Abstract
Candida albicans resides as a commensal within the oral mucosa but becomes pathogenic when epithelial or immune equilibrium collapses. Neutrophils constitute the dominant effector population controlling this transition, integrating pathogen- and host-derived cues into a coordinated antimicrobial program. Fungal β-glucan recognition through Dectin-1 and complement receptor 3 (CR3) activates spleen tyrosine kinase (SYK)–phosphoinositide 3-kinase (PI3K)–extracellular signal-regulated kinase (ERK) pathways and drives microtubule-associated protein 1 light chain 3B-II (LC3B-II) accumulation and NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome assembly, thereby coupling phagocytosis with oxidative burst and neutrophil extracellular trap (NET) formation. Caspase recruitment domain-containing protein 9 (CARD9)-dependent interleukin (IL)-17 and tumor necrosis factor-α (TNF-α) circuits sustain chemokine (C-X-C motif) ligand 1/2 (CXCL1/2)-directed neutrophil recruitment and granulocyte colony-stimulating factor (G-CSF)-mediated granulopoiesis, while tissue matrix components determine site-specific antigen handling and NETosis thresholds. Hypha-restricted peptide toxin Candidalysin links epithelial injury to NLRP3 activation and release of IL-1β, IL-6, and G-CSF, establishing an oropharyngeal candidiasis (OPC)-specific neutrophil regulatory loop critical for pathogen clearance but also for mucosal inflammation. Conversely, fungal morphogenesis, biofilm organization, and metabolic rewiring dampen reactive oxygen species (ROS) generation and promote immune tolerance and drug resistance. Clinically, G-/granulocyte-macrophage colony-stimulating factor (GM-CSF) adjuvants and G-CSF-mobilized granulocyte transfusion offer context-dependent benefits yet pose toxicity risks, underscoring the need for precise intervention in neutrophil activation. Advances in single-cell and spatial multi-omics approaches are uncovering the metabolic and functional heterogeneity of neutrophils within mucosal environments, providing mechanistic insight for targeted immunomodulation.
1 Introduction
Oral candidiasis is one of the most common human fungal infections, primarily caused by the opportunistic pathogen Candida albicans. Studies have shown that in immunocompetent populations, approximately 45% may be colonized by Candida albicans, while in immunocompromised patients, such as HIV-infected individuals and organ transplant recipients, the risk of infection significantly increases (1). More importantly, oral infections can become a crucial source of deep and disseminated infections, particularly in severely immunocompromised patients (2).
As one of the most important effector cells in the innate immune system, neutrophils play a vital role in resisting Candida infections. Research indicates that deficiencies in neutrophil count or function are closely associated with infection occurrence and prognosis (3). Recent studies reveal that neutrophils participate in anti-Candida immune responses through multiple mechanisms, including phagocytosis, release of reactive oxygen species (ROS), and formation of neutrophil extracellular traps (NETs) (4, 5).
Notably, neutrophil activation and function exhibit distinct tissue specificity. Studies have found that at different infection sites, neutrophils are recruited and activated through specific signaling pathways and display unique functional characteristics (6–10). However, Candida albicans has evolved complex immune evasion strategies. Studies show that clinical isolates can interfere with neutrophil function through various mechanisms, including biofilm formation, morphological transition regulation, and secretion of specific factors (11–14). Particularly in biofilm-associated infections, Candida not only demonstrates enhanced immune evasion capabilities but often exhibits significant antifungal drug resistance. Recent research has also discovered that Candida albicans can influence neutrophil metabolic reprogramming, thereby interfering with their immune function (15).
Neutrophil responses to Candida albicans are dynamic and highly context dependent, particularly within the oral mucosa, where epithelial integrity, microbial composition, and local immune tone collectively shape host resistance. Acting as both indispensable effectors and potential mediators of tissue injury, neutrophils mount rapid antifungal activity while at times amplifying inflammation and collateral damage. Recent evidence indicates that their roles extend beyond direct fungal killing to encompass metabolic coordination, antigen presentation, and reciprocal communication with epithelial cells that influence mucosal immune homeostasis (16, 17). Distinguishing protective from pathological neutrophil responses remains challenging because of spatial heterogeneity within mucosal tissues, the limited translational relevance of existing murine models, and the complexity of microbial–immune interactions (16). Increasing use of single-cell and spatial multi-omics approaches has begun to define context-specific neutrophil programs and their evolution during Candida infection (18, 19). At the same time, therapeutic strategies targeting neutrophil metabolism, reactive oxygen species production, or extracellular trap clearance are showing promise in enhancing antifungal efficacy while limiting tissue injury (20). Collectively, these advances provide a framework for developing precision immunotherapies that strengthen epithelial barrier defense and restore immune balance in oropharyngeal and other mucosal forms of candidiasis.
Elucidating the mechanisms of neutrophil function in anti-Candida infections is crucial for developing new therapeutic strategies. Currently, individualized treatment options for patients with immune dysfunction remain limited. With increasing drug resistance, traditional antifungal treatments face severe challenges (21). Developing immunomodulatory strategies targeting enhanced neutrophil function has become an important research direction in this field. This article will systematically review the progress in molecular mechanism research of neutrophil involvement in anti-Candida infections, focusing on their regulatory networks in different tissue microenvironments, and discuss the translational prospects of related research in clinical applications (Figure 1).
Figure 1
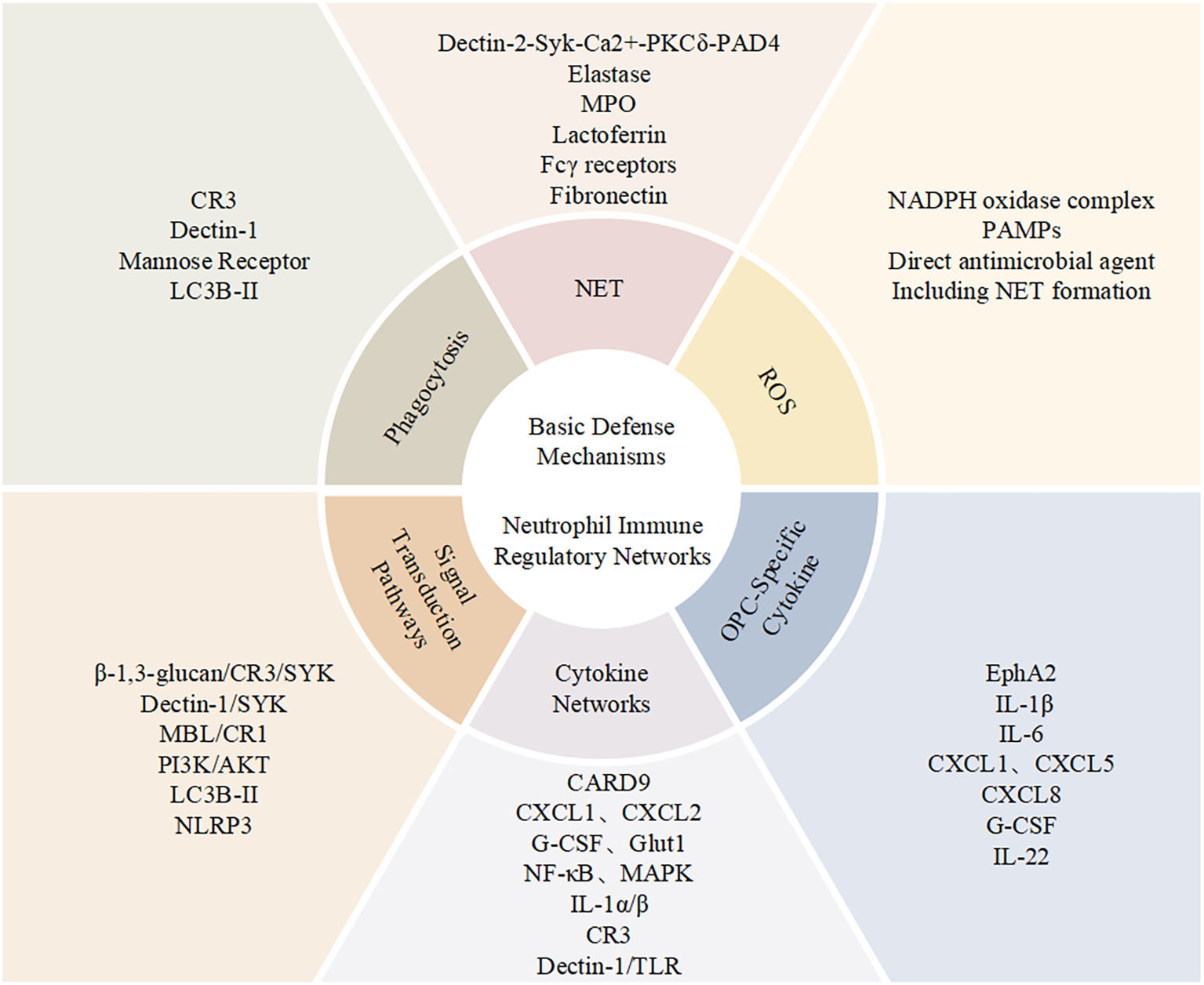
Neutrophil functional network in antifungal immunity. Schematic overview of neutrophil-mediated immune mechanisms during Candida infection, summarizing key molecular interactions, effector responses, and signaling pathways, together with a panel of OPC-related cytokines.
2 Basic defense mechanisms of neutrophils
Neutrophils are key innate immune cells in defending against Candida albicans infections, exerting antifungal effects through a complex and precise defense network. This defense system primarily comprises three major effector mechanisms: phagocytosis, neutrophil extracellular trap (NET) formation, and reactive oxygen species (ROS) production. These mechanisms work in coordination and close cooperation to form an important barrier against Candida infections (4, 22, 23).
In terms of phagocytosis, neutrophils rely on various pattern recognition receptors (PRRs) to recognize pathogen-associated molecular patterns of Candida albicans (24). Complement receptor 3 (CR3, consisting of CD11b/CD18) recognizes β-glucans through its unique lectin-like domain (25), Dectin-1 specifically recognizes β-1,3-glucans, while mannose receptors recognize cell wall mannan components (26). Neutrophil phagocytosis of Candida exhibits marked functional heterogeneity, encompassing at least two complementary effector programs. The first is a phagocytic–oxidative pathway coupled to the respiratory burst: upon activation, the NADPH oxidase complex assembles at the plasma and phagosomal membranes to generate superoxide, which is rapidly dismutated to H2O2; myeloperoxidase (MPO) then converts H2O2 to hypochlorous acid (HOCl) and, through reactions with amines, forms chloramines with additional microbicidal activity, thereby achieving potent oxidative killing within phagosomes and at adjacent extracellular sites (27–29). The second is a recently established extracellular mechanism—NETs: during the specialized program NETosis, chromatin decondenses and the nuclear and granule membranes are reorganized, culminating in the release of an expanded DNA–histone meshwork whose surface is decorated with microbicidal factors derived from granules and cytosol, including serine proteases and antimicrobial peptides (e.g., calprotectin). This enables the capture, confinement, and direct damage of both yeast and hyphal forms of Candida; NETs are particularly advantageous when pathogen burden is high, hyphae/biofilms are formed, or targets are poorly phagocytosed (30, 31). Functionally, NETosis corresponds to a “rapid-release” phenotype that establishes an immediate extracellular barrier within a short time, whereas the respiratory burst–phagocytic pathway provides sustained, primarily intracellular killing. Together, these programs coordinate across time and space to constitute a dual, complementary defense against Candida.
NET formation represents another crucial defense mechanism, with its structure composed of decondensed chromatin scaffolds and various antimicrobial proteins (such as elastase, MPO, and lactoferrin) (32). Studies have demonstrated two parallel but interconnected pathways for NET formation: NADPH oxidase-dependent and independent pathways. The former operates primarily through Fcγ receptors and protein kinase C (PKC), requiring ROS participation, while the latter proceeds through the Dectin-2-SYK-Ca2+-PKCδ-protein arginine deiminase 4 (PAD4) signaling cascade (33). Particularly noteworthy is the important regulatory role of extracellular matrix fibronectin (FN) in NET release, providing new insights into how tissue microenvironments modulate immune responses (34). Furthermore, hyphal forms of fungal filaments induce stronger NET release compared to yeast forms, reflecting the host immune system’s differential response to pathogens at various invasion stages (32).
The generation of reactive oxygen species (ROS) constitutes the third fundamental defense mechanism. Studies have demonstrated that neutrophils predominantly produce ROS through the activation of the NADPH oxidase complex, a process essential for antifungal immunity (35). Indeed, multiple stimuli can initiate NADPH oxidase complex activation, including phagocytic processes and the recognition of pathogen-associated molecular patterns (PAMPs) (36). Significantly, ROS demonstrates bifunctional properties in antifungal immunity: it serves both as a direct antimicrobial agent through oxidative damage and as a critical signaling molecule to regulate various immune processes, including NET formation (35, 37, 38).
3 Neutrophil immune regulatory networks
Candida albicans infection can initiate a multi-layered immune regulatory network: as central effectors, neutrophils integrate signals from Dectin-1/PRR-SYK, integrins, and complement/FcγR. Through canonical kinase and inflammasome pathways, neutrophils spatiotemporally coordinate the progression of phagocytosis, oxidative burst, and NETs. Meanwhile, cytokine networks including IL-17, IL-1, and TNF shape granulopoiesis, recruitment, and effector thresholds, ensuring precise defense against mucosal and invasive infections (39–42).
At the molecular level, these defense mechanisms are controlled by sophisticated regulatory networks. The β-1,3-glucan/CR3/SYK axis (39), similarly, the Dectin-1/SYK pathway (40), Mannan-binding lectin (MBL)/complement receptor 1 (CR1, CD35) signaling system (26) constitute essential molecular foundations of antifungal immunity, participating in Candida albicans recognition and signal transduction. These pathways regulate neutrophil functions, including phagocytosis, autophagy, and the production and regulation of ROS, through classical signaling cascades (such as PI3K/AKT and ERK (34)) and the newly discovered SYK-dependent LC3B-II accumulation mechanism (39). Furthermore, these molecular pathways activate key transcriptional regulatory pathways, such as NOD-like receptor signaling pathways, particularly the NLRP3 inflammasome (35), which further amplifies the immune response. These upstream signaling events and molecular regulation provide a solid foundation for the downstream cytokine network (26). Notably, these molecular recognition pathways exhibit distinct functional hierarchies in different infection contexts.
Neutrophil responses to Candida albicans differ markedly between systemic and mucosal tissues, shaped by distinct receptor repertoires and microenvironmental cues. In systemic candidiasis, recognition of fungal β-glucans by the C-type lectin receptor Dectin-1 (encoded by Clec7a) is pivotal for the activation of SYK–CARD9–NF-κB signaling, which induces pro-inflammatory cytokines such as TNF and IL-1β and promotes the generation of reactive oxygen species critical for fungal control (43, 44). Accordingly, Dectin-1-deficient mice are unable to restrict systemic fungal dissemination (45). In contrast, during oropharyngeal candidiasis, epithelial recognition of C. albicans depends primarily on EphA2- and TLR-driven pathways that coordinate early neutrophil recruitment (16). While Dectin-1 expression on resident oral macrophages contributes to immune signaling, its loss does not impair immediate neutrophil infiltration (46). At later stages, the adaptor CARD9 becomes indispensable for sustaining IL-17-mediated mucosal immunity and long-term protection. CARD9-deficient mice—and patients carrying deleterious CARD9 variants—exhibit susceptibility to chronic mucocutaneous candidiasis due to impaired Th17 cytokine responses and persistent fungal colonization (40, 47). These findings demonstrate that Dectin-1 governs early innate sensing of fungal β-glucans, whereas CARD9 serves as the essential signaling node linking pattern recognition to durable IL-17-dependent mucosal protection.
Molecular recognition and signal transduction processes often work in conjunction with cytokine networks to jointly amplify the effector functions of neutrophils in the inflammatory environment. Among these, the IL-17 signaling pathway is a core component of anti-Candida immunity, exhibiting multiple functions (41, 42). Candida albicans infection triggers Dectin-1 to activate dendritic cells and macrophages through caspase recruitment domain 9 (CARD9)-dependent signaling, which subsequently induces IL-17 production (48). The effects of IL-17 release regulatory signals through two major pathways: firstly, by inducing the secretion of chemokines such as C-X-C motif chemokine ligand 1 (CXCL1) and C-X-C motif chemokine ligand 2 (CXCL2) to form chemotactic gradients that recruit neutrophils to infection sites (49, 50); secondly, by promoting the production of granulocyte colony-stimulating factor (G-CSF) to enhance neutrophil generation and activation(32). Both genetic and acquired defects leading to IL-17 signaling abnormalities are directly associated with neutrophil dysfunction, representing one of the key pathogenic mechanisms for susceptibility to mucosal candidiasis (51). Beyond classical Th17-driven adaptive immunity, early innate IL-17 responses have emerged as key determinants of neutrophil recruitment during oropharyngeal candidiasis (OPC). In the oral mucosa, innate-like lymphocytes—including γδ T cells, natural Th17 cells, and group 3 innate lymphoid cells (ILC3s)—produce IL-17A and IL-17F within hours of C. albicans exposure, preceding conventional Th17 polarization (52, 53). These cells respond to epithelial-derived IL-1β and IL-23, stimulating G-CSF release and neutrophil-attracting chemokines CXCL1 and CXCL5 (54). Mice lacking γδ T cells or ILC3s exhibit delayed neutrophil infiltration and increased fungal burden (49, 55), highlighting the importance of the innate IL-17 axis in the early containment of infection. At the same time, epithelial sensing of pathogen-derived virulence factors provides an additional layer of neutrophil activation. Among these, the hypha-associated peptide toxin Candidalysin—encoded by ECE1—serves as a crucial epithelial trigger bridging fungal morphogenesis with host inflammation (56, 57). Upon hyphal contact, Candidalysin induces epithelial membrane damage and Ca²+ influx, activating EGFR–MAPK and NLRP3 inflammasome signaling, followed by IL-1β, IL-6, and G-CSF release that drives neutrophil recruitment (58, 59). In corticosteroid-induced OPC models, C. albicans ece1Δ/Δ mutants lacking Candidalysin fail to elicit these cytokines, resulting in impaired neutrophil infiltration and attenuated disease (56). Together, these findings establish Candidalysin as a central virulence determinant linking epithelial injury, cytokine induction, and neutrophil activity—thereby integrating fungal morphogenesis with innate and adaptive antifungal responses in the oral mucosa.
Furthermore, the TNF-α signaling pathway plays an equally important role in immune defense against Candida albicans (60). Following recognition of Candida albicans through pattern recognition receptors (such as Dectin-1), neutrophils release TNF-α, which acts on surrounding tissues through autocrine and paracrine mechanisms, activating NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways. This further enhances neutrophil effector functions, including increased phagocytic rate, enhanced ROS production through NADPH oxidase, and stimulation of NET (neutrophil extracellular trap) formation (61). The TNF-α signaling pathway forms a synergistic network with the IL-17 pathway, jointly regulating neutrophil-driven antipathogen effector functions.
IL-1α and IL-1β from the IL-1 family also play crucial roles in neutrophil regulation. For instance, in early infection, Candida-stimulated keratinocytes release IL-1α, which stimulates local endothelial cells to secrete G-CSF, thereby establishing a “tissue-blood axis” between local tissues and bone marrow for rapid mobilization of neutrophil proliferation and migration. Additionally, IL-1-related signaling can precisely control neutrophil recruitment and directed migration through regulation of chemokine CXCL1/2 expression (62).
Moreover, some emerging cytokines further enhance neutrophil functions by working in conjunction with traditional mechanisms. For instance, IL-33 not only enhances phagocytic capacity by upregulating complement receptor CR3 expression but also increases neutrophil fungicidal activity through elevated ROS generation(41). IL-33 further modulates the Dectin-1/TLR signaling mechanism, promoting CXCL1/2 secretion, which strengthens neutrophil migration and activation(41). Animal experiments have demonstrated that IL-33 pretreatment significantly reduces Candida albicans infection-related mortality, indicating its potential clinical application value (63).
Beyond classic molecular recognition and cytokine networks, recent research has revealed novel regulatory mechanisms of neutrophils. For instance, NADPH oxidase not only serves as the primary pathway for ROS generation but also participates in more complex immune responses through signal regulation functions. Similarly, neutrophils’ adaptive adjustment of glucose metabolism plays a crucial supporting role in their efficacy. For example, the key role of glucose transporter-1 (Glut1) in neutrophil local energy metabolism contributes to efficient immune responses in the lesion microenvironment (26). These new functions integrate with classical receptor mechanisms (such as Dectin-1/SYK and CR3/SYK axes) to further enhance the flexibility and adaptability of neutrophils in anti-Candida immunity (39).
4 Current clinical treatment strategies
Current clinical treatment for Candida infections primarily encompasses two strategies: antifungal drug therapy and immune function modulation. Antifungal drugs mainly include azoles, echinocandins, and polyenes. Echinocandins (such as caspofungin), which inhibit β-1,3-D-glucan synthase in fungal cell walls, are the first-line treatment for invasive candidiasis in neutropenic patients (64, 65). For non-neutropenic patients, fluconazole remains the empirical treatment of choice due to its favorable bioavailability and safety profile (66). Polyene drugs (such as amphotericin B) are primarily used for severe and refractory infections, though their application is limited by significant nephrotoxicity (67).
However, antifungal therapy faces significant challenges. Clinical studies have shown that among patients with prolonged azole exposure and during hospital outbreaks—particularly those involving fluconazole−resistant Candida parapsilosis—clinical isolates often exhibit elevated resistance to fluconazole, itraconazole, and ketoconazole (68–70). Biofilm formation further compromises therapy by limiting drug penetration and conferring marked tolerance, leading to higher risks of treatment failure and relapse than in non−biofilm infections (71, 72). Mechanistically, Candida biofilms suppress neutrophil extracellular trap (NET) release and blunt ROS−dependent killing; these effects are conserved across clinical isolates with clear strain−dependent variation in ROS and NET responses (38, 73). In addition, clinically relevant drug–drug interactions and hepatorenal toxicities constrain antifungal selection and dosing, complicating management (71, 74).
Immune function modulation therapy primarily focuses on hematopoietic growth factor treatment and cytokine regulation (75, 76). G−CSF is the most commonly used immunomodulator in clinical practice, promoting neutrophil production and release while enhancing chemotaxis and phagocytic functions (77, 78). In neutropenic patients with invasive candidiasis, adjunctive use of G−CSF or G−CSF–mobilized granulocyte transfusions alongside antifungal therapy has been associated with improved clearance and survival, although the quality of evidence is low and recommendations are conditional (79, 80). Granulocyte−macrophage colony−stimulating factor (GM−CSF) has shown promising results as adjunctive therapy in refractory Candida infections by augmenting neutrophil phagocytic and fungicidal activity, including pilot and cohort studies of sargramostim in fluconazole−refractory disease and refractory invasive fungal infections (81–83).
Immune function modulation therapy primarily includes hematopoietic growth factor support and cytokine−pathway targeting; G−CSF, produced by endothelial and other stromal/immune cells, promotes neutrophil production and release while enhancing chemotaxis and phagocytic functions (80, 84). In invasive candidiasis with profound or prolonged neutropenia, clinicians may add G−CSF to antifungals to hasten neutrophil recovery, and consider GM−CSF (sargramostim) in selected refractory cases to augment phagocyte function; observational cohorts report improved responses with GM−CSF, although evidence quality remains low and use is case−selected (80, 85, 86). In practice, G−CSF (filgrastim) is administered by subcutaneous or intravenous injection once daily until a higher absolute neutrophil count (ANC) recovery, pegfilgrastim is given as a single subcutaneous dose in prophylaxis contexts, and GM−CSF (sargramostim) is typically delivered by subcutaneous injection in short adjunctive courses; in persistent, severe neutropenia, granulocyte transfusions from G−CSF–mobilized donors can serve as a short−term bridge while definitive antifungals and source control proceed (80, 84, 86). At mucosal surfaces, IL−17 and IL−22 cooperate to reduce oral fungal burden and enhance host defense by inducing epithelial antimicrobial programs, including S100A8/A9 and the chemokine CCL20, although contributions are context−dependent (53, 87, 88). IL−33 primes neutrophils for antifungal activity by tuning TLR and Dectin−1 signaling, promoting C-X-C motif chemokine receptor 2 (CXCR2)−axis chemokine responses, upregulating complement receptor 3 (CR3), and enhancing ROS−dependent killing in experimental models (63, 89, 90). These cytokines currently inform pathogenesis rather than routine therapy; recombinant IL−17, IL−22, or IL−33 are not established treatments for candidiasis outside research settings, so clinical immunomodulation in candidiasis relies on G−/GM−CSF alongside optimized antifungal therapy and source control (80, 88).
However, immunomodulatory therapy has limitations. Neutrophils can actively tune inflammation via neutrophil serine proteases (neutrophil elastase, proteinase−3, cathepsin G), which proteolytically process and can degrade pro−inflammatory cytokines such as interleukin-1 β (IL-1β) and tumor necrosis factor α (TNF-α); this immune “fine−tuning” helps explain inter−patient heterogeneity in responses to cytokine− or growth factor–based therapies (91, 92). Clinically, growth−factor–based immunotherapy is constrained by adverse effects: G−CSF commonly causes bone pain and leukocytosis; rare but serious complications include splenic rupture and capillary−leak phenomena, warranting careful dosing and monitoring, especially in patients at pulmonary risk (93, 94). GM−CSF (sargramostim) can induce flu−like symptoms, fever, edema, injection−site reactions, and leukocytosis, with dose−related increases in adverse events reported and risks detailed in regulatory labeling, including hypersensitivity and fluid retention in susceptible patients (95). More broadly, augmenting myeloid cytokine signaling (for example, with GM−CSF) can be a double−edged sword that skews inflammatory milieus and, context−dependently, may exacerbate disease processes, underscoring the need for individualized risk–benefit assessment and close clinical monitoring when using these agents as adjuncts to antifungal therapy (96). Recent research has revealed several important advances. The β-1,3-glucan/CR3/SYK pathway-dependent LC3B-II accumulation can enhance neutrophil fungicidal activity (97, 98). Single-cell sequencing has identified neutrophil subpopulations with distinct functional characteristics (17, 99, 100). Additionally, studies have confirmed that neutrophils upregulate glucose metabolism through selective expression of glucose transporter Glut1 to meet antifungal demands (40, 101, 102). These findings provide important clues for developing novel therapeutic strategies, although their clinical application value still requires further validation. Building on these advances, recent single-cell and spatial multi-omics investigations have provided unprecedented insight into the transcriptional and functional diversity of neutrophils residing in the oral and periodontal mucosa. These studies reveal that neutrophils are not a uniform antimicrobial population but instead encompass transcriptionally, metabolically, and developmentally distinct subsets that adapt to specific tissue niches and phases of inflammation.
5 Neutrophil heterogeneity and single-cell insights
Recent advances in single-cell and spatial multi-omics technologies have profoundly reshaped the current view of neutrophil heterogeneity in the oral mucosa (19). Rather than representing a uniform antimicrobial population, neutrophils display extensive transcriptional, metabolic, and functional diversity that reflects their adaptation to specific mucosal niches and inflammatory contexts. In human gingival tissue, single-cell RNA sequencing delineates at least two major neutrophil trajectories: (i) tissue-resident neutrophils, which persist under steady-state conditions and express high levels of survival and regulatory molecules such as BCL2A1 and IL1RN, relying primarily on oxidative phosphorylation; and (ii) inflammation-recruited neutrophils, characterized by enhanced glycolytic flux and elevated expression of NADPH oxidase subunits, enabling vigorous reactive oxygen species (ROS) production and robust NETosis (103). This metabolic divergence defines distinct effector thresholds—resident cells contribute to epithelial homeostasis through restrained ROS and NET release, whereas recruited neutrophils generate strong fungicidal activity at the expense of potential collateral tissue injury (103, 104). In chronically inflamed oral lesions, polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC)–like cells with immunosuppressive transcriptional signatures (ARG1, S100A8/A9, PD-L1) have been identified, acting to curtail excessive inflammation and support tissue repair (105, 106). Trajectory analyses further indicate that emergency granulopoiesis driven by G-CSF signaling contributes to the emergence of these subsets, which exhibit metabolic and phenotypic plasticity—transitioning between antimicrobial and suppressive states in response to local cytokine and metabolic cues (105). The integration of single-cell transcriptomics with metabolic and spatial profiling thus reveals a continuum of neutrophil differentiation along a protective–regulatory–pathogenic axis. This multidimensional landscape provides a mechanistic basis for understanding how neutrophil diversity governs NETosis and ROS thresholds within oral tissues and informs precision therapeutic approaches that modulate neutrophil metabolism and differentiation to sustain antifungal defense while minimizing mucosal injury.
6 Conclusions and future directions
Neutrophils represent pivotal yet paradoxical regulators of oral mucosal immunity, acting as both essential effectors in antifungal defense and potential mediators of inflammatory injury. Recent findings demonstrate that their functions in Candida-associated infection extend beyond pathogen killing to encompass antigen presentation, metabolic cross−talk, and epithelial crosstalk that determine infection outcomes (107, 108). However, distinguishing protective from pathological neutrophil programs in the oral environment remains challenging due to spatial and temporal heterogeneity, limited model fidelity, and the complexity of local microbial–immune interactions (103). Integrative multi−omics and single−cell profiling are expected to delineate these context−dependent neutrophil trajectories (109, 110), while interventions targeting metabolic rewiring, ROS modulation, or NET clearance are emerging as strategies to enhance antifungal efficacy without exacerbating mucosal injury (111, 112). Translating these mechanistic insights into precise immunotherapies that reinforce barrier protection while restraining collateral inflammation will be crucial for future management of oral epithelial disease and periodontitis. Although considerable progress has been made in uncovering how neutrophil responses are regulated during Candida infection, important gaps remain when linking these findings to human biology. Much of our understanding comes from murine OPC models—most commonly cortisone-induced or Card9-deficient systems—that, while highly informative mechanistically, only partially recapitulate the architecture, microbiota, and immunoregulatory environment of the human oral mucosa (113, 114). Differences in epithelial keratinization, salivary composition, and microbial ecology all shape neutrophil recruitment and antifungal thresholds (115, 116). As a result, insights from animal studies regarding pattern-recognition receptor hierarchies (for example, limited Dectin-1 dependence), cytokine interactions (IL-17, IL-33), or metabolic remodeling may not fully reflect human physiology (117, 118).
In humans, OPC in otherwise healthy individuals is typically mild or self-resolving, suggesting a balanced relationship between C. albicans and epithelial-neutrophil homeostasis . Clinical disease tends to emerge under defined immunologic vulnerabilities—such as HIV infection, iatrogenic immunosuppression, or monogenic CARD9 deficiency—highlighting the challenge of extrapolating murine data to the wider population. Variation in fungal strain traits, epithelial signaling capacity, and the surrounding microbiome further complicates translation (119–121).
Bridging these differences will require experimental systems that more closely model human tissues, including three-dimensional oral mucosal co-cultures, microfluidic infection platforms, and longitudinal immune profiling in patient cohorts. Integrating these approaches with single-cell and spatial multi-omics analyses will be key to validating mechanisms defined in rodents and to developing precision strategies that harness neutrophil function against mucosal Candida infection (122–126).
Statements
Author contributions
YX: Conceptualization, Funding acquisition, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. RC: Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing, Conceptualization, Validation. WL: Investigation, Methodology, Visualization, Writing – review & editing. WY: Funding acquisition, Investigation, Resources, Validation, Visualization, Writing – review & editing. CC: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by grants from the National Cancer Institute (NCI) and the National Institute of Dental and Craniofacial Research (NIDCR), National Institutes of Health (R01CA288366 and R21DE033128) to CC and (K08DE035170) to WY. This work was also supported by grants from the Yunnan Young and Middle-aged Academic and Technical Leaders Reserve Talent Project (202205AC160067), the Xingdian Talent Support Program (XDYC-MY-2022-0050), and the Kunming Medical University Team for Diagnosis and Treatment of Complex Craniofacial Malformations (2024XKTDTS08) to YX.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor TF declared a past co-authorship with the author CC.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
- AKT
Protein Kinase B
- ANC
Absolute Neutrophil Count
- CARD9
Caspase Recruitment Domain-containing Protein 9
- CCL20
C-C Motif Chemokine Ligand 20
- Clec7a
C-type Lectin Domain Family 7 Member A
- CR1
Complement Receptor 1
- CR3
Complement Receptor 3 (CD11b/CD18)
- CXCL1
C-X-C Motif Chemokine Ligand 1
- CXCL2
C-X-C Motif Chemokine Ligand 2
- CXCL5
C-X-C Motif Chemokine Ligand 5
- CXCR2
C-X-C Motif Chemokine Receptor 2
- ECE1
Extent of Cell Elongation 1
- EGFR
Epidermal Growth Factor Receptor
- ERK
Extracellular Signal-regulated Kinase
- G-CSF
Granulocyte Colony-stimulating Factor
- Glut1
Glucose Transporter 1
- GM-CSF
Granulocyte-macrophage Colony-stimulating Factor
- H2O2
Hydrogen Peroxide
- HIV
Human Immunodeficiency Virus
- HOCl
Hypochlorous Acid
- IL-1α
Interleukin-1 Alpha
- IL-1β
Interleukin-1 Beta
- IL-6
Interleukin-6
- IL-17
Interleukin-17
- IL-17A
Interleukin-17A
- IL-17F
Interleukin-17F
- IL-22
Interleukin-22
- IL-23
Interleukin-23
- IL-33
Interleukin-33
- IL1RN
Interleukin-1 Receptor Antagonist
- ILC3s
Group 3 Innate Lymphoid Cells
- LC3B-II
Microtubule-associated Protein 1 Light Chain 3B-II
- MAPK
Mitogen-activated Protein Kinase
- MBL
Mannan-binding Lectin
- MPO
Myeloperoxidase
- NET
Neutrophil Extracellular Trap
- NETosis
Neutrophil Extracellular Trap Formation
- NF-κB
Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells
- NLRP3
NOD-like Receptor Family Pyrin Domain-containing 3
- NOD
Nucleotide-binding Oligomerization Domain
- OPC
Oropharyngeal Candidiasis
- PAD4
Protein Arginine Deiminase 4
- PAMPs
Pathogen-associated Molecular Patterns
- PI3K
Phosphoinositide 3-kinase
- PKC
Protein Kinase C
- PKCδ
Protein Kinase C Delta
- PRRs
Pattern Recognition Receptors
- ROS
Reactive Oxygen Species
- S100A8/A9
S100 Calcium-binding Protein A8/A9
- SYK
Spleen Tyrosine Kinase
- Th17
T Helper 17 Cells
- TLR
Toll-like Receptor
- TNF-α
Tumor Necrosis Factor-alpha.
References
1
Fortuna MC Garelli V Pranteda G Carlesimo M D’Arino A Rossi A . Scalp infection by candida albicans in an immunocompetent patient: A description of a rare case. J Chemother. (2018) 30:316–7. doi: 10.1080/1120009X.2018.1518200
2
Murray BA . Mouth infection as a source of systemic disease. Can Med Assoc J. (1918) 8:988–92.
3
Estcourt LJ Stanworth S Doree C Blanco P Hopewell S Trivella M et al . Granulocyte transfusions for preventing infections in people with neutropenia or neutrophil dysfunction. Cochrane Database Syst Rev. (2015) 2015:CD005341. doi: 10.1002/14651858.CD005341.pub3
4
He Y Liu J Chen Y Yan L Wu J . Neutrophil extracellular traps in candida albicans infection. Front Immunol. (2022) 13:913028. doi: 10.3389/fimmu.2022.913028
5
Johnson CJ Nett JE . Examining neutrophil-candida auris interactions with human neutrophils ex vivo. Methods Mol Biol. (2022) 2517:243–50. doi: 10.1007/978-1-0716-2417-3_19
6
Immler R Simon SI Sperandio M . Calcium signalling and related ion channels in neutrophil recruitment and function. Eur J Clin Invest. (2018) 48 Suppl 2:e12964. doi: 10.1111/eci.12964
7
Kaieda S Kinoshita T Chiba A Miyake S Hoshino T . Il-18 receptor-alpha signalling pathway contributes to autoantibody-induced arthritis via neutrophil recruitment and mast cell activation. Mod Rheumatol. (2024) 34:500–8. doi: 10.1093/mr/road043
8
Mocsai A Walzog B Lowell CA . Intracellular signalling during neutrophil recruitment. Cardiovasc Res. (2015) 107:373–85. doi: 10.1093/cvr/cvv159
9
Presicce P Park CW Senthamaraikannan P Bhattacharyya S Jackson C Kong F et al . Il-1 signaling mediates intrauterine inflammation and chorio-decidua neutrophil recruitment and activation. JCI Insight. (2018) 3:e98306. doi: 10.1172/jci.insight.98306
10
Singhal A Dhankani P Gupta KL Mazumder J R A Dikshit M et al . Rho signaling inhibition mitigates lung injury via targeting neutrophil recruitment and selectin-akt signaling. Biochim Biophys Acta Mol Cell Res. (2021) 1868:119122. doi: 10.1016/j.bbamcr.2021.119122
11
Ermert D Niemiec MJ Rohm M Glenthoj A Borregaard N Urban CF . Candida albicans escapes from mouse neutrophils. J Leukoc Biol. (2013) 94:223–36. doi: 10.1189/jlb.0213063
12
Perry AM Hernday AD Nobile CJ . Unraveling how candida albicans forms sexual biofilms. J Fungi (Basel). (2020) 6:14. doi: 10.3390/jof6010014
13
Prasad P Tippana M . Morphogenic plasticity: the pathogenic attribute of candida albicans. Curr Genet. (2023) 69:77–89. doi: 10.1007/s00294-023-01263-5
14
Khamzeh A Dahlstrand Rudin A Venkatakrishnan V Stylianou M Sanchez Klose FP Urban CF et al . High levels of short-chain fatty acids secreted by candida albicans hyphae induce neutrophil chemotaxis via free fatty acid receptor 2. J Leukoc Biol. (2024) 115:536–46. doi: 10.1093/jleuko/qiad146
15
Reales-Calderon JA Tso GHW Tan ASM Hor PX Bohme J Teng KWW et al . Gut-evolved candida albicans induces metabolic changes in neutrophils. Front Cell Infect Microbiol. (2021) 11:743735. doi: 10.3389/fcimb.2021.743735
16
Pellon A Sadeghi Nasab SD Moyes DL . New insights in candida albicans innate immunity at the mucosa: toxins, epithelium, metabolism, and beyond. Front Cell Infect Microbiol. (2020) 10:81. doi: 10.3389/fcimb.2020.00081
17
Zhou Y Cheng L Lei YL Ren B Zhou X . The interactions between candida albicans and mucosal immunity. Front Microbiol. (2021) 12:652725. doi: 10.3389/fmicb.2021.652725
18
G M Ravi SSS Maheswary D Leela KV Harikumar Lathakumari R K S L . Role of candida albicans in chronic inflammation and the development of oral squamous cell carcinoma. Cancer Pathog Ther. (2025) 3:402–10. doi: 10.1016/j.cpt.2025.03.002
19
Zhang F Xia Y Su J Quan F Zhou H Li Q et al . Neutrophil diversity and function in health and disease. Signal Transduct Target Ther. (2024) 9:343. doi: 10.1038/s41392-024-02049-y
20
Juszczak M Brankiewicz A Zawrotniak M Rapala-Kozik M . The hypoxic microenvironment of candida albicans biofilms shapes neutrophil responses. Front Immunol. (2025) 16:1547559. doi: 10.3389/fimmu.2025.1547559
21
Yang F Gritsenko V Slor Futterman Y Gao L Zhen C Lu H et al . Tunicamycin potentiates antifungal drug tolerance via aneuploidy in candida albicans. mBio. (2021) 12:e0227221. doi: 10.1128/mBio.02272-21
22
Gresnigt MS Joosten LA Verschueren I van der Meer JW Netea MG Dinarello CA et al . Neutrophil-mediated inhibition of proinflammatory cytokine responses. J Immunol. (2012) 189:4806–15. doi: 10.4049/jimmunol.1103551
23
Zawrotniak M Juszczak M Rapala-Kozik M . Release of neutrophil extracellular traps in response to candida albicans yeast, as a secondary defense mechanism activated by phagocytosis. Yeast. (2023) 40:349–59. doi: 10.1002/yea.3842
24
Bojang E Ghuman H Kumwenda P Hall RA . Immune sensing of candida albicans. J Fungi (Basel). (2021) 7:119. doi: 10.3390/jof7020119
25
O’Brien XM Reichner JS . Neutrophil integrins and matrix ligands and net release. Front Immunol. (2016) 7:363. doi: 10.3389/fimmu.2016.00363
26
Li D Dong B Tong Z Wang Q Liu W Wang Y et al . Mbl-mediated opsonophagocytosis of candida albicans by human neutrophils is coupled with intracellular dectin-1-triggered ros production. PloS One. (2012) 7:e50589. doi: 10.1371/journal.pone.0050589
27
Segal BH Grimm MJ Khan AN Han W Blackwell TS . Regulation of innate immunity by nadph oxidase. Free Radic Biol Med. (2012) 53:72–80. doi: 10.1016/j.freeradbiomed.2012.04.022
28
Fang FC . Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. (2004) 2:820–32. doi: 10.1038/nrmicro1004
29
Miramon P Kasper L Hube B . Thriving within the host: candida spp. Interactions with phagocytic cells. Med Microbiol Immunol. (2013) 202:183–95. doi: 10.1007/s00430-013-0288-z
30
Urban CF Ermert D Schmid M Abu-Abed U Goosmann C Nacken W et al . Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PloS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
31
Urban CF Reichard U Brinkmann V Zychlinsky A . Neutrophil extracellular traps capture and kill candida albicans yeast and hyphal forms. Cell Microbiol. (2006) 8:668–76. doi: 10.1111/j.1462-5822.2005.00659.x
32
Kenno S Perito S Mosci P Vecchiarelli A Monari C . Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. Albicans morphotypes. Front Microbiol. (2016) 7:879. doi: 10.3389/fmicb.2016.00879
33
Wu SY Weng CL Jheng MJ Kan HW Hsieh ST Liu FT et al . Candida albicans triggers nadph oxidase-independent neutrophil extracellular traps through dectin-2. PloS Pathog. (2019) 15:e1008096. doi: 10.1371/journal.ppat.1008096
34
Byrd AS O’Brien XM Johnson CM Lavigne LM Reichner JS . An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to candida albicans. J Immunol. (2013) 190:4136–48. doi: 10.4049/jimmunol.1202671
35
Hogan D Wheeler RT . The complex roles of nadph oxidases in fungal infection. Cell Microbiol. (2014) 16:1156–67. doi: 10.1111/cmi.12320
36
Nunes P Demaurex N Dinauer MC . Regulation of the nadph oxidase and associated ion fluxes during phagocytosis. Traffic. (2013) 14:1118–31. doi: 10.1111/tra.12115
37
Swidergall M Solis NV Wang Z Phan QT Marshall ME Lionakis MS et al . Epha2 is a neutrophil receptor for candida albicans that stimulates antifungal activity during oropharyngeal infection. Cell Rep. (2019) 28:423–33 e5. doi: 10.1016/j.celrep.2019.06.020
38
Kernien JF Johnson CJ Nett JE . Conserved inhibition of neutrophil extracellular trap release by clinical candida albicans biofilms. J Fungi (Basel). (2017) 3:49. doi: 10.3390/jof3030049
39
Li D Bai C Zhang Q Li Z Shao D Li X . Beta-1,3-glucan/cr3/syk pathway-dependent lc3b-ii accumulation enhanced the fungicidal activity in human neutrophils. J Microbiol. (2019) 57:263–70. doi: 10.1007/s12275-019-8298-1
40
Li DD Jawale CV Zhou C Lin L Trevejo-Nunez GJ Rahman SA et al . Fungal sensing enhances neutrophil metabolic fitness by regulating antifungal glut1 activity. Cell Host Microbe. (2022) 30:530–44 e6. doi: 10.1016/j.chom.2022.02.017
41
Mosci P Gabrielli E Luciano E Perito S Cassone A Pericolini E et al . Involvement of il-17a in preventing the development of deep-seated candidiasis from oropharyngeal infection. Microbes Infect. (2014) 16:678–89. doi: 10.1016/j.micinf.2014.06.007
42
Trautwein-Weidner K Gladiator A Nur S Diethelm P LeibundGut-Landmann S . Il-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. (2015) 8:221–31. doi: 10.1038/mi.2014.57
43
Mata-Martinez P Bergon-Gutierrez M Del Fresno C . Dectin-1 signaling update: new perspectives for trained immunity. Front Immunol. (2022) 13:812148. doi: 10.3389/fimmu.2022.812148
44
Gringhuis SI Kaptein TM Remmerswaal EBM Drewniak A Wevers BA Theelen B et al . Fungal sensing by dectin-1 directs the non-pathogenic polarization of T(H)17 cells through balanced type I ifn responses in human dcs. Nat Immunol. (2022) 23:1735–48. doi: 10.1038/s41590-022-01348-2
45
Brown GD . Dectin-1: A signalling non-tlr pattern-recognition receptor. Nat Rev Immunol. (2006) 6:33–43. doi: 10.1038/nri1745
46
Yu Y Wang RR Miao NJ Tang JJ Zhang YW Lu XR et al . Pd-L1 negatively regulates antifungal immunity by inhibiting neutrophil release from bone marrow. Nat Commun. (2022) 13:6857. doi: 10.1038/s41467-022-34722-7
47
Drummond RA Franco LM Lionakis MS . Human card9: A critical molecule of fungal immune surveillance. Front Immunol. (2018) 9:1836. doi: 10.3389/fimmu.2018.01836
48
Whibley N Jaycox JR Reid D Garg AV Taylor JA Clancy CJ et al . Delinking Card9 and Il-17: Card9 Protects against Candida Tropicalis Infection through a Tnf-Alpha-Dependent, Il-17-Independent Mechanism. J Immunol. (2015) 195:3781–92. doi: 10.4049/jimmunol.1500870
49
Huppler AR Verma AH Conti HR Gaffen SL . Neutrophils do not express il-17a in the context of acute oropharyngeal candidiasis. Pathogens. (2015) 4:559–72. doi: 10.3390/pathogens4030559
50
Goupil M Cousineau-Cote V Aumont F Senechal S Gaboury L Hanna Z et al . Defective il-17- and il-22-dependent mucosal host response to candida albicans determines susceptibility to oral candidiasis in mice expressing the hiv-1 transgene. BMC Immunol. (2014) 15:49. doi: 10.1186/s12865-014-0049-9
51
Zhang C Chen J Wang H Chen J Zheng MJ Chen XG et al . Il-17 exacerbates experimental autoimmune prostatitis via cxcl1/cxcl2-mediated neutrophil infiltration. Andrologia. (2022) 54:e14455. doi: 10.1111/and.14455
52
Dos Santos Dias L Lionakis MS . Il-17: A critical cytokine for defense against oral candidiasis. J Immunol. (2024) 213:1049–51. doi: 10.4049/jimmunol.2400510
53
Verma AH Richardson JP Zhou C Coleman BM Moyes DL Ho J et al . Oral epithelial cells orchestrate innate type 17 responses to candida albicans through the virulence factor candidalysin. Sci Immunol. (2017) 2:eaam8834. doi: 10.1126/sciimmunol.aam8834
54
Brown MA Weinberg RB . Mast cells and innate lymphoid cells: underappreciated players in cns autoimmune demyelinating disease. Front Immunol. (2018) 9:514. doi: 10.3389/fimmu.2018.00514
55
Yang C Kwon DI Kim M Im SH Lee YJ . Commensal microbiome expands tgammadelta17 cells in the lung and promotes particulate matter-induced acute neutrophilia. Front Immunol. (2021) 12:645741. doi: 10.3389/fimmu.2021.645741
56
Moyes DL Wilson D Richardson JP Mogavero S Tang SX Wernecke J et al . Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. (2016) 532:64–8. doi: 10.1038/nature17625
57
Engku Nasrullah Satiman EAF Ahmad H Ramzi AB Abdul Wahab R Kaderi MA Wan Harun WHA et al . The role of candida albicans candidalysin ece1 gene in oral carcinogenesis. J Oral Pathol Med. (2020) 49:835–41. doi: 10.1111/jop.13014
58
Swidergall M Khalaji M Solis NV Moyes DL Drummond RA Hube B et al . Candidalysin is required for neutrophil recruitment and virulence during systemic candida albicans infection. J Infect Dis. (2019) 220:1477–88. doi: 10.1093/infdis/jiz322
59
Willems HME Lowes DJ Barker KS Palmer GE Peters BM . Comparative analysis of the capacity of the candida species to elicit vaginal immunopathology. Infect Immun. (2018) 86:e00527-18. doi: 10.1128/IAI.00527-18
60
Negoro PE Xu S Dagher Z Hopke A Reedy JL Feldman MB et al . Spleen tyrosine kinase is a critical regulator of neutrophil responses to candida species. mBio. (2020) 11:e02043-19. doi: 10.1128/mBio.02043-19
61
Langereis JD Raaijmakers HA Ulfman LH Koenderman L . Abrogation of nf-kappab signaling in human neutrophils induces neutrophil survival through sustained P38-mapk activation. J Leukoc Biol. (2010) 88:655–64. doi: 10.1189/jlb.0809544
62
Altmeier S Toska A Sparber F Teijeira A Halin C LeibundGut-Landmann S . Il-1 coordinates the neutrophil response to C. Albicans in the oral mucosa. PloS Pathog. (2016) 12:e1005882. doi: 10.1371/journal.ppat.1005882
63
Le HT Tran VG Kim W Kim J Cho HR Kwon B . Il-33 priming regulates multiple steps of the neutrophil-mediated anti-candida albicans response by modulating tlr and dectin-1 signals. J Immunol. (2012) 189:287–95. doi: 10.4049/jimmunol.1103564
64
Zeng H Wu Z Yu B Wang B Wu C Wu J et al . Network meta-analysis of triazole, polyene, and echinocandin antifungal agents in invasive fungal infection prophylaxis in patients with hematological Malignancies. BMC Cancer. (2021) 21:404. doi: 10.1186/s12885-021-07973-8
65
Heredia MY Gunasekaran D Ikeh MAC Nobile CJ Rauceo JM . Transcriptional regulation of the caspofungin-induced cell wall damage response in candida albicans. Curr Genet. (2020) 66:1059–68. doi: 10.1007/s00294-020-01105-8
66
Silling G Fegeler W Roos N Essink M Buchner T . Early empiric antifungal therapy of infections in neutropenic patients comparing fluconazole with amphotericin B/flucytosine. Mycoses. (1999) 42 Suppl 2:101–4. doi: 10.1111/j.1439-0507.1999.tb00023.x
67
Schlottfeldt Fdos S Fernandes SM Martins DM Cordeiro P Fonseca CD Watanabe M et al . Prevention of amphotericin B nephrotoxicity through use of phytotherapeutic medication. Rev Esc Enferm USP. (2015) 49 Spec No:74–9. doi: 10.1590/S0080-623420150000700011
68
Daneshnia F de Almeida Junior JN Ilkit M Lombardi L Perry AM Gao M et al . Worldwide emergence of fluconazole-resistant candida parapsilosis: current framework and future research roadmap. Lancet Microbe. (2023) 4:e470–e80. doi: 10.1016/S2666-5247(23)00067-8
69
Thomaz DY de Almeida JN Jr. Sejas ONE Del Negro GMB Carvalho G Gimenes VMF et al . Environmental clonal spread of azole-resistant candida parapsilosis with erg11-Y132f mutation causing a large candidemia outbreak in a Brazilian cancer referral center. J Fungi (Basel). (2021) 7:259. doi: 10.3390/jof7040259
70
Fekkar A Blaize M Bougle A Normand AC Raoelina A Kornblum D et al . Hospital outbreak of fluconazole-resistant candida parapsilosis: arguments for clonal transmission and long-term persistence. Antimicrob Agents Chemother. (2023) 95:e02036-20. doi: 10.1128/AAC.02036-20
71
Ramage G Borghi E Rodrigues CF Kean R Williams C Lopez-Ribot J . Our current clinical understanding of candida biofilms: where are we two decades on? APMIS. (2023) 131:636–53. doi: 10.1111/apm.13310
72
Atiencia-Carrera MB Cabezas-Mera FS Tejera E MaChado A . Prevalence of biofilms in candida spp. Bloodstream infections: A meta-analysis. PloS One. (2022) 17:e0263522. doi: 10.1371/journal.pone.0263522
73
Shankar M Lo TL Traven A . Natural variation in clinical isolates of candida albicans modulates neutrophil responses. mSphere. (2020) 5:e00501-20. doi: 10.1128/mSphere.00501-20
74
Johnson CJ Cabezas-Olcoz J Kernien JF Wang SX Beebe DJ Huttenlocher A et al . The extracellular matrix of candida albicans biofilms impairs formation of neutrophil extracellular traps. PloS Pathog. (2016) 12:e1005884. doi: 10.1371/journal.ppat.1005884
75
Lionakis MS Drummond RA Hohl TM . Immune responses to human fungal pathogens and therapeutic prospects. Nat Rev Immunol. (2023) 23:433–52. doi: 10.1038/s41577-022-00826-w
76
Casadevall A Pirofski LA . Adjunctive immune therapy for fungal infections. Clin Infect Dis. (2001) 33:1048–56. doi: 10.1086/322710
77
Desai JV Lionakis MS . The role of neutrophils in host defense against invasive fungal infections. Curr Clin Microbiol Rep. (2018) 5:181–9. doi: 10.1007/s40588-018-0098-6
78
Roilides E Farmaki E . Granulocyte colony-stimulating factor and other cytokines in antifungal therapy. Clin Microbiol Infect. (2001) 7 Suppl 2:62–7. doi: 10.1111/j.1469-0691.2001.tb00011.x
79
West KA Gea-Banacloche J Stroncek D Kadri SS . Granulocyte transfusions in the management of invasive fungal infections. Br J Haematol. (2017) 177:357–74. doi: 10.1111/bjh.14597
80
Pappas PG Kauffman CA Andes DR Clancy CJ Marr KA Ostrosky-Zeichner L et al . Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of america. Clin Infect Dis. (2016) 62:e1–50. doi: 10.1093/cid/civ933
81
Chen TK Batra JS Michalik DE Casillas J Patel R Ruiz ME et al . Recombinant human granulocyte-macrophage colony-stimulating factor (Rhu gm-csf) as adjuvant therapy for invasive fungal diseases. Open Forum Infect Dis. (2022) 9:ofac535. doi: 10.1093/ofid/ofac535
82
Vazquez JA Hidalgo JA De Bono S . Use of sargramostim (Rh-gm-csf) as adjunctive treatment of fluconazole-refractory oropharyngeal candidiasis in patients with aids: A pilot study. HIV Clin Trials. (2000) 1:23–9. doi: 10.1310/LF5T-WYY7-0U3E-G8BQ
83
van de Veerdonk FL Kullberg BJ Netea MG . Adjunctive immunotherapy with recombinant cytokines for the treatment of disseminated candidiasis. Clin Microbiol Infect. (2012) 18:112–9. doi: 10.1111/j.1469-0691.2011.03676.x
84
Mehta HM Malandra M Corey SJ . G-csf and gm-csf in neutropenia. J Immunol. (2015) 195:1341–9. doi: 10.4049/jimmunol.1500861
85
Chu S McCormick TS Lazarus HM Leal LO Ghannoum MA . Invasive fungal disease and the immunocompromised host including allogeneic hematopoietic cell transplant recipients: improved understanding and new strategic approach with sargramostim. Clin Immunol. (2021) 228:108731. doi: 10.1016/j.clim.2021.108731
86
Safdar A Rodriguez G Zuniga J Al Akhrass F Georgescu G Pande A . Granulocyte macrophage colony-stimulating factor in 66 patients with myeloid or lymphoid neoplasms and recipients of hematopoietic stem cell transplantation with invasive fungal disease. Acta Haematol. (2013) 129:26–34. doi: 10.1159/000342121
87
Onishi RM Gaffen SL . Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. (2010) 129:311–21. doi: 10.1111/j.1365-2567.2009.03240.x
88
Valeri M Raffatellu M . Cytokines il-17 and il-22 in the host response to infection. Pathog Dis. (2016) 74:ftw111. doi: 10.1093/femspd/ftw111
89
Rostan O Arshad MI Piquet-Pellorce C Robert-Gangneux F Gangneux JP Samson M . Crucial and diverse role of the interleukin-33/st2 axis in infectious diseases. Infect Immun. (2015) 83:1738–48. doi: 10.1128/IAI.02908-14
90
Park SJ Cho HR Kwon B . Roles of il-33 in resistance and tolerance to systemic candida albicans infections. Immune Netw. (2016) 16:159–64. doi: 10.4110/in.2016.16.3.159
91
Afonina IS Muller C Martin SJ Beyaert R . Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
92
Pham CT . Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol. (2008) 40:1317–33. doi: 10.1016/j.biocel.2007.11.008
93
Qu Y Zuo L Zhang S Zhou W Chen R . Comparative analysis of adverse events linked to peg-rhg-csf and rhg-csf in real-world settings: disproportionate examination of the us food and drug administration (Fda) adverse event reporting system (Faers) database. Clin Ther. (2025) 47:624–30. doi: 10.1016/j.clinthera.2025.04.017
94
Link H . Current state and future opportunities in granulocyte colony-stimulating factor (G-csf). Support Care Cancer. (2022) 30:7067–77. doi: 10.1007/s00520-022-07103-5
95
Olson KE Namminga KL Lu Y Schwab AD Thurston MJ Abdelmoaty MM et al . Safety, tolerability, and immune-biomarker profiling for year-long sargramostim treatment of parkinson’s disease. EBioMedicine. (2021) 67:103380. doi: 10.1016/j.ebiom.2021.103380
96
Kumar A Taghi Khani A Sanchez Ortiz A Swaminathan S . Gm-csf: A double-edged sword in cancer immunotherapy. Front Immunol. (2022) 13:901277. doi: 10.3389/fimmu.2022.901277
97
Chen Y Yi H Liao S He J Zhou Y Lei Y . Lc3b: A microtubule-associated protein influences disease progression and prognosis. Cytokine Growth Factor Rev. (2025) 81:16–26. doi: 10.1016/j.cytogfr.2024.11.006
98
Ahmad F Ahmad S Srivastav AK Upadhyay TK Husain A Khubaib M et al . Beta-glucan signalling stimulates nox-2 dependent autophagy and lc-3 associated autophagy (Lap) pathway. Int J Biol Macromol. (2024) 282:136520. doi: 10.1016/j.ijbiomac.2024.136520
99
Salcher S Sturm G Horvath L Untergasser G Kuempers C Fotakis G et al . High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. Cancer Cell. (2022) 40:1503–20 e8. doi: 10.1016/j.ccell.2022.10.008
100
Nishide M Nishimura K Matsushita H Kawada S Shimagami H Metsugi S et al . Neutrophil single-cell analysis identifies a type ii interferon-related subset for predicting relapse of autoimmune small vessel vasculitis. Nat Commun. (2025) 16:3581. doi: 10.1038/s41467-025-58550-7
101
Sun W Xu J Li S Zhao Y Fu J Di L et al . Glut1-mediated hmgb1 O-glcnacylation drives hyperglycemia-induced neutrophil extracellular trap networks formation via tlr4 signaling and exacerbates fibroblast inflammation. Sci Rep. (2025) 15:18853. doi: 10.1038/s41598-025-03642-z
102
Lika J Fan J . Carbohydrate metabolism in supporting and regulating neutrophil effector functions. Curr Opin Immunol. (2024) 91:102497. doi: 10.1016/j.coi.2024.102497
103
Kim TS Moutsopoulos NM . Neutrophils and neutrophil extracellular traps in oral health and disease. Exp Mol Med. (2024) 56:1055–65. doi: 10.1038/s12276-024-01219-w
104
Ando Y Tsukasaki M Huynh NC Zang S Yan M Muro R et al . The neutrophil-osteogenic cell axis promotes bone destruction in periodontitis. Int J Oral Sci. (2024) 16:18. doi: 10.1038/s41368-023-00275-8
105
Shen J Lin H Mo K Liang Z Zhang Y Quan H et al . Bidirectional roles of neutrophil extracellular traps in oral microbiota carcinogenesis: A systematic review. Transl Oncol. (2025) 56:102361. doi: 10.1016/j.tranon.2025.102361
106
Wang Y Li J Chen R Xu Q Wang D Mao C et al . Emerging concepts in mucosal immunity and oral microecological control of respiratory virus infection-related inflammatory diseases. Microbiol Res. (2024) 289:127930. doi: 10.1016/j.micres.2024.127930
107
Wigerblad G Kaplan MJ . Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat Rev Immunol. (2023) 23:274–88. doi: 10.1038/s41577-022-00787-0
108
Weindl G Wagener J Schaller M . Interaction of the mucosal barrier with accessory immune cells during fungal infection. Int J Med Microbiol. (2011) 301:431–5. doi: 10.1016/j.ijmm.2011.04.011
109
Thind MK Uhlig HH Glogauer M Palaniyar N Bourdon C Gwela A et al . A metabolic perspective of the neutrophil life cycle: new avenues in immunometabolism. Front Immunol. (2023) 14:1334205. doi: 10.3389/fimmu.2023.1334205
110
Ward RA Aghaeepour N Bhattacharyya RP Clish CB Gaudilliere B Hacohen N et al . Harnessing the potential of multiomics studies for precision medicine in infectious disease. Open Forum Infect Dis. (2021) 8:ofab483. doi: 10.1093/ofid/ofab483
111
Rizo-Tellez SA Filep JG . Beyond host defense and tissue injury: the emerging role of neutrophils in tissue repair. Am J Physiol Cell Physiol. (2024) 326:C661–C83. doi: 10.1152/ajpcell.00652.2023
112
Hageb A Farjia M Osei-Sarpong C Silvestre-Roig C . Ontogenetic drivers of neutrophil heterogeneity. Exp Hematol. (2025) 151:104863. doi: 10.1016/j.exphem.2025.104863
113
Bishu S Hernandez-Santos N Simpson-Abelson MR Huppler AR Conti HR Ghilardi N et al . The adaptor card9 is required for adaptive but not innate immunity to oral mucosal candida albicans infections. Infect Immun. (2014) 82:1173–80. doi: 10.1128/IAI.01335-13
114
Edgerton M Rojas I Kumar R Li R Salvatori O Abrams S et al . Neutrophil swarms containing myeloid-derived suppressor cells are crucial for limiting oral mucosal infection by C. Albicans. Res Sq. (2023). doi: 10.21203/rs.3.rs-3346012/v1
115
Vinh DC . The molecular immunology of human susceptibility to fungal diseases: lessons from single gene defects of immunity. Expert Rev Clin Immunol. (2019) 15:461–86. doi: 10.1080/1744666X.2019.1584038
116
Naglik JR Fidel PL Jr. Odds FC . Animal models of mucosal candida infection. FEMS Microbiol Lett. (2008) 283:129–39. doi: 10.1111/j.1574-6968.2008.01160.x
117
Drummond RA Lionakis MS . Mechanistic insights into the role of C-type lectin receptor/card9 signaling in human antifungal immunity. Front Cell Infect Microbiol. (2016) 6:39. doi: 10.3389/fcimb.2016.00039
118
Al-Kenani HQM Shaheed OM . Evaluation of some immunological markers in co-infection of covid-19 with thrush candidiasis. Rev Assoc Med Bras (1992). (2024) 70:e20230845. doi: 10.1590/1806-9282.20230845
119
Vila T Sultan AS Montelongo-Jauregui D Jabra-Rizk MA . Oral candidiasis: A disease of opportunity. J Fungi (Basel). (2020) 6:15. doi: 10.3390/jof6010015
120
Ji C Yang Z Zhong X Xia J . The role and mechanism of card9 gene polymorphism in diseases. BioMed J. (2021) 44:560–6. doi: 10.1016/j.bj.2020.12.006
121
Brown GD Denning DW Gow NA Levitz SM Netea MG White TC . Hidden killers: human fungal infections. Sci Transl Med. (2012) 4:165rv13. doi: 10.1126/scitranslmed.3004404
122
Shang L Deng D Krom BP Gibbs S . Oral host-microbe interactions investigated in 3d organotypic models. Crit Rev Microbiol. (2024) 50:397–416. doi: 10.1080/1040841X.2023.2211665
123
Last A Maurer M A SM M SG Hube B . In vitro infection models to study fungal-host interactions. FEMS Microbiol Rev. (2021) 45:fuab005. doi: 10.1093/femsre/fuab005
124
Baxter CG Moore CB Jones AM Webb AK Denning DW . Ige-mediated immune responses and airway detection of aspergillus and candida in adult cystic fibrosis. Chest. (2013) 143:1351–7. doi: 10.1378/chest.12-1363
125
Moreno-Velasquez SD Perez JC . Imaging and quantification of mrna molecules at single-cell resolution in the human fungal pathogen candida albicans. mSphere. (2021) 6:e0041121. doi: 10.1128/mSphere.00411-21
126
Sulyanto RM Beall CJ Ha K Montesano J Juang J Dickson JR et al . Fungi and bacteria occupy distinct spatial niches within carious dentin. PloS Pathog. (2024) 20:e1011865. doi: 10.1371/journal.ppat.1011865
Summary
Keywords
neutrophil, oral candidiasis, Candida albicans, neutrophil extracellular trap (NET), immune regulatory networks
Citation
Xu Y, Cheng R, Li W, Yu W and Chen C (2025) Molecular mechanisms of neutrophil regulatory network in anti-Candida infection. Front. Immunol. 16:1716645. doi: 10.3389/fimmu.2025.1716645
Received
30 September 2025
Accepted
31 October 2025
Published
18 November 2025
Volume
16 - 2025
Edited by
Takao Fukuda, Kyushu University, Japan
Reviewed by
Tetsuhiro Kajikawa, Tohoku University, Japan
Weiwei Deng, Third Affiliated Hospital of Sun Yat-sen University, China
Updates
Copyright
© 2025 Xu, Cheng, Li, Yu and Chen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chider Chen, chenc10@upenn.edu
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.