 Michael T. Leonard1
Michael T. Leonard1 Austin G. Davis-Richardson1
Austin G. Davis-Richardson1 Alexandria N. Ardissone1
Alexandria N. Ardissone1 Kaisa M. Kemppainen1Jennifer C. Drew1Jorma Ilonen2,3
Kaisa M. Kemppainen1Jennifer C. Drew1Jorma Ilonen2,3 Mikael Knip4,5,6Olli Simell7
Mikael Knip4,5,6Olli Simell7 Jorma Toppari7Riitta Veijola8Heikki Hyöty9
Jorma Toppari7Riitta Veijola8Heikki Hyöty9 Eric W. Triplett1*
Eric W. Triplett1*- 1Department of Microbiology and Cell Science, Institute of Food and Agricultural Sciences, University of Florida, Gainesville, FL, USA
- 2Department of Clinical Microbiology, University of Eastern Finland, Kuopio, Finland
- 3Immunogenetics Laboratory, University of Turku, Turku, Finland
- 4Children's Hospital, University of Helsinki and Helsinki University Central Hospital, Helsinki, Finland
- 5Diabetes and Obesity Research Program, University of Helsinki, Helsinki, Finland
- 6Department of Pediatrics, Tampere University Hospital, Tampere, Finland
- 7Department of Pediatrics, Turku University Hospital, University of Turku, Turku, Finland
- 8Department of Pediatrics, Oulu University Hospital, University of Oulu, Oulu, Finland
- 9School of Medicine, University of Tampere, Tampere, Finland
Despite the large interest in the human microbiome in recent years, there are no reports of bacterial DNA methylation in the microbiome. Here metagenomic sequencing using the Pacific Biosciences platform allowed for rapid identification of bacterial GATC methylation status of a bacterial species in human stool samples. For this work, two stool samples were chosen that were dominated by a single species, Bacteroides dorei. Based on 16S rRNA analysis, this species represented over 45% of the bacteria present in these two samples. The B. dorei genome sequence from these samples was determined and the GATC methylation sites mapped. The Bacteroides dorei genome from one subject lacked any GATC methylation and lacked the DNA adenine methyltransferase genes. In contrast, B. dorei from another subject contained 20,551 methylated GATC sites. Of the 4970 open reading frames identified in the GATC methylated B. dorei genome, 3184 genes were methylated as well as 1735 GATC methylations in intergenic regions. These results suggest that DNA methylation patterns are important to consider in multi-omic analyses of microbiome samples seeking to discover the diversity of bacterial functions and may differ between disease states.
Introduction
DNA adenosine methylation (Dam methylation) of the 5′-GATC-3′ motif in bacterial genomes, particularly in Escherichia coli, has been well characterized for decades (Marinus, 1987; Barras and Marinus, 1989; Marinus and Casadesus, 2009). Of all of the DNA methylation systems known in bacteria, GATC methylation appears to have the highest impact on gene expression (Barras and Marinus, 1989). The biochemistry of the adenosine methylation reaction catalyzed by bacterial DNA adenine methyltransferase (DamMT) is also well characterized (Thielking et al., 1997; Urig et al., 2002). As DamMT mutants are not lethal, the phenotype of DamMT mutants in several bacteria have shown that methylation of the GATC motif affects gene expression of many processes including chromosome replication, mismatch repair, and nucleoid structure (Løbner-Olesen et al., 2005). The GATC methylation system has been shown to increase virulence in Salmonella enterica serovar Typhimurium, Vibrio cholerae, Yersinia pestis, and Yersinia pseudotuberculosis (Heithoff et al., 1999; Julio et al., 2001, 2002; Robinson et al., 2005). As a result, significant efforts have been made in recent years to design antibiotics that inhibit DamMT (Mashhoon et al., 2004, 2006; Hobley et al., 2012; McKelvie et al., 2013).
Despite the enormous interest in recent years to characterize the bacterial diversity of the human microbiome, particularly as it relates to gut diseases, the role that DNA methylation plays in function of the microbiome remains unknown. The PacBio RS II system is capable of detecting DNA methylation through analysis of polymerase kinetics (Flusberg et al., 2010; Clark et al., 2012; Fang et al., 2012; Murray et al., 2012). This technology was used here to discover the profound differences in the extent of Dam methylation in one dominant bacterial species in the gut between two children at high genetic risk for type 1 diabetes and 1 year prior to the development of type 1 diabetes autoimmunity in one of these children.
Materials and Methods
The stool samples used here were collected by the Finnish Type 1 Diabetes Prediction and Prevention Study (DIPP) (Kukko et al., 2005). Newborns were screened for high-risk HLA-DR and HLA-DQ genotypes using a previously described method (Kukko et al., 2005). Stool samples were collected by the subjects' parents at home and mailed to the DIPP Virus Laboratory for virology in Tampere, Finland, where they were stored at −80°C. Detection of beta-cell autoimmunity was done as described in Parikka et al. (2012). Sample 105 was collected from the subject at 13.5 months of age. This subject became autoimmune for type 1 diabetes at 15.1 months of age. Sample 439 was collected from a subject who remained healthy at 3.3 months of age. Both subjects were genetically at high risk for type 1 diabetes given their HLA genotype.
In this study, DNA extraction and 16S rRNA amplification, sequencing, and analysis was done as described previously (Fagen et al., 2012) except the Qiagen AllPrep DNA/RNA/Protein Mini Kit (QIAGEN) was used to extract DNA, RNA, and protein from stool. Based on the 16S rRNA results, two samples were chosen for long-read Pacific Biosciences sequencing based on the high relative abundance of this organism was 63.7 and 47.9% from samples 105 and 439, respectively. These samples, 105 and 439, were collected from children who became autoimmune or remained healthy, respectively.
Pacific Biosciences (PacBio RS II system) library construction and sequencing was done by the University of Florida's Interdisciplinary Center for Biotechnology Research. Prior to sequencing, a PacBio library was made with SMRTbell Adaptors. The Bacteroides dorei genome was assembled to closure from sample 105 after obtaining eight SMRT cells of sequence data. A total of 1,502,920 reads and 1,860,712,096 bases were obtained, with a mean read length of 2706 bp. Average read quality was 0.848. The initial Pacbio reads were error corrected using the Pacbio RS_PreAssembler.1 module (Koren et al., 2012) with minimum subread length of 400 bp, minimum read quality 0.60, and minimum seed read length of 3800 bp. The error correction process yielded 47,654 reads of 2378 bp average length. Reads were binned according to coverage reported by the Pacbio RS_PreAssembler.1 protocol, filtering out reads with lower than 200× coverage. A set of 27 contigs was assembled directly from the binned reads using SPAdes assembler v3.0 (Bankevich et al., 2012). A single scaffold was obtained by detecting overlaps with Mauve 2.3.1 (Darling et al., 2010) and manually assembling the remaining contigs. The initial genome assembly was further refined using the Pacbio RS_Resequencing.1 module with Quiver consensus calling. The final, circular genome consists of 5,726,633 bp and an overall GC content of 42.0%.
The complete B. dorei genome from sample 439 metageomic DNA was closed in the same manner as described above for the 105 sample. The 439 closed genome was significantly smaller than the 105 genome with 5,243,219 bp. Genome annotation for both genomes was done by using the NCBI Prokaryotic Genome Annotation pipeline (Angiuoli et al., 2008), which relies on GeneMarkS+ for gene prediction (Besemer et al., 2001). The NCBI accession numbers for the closed 105 B. dorei genome and the 439 B. dorei genome are CP007619 and CP008741, respectively. Two-way average nucleotide identities between pairs of genomes were determined as described by Goris et al. (2007).
Methylation patterns obtained from the PacBio data were recovered from both genomes using the assembled genomes as a reference. Methylation data was extracted from the full set of sequence data using Kinetics Tools. This tool utilizes the P_ModificationDetection module in SMRT Portal, which is utilized by the RS Modification Detection as well as the RS Modifications and Motif Detection protocol. The Motif Detection protocol generated motifs by comparing methylation patterns to genomic context.
Results
PacBio sequencing and assembly from 105 and 439 metagenomic DNA resulted in two closed Bacteroides dorei genomes of 5,726,633 and 5,243,219 bp, respectively, The 105 and 439 genomes assembled to an average coverage across each genome of 306.72- and 249.47-fold, respectively. The 16S rRNA and whole genome average nucleotide identities were compared between the 105 and 439 genomes as well as two reference genomes, the nearly complete genome of B. dorei DSM 17855 and the complete genome of B. vulgatus 8482 (Tables 1, 2). These data show that the 105 and 439 genomes are far more closely related to B. dorei than B. vulgatus.
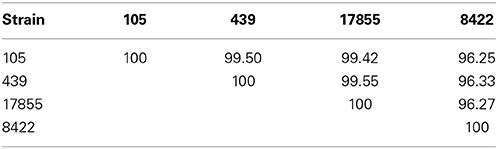
Table 1. Percent two-way average nucleotide identities (ANI) at the whole genome level between the B. dorei 105 and 439 genomes described here and the nearly complete genome of B. dorei DSM 17855 and the complete genome of B. vulgatus ATCC 8482.
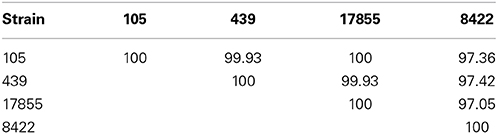
Table 2. Percent 16S rRNA identities between the B. dorei 105 and 439 genomes described here and the nearly complete genome of B. dorei DSM 17855 and the complete genome of B. vulgatus ATCC 8482.
Methylated GATC motifs were only found in the B. dorei from the 105 sample and were completely absent in the B. dorei contigs obtained for sample 439 (Table 3). There were 49,007 total methylations detected in B. dorei 105, and 38,203 total methylations detected in B. dorei 439. Of these, 14,322 and 24,770 methylation sites for the 105 and 439 genomes, respectively, could not be clustered to any motif. In the B. dorei genome from the 105 sample, 20,551 GATC methylations and one copy of the DamMT gene were identified. Of 20,554 GATC sites in the 105 genome, only 3 were not detected as methylated. In contrast, the B. dorei genome of 5,243,219 bp from sample 439 lacked a DamMT gene and no GATC motif methylations were detected among the 18,908 GATC sites identified.
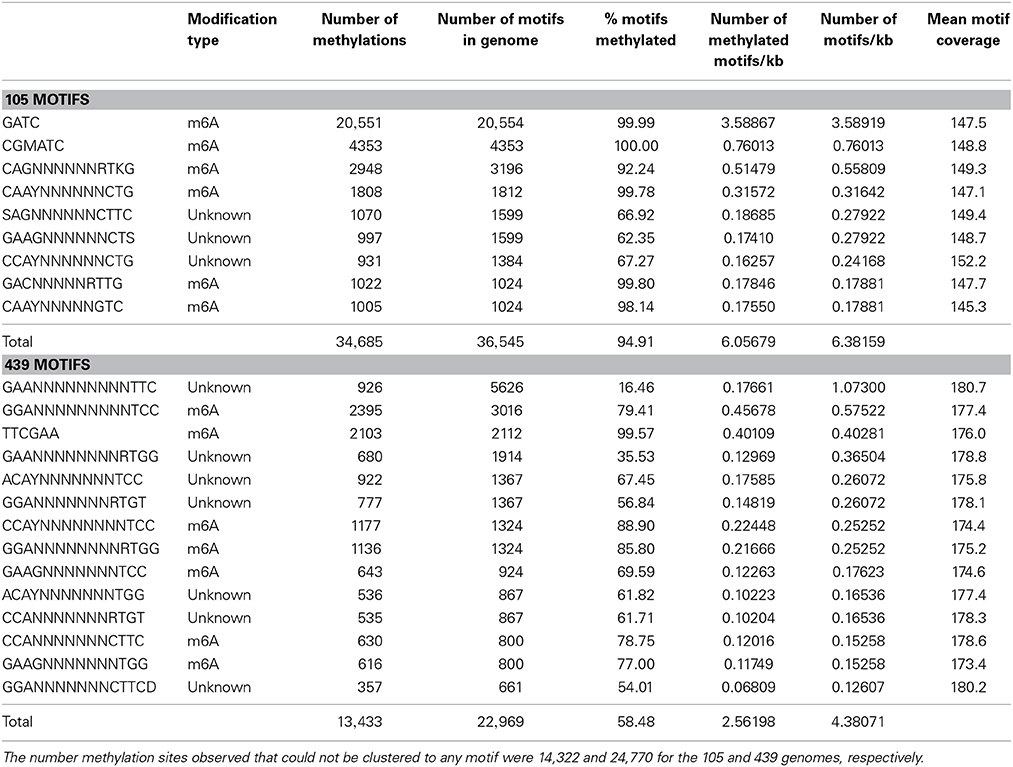
Table 3. Methylated motif analysis of the 105 and 439 genomes.
A total of 5,107,008 bp or 89% of the B. dorei 105 genome codes directly for ORFs. Of the 20,551 methylated GATC sties found in B. dorei 105, 18,816 were found within 3184 of the 4970 annotated open reading frames. Of the 1735 methylated GATC sites identified between ORFs many are likely within promoter regions. Thus, there are 2.80 GATC methylation sites per kbp of non-coding genome space which is slightly lower than the 3.68 GATC methylation sites per kbp of the coding region. This suggests that the evolutionary pressure to maintain GATC sites is stronger in coding regions compared to non-coding regions.
Of the 245 SEED subsystems identified by RAST annotation of the complete B. dorei 105 genome, only 13 subsystems had no GATC methylation sites (Table S1). Those include purine utilization, carbon starvation, copper homeostasis, gentisate degradation, salicylate, and gentisate catabolism.
The remaining 232 subsystems were methylated at nearly every GATC site. The Ton and Tol transport systems which provide energy for transport across the outer membrane in Gram negative bacteria was the most highly GATC methylated subsystem, accounting for 6.7% of all GATC methylations and 5.5% of the genome. Transport plays a crucial role in the heterotrophic bacteria that reside in the gut.
Other motifs in both genomes were detected in this analysis as well (Table 3). About 8.6 and 35.2% of all of the motifs detected in the 105 and 439 genomes, respectively, cannot be characterized as to the position of modification on the adenosine ring. The remaining motifs were all methylations of the 6-position of the adenosine ring and less abundant than the GATC modification. Excluding the GATC motif, there are 15,991 motifs remaining in the 105 genome, of which 14,134 (88.4%) are modified. In the 439 genome, there are 22,969 motifs detected, however, only 13,433 (58.5%) are modified. Thus, even non-GATC adenosine methylation sites are much more likely to be methylated in the 105 genome compared to the 439 genome. The phenotypic effects of these methylations are unknown.
As the primary objective of this work was to obtain closed B. dorei genomes and examine GATC methylations, the libraries were not prepared in a way that would allow detection of 5-methylcytosine modifications. Both the 105 and 439 genomes each have genes that code for putative DNA cytosine methyltransferases. Hence, we predict that 5′-cytosine methylation is normal in both the 105 and 439 samples.
This is in contrast to the Dam methyltransferase gene, which is only present in the 105 genome. In the 105 genome, a single Dam methyltransferase gene was annotated that is predicted to recognize the GATC motif (Figure 1). This gene is within a bacterial prophage region that is 47,617 bp in length (between bases 1,014,516 and 1,062,132 bp in the genome), making it similar to other bacteriophage orphan DNA methyltransferases found in bacteria (Murphy et al., 2013).
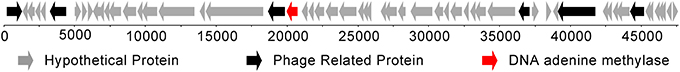
Figure 1. Location of the DNA adenine methyltransferase (in red) within a bacteriophage present in the Bacteroides dorei genome.
Another interesting observation is that of all of the methylation motifs observed in these two genomes, none is methylated in both genomes. This suggests that the primary source of methyltransferases in these genomes is through lateral transfer, often from phage. Nevertheless, the GATC motif methylation and abundance is the primary difference between the two genomes.
Discussion
Given that nearly all subsystems can be virtually completely methylated in one strain of a dominant gut bacterium and completely unmethylated in another strain of the same species from a different subject, suggests that epigenetics may be a crucial process regulating the metabolic potential of heterotrophic bacteria in the human gut. The observation that similar species reside in the gut of two individuals is not sufficient to propose that the metabolic capabilities of the two microbiomes are similar. This work suggests that future microbiome studies should consider the methylome when describing the bacterial diversity in the gut. Such analyses are no longer difficult given the latest sequencing technologies.
Although methylation can lead to up-regulation or down-regulation of a gene (Marinus and Casadesus, 2009), it is impossible to say whether the GATC motif affects the expression of B. dorei genes in the gut. However, the sheer number of GATC methylations found in one of these genomes suggests that GATC methylation is worthy of analysis in future investigations. It is likely that gene expression varies widely between a strain that is fully GATC methylated vs. another that is not. Unfortunately, unlike 16S rRNA, DamMT genes are not sufficiently conserved at the nucleotide level to allow rapid PCR analyses of this gene in metagenomic samples.
The effect of Dam methylation on gut colonization and survival may be significant. For example, Dam methylation of various transport systems affects many processes including import of nutrients, energy transfer, and antibiotic resistance conferred by antibiotic efflux. All of these are important to gut colonization and survival. A Dam mutant of Haemophilus influenzae is more susceptible to many antibiotics including erythromycin, tetracycline, kanamycin, and spectinomycin (Sanchez et al., 1997). In most cases, the H. influenzae Dam mutant showed not only a lower minimum inhibitory concentration, but also increased susceptibility to higher concentrations of antibiotics (Zaleski and Piekarowicz, 2004) suggesting that Dam methylation has a role in upregulating expression of efflux pumps and ultimately antibiotic resistance.
GATC methylation has also been implicated in pathogenesis by bacteria. DamMT mutants in several species have shown attenuated pathogenesis (Heithoff et al., 1999; Julio et al., 2001, 2002; Watson et al., 2004; Robinson et al., 2005; Balbontin et al., 2006; Mehling et al., 2007; Kim et al., 2008; Murphy et al., 2008) and as GATC methylation is involved in many fundamental bacterial processes including, but not limited to, replication, DNA repair, transcription, and LPS composition (Marinus and Casadesus, 2009), it is not surprising that DamMT deficient strains would be impaired in their ability to interact with eukaryotic hosts. Of course, given that have only two samples were examined here, no estimate of the effects of gut bacterial GATC methylation on type 1 diabetes autoimmunity outcome can be made.
GATC methylation in the B. dorei 105 genome appears to be the result of a single orphan DNA adenine methyltransferase gene embedded within a bacterial prophage of unknown origin. These orphan DamMTases have been reported to protect phage DNA that include the GATC motif from restriction (Kossykh et al., 1995; Miller et al., 2003). That may be the case here as the frequency of GATC motifs within the DamMT-containing bacteriophage region is 1.7 times higher than in the 105 genome overall with 6.09 and 3.59 GATC motifs per kb in the prophage and whole genome, respectively.
These orphan DamMT-containing bacteriophage can also regulate lysogeny by the bacteriophage (Murphy et al., 2008). Until this strain is cultured and a mutant is made that lacks DamMTase activity, the phenotype(s) conferred by this gene will remain unknown.
How GATC methylation affects microbiome function is unknown. But given the many bacterial processes affected by GATC methylation, environmental shaping of bacterial genomes through environmental exposures may influence the role of the microbiome in gut diseases. The surprising aspect of this work is that a single dominant species in the gut of these two subjects, B. dorei, can vary so radically in methylation patterns simply because of the absence of the DamMT gene in B. dorei in one of these subjects. Current and future metagenomic datasets from the gut microbiome should be mined for DamMT genes and GATC methylation sites to determine their abundance and taxonomic breadth.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was generously supported by grants from JDRF, the Juvenile Diabetes Research Foundation (www.jdrf.org) (Project 17-2011-266 to Eric W. Triplett) and the National Science Foundation, Division of Undergraduate Education: Course, Curriculum and Laboratory Instruction CCLI Phase 2 (DUE-0920151 to Eric W. Triplett).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00361/abstract
Table S1. This is an excel file showing the number of GATC and total methylations in the subsystems identified in the 105 genome.
References
Angiuoli, S. V., Gussman, A., Klimke, W., Cochrane, G., Field, D., Garrity, G., et al. (2008). Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS 12, 137–141. doi: 10.1089/omi.2008.0017
Balbontin, R., Rowley, G., Pucciarelli, M. G., Lopez-Garrido, J., Wormstone, Y., Lucchini, S., et al. (2006). DNA adenine methylation regulates virulence gene expression in Salmonella enterica serovar typhimurium. J. Bacteriol. 188, 8160–8168. doi: 10.1128/JB.00847-06
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barras, F., and Marinus, M. G. (1989). The great GATC: DNA methylation in E. coli. Trends Genet. 5, 139–143. doi: 10.1016/0168-9525(89)90054-1
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS+ is an extension of the GeneMarkS ab initio gene finding program. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Clark, T. A., Murray, I. A., Morgan, R. D., Kislyuk, A. O., Spittle, K. E., Boitano, M., et al. (2012). Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 40, e29. doi: 10.1093/nar/gkr1146
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLOS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Fagen, J. R., Giongo, A., Brown, C. T., Davis-Richardson, A. G., Gano, K. A., and Triplett, E. W. (2012). Characterization of the relative abundance of the citrus pathogen Ca. Liberibacter asiaticus in the microbiome of its insect vector, Diaphorina citri, using high throughput 16S rRNA sequencing. Open Microbiol. J. 6, 29–33. doi: 10.2174/1874285801206010029
Fang, G., Munera, D., Friedman, D. I., Mandlik, A., Chao, M. C., Banerjee, O., et al. (2012). Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 30, 1232–1242. doi: 10.1038/nbt.2432
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–467. doi: 10.1038/nmeth.1459
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Heithoff, D. M., Sinsheimer, R. L., Low, D. A., and Mahan, M. J. (1999). An essential role for DNA adenine methylation in bacterial virulence. Science 284, 967-970. doi: 10.1126/science.284.5416.967
Hobley, G., McKelvie, J. C., Harmer, J. E., Howe, J., Oyston, P. C. F., and Roach, P. L. (2012). Development of rationally designed DNA N6 adenine methyltransferase inhibitors. Bioorg. Med. Chem. Lett. 22, 3079–3082. doi: 10.1016/j.bmcl.2012.03.072
Julio, S. M., Heithoff, D. M., Provenzano, D., Klose, K. E., Sinsheimer, R. L., Low, D. A., et al. (2001). DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect. Immun. 6, 7610–7615. doi: 10.1128/IAI.69.12.7610-7615.2001
Julio, S. M., Heithoff, D. M., Sinsheimer, R. L., Low, D. A., and Mahan, M. J. (2002). DNA adenine methylase overproduction in Yersinia pseudotuberculosis alters YopE expression and secretion and host immune responses to infection. Infect. Immun. 70, 1006–1009. doi: 10.1128/IAI.70.2.1006-1009.2002
Kim, J. S., Li, J., Barnes, I. H. A., Baltzegar, D. A., Pajaniappan, M., Cullen, T. W., et al. (2008). Role of the Campylobacter jejuni Cj1461 DNA methyltransferase in regulating virulence characteristics. J. Bacteriol. 190, 6524–6529. doi: 10.1128/JB.00765-08
Koren, S., Schatz, M. C., Walenz, B. P., Martin, J., Howard, J. T., Ganapathy, G., et al. (2012). Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700. doi: 10.1038/nbt.2280
Kossykh, V. G., Schlagman, S. L., and Hattman, S. (1995). Phage T4 DNA [N6-adenine]methyltransferase. Overexpression, purification, and characterization. J. Biol. Chem. 270, 14389–14393. doi: 10.1074/jbc.270.24.14389
Kukko, M., Kimpimäki, T., Korhonen, S., Kupila, A., Simell, S., Veijola, R., et al. (2005). Dynamics of diabetes-associated autoantibodies in young children with increased genetic risk of Type 1 diabetes recruited from the general population. J. Clin. Endocrinol. Metab. 90, 2712–2717. doi: 10.1210/jc.2004-1371
Løbner-Olesen, A., Skovgaard, O., and Marinus, M. G. (2005). Dam methylation: coordinating cellular processes. Curr. Opin. Microbiol. 8, 154–160. doi: 10.1016/j.mib.2005.02.009
Marinus, M. G. (1987). DNA methylation in Escherichia coli. Annu. Rev. Genet. 21, 113–131. doi: 10.1146/annurev.ge.21.120187.000553
Marinus, M. G., and Casadesus, J. (2009). Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol. Rev. 33, 488–503. doi: 10.1111/j.1574-6976.2008.00159.x
Mashhoon, N., Carroll, M., Pruss, C., Eberhard, J., Ishikawa, S., Estabrook, R. A., et al. (2004). Functional characterization of Escherichia coli DNA adenine methyltransferase, a novel target for antibiotics. J. Biol. Chem. 279, 52075–52081. doi: 10.1074/jbc.M408182200
Mashhoon, N., Pruss, C., Carroll, M., Johnson, P. H., and Reich, N. O. (2006). Selective inhibitors of bacterial DNA adenine methyltransferases. J. Biomol. Screen 11, 497–510. doi: 10.1177/1087057106287933
McKelvie, J. C., Richards, M. I., Harmer, J. E., Milne, T. S., Roach, P. L., and Oyston, P. C. F. (2013). Inhibition of Yersinia pestis DNA adenine methyltransferase in vitro by a stibonic acid compound: identification of a potential novel class of antimicrobial agents. Brit. J. Pharmacol. 168, 172–188. doi: 10.1111/j.1476-5381.2012.02134.x
Mehling, J. S., Lavender, H., and Clegg, S. (2007). A Dam methylation mutant of Klebsiella pneumoniae is partially attenuated. FEMS Microbiol. Lett. 268, 187–193. doi: 10.1111/j.1574-6968.2006.00581.x
Miller, E. S., Kutter, E., Mosig, G., Arisaka, F., Kunisawa, T., and Ruger, W. (2003). Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156. doi: 10.1128/MMBR.67.1.86-156.2003
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A., and van Sinderen, D. (2013). Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 79, 7547–7555. doi: 10.1128/AEM.02229-13
Murphy, K. C., Ritchie, J. M., Waldor, M. K., Løbner-Olesen, A., and Marinus, M. G. (2008). Dam methyltransferase is required for stable lysogeny of the Shiga toxin (Stx2)-encoding bacteriophage 933W of enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol. 190, 438–441. doi: 10.1128/JB.01373-07
Murray, I. A., Clark, T. A., Morgan, R. D., Boitano, M., Anton, B. P., Luong, K., et al. (2012). The methylomes of six bacteria. Nucleic Acids Res. 40, 11450–11462. doi: 10.1093/nar/gks891
Parikka, V., Näntö-Salonen, K., Saarinen, M., Simell, T., Ilonen, J., Hyoty, H., et al. (2012). Early seroconversion and rapidly increasing autoantibody concentrations predict prepubertal manifestation of type 1 diabetes in children at genetic risk. Diabetologia 55, 1926–1936. doi: 10.1007/s00125-012-2523-3
Robinson, V. L., Oyston, P. C., and Titball, R. W. (2005). A dam mutant of Yersinia pestis is attenuated and induces protection against plaque. FEMS Microbiol. Lett. 252, 251–256. doi: 10.1016/j.femsle.2005.09.001
Sanchez, L., Pan, W., Vinas, M., and Nikaido, H. (1997). The acrAB homolog of Haemophilus influenzae codes for a functional multidrug efflux pump. J. Bacteriol. 179, 6855–6857.
Thielking, V., DuBois, S., Eritja, R., and Guschlbauer, W. (1997). Dam methyltransferase from Escherichia coli: kinetic studies using modified DNA oligomers: nonmethylated substrates. Biol. Chem. 378, 407–415.
Urig, S., Gowher, H., Hermann, A., Beck, C., Fatemi, M., Humeny, A., et al. (2002). The Escherichia coli dam DNA methyltransferase modifies DNA in a highly processive reaction. J. Mol. Biol. 319, 1085–1096. doi: 10.1016/S0022-2836(02)00371-6
Watson, M. E. Jr., Jarisch, J., and Smith, A. L. (2004). Inactivation of deoxyadenosine methyltransferase (dam) attenuates Haemophilus influenzae virulence. Mol. Microbiol. 53, 651–664. doi: 10.1111/j.1365-2958.2004.04140.x
Keywords: DNA adenine methyltransferase, GATC, metagenomics, genomics, epigenetics
Citation: Leonard MT, Davis-Richardson AG, Ardissone AN, Kemppainen KM, Drew JC, Ilonen J, Knip M, Simell O, Toppari J, Veijola R, Hyöty H and Triplett EW (2014) The methylome of the gut microbiome: disparate Dam methylation patterns in intestinal Bacteroides dorei. Front. Microbiol. 5:361. doi: 10.3389/fmicb.2014.00361
Received: 08 May 2014; Accepted: 26 June 2014;
Published online: 17 July 2014.
Edited by:
Scott Norman Peterson, Sanford-Burnham Medical Research Institute, USAReviewed by:
William C. Nelson, University of Southern California, USAAlex Y. Strongin, Sanford-Burnham Medical Research Institute, USA
Copyright © 2014 Leonard, Davis-Richardson, Ardissone, Kemppainen, Drew, Ilonen, Knip, Simell, Toppari, Veijola, Hyöty and Triplett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric W. Triplett, Department of Microbiology and Cell Science, Institute of Food and Agricultural Sciences, University of Florida, 1355 Museum Rd., Gainesville, FL 32611-0700, USA e-mail:ZXd0QHVmbC5lZHU=