 Yuhui Wang1,2,3†
Yuhui Wang1,2,3† Bing Wang4†
Bing Wang4† Shuhan Liu3Yinuo Chen3Shimei Zhang1Lifang Bu3Wenjing Zhu5*
Shuhan Liu3Yinuo Chen3Shimei Zhang1Lifang Bu3Wenjing Zhu5* Xinlin Liu1,2,3*
Xinlin Liu1,2,3* Peng Sun1*
Peng Sun1*- 1Department of Hepatobiliary and Pancreatic Surgery, The Affiliated Hospital of Qingdao University, Qingdao, China
- 2Qingdao Cancer Institute , Qingdao, China
- 3Qingdao Medical College, Qingdao University, Qingdao, China
- 4Biomedical Center of Qingdao University , Qingdao, China
- 5Medical Research Department, Qingdao Hospital, University of Health and Rehabilitation Sciences (Qingdao Municipal Hospital), Qingdao, China
Immunoglobulin M (IgM) antibodies are gaining renewed attention as next-generation platforms for cancer immunotherapy. Compared with IgG, IgM exhibits distinct biological advantages, including higher avidity from multivalent binding, potent complement activation, and enhanced recognition of heterogeneous tumor antigens within immunosuppressive microenvironments. These attributes position IgM as a promising candidate for solid tumor therapy, despite the absence of currently approved IgM-based therapeutics. Recent advances in genetic engineering, antibody design, and protein manufacturing have enabled the generation of diverse IgM formats—ranging from monoclonal and bispecific constructs to engineered IgM derivatives—demonstrating substantial antitumor potential in preclinical and early translational studies. Nonetheless, clinical development faces persistent challenges, including short serum half-life, restricted tumor penetration, structural and biophysical complexity, and scalability of production. In this review, we discuss the structure and biology of IgM, highlight progress in developing novel IgM-based antibody formats for solid tumors, and critically examine the key translational barriers and future opportunities. Together, these insights underscore the therapeutic promise of IgM and chart a path toward its integration into the next generation of antibody-based cancer immunotherapies.
1 Introduction
Immunoglobulins (Igs) are essential glycoproteins that play a central role in the adaptive immune system and are synthesized by B lymphocytes and plasma cells. Humans have five major immunoglobulin isotypes: IgA, IgD, IgE, IgG, and IgM. Each isotype, including its subclasses, exhibits distinct structural and functional characteristics. Among these, IgG is the most abundant serum isotype and has become a cornerstone of cancer therapy due to its unique structural and functional properties (1). IgG antibodies demonstrate high target specificity, thereby enhancing therapeutic safety. Furthermore, IgG mediates immune responses via multiple mechanisms, such as antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) (1–4). These mechanisms have revolutionized oncology, enabling the development of targeted therapies such as immune checkpoint inhibitors and antibody–drug conjugates (5).
However, IgG therapies face several limitations, including low avidity for antigens with low density or weak affinity as a result of their bivalency (6), restricted penetration into solid tumors, and a limited capacity for potent complement-mediated lysis. These limitations have stimulated interest in alternative isotypes, particularly immunoglobulin M (IgM). IgM antibodies have previously been explored in infectious and autoimmune diseases, where they enhanced pathogen clearance and immune regulation (7). These findings laid the groundwork for their development in cancer. IgM possesses ten binding sites, conferring higher binding avidity than IgG antibodies targeting the same epitope (8). This property enables IgM to bind effectively to low-density or weakly expressed tumor-associated antigens, thereby overcoming a key limitation of IgG. Its pentameric architecture further promotes potent complement activation and direct lysis of tumor cells (9). These functional advantages underscore the potential of developing novel antibody therapies based on IgM. Such therapies may overcome the shortcomings of IgG and provide a promising avenue for the effective treatment of solid tumors (10). In this review, we summarize the structural and biological features of IgM, outline recent advances in IgM-based therapeutic antibodies for solid tumor therapy, discuss major challenges such as short half-life, limited tumor penetration, and manufacturing complexity, and offer perspectives on future directions.
2 IgM structure and biology
Immunoglobulins are proteins produced by immune cells, constituting an essential component of the immune system. They consist of two heavy chains (HCs) and two light chains (LCs). According to the type of heavy chain, immunoglobulins are classified into five isotypes (IgA, IgD, IgE, IgG, and IgM) (11, 12). The heavy and light chains are linked through disulfide bonds to form a Y-shaped structure (13). At the Y-shaped junction, one or more disulfide bonds are typically connected to the heavy chains, forming the hinge region that permits independent movement of the Fab arms and confers relative flexibility between Fab and Fc regions (14). Such a hinge structure is absent in IgM and IgE. The N-terminal region of the immunoglobulin is designated as the variable region and comprises three complementarity-determining regions (CDRs) capable of directly binding antigens, whereas the C-terminal part of the heavy chain is termed the constant region. Most immunoglobulins contain three constant domains (Cμ1–Cμ3), whereas IgM and IgE contain four (Cμ1–Cμ4). Each class is defined by a distinct heavy chain constant region structure that determines its effector functions and biological properties (15). Immunoglobulins are found in plasma and on B-cell surfaces. IgD, IgE, and IgG occur as monomers, while IgA is most commonly present as dimers. IgM exists as a monomer on B-cell surface but polymerizes into either a J chain–containing pentamer or a J chain–independent hexamer, with the pentameric form predominating in humans (16, 17). (An overview of the five immunoglobulin isotypes and the detailed architecture of IgM are presented in Figure 1).
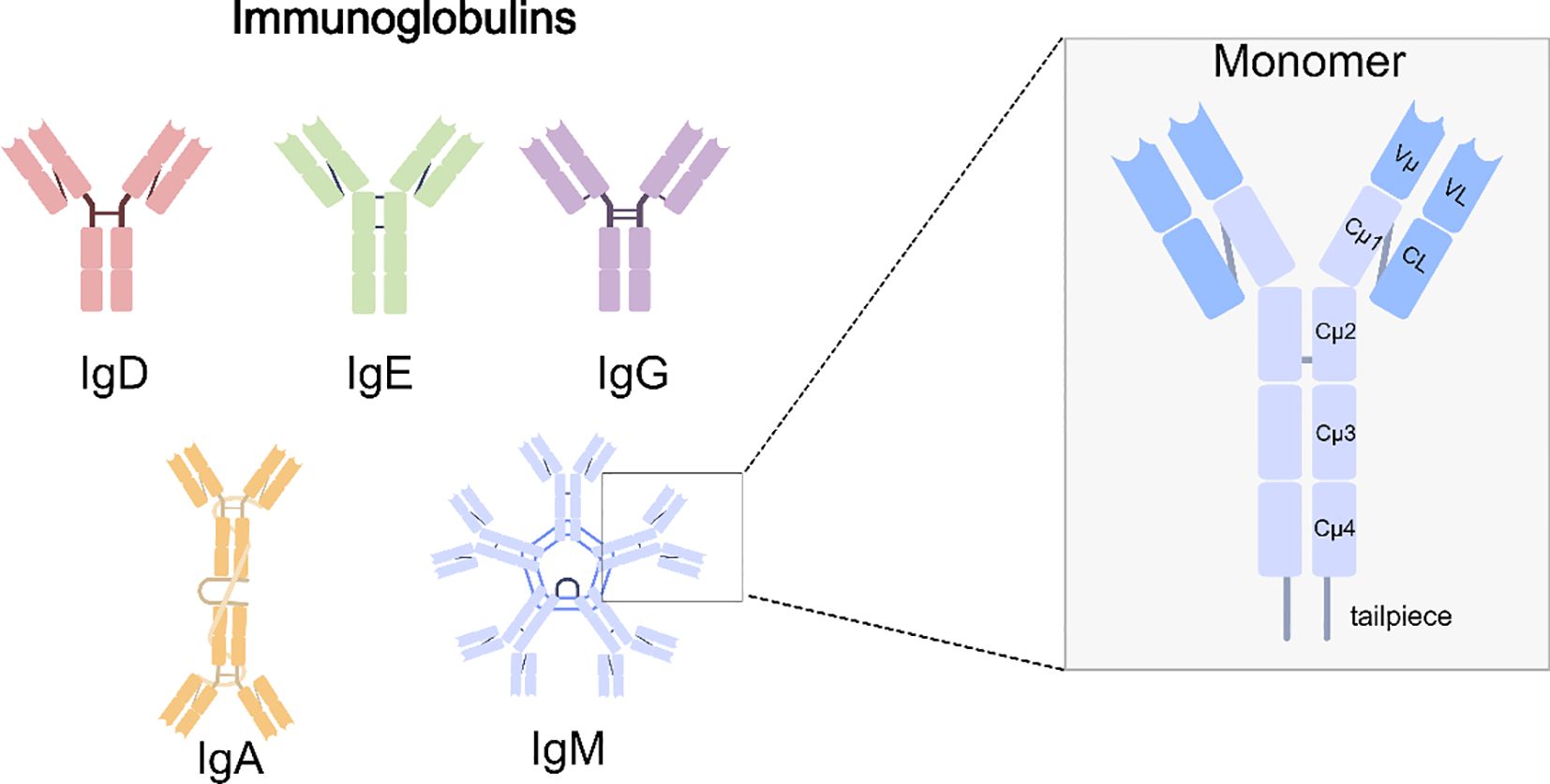
Figure 1. Human immunoglobulin isotypes and IgM structure. Schematic representation of the five major immunoglobulin classes. Among them, IgM is secreted predominantly as a pentamer, conferring ten antigen-binding sites and high avidity. The right panel depicts the IgM monomer, highlighting the variable domains (Vμ, VL), constant domains (Cμ1–Cμ4, CL), and the tailpiece that is essential for multimerization.
IgM is the first antibody isotype generated during the humoral immune response and plays a critical role in mucosal immunity, together with IgA. The IgM light chain comprises ~220 amino acids, whereas the heavy chain consists of ~576 amino acids. The C-terminus of IgM heavy chain contains tailpieces comprising an 18-amino-acid peptide sequence (18). These tailpieces interact with one another, a process essential for IgM polymerization and assembly with the J chain (19). The J chain, a 137-amino acid polypeptide, is an essential component of polymeric IgM and joins two IgM-Fc molecules to stabilize the pentamer. Additionally, it facilitates IgM transport through interaction with polymeric immunoglobulin receptors (pIgR) (20, 21).
Advances in cryo-electron microscopy (cryo-EM) have yielded new insights into IgM structure. Contrary to the previously hypothesized pentagon, single-particle negative-stain electron microscopy revealed that the IgM pentamer adopts an asymmetric pentagon with a pronounced gap (18, 21, 22). High-resolution cryo-EM demonstrated that the pentameric core is an asymmetric, disc-shaped Fc ring formed by the constant regions (Cμ2–Cμ4) of ten μ chains interlaced by disulfide bonds (23). IgM possesses an asymmetric, rigid core formed by the Cμ4 and Cμ3 constant regions and the J chain, with the Fab and Cμ2 domains rotating as a unit around a hinge located at the Cμ3/Cμ2 interface. This structural feature is likely associated with multivalent binding of surface-associated antigens and the activation of the complement pathway (24). The Fc ring is asymmetric and relatively rigid, stabilized by the J chain, whereas the Fab arms exhibit wide mobility in their connection to the Fc ring via the hinge region (24). This architecture enables IgM to bind multiple antigenic epitopes and may facilitate multivalent engagement with tumor-associated antigens (25). Li et al. demonstrated that Fcμ receptor (FcμR) binds specifically to the side of the IgM pentamer rather than in a random manner, and a single IgM pentamer can simultaneously bind up to four FcμR molecules. Moreover, the FcμR binding sites overlap with those of pIgR, suggesting mutually exclusive binding, thereby providing a structural basis for understanding IgM selection in distinct physiological pathways (26). These structural insights further indicate competition between FcμR and pIgR for binding sites, thereby modulating IgM transport and functional pathways (26, 27). Collectively, these observations suggest that IgM exerts potent complement-dependent cytotoxicity (CDC) and may additionally regulate immune balance via receptor-mediated mechanisms.
IgM functions as a critical first line of adaptive immune defense. Its unique structure confers high avidity, enabling efficient pathogen aggregation and toxin neutralization. Early studies showed that IgM activates complement to mediate immune responses (28). More recent studies have revealed that, beyond complement activation, IgM functions through alternative pathways. For example, in solid tumors, IgM may regulate the immune response via non-complement-dependent mechanisms, such as FcμR-mediated pathways (29). Furthermore, although IgM has a larger molecular size than IgG, recent studies indicate that IgM has better relative distribution and selective accumulation in inflamed and tumor tissues due to the extravasation through leaky vasculature and subsequent inflammatory cell-mediated sequestration (ELVIS) phenomenon and the enhanced permeability and retention (EPR) effect (30). These characteristics underscore the promise of IgM antibodies as therapeutic agents in cancer immunotherapy, particularly in the treatment of solid tumors.
3 Therapeutic IgM formats in solid tumors
This section outlines the major types of IgM antibodies investigated in tumor therapy and summarizes their current research and clinical status. (Representative antitumor mechanisms of monoclonal, bispecific, and engineered IgM antibodies are presented in Figure 2).
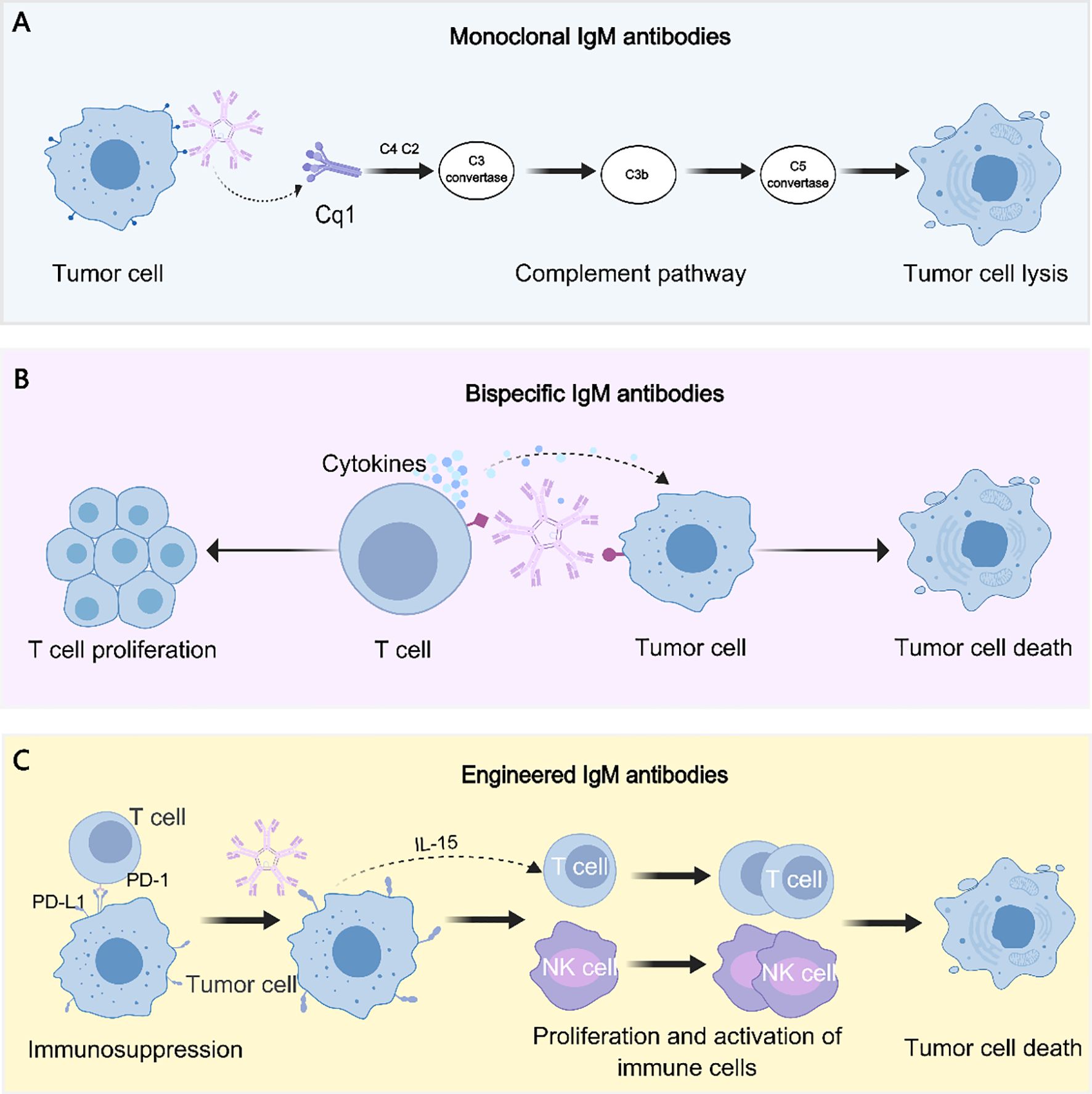
Figure 2. Antitumor mechanisms of IgM antibody formats. (A) Monoclonal IgM antibodies mediate tumor cell lysis primarily through potent activation of the classical complement pathway. (B) Bispecific IgM antibodies concurrently engage tumor-associated antigens and T cells, thereby promoting cytokine release, immune synapse formation, and tumor cell cytotoxicity. (C) Engineered IgM antibodies are designed to overcome immunosuppression (e.g., targeting the PD-1/PD-L1 axis) and to stimulate proliferation and activation of effector immune cells, such as T cells and NK cells, ultimately inducing tumor cell death.
3.1 Natural IgM
Natural IgM antibodies are primarily secreted by peritoneal B1 B cells and have the capacity to recognize and bind self-antigens. They play critical roles in both immunity and autoimmunity (31). Their polyreactivity and broad specificity enable recognition of pathogen-associated molecular patterns, apoptotic debris, and tumor-associated antigens (31, 32). Mechanistically, natural IgM mediates antitumor activity through two principal pathways. First, it strongly activates the classical complement cascade, inducing CDC and facilitating opsonization of tumor cells (33–35). Second, natural IgM can signal through the FcμR, shaping adaptive immune responses by influencing T- and B-cell cross-talk (36, 37). Together, these mechanisms provide a multifaceted defense against malignant transformation.
Early work demonstrated that IgM antibodies against ganglioside GT1b significantly suppressed Ehrlich solid tumor growth, establishing one of the first links between natural IgM and direct tumor inhibition (38). In breast cancer, natural and adaptive IgM antibodies recognize aberrant glycan structures such as mucins, facilitating immune clearance of transformed cells and preventing tumor progression (39). For example, Atif et al. demonstrated that natural IgM is indispensable for early neoantigen recognition and the activation of adaptive immunity (40). It initiates a cascade of signaling events between monocytes and dendritic cells through immune complex formation, ultimately leading to the activation of CD8+ T cells and the induction of cytotoxic responses. This dual role has been validated in two cancer models, urethane-induced tumor and melanoma, underscoring its contribution not only as an innate defense molecule but also as a critical initiator of antitumor immunity (40). These findings suggest novel opportunities for immunotherapy. Natural IgM exhibits strong avidity for repetitive antigens and mediates potent CDC, features that have inspired the design of engineered IgM molecules.
Its unique ability to recognize weakly expressed or structurally altered tumor antigens provides a conceptual foundation for engineering therapeutic IgM molecules inspired by natural prototypes. By leveraging these natural effector mechanisms, engineered IgM antibodies may overcome the limitations of IgG-based antibodies, particularly in targeting heterogeneous and weakly expressed tumor antigens.
3.2 Monoclonal IgM antibodies
Monoclonal IgM(mIgM) antibodies are fully human or humanized IgM molecules engineered to bind specific tumor-associated antigens with high affinity. Owing to their multivalent structure, mIgM antibodies can simultaneously engage multiple epitopes with strong avidity. Unlike IgG, which binds only two antigen sites, IgM can effectively target weakly expressed or heterogeneous antigens, making it especially valuable for solid tumor therapy. Recent studies have highlighted unique tumor-killing mechanisms mediated by IgM. In some situations, IgM can induce non-canonical, complement-independent cytotoxicity, including receptor-interacting serine/threonine-protein kinase(RIPK)-independent necroptosis and lipoptosis through lipid accumulation pathways, which are unique pathways that IgG antibodies don’t possess. For example, experimental evidence demonstrated that only IgM antibodies, especially clone M6-1D4, significantly reduce the viability of hepatocellular carcinoma (HCC) cell lines by inducing RIPK-independent necroptosis, while the IgG antibodies were ineffective (41). PAT-SM6 can induce lipoptosis via GRP78–LDL complex internalization (42). These findings emphasize the distinctive advantages of IgM over IgG in solid tumors.
Several mIgM antibodies have shown encouraging preclinical and early clinical potential. IGM-8444 (Aplitabart), although molecularly engineered to enhance DR5 clustering and agonistic signaling, remains a monospecific IgM antibody and is therefore discussed within the monoclonal IgM category (Table 1). Preclinical studies revealed that IGM-8444 binds DR5 with high affinity and induces potent cytotoxicity compared with IgG agonists (8). In Colo205 cells, IGM-8444 was more than 10,000-fold more potent than anti-DR5 IgG. Importantly, it exhibited no hepatotoxicity at concentrations up to 500 μg/mL, whereas TNF-related apoptosis-inducing ligand (TRAIL) induced toxicity with an IC50 of 0.04 μg/mL. Broad screening across 190 cancer cell lines representing 15 solid and 5 hematological tumors showed strong responses in 25 cell lines (IC50 < 2 ng/mL), moderate responses in 75, and weak responses in 90. Combination studies further demonstrated synergistic activity with chemotherapy agents and the BCL-2 inhibitor ABT-199, without additional hepatotoxicity. In vivo, IGM-8444 inhibited tumor growth in a dose-dependent manner and achieved complete remission in the gastric PDX model. Collectively, the multivalent structure and efficient cross-linking ability of IGM-8444 address key limitations of IgG-based agonists, providing a promising approach for DR5-targeted therapy.
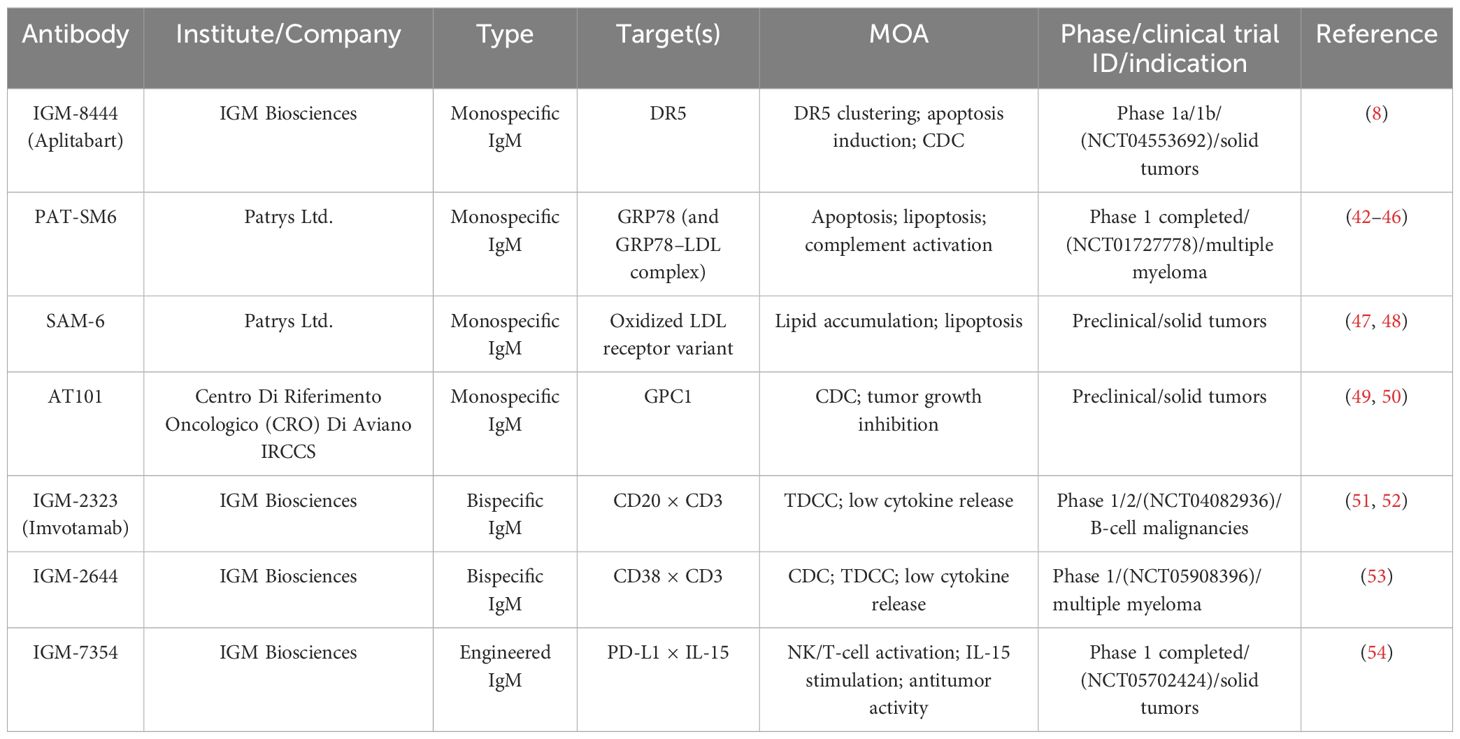
Table 1. Summary of therapeutic IgM antibodies investigated in solid tumors.
Another well-studied candidate is PAT-SM6, a human IgM monoclonal antibody targeting a cancer-specific isoform of glucose-regulated protein 78 (GRP78), with additional binding to low-density lipoprotein (LDL) complexes through GRP78-mediated interactions (43) (Table 1). GRP78 is aberrantly expressed on the surface of various solid and brain tumors and is implicated in cancer progression (55, 56). PAT-SM6 exerts anticancer activity through apoptosis, proliferation inhibition, CDC, and the unique mechanism termed lipoptosis (42–44). Preclinical studies showed that selective cytotoxicity against melanoma, pancreatic cancer, and multiple myeloma cells while sparing normal tissues. In a Phase 1 trial with 12 heavily pretreated patients with relapsed or refractory multiple myeloma, PAT-SM6 achieved stable disease (SD) in 33.3% of patients, but no partial or complete responses were observed (45, 46, 57). By contrast, SAM-6, another IgM antibody derived from the same research group, specifically recognizes an oxidized LDL receptor variant expressed on malignant cells and induces apoptosis through lipid accumulation (lipoptosis) (47, 48). However, SAM-6 has not yet entered clinical trials; its development remains at the preclinical stage.
Another promising monoclonal antibody is AT101, a complement-fixing mouse IgM that targets glypican-1 (GPC1) (Table 1). GPC1 is a cell surface proteoglycan that is highly expressed in pancreatic ductal adenocarcinoma (PDAC) tumor tissues but shows little to no expression in normal pancreatic tissue or chronic pancreatitis (49). It is associated with several growth factors that promote cancer cell proliferation, angiogenesis, and metastasis. AT101 is capable of selectively triggering complement activation and promoting the recruitment of immune effector cells within the tumor microenvironment (TME). In an experiment, it was proven that AT101 can effectively inhibit tumor growth and prolong survival in PDAC xenograft models (50). The data indicate that the average survival time of mice in the AT101 group was significantly longer than that of the control group. Among the mice treated with AT101, most had a reduction in tumor mass, and one achieved complete tumor remission. Moreover, no toxicity was observed in the mice that received multiple injections of AT101. However, AT101 remains in preclinical development, and the critical step in clinical translation will be humanization of the antibody.
Despite these advances, major challenges remain for monoclonal IgM development. Their large molecular size(900–950 kDa for pentamers and 1050–1150 kDa for hexamers), limited stability, and short pharmacokinetic half-life complicates large-scale production and purification (58). Furthermore, most mIgM-based therapies are still in preclinical or early clinical stages, and further optimization, including combination strategies, will be essential to realize their full therapeutic potential.
3.3 Bispecific IgM antibodies
The treatment of solid tumors remains highly challenging because of the complexity of the immunosuppressive tumor microenvironment, the heterogeneity of antigen expression, and the limited penetration of large-molecule antibodies into tumor tissues. While monoclonal antibodies provide clinical benefit, their effectiveness is often constrained under those conditions. Bispecific antibodies (bsAbs) have emerged as a representative innovative therapeutic strategy (59, 60). In the bispecific antibodies for treating solid tumors, IgG plays a dominant role due to its longer half-life and efficient immune function. However, their bivalency and limited Fc-mediated clustering often constrain activity in low-antigen-density tumors, motivating the exploration of multivalent alternatives such as IgM (61). Recently, the development of bispecific IgM (bsIgM) antibodies has attracted growing attention, extending beyond infectious diseases to cancer therapy. Although research is still in its early stages, the structural and functional properties of bsIgMs make them a promising approach for overcoming the limitations of existing antibody formats. BsIgMs combine the multivalency of IgM, which has ten antigen-binding sites, with the bispecificity function, and can simultaneously bind to tumor antigens and immune cell markers. This dual capacity provides a distinctive platform for solid tumor treatment. Their high avidity enables effective binding to low-density tumor antigens, and the pentameric structure enhances immune effector activation via complement and Fc receptors (9, 16, 62).
Compared with bispecific IgGs, bsIgMs have demonstrated superior biological activity. For instance, IgM-2323 (Imvotamab), a CD20×CD3 bsIgM, displayed 100-fold higher binding activity to CD20 than IgG-based T cell bispecifics, mediated CDC at levels 100-fold greater, and induced highly potent T cell-dependent cytotoxicity (TDCC) (51) (Table 1). In a Phase 1/1b clinical trial (NCT04082936) in relapsed or refractory non-Hodgkin lymphoma (R/R NHL), objective responses were observed in 11 of 38 evaluable patients (29%), including 8complete responses (21%). Notably, activity was seen even in heavily pretreated patients, including those who had undergone CAR-T therapy (52). Based on the encouraging results of IGM-2323 (imvotamab), IGM Biosciences developed a novel CD38×CD3 bispecific IgM T cell engager, IGM-2644 (Table 1). It has 10 binding sites for human CD38, and a single anti-CD3 scFv fused to the joining (J) chain. Previous clinical studies have already demonstrated that IGM-2644 exhibits dual CDC and TDCC mechanisms and demonstrates activity against daratumumab-resistant tumor cells. In addition, IGM-2644 also demonstrated reduced T cell fratricide compared to bispecific IgGs (53). Currently, IGM-2644 has an ongoing Phase 1 clinical trial (NCT05908396) for relapsed/refractory multiple myeloma. However, despite this encouraging activity, IGM Biosciences announced in January 2025 that it would terminate all cancer-related pipelines following the failure to achieve expected outcomes and difficulties in strategic development. This result underscores the significant translational challenges facing bsIgMs development. Although preclinical studies indicated potent antitumor activity and reduced cytokine release in vitro and in murine models, these findings did not translate consistently into clinical efficacy. The experience with IGM-2323 and IGM-2644 highlights the urgent need to design safer and more effective bsIgM formats.
Despite their promise, bsIgMs face multiple challenges related to structure, manufacturing, and translation. The large pentameric structure of IgM complicates protein folding, stability, and purification, resulting in low yields and batch variability (63). Maintaining high affinity at both binding sites adds further complexity to structural design and production. Additionally, IgM antibodies have relatively short half-lives compared with IgG formats (64), and their large size can hinder penetration and distribution within solid tumors, particularly in dense or immune-excluded tissues. Safety concerns, including immunogenicity and the risks of cytokine release, necessitate cautious dose escalation and rigorous clinical monitoring (54, 65).
3.4 Engineered IgM formats
Engineered IgM antibodies are designed to overcome the intrinsic limitations of natural IgM by introducing genetic or structural modifications. These engineered formats leverage the multivalency and immune-activating potential of IgM to enhance tumor targeting, particularly for low-density or heterogeneous antigens. Early studies demonstrated that IgM could serve as an efficient drug carrier. For example, methotrexate-conjugated IgM retained full antigen-binding activity and achieved superior antitumor efficacy in vivo compared with free drug or non-specific conjugates (66). Similarly, IgM-based radioimmunoconjugates labeled with α-particle emitters show highly potent and antigen-specific cytotoxicity in vitro and in vivo, with only a few isotopes per cell sufficient to induce growth inhibition (67).
A representative example is IGM-7354, developed by IGM Biosciences (Table 1). This antibody binds multiple PD-L1 receptors while simultaneously trans-presenting a single IL15/IL15Rα complex via the j-chain to activate NK and CD8+ T cells both in vitro and in vivo. Preclinical studies demonstrated that IGM-7354 exhibits high binding avidity, promotes NK and CD8+ T-cell proliferation, and inhibits tumor growth in PD-L1+ triple-negative breast cancer models. It also showed potent single-agent activity in xenograft models, enhanced antitumor effects in combination with ADCC-capable antibodies or CAR T cells, and robust immune activation in cynomolgus monkeys. Based on these data, IGM-7354 entered a Phase 1 clinical trial (NCT05702424) for advanced solid tumors (54). Other engineered IgM molecules, such as IGM-8444, further highlight the capacity of multivalent formats to improve death receptor clustering and amplify apoptosis signaling (8).
Beyond immune checkpoint targeting, other engineered IgM formats are being explored. For instance, the IgM-based T-cell engagers have been designed to activate T cells and induce their killing effect on tumor cells through simultaneously targeting tumor antigens and T-cell receptors (38). Compared with traditional IgG-based bispecific antibodies, IgM-based designs may have higher stability and lower immunogenicity, thereby reducing treatment-related adverse reactions. IgM antibodies have long faced challenges in ADC development due to their high molecular weight, polymeric structure, and a large number of glycosylation sites, but the emergence of chemoenzymatic methods has provided a new platform for the development of IgM-ADCs (68). Recent advances include conditionally activated anti-IgM ADCs. The antibody is shielded by an IgM domain and becomes exposed only in the protease-rich TME. This strategy prevented off-target binding to soluble or normal B cell–expressed IgM, while allowing efficient MMAE-mediated cytotoxicity against malignant IgM+ lymphoma cells after activation (69). These findings highlight the diverse strategies of engineered IgM, from T-cell engagers to conditionally activated ADCs, underscoring its therapeutic versatility.
Engineered IgM antibodies provide several advantages compared with IgG or other formats. Their multivalency confers high avidity, enabling efficient binding even to targets expressed at low antigen density within the tumor environment. Although engineered IgMs demonstrate improved stability, extended half-life, and enhanced delivery efficiency compared with natural IgM, significant hurdles remain. From a manufacturing perspective, due to the large molecular size and complex quaternary structure of IgM expression, assembly, and purification often lead to low yields and batch variability. Pharmacokinetically, IgM molecules display rapid systemic clearance and limited tissue penetration, creating a need to balance half-life extension with tumor accessibility. In addition, the multivalency of IgM may increase risks of unwanted complement activation, off-target immune responses, or cytokine release, particularly at high doses or in multifunctional constructs. Advances in protein engineering, optimization of bioprocess, and carefully designed clinical trials will be critical to realize the therapeutic potential of engineered IgM antibodies.
4 Challenges and perspectives
Immunoglobulin M (IgM) antibodies are re-emerging as a promising therapeutic modality for solid tumors. Although notable advances have been made in IgM research, design, and structural characterization, several unmet needs remain. Importantly, current IgM studies are still at an early stage, and more reliable preclinical models are required to predict and evaluate efficacy, toxicity, and pharmacokinetics before translation into human clinical trials.
One of the most significant challenges is the short half-life of IgM (9). In 1964, Barth et al. reported that the half-life of IgM was 5.1 days, whereas IgG antibodies exhibit a half-life of up to 21 days or longer (70, 71). This discrepancy is largely attributable to the neonatal Fc receptor (FcRn), which binds endogenous IgG, protecting it from lysosomal degradation and recycling it back into circulation (72). IgM, however, does not undergo this protective pathway. Engineering IgM with FcRn-binding domains (73–76), albumin-fusion motifs, or protective approaches such as liposomal encapsulation or PEGylation (77) has shown promise in extending its circulation time.
In addition to advances in antibody engineering, a deeper understanding of Fc receptor (FcR) biology is essential for optimizing IgM-based therapeutics. FcRs are immune receptors that bind to the Fc region of Igs and play central roles in antibody effector functions (78). While IgG primarily exerts its effects through Fc gamma receptors (FcγRs) to mediate cytotoxic and phagocytic responses, IgM interacts mainly with the complement system and FcμR. Extensive research has focused on FcγRs, which display distinct expression patterns across immune effector cells, including macrophages, dendritic cells, NK cells, neutrophils, and B cells, where they regulate ADCC, phagocytosis, and cytokine production (79). Activating receptors such as FcγRI (CD64), FcγRIIA (CD32A), and FcγRIIIA (CD16A) promote immune activation, whereas the inhibitory FcγRIIB (CD32B) counterbalances these signals to maintain immune homeostasis (80). Understanding this bidirectional regulation provides valuable insight into the rational design of IgM-based therapeutic strategies. FcμR specifically binds to the Fc region of pentameric or hexameric IgM with high affinity, modulating B- and T-cell responses and contributing to immune homeostasis (62). However, its precise role in regulating IgM-mediated antitumor immunity remains largely unexplored, representing a critical frontier for the clinical translation of IgM-based therapeutic approaches.
Despite its multivalency and strong binding avidity, IgM’s large molecular size restricts penetration into dense, stromal-rich tumors. Furthermore, TME features such as elevated interstitial fluid pressure, hypoxia, and acidic pH may impair IgM stability and activity (81, 82). While potent complement activation by IgM can induce tumor cell lysis, it may also amplify pro-inflammatory signaling, thereby exacerbating TME dysfunction (83). Future studies are needed to better elucidate the interaction between IgM and TME, which may enable more precise strategies for tumor targeting. Manufacturability and stability represent additional barriers. The structural complexity of IgM complicates large-scale production and reduces biophysical stability during formulation (9). Encouragingly, advances in related fields have brought new opportunities for IgM development. The concept of developability, which has been critical in the optimization of IgG antibodies (84–86), may similarly help identify superior IgM candidates and streamline drug development. In addition, progress in computational technologies is likely to facilitate the discovery of IgM molecules with enhanced biophysical and pharmacological properties (87). Optimizing expression hosts, applying glycoengineering, and employing machine learning–based developability screening could significantly improve IgM yield and formulation stability.
Recent advances in antibody engineering, expression systems, and bioprocess optimization have begun to address these limitations (58). The future success of IgM therapies for solid tumors will depend on continued progress in antibody engineering, translational biology, and clinical development. With deeper insights into IgM biology and the emergence of innovative formats, improved strategies are expected to overcome current challenges, thereby accelerating the translation of IgM-based therapeutics into clinical trials and ultimately providing new hope for patients with solid tumors.
Author contributions
YW: Conceptualization, Writing – original draft, Writing – review & editing. BW: Supervision, Writing – review & editing. SL: Writing – review & editing. YC: Writing – review & editing. SZ: Writing – review & editing. LB: Writing – review & editing. WZ: Funding acquisition, Resources, Writing – review & editing. XL: Funding acquisition, Resources, Writing – review & editing. PS: Conceptualization, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Shandong Province Grants ZR202111120048 (Wenjing Zhu), ZR2022QH201 (Xinlin Liu) and ZR2024MC119 (Xinlin Liu), 2022 Shinan District Science and Technology Plan Project Grants 2023-2-015-YY (Wenjing Zhu), Development of innovative medical devices for pediatric ophthalmology based on machine vision and eye tracking technology Grants 24-1-5-yqpy-23-qy (Wenjing Zhu), the role and mechanism of SnoRD14E-PBX3 axis in regulating the progression of lung adenocarcinoma Grants 82473113 (Wenjing Zhu), the National Natural Science Foundation of China Grants 32300788 (Xinlin Liu).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kumar M, Jalota A, Sahu SK, and Haque S. Therapeutic antibodies for the prevention and treatment of cancer. J Biomed Sci. (2024) 31:6. doi: 10.1186/s12929-024-00996-w
2. Seledtsov VI, Seledtsova GV, Darinskas A, and von Delwig A. Low antibody dosing in cancer therapy: targeted cytotoxicity combined with anti-tumour immunostimulation. Int J Mol Sci. (2025) 26:7724. doi: 10.3390/ijms26167724
3. Liu X, Song Y, Cheng P, Liang B, and Xing D. Targeting HER2 in solid tumors: Unveiling the structure and novel epitopes. Cancer Treat Rev. (2024) 130:102826. doi: 10.1016/j.ctrv.2024.102826
4. Chen S, Lin J, Yang Z, Wang Y, Wang Q, Wang D, et al. TRIM24-mediated K27-linked ubiquitination of ULK1 alleviates energy stress-induced autophagy and promote prostate cancer growth in the context of SPOP mutation. Cell Death Differ. (2025). doi: 10.1038/s41418-025-01582-9
5. Liu X, Ma L, Li J, Sun L, Yang Y, Liu T, et al. Trop2-targeted therapies in solid tumors: advances and future directions. Theranostics. (2024) 14:3674–92. doi: 10.7150/thno.98178
6. Zuckier LS, Berkowitz EZ, Sattenberg RJ, Zhao QH, Deng HF, and Scharff MD. Influence of affinity and antigen density on antibody localization in a modifiable tumor targeting model. Cancer Res. (2000) 60:7008–13.
7. Fereidan-Esfahani M, Nayfeh T, Warrington A, Howe CL, and Rodriguez M. IgM natural autoantibodies in physiology and the treatment of disease. Methods Mol Biol (Clifton N.J.). (2019) 1904:53–81. doi: 10.1007/978-1-4939-8958-4_3
8. Wang BT, Kothambawala T, Wang L, Matthew TJ, Calhoun SE, Saini AK, et al. Multimeric anti-DR5 igM agonist antibody IGM-8444 is a potent inducer of cancer cell apoptosis and synergizes with chemotherapy and BCL-2 inhibitor ABT-199. Mol Cancer Ther. (2021) 20:2483–94. doi: 10.1158/1535-7163.Mct-20-1132
9. Keyt BA, Baliga R, Sinclair AM, Carroll SF, and Peterson MS. Structure, Function, and Therapeutic Use of IgM Antibodies. Antibodies (Basel). (2020) 9:53. doi: 10.3390/antib9040053
10. Paul S, Konig MF, Pardoll DM, Bettegowda C, Papadopoulos N, Wright KM, et al. Cancer therapy with antibodies. Nat Rev Cancer. (2024) 24:399–426. doi: 10.1038/s41568-024-00690-x
11. Wan H, Zhang J, Ding Y, Wang H, and Tian G. Immunoglobulin classification based on FC* and GC* Features. Front Genet. (2021) 12:827161. doi: 10.3389/fgene.2021.827161
12. Schroeder HW Jr. and Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. (2010) 125:S41–52. doi: 10.1016/j.jaci.2009.09.046
13. JM W and DR B. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol. (2004) 4:89–99. doi: 10.1038/nri1266
14. Robert H, Deisenhofer J, Peter MC, Masaaki M, and Walter P. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature. (1976) 264:415–20. doi: 10.1038/264415a0
15. SK R. & K, H. Immunoglobulin heavy chain constant regions regulate immunity and tolerance to idiotypes of antibody variable regions. Proc Natl Acad Sci United States America. (2002) 99:7588–93. doi: 10.1073/pnas.052150899
16. Buchner J, Sitia R, and Svilenov HL. Understanding igM structure and biology to engineer new antibody therapeutics. BioDrugs. (2025) 39:347–57. doi: 10.1007/s40259-025-00720-6
17. Matsumoto ML. Molecular mechanisms of multimeric assembly of igM and igA. Annu Rev Immunol. (2022) 40:221–47. doi: 10.1146/annurev-immunol-101320-123742
18. Pan S, Manabe N, and Yamaguchi Y. 3D structures of igA, igM, and components. Int J Mol Sci. (2021) 22:12776. doi: 10.3390/ijms222312776
19. Pasalic D, Weber B, Giannone C, Anelli T, Müller R, Fagioli C, et al. A peptide extension dictates IgM assembly. Proc Natl Acad Sci U.S.A. (2017) 114:E8575–84. doi: 10.1073/pnas.1701797114
20. Su Q, Chen M, Shi Y, Zhang X, Huang G, Huang B, et al. Cryo-EM structure of the human IgM B cell receptor. Science. (2022) 377:875–80. doi: 10.1126/science.abo3923
21. Li Y, Wang G, Li N, Wang Y, Zhu Q, Chu H, et al. Structural insights into immunoglobulin M. Science. (2020) 367:1014–7. doi: 10.1126/science.aaz5425
22. Hiramoto E, Tsutsumi A, Suzuki R, Matsuoka S, Arai S, Kikkawa M, et al. The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci Adv. (2018) 4:eaau1199. doi: 10.1126/sciadv.aau1199
23. Nikit K, Christopher PA, Claudio C, and Matsumoto M. Structure of the human secretory immunoglobulin M core. Structure (London England: 1993). (2021) 29:564–71.e563. doi: 10.1016/j.str.2021.01.002
24. Chen Q, Menon R, Calder LJ, Tolar P, and Rosenthal PB. Cryomicroscopy reveals the structural basis for a flexible hinge motion in the immunoglobulin M pentamer. Nat Commun. (2022) 13:6314. doi: 10.1038/s41467-022-34090-2
25. Czajkowsky DM and Shao Z. The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. Proc Natl Acad Sci United States America. (2009) 106:14960–5. doi: 10.1073/pnas.0903805106
26. Li Y, Shen H, Zhang R, Ji C, Wang Y, Su C, et al. Immunoglobulin M perception by fcμR. Nature. (2023) 615:907–12. doi: 10.1038/s41586-023-05835-w
27. Chen Q, Menon RP, Masino L, Tolar P, and Rosenthal PB. Structural basis for Fc receptor recognition of immunoglobulin M. Nat Struct Mol Biol. (2023) 30:1033–9. doi: 10.1038/s41594-023-00985-x
28. Sharp TH, Boyle AL, Diebolder CA, Kros A, Koster AJ, and Gros P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc Natl Acad Sci United States America. (2019) 116:11900–5. doi: 10.1073/pnas.1901841116
29. Rutemark C, Alicot E, Bergman A, Ma M, Getahun A, Ellmerich S, et al. Requirement for complement in antibody responses is not explained by the classic pathway activator IgM. Proc Natl Acad Sci U.S.A. (2011) 108:E934–942. doi: 10.1073/pnas.1109831108
30. Aldayel AM, Bosaeed M, Almansour S, Alharbi NK, Alenazi M, Aljami HA, et al. IgM has a better relative distribution in inflammation sites and tumor tissues than IgG. J Nanobiotechnology. (2025) 23:253. doi: 10.1186/s12951-025-03213-4
31. Ehrenstein MR and Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. (2010) 10:778–86. doi: 10.1038/nri2849
32. Vollmers HP and Brändlein S. The “early birds”: natural IgM antibodies and immune surveillance. Histol histopathology. (2005) 20:927–37. doi: 10.14670/hh-20.927
33. Hunt KK, Shibata M, Gupta RK, and Morton DL. Complement-dependent lysis of tumor cells by a baboon IgM antibody to a tumor-associated antigen. Cancer immunology immunotherapy: CII. (1992) 34:377–82. doi: 10.1007/bf01741747
34. Ollert MW, David K, Schmitt C, Hauenschild A, Bredehorst R, Erttmann R, et al. Normal human serum contains a natural IgM antibody cytotoxic for human neuroblastoma cells. Proc Natl Acad Sci U.S.A. (1996) 93:4498–503. doi: 10.1073/pnas.93.9.4498
35. Vollmers HP and Brandlein S. Death by stress: natural IgM-induced apoptosis. Methods findings Exp Clin Pharmacol. (2005) 27:185–91. doi: 10.1358/mf.2005.27.3.890876
36. Kubagawa H, Clark C, Skopnik CM, Mahmoudi Aliabadi P, Al-Qaisi K, Teuber R, et al. Physiological and pathophysiological roles of igM fc receptor (FcµR) isoforms. Int J Mol Sci. (2023) 24:5728. doi: 10.3390/ijms24065728
37. Bohn J, Roggenbuck D, Settmacher U, Docke W, Volk HD, Von Baehr R, et al. Binding of natural human IgM auto-antibodies to human tumor cell lines and stimulated normal T lymphocytes. Immunol Lett. (1994) 39:187–94. doi: 10.1016/0165-2478(94)90106-6
38. Saha S and Mondal S. Suppression of Ehrlich subcutaneous solid tumor growth by immunization with ganglioside GT1b of its origin, its IgM antibody or anti-idiotype antibody. J Exp Clin Cancer Res. (2001) 20:75–84.
39. Díaz-Zaragoza M, Hernández-Ávila R, Viedma-Rodríguez R, Arenas-Aranda D, and Ostoa-Saloma P. Natural and adaptive IgM antibodies in the recognition of tumor-associated antigens of breast cancer (Review). Oncol Rep. (2015) 34:1106–14. doi: 10.3892/or.2015.4095
40. Atif SM, Gibbings SL, Redente EF, Camp FA, Torres RM, Kedl RM, et al. Immune surveillance by natural igM is required for early neoantigen recognition and initiation of adaptive immunity. Am J Respir Cell Mol Biol. (2018) 59:580–91. doi: 10.1165/rcmb.2018-0159OC
41. Pomlok K, Pata S, Kulaphisit M, Pangnuchar R, Wipasa J, Smith DR, et al. An IgM monoclonal antibody against domain 1 of CD147 induces non-canonical RIPK-independent necroptosis in a cell type specific manner in hepatocellular carcinoma cells. Biochim Biophys Acta Mol Cell Res. (2022) 1869:119295. doi: 10.1016/j.bbamcr.2022.119295
42. Kaufmann GF, Rosenes Z, Mok Y-F, Yang S, Griffin MDW, Mulhern TD, et al. Simultaneous binding of the anti-cancer IgM monoclonal antibody PAT-SM6 to low density lipoproteins and GRP78. PloS One. (2013) 8:e61239. doi: 10.1371/journal.pone.0061239
43. Hensel F, Eckstein M, Rosenwald A, and Brandlein S. Early development of PAT-SM6 for the treatment of melanoma. Melanoma Res. (2013) 23:264–75. doi: 10.1097/CMR.0b013e328362cbc8
44. Pizzo SV, Rosenes Z, Mulhern TD, Hatters DM, Ilag LL, Power BE, et al. The anti-cancer IgM monoclonal antibody PAT-SM6 binds with high avidity to the unfolded protein response regulator GRP78. PloS One. (2012) 7:e44927. doi: 10.1371/journal.pone.0044927
45. Han Z, Rasche L, Duell J, Morgner C, Chatterjee M, Hensel F, et al. The natural human IgM antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PloS One. (2013) 8:e63414. doi: 10.1371/journal.pone.0063414
46. Rasche L, Menoret E, Dubljevic V, Menu E, Vanderkerken K, Lapa C, et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma - results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica. (2015) 100:377–84. doi: 10.3324/haematol.2014.117945
47. Brandlein S, Rauschert N, Rasche L, Dreykluft A, Hensel F, Conzelmann E, et al. The human IgM antibody SAM-6 induces tumor-specific apoptosis with oxidized low-density lipoprotein. Mol Cancer Ther. (2007) 6:326–33. doi: 10.1158/1535-7163.MCT-06-0399
48. Fugmann SD, Hasegawa H, Wang S, Kast E, Chou H-T, Kaur M, et al. Understanding the biosynthesis of human IgM SAM-6 through a combinatorial expression of mutant subunits that affect product assembly and secretion. PloS One. (2024) 19:e0291568. doi: 10.1371/journal.pone.0291568
49. Kleeff J, Ishiwata T, Kumbasar A, Friess H, Buchler MW, Lander AD, et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest. (1998) 102:1662–73. doi: 10.1172/jci4105
50. Busato D, Capolla S, Durigutto P, Mossenta M, Bozzer S, Sblattero D, et al. A novel complement-fixing IgM antibody targeting GPC1 as a useful immunotherapeutic strategy for the treatment of pancreatic ductal adenocarcinoma. J Transl Med. (2023) 21:864. doi: 10.1186/s12967-023-04745-9
51. Baliga R, Li K, Manlusoc M, Hinton P, Ng D, Tran M, et al. High avidity igM-based CD20xCD3 bispecific antibody (IGM-2323) for enhanced T-cell dependent killing with minimal cytokine release. Blood. (2019) 134:1574–4. doi: 10.1182/blood-2019-131650
52. Budde E, Gopal AK, Kim WS, Flinn IW, Cheah CYY, Nastoupil L, et al. A phase 1 dose escalation study of igm-2323, a novel anti-CD20 x anti-CD3 igM T cell engager (TCE) in patients with advanced B-cell Malignancies. Blood. (2021) 138:132–2. doi: 10.1182/blood-2021-153355
53. Li K, Yun R, Chai M, Yakkundi P, Rosete R, Li G, et al. Igm-2644, a novel CD38xCD3 bispecific igM T cell engager demonstrates potent efficacy on myeloma cells with an improved preclinical safety profile. Blood. (2022) 140:6010–1. doi: 10.1182/blood-2022-159205
54. Desbois M, Giffon T, Yakkundi P, Denson CR, Sekar K, Hart KC, et al. IGM-7354, an immunocytokine with IL15 fused to an anti-PD-L1 igM, induces NK and CD8+ T cell-mediated cytotoxicity of PD-L1-positive tumor cells. Cancer Immunol Res. (2025) 13:1172–89. doi: 10.1158/2326-6066.CIR-24-0937
55. Ibanez J, Hebbar N, Thanekar U, Yi Z, Houke H, Ward M, et al. GRP78-CAR T cell effector function against solid and brain tumors is controlled by GRP78 expression on T cells. Cell Rep Med. (2023) 4:101297. doi: 10.1016/j.xcrm.2023.101297
56. Farshbaf M, Khosroushahi AY, Mojarad-Jabali S, Zarebkohan A, Valizadeh H, Walker PR, et al. Cell surface GRP78: An emerging imaging marker and therapeutic target for cancer. J Control Release. (2020) 328:932–41. doi: 10.1016/j.jconrel.2020.10.055
57. Rasche L, Duell J, Castro IC, Dubljevic V, Chatterjee M, and Knop S. A GRP78-directed monoclonal antibody recaptures response in refractory multiple myeloma with extramedullary involvement. Clin Cancer Res. (2016) 22:4341–9. doi: 10.1158/1078-0432.CCR-15-3111
58. Chan AC, Martyn GD, and Carter PJ. Fifty years of monoclonals: the past, present and future of antibody therapeutics. Nat Rev Immunol. (2025) 25:745–65. doi: 10.1038/s41577-025-01207-9
59. Blanco B, Dominguez-Alonso C, and Alvarez-Vallina L. Bispecific immunomodulatory antibodies for cancer immunotherapy. Clin Cancer Res. (2021) 27:5457–64. doi: 10.1158/1078-0432.CCR-20-3770
60. Liguori L, Polcaro G, Nigro A, Conti V, Sellitto C, Perri F, et al. Bispecific antibodies: A novel approach for the treatment of solid tumors. Pharmaceutics. (2022) 14. doi: 10.3390/pharmaceutics14112442
61. Hart KC, Hinton PR, Manlusoc M, Carlin KB, Schneider S, Kotturi MF, et al. An engineered IgM antibody targeting CD20 has enhanced complement-dependent cytotoxicity compared with an IgG. Exp Hematol. (2025) 152:105250. doi: 10.1016/j.exphem.2025.105250
62. Hiromi K, Kazuhito H, Naganari O, Shimon S, Andreas R, Fritz M, et al. Functional roles of the igM fc receptor in the immune system. Front Immunol. (2019) 10:945. doi: 10.3389/fimmu.2019.00945
63. Amash A, Volkers G, Farber P, Griffin D, Davison KS, Goodman A, et al. Developability considerations for bispecific and multispecific antibodies. MAbs. (2024) 16:2394229. doi: 10.1080/19420862.2024.2394229
64. Condado-Morales I, Dingfelder F, Waibel I, Turnbull OM, Patel B, Cao Z, et al. A comparative study of the developability of full-length antibodies, fragments, and bispecific formats reveals higher stability risks for engineered constructs. MAbs. (2024) 16:2403156. doi: 10.1080/19420862.2024.2403156
65. Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. (2018) 9:86–120. doi: 10.1007/s13238-017-0457-8
66. Persiani S, Ballou B, Shen WC, Ryser HJ, Reiland JM, and Hakala TR. In vivo antitumor effect of methotrexate conjugated to a monoclonal IgM antibody specific for stage-specific embryonic antigen-1, on MH-15 mouse teratocarcinoma. Cancer Immunol Immunother. (1989) 29:167–70. doi: 10.1007/BF00199991
67. Macklis RM, Kinsey BM, Kassis AI, Ferrara JL, Atcher RW, Hines JJ, et al. Radioimmunotherapy with alpha-particle-emitting immunoconjugates. Science. (1988) 240:1024–6. doi: 10.1126/science.2897133
68. Moh ESX, Sayyadi N, and Packer NH. Chemoenzymatic glycan labelling as a platform for site-specific IgM-antibody drug conjugates. Analytical Biochem. (2019) 584:113385. doi: 10.1016/j.ab.2019.113385
69. Schoenfeld K, Harwardt J, Habermann J, Elter A, and Kolmar H. Conditional activation of an anti-IgM antibody-drug conjugate for precise B cell lymphoma targeting. Front Immunol. (2023) 14:1258700. doi: 10.3389/fimmu.2023.1258700
70. Barth WF, Wochner RD, Waldmann TA, and Fahey JL. Metabolism of human gamma macroglobulins. J Clin Invest. (1964) 43:1036–48. doi: 10.1172/JCI104987
71. Vieira P and Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol. (1988) 18:313–6. doi: 10.1002/eji.1830180221
72. Dalakas MC and Spaeth PJ. The importance of FcRn in neuro-immunotherapies: From IgG catabolism, FCGRT gene polymorphisms, IVIg dosing and efficiency to specific FcRn inhibitors. Ther Adv Neurol Disord. (2021) 14:1756286421997381. doi: 10.1177/1756286421997381
73. Lim S, Chung HJ, Oh YJ, Hinterdorfer P, Myung SC, Seo YJ, et al. Modification of Fc-fusion protein structures to enhance efficacy of cancer vaccine in plant expression system. Plant Biotechnol J. (2025) 23:960–82. doi: 10.1111/pbi.14552
74. Deng W, Zhao Z, Zou T, Kuang T, and Wang J. Research advances in fusion protein-based drugs for diabetes treatment. Diabetes Metab Syndr Obes. (2024) 17:343–62. doi: 10.2147/DMSO.S421527
75. Booth BJ, Ramakrishnan B, Narayan K, Wollacott AM, Babcock GJ, Shriver Z, et al. Extending human IgG half-life using structure-guided design. MAbs. (2018) 10:1098–110. doi: 10.1080/19420862.2018.1490119
76. Foss S, Sakya SA, Aguinagalde L, Lustig M, Shaughnessy J, Cruz AR, et al. Human IgG Fc-engineering for enhanced plasma half-life, mucosal distribution and killing of cancer cells and bacteria. Nat Commun. (2024) 15:2007. doi: 10.1038/s41467-024-46321-9
77. Wang H, Lin S, Wu X, Jiang K, Lu H, and Zhan C. Interplay between liposomes and igM: principles, challenges, and opportunities. Adv Sci (Weinh). (2023) 10:e2301777. doi: 10.1002/advs.202301777
78. Bruhns P and Jönsson F. Mouse and human FcR effector functions. Immunol Rev. (2015) 268:25–51. doi: 10.1111/imr.12350
79. Galvez-Cancino F, Simpson AP, Costoya C, Matos I, Qian D, Peggs KS, et al. Fcgamma receptors and immunomodulatory antibodies in cancer. Nat Rev Cancer. (2024) 24:51–71. doi: 10.1038/s41568-023-00637-8
80. Nimmerjahn F and Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
81. Xiao Y and Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
82. Jin MZ and Jin WL. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. (2020) 5:166. doi: 10.1038/s41392-020-00280-x
83. Magrini E, Minute L, Dambra M, and Garlanda C. Complement activation in cancer: Effects on tumor-associated myeloid cells and immunosuppression. Semin Immunol. (2022) 60:101642. doi: 10.1016/j.smim.2022.101642
84. Waight AB, Prihoda D, Shrestha R, Metcalf K, Bailly M, Ancona M, et al. A machine learning strategy for the identification of key in silico descriptors and prediction models for IgG monoclonal antibody developability properties. MAbs. (2023) 15:2248671. doi: 10.1080/19420862.2023.2248671
85. Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, and Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. (2015) 104:1885–98. doi: 10.1002/jps.24430
86. Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, et al. Predicting antibody developability profiles through early stage discovery screening. mAbs. (2020) 12:1743053. doi: 10.1080/19420862.2020.1743053
Keywords: IgM, immunotherapy, antibody therapy, solid tumor, clinical translations
Citation: Wang Y, Wang B, Liu S, Chen Y, Zhang S, Bu L, Zhu W, Liu X and Sun P (2025) Harnessing IgM for solid tumor therapy: biology, engineering advances, and translational challenges. Front. Immunol. 16:1712344. doi: 10.3389/fimmu.2025.1712344
Received: 24 September 2025; Accepted: 23 October 2025;
Published: 05 November 2025.
Edited by:
Yuanzhi Chen, Xiamen University, ChinaReviewed by:
Akram N. Salah, Ain Shams University Faculty of Pharmacy Microbiology and Immunology, EgyptHui Sun, Tianfu Jincheng Laboratory, China
Chaolong Lin, Xiamen University, China
Copyright © 2025 Wang, Wang, Liu, Chen, Zhang, Bu, Zhu, Liu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Sun, cHN1bjFAcWR1LmVkdS5jbg==; Xinlin Liu, aHVhemhvbmdseGxAMTYzLmNvbQ==; Wenjing Zhu, emh1d2pAdW9yLmVkdS5jbg==
†These authors have contributed equally to this work