 Tatsuhiko Hoshino1
Tatsuhiko Hoshino1 Yuki Morono1
Yuki Morono1 Takeshi Terada2
Takeshi Terada2 Hiroyuki Imachi3
Hiroyuki Imachi3 Timothy G. Ferdelman4
Timothy G. Ferdelman4 Fumio Inagaki1*
Fumio Inagaki1*- 1 Geomicrobiology Group, Kochi Institute for Core Sample Research, Japan Agency for Marine-Earth Science and Technology, Nankoku, Kochi, Japan
- 2 Marine Works Japan Ltd., Yokohama, Japan
- 3 Extremobiosphere Research Program, Institute of Biogeosciences, Japan Agency for Marine-Earth Science and Technology, Yokosuka, Japan
- 4 Biogeochemistry Group, Max Planck Institute for Marine Microbiology, Bremen, Germany
Subseafloor sedimentary environments harbor remarkably diverse microbial communities. However, it remains unknown if the deeply buried fossils in these sediments play ecological roles in deep microbial habitats, or whether the microbial communities inhabiting such fossils differ from those in the surrounding sediment matrix. Here we compare the community structures of subseafloor microbes in cold-water coral carbonates (Madrepora oculata and Lophelia pertusa) and the clay matrix. Samples were obtained from the Challenger Mound in the Porcupine Seabight at Site U1317 Hole A during the Integrated Ocean Drilling Program Expedition 307. DNA was extracted from coral fossils and the surrounding sedimentary matrix at 4, 20, and 105 m below the seafloor. 16S rRNA genes of Bacteria and Archaea were amplified by PCR, and a total of 213,792 16S rRNA gene-tagged sequences were analyzed. At the phylum level, dominant microbial components in both habitats consisted of Proteobacteria, Firmicutes, Nitrospirae, Chloroflexi, and Miscellaneous Crenarchaeota Group (MCG) at all three of the depths examined. However, at the genus and/or species level (similarity threshold 97.0%), the community compositions were found to be very different, with 69–75 and 46–57% of bacterial and archaeal phylotypes not overlapping in coral fossils and the clay matrix, respectively. Species richness analysis revealed that bacterial communities were generally more diverse than archaea, and that the diversity scores of coral fossils were lower than those in sediment matrix. However, the evenness of microbial communities was not significantly different in all the samples examined. No eukaryotic DNA sequences, such as 18S rRNA genes, were obtained from the corals. The findings suggested that, even at the same or similar depths, the sedimentological characteristics of a habitat are important factors affecting microbial diversity and community structure in deep subseafloor sedimentary habitats.
Introduction
During the past decade, numerous studies to infer microbial diversity using 16S rRNA gene-clone libraries have been undertaken in subseafloor sedimentary habitats that have been explored by scientific ocean drilling. These molecular ecological surveys have demonstrated that phylogenetically diverse bacteria and archaea are present, even in deep and old subseafloor sediments, and that these taxa are generally physiologically distinct from known isolates obtained at the surface (e.g., Inagaki et al., 2006; Teske, 2006; Webster et al., 2006). In most cases, core samples used for molecular analysis are 10-cm (or less) whole-round cores (i.e., without half-splitting), which are immediately frozen onboard. Since this sampling scheme minimizes potential microbiological contamination of the innermost core region, microbiologists are able to extract bulk nucleic acids from indigenous microbial cells for molecular ecological studies. However, although 10 cm whole-round core samples may represent a certain period of sedimentological history, little is known about how the concealed sedimentological characteristics and physical properties affect microbial diversity and community structure in such a fine-scaled habitat. For example, a previous study revealed that microbial diversity in hemipelagic clay and volcanic ash layers of a core from the Sea of Okhotsk supported very different bacterial and archaeal subseafloor microbial communities (Inagaki et al., 2003). In addition, differences in paleoenvironmental settings, such as depositional rates, affect the hydrological regimes of sediments resulting in stratification of different microbial communities at different depths (Beck et al., 2011).
During the Integrated Ocean Drilling Program (IODP) Expedition 307, cold-water coral reefs buried in Pleistocene sediments were recovered from Site U1317 on the Challenger Mound in the Porcupine Seabight located in the southwest of Ireland (Ferdelman et al., 2006). The cored samples provided an unprecedented opportunity to study how buried fossil corals (i.e., carbonate skeletons), which constitute sedimentologically distinct habitat compared to the surrounding matrix, affect subseafloor microbial communities. The upper sedimentary unit at Site U1317 was primarily composed of dead, deep-sea cold-water coral matter derived from species such as Madrepora oculata and Lophelia pertusa, that had been embedded in a clay-type matrix (De Mol et al., 2007). Previous microbiological studies of living Lophelia corals on the seafloor showed that microbial communities associated with Lophelia are distinct from those in the surrounding sediment and seawater, and that this was likely because the living corals provided the microbes with specific nutrients and niches (Schöttner et al., 2009). However, it is not currently known how the burial of coral reefs and its diagenetic processes (e.g., carbonate dissolution) affect the microbial diversity or community structure of microbes in subseafloor sediments.
Microscopic and quantitative molecular analyses of the Pleistocene sediment matrix at Site U1317 revealed that the site harbors relatively low numbers of microbial cells (∼106 cell/cm3; Webster et al., 2009). The composition of bacterial communities was previously examined at three different depths by denaturing gradient gel electrophoresis (DGGE). The few resulting DGGE-fragments were subjected to nested-PCR analysis, which revealed the presence of members of Beta-, Gamma-, Deltaproteobacteria, Chloroflexi, and Actinobacteria (Webster et al., 2009). Tracer incubation experiments (e.g., methanogenesis and thymidine incorporation) indicated the presence of metabolically active microbial components that mediate biogeochemical cycles in the carbonate mound (Webster et al., 2009), albeit at very low rates for continental margin setting.
In this study, we extracted DNA from the coral carbonate structure and the surrounding clay matrix at three different depths at Site U1317, and examined the community structures of the associated bacterial and archaeal communities by statistically analyzing 454-pyrosequenced 16S rRNA gene fragments. We also attempted PCR amplification of 18S rRNA genes to determine whether the coral DNA fragments buried in the ancient sedimentary layers had been preserved or not. Based on the molecular results obtained from two distinct sedimentary habitats at almost the same depth and age, the potential impacts of sedimentological characteristics on subseafloor microbial communities were also discussed.
Materials and Methods
Site Description
We examined the coral fossils contained in deep-frozen, whole-round cores obtained 4, 20, and 105 meters below the seafloor (mbsf; sample codes: 1H-3, 3H-3, and 12H-3, respectively). Core samples were obtained as 10 cm whole-round cores from Site U1317 (51°22.8′N, 11°43.1′W) at the Challenger Mound in the Porcupine Seabight during IODP Expedition 307 (Ferdelman et al., 2006). Total organic carbon content in the clay matrix of 1H-3, 3H-3, and 12H-3 were 0.06, 0.21, and 0.31%, respectively. Microbial cell counts, as well as some other biogeochemical and paleontological characteristics, such as sulfate and methane concentrations and sedimentation ages, have been reported elsewhere (Ferdelman et al., 2006; Kano et al., 2007; Webster et al., 2009). After core recovery on the drilling vessel JOIDES Resolution, the whole-round cores were immediately placed in a deep freezer and kept at −80°C until laboratory use. X-ray computed tomography (CT) showed that the cores contained intact carbonate skeletons of deep-sea cold-water corals, such as L. pertusa and M. oculata (De Mol et al., 2007; Figures 1A,B); the seafloor sediments of the Challenger Mound have few to no modern living corals (Foubert et al., 2005).
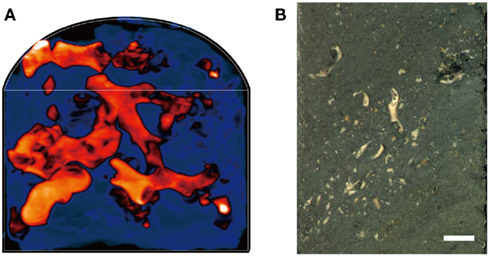
Figure 1. An example of core samples that contain coral fossils. (A) A 3D X-ray CT scan image of a half-round core sample from 20 mbsf (Sample code: 3H-3). Orange-red color indicates the buried coral fossils, while dark blue indicate the sediment matrix. (B) A photo image of half-round core surface (3H-3). Scale bar: 1 cm.
X-ray Computed Tomography
X-ray CT scanning was performed using PRATICO X-ray CT Scanner (Hitachi Medical Corporation, Tokyo, Japan) with a resolution of 0.5 mm in the scanned cross-section. Obtained images from half-round cores were reconstructed to 3D transmission images using the OsiriX software1.
Sub-Sampling Procedure and DNA Extraction
The frozen whole-round cores were first aseptically cut without sample melt using an electric band saw system in a clean booth equipped with two HEPA-filter units (Masui et al., 2009). To avoid potential contamination, the surface of the core was carefully removed with sterile spatulas in a laminar-flow clean bench. Approximately 10 g of dead coral material (referred to hereafter as “coral fossils”) were carefully removed from the clayish sedimentary matrix of each core sample using sterile tweezers, placed in 30 ml phosphate buffered saline (pH 8.0), and then washed at least three times with shaking (Figure 1). Samples of the sediment matrix were also collected from the same sample using sterile spatulas. Subsequently, 2 g (wet weight) of the coral fossils or sediment matrix were separately placed in a sterile plastic tube containing several autoclaved metal beads before being mechanically crushed by shaking (Shake Master, Bio Medical Science, Tokyo, Japan). Whole DNA was then extracted using a Power Max Soil DNA Isolation Kit (MoBIO Lab. Inc., CA, USA) according to the manufacturer’s instructions. The concentration of the extracted DNA was measured using a Quanti-iT DNA assay kit (Invitrogen, CA, USA), and the DNA samples were stored at −20°C until further use.
PCR Amplification and Pyrosequencing
PCR amplification of 16S RNA gene fragments was conducted with the primers EUB27F (Frank et al., 2008) and EUB338Rmix (I: 5′-ACTCCTACGGGAGGCAGC-3′, II: 5′-ACACCTACGGGTGGoCTGC-3′, III: 5′-ACACCTACGGGTGGCAGC-3′; Amann et al., 1990) for bacteria, and ARC21F (5′-TTCCGGTTGATCCYGCCGGA-5′; DeLong, 1992) and ARC912R (I: 5′-CCCCCGCCAATTCCTTTAA-3′, II: 5′-CCCCCGTCAATTCCTTCAA-3′, III: 5′-CCCCCGCCAATTTCTTTAA-3′; Miyashita et al., 2009) for archaea. To amplify the archaeal sequences belonged to SAGMEG-1 and Korarchaeota, which sequences are potentially biased by the previously used primer set (i.e., ARC21F and ARC912R-I), we mixed two new primers (i.e., ARC912R-II and III) for the PCR reaction used in this study. The used PCR conditions were initial denaturation at 98°C for 30 s followed by 35–38 cycles of 98°C for 10 s, 50°C for 15 s, and 68°C for 30 s for bacterial 16S rRNA genes; 31–34 cycles of 98°C for 10 s, 50°C for 15s, and 68°C for 60s for archaeal 16S rRNA genes. The number of PCR cycles was determined by real-time PCR, for which the amplification curve did not reach a plateau at the cycle number used in this study. For sequencing using a GS FLX pyrosequencer (454 Life Sciences, Branford, CT, USA), another amplification (six cycles) was performed using primers [EUB27F and EUB338Rmix for bacteria, and UNIV530F (I: 5′-GTGCCAGCMGCCGCGG-3′, II: 5′-GTGTCAGCCGCCGCGG-3′) and ARC912R for archaea] with 454 FLX Titanium adapters A, B, and a six-base sample identifier tag. Purification of the amplified product, quality checks, and sequencing with the GS FLX pyrosequencer were conducted by TaKaRa Bio Inc. (Shiga, Japan). All the sequences obtained in this study have been deposited in the DDBJ Sequence Read Archive (accession numbers DRS001093 through DRS001103).
Sequences Analyses
All of the reads, including sample identifier tags and primer sequences, were first processed with the Pipeline Initial Process2, which is part of the Ribosomal Database Project (Cole et al., 2009). Parameters for the Pipeline Initial Process are; forward primer max edit distance: 2, max number of N’s: 0, minimum average exp. quality score: 20, reverse primer max edit distance: 0, min sequence length: 150. Reads that did not match the tags and primer sequences were also eliminated through this process. Taxonomic classification of each processed read was then assigned by BLAST analysis with a customized computer script using the ARB SILVA sequence package (Pruesse et al., 2007) as the database.
Statistical Analysis of Microbial Community Structure
Statistical analysis of the 16S rRNA gene-primer-tagged sequence data was performed using the Mothur Utility package (Schloss et al., 2009). Operational taxonomic units (OTU) were calculated using a 97% sequence similarity cutoff, and the Chao-1 estimator (Chao, 1987) and Shannon diversity index (Krebs, 1989) were also calculated. To evaluate the evenness of community structure, we calculated Pielou’s evenness index (J′) using the equation (Pielou, 1966)
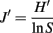
where, H′ is the number derived from the Shannon diversity index and S is the total number of OTUs. The difference in bacterial and archaeal community composition was assessed by the P-test (Martin, 2002). To evaluate similarity between microbial communities, Sørensen’s similarity coefficient (Sørensen, 1957) was calculated as follows:
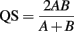
where, A and B are the number of OTUs observed in communities A and B, respectively, and AB is the number of OTUs occurring in both of the communities. Consequently, QS corresponds to the ratio of the number of shared OTUs between communities A and B to the sum of the total number of OTUs in each community.
Results and Discussion
Phylum-Level Microbial Community Compositions
Whole DNA was extracted from the coral fossil and sediment matrix samples collected from three different depths (4, 20, and 105 mbsf). Using a commercial DNA extraction kit, we obtained PCR-amplifiable DNA from 1 g of coral fossil material at yields of 143, 80, and 78 ng from of the fossils from the 1H-3C, 3H-3C, and 12H-3C samples, respectively. Markedly higher DNA yields were obtained concentrations of at 980 and 348 ng from the 1H-3M and 3H-3M samples, respectively. However, we did not obtain any measurable or PCR-amplifiable DNA from the clay matrix sample from 12H-3M at 105 mbsf, which is not expected from the cell count result that all of the depths contained the order of 106 cells and did not show significant difference (Webster et al., 2009). It is not apparent why, but possibly because of inefficient lysis of the microbes in the sample or fragmentation of extracted DNA, which are often the problem in DNA extraction from the environmental sample. Primer-tagged PCR of bacterial and archaeal 16S rRNA genes resulted in successful amplification of the target gene sequences. No amplification was observed in the negative control samples (i.e., DNA extracts performed without substrate samples). Approximately 20,000 reads from each sample were obtained by 454-sequencing (i.e., 213,792 fragment reads in total) and then used for the comparative statistical analysis of microbial community structure of the different subseafloor habitats.
Phylogenetic clustering analysis of primer-tagged bacterial 16S rRNA gene fragments showed that the dominant bacterial taxa belonged to Proteobacteria, Actinobacteria, Candidate division OD1, candidate division OP1, Candidate division OP11, Chloroflexi, Firmicutes, and Nitrosopirae (Figure 2A. Sequences related to Chloroflexi were identified as the dominant bacterial components in all the samples examined, accounting for 20–46% of all the bacterial 16S rRNA gene-tagged sequences (Figure 2A). These bacterial sequences have frequently been detected in the organic-rich sediments at the continental margins (e.g., off Peru, Nankai Trough), suggesting that the Challenger Mound may support an heterotrophic microbial ecosystem, even though the microbial populations and activities, as well as total organic matter concentrations, are all relatively low (Ferdelman et al., 2006; Webster et al., 2009). Interestingly, the sequences of Candidate division JS1 and the Planctomycetes, both of which are generally widespread in organic-rich subseafloor sediments (Inagaki et al., 2003, 2006; Webster et al., 2004), were rarely detected in the collected samples. Members of gamma Proteobacteria which were reported to be a major component of microbial community associated with living L. pertusa (Neulinger et al., 2008; Kellogg et al., 2009) and highly dominant in metabolically active portion of a microbial community colonizing dead L. pertusa (Yakimov et al., 2006) did not constitute large fraction in this study.
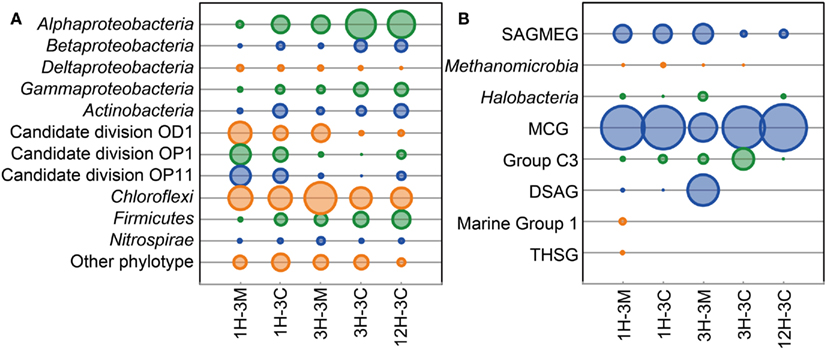
Figure 2. Microbial community compositions in the buried coral fossil carbonates and the clay matrices. Phylogenetic clustering analysis of (A) bacterial and (B) archaeal communities is based on the 16S rRNA gene-tagged sequences. The area of each bubble represents the phylotype frequency in the sample examined.
At the phylum level, archaeal community compositions were generally simpler than that observed in the bacterial communities. The members of the Miscellaneous Crenarchaeotic Group (MCG; Inagaki et al., 2003) were the most commonly detected archaeal phylotypes in all of the samples, representing 80% of the 16S rRNA gene-tagged-archaeal sequences obtained (Figure 2B). Some of the sequences were affiliated to the South African Gold Mine Euryarchaeotic Group (SAGMEG) and the Deep-Sea Archaeal Group [DSAG; alternatively classified as Marine Benthic Group D (MBG-D)]. Marine Group I archaea were only detected from the sediment matrix of 1H-3. The sequences of potential methanogens within the Methanomicrobia were detected from 1H-3 and 3H-3 as relatively minor archaeal components.
Similarities between Communities in Different Habitats
Classifications generated using the 16S rRNA gene-tagged sequences revealed that the community compositions of the coral fossil and the clay matrix samples were generally similar at the phylum- and class-levels (Figure 2). We therefore analyzed the data further using the P-test (Martin, 2002) and Sørensen’s similarity coefficient. Interestingly, all of the P-values obtained were less than 0.01, implying that microbial communities from any different habitats and depths were distinct from each other; except for samples 1H-3M and 3H-3C for which the difference was not significant (P > 0.1). Sørencen’s index values (QS) among bacterial communities ranged from 0.15 to 0.36, indicating that even though bacterial communities were similar at the phylum level, the bacterial community compositions are very different at the genus- or species level (Figure 3A). Similarly, QS values among archaeal communities ranged from 0.29 to 0.54, which were slightly higher than those obtained for bacteria (Figure 3B). Even when the community compositions of the coral fossils and matrices from the same depth were compared, the QS values are unexpectedly low: e.g., QS values among bacterial communities in coral fossils and matrices from 4 to 20 mbsf were 0.31 and 0.25, respectively. The highest QS value for bacteria was obtained from 3H-3C (4 mbsf) and 12H-3C (105 mbsf), which were derived from the spatially distinct subseafloor habitat. This finding suggests that the carbonate coral fossils harbor microbial communities that are more similar to each other than the communities in the matrix from the same depth (i.e., QS = 0.25 for 3H-3M and 3H-3C, 0.31 for 1H-3M and 1H-3C). The coral fossils also harbor more shared archaeal components than the clay matrix (i.e., QS = 0.47 for 1H-3C and 3H-3C, 0.44 for 1H-3C and 12H-3C, and 0.41 for 3H-3C and 12H-3C). These finding provide conclusive statistical evidence of microbial diversity differences in sedimentologically distinct microbial habitats, and that deeply buried carbonate fossils constitute an ecologically significant habitat for unique microbial components.
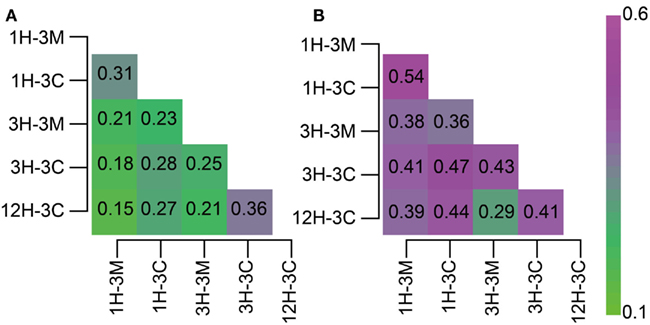
Figure 3. Heat map matrix of Sørenson index values among 16S rRNA gene-tagged sequence libraries. The index score indicates the similarity between two (A) bacterial or (B) archaeal communities.
Comparison of Community Richness and Evenness
We examined the structure of bacterial and archaeal communities using a total of 213,792 16S rRNA gene-tagged sequences (119,785 and 94,007 for bacteria and archaea, respectively: Figure 4A). The number of OTUs determined by 3% sequence divergence (Figure 4A), as well as the Chao-1 estimator (Figure 4B), clearly showed that bacterial communities were generally more diverse than archaeal communities. The results showed that Chao-1 estimates of the bacterial communities in matrix samples (i.e., 1H-3M and 3H3-M) had over 3,000 OTUs, whereas the Chao estimates of coral fossils (i.e., 1H-3C, 3H-3C, and 12H-3C) were approximately 50% of the diversity richness (Figure 4B). A similar trend was also observed in the community structure of the archaea. Taken together, these findings indicate that microbial communities inhabiting the buried coral carbonate fossils were less diverse than those in the clay matrix. Interestingly, the diversity of the microbial communities in the Porcupine Mound subseafloor habitat was very similar between different depth, and no decrease of the diversity along the depths was observed (Figure 4B).
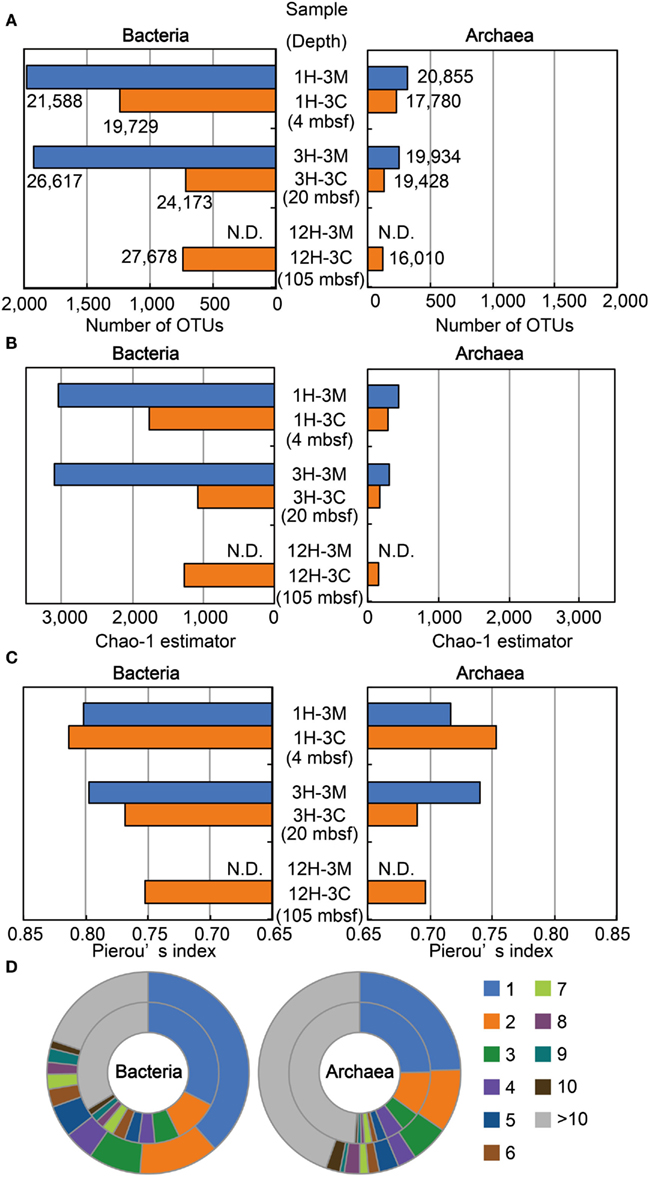
Figure 4. Microbial diversity and community structure estimated by OTU-based analysis of 16S rRNA gene-tagged sequences. (A) The number of OTUs defined as a group of sequences with less than 3% sequence divergence. The number of tag sequences obtained by 454-pyrosequencing is shown adjacent to the bars. (B) Chao-1 estimator for estimating community richness. (C) Pierou’s index for assessing evenness of the community. (D) Phylotype frequency for total bacterial (left) and archaeal (right) communities. The outer and inner circles indicate the community from clay matrices and coral fossils, respectively. The number of tag sequences classified as the same phylotype (OTU) is indicated by color as shown in the right legend. Blue and orange bars in (A–C) indicate sediment matrix and coral fossil, respectively. N.D., Not determined.
Results obtained for Pielou’s evenness index (J′) and the phylotype frequency are shown in Figures 4C,D, respectively. The evenness scores of bacterial and archaeal communities ranged from 0.75 to 0.81 and 0.69 to 0.75, respectively (Figure 4C). These data indicate that the relatively high evenness of microbial communities are observed in all of the samples examined, and that the phylotypes of bacterial communities are more evenly distributed than those of archaeal communities. The Pielou’s index data were highly consistent with the ratios obtained for phylotype frequency (Figure 4D). For example, the ratios of singletons in the bacterial 16S rRNA gene-tagged sequence data were higher than those obtained for archaeal tag sequences, even though the frequency patterns of the obtained OTUs in coral fossils and clay matrices were relatively similar to each other. These analyses of community structure using measures of diversity and evenness consistently suggest that habitability of the detected microbial components might be relatively evenly supported by geochemical and/or geophysical constraints.
Eukaryotic DNA Amplification
In order to determine whether coral DNA could be preserved in sediments over a geological timescale, we attempted to amplify eukaryotic 18S rRNA genes from fossil coral and clays. Although no eukaryotic DNA fragments were obtained, several archaeal 16S rRNA genes were amplified (data not shown), most likely due to the mismatched primer annealing. These results suggested that the buried eukaryotic DNA had been almost completely degraded and utilized by indigenous microbes in the sediments. The data also indicate that the presence of eukaryotes living below the seafloor is unlikely in the environment examined, or if any eukaryotes were present, the populations are either below detectable limits or not detected with the used primers.
Conclusion and Prospects
In this study, we characterized the structure of microbial communities in different sedimentary habitats of the Porcupine Mound. We found that the buried coral carbonate fossils harbor microbial communities that differ from those in the sediment matrices. Bacterial communities were generally more diverse than archaeal communities in all substrate types and depths. The species richness of microbial communities in the sediment matrix was higher than that in the coral fossil substrates, while evenness was relatively high for all of the samples examined. These results suggested that the Pleistocene coral sediments in the Porcupine Mound represent unique subseafloor microbial habitat and, even at the same or similar depths, the characteristics of the sediments were shown to affect microbial diversity and community structure. However, it remains to be determined whether fossil coral-associated microbial components play an ecological role in carbonate diagenesis. Similarly, since the role of geochemical factors and/or geophysical constraints on niche preference by subseafloor microbial communities remains unknown. These factors should be clarified through scientific ocean drilling in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Ms. Sayo Hashimoto for technical assistance and Dr. Takayuki Tomiyama for X-CT imaging of core samples. Samples and photo images used in this study were provided by the Integrated Ocean Drilling Program (IODP) Expedition 307. This work is supported in part by the JSPS Strategic Fund for Strengthening Leading-Edge Research and Development (to JAMSTEC), and the JSPS Funding Program for Next Generation World-Leading Researchers (NEXT Program; to Fumio Inagaki).
Footnotes
References
Amann, R. I., Binder, B. J., Olson, R. J., Chisholm, S. W., Devereux, R., and Stahl, D. A. (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925.
Beck, M., Riedel, T., Graue, J., Köster, J., Kowalski, N., Wu, C. S., Wegener, G., Lipsewers, Y., Freund, H., Böttcher, M. E., Brumsack, H.-J., Cypionka, H., Rullkötter, J., and Engelen, B. (2011). Imprint of past and present environmental conditions on microbiology and biogeochemistry of coastal quaternary sediments. Biogeosciences 8, 55–68.
Chao, A. (1987). Estimating the population size for capture-recapture data with unequal catchability. Biometrics 43, 783–791.
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., Marsh, T., Garrity, G. M., and Tiedje, J. M. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145.
De Mol, B., Kozachenko, M., Wheeler, A., Alvares, H., Henriet, J. P., and Olu-Le Roy, K. (2007). Thérèse Mound: a case study of coral bank development in the Belgica Mound Province, Porcupine Seabight. Int. J. Earth Sci. 96, 103–120.
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 685–689.
Ferdelman, T. G., Kano, A., Williams, T., Henriet, J. P., and the Expedition 307 Scientists. (2006). “Modern carbonate mounds: Porcupine drilling,” in Proceedings of the Integrated Ocean Drilling Program, Volume 307 Expedition Reports. Washington, DC: IODP Management International. doi:10.2204/iodp.proc.307.102.2006
Foubert, A., Beck, T., Wheeler, A. J., Opderbecke, J., Grehan, A., Klages, M., Thiede, J., Henriet, J.-P., and the Polar-stern ARK-XIX/3A Shipboard Party. (2005). “New view of the Belgica Mounds, Porcupine Seabight, NE Atlantic: preliminary results from the Polarstern ARK-XIX/3a ROV cruise,” in Cold Water Corals and Ecosystems, eds A. Freiwald and J. M. Roberts (Heidelberg: Springer-Verlag), 403–415.
Frank, J. A., Reich, C. I., Sharma, S., Weisbaum, J. S., Wilson, B. A., and Olsen, G. J. (2008). Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74, 2461–2470.
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F. S., Nealson, K. H., Horikoshi, K., D’Hondt, S., and Jørgensen, B. B. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820.
Inagaki, F., Suzuki, M., Takai, K., Oida, H., Sakamoto, T., Aoki, K., Nealson, K. H., and Horikoshi, K. (2003). Microbial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk. Appl. Environ. Microbiol. 69, 7224-7235
Kano, A., Ferdelman, T. G., Williams, T., Henriet, J. P., Ishikawa, T., Kawagoe, N., Takashima, C., Kakizaki, Y., Abe, K., Sakai, S., Browning, E. L., Li, X., and Integrated Ocean Drilling Program Expedition 307 Scientists. (2007). Age constraints on the origin and growth history of a deep-water coral mound in the northeast Atlantic drilled during Integrated Ocean Drilling Program Expedition 307. Geology 35, 1051–1054.
Kellogg, C. A., Lisle, J. T., and Galkiewicz, J. P. (2009). Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the northeastern Gulf of Mexico. Appl. Environ. Microbiol. 75, 2294–2303.
Martin, A. P. (2002). Phylogenetic approaches for describing and comparing the diversity of microbial communities. Appl. Environ. Microbiol. 68, 3673–3682.
Masui, N., Morono, Y., and Inagaki, F. (2009). Bio-archive core storage and subsampling procedure for subseafloor molecular biological research. Sci. Drill. 8, 35–37.
Miyashita, A., Mochimaru, H., Kazama, H., Ohashi, A., Yamaguchi, T., Nunoura, T., Horikoshi, K., Takai, K., and Imachi, H. (2009). Development of 16S rRNA gene-targeted primers for detection of archaeal anaerobic methanotrophs (ANMEs). FEMS Microbiol. Lett. 297, 31–37.
Neulinger, S. C., Jarnegren, J., Ludvigsen, M., Lochte, K., and Dullo, W. C. (2008). Phenotype-specific bacterial communities in the cold-water coral Lophelia pertusa (Scleractinia) and their implications for the coral’s nutrition, health, and distribution. Appl. Environ. Microbiol. 74, 7272–7285.
Pielou, E. C. (1966). Species-diversity and pattern-diversity in the study of ecological succession. J. Theor. Biol. 10, 370–383.
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., and Glockner, F. O. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J., and Weber, C. F. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541.
Schöttner, S., Hoffmann, F., Wild, C., Rapp, H. T., Boetius, A., and Ramette, A. (2009). Inter- and intra-habitat bacterial diversity associated with cold-water corals. ISME J. 3, 756-759
Sørensen, T. (1957). A method of establishing groups of equal amplitude in plant sociology based on similarity of species and its application to analyses of the vegetation on Danish commons. Biologiske Skrifter/Kongelige Danske Videnskabernes Selskab 5, 1–34
Teske, A. P. (2006). Microbial communities of deep marine subsurface sediments: molecular and cultivation surveys. Geomicrobiol. J. 23, 357–368.
Webster, G., Blazejak, A., Cragg, B. A., Schippers, A., Sass, H., Rinna, J., Tang, X., Mathes, F., Ferdelman, T. G., Fry, J. C., Weightman, A. J., and Parkes, R. J. (2009). Subsurface microbiology and biogeochemistry of a deep, cold-water carbonate mound from the Porcupine Seabight (IODP Expedition 307). Environ. Microbiol. 11, 239–257.
Webster, G., Parkes, R. J., Cragg, B. A., Newberry, C. J., Weightman, A. J., and Fry, J. C. (2006). Prokaryotic community composition and biogeochemical processes in deep subseafloor sediments from the Peru Margin. FEMS Microbiol. Ecol. 58, 65–85.
Webster, G., Parkes, R. J., and Weightman, A. (2004). Widespread occurrence of a novel division of bacteria identified by 16S rRNA gene sequences originally found in deep marine sediments. Appl. Environ. Microbiol. 70, 5708–5713.
Yakimov, M. M., Cappello, S., Crisafi, E., Tursi, A., Savini, A., Corselli, C., Scarfi, S., and Giuliano, L. (2006). Phylogenetic survey of metabolically active microbial communities associated with the deep-sea coral Lophelia pertusa from Apulian plateau, Central Mediterranean Sea. Deep Sea Res. Part I. 53, 62–75.
Keywords: 16S rRNA gene, tag sequence, deep subseafloor biosphere, community structure, coral fossils
Citation: Hoshino T, Morono Y, Terada T, Imachi H, Ferdelman TG and Inagaki F (2011) Comparative study of subseafloor microbial community structures in deeply buried coral fossils and sediment matrices from the Challenger Mound in the Porcupine Seabight. Front. Microbio. 2:231. doi: 10.3389/fmicb.2011.00231
Received: 19 August 2011;
Paper pending published: 06 September 2011;
Accepted: 01 November 2011;
Published online: 17 November 2011.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
Doug Bartlett, Scripps Institution of Oceanography, USAKarine Alain, Centre National de la Recherche Scientifique, France
Copyright: © 2011 Hoshino, Morono, Terada, Imachi, Ferdelman and Inagaki. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Fumio Inagaki, Geomicrobiology Group, Kochi Institute for Core Sample Research, Japan Agency for Marine-Earth Science and Technology, Nankoku, Kochi 783-8502, Japan. e-mail:aW5hZ2FraUBqYW1zdGVjLmdvLmpw