Introduction
Fish in aquatic environments generally obtain oxygen from the water. Mud loaches inhabit muddy swamps, ponds, and rice fields subject to periodic drying. The respiratory systems of freshwater fish subject to drought have adapted to enable cutaneous/air respiration via other organs. Loaches can breathe in water or soil depending on the dissolved oxygen content (Park et al., 2001). Mud loach aquaculture in freshwater is common in South Korea, Taiwan, China, and Japan. The mud loach Misgurnus mizolepis belongs to the family Cobitidae and is widely used in basic biological research. It can live in soil and water and survive in human wastewater, such as ditches and septic tanks; it is also useful for harvesting antimicrobial peptides. Controlling disease outbreaks in aquaculture systems, such as the 2012 Aeromonas sobria outbreak that caused 61% mortality in 2 days, is a major challenge. This devastated fisheries aquaculture production; the production in the previous 5 years averaged 766 tons and was valued at $ 7.2 M (https://www.kostat.go.kr/). Considering the above, and to expand genetic research to preserve this species, we generated a draft genome for Misgurnus mizolepis. Presently, only the mitochondrial genome of this species is available (Lee, 2016) and no nuclear genomes for the family Cobitidae have been reported.
Value of the Data
This M. mizolepis genome is the first reference genome for molecular studies in the family Cobitidae. It should be useful for comparative analyses among or within species in the genus Misgurnus or closely related genera in the family Cobitidae, and could enhance the genome selection process in molecular breeding.
Materials and Methods
Sampling and Genomic DNA and RNA Preparation
Four 1-year-old wild M. mizolepis were supplied by Inland Aquaculture Research Center, National Institute Fisheries Science (NIFS), Changwon, South Korea in April 2019, at Buk-myeon, Jeongeup, South Korea. Abdominal muscle tissue was removed aseptically from one specimen as per the NIFS ethical committee provided instruction (2018-NIFS-IACUC-03) and dipped in liquid nitrogen for genomic DNA (gDNA) and RNA preparation; liver, abdominal muscle, and brain tissues were taken from the other three specimens for RNA extraction. The DNA and RNA isolation and sequencing were conducted by DNALink (Seoul, South Korea).
Genomic DNA and Transcriptome Sequencing
The gDNA and RNA were isolated from the samples by the DNeasy Animal Mini Kit and RNeasy Animal Mini Kit (QIAGEN, Hilden, Germany), respectively. The isolated gDNA sequenced with PacBio Sequel platform (Pacific Biosciences of California, Menlo Park, CA, USA), by capturing a 240-min movies for each SMRT cell. The RNA from the same individual was converted into cDNA using the SMARTer PCR cDNA Synthesis kit and subjected for the above steps for SMRTbell™ library preparation (except for fragmentation), and then sequenced with the PacBio Sequel platform. Similarly, another portion of the isolated gDNA and RNA from three different tissue samples of the three biological replicates was used to prepare sequencing libraries with the stranded Illumina paired-end (PE) protocol, using the TruSeq Nano DNA Prep Kit and TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA), respectively. The Illumina NovaSeq6000 sequence machine used with desire size of DNA and RNA fragments.
Sequencing Read Preprocessing and Genome Size Estimation
The DNA and mRNA sequences from illumina sequencer were subjected to pre-processing steps involving adapter and quality trimming (Q20), with subsequent contaminant removal for DNA sequences. The adapter and quality trimming processes were conducted using Trimmomatic-0.32 functions (Bolger et al., 2014), and microbial contaminants were removed using Bowtie2 with specific in-house database constructed for bacterial, viral, and marine metagenomes. The processed DNA sequences from the Paired end library were subjected to genome size estimation using the k-mer based method (Shin et al., 2018). The k-mer frequencies (k-mer size = 21) were received by Jellyfish v2.0 (Marçais and Kingsford, 2011) and calculated using the below formulas: Genome Coverage Depth (CD) = (k-mer CD) × Average Read Length (ARL)/(ARL – k-mer size + 1) and Genome size = Total Base Number/Genome CD.
De-novo Genome Assembly and Scaffolding
Error correction for the complete sequence processed with SMRT Analysis v2.3, and imported into a diploid-aware FALCON (Chin et al., 2016) genome assembler used to assemble long contigs from the PacBio reads. Additionally, assembled contigs subjected to polishing by Quiver method to reduce the base call errors (Chin et al., 2013). Further, contigs were used to assess the genome completeness with BUSCO v5.0 (Simão et al., 2015). The reference BUSCO dataset was actinopterygii_odb10. The quality of the assembled genome was assessed by short-read mapping to the draft assembly with Bowtie2. Finally, the assembled contigs were scaffolded based on 25 chromosomes of the stone loach Triplophysa tibetana genome (GCA_008369825.1), which belongs to suborder Cobitoidei, using RagTag v2.0.1 (Alonge et al., 2019). The unknown sequences between contigs in a scaffold were filled with 100 bp of N.
De-novo Repeats Identification Process
Repeat regions in assembled genome were predicted using the de-novo method and categorized into repeat subclasses; de novo repeats estimation for M. mizolepis was conducted using RepeatModeler, which incorporated with methods as RECON (Bao and Eddy, 2002), RepeatScout (Price et al., 2005), and TRF (Benson, 1999). Those modeled repeats were sub-categorized using the Repbase v20.08 database as a reference (Bao et al., 2015), and the repeats were masked using RepeatMasker v4.0.5 with RMBlastn v2.2.27+.
Gene-Prediction and Annotation
Genes from the genome of M. mizolepis, predicted by an gene prediction pipeline developed in house, which incorporated an evidence-based gene modeler, an ab-initio gene modeler, and a consensus gene modeler. After gene prediction, functional annotation was conducted for the consensus genes. Initially, sequenced transcriptomes from the Illumina Novaseq6000 were mapped to the draft repeat-masked draft genome using STAR (Dobin et al., 2013) and assembled transcriptome used for genome-guided Trinity (Grabherr et al., 2011; Haas et al., 2013). Full-length transcript sequences were also generated from high-fidelity PacBio Sequel cDNA sequences using IsoSeq3 (Pacific Biosciences of California, Menlo Park, CA, USA). The de novo assembled transcriptome and full-length transcript sequences were then subjected to the following steps. To train the ab-initio and evidence-based gene models, which include Exonerate (Slater and Birney, 2005) and AUGUSTUS (Stanke et al., 2006), with several genomes were used for gene prediction (Supplementary Table 4). Finally, the transcripts and predicted evidence-based gene and ab-initio models were subjected to a “consensus gene modeler” to produce the final gene and transcript models. The consensus transcripts were subjected to functional annotation using biological databases (NCBI-NR, Swiss-Prot, Gene Ontology, and KEGG databases) using BLAST+ v2.6, OmicsBox v1.4 and Trinotate v3.2 (Bryant et al., 2017).
Gene Expression Profiling
The pre-processed RNA-Seq reads from the liver, muscle, and brain tissues of three biological replicates were mapped to the coding sequences of the predicted genes using Salmon v1.4 (Patro et al., 2017). Genes with NumReads (estimated read counts) values greater than five and transcript per million (TPM) values > 0.3 in one or more tissue-specific group(s) were counted as expressed. Differentially expressed genes (DEGs) were identified using edgeR v3.30 in the TCC v1.28 R package (Robinson et al., 2010; Sun et al., 2013), with a threshold of 2 for log2 fold-change values and 0.05 for false-discovery rates (FDRs) in the pairwise control-case comparisons.
Mitochondrial Genome Assembly and Annotation
Pre-processed DNA short reads, including organelle sequence reads, were used to assemble the M. mizolepis mitochondrial genome (mitogenome) using NOVOPlasty v4.2 (Dierckxsens et al., 2017), assisted by the reference M. mizolepis mitogenome sequence (NC_038151.1). The mito-genome annotated with MitoAnnotator from MitoFish database (Iwasaki et al., 2013).
Preliminary Analysis
Initially, the M. mizolepis genome was estimated to be 974.4 MB (Figure 1A), with 49.6 GB of short read sequences (Table 1A; Supplementary Tables 1, 2); 1.112 GB of the representative contigs were de-novo assembled from 96.2 GB of error-corrected long read sequences (Supplementary Tables 2, 3). Then, the de-novo assembled contigs were scaffolded into 135 scaffolds of the draft genome, with a scaffold length N50 of 41,826,286 bases and an average scaffold length of 8,237,736.20 bases (Table 1B; Figure 1B). In total, 574 MB (i.e., 51.65%) of the draft genome was covered by repeats, in which DNA elements dominated (i.e., 28.09%) (Table 1B; Figure 1C; Supplementary Table 5). First, 99.33% of the pre-processed whole-genome sequencing reads, and an average of 81.82% of the pre-processed RNA-Seq reads, were mapped on the draft genome (Supplementary Table 1; Supplementary Figure 2). In total, 43,153 genes predicted in the genome, with an average size of 10,169.51 bases and a 95.8% complete BUSCO score (Tables 1B,C and Figure 1D). Ultimately, 33,326 genes had homologous sequences in GenBank and 31,338 had Gene Ontology annotations (Table 1D). Of the 43,153 genes, 24,699 were found to be expressed and 13,385 were DEGs (Supplementary Table 6; Supplementary Figure 3). The mitogenome was assembled into a complete circular sequence of 16,570 bases, annotated with 13 protein-coding genes, 22 tRNA genes, and 2 rRNA genes (Greiner et al., 2019) (Supplementary Table 7; Supplementary Figure 4). The complete workflow used in this study is shown in Supplementary Figure 1. This is the first genome assembly for the family Cobitidae. Due to the lack of genomic knowledge of this lineage, most of the NCBI-NR BLAST annotations overlapped with the proteome of the closely related suborder Cyprinoidei, which have well-established genomic profiles (Figure 1E). Distinct HoxBb cluster duplication was inferred in the M. mizolepis genome, but was not found in most genomes of closely related teleosts, including zebra fish (Danio rerio) (Hoegg et al., 2007; Henkel et al., 2012) (Figure 1F). This first genome assembly for the family Cobitidae can be used to elucidate additional genomic features to better understand this lineage, and provides new insight for comparative genomic studies of teleosts.
Figure 1
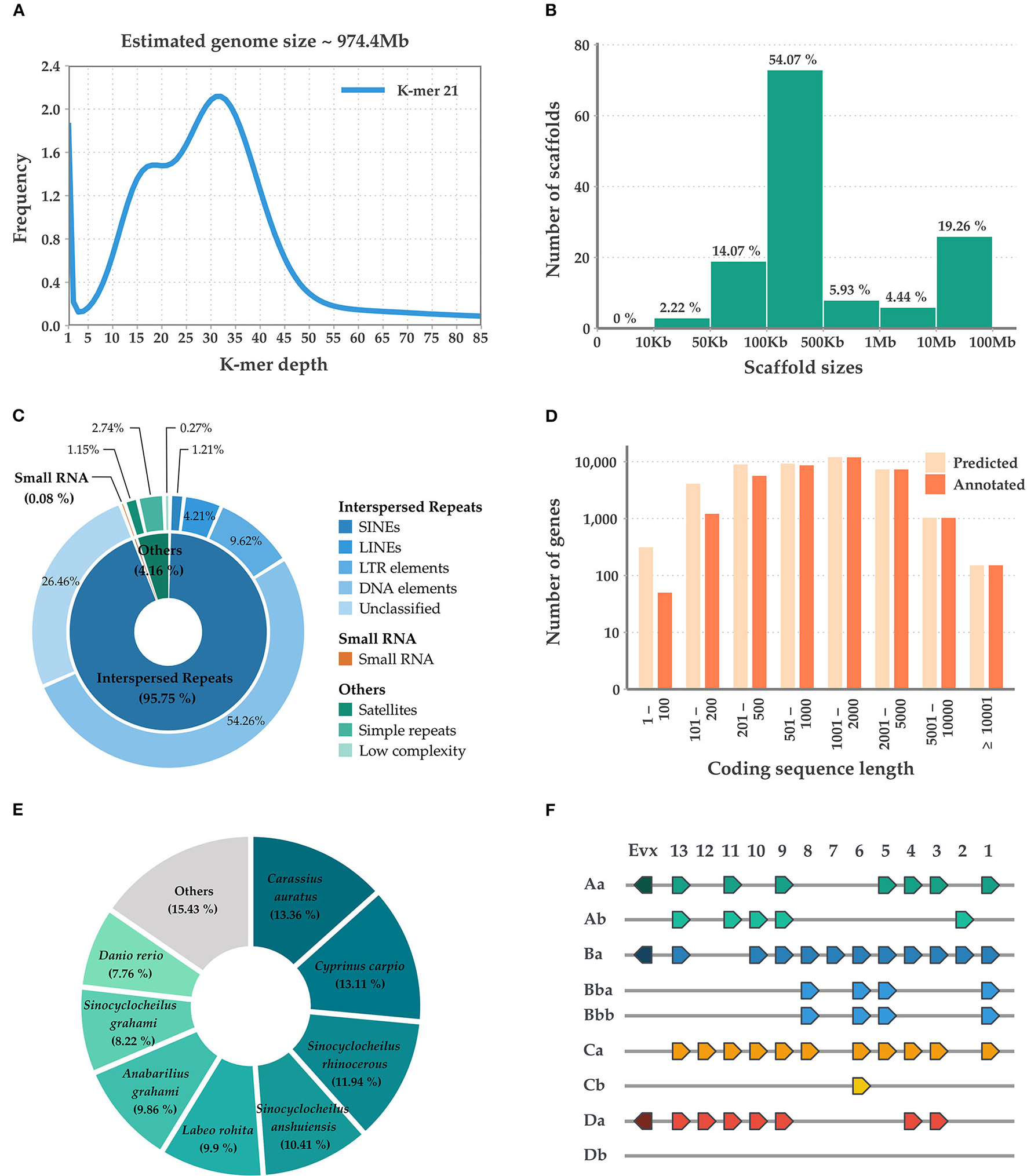
Summary of the sequencing, assembly, and annotation of the Misgurnus mizolepis draft genome. (A)K-mer-based genome size estimation; (B) Length distribution of the assembled scaffolds; (C)De-novo repeat prediction for three classes (inner circle) and their sub-classes (outer circle); (D) Length distribution of predicted, functionally annotated genes; (E) Species distribution of the top NCBI-NR BLAST hits; and (F) Hox gene clusters.
Table 1
| (A) Sequencing | |
| Illumina short-read yield | 49,615,230,708 bp |
| Pre-processed short-read data | 38,976,952,577 bp |
| PacBio long-read yield | 105,939,920,101 bp |
| High-quality subread data | 96,172,405,713 bp |
| (B) Assembly and scaffolding | |
| No. of scaffolds | 135 |
| Total bases | 1,112,094,387 bp |
| Average length | 8,237,736.20 bp |
| Minimum length | 37,458 bp |
| Maximum length | 77,600,393 bp |
| N50 | 41,826,286 bp |
| Ns | 43,700 bp (0.00%) |
| GC ratio | 38.07% |
| Repeats | 574,403,339 bp (51.65%) |
| Complete BUSCO (Actinopterygii_odb10) | 3,487 (95.8%) |
| (C) Gene prediction | |
| No. of genes | 43,153 |
| Average gene length | 10,169.51 bp |
| Genome coverage | 39.46% |
| Exon/gene | 7.07 |
| Average exon length | 190.31 bp |
| Average intron length | 1,454.05 bp |
| (D) Annotations | |
| NCBI nr BLAST hits | 33,326 |
| UniProt BLAST hits | 29,212 |
| Gene ontology hits | 31,338 |
| KEGG orthology hits | 26,665 |
| EggNOG hits | 24,685 |
| Pfam hits | 26,036 |
| SignalP hits | 3,578 |
| TmHMM hits | 8,362 |
| No annotation hits | 7,287 |
Summary of the sequencing for annotation of the Misgurnus mizolepis draft genome.
Funding
This work contributed by the Collaborative Genome Program of the Korea Institute of Marine Science and Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (MOF) (No. 2018043) and the National Institute of Fisheries Science (R2021041).
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was reviewed and approved by National Institute Fisheries Science (2018-NIFS-IACUC-03). Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
YS and J-HJ: genome assembly and annotations. YS, G-HS, and B-HN: manuscript preparation. EN and HK: sampling and sequencing. B-HN: funding acquisition and modeling. EK and J-HJ: data curation. Y-OK: investigation. All authors contributed to the article and approved the submitted version.
Conflict of interest
YS, J-HJ, and G-HS were employed by Insilicogen. Inc. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.799148/full#supplementary-material
References
1
Alonge M. Soyk S. Ramakrishnan S. Wang X. Goodwin S. Sedlazeck F. J. et al . (2019). RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol.20:224. 10.1186/s13059-019-1829-6
2
Bao W. Kojima K. K. Kohany O. (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA6:11. 10.1186/s13100-015-0041-9
3
Bao Z. Eddy S. R. (2002). Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res.12, 1269–1276. 10.1101/gr.88502
4
Benson G.. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res.27, 573–580. 10.1093/nar/27.2.573
5
Bolger A. M. Lohse M. Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics30, 2114–2120. 10.1093/bioinformatics/btu170
6
Bryant D. M. Johnson K. DiTommaso T. Tickle T. Couger M. B. Payzin-Dogru D. et al . (2017). A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep.18, 762–776. 10.1016/j.celrep.2016.12.063
7
Chin C.-S. Alexander D. H. Marks P. Klammer A. A. Drake J. Heiner C. et al . (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods10:563. 10.1038/nmeth.2474
8
Chin C. S. Peluso P. Sedlazeck F. J. Nattestad M. Concepcion G. T. Clum A. et al . (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods13, 1050–1054. 10.1038/nmeth.4035
9
Dierckxsens N. Mardulyn P. Smits G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res.45:e18. 10.1093/nar/gkw955
10
Dobin A. Davis C. A. Schlesinger F. Drenkow J. Zaleski C. Jha S. et al . (2013). STAR: ultrafast universal RNA-Seq aligner. Bioinformatics29, 15–21. 10.1093/bioinformatics/bts635
11
Grabherr M. G. Haas B. J. Yassour M. Levin J. Z. Thompson D. A. Amit I. et al . (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol.29, 644–652. 10.1038/nbt.1883
12
Greiner S. Lehwark P. Bock R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res.47, W59–W64. 10.1093/nar/gkz238
13
Haas B. J. Papanicolaou A. Yassour M. Grabherr M. Blood P. D. Bowden J. et al . (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc.8, 1494–1512. 10.1038/nprot.2013.084
14
Henkel C. V. Burgerhout E. de Wijze D. L. Dirks R. P. Minegishi Y. Jansen H. J. et al . (2012). Primitive duplicate Hox clusters in the European eel's genome. PLoS ONE7:e32231. 10.1371/journal.pone.0032231
15
Hoegg S. Boore J. L. Kuehl J. V. Meyer A. (2007). Comparative phylogenomic analyses of teleost fish Hox gene clusters: lessons from the cichlid fish Astatotilapia burtoni. BMC Genomics8:317. 10.1186/1471-2164-8-317
16
Iwasaki W. Fukunaga T. Isagozawa R. Yamada K. Maeda Y. Satoh T. P. et al . (2013). MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol.30, 2531–2540. 10.1093/molbev/mst141
17
Lee S.. (2016). Complete mitochondrial genome of the mud loach Misgurnus mizolepis (Cypriniformes, Cobitidae) and its phylogenetic position in the Cypriniformes. Mitochondrial DNA Part B1, 839–840. 10.1080/23802359.2016.1247675
18
Marçais G. Kingsford C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics27, 764–770. 10.1093/bioinformatics/btr011
19
Park J. Y. Kim I. S. Kim S. Y. (2001). Morphology and histochemistry of the skin of the mud loach, Misgurnus mizolepis, in relation to cutaneous respiration. Korean J. Biol. Sci.5, 303–308. 10.1080/12265071.2001.9647619
20
Patro R. Duggal G. Love M. I. Irizarry R. A. Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods14, 417–419. 10.1038/nmeth.4197
21
Price A. L. Jones N. C. Pevzner P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics21, i351–i358. 10.1093/bioinformatics/bti1018
22
Robinson M. D. McCarthy D. J. Smyth G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics26, 139–140. 10.1093/bioinformatics/btp616
23
Shin G.-H. Shin Y. Jung M. Hong J.-M. Lee S. Subramaniyam S. et al . (2018). First draft genome for red sea bream of family sparidae. Front. Genet.9:643. 10.3389/fgene.2018.00643
24
Simão F. A. Waterhouse R. M. Ioannidis P. Kriventseva E. V. Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics31, 3210–3212. 10.1093/bioinformatics/btv351
25
Slater G. S. C. Birney E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinform.6:31. 10.1186/1471-2105-6-31
26
Stanke M. Schöffmann O. Morgenstern B. Waack S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform.7, 62–62. 10.1186/1471-2105-7-62
27
Sun J. Nishiyama T. Shimizu K. Kadota K. (2013). TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinform.14:219. 10.1186/1471-2105-14-219
Summary
Keywords
Cobitidae, fish, genome, mud loach, Misgurnus mizolepis
Citation
Shin Y, Noh ES, Jeon J-H, Shin G-H, Kim EM, Kim Y-O, Kim H, Jung H and Nam B-H (2022) First Draft Genome of a Mud Loach (Misgurnus mizolepis) in the Family Cobitidae. Front. Mar. Sci. 8:799148. doi: 10.3389/fmars.2021.799148
Received
21 October 2021
Accepted
29 November 2021
Published
03 January 2022
Volume
8 - 2021
Edited by
Stephen Allen Smith, Virginia Tech, United States
Reviewed by
Sonia Andrade, University of São Paulo, Brazil; Khor Waiho, University of Malaysia Terengganu, Malaysia
Updates
Copyright
© 2022 Shin, Noh, Jeon, Shin, Kim, Kim, Kim, Jung and Nam.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo-Hye Nam nambohye@korea.kr
†These authors have contributed equally to this work
This article was submitted to Marine Fisheries, Aquaculture and Living Resources, a section of the journal Frontiers in Marine Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.