic fatigue syndrome with serum autoantibody against muscarinic cholinergic receptor. PloS One. (2012) 7:e51515. doi: 10.1371/JOURNAL.PONE.0051515
276. Gravelsina S, Vilmane A, Svirskis S, Rasa-Dzelzkaleja S, Nora-Krukle Z, Vecvagare K, et al. Biomarkers in the diagnostic algorithm of myalgic encephalomyelitis/chronic fatigue syndrome. Front Immunol. (2022) 13:928945. doi: 10.3389/FIMMU.2022.928945
277. Seibert FS, Stervbo U, Wiemers L, Skrzypczyk S, Hogeweg M, Bertram S, et al. Severity of neurological Long-COVID symptoms correlates with increased level of autoantibodies targeting vasoregulatory and autonomic nervous system receptors. Autoimmun Rev. (2023) 22:103445. doi: 10.1016/J.AUTREV.2023.103445
278. Pavlov VA and Tracey KJ. The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat Rev Endocrinol. (2012) 8:743–54. doi: 10.1038/NRENDO.2012.189
279. Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. (2007) 117:289–96. doi: 10.1172/JCI30555
280. Palma JA, Gupta A, Sierra S, Gomes I, Balgobin B, Norcliffe-Kaufmann L, et al. Autoantibodies blocking M3 muscarinic receptors cause postganglionic cholinergic dysautonomia. Ann Neurol. (2020) 88:1237–43. doi: 10.1002/ANA.25882
281. Teven S, Ernino V, Ow HAL, Ealey ODF, Ohn J, Tewart DS, et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. New Engl J Med. (2000) 343:847–55. doi: 10.1056/NEJM200009213431204
282. McKeon A, Lennon VA, Lachance DH, Fealey RD, and Pittock SJ. Ganglionic acetylcholine receptor autoantibody: oncological, neurological, and serological accompaniments. Arch Neurol. (2009) 66:735–41. doi: 10.1001/ARCHNEUROL.2009.78
283. Abe S, Tsuboi H, Kudo H, Asashima H, Ono Y, Honda F, et al. M3 muscarinic acetylcholine receptor–reactive Th17 cells in primary Sjögren’s syndrome. JCI Insight. (2020) 5:e135982. doi: 10.1172/JCI.INSIGHT.135982
284. Mona M, Mondello S, Hyon JY, Saleh W, Han K, Lee HJ, et al. Clinical usefulness of anti-muscarinic type 3 receptor autoantibodies in patients with primary Sjögren’s syndrome. Clin Exp Rheumatol. (2021) 39:795–803. doi: 10.55563/clinexprheumatol/gy6udz
285. Fedorowski A and Sutton R. Autonomic dysfunction and postural orthostatic tachycardia syndrome in post-acute COVID-19 syndrome. Nat Rev Cardiol. (2023) 20:281–2. doi: 10.1038/S41569-023-00842-W;SUBJMETA
286. Schlömer E, Stein E, Kedor C, Rust R, Brock A, Wittke K, et al. Pyridostigmine improves hand grip strength in patients with myalgic encephalomyelitis/chronic fatigue syndrome. Front Neurosci. (2025) 19:1637838. doi: 10.3389/FNINS.2025.1637838
287. Kanjwal K, Karabin B, Sheikh M, Elmer L, Kanjwal Y, Saeed B, et al. Pyridostigmine in the treatment of postural orthostatic tachycardia: a single-center experience. Pacing Clin Electrophysiol. (2011) 34:750–5. doi: 10.1111/J.1540-8159.2011.03047.X
288. Kichloo A, Aljadah M, Grubb B, and Kanjwal K. Management of postural orthostatic tachycardia syndrome in the absence of randomized controlled trials. J Innov Cardiac Rhythm Manage. (2021) 12:4607–12. doi: 10.19102/ICRM.2021.120705
289. Joseph P, Pari R, Miller S, Warren A, Stovall MC, Squires J, et al. Neurovascular dysregulation and acute exercise intolerance in myalgic encephalomyelitis/chronic fatigue syndrome: A randomized, placebo-controlled trial of pyridostigmine. Chest. (2022) 162:1116–26. doi: 10.1016/j.chest.2022.04.146
290. Wiest R, Moleda L, Zietz B, Hellerbrand C, Schölmerich J, and Straub R. Uncoupling of sympathetic nervous system and hypothalamic-pituitary-adrenal axis in cirrhosis. J Gastroenterol Hepatol. (2008) 23:1901–8. doi: 10.1111/J.1440-1746.2008.05456.X
291. Straub RH, Herfarth H, Falk W, Andus T, and Schölmerich J. Uncoupling of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis in inflammatory bowel disease? J Neuroimmunol. (2002) 126:116–25. doi: 10.1016/S0165-5728(02)00047-4
292. Mousa SA, Dehe L, Aboryag N, Shaqura M, Beyer A, Schäfer M, et al. Identification of glucocorticoid receptors as potential modulators of parasympathetic and sympathetic neurons within rat intracardiac ganglia. Front Neuroanat. (2022) 16:902738/BIBTEX. doi: 10.3389/FNANA.2022.902738/BIBTEX
293. Steere AC, Dwyer E, and Winchester R. Association of chronic lyme arthritis with HLA-DR4 and HLA-DR2 alleles. New Engl J Med. (1990) 323:219–23. doi: 10.1056/NEJM199007263230402
294. Steere AC, Klitz W, Drouin EE, Falk BA, Kwok WW, Nepom GT, et al. Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J Exp Med. (2006) 203:961–71. doi: 10.1084/JEM.20052471
295. Läderach F and Münz C. Epstein barr virus exploits genetic susceptibility to increase multiple sclerosis risk. Microorganisms. (2191) 2021:9:2191. doi: 10.3390/MICROORGANISMS9112191
296. Soldan SS and Lieberman PM. Epstein–Barr virus and multiple sclerosis. Nat Rev Microbiol. (2023) 21:51–64. doi: 10.1038/S41579-022-00770-5;SUBJMETA
297. Baazim H, Schweiger M, Moschinger M, Xu H, Scherer T, Popa A, et al. CD8+ T cells induce cachexia during chronic viral infection. Nat Immunol. (2019) 20:701. doi: 10.1038/S41590-019-0397-Y
298. Thakur A, Mikkelsen H, and Jungersen G. Intracellular pathogens: host immunity and microbial persistence strategies. J Immunol Res. (2019) 2019:1356540. doi: 10.1155/2019/1356540
299. Schradin C, Jaeggi AV, and Criscuolo F. Quick guide to evolutionary medicine in neuroimmunomodulation: why “Evolved for the benefit of the species” Is not a valid argument. Neuroimmunomodulation. (2024) 31:66–77. doi: 10.1159/000538294
300. Abadie V, Sollid LM, Barreiro LB, and Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol. (2011) 29:493–525. doi: 10.1146/ANNUREV-IMMUNOL-040210-092915/CITE/REFWORKS
301. Straub RH. Evolutionary medicine and chronic inflammatory state—known and new concepts in pathophysiology. J Mol Med (Berl). (2012) 90:523. doi: 10.1007/S00109-012-0861-8
302. Straub RH and Schradin C. Chronic inflammatory systemic diseases: An evolutionary trade-off between acutely beneficial but chronically harmful programs. Evol Med Public Health. (2016) 2016:37–51. doi: 10.1093/EMPH/EOW001
303. Nordin J, Ameur A, Lindblad-Toh K, Gyllensten U, and Meadows JRS. SweHLA: the high confidence HLA typing bio-resource drawn from 1000 Swedish genomes. Eur J Hum Genet. (2020) 28:627–35. doi: 10.1038/S41431-019-0559-2;SUBJMETA=114,208,248,250,631;KWRD=COMPUTATIONAL+BIOLOGY+AND+BIOINFORMATICS,IMMUNOGENETICS
304. Austad SN and Hoffman JM. Is antagonistic pleiotropy ubiquitous in aging biology? Evol Med Public Health. (2018) 2018:287–94. doi: 10.1093/EMPH/EOY033
305. Crespi BJ and Go MC. Diametrical diseases reflect evolutionary-genetic tradeoffs: Evidence from psychiatry, neurology, rheumatology, oncology and immunology. Evol Med Public Health. (2015) 2015:216. doi: 10.1093/EMPH/EOV021
306. Rook GAW, Lowry CA, and Raison CL. Microbial “Old Friends”, immunoregulation and stress resilience. Evol Med Public Health. (2013) 2013:46–64. doi: 10.1093/EMPH/EOT004
307. Semmes EC, Hurst JH, Walsh KM, and Permar SR. Cytomegalovirus as an immunomodulator across the lifespan. Curr Opin Virol. (2020) 44:112–20. doi: 10.1016/J.COVIRO.2020.07.013
308. Olivieri B, Betterle C, and Zanoni G. Vaccinations and autoimmune diseases. Vaccines. (2021) 9:815. doi: 10.3390/VACCINES9080815
309. Kunnumakkara AB, Shabnam B, Girisa S, Harsha C, Banik K, Devi TB, et al. Inflammation, nf-κb, and chronic diseases: How are they linked? Crit Rev Immunol. (2020) 40:1–39. doi: 10.1615/CRITREVIMMUNOL.2020033210
310. Fulop T, Larbi A, Pawelec G, Khalil A, Cohen AA, Hirokawa K, et al. Immunology of aging: the birth of inflammaging. Clin Rev Allergy Immunol. (2021) 64:2 2021. doi: 10.1007/S12016-021-08899-6
311. Sharma Y, Arora M, and Kumud B. The potential of immunomodulators in shaping the future of healthcare. . Discover Med. (2024) 1:1 2024. doi: 10.1007/S44337-024-00029-3
312. ElAbd H, Bacher P, Tholey A, Lenz TL, and Franke A. Challenges and opportunities in analyzing and modeling peptide presentation by HLA-II proteins. Front Immunol. (2023) 14:1107266/BIBTEX. doi: 10.3389/FIMMU.2023.1107266/BIBTEX
313. Shapiro IE and Bassani-Sternberg M. The impact of immunopeptidomics: From basic research to clinical implementation. Semin Immunol. (2023) 66:101727. doi: 10.1016/J.SMIM.2023.101727
314. Nilsson JB, Kaabinejadian S, Yari H, Kester MGD, van Balen P, Hildebrand WH, et al. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning. Sci Adv. (2023) 9:eadj6367. doi: 10.1126/SCIADV.ADJ6367
315. Nilsson JB and Nielsen M. The journey towards complete and accurate prediction of HLA antigen presentation. ImmunoInformatics. (2024) 13:100032. doi: 10.1016/J.IMMUNO.2024.100032
316. Dobson CS, Reich AN, Gaglione S, Smith BE, Kim EJ, Dong J, et al. Antigen identification and high-throughput interaction mapping by reprogramming viral entry. Nat Methods. (2022) 19:449–60. doi: 10.1038/s41592-022-01436-z
317. Kohlgruber AC, Dezfulian MH, Sie BM, Wang CI, Kula T, Laserson U, et al. High-throughput discovery of MHC class I- and II-restricted T cell epitopes using synthetic cellular circuits. Nat Biotechnol. (2024) 43:623–34. doi: 10.1038/s41587-024-02248-6
318. Sarkanen T, Alakuijala A, Julkunen I, and Partinen M. Narcolepsy associated with pandemrix vaccine. Curr Neurol Neurosci Rep. (2018) 18:1–10. doi: 10.1007/S11910-018-0851-5/METRICS
319. Jiang CY, Zhao L, Green MD, Ravishankar S, Towlerton AMH, Scott AJ, et al. Class II HLA-DRB4 is a predictive biomarker for survival following immunotherapy in metastatic non-small cell lung cancer. Sci Rep. (2024) 14:1–11. doi: 10.1038/s41598-023-48546-y
320. Abed A, Law N, Calapre L, Lo J, Bhat V, Bowyer S, et al. Human leucocyte antigen genotype association with the development of immune-related adverse events in patients with non-small cell lung cancer treated with single agent immunotherapy. Eur J Cancer. (2022) 172:98–106. doi: 10.1016/J.EJCA.2022.05.021
321. Chang JQ, Guo Y, Yuan WJ, Chen YM, Liu BW, Li WT, et al. HLA-DR+ Tumor cells show an association with a distinct immune microenvironment and CD8+ T-cell exhaustion in HBV-associated hepatocellular carcinoma. Advanced Sci. (2025) 12:e02979. doi: 10.1002/ADVS.202502979
322. Al-Motwee S, Jawdat D, Jehani GS, Anazi H, Shubaili A, Sutton P, et al. Association of HLA-DRB1*15 and HLA-DQB1* 06 with SLE in saudis. Ann Saudi Med. (2013) 33:229–34. doi: 10.5144/0256-4947.2013.229
323. Liphaus BL, Kiss MHB, and Goldberg AC. HLA-DRB1 alleles in juvenile-onset systemic lupus erythematosus: Renal histologic class correlations. Braz J Med Biol Res. (2007) 40:591–7. doi: 10.1590/S0100-879X2007000400019
324. Shimane K, Kochi Y, Suzuki A, Okada Y, Ishii T, Horita T, et al. An association analysis of HLA-DRB1 with systemic lupus erythematosus and rheumatoid arthritis in a Japanese population: effects of *09:01 allele on disease phenotypes. Rheumatology. (2013) 52:1172–82. doi: 10.1093/rheumatology/kes427
325. Morris DL, Taylor KE, Fernando MMA, Nititham J, Alarcón-Riquelme ME, Barcellos LF, et al. Unraveling multiple MHC gene associations with systemic lupus erythematosus: Model choice indicates a role for HLA alleles and non-HLA genes in europeans. Am J Hum Genet. (2012) 91:778–93. doi: 10.1016/j.ajhg.2012.08.026
326. Thomas NJ, Dennis JM, Sharp SA, Kaur A, Misra S, Walkey HC, et al. DR15-DQ6 remains dominantly protective against type 1 diabetes throughout the first five decades of life. Diabetologia. (2021) 64:2258–65. doi: 10.1007/S00125-021-05513-4
327. Pugliese A, Boulware D, Yu L, Babu S, Steck AK, Becker D, et al. HLA-DRB1*15:01-DQA1*01:02-DQB1*06:02 haplotype protects autoantibody-positive relatives from type 1 diabetes throughout the stages of disease progression. Diabetes. (2016) 65:1109–19. doi: 10.2337/DB15-1105
328. Pahkuri S, Katayama S, Valta M, Nygård L, Knip M, Kere J, et al. The effect of type 1 diabetes protection and susceptibility associated HLA class II genotypes on DNA methylation in immune cells. HLA. (2024) 103:e15548. doi: 10.1111/TAN.15548
329. Hajjej A, Almawi WY, Stayoussef M, Arnaiz-Villena A, Hattab L, and Hmida S. Association of HLA-DRB1 and -DQB1 alleles with type 1 (autoimmune) diabetes in African Arabs: systematic review and meta-analysis. Immunol Invest. (2019) 48:130–46. doi: 10.1080/08820139.2018.1493498
330. Cobb BL, Lessard CJ, Harley JB, and Moser KL. Genes and sjögren’s syndrome. Rheumatic Dis Clinics North America. (2008) 34:847–68. doi: 10.1016/j.rdc.2008.08.003
331. Guggenbuhl P, Jean S, Jego P, Grosbois B, Chalès G, Semana G, et al. Primary Sjogren’s syndrome: Role of the HLA-DRB1 0301–1501 heterozygotes. J Rheumatol. (1998) 25:900–5.
332. Hollenbach JA and Oksenberg JR. The immunogenetics of multiple sclerosis: A comprehensive review. J Autoimmun. (2015) 64:13–25. doi: 10.1016/j.jaut.2015.06.010
333. Lünemann JD, Jelčić I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2. J Exp Med. (2008) 205:1763–73. doi: 10.1084/jem.20072397
334. Smith J, Fritz EL, Kerr JR, Cleare AJ, Wessely S, and Mattey DL. Association of chronic fatigue syndrome with human leucocyte antigen class II alleles. J Clin Pathol. (2005) 58:860–3. doi: 10.1136/jcp.2004.022681
335. Underhill JA, Mahalingam M, Peakman M, and Wessely S. Lack of association between HLA genotype and chronic fatigue syndrome. Eur J Immunogenetics. (2001) 28:425–8. doi: 10.1046/j.1365-2370.2001.00235.x
336. Maniaol AH, Elsais A, Lorentzen ÅR, Owe JF, Viken MK, Sæther H, et al. Late onset myasthenia gravis is associated with HLA DRB1*15:01 in the norwegian population. PloS One. (2012) 7:e36603. doi: 10.1371/journal.pone.0036603
337. Misra MK, Damotte V, and Hollenbach JA. The immunogenetics of neurological disease. Immunology. (2018) 153:399–414. doi: 10.1111/imm.12869
338. Fujiya A, Ochiai H, Mizukoshi T, Kiyota A, Shibata T, Suzuki A, et al. Fulminant type 1 diabetes mellitus associated with a reactivation of Epstein-Barr virus that developed in the course of chemotherapy of multiple myeloma. J Diabetes Investig. (2010) 1:286–9. doi: 10.1111/j.2040-1124.2010.00061.x
339. Sikorova K, Osoegawa K, Kocourkova L, Strnad A, Petrkova J, Fernández-Viña MA, et al. Association between sarcoidosis and HLA polymorphisms in a Czech population from Central Europe: focus on a relationship with clinical outcome and treatment. Front Med (Lausanne). (2023) 10:1094843/BIBTEX. doi: 10.3389/FMED.2023.1094843/BIBTEX
340. Voorter CEM, Drent M, and Van Den Berg-Loonen EM. Severe pulmonary sarcoidosis is strongly associated with the haplotype HLA-DQB1*0602–DRB1*150101. Hum Immunol. (2005) 66:826–35. doi: 10.1016/J.HUMIMM.2005.04.003
341. Nishino S and Kanbayashi T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications in the hypothalamic hypocretin/orexin system. Sleep Med Rev. (2005) 9:269–310. doi: 10.1016/j.smrv.2005.03.004
342. Morris DL, Taylor KE, Fernando MMA, Nititham J, Alarcón-Riquelme ME, Barcellos LF, et al. Unraveling multiple MHC gene associations with systemic lupus erythematosus: Model choice indicates a role for HLA alleles and non-HLA genes in europeans. Am J Hum Genet. (2012) 91:778–93. doi: 10.1016/j.ajhg.2012.08.026
343. Pociot F and Lernmark Å. Genetic risk factors for type 1 diabetes. Lancet. (2016) 387:2331–9. doi: 10.1016/S0140-6736(16)30582-7
344. Burrack AL, Martinov T, and Fife BT. T cell-mediated beta cell destruction: Autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol (Lausanne). (2017) 8:343. doi: 10.3389/fendo.2017.00343
345. Camarca ME, Mozzillo E, Nugnes R, Zito E, Falco M, Fattorusso V, et al. Celiac disease in type 1 diabetes mellitus. Ital J Pediatr. (2012) 38:10. doi: 10.1186/1824-7288-38-10
346. Smigoc Schweiger D, Mendez A, Kunilo Jamnik S, Bratanic N, Bratina N, Battelino T, et al. High-risk genotypes HLA-DR3-DQ2/DR3-DQ2 and DR3-DQ2/DR4-DQ8 in co-occurrence of type 1 diabetes and celiac disease. Autoimmunity. (2016) 49:240–7. doi: 10.3109/08916934.2016.1164144
347. Liu E, Lee H-S, Aronsson CA, Hagopian WA, Koletzko S, Rewers MJ, et al. Risk of pediatric celiac disease according to HLA haplotype and country. New Engl J Med. (2014) 371:42–9. doi: 10.1056/nejmoa1313977
348. Zeitlin AA, Heward JM, Newby PR, Carr-Smith JD, Franklyn JA, Gough SCL, et al. Analysis of HLA class II genes in Hashimoto’s thyroiditis reveals differences compared to Graves’ disease. Genes Immun. (2008) 9:358–63. doi: 10.1038/gene.2008.26
349. El-Ahwal L and AbdEL-Bar E. The frequency of HLA DRB1-DQB1 alleles in autoimmune type 1 diabetes with or without autoimmune thyroid disease. Tanta Med J. (2015) 43:66. doi: 10.4103/1110-1415.158055
350. Cobb BL, Lessard CJ, Harley JB, and Moser KL. Genes and sjögren’s syndrome. Rheumatic Dis Clinics North America. (2008) 34:847–68. doi: 10.1016/j.rdc.2008.08.003
351. Guggenbuhl P, Jean S, Jego P, Grosbois B, Chalès G, Semana G, et al. Primary Sjogren’s syndrome: Role of the HLA-DRB1 0301–1501 heterozygotes. J Rheumatol. (1998) 25:900–5.
352. Zagoriti Z, Kambouris ME, Patrinos GP, Tzartos SJ, and Poulas K. Recent advances in genetic predisposition of myasthenia gravis. BioMed Res Int. (2013) 2013:404053. doi: 10.1155/2013/404053
353. Hjelmström P, Giscombe R, Lefvert AK, Pirskanen R, Kockum I, Landin-Olsson M, et al. Different HLA-DQ are positively and negatively associated in swedish patients with myasthenia gravis. Autoimmunity. (1995) 22:59–65. doi: 10.3109/08916939508995300
354. Maniaol AH, Elsais A, Lorentzen ÅR, Owe JF, Viken MK, Sæther H, et al. Late onset myasthenia gravis is associated with HLA DRB1*15:01 in the norwegian population. PloS One. (2012) 7:e36603. doi: 10.1371/journal.pone.0036603
355. Husebye E and Løvås K. Pathogenesis of primary adrenal insufficiency. Best Pract Res Clin Endocrinol Metab. (2009) 23:147–57. doi: 10.1016/j.beem.2008.09.004
356. Skinningsrud B, Lie BA, Lavant E, Carlson JA, Erlich H, Akselsen HE, et al. Multiple loci in the HLA complex are associated with addison’s disease. J Clin Endocrinol Metab. (2011) 96:E1703–8. doi: 10.1210/JC.2011-0645
357. Oliveira LC, Porta G, Marin MLC, Bittencourt PL, Kalil J, and Goldberg AC. Autoimmune hepatitis, HLA and extended haplotypes. Autoimmun Rev. (2011) 10:189–93. doi: 10.1016/J.AUTREV.2010.09.024
358. Hyun JW, Kim S, Moon J, Park NY, Kang YR, Kim KH, et al. HLA association with AQP4-igG-positive neuromyelitis optica spectrum disorder in the korean population. Neurol Neuroimmunol Neuroinflamm. (2025) 12:e200366. doi: 10.1212/NXI.0000000000200366
359. Luckey D, Bastakoty D, and Mangalam AK. Role of HLA class II genes in susceptibility and resistance to multiple sclerosis: Studies using HLA transgenic mice. J Autoimmun. (2011) 37:122–8. doi: 10.1016/j.jaut.2011.05.001
360. Pociot F and Lernmark Å. Genetic risk factors for type 1 diabetes. Lancet. (2016) 387:2331–9. doi: 10.1016/S0140-6736(16)30582-7
361. Burrack AL, Martinov T, and Fife BT. T cell-mediated beta cell destruction: Autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol (Lausanne). (2017) 8:343. doi: 10.3389/fendo.2017.00343
362. Camarca ME, Mozzillo E, Nugnes R, Zito E, Falco M, Fattorusso V, et al. Celiac disease in type 1 diabetes mellitus. Ital J Pediatr. (2012) 38:10. doi: 10.1186/1824-7288-38-10
363. Smigoc Schweiger D, Mendez A, Kunilo Jamnik S, Bratanic N, Bratina N, Battelino T, et al. High-risk genotypes HLA-DR3-DQ2/DR3-DQ2 and DR3-DQ2/DR4-DQ8 in co-occurrence of type 1 diabetes and celiac disease. Autoimmunity. (2016) 49:240–7. doi: 10.3109/08916934.2016.1164144
364. Liu E, Lee H-S, Aronsson CA, Hagopian WA, Koletzko S, Rewers MJ, et al. Risk of pediatric celiac disease according to HLA haplotype and country. New Engl J Med. (2014) 371:42–9. doi: 10.1056/nejmoa1313977
365. Trier N, Izarzugaza J, Chailyan A, Marcatili P, and Houen G. Human MHC-II with shared epitope motifs are optimal epstein-barr virus glycoprotein 42 ligands—Relation to rheumatoid arthritis. Int J Mol Sci. (2018) 19:317. doi: 10.3390/ijms19010317
366. Zeitlin AA, Heward JM, Newby PR, Carr-Smith JD, Franklyn JA, Gough SCL, et al. Analysis of HLA class II genes in Hashimoto’s thyroiditis reveals differences compared to Graves’ disease. Genes Immun. (2008) 9:358–63. doi: 10.1038/gene.2008.26
367. El-Ahwal L and AbdEL-Bar E. The frequency of HLA DRB1-DQB1 alleles in autoimmune type 1 diabetes with or without autoimmune thyroid disease. Tanta Med J. (2015) 43:66. doi: 10.4103/1110-1415.158055
368. Stassen PM, Cohen-Tervaert JW, Lems SPM, Hepkema BG, Kallenberg CGM, and Stegeman CA. HLA-DR4, DR13(6) and the ancestral haplotype A1B8DR3 are associated with ANCA-associated vasculitis and Wegener’s granulomatosis. Rheumatology. (2009) 48:622–5. doi: 10.1093/RHEUMATOLOGY/KEP057
369. Bettencourt A, Carvalho C, Leal B, Brás S, Lopes D, Martins Da Silva A, et al. The protective role of HLA-DRB1∗13 in autoimmune diseases. J Immunol Res. (2015) 2015:948723. doi: 10.1155/2015/948723
370. Kawasaki A, Hasebe N, Hidaka M, Hirano F, Sada KE, Kobayashi S, et al. Protective role of HLA-DRB1*13:02 against microscopic polyangiitis and MPO-ANCA-positive vasculitides in a Japanese population: A case-control study. PloS One. (2016) 11:e0154393. doi: 10.1371/JOURNAL.PONE.0154393
371. Furukawa H, Oka S, Tsuchiya N, Shimada K, Hashimoto A, Tohma S, et al. The role of common protective alleles HLA-DRB1*13 among systemic autoimmune diseases. Genes Immun. (2017) 18:1–7. doi: 10.1038/GENE.2016.40
372. Mattey DL, Hassell AB, Plant MJ, Cheung NT, Dawes PT, Jones PW, et al. The influence of HLA-DRB1 alleles encoding the DERAA amino acid motif on radiological outcome in rheumatoid arthritis. Rheumatol (Oxford). (1999) 38:1221–7. doi: 10.1093/RHEUMATOLOGY/38.12.1221
Keywords: autoimmunity, HLA class II, DRB1, DQB1, cytokines, immune tolerance, evolutionary genetics, pathogen selection
Citation: Ruiz-Pablos M, Paiva B and Zabaleta A (2025) The origin of autoimmune diseases: is there a role for ancestral HLA-II haplotypes in immune hyperactivity. Front. Immunol. 16:1710571. doi: 10.3389/fimmu.2025.1710571
Received: 22 September 2025; Accepted: 17 November 2025; Revised: 05 November 2025;
Published: 04 December 2025.
Edited by:
Aya Nambu, Boston Children’s Hospital and Harvard Medical School, United StatesReviewed by:
Laura Santambrogio, Cornell University, United StatesBakrim Saad, Université Ibn Zohr, Morocco
Copyright © 2025 Ruiz-Pablos, Paiva and Zabaleta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Ruiz-Pablos, bWFucnVpcGFAZ21haWwuY29t; Aintzane Zabaleta, YXphYmFsZXRhYUB1bmF2LmVz