
 Isaac D. Wagner
Isaac D. Wagner
- Department of Biology, Center for Life in Extreme Environments, Portland State University, Portland, OR, USA
Cultivation-independent studies have shown that taxa belonging to the “deep-sea hydrothermal vent euryarchaeota 2” (DHVE2) lineage are widespread at deep-sea hydrothermal vents. While this lineage appears to be a common and important member of the microbial community at vent environments, relatively little is known about their overall distribution and phylogenetic diversity. In this study, we examined the distribution, relative abundance, co-occurrence patterns, and phylogenetic diversity of cultivable thermoacidophilic DHVE2 in deposits from globally distributed vent fields. Results of quantitative polymerase chain reaction assays with primers specific for the DHVE2 and Archaea demonstrate the ubiquity of the DHVE2 at deep-sea vents and suggest that they are significant members of the archaeal communities of established vent deposit communities. Local similarity analysis of pyrosequencing data revealed that the distribution of the DHVE2 was positively correlated with 10 other Euryarchaeota phylotypes and negatively correlated with mostly Crenarchaeota phylotypes. Targeted cultivation efforts resulted in the isolation of 12 axenic strains from six different vent fields, expanding the cultivable diversity of this lineage to vents along the East Pacific Rise and Mid-Atlantic Ridge. Eleven of these isolates shared greater than 97% 16S rRNA gene sequence similarity with one another and the only described isolate of the DHVE2, Aciduliprofundum boonei T469T. Sequencing and phylogenetic analysis of five protein-coding loci, atpA, EF-2, radA, rpoB, and secY, revealed clustering of isolates according to geographic region of isolation. Overall, this study increases our understanding of the distribution, abundance, and phylogenetic diversity of the DHVE2.
Introduction
The diversity of Archaea associated with marine hydrothermal vent habitats is unrivaled in any other ecosystem on Earth (Auguet et al., 2009). Much of this diversity resides within the Euryarchaeota where numerous cultivated and uncultivated lineages appear to be endemic to the deep-sea. One such lineage is the “deep-sea hydrothermal vent euryarchaeota 2” (DHVE2; Takai and Horikoshi, 1999). Previously, our knowledge of the distribution and diversity of the DHVE2 was based primarily on cultivation-independent assessments (Table A1 in Appendix). These studies established that the DHVE2 are widespread in marine hydrothermal environments and can account for up to 15% of the archaeal 16S rRNA gene sequences, suggesting that they are important members of deep-sea hydrothermal ecosystems (Reysenbach et al., 2006). More recently, 16S rRNA gene cloning studies have shown that certain types of deep-sea vent mineral deposits, namely horizontal flanges (shelf-like structures that form in some hydrothermal systems, Figure 1, Tivey, 2007), can harbor an even greater proportion of DHVE2 16S rRNA gene sequences (Nunoura and Takai, 2009).

Figure 1. Photograph of deep-sea hydrothermal vent mineral deposits from the Eastern Lau Spreading Center. A series of horizontal flanges are shown in the foreground while two vertical chimneys can be seen in the background. Photo was taken from ROV Jason II (courtesy of Woods Hole Oceanographic Institution).
The first cultured representative of the DHVE2, Aciduliprofundum boonei T469T, was the first obligate thermoacidophile isolated from deep-sea vents despite the low pH of most hydrothermal fluids (pH 2.8–4.5) and predictions of acidic microhabitats within the walls of vent deposits (Tivey, 2004; Reysenbach et al., 2006). Acidic habitats are generated in vent deposits by conductive cooling of end-member fluids or by transport of hydrothermal fluids outward across deposit walls by diffusion (Tivey, 2004). However, when hydrothermal fluids and seawater mix, either in the subsurface or by advection across deposit walls, neutrality of the fluids is quickly reached resulting in most marine hydrothermal vent habitats being circumneutral. Therefore, the pH of end-member fluids and the degree of fluid mixing within an individual deposit are likely important factors in controlling the distribution and abundance of thermoacidophilic DHVE2 both within and between vent fields. Factors that influence the pH of hydrothermal fluids at the vent field scale include the presence of organic sediments, which increases the pH of fluids as at the Guaymas Basin (GB), and inputs of magmatic volatiles as observed in the low pH fluids of the Mariner vent site along the Eastern Lau Spreading Center (ELSC, Tivey, 2007). Furthermore, fluid mixing styles can be influenced by the type of vent deposit as hydrothermal fluids associated with horizontal flanges are conductively cooled with little or no mixing of seawater, while the mixing styles of vertical chimney deposits are much more variable (Tivey, 2004). Taken together, these factors suggest that thermoacidophilic niches in hydrothermal vent deposits vary not only across vent fields but also within any individual vent field.
Geologic processes such as significant breaks in ridge axes (i.e., transform faults) or hotspots in hydrothermal activity can provide barriers for dispersal and hence influence diversification and speciation. Such distribution patterns influenced by geologic processes have been shown to play a significant role in the biogeographical patterns of deep-sea hydrothermal vent fauna (Van Dover et al., 2002), but whether similar factors influence microbial distribution patterns is less clear. Microbial biogeographical diversity patterns have been reported for terrestrial geothermal springs (Papke et al., 2003; Whitaker et al., 2003; Takacs-Vesbach et al., 2008; Wagner, 2010), yet there are relatively few studies that report the biogeography of microorganisms at deep-sea hydrothermal vents (Holden et al., 2001; Huber et al., 2006; Flores et al., 2011b). Here, we investigated the occurrence, abundance, and phylogenetic diversity of the DHVE2 in hydrothermal vent deposits from several geochemically distinct and spatially distant vent fields to explore whether the distribution patterns of this lineage are influenced by geographic separation.
Materials and methods
Sample Collection
Deep-sea hydrothermal mineral deposits were collected with the HOV Alvin or ROV Jason II during research cruises to the East Pacific Rise (EPR) in 2007, the Mid-Atlantic Ridge (MAR) in 2008, the ELSC in 2009, and the GB in 2009. Once shipboard, individual samples were processed and stored anaerobically as previously described (Götz et al., 2002; Moussard et al., 2004; Reysenbach et al., 2006).
Quantitative Polymerase Chain Reaction
DNA from environmental samples was extracted from homogenized mineral deposits [≈1.6–3.2 g (w/w)] using the Ultra Clean Soil DNA Isolation Kit (MO BIO Laboratories) according to the modified protocol of Reysenbach et al. (2006). Quantitative PCR (qPCR) was performed according to manufacturer’s instructions using the Quantitect SYBR green PCR kit (Qiagen, Inc., Valencia, CA, USA) and 0.8 μM final primer concentrations, with melting curves run at the end of each reaction to ensure product specificity. Primers and thermocycling conditions were followed according to Reysenbach et al. (2006). Standard curves (107–1010 gene copies/μl) for both Archaea and the DHVE2 were generated from a plasmid containing the nearly full-length 16S rRNA gene sequence of A. boonei T469T. All standards and samples were run in at least duplicate reactions. Gene copy numbers presented were averaged across replicates and normalized by the amount of material in grams (w/w) extracted.
Local Similarity Analysis
The variable region 4 (V4) of archaeal 16S rRNA genes from 57 deposits from the MAR (Flores et al., 2011a), ELSC (Flores et al., in revision), and GB (Reysenbach, unpublished data) were amplified, pyrosequenced, trimmed, aligned, and clustered at 97% sequence similarity as previously described (Flores et al., 2011a). DHVE2 operational taxonomic units (OTUs) were identified by performing BLAST searches (Altschul et al., 1990) using representative sequences of each OTU against the 16S rRNA gene of A. boonei T469T, selecting OTUs with greater than 95% sequence similarity to A. boonei T469T and manually aligning sequences and generating phylogenetic trees in a custom ARB database (Ludwig et al., 2004). For local similarity analysis (LSA), a technique that explores co-varying relationships of microbial species (or OTUs) to one another (Ruan et al., 2006), all OTUs with greater than 100 sequences, including three DHVE2 OTUs, were normalized and imported into the LSA compute tool (http://meta.cmb.usc.edu/). Results of LSA were trimmed to include only the two most prevalent DHVE2 OTUs (ID no.’s DHVE2-727 and DHVE2-1148), and other OTUs that were positively and negatively correlated (P < 0.05). Visualization of the resulting interaction network was performed using Cytoscape (Shannon et al., 2003). Correlated OTUs were classified to the lowest taxonomic level that had a bootstrap value of ≥50% (Claesson et al., 2009) using the RDP-classifier (Wang et al., 2007). Pyrosequencing data is available for download from the MG-RAST server (Meyer et al., 2008) or by contacting the corresponding author.
Enrichment Culturing, Isolation, and Phylogenetic Analysis
The anaerobic medium used for enrichments and isolation was identical to that used by Reysenbach et al. (2006) in the isolation of the thermoacidophile A. boonei T469T. Pure cultures were obtained through multiple serial dilutions, and culture purity was confirmed by sequencing of the 16S rRNA gene. Genomic DNA was extracted from isolated cultures using the DNeasy Tissue Kit (Qiagen) following the manufacturer’s protocol. The 16S rRNA gene of each isolate was amplified, purified, and sequenced as described previously (Reysenbach et al., 2006). Nearly complete 16S rRNA gene sequences were assembled using the software SeqMan (DNASTAR, Inc.), compared to the NCBI non-redundant database using BLAST (Altschul et al., 1990), and aligned in ARB (Ludwig et al., 2004) according to secondary structure constraints. Neighbor-joining (Olsen correction, 500 bootstrap replicates) and maximum-likelihood (default parameters, 100 bootstrap replicates) analyses were conducted in ARB (Ludwig et al., 2004) and MEGA V 5.04 (Tamura et al., 2011), respectively, using only unambiguous nucleotide positions for a diversity of Archaea (789 nt).
Multi-Locus Sequence Analysis
We used multi-locus sequence analysis (MLSA; Gevers et al., 2005) to further examine the phylogenetic divergence among isolates. The protein-coding genes chosen for MLSA were: DNA repair and recombination protein RadA, radA; ATP synthase, A subunit, atpA; DNA-directed RNA polymerase subunit B, rpoB; translation elongation factor aEF-2, EF-2; and preprotein translocase, SecY subunit, secY. These genes were chosen because they were previously used to investigate the relationships between taxa within the Halobacteriales (Papke et al., 2011). The protein-coding genes were distributed around the genome of A. boonei T469T (Table A2 in Appendix).
Primers for the PCR amplification and sequencing of the protein-coding loci were designed based on the protein-coding genes in A. boonei T469T (NCBI # PRJNA43333), and related Thermoplasmatales with sequenced genomes; Thermoplasma acidophilum (Accession: PRJNA61573), Thermoplasma volcanium (NCBI # PRJNA35129), Picrophilus torridus (NCBI # PRJNA36697), and Ferroplasma acidarmanus (NCBI # PRJNA35151). Initial PCR primers were designed by using the oligonucleotide design tool from IDT SciTools (Integrated DNA Technologies), and then modified based on the nucleotide and amino acid conservation at potential primer regions (Table 1). Primers were supplied by Invitrogen (Life Technologies). Thermocycler conditions for the amplification of the protein-coding loci were 94°C for 2 min; 30 cycles of: 94°C for 45 s, annealing temperature (Table 1) for 45 s, 72°C for extension; then 72°C for 5 min. Annealing temperatures were optimized for each primer set (data not shown). PCR products were purified using the UltraClean PCR Clean-Up Kit (MO BIO Laboratories) according to manufacturer’s instructions. Purified PCR products were used as templates for Sanger sequencing reactions. Electropherograms of the protein-coding loci sequencing reads were analyzed and assembled using the software SeqMan (DNASTAR, Inc.).
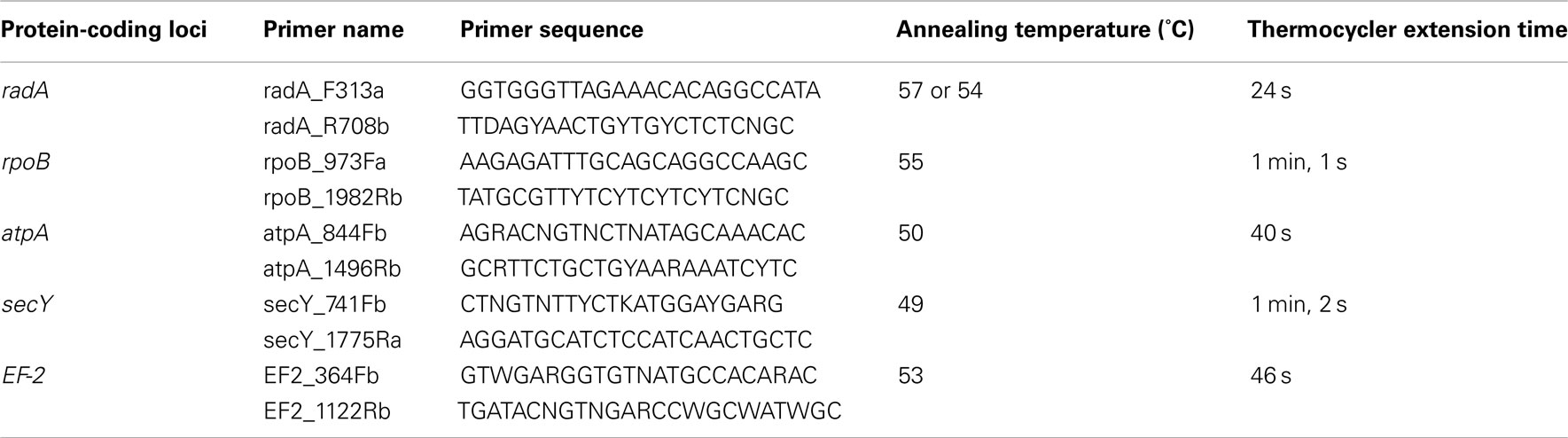
Table 1. Primers and thermocycler conditions for the amplification of protein-coding loci from DHVE2 isolates.
Nucleotide polymorphism and DNA sequence variation of the protein-coding loci were calculated using the software DnaSP v 5.10.01 (Librado and Rozas, 2009) and MEGA v 5.04 (Tamura et al., 2011). Metrics calculated were: G + C mol%; number of nucleotide sequence alleles, na; the number of polymorphic nucleotide sites, Snt; the total number of mutations, eta; the nucleotide diversity, Pi; the number of inferred primary protein sequence alleles, npp; and the number of polymorphic amino acid residues, Spp. Synonymous and non-synonymous positions and substitutions were examined using DnaSP v 5.10.01 (Librado and Rozas, 2009). MEGA v5.04 (Tamura et al., 2011) was used to evaluate models for nucleotide substitution for each protein-coding locus and to construct phylogenetic trees. The model having the lowest goodness-of-fit Bayesian Information Criterion (BIC) value was used to generate a maximum-likelihood bootstrap consensus tree based on 100 replicates. The initial tree for the maximum-likelihood analysis was constructed automatically and the Nearest-Neighbor-Interchange heuristic search method was used to search for topologies that fit the data better. In all analyses of sequence diversity or phylogeny, the sequence length homologous among all isolates was utilized.
Results and Discussion
Occurrence and Relative Abundance of the DHVE2
To determine the occurrence and relative abundance of the DHVE2 in deposits from geologically distinct vent fields, archaeal and DHVE2 16S rRNA genes were quantified using qPCR. Archaeal 16S rRNA genes were successfully amplified from 130 samples. Deposits from Tui Malila along the ELSC had, on average, the lowest archaeal copy number [8.35 × 104 copies/g (w/w)] while TAG along the MAR had the highest archaeal copy number [9.78 × 107 copies/g (w/w); Table 2]. Although these values cover a wide range, they are similar to archaeal abundances that have been reported from other hydrothermal vent deposits (Takai et al., 2001; Schrenk et al., 2003; Nakagawa et al., 2005; Zhou et al., 2009). Using group specific primers, the DHVE2 were observed at all vent fields but in only 60% (78/130) of samples analyzed. At individual vent fields, the DHVE2 were most frequently observed at Mariner (80% of samples), EPR (77.8%), and TAG (75%). In contrast, they were detected in less than 50% of samples from Tui Malila (37.5%), TowCam (42.9%), and the GB (48.1%; Table 2). While their occurrence varied within an individual vent field, these results clearly illustrate the ubiquity of the DHVE2 at deep-sea vents and suggest that differences in the geological properties that influence end-member fluid pH over the ranges we examined do not inhibit colonization by the DHVE2 at these vent sites. For example, the end-member fluid pH at Mariner is around 2.5 while at GB the fluids are around pH 4.5. Yet niches are still available for colonization of the DHVE2 at both sites. Assuming all members of the DHVE2 are thermoacidophilic, then thermoacidophily is a common ecological strategy in deep-sea hydrothermal ecosystems.
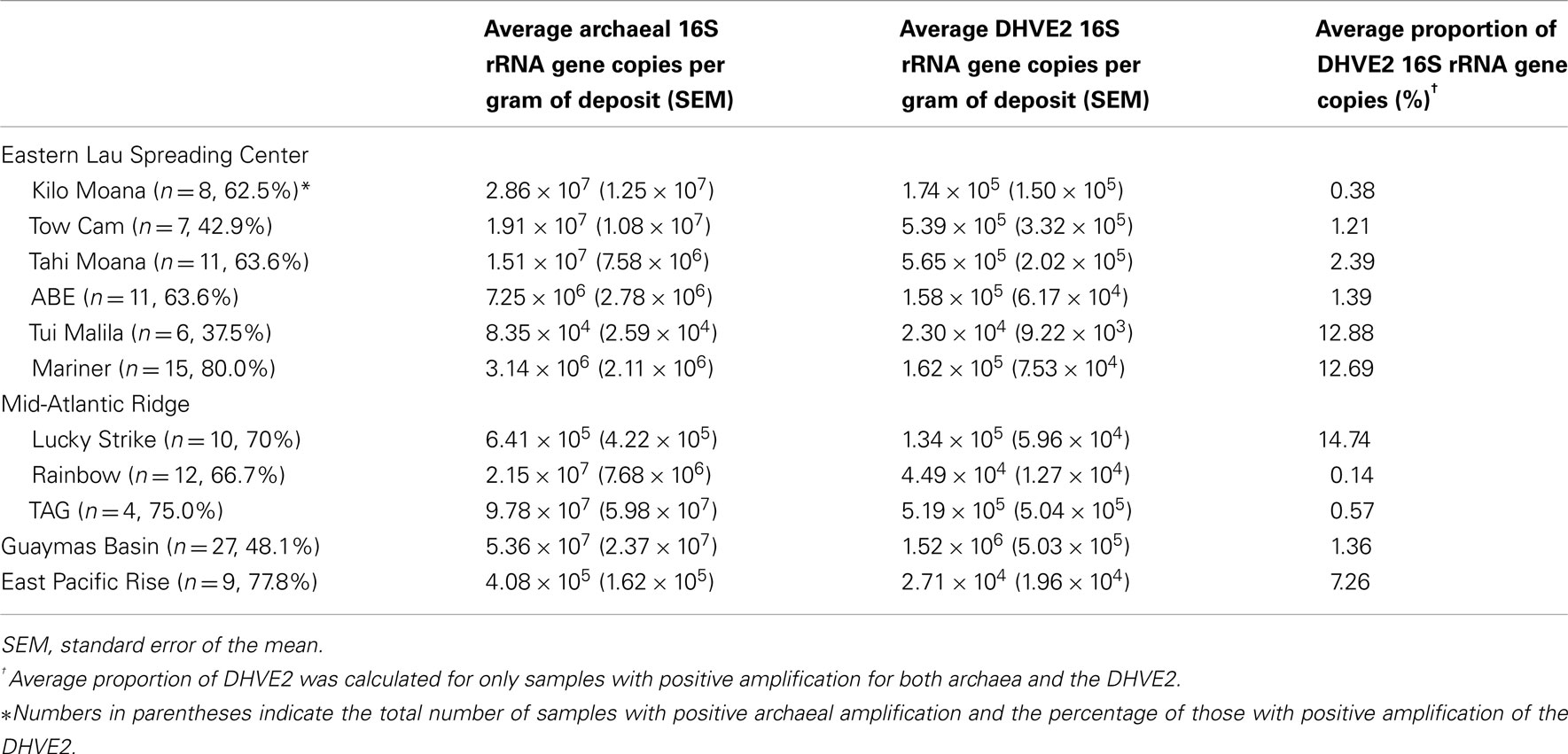
Table 2. Results of qPCR assays to determine the occurrence and relative abundance of the DHVE2 in hydrothermal vent deposits collected between 2006 and 2009 from several different vent fields.
In samples where the DHVE2 were not detected, the average archaeal abundance was significantly lower, at 3.64 × 105 copies/g (w/w), than in deposits where the DHVE2 were observed, which averaged 3.34 × 107 copies/g (P = 0.002, one-tailed T-test; data not shown). Additionally, deposits where the DHVE2 were absent were typically newly formed, thin-walled structures without an obvious biofilm on the exterior of the deposit. Previous work demonstrated that, while Archaea are typically the initial colonizers of newly formed vent deposits, they are primarily autotrophic with later colonization by heterotrophic Archaea and Bacteria (Page et al., 2008). Consequently, the occurrence of the DHVE2 in an individual deposit may be dependent upon the presence of a mature microbial community from which to scavenge fermentable peptides (Reysenbach and Flores, 2008). Older deposits also generally have more defined walls and fluid conduits that would help isolate the hydrothermal fluids from seawater allowing for less mixing of seawater and more conductive cooling of the fluids.
Quantitative PCR was also used to determine the proportion of the DHVE2 in the archaeal communities. Within individual vent fields, the average percentage of DHVE2 16S rRNA gene copies in the archaeal community ranged from 0.14% at Rainbow to 14.74% at Lucky Strike. Deposits from Mariner (12.69%), Tui Malila (12.88%), and EPR (7.26%) also had, on average, a high percentage of DHVE2 sequences within their archaeal communities (Table 2). This level of relative abundance is in agreement with previous reports (Reysenbach et al., 2006; Nunoura and Takai, 2009) and implies that, when the DHVE2 are present, they can be a significant component of the archaeal community. However, it is difficult to compare their relative abundance to other archaeal groups from previous studies because of differences in the methods employed to determine abundances.
In general, individual samples having the highest proportion of DHVE2 gene copies were typically flanges (Figure 2), although some exceptions were noticed (e.g., chimney sample EPR07-75). As fluids in flanges are conductively cooled with little seawater mixing, these fluids remain acidic as they cool to habitable temperatures and percolate vertically through the structure generating relatively larger thermoacidic zones than predicted in vertical chimney structures. Similar situations could conceivably develop in thin-walled, chalcopyrite-lined chimneys (like Mariner-1652). In a recent study, the DHVE2 accounted for nearly 46% of the archaeal 16S rRNA gene diversity associated with a sample from a flange deposit from the Yonaguni Knoll IV hydrothermal field in the western Pacific Ocean (Nunoura and Takai, 2009). Our data further support the observation that flanges may be “hotspots” for the DHVE2 and likely, other thermoacidophiles.
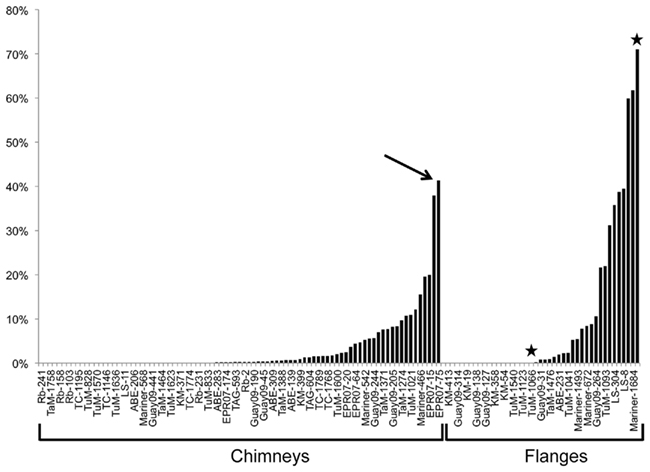
Figure 2. Percentage of DHVE2 16S rRNA gene sequences in the archaeal communities of hydrothermal vent deposits from several different vent fields as determined using qPCR. →, Indicates a chimney sample with a high proportion of DHVE2 sequences while ⋆ indicates two samples collected from a single flange deposit and illustrates the spatial heterogeneity of the DHVE2. Abbreviations are: Rb, Rainbow; TaM, Tahi Moana; TC, Tow Cam; TuM, Tui Malila; LS, Lucky Strike; Guay09, Guaymas Basin 2009; KM, Kilo Moana; EPR07, East Pacific Rise 2007.
Spatial variability on and in a deposit can be shaped by a number of factors including heterogeneous wall thickness, deposit mineralogy, and fluid flow rate. For most of the deposits collected and analyzed in this study, only the outer few millimeters were sampled, as this is where the majority of microorganisms are detected (Takai et al., 2001; Schrenk et al., 2003; Nakagawa et al., 2005; Kormas et al., 2006; Nunoura and Takai, 2009). However, for some of the deposits, we sampled exterior and interior sections of chimneys and for flanges, different spatial areas on the top, bottom and edge. As a result of this sampling strategy, we have paired samples from a few deposits that illustrate the spatial variability of the DHVE2 associated with individual deposits. Spatial variability on a deposit was best illustrated by a flange structure collected from the Tui Malila vent field along the ELSC. On the bottom of this deposit, the DHVE2 comprised over 70% of the archaeal 16S rRNA gene sequences detected (Tui Malila-1059) while they were undetectable on the edge (Tui Malila-1066; Figure 2).
Co-Occurrence Patterns of the DHVE2 with Other Archaea
In previous studies, barcoded pyrosequencing was used to characterize the archaeal communities of numerous vent deposits from geochemically and geographically distinct hydrothermal vent fields MAR (Flores et al., 2011a), ELSC (Flores et al., in revision) and GB (Reysenbach, unpublished data). These large data sets provided an opportunity to examine the co-occurrence of the DHVE2 with other archaeal lineages using LSA. In total, three OTUs were identified as the DHVE2 (ID no.’s DHVE2-727, DHVE2-1148, and DHVE2-1137) and contained 3557, 1389, and 120 sequences, respectively (Table 3). DHVE2-727 and DHVE2-1148 were present in 89 and 65% of samples, respectively, while DHVE2-1137 was present in only 10% of samples. Using LSA, we found that the occurrence of DHVE2-727 and DHVE2-1148 were positively correlated with one another but not with DHVE2-1137 (Figure 3). Due to the low abundance, relatively rare occurrence and lack of correlation with the other DHVE2, results of LSA including DHVE2-1137 are not presented.
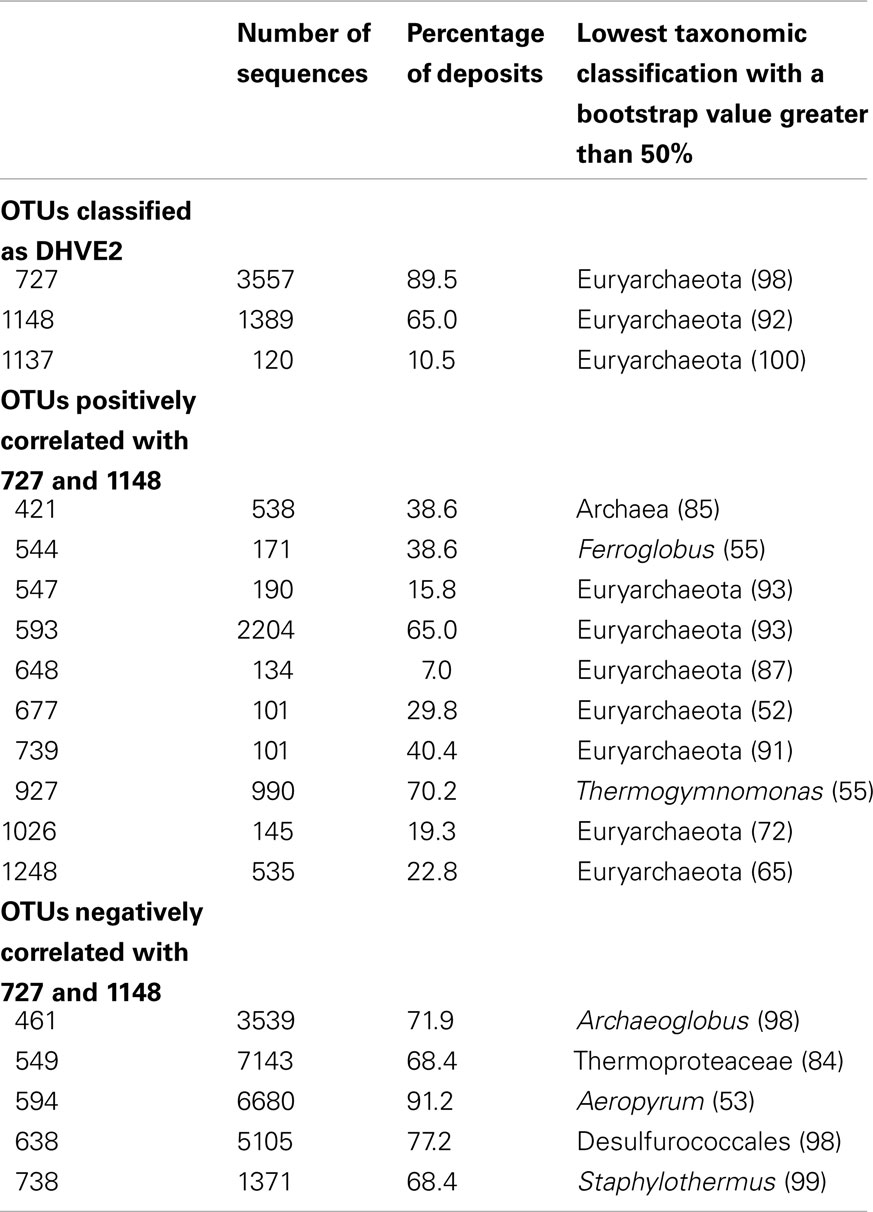
Table 3. Results of local similarity analysis illustrating the co-occurrence of the DHVE2 with other archaeal OTUs.
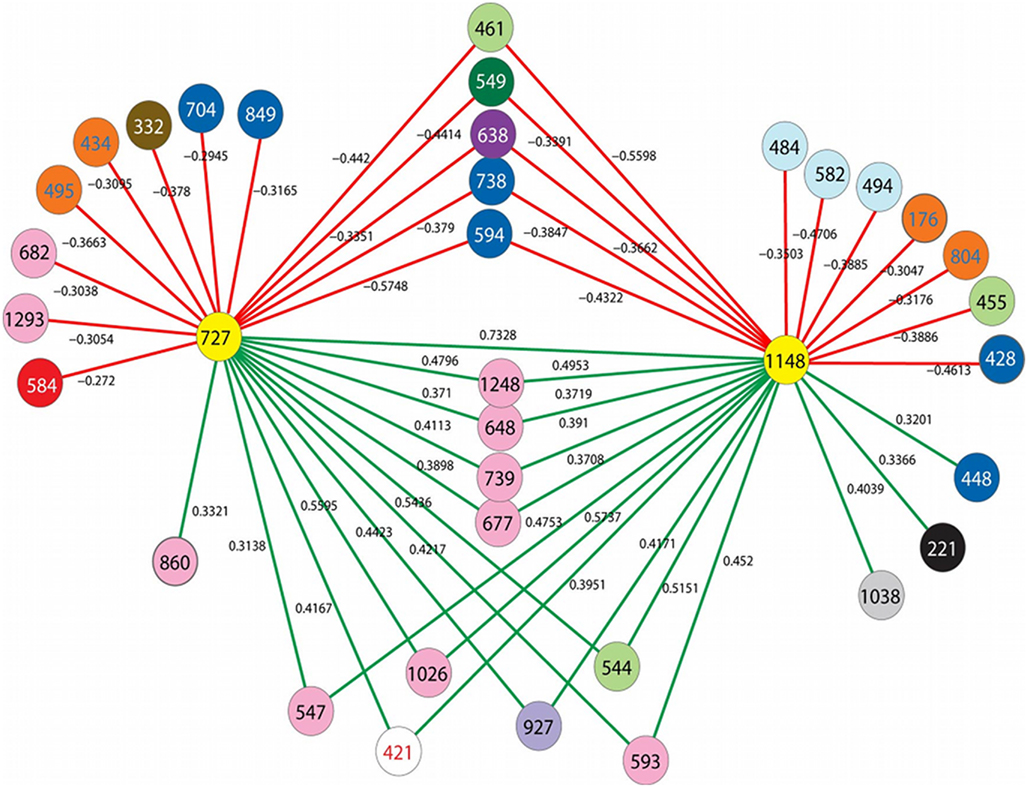
Figure 3. Results of local similarity analysis showing OTUs that are negatively (red lines) and positively (green lines) correlated with the DHVE2 (yellow circles). Circles represent OTUs and are colored according to family level taxonomic classification by the RDP-classifier when possible. Color key is as follows: white, unclassified Archaea; pink, unclassified Euryarchaeota; red, unclassified Crenarchaeota; orange, Nanoarchaeota; purple, unclassified Desulfurococcales; lavender, Thermoplasmatales; light green, Archaeoglobaceae; light blue, Methanococcaceae; gray, Thermococcaceae; brown, Thermofilaceae; black, Pyrodictaceae; dark blue, Desulfurococcaceae; dark green, Thermoproteaceae.
The majority (9/10) of positively correlated OTUs were Euryarchaeota (Figure 3; Table 3), but most (7 of 9) could not be classified below the phylum making it difficult to speculate on potential ecological relationships between these OTUs and the DHVE2. Some may be involved in syntrophic relationships with the DHVE2 as has been proposed for the fermentative Thermococcales (Bonch-Osmolovskaya and Stetter, 1991; Rinker and Kelly, 2000). Others may share the acidophilic strategy with the DHVE2 but utilize different carbon and/or energy sources that would allow all to co-exist in the same biotope. For example, the two OTUs that could be identified below the phylum level were related to Thermogymnomonas and Ferroglobus (Table 3). Described species of these two genera have somewhat complementary non-competing lifestyles to the DHVE2 representative, the peptide-utilizing anaerobic acidophile, A. boonei T469T. Thermogymnomonas is a an aerobic sugar-utilizing thermoacidophile (Itoh et al., 2007) and therefore would occupy a slightly different acidic niche than A. boonei T469T. The only described Ferroglobus species, F. placidus uses a range of electron donors and acceptors, and can reduce (Tor et al., 2001) and oxidize (Hafenbradl et al., 1996) iron, which could provide relatives of this OTU metabolic flexibility in the dynamic hydrothermal environment. Because iron solubility is greater at lower pH, the acidic niche may favor these potential iron oxidizers.
In contrast to the positively correlated OTUs, all negatively correlated OTUs could be confidently classified to at least the order level (Figure 3; Table 3). Some of the negatively correlated OTUs were related to the thermophilic neutrophiles, Archaeoglobus (Stetter, 1988; Burggraf et al., 1990; Beeder et al., 1994) and Aeropyrum (Sako et al., 1996). Since no known acidophilic members belong to these genera, the negatively correlated OTUs may require different physical conditions (e.g., neutral pH, more oxidizing conditions) that do not permit anaerobic thermoacidophiles to grow. Thus, the positively correlated OTUs may share the same acidophilic strategy of the DHVE2 and are able to co-exist because of different carbon and/or energy requirements, while the negatively correlated OTUs may require different physical conditions.
Phylogenetic Diversity of Cultured DHVE2
To expand the cultured diversity of the DHVE2, numerous enrichment cultures targeting thermoacidophiles were initiated from samples collected in 2006 to 2009. In total, 12 axenic DHVE2 isolates were obtained with 4 from the MAR (2 from Lucky Strike, 1 from Rainbow, 1 from TAG), 6 from the ELSC (1 from Tui Malila, 5 from Mariner), and 2 from the EPR (Table A3 in Appendix). All isolates grew well (overnight growth) under anaerobic, thermoacidophilic conditions but optimal growth conditions were not determined. No isolates were obtained from the GB despite the presence of similar sequences in the pyrosequencing dataset (Table 2) and detection of the DHVE2 (by amplification with DHVE2 specific PCR primers) in two enrichment cultures (data not shown). Manipulating the pH, temperature, and organic substrates in order to isolate the DHVE2 from GB samples were unsuccessful as Thermococcus species typically outgrew the DHVE2.
With the exception of strain Lau09-1128, all isolates shared greater than 97% 16S rRNA gene sequence similarity with one another and A. boonei T469T (Figure 4). The high 16S rRNA sequence similarity is comparable to the diversity reported for Thermococcus species from different vent fields (e.g., Huber et al., 1995, 2006; Canganella et al., 1998; Holden et al., 2001) but quite different from deep-sea vent thermoacidophilic Deltaproteobacteria, which were clearly differentiated based on their 16S rRNA gene sequences (Flores et al., 2011b). Despite the overall high 16S rRNA gene sequence similarity of all cultured DHVE2, the isolates nonetheless clustered together based on the vent field of isolation (Figure 4). Other thermophilic archaeal lineages, most notably the Sulfolobales (Whitaker et al., 2003; Reno et al., 2009) and Thermococcales (Huber et al., 2006), did not exhibit such clear biogeographical separation based only on 16S rRNA gene sequences and required MLSA to resolve the biogeographical relationships amongst strains.
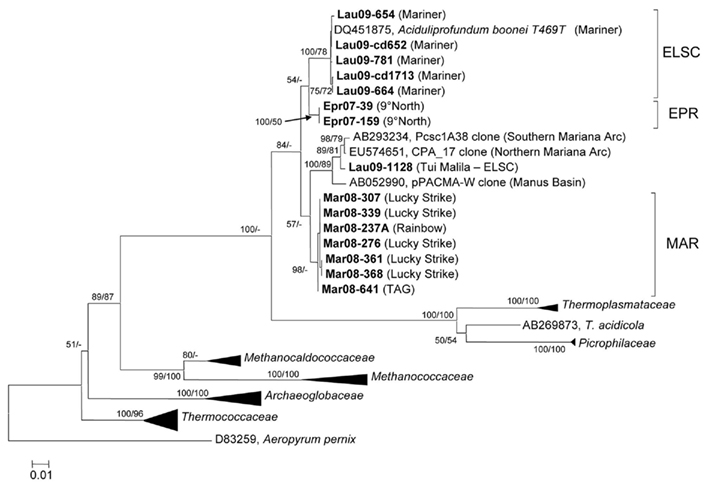
Figure 4. Neighbor-joining tree based on 16S rRNA gene sequence comparisons of novel DHVE2 isolates and other archaeal families. Note that strains MAR08-276, MAR08-307, and MAR08-361 were not included in MLSA analysis as ambiguities were observed in protein-coding genes but not in 16S rRNA gene sequences. Bootstrap percentages above 50% are shown for the neighbor-joining analysis (based on 500 replicates) and for the maximum-likelihood analysis (based on 100 replicates). The phylogenetic tree was generated considering only unambiguously aligned nucleotide positions for a diversity of Archaea (n = 789). New isolates are shown in bold. The scale bar represents 0.01 changes per nucleotide position. All 16S rRNA gene sequences from the DHVE2 isolates were deposited in the European Nucleotide Archive database under accession numbers FR865176 to FR865190.
Multi-Locus Sequence Analysis
Likewise, we used MLSA to further assess the phylogenetic divergence between the DHVE2 isolates. Protein-coding loci were successfully amplified and sequenced from most, but not all of the 12 isolates. In the MLSA of members of the Halobacteriales, Papke et al. (2011) were similarly unable to obtain amplification from all strains. In our study, the radA and EF-2, loci were not obtained from strains Lau09-1128, EPR-159, and EPR-39, while secY could not be amplified from strain Lau09-1128. Subsequent phylogenetic analyses revealed that all sequences from strain Lau09-1128 were quite divergent (Figures 5A,B). All protein-coding loci sequences were submitted to GenBank (EF-2, JN375640 to JN375648; atpA, JN375649 to JN375660; secY, JN375661 to JN375671; rpoB, JN375672 to JN375682; radA, JN375683 to JN375692).
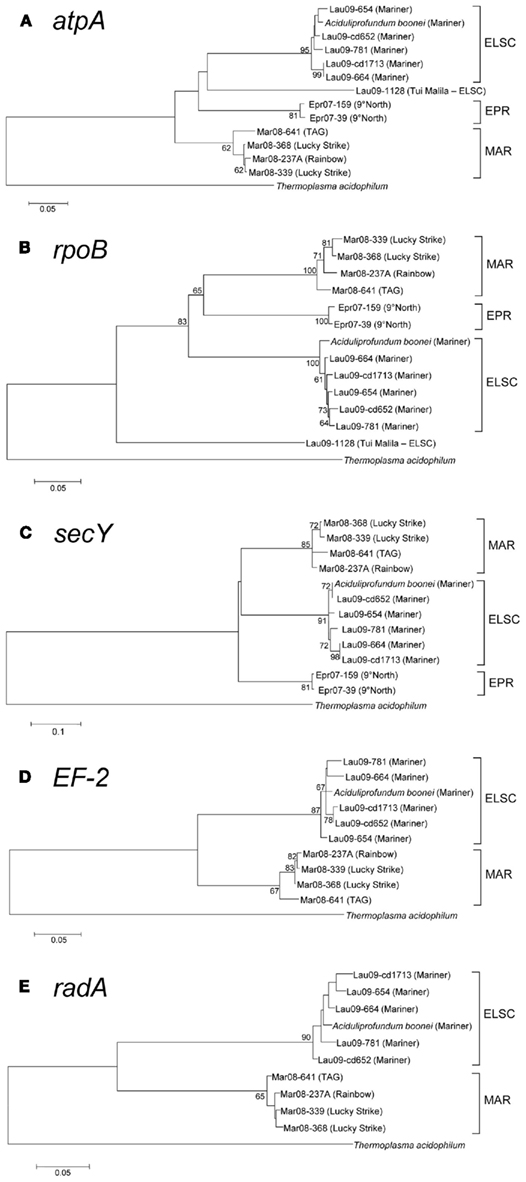
Figure 5. Maximum-likelihood bootstrapped phylogenetic trees of various protein-coding genes from DHVE2 isolates. (A) atpA consensus tree constructed using the Kimura 2-parameter model (Kimura, 1980) taking into account a gamma distribution for the substitution rate. (B) rpoB consensus tree constructed using the Tamura-Nei model (Tamura and Nei, 1993). (C) secY consensus tree generated using Tamura 3-parameter model (Tamura, 1992). (D) EF-2 consensus tree generated as in (C). (E) radA consensus tree generated as in (A). Bootstrap values greater than 60 are shown at nodes in each tree. Branch lengths are the number of substitutions per site. Gene sequences from Thermoplasma acidophilum were used as outgroups in all trees [Gene ID’s (A) = 1455676, (B) = 1456006, (C) = 1456739, (D) = 1456055, (E) = 1456613].
Remarkably high variation between geographic regions within the protein-encoding genes was observed (Table 4). The average nucleotide p-distance values, the proportion of nucleotide sites at which two sequences being compared, are different considering sequences from strains from different geographic regions (Π), varied from 0.19 to 0.25 for the different protein-coding loci (Table 5). The between-region Π values of 0.009–0.070 were observed in the MLSA study of Sulfolobus isolates with ≥99.8% 16S rRNA gene sequence similarity from terrestrial geothermal hot springs of Iceland, North America, and Kamchatka, Russia (Whitaker et al., 2003). There were a total of 983 variable nucleotide sites over 3366 bp from five protein-coding loci examined (Table 4). If the radA and EF-2 loci from strains EPR-159 and EPR-39 had been obtained the total number of variable nt sites would likely be even greater. By comparison, within 78 Sulfolobus strains, there were 124 variable nucleotide sites over 4111 bp from eight protein-coding loci (Whitaker et al., 2003), and among 106 Thermoanaerobacter uzonensis strains with ≥98% 16S rRNA sequence similarity, there were 145 variable nucleotide positions over a total of 8005 nt sites across eight protein-coding loci (Wagner, 2010). Thus, while the 16S rRNA similarity between all DHVE2 isolates (except strain Lau09-1128) is high enough that they would likely be considered the same species, they are extremely divergent considering several protein-coding loci and may actually represent different species.
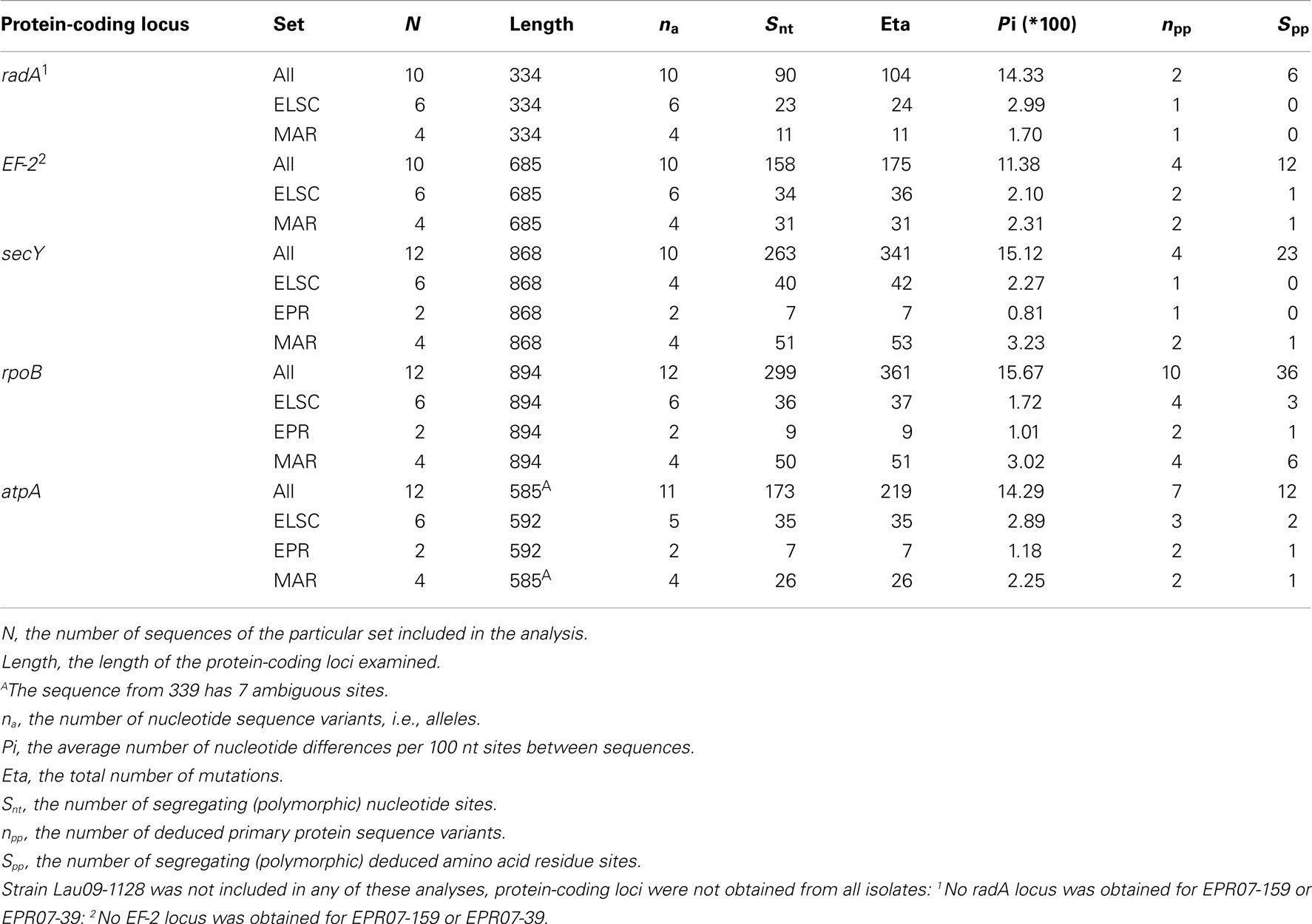
Table 4. DNA polymorphism and nucleotide sequence characteristics of the five protein-coding loci examined from the DHVE2 isolates.
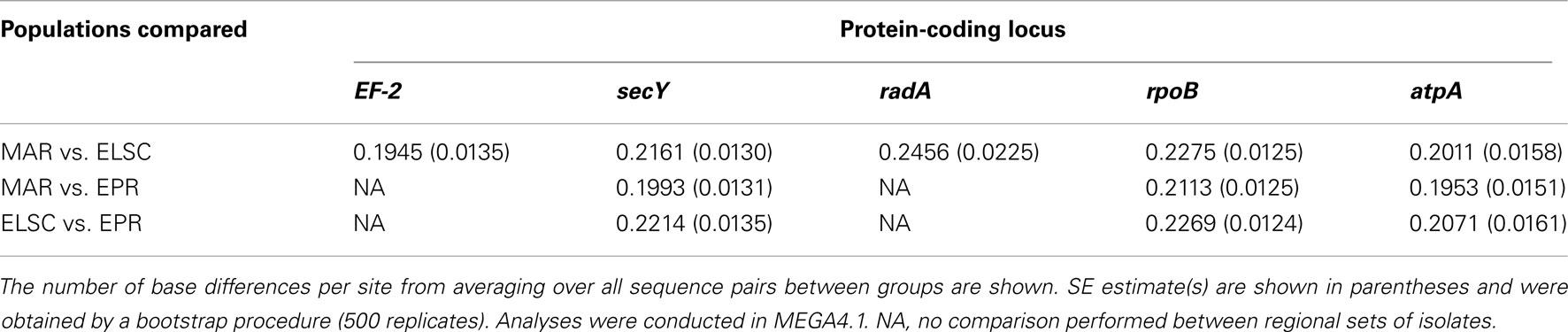
Table 5. Nucleotide divergence between DHVE2 isolates from different regions.
There were consistent differences in the G + C% content of the protein-coding loci from the DHVE2 isolates from different regions with the G + C% values from the ELSC < EPR < MAR (Figure 6). For example, for the rpoB locus the average G + C% for isolates were 42.3% for the ELSC, 45% for EPR, and 49.1% for MAR. Differences in the GC content among related strains have been reported in previous studies and have been attributed to reductive evolution and gene loss (Rocap et al., 2003). While it is possible that gene loss or gain may have influenced the overall nucleotide composition among the DHVE2 isolates, it is more likely that the GC content differences observed between the DHVE2 isolates are due to regional codon usage bias differences (Ermolaeva, 2001). This view is supported by the high proportion of synonymous nucleotide substitutions relative to the number of possible synonymous nucleotide positions that were observed in the pairwise analyses of the protein-coding loci from DHVE2 strains from different regions (Figure 7). For example, considering the radA locus (334 bp length) from the ELSC and MAR isolates, there are an average of 77.7 possible silent nucleotide substitution sites and an average of 67.1 actual silent nucleotide substitutions among the 20 pairwise comparisons (Figure 7A). Considering the same radA locus, there are only six amino acid residue differences (Table 4).
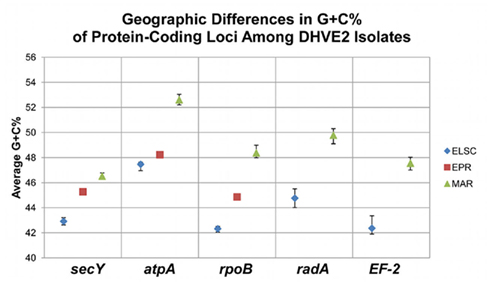
Figure 6. Regional G + C% differences among protein-coding loci from DHVE2 isolates. Average shown, bars indicate range of observed values.
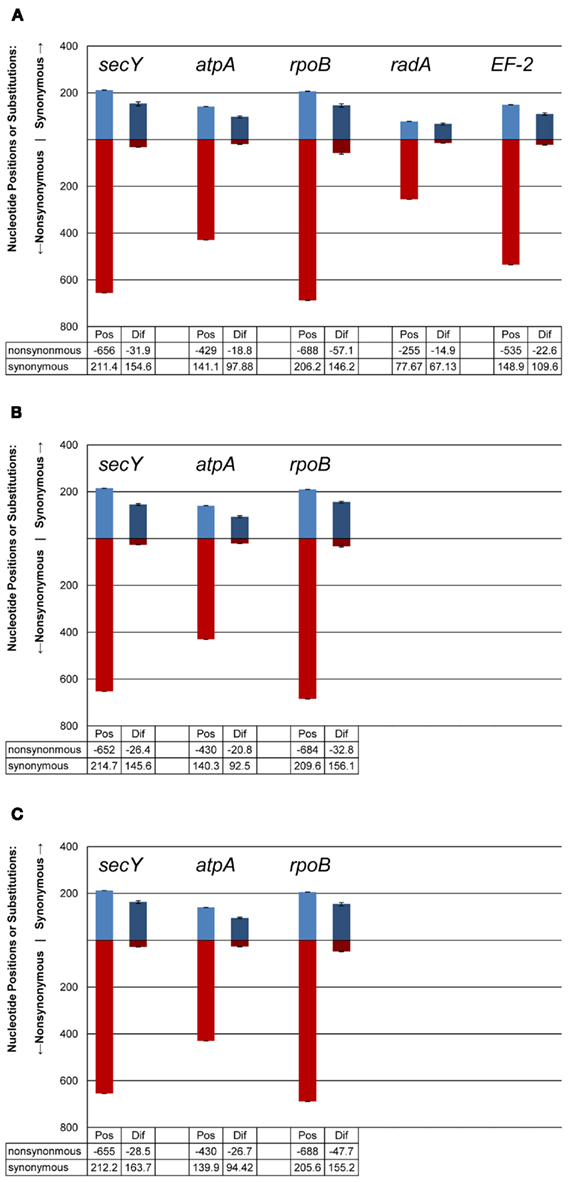
Figure 7. Synonymous and non-synonymous nucleotide positions and substitutions in protein-coding loci among DHVE2 isolates from different regions. (A) Isolates from the MAR compared to isolates from the ELSC (B) isolates from the ELSC compared to isolates from the East Pacific Rise; (C) isolates from the MAR compared to the EPR. Mean values shown on graph, range given with bars. Abbreviations are: Pos, possible synonymous or non-synonymous nucleotide positions; Dif, actual synonymous or non-synonymous differences.
The slowly evolving 16S rRNA gene, as well as all of the protein-coding loci, revealed regional clustering patterns for the DHVE2 isolates (Figures 4 and 5) suggesting an early origin of the differentiation between ELSC, EPR, and MAR populations. Although we only had a limited number of isolates, these observations suggest divergent evolution of geographically isolated DHVE2 populations, i.e., allopatry (Whitaker, 2006). Additionally, some evidence for biogeographical patterns within oceanic regions was also noted. For example, most of the protein-coding loci from strain MAR-641, obtained from the TAG vent field of the MAR, are phylogenetically different from the other strains obtained from the Lucky Strike and Rainbow vent sites also along the MAR (Figure 5). Intra-regional differences have been observed in other studies of the spatial diversity patterns of microorganisms from thermal environments (Petursdottir et al., 2000; Hreggvidsson et al., 2006; Takacs-Vesbach et al., 2008; Wagner, 2010). With additional DHVE2 strains and genomes, the global patterns of diversity of the DHVE2 could be explored in greater detail. Based on the variation observed in the protein-coding genes, which are part of the core genome for this lineage, extensive differences in the variable genome (Tettelin et al., 2008) are also expected.
Conclusion
Results from this study show that the DHVE2 are ubiquitous in deep-sea hydrothermal environments and tend to co-occur with other Euryarchaeota. Phylogenetic analyses of the 16S rRNA genes and protein-coding loci from 12 different DHVE2 isolates revealed clear biogeographical clustering patterns indicative of allopatric speciation. Assuming that all DHVE2 are thermoacidophilic, then thermoacidophily is an important physiological strategy for some microorganisms in these ecosystems. Factors that seemed to influence the occurrence and abundance of the DHVE2 within an individual vent field include the age of the vent deposit (as this is indicative of the maturity of the microbial community, Page et al., 2008), fluid mixing style, and type of vent structure (chimney vs. flange). Other factors like deposit mineralogy, grazing by eukaryotes, and viruses may also be influencing the biogeography of the DHVE2 but were not examined as part of this study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thanks to all crew members of the R/V Atlantis, R/V Thomas G. Thompson, R/V Roger Revelle, HOV Alvin, and ROV Jason II for help collecting samples. Also, thanks to Isabel Ferrera, Julie Kirshtein, Kristen Myers, Josh Steinberg and Anna Perevalova for help extracting nucleic acids from some samples used in this study. We would also like to thank two anonymous reviewers for their thoughtful and thorough reviews. Funding for this research was provided by the US-National Science Foundation through an IGERT fellowship to Gilberto E. Flores, and by grants OCE-0728391 and OCE-0937404 to Anna-Louise Reysenbach.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Auguet, J. C., Barberan, A., and Casamayor, E. O. (2009). Global ecological patterns in uncultured archaea. ISME J. 4, 182–190.
Beeder, J., Nilsen, R. K., Rosnes, J. T., Torsvik, T., and Lien, T. (1994). Archaeoglobus fulgidus isolated from hot North Sea oil field waters. Appl. Environ. Microbiol. 60, 1227.
Bonch-Osmolovskaya, E. A., and Stetter, K. O. (1991). Interspecies hydrogen transfer in cocultures of thermophilic archaea. Syst. Appl. Microbiol. 14, 205–208.
Burggraf, S., Jannasch, H., Nicolaus, B., and Stetter, K. (1990). Archaeoglobus profundus sp. nov., represents a new species within the sulfate-reducing archaebacteria. Syst. Appl. Microbiol. 13, 24–28.
Canganella, F., Jones, W. J., Gambacorta, A., and Antranikian, G. (1998). Thermococcus guaymasensis sp. nov. and Thermococcus aggregans sp. nov., two novel thermophilic archaea isolated from the Guaymas Basin hydrothermal vent site. Int. J. Syst. Evol. Microbiol. 48, 1181–1185.
Claesson, M. J., O’Sullivan, O., Wang, Q., Nikkila, J., Marchesi, J. R., Smidt, H., De Vos, W. M., Ross, R. P., and O’Toole, P. W. (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 4, e6669. doi:10.1371/journal.pone.0006669
Flores, G. E., Campbell, J. H., Kirshtein, J. D., Meneghin, J., Podar, M., Steinberg, J. I., Seewald, J. S., Tivey, M. K., Voytek, M. A., Yang, Z. K., and Reysenbach, A. L. (2011a). Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge. Environ. Microbiol. 13, 2158–2171.
Flores, G. E., Hunter, R. C., Liu, Y., Mets, A., Schouten, S., and Reysenbach, A. L. (2011b). Hippea jasoniae sp. nov. and Hippea alviniae sp. nov., thermoacidophilic Deltaproteobacteria isolated from deep-sea hydrothermal vent deposits. Int. J. Syst. Evol. Microbiol. doi:10.1099/ijs.0.033001-0
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., Feil, E. J., Stackebrandt, E., Van De Peer, Y., Vandamme, P., and Thompson, F. L. (2005). Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739.
Götz, D., Banta, A., Beveridge, T. J., Rushdi, A. I., Simoneit, B. R., and Reysenbach, A. L. (2002). Persephonella marina gen. nov., sp. nov. and Persephonella guaymasensis sp. nov., two novel, thermophilic, hydrogen-oxidizing microaerophiles from deep-sea hydrothermal vents. Int. J. Syst. Evol. Microbiol. 52, 1349–1359.
Hafenbradl, D., Keller, M., Dirmeier, R., Rachel, R., Roßnagel, P., Burggraf, S., Huber, H., and Stetter, K. O. (1996). Ferroglobus placidus gen. nov., sp. nov., a novel hyperthermophilic archaeum that oxidizes Fe2+ at neutral pH under anoxic conditions. Arch. Microbiol. 166, 308–314.
Hoek, J., Banta, A., Hubler, F., and Reysenbach, A. (2003). Microbial diversity of a sulphide spire located in the Edmond deep-sea hydrothermal vent field on the Central Indian Ridge. Geobiology 1, 119–127.
Holden, J. F., Takai, K., Summit, M., Bolton, S., Zyskowski, J., and Baross, J. A. (2001). Diversity among three novel groups of hyperthermophilic deep-sea Thermococcus species from three sites in the northeastern Pacific Ocean. FEMS Microbiol. Ecol. 36, 51–60.
Hreggvidsson, G. O., Skirnisdottir, S., Smit, B., Hjorleifsdottir, S., Marteinsson, V. T., Petursdottir, S., and Kristjansson, J. K. (2006). Polyphasic analysis of Thermus isolates from geothermal areas in Iceland. Extremophiles 10, 563–575.
Huber, J. A., Butterfield, D. A., and Baross, J. A. (2002). Temporal changes in archaeal diversity and chemistry in a mid-ocean ridge subseafloor habitat. Appl. Environ. Microbiol. 68, 1585–1594.
Huber, J. A., Butterfield, D. A., and Baross, J. A. (2006). Diversity and distribution of subseafloor Thermococcales populations in diffuse hydrothermal vents at an active deep-sea volcano in the northeast Pacific Ocean. J. Geophys. Res. 111, G04016.
Huber, R., Stöhr, J., Hohenhaus, S., Rachel, R., Burggraf, S., Jannasch, H. W., and Stetter, K. O. (1995). Thermococcus chitonophagus sp. nov., a novel, chitin-degrading, hyperthermophilic archaeum from a deep-sea hydrothermal vent environment. Arch. Microbiol. 164, 255–264.
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F. S., Nealson, K. H., Horikoshi, K., D’Hondt, S., and Jorgensen, B. B. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820.
Itoh, T., Yoshikawa, N., and Takashina, T. (2007). Thermogymnomonas acidicola gen. nov., sp. nov., a novel thermoacidophilic, cell wall-less archaeon in the order Thermoplasmatales, isolated from a solfataric soil in Hakone, Japan. Int. J. Syst. Evol. Microbiol. 57, 2557–2561.
Kato, S., Takano, Y., Kakegawa, T., Oba, H., Inoue, K., Kobayashi, C., Utsumi, M., Marumo, K., Kobayashi, K., Ito, Y., Ishibashi, J., and Yamagishi, A. (2010). Biogeography and biodiversity in sulfide structures of active and inactive vents at deep-sea hydrothermal fields of the Southern Mariana Trough. Appl. Environ. Microbiol. 76, 2968–2979.
Kimura, H., Mori, K., Tashiro, T., Kato, K., Yamanaka, T., Ishibashi, J. I., and Hanada, S. (2010). Culture-independent estimation of optimal and maximum growth temperatures of archaea in subsurface habitats based on the G plus C content in 16S rRNA gene sequences. Geomicrobiol. J. 27, 114–122.
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120.
Kormas, K. A., Tivey, M. K., Von Damm, K., and Teske, A. (2006). Bacterial and archaeal phylotypes associated with distinct mineralogical layers of a white smoker spire from a deep-sea hydrothermal vent site (9 degrees N, East Pacific Rise). Environ. Microbiol. 8, 909–920.
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451.
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, Buchner, A., Lai, T., Steppi, S., Jobb, G., Forster, W., Brettske, I., Gerber, S., Ginhart, A. W., Gross, O., Grumann, S., Hermann, S., Jost, R., Konig, A., Liss, T., Lussmann, R., May, M., Nonhoff, B., Reichel, B., Strehlow, R., Stamatakis, A., Stuckmann, N., Vilbig, A., Lenke, M., Ludwig, T., Bode, A., and Schleifer, K. H. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371.
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E. M., Kubal, M., Paczian, T., Rodriguez, A., Stevens, R., Wilke, A., Wilkening, J., and Edwards, R. A. (2008). The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386. doi:10.1186/1471-2105-9-386
Moussard, H., Henneke, G., Moreira, D., Jouffe, V., Lopez-Garcia, P., and Jeanthon, C. (2006a). Thermophilic lifestyle for an uncultured archaeon from hydrothermal vents: evidence from environmental genomics. Appl. Environ. Microbiol. 72, 2268–2271.
Moussard, H., Moreira, D., Cambon-Bonavita, M., López-García, P., and Jeanthon, C. (2006b). Uncultured Archaea in a hydrothermal microbial assemblage: phylogenetic diversity and characterization of a genome fragment from a euryarchaeote. FEMS Microbiol. Ecol. 57, 452–469.
Moussard, H., L’Haridon, S., Tindall, B. J., Banta, A., Schumann, P., Stackebrandt, E., Reysenbach, A. L., and Jeanthon, C. (2004). Thermodesulfatator indicus gen. nov., sp. nov., a novel thermophilic chemolithoautotrophic sulfate-reducing bacterium isolated from the Central Indian Ridge. Int. J. Syst. Evol. Microbiol. 54, 227–233.
Nakagawa, S., Takai, K., Inagaki, F., Chiba, H., Ishibashi, J., Kataoka, S., Hirayama, H., Nunoura, T., Horikoshi, K., and Sako, Y. (2005). Variability in microbial community and venting chemistry in a sediment-hosted backarc hydrothermal system: impacts of subseafloor phase-separation. FEMS Microbiol. Ecol. 54, 141–155.
Nakagawa, T., Takai, K., Suzuki, Y., Hirayama, H., Konno, U., Tsunogai, U., and Horikoshi, K. (2006). Geomicrobiological exploration and characterization of a novel deep-sea hydrothermal system at the TOTO caldera in the Mariana Volcanic Arc. Environ. Microbiol. 8, 37–49.
Nercessian, O., Reysenbach, A. L., Prieur, D., and Jeanthon, C. (2003). Archaeal diversity associated with in situ samplers deployed on hydrothermal vents on the East Pacific Rise (13 degrees N). Environ. Microbiol. 5, 492–502.
Nunoura, T., Oida, H., Nakaseama, M., Kosaka, A., Ohkubo, S. B., Kikuchi, T., Kazama, H., Hosoi-Tanabe, S., Nakamura, K., Kinoshita, M., Hirayama, H., Inagaki, F., Tsunogai, U., Ishibashi, J., and Takai, K. (2010). Archaeal diversity and distribution along thermal and geochemical gradients in hydrothermal sediments at the Yonaguni Knoll IV hydrothermal field in the Southern Okinawa trough. Appl. Environ. Microbiol. 76, 1198–1211.
Nunoura, T., and Takai, K. (2009). Comparison of microbial communities associated with phase-separation-induced hydrothermal fluids at the Yonaguni Knoll IV hydrothermal field, the Southern Okinawa Trough. FEMS Microbiol. Ecol. 67, 351–370.
Page, A., Tivey, M. K., Stakes, D. S., and Reysenbach, A. L. (2008). Temporal and spatial archaeal colonization of hydrothermal vent deposits. Environ. Microbiol. 10, 874–884.
Papke, R. T., Ramsing, N. B., Bateson, M. M., and Ward, D. M. (2003). Geographical isolation in hot spring cyanobacteria. Environ. Microbiol. 5, 650–659.
Papke, R. T., White, E., Reddy, P., Weigel, G., Kamekura, M., Minegishi, H., Usami, R., and Ventosa, A. (2011). A multilocus sequence analysis (MLSA) approach to Halobacteriales phylogeny and taxonomy. Int. J. Syst. Evol. Microbiol. 61, 2984–2995.
Petursdottir, S. K., Hreggvidsson, G. O., Da Costa, M. S., and Kristjansson, J. K. (2000). Genetic diversity analysis of Rhodothermus reflects geographical origin of the isolates. Extremophiles 4, 267–274.
Reno, M. L., Held, N. L., Fields, C. J., Burke, P. V., and Whitaker, R. J. (2009). Biogeography of the Sulfolobus islandicus pan-genome. Proc. Natl. Acad. Sci. U.S.A. 106, 8605–8610.
Reysenbach, A. L., and Flores, G. E. (2008). Electron microscopy encounters with unusual thermophiles helps direct genomic analysis of Aciduliprofundum boonei. Geobiology 6, 331–336.
Reysenbach, A. L., Liu, Y., Banta, A. B., Beveridge, T. J., Kirshtein, J. D., Schouten, S., Tivey, M. K., Von Damm, K. L., and Voytek, M. A. (2006). A ubiquitous thermoacidophilic archaeon from deep-sea hydrothermal vents. Nature 442, 444–447.
Reysenbach, A. L., Longnecker, K., and Kirshtein, J. (2000). Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a Mid-Atlantic Ridge hydrothermal vent. Appl. Environ. Microbiol. 66, 3798–3806.
Rinker, K. D., and Kelly, R. M. (2000). Effect of carbon and nitrogen sources on growth dynamics and exopolysaccharide production for the hyperthermophilic archaeon Thermococcus litoralis and bacterium Thermotoga maritima. Biotechnol. Bioeng. 69, 537–547.
Rocap, G., Larimer, F. W., Lamerdin, J., Malfatti, S., Chain, P., Ahlgren, N. A., Arellano, A., Coleman, M., Hauser, L., Hess, W. R., Johnson, Z. I., Land, M., Lindell, D., Post, A. F., Regala, W., Shah, M., Shaw, S. L., Steglich, C., Sullivan, M. B., Ting, C. S., Tolonen, A., Webb, E. A., Zinser, E. R., and Chisholm, S. W. (2003). Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424, 1042–1047.
Ruan, Q., Dutta, D., Schwalbach, M. S., Steele, J. A., Fuhrman, J. A., and Sun, F. (2006). Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 22, 2532–2538.
Sako, Y., Nomura, N., Uchida, A., Ishida, Y., Morii, H., Koga, Y., Hoaki, T., and Maruyama, T. (1996). Aeropyrum pernix gen. nov., sp. nov., a novel aerobic hyperthermophilic archaeon growing at temperatures up to 100 degrees C. Int. J. Syst. Bacteriol. 46, 1070–1077.
Schrenk, M. O., Kelley, D. S., Delaney, J. R., and Baross, J. A. (2003). Incidence and diversity of microorganisms within the walls of an active deep-sea sulfide chimney. Appl. Environ. Microbiol. 69, 3580–3592.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., Amin, N., Schwikowski, B., and Ideker, T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504.
Stetter, K. (1988). Archaeoglobus fulgidus gen. nov., sp. nov.: a new taxon of extremely thermophilic archaebacteria. Syst. Appl. Microbiol. 10, 172–173.
Stott, M. B., Saito, J. A., Crowe, M. A., Dunfield, P. F., Hou, S., Nakasone, E., Daughney, C. J., Smirnova, A. V., Mountain, B. W., Takai, K., and Alam, M. (2008). Culture-independent characterization of a novel microbial community at a hydrothermal vent at Brothers Volcano, Kermadec Arc, New Zealand. J. Geophys. Res. 113, B08S06.
Takacs-Vesbach, C., Mitchell, K., Jackson-Weaver, O., and Reysenbach, A. L. (2008). Volcanic calderas delineate biogeographic provinces among Yellowstone thermophiles. Environ. Microbiol. 10, 1681–1689.
Takai, K., and Horikoshi, K. (1999). Genetic diversity of archaea in deep-sea hydrothermal vent environments. Genetics 152, 1285–1297.
Takai, K., Komatsu, T., Inagaki, F., and Horikoshi, K. (2001). Distribution of archaea in a black smoker chimney structure. Appl. Environ. Microbiol. 67, 3618–3629.
Tamura, K. (1992). Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol. Biol. Evol. 9, 678–687.
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. doi:10.1093/molbev/msr121
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477.
Tivey, M. K. (2004). “Environmental conditions within active seafloor vent structures: sensitivity to vent fluid composition and fluid flow,” in Subseafloor Biosphere at Mid-Ocean Ridges, eds W. Wilcock, C. Cary, E. Delong, D. Kelley, and J. Baross (Washington, DC: American Geophysical Union), 137–152.
Tivey, M. K. (2007). Generation of seafloor hydrothermal vent fluids and associated mineral deposits. Oceanography 20, 50–65.
Tor, J. M., Kashefi, K., and Lovley, D. R. (2001). Acetate oxidation coupled to Fe (III) reduction in hyperthermophilic microorganisms. Appl. Environ. Microbiol. 67, 1363–1365.
Van Dover, C. L., German, C. R., Speer, K. G., Parson, L. M., and Vrijenhoek, R. C. (2002). Evolution and biogeography of deep-sea vent and seep invertebrates. Science 295, 1253–1257.
Wagner, I. D. (2010). Novel Anaerobic Thermophilic Bacteria; Intraspecies Heterogeneity and Biogeography of Thermoanaerobacter Isolates from the Kamchatka Peninsula, Russian Far East. Ph.D. dissertation, University of Georgia, Athens.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267.
Whitaker, R. J. (2006). Allopatric origins of microbial species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1975–1984.
Whitaker, R. J., Grogan, D. W., and Taylor, J. W. (2003). Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301, 976–978.
Zhou, H., Li, J., Peng, X., Meng, J., Wang, F., and Ai, Y. (2009). Microbial diversity of a sulfide black smoker in main endeavour hydrothermal vent field, Juan de Fuca Ridge. J. Microbiol. 47, 235–247.
Appendix
Table A1. Summary of 16S rRNA gene sequences previously detected in marine hydrothermal environments.

Table A2. Location of the protein-coding loci in the genome of A. boonei T469T.
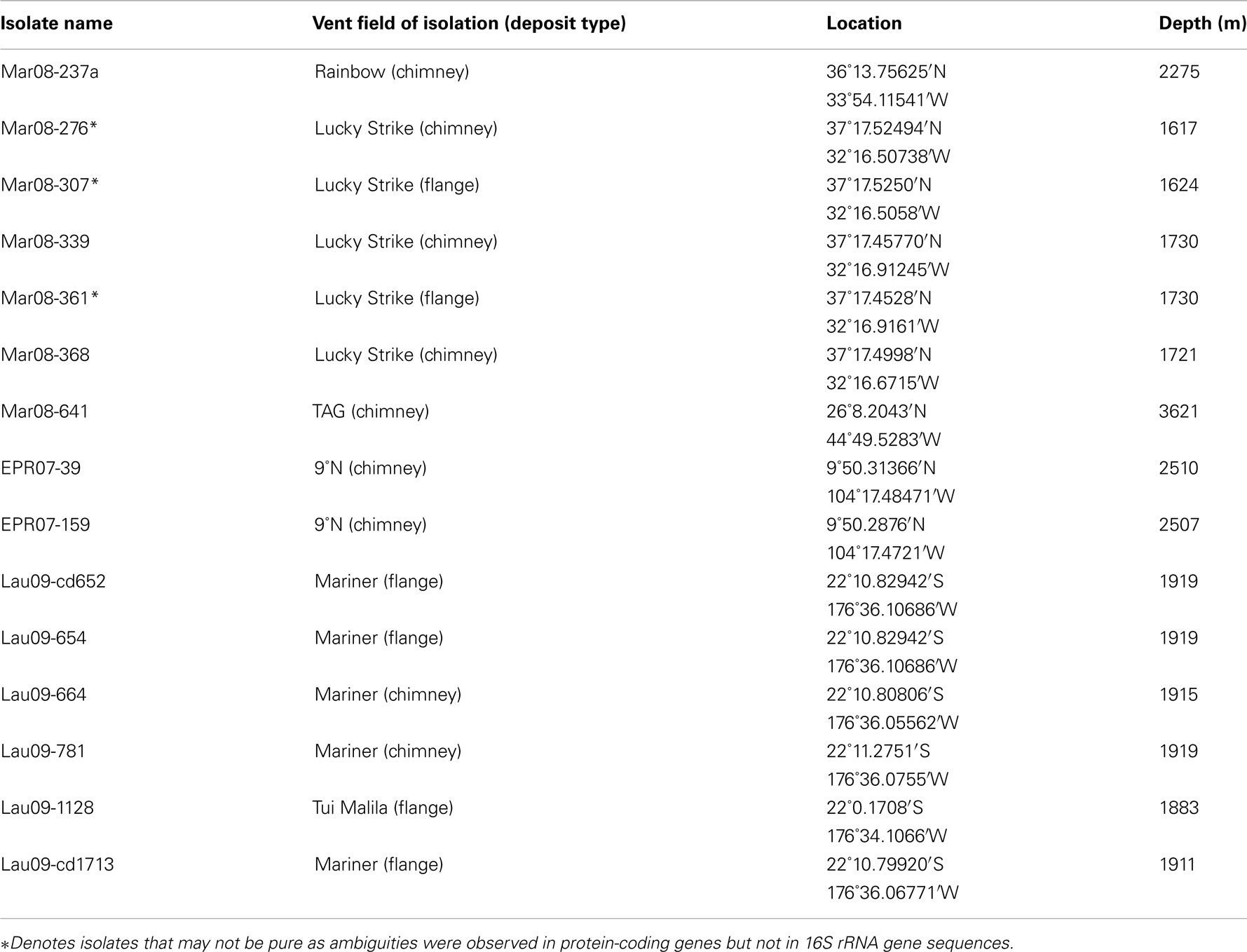
Table A3. Hydrothermal vent deposits from which new DHVE2 isolates were obtained.
Keywords: archaea, hydrothermal vents, deep-sea, multi-locus sequence analysis, biogeography, acidophile, thermophile
Citation: Flores GE, Wagner ID, Liu Y and Reysenbach A-L (2012) Distribution, abundance, and diversity patterns of the thermoacidophilic “deep-sea hydrothermal vent euryarchaeota 2”. Front. Microbio. 3:47. doi: 10.3389/fmicb.2012.00047
Received: 06 November 2011;
Paper pending published: 06 December 2011;
Accepted: 30 January 2012;
Published online: 20 February 2012.
Edited by:
Kirsten Silvia Habicht, University of Southern Denmark, DenmarkReviewed by:
Kuk-Jeong Chin, Georgia State University, USAElizaveta Bonch-Osmolovskyaya, Winogradsky Institute of Microbiology Russian Academy of Sciences, Russia
Copyright: © 2012 Flores, Wagner, Liu and Reysenbach. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Anna-Louise Reysenbach, Department of Biology, Center for Life in Extreme Environments, Portland State University, PO Box 751, Portland, OR 97207-0751, USA. e-mail:cmV5c2VuYmFjaGFAcGR4LmVkdQ==
†Present address: Gilberto E. Flores, Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO 80309, USA.