 Fabai Wu
Fabai Wu Erwin Van Rijn
Erwin Van Rijn Juan E. Keymer
Juan E. Keymer Cees Dekker
Cees Dekker- Department of Bionanoscience, Kavli Institute of Nanoscience, Delft University of Technology, Delft, Netherlands
Studies of the spatiotemporal protein dynamics within live bacterial cells impose a strong demand for multi-color imaging. Despite the increasingly large collection of fluorescent protein (FP) variants engineered to date, only a few of these were successfully applied in bacteria. Here, we explore the performance of recently engineered variants with the blue (TagBFP), orange (TagRFP-T, mKO2), and far-red (mKate2) spectral colors by tagging HU, LacI, MinD, and FtsZ for visualizing the nucleoid and the cell division process. We find that, these FPs outperformed previous versions in terms of brightness and photostability at their respective spectral range, both when expressed as cytosolic label and when fused to native proteins. As this indicates that their folding is sufficiently fast, these proteins thus successfully expand the applicable spectra for multi-color imaging in bacteria. A near-infrared protein (eqFP670) is found to be the most red-shifted protein applicable to bacteria so far, with brightness and photostability that are advantageous for cell-body imaging, such as in microfluidic devices. Despite the multiple advantages, we also report the alarming observation that TagBFP directly interacts with TagRFP-T, causing interference of localization patterns between their fusion proteins. Our application of diverse FPs for endogenous tagging provides guidelines for future engineering of fluorescent fusions in bacteria, specifically: (1) The performance of newly developed FPs should be quantified in vivo for their introduction into bacteria; (2) spectral crosstalk and inter-variant interactions between FPs should be carefully examined for multi-color imaging; and (3) successful genomic fusion to the 5′-end of a gene strongly depends on the translational read-through of the inserted coding sequence.
Introduction
The use of fluorescent proteins (FPs) has greatly advanced our understanding of the subcellular architecture of bacteria. Soon after the first cloning of the green fluorescence protein (gfp) gene from Aequorea victoria (Prasher et al., 1992) and its first application as fluorescence marker in vivo (Chalfie et al., 1994), it was successfully adopted to visualize essential proteins involved in cell division and division-site selection in bacteria, such as FtsZ/FtsA (Ma et al., 1996) and MinE/MinD/MinC (Raskin and de Boer, 1997, 1999; Hu and Lutkenhaus, 1999). The results from these studies planted the significant notion that the intracellular environment of bacteria is not only structured, but also extremely dynamic. Other hallmarks include cytoskeletal filaments responsible for cell shape maintenance (Jones et al., 2001; Ausmees et al., 2003; Domínguez-Escobar et al., 2011; Garner et al., 2011; van Teeffelen et al., 2011), and polarly localized proteins involved in chemotaxis, virulence and metabolism (Sourjik and Berg, 2000; Charles et al., 2001; Lindner et al., 2008; Li and Young, 2012). Recently, several large fluorescent-fusion libraries have been constructed for Escherichia coli and Caulobacter crescentus, allowing genome-scale, quantitative studies of protein localization, which is especially powerful when accompanied by the development of a quantitative analysis toolbox (Kitagawa et al., 2006; Werner et al., 2009; Taniguchi et al., 2010; Kuwada et al., 2015).
Two decades of efforts have expanded the spectrum of FPs to a full range, showing strong promise for multi-color imaging. However, the application of many FPs for live cell imaging of bacteria have been hindered by various factors. Most prominently, the fast synthesis and degradation of protein in bacteria (in contrast to eukaryotes) demands fast-folding of FPs. This is clearly indicated by the fact that mutations improving the folding properties of EGFP generation (which resulted in SBFP2, SCPF3A, SGFP2, and SYFP2), enhanced their effective brightness by several folds in bacteria, while this was much less the case when expressed in mammalian cells (Kremers et al., 2006, 2007). Also, the orange FPs mOrange and mKO were not visible in live E. coli due to slow maturation, with the latter only visible after an overnight incubation after fixation (Alexeeva et al., 2010). The fast-folding properties of some FPs such as Venus, mCherry, and sfGFP have shown advantages for functional fusions (Osawa and Erickson, 2005; Bendezú et al., 2009; Dinh and Bernhardt, 2011), and have been broadly used. On the other hand, these fast-folding FPs do not always provide the native protein localization patterns (Landgraf et al., 2012). Furthermore, the degree of oligomerization, the brightness, the photostability, as well as the spectral separation between FPs are properties no less essential for successful capture of native events at the demanded spatial and temporal resolution. However, thus far, these factors have been barely quantified in live bacteria.
Here, we set out to expand the spectrum of FPs for live cell imaging in E. coli by labeling the cytosol, the nucleoid, as well as the division proteins. HU-2, encoded by hupA gene, is a subunit of nucleoid-associated proteins HU, which serves as a marker for the nucleoid that was previously shown to colocalize with DAPI when fused to GFP (Wery et al., 2001). Fluorescently labeled LacI has been used as an operator-repressor system to label specific genomic loci by targeting repeated lacO sequences (Lau et al., 2003). Combining the HU-2 and LacI labels allows us to localize a genomic locus in the context of the global nucleoid structure. For imaging division, we focus on FtsZ proteins, which polymerize to initiate a cytokinetic ring, and whose localization is regulated by the nucleoid and MinCDE proteins (Ma et al., 1996; Hu and Lutkenhaus, 1999; Raskin and de Boer, 1999; Bailey et al., 2014; Du and Lutkenhaus, 2014). To visualize the latter, we constructed a fusion gene at the endogenous minD locus for expressing sfGFP-MinD proteins, which oscillate between the two cell halves of the rod-shape E. coli and form a time-averaged concentration gradient that have a maxima at the cell poles and minimum at the mid-cell (Raskin and de Boer, 1999).
Expanding from the FPs derived from jellyfish A. victoria, we examine the performances of the monomeric FPs at the blue (TagBFP), orange (TagRFP-T and mKO2), far red (mKate2), and near-infrared (dimeric eqFP670) spectral colors. All FPs used in this study are listed in Table 1. The latter proteins are derivatives of the FPs from the sea anemone Entacmaea quadricolor and Fungia concinna, known for their brightness, photostability, and relatively fast maturation (Shaner et al., 2008; Subach et al., 2008; Shcherbo et al., 2009, 2010; Sun et al., 2009; Morozova et al., 2010). By quantitatively comparing the brightness, photostability, and spectral properties of these proteins to other proteins with the same spectral colors, we find that these proteins function well as cytosolic labels and/or C-terminal tags, and that they provide strong advantages in brightness, photostability, and spectral separation compared to other FPs that are currently in use for bacteria. Furthermore, our approach to tag the minD gene at its endogenous locus revealed a detrimental effect of the coding sequence on N-terminal fusions. Finally, we combined these FP tags in bacterial strains to assess their suitability for multi-color imaging. Together, Our data provide guidelines for an optimal strategy in choosing new FPs for multi-color imaging in bacteria.
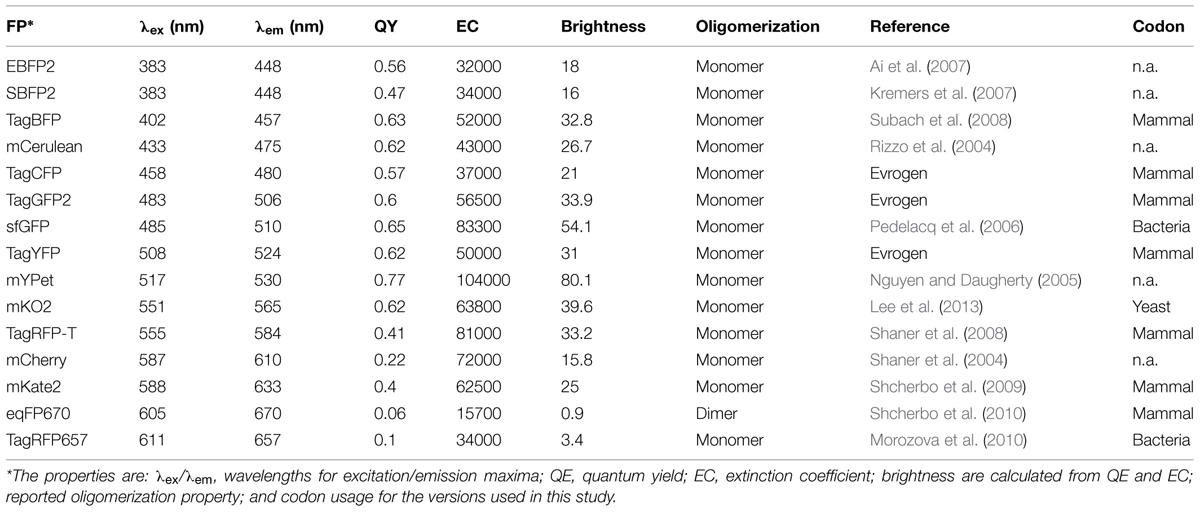
TABLE 1. Properties of the fluorescent proteins (FPs) used in this study.
Materials and Methods
Plasmid and Strain Construction
The plasmids were constructed using Gateway cloning kit (Invitrogen, catalog # 11789013, 11791019) and Infusion EcoDry kits (Clonetech, catalog # 638912), Coding sequences of tagYFP, tagGFP2, tagRFP-T, ebfp2, tagBFP, hupA, minDE, and aph frt were respectively amplified from the pTagYFP-C1, pTagGFP2-C1, pTagRFP-T, pBAD-EBFP, pTagBFP-C1, W3110 genome, W3110 genome, and pKD13, and inserted into the Gateway entry vectors through BP reactions as described in the Gateway protocol. These and previously described entry vectors in Wu et al. (2015) were then combined through Gateway LR reaction to produce destination vectors pERB006, pFWB007, pBVS32, pFWB009, pBVS36, and pBVS37. For constructing pFWM006, we amplified the backbone of pBVS3 with Plac and aph, the hupA fragment from pFWB006, and the mKO2 fragment from pyomKO2, and combined these into one plasmid through a three-fragments Infusion reaction. For construction of pFWM007, we amplified the backbone of pBVS3 with Plac and aph, the hupA fragment from pFWB006, and the sBFP2 fragment from pSBFP2-C1, and combined these into one plasmid through a three-fragment Infusion reaction. Plasmid pFWZ7 was constructed through an ligation reaction (Infusion kit) with four PCR-amplified fragments, which were the backbone of pFB174 with arabinose promoter and chloramphenicol resistance gene, an ftsZ (5′ sequence, 1–999 bp) fragment with an 18 base overhang coding the flexible linker GSGSGS, a GGSGSS flexible linker plus ftsZ (3′ sequence, 991–1052 bp) plus aph frt sequence amplified from strain FW1370, two synthesized oligos containing the tetracysteine (TC) peptide coding sequence and the two flanking flexible linkers. The TC coding sequence was then replaced by tagRFP-T, sfGFP, and tagCFP to produce plasmid pFWZ4, pFWZ5, and pFWZ6 through two-fragment Infusion reactions. pFWZ0 was constructed through Gibson assembly of pFB174 backbone and the ftsZ::aph frt sequence amplified from strain FW1370. All plasmids are listed in Table 2.

TABLE 2. Plasmids used in this study.
The genomic insertions were constructed using λ Red recombination (Datsenko and Wanner, 2000) and shuffled between strains using P1 transduction as described previously (Wu et al., 2015). The PCR fragments from plasmids pERB006, pFWM007, pFWB006, pBVS32, pFW009, pFWM006, pBVS3, and pBVS4 were electroporated into the electro-competent cells of W3110 containing pKD46, to result in strains FW1722, FW1951 and FW1344, FW1401, FW1464, FW2455, FW1462, FW1534. The PCR fragments of pFWM007, pFWM006, and pFWB019 were amplified to replace the mCherry in strain RRL189 through λ Red recombination to result in strains FW1965, FW2417, and FW2450. The PCR fragments of pBVS3, pBVS36, and pBVS37 were used to replace the ΔminD minE::cat sacB in strain FW1363 through Red recombination to result in strains FW1480, FW1248, FW1393. Note that in strain FW1363 the minC gene is intact. The above listed strains were cured of kanamycin resistance using pCP20 as described in Datsenko and Wanner (2000), and listed in Table 2. To confirm the functionality of YFP-MinD, Plac::yfp-minDE was transduced from FW1462 into FW1363 to yield strain FW1463. For multi-color imaging, hupA-tagBFP from FW1344 was transduced into strain FW1554 to yield 1561; ΔleuB::eqFP670 was transduced from FW1489 into FW1554 to yield FW1559; ΔleuB::tagRFP-T was transduced from FW1401 into FW1359 to yield FW1406; hupA-mYPet from FW1551 was transduced into a strain with aph cured from FW1965 to yield FW2480; Plac::yfp-minDE was transduced from FW1462 into FW1459 to yield strain FW1503; pFWZ4 was transformed into strain FW1559. All strains used are listed in Table 3.

TABLE 3. Bacterial strains used in this study.
Growth Conditions
For genetic engineering, E. coli cells were incubated in Lysogeny broth (LB) supplemented, when required, with 100 μg/ml ampicillin (Sigma-Aldrich), 50 μg/ml kanamycin (Sigma-Aldrich), or 34 μg/ml chloramphenicol (Sigma-Aldrich) for plasmid selection, or with 25 μg/ml kanamycin, 20 μg/ml chloramphenicol, or 0.2% sucrose for selection of the genomic insertions of gene cassettes. For imaging strains with fluorescent foci with LacI fusions, we grew cells in liquid M9 minimum medium (Fluka Analytical) supplemented with 2 mM MgSO4, 0.1 mM CaCl2, 0.4% glycerol (Sigma-Aldrich), and 0.01% protein hydrolysate amicase (PHA; Fluka Analytical). For imaging other strains, we grew cells either in liquid M9 minimum medium supplemented with 2 mM MgSO4, 0.1 mM CaCl2, 0.4% glucose (Sigma-Aldrich), and 0.25% PHA, or in LB medium. For imaging, overnight cultures were back diluted into the fresh medium described above to an OD (600 nm, same below) of 0.01 in falcon tubes until an OD of 0.4–0.6 for M9 medium with 0.25% PHA and LB, and OD of 0.1 for M9 medium with 0.01% PHA. The growth conditions for the FtsZ complementation assay are as described in Osawa and Erickson (2005). 0.002% arabinose was used for the induction of ectopic FtsZswTagRFP-T fusion from the plasmids in the presence of the endogenous ftsZ.
Microscopy
Fluorescence imaging was carried out using Nikon Ti-E microscope with 100X CFI Apo TIRF objective with an NA of 1.49. All fluorescent probes were excited using a Nikon Intensilight, except for the photo-bleaching of lacI foci, which was excited by SpectraX LED single-spectrum light sources (Lumencor) with SpectraX filter sets (Lumencor). For imaging with Nikon Intensilight, the λex/λbs/λem wavelengths of the filter cubes are as follows: SBFP2 and EBFP2 (363–391/425/435–485 nm), TagBFP (395–415/420/435–485 nm), mCerulean (426–446/455/460–500 nm), sfGFP (450–490/495/500–550 nm), mYpet and FlAsH (490–510/515/520–550 nm), TagRFP-T and mKO2 (530–560/562/570–620 nm), mCherry and mKate2 (540–580/585/592–668 nm), eqFP670 (589–625/649/655–1200 nm). For imaging with SpectraX, the excitation filters for orange and red proteins are respectively 555/25 nm (center/width, same below) and 575/22 nm. The multiband emission filters are respectively 435/26 – 510/40 – 595/40 – 705/72 nm, and 465/25 – 545/30 – 630/60 nm. The fluorescence signal was recorded by an Andor EMCCD camera (iXon Ultra 885), with an EM gain of 100. While our emission filter for the eqFP670 extends to the infrared region, eqFP670 does not fluoresce beyond 850 nm, which is well-within the detection range for most EMCCD cameras, including the one used in this study.
Image Analysis
Analysis of fluorescent microscopy images was carried out using Matlab with our customized programs. The background intensity was subtracted for all images individually. For identification of fluorescent LacI foci or nucleoid, the images were Gaussian blurred for subtraction, and a threshold for the expected object size and intensity was applied to eliminate noise. The intensities of the identified objects were then collected for statistics. For the photobleaching data, the mean per-pixel intensity of each nucleoid was calculated independently. To compare the photostability of more than two FPs in the main figure, the total intensities of all identified objects were summed and divided by the total number of pixel for all found objects in the first image, in order to take the completely bleached objects at the later stage of the bleaching period into account. In the latter case, the sum fluorescence intensity of the FPs in individual objects was plotted only in the supplementary figures. The standard deviation values of the intensities were not shown in the main figures for the convenience of display, and are instead plotted in supplementary figures. The matlab scripts used for these measurements can be found at http://ceesdekkerlab.tudelft.nl/downloads/.
The signal-to-noise ratio (SNR) of the cytosolic FPs is calculated using SNRcell = (Icell-Ibg)/SDbg. We use the standard deviation value of the cell-free region as a measure of background noise (SDbg). We use the difference between the mean intensity of a cell (Icell) and the mean background intensity (Ibg) as a measure of signal. The mean and SD values of the SNR calculated from all cells are used for plotting.
Results
A Bright Blue Fluorescent Protein for Multi-Color Imaging in Bacteria
The blue variants of the fluorescent proteins (BFPs) have seen few applications in bacteria due to their low brightness and short excitation wavelength (see Table 1). In principle, if a BFP would be sufficiently bright, it can be imaged with excitation light that is weak enough to avoid photodamage to the cells. It was recently reported that the purified monomeric mTagBFP (commercial name TagBFP) has a brightness that is similar to EGFP and 1.8 times that of EBFP2 (Subach et al., 2008). To examine its performance in bacteria, we first transformed a high-copy plasmid carrying the tagBFP gene under a T3 promoter into an E. coli strain, and indeed we observed bright blue fluorescence upon excitation through a customized filter set in a Nikon Intensilight. Furthermore, we engineered a new construct into the leuB locus in the E. coli genome, yielding strain FW1268, where the expression of tagBFP gene is driven by a synthetic constitutive promoter and a synthetic ribosome-binding site (RBS). The fluorescence signal of the expressed TagBFP is indeed sufficient for full cell labeling of bacteria (Figure 1A), indicating that TagBFP may have the brightness and maturation rate suitable for labeling proteins expressed at their endogenous level.
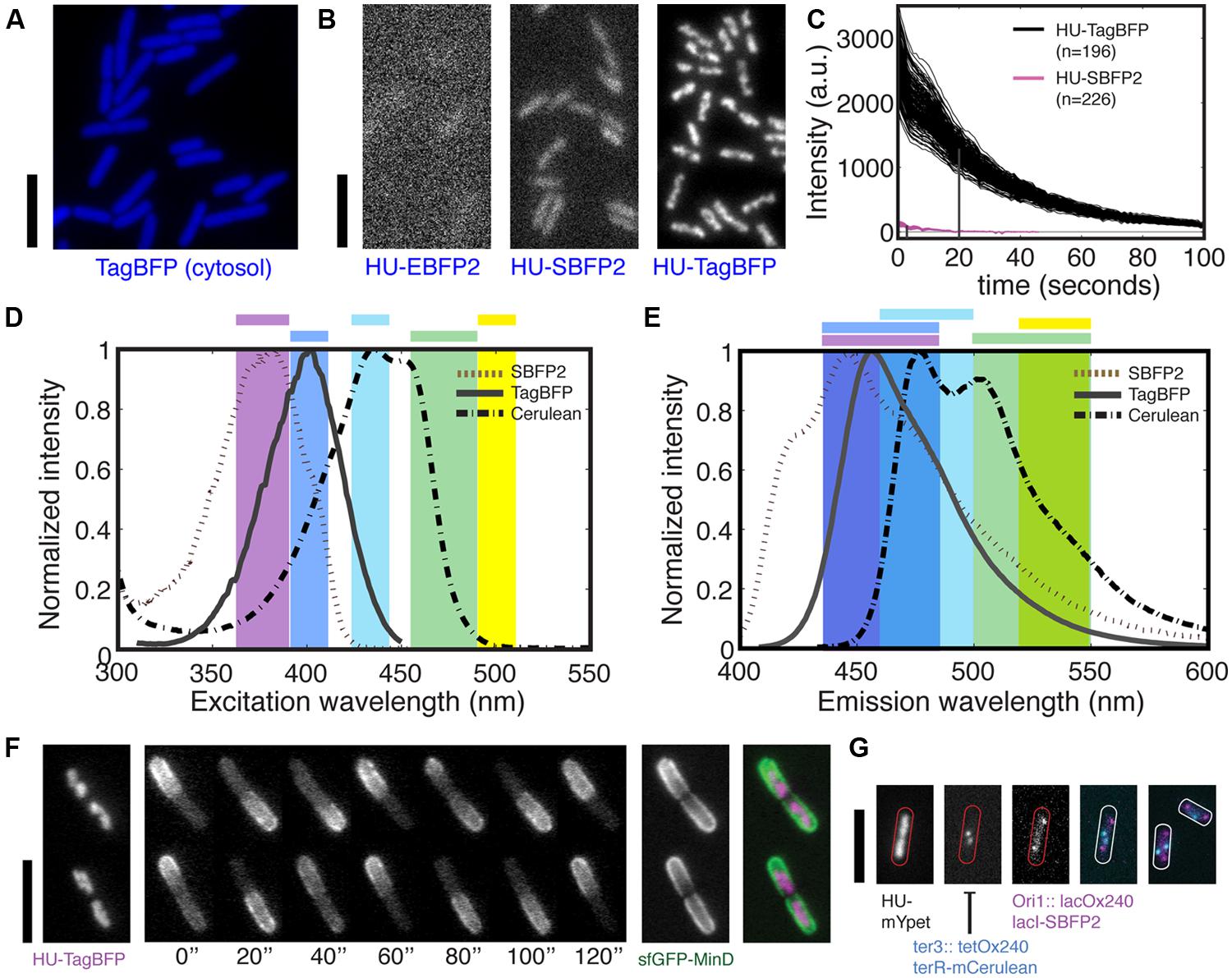
FIGURE 1. Bright blue fluorescent proteins for multi-color imaging in bacteria. (A) TagBFP is bright when expressed from a genomic copy under a constitutive promoter for cytosolic label in live Escherichia coli cell. (B) Fluorescent images of HU-2 tagged by three blue fluorescent protein (BFP) variants in live E. coli during exponential growth. (C) Brightness and photostability of HU-TagBFP and HU-SBFP2 in live E. coli cells under the constant exposure to the Nikon Intensilight with the respective filter sets. Each line represents the average intensity of one nucleoid. The index n indicates the numbers of nucleoids examined. The vertical lines indicate the bleaching halftime. (D) Excitation spectra of SBFP2, TagBFP, and Cerulean (a CFP), and the ranges of bandpass filters (shown in color) used for the excitation of blue, cyan, green, and yellow FPs. Note that different filters were used for SBFP2 and TagBFP due to their different excitation peak. (E) Emission spectra of SBFP2, TagBFP, and Cerulean, and the ranges of bandpass filters used for collecting the fluorescent light emitted by blue, cyan, green, and yellow FPs. (F) Endogenous fusions HU-TagBFP and sfGFP-MinD are combined for dual-color imaging. From left to right: a HU-TagBFP image, an sfGFP-MinD time series, a per-pixel standard deviation (SD) image of sfGFP-MinD calculated over time, and a false-color overlay of HU2-TagBFP and sfGFP-MinD. (G) Three-colors imaging of chromosome and chromosomal loci using HU2 label and two operator-repressor systems. Shown are three individual images followed by an overlay of the latter two, and an extra example. All scale bars indicate 5 μm.
To compare the performance of TagBFP to other BFP variants as fluorescent tags in bacteria, we fused either tagBFP, eBFP2, or sBFP2 genes to the 3′-end of the endogenous hupA gene, and examined their performance (Figures 1B,C). For imaging, we used two customized filter sets for the different excitation peaks of the EBFP2/SBFP2 (379 nm) and TagBFP (402 nm; Figure 1D), and the same emission filter (Figure 1E). Importantly, direct FP fusions to the C-termini of genes at their endogenous loci have been confirmed to maintain the endogenous expression level of these genes (Taniguchi et al., 2010).
HU-TagBFP outperforms HU-EBFP2 and HU-SBFP2 to a surprising extent in live bacteria, as shown in Figure 1B. It shows a 20 times higher fluorescence intensity than HU-SBFP2, showing clear chromosome morphologies for 200 ms exposure to half of the maximum excitation intensity provided by Nikon Intensilight. Note that the intensities of the Intensilight do vary for the different spectral range. Unexpectedly, HU-SBFP2 was threefold brighter in the stationary growth phase, despite that HU-2 was shown to be four times more abundant in the exponential phase (Ali Azam et al., 1999). Such an increase in brightness in the stationary phase was not observed in any of the other HU-2 fluorescent fusions. We hypothesize that the increase in brightness is due to a slower turnover of HU-SBFP2 proteins at stationary phase, allowing the proteins to successfully mature and fluoresce. Next to their higher brightness, the HU-TagBFP also shows superior photostability. The above constructs were used to compare the photostability of HU-TagBFP and HU-SBFP2 under the constant exposure to the same excitation light as imaged for Figure 1B. HU-TagBFP showed a bleaching half-time of 18 s, in contrast to the 6-s half-time for HU-SBFP2, see Figure 1C. In other words, the fluorescence signal of HU-TagBFP would only drop 50% after acquiring 90 images with the settings for Figure 1B. The initial intensity of the 196 nucleoids is measured to equal 2671 ± 354 (mean ± SD, a.u.), i.e., the HU-TagBFP concentration has a standard deviation that is only 13% of the mean value across the cell population during exponential growth in LB.
TagBFP’s excitation/emission spectra are well-separable from green and yellow fluorescent proteins (GFPs and YFPs) for multi-color imaging (Figures 1D,E). Taking advantage of the narrow excitation profile of TagBFP and its excitation peak at 402 nm, we customized a filter set to maximize the excitation efficiency and to collect the majority of the emitted light (Figures 1D,E). This filter set can be combined with regular commercial filter sets for GFP or YFP for two-colors imaging. Here, we illustrate such a combination by transducing the endogenous hupA-tagBFP fusion construct into a strain with a bright endogenous sfGFP-minD fusion (for details see Figure 4), yielding strain FW1561. Shown in Figure 1F, these two fluorescent probes successfully captured both the localizations of the nucleoids and the MinD oscillations, which together define the mid-cell for the localization of the cytokinesis machinery.
A potential combination of BFPs with cyan fluorescent proteins (CFPs) can further increase the options for multi-color imaging. Despite the higher brightness of TagBFP compared to SBFP2, it has more spectral overlap with CFP and thus is found less suitable for this application. As shown in Figures 1D,E, SBFP2 can be well-separated from common CFPs through customized filters, whereas the overlap between TagBFP and the CFPs is larger. Note that our current emission filter range for SBFP2 can be further adjusted to a 410–460 nm range to avoid crosstalk with CFPs. The combination of SBFP2 and SCFP3A was previously shown in Hela cells, where SBFP2 was highly expressed in the cytosol (Kremers et al., 2007). However, the limited subsequent usage in bacteria and our not fully satisfactory results for the HU-SBFP2 fusion lowered our expectations for the extent of its applications. Nevertheless, we suggest that, SBFP2 may find its application in protein co-localization studies if the morphology is not overly complicated. Here, for example, we replaced the mCherry in a LacI fusion (strain RRL189 Reyes-Lamothe et al., 2008), resulting in a LacI-SBFP2 fusion that targets the 240 lacO repeats in the origin region of the E. coli genome (strain FW1965). In this strain, the expression of the LacI-SBFP2 is constitutive. We found that it can be co-imaged with ter foci label TetR-mCerulean (cyan) and chromosome label HupA-mYpet (yellow; strain FW2480) (Figure 1G). Notably, the LacI-SBFP2 foci are much dimmer than the original LacI-mCherry fusion, requiring 2-s exposure time with Nikon Intensilight, thus less suitable for time-lapse imaging in dynamics studies. Alternatively, Everogen has produced TagCFP (Ex/Em = 457/480 nm) which can be combined with TagBFP, but we did not persue this route due to its potential spectral crosstalk with YFPs, and that our FtsZ-TagCFP fusion did not fluoresce.
Bright and Photostable Fluorescent Proteins in the Orange-Red, Far-Red, and Near-Infrared Spectral Range
By applying the same procedure above to characterize the performance of TagRFP-T and eqFP670 (commercial name NirFP) as cytosolic labels, we found that they perform superiorly in brightness and photostability for the orange and near-infrared spectral range, respectively (Figure 2A). Despite the fact that these two proteins are excited with long-wavelength and hence less phototoxic compared to GFP, frequent time-lapse imaging of strains FW1459 and FW2095, with the same setting for Figure 2A at a frame rate of 5 s for 5 min did lead to cell growth arrest although no significant photobleaching was observed (data not shown), indicating that the photostability is not a limiting factor for imaging live cells using these FPs. Note that this is an overexposure test for whether the photostability is sufficiently high when imaging is carried out at a lower rate where photodamage is avoided. The trade-off between photodamage, fluorescence signal, and temporal resolution requires moderation of the settings to specific cases.
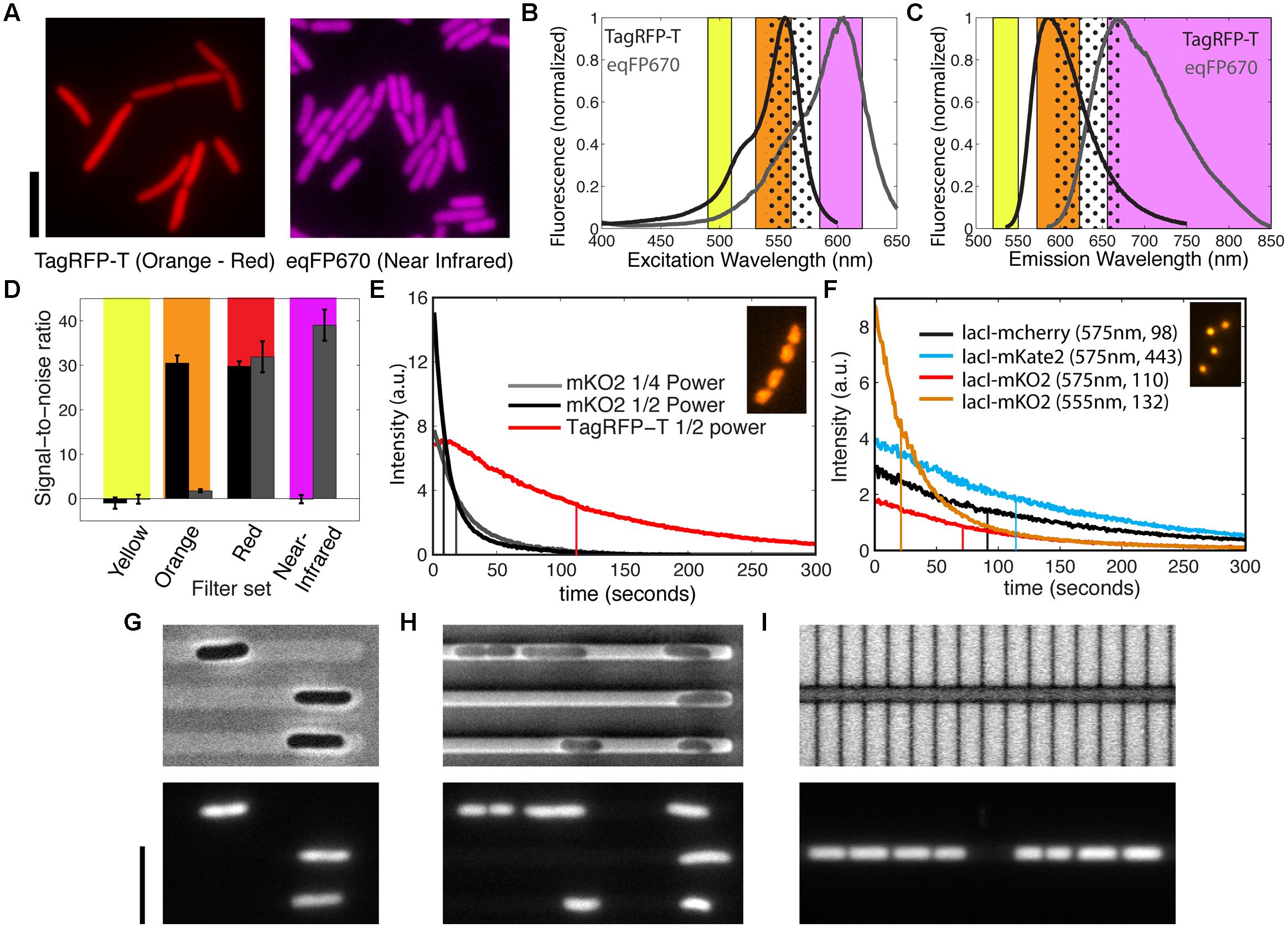
FIGURE 2. Bright and photostable fluorescent proteins in the orange-red, far-red, and near-infrared spectral range. (A) E. coli showing the fluorescence of cytosolic TagRFP-T (using orange filter set) and cytosolic eqFP670 (using near-infrared filter set) when expressed from a genomic copy under a constitutive promoter for cell body label in live E. coli. Scale bars, 5 μm. (B,C) Excitation and emission spectra of TagRFP-T and eqFP670, and the ranges of bandpass filters for the excitation and emission at yellow, orange, far-red, and near-infrared spectral range. These spectral ranges are as specified in the methods section for mYpet, TagRFP-T, mCherry, and eqFP670. The dotted regions indicate far-red filters. (D) Signal-to-noise ratios of TagRFP-T (black bar) and eqFP670 (gray bar) expressed in the strains shown in (A), when imaged with different filter cubes shown in (B,C). Here, the noise is defined by the standard deviation of the background intensity, and the signal is the difference between the fluorescence intensity of the cell and the mean background intensity. (E) Photobleaching of HU-mKO2 and HU-TagRFP-T in live E. coli cells under the constant exposure to the Nikon Intensilight at different power as indicated with the orange filter set. Inset shows a false-color image of nucleoids labeled by HU-TagRFP-T. Vertical lines indicate the bleaching halftime. (F) Photobleaching of chromosomal ori foci (Ori1::lacOx240) labeled by LacI-mcherry, LacI-mKate2, and LacI-mKO2 under the constant exposure to the SpectraX LED light source (1/4 power) with far-red or orange filter sets. The intensity values are the mean intensities of all the loci, i.e., the total intensity of detected loci divided by the initial number of detected loci. The indicated numbers for each probe are, respectively, the excitation wavelength and the number of spots measured. The inset shows a fluorescence image of the lacI-mKO2 foci. Vertical lines indicate the bleaching halftime. (G–I) Effect of microstructures made from different materials on the single-cell identifications in bright-field imaging (top panels) and using fluorescent microscopy of cytosolic labels (bottom panels). (G) E. coli cells in agarose-based microstructures, as described in Takeuchi et al. (2005). (H) E. coli cells between PDMS structures and an agarose pad, as described in Wu et al. (2015). (I) E. coli cells between silicon structures and PDMS, as described in Männik et al. (2012). The E. coli cells in (G–I) are strain BN1590 and fluorescent imaging was done in the near-infrared channel. The bright-field images in (G,H) were obtained through phase-contrast microscopy, whereas in (I) it was a regular wide-field image obtained through reflective light. Scale bar is 5 μm.
Notably, despite the fact that the near-infrared dimer eqFP670 was reported to have a brightness that is only 8% of EGFP as well as a modest maturation speed (Shcherbo et al., 2010), we were able to visualize the cells using either a regular far-red filter set or a customized near-infrared filter set (Figures 2B,C). This is largely owing to the almost full collection of far-red emission lights, while a smaller part of the emitted light is collected for the other proteins to avoid cross-talk. By contrast, a monomeric derivative TagRFP657 (excitation/emission peaks at 611/657 nm), which was produced for flow cytometer applications is not visible in our constructs even when placed under either the same synthetic promoter as above or a T7 promoter in a plasmid with pBR322 origin (pERB004 and pERB005). This agrees with the invisibility of TagRFP657 in a previous attempt in yeast (Lee et al., 2013). These facts thus make eqFP670 the most red-shifted protein applicable to bacteria so far, although we note that it’s dimeric property limits its application for protein fusions.
TagRFP-T and eqFP670 have the spectral properties that allow multi-color imaging at the red-shifted spectral range. While TagRFP-T has been reported as a red FP, its excitation and emission spectra are more blue shifted compared to conventional RFPs such as mCherry (Shaner et al., 2008). By comparing the spectral data of TagRFP-T and eqFP670, we propose that TagRFP-T can be imaged solely at the orange spectral range for excellent separation from GFPs/YFPs as well as from eqFP670, provided that latter is imaged at the near-infrared spectral range (Figures 2B,C). These customized filter sets allow us to measure the relative brightness of the two proteins in live cells (as shown in Figure 2A) from yellow to near-infrared spectral region (Figure 2D). Both TagRFP-T and eqFP670 are invisible with a YFP filter, while they show an equal brightness when imaged with a regular far-red filter used for mCherry. As expected from the spectral data, TagRFP-T does not show any bleed-through into the near-infrared spectral range. In the orange spectral range, TagRFP-T exhibits an over 10 times higher signal-to-noise ratio than the small bleed-through signal from eqFP670. The bleed-through from the eqFP670 into the orange spectral range is difficult to avoid due to the long tail of its excitation spectrum into the shorter wavelength. From these data, we conclude that the combination of TagRFP-T and eqFP670 is excellent for multi-color imaging at the orange and near-infrared spectral range.
As TagRFP-T was reported to be the most photostable FP at the red-shifted spectrum (Shaner et al., 2008), we set out to examine its performance as a fluorescent tag in live bacteria. We fused the 3′-end of hupA gene to tagRFP-T as well as to another recently reported FP gene, mKO2, which is expected to encode a bright, fast-folding orange/red FP (Figure 2E). Indeed, these two fluorescent probes show excellent brightness as an HU-2 tag in live bacteria, clearly improving the performance of orange proteins mKO and mOrange, which were previously shown to be invisible in live cells as fluorescent tags to FtsZ (Alexeeva et al., 2010). Under the same microscope and camera settings, HU-mKO2 appear to be 2.2 times as bright as TagRFP-T, but 15 times less photostable (Figure 2E, Supplementary Figure S1). Even when the exposure of HU-mKO2 was tuned to result in the same initial intensity as HU-TagRFP-T, it showed a 17-s bleaching half-time, in contrast to the 102 s for TagRFP-T. Thus, comparing these two proteins, mKO2 is more suitable for imaging proteins at low abundance, whereas TagRFP-T is more suitable for long-term time-lapse imaging.
Besides TagRFP-T and eqFP670, a far-red monomeric FP mKate2 was derived from the same origin. It showed more emission at the far-red spectral range and 57% higher brightness than mCherry (Shcherbo et al., 2009). Its photostability was shown to be similar to mCherry for purified proteins and 2.5-fold enhanced when expressed in yeast after codon optimization (Shcherbo et al., 2009; Lee et al., 2013). Here, we compare its brightness and photostability in bacteria by replacing the lacI-mCherry construct in strain RRL189 (see above for LacI-SBFP2 fusion) with lacI-mKate2. Shown in Figure 2F, the average intensity of the fluorescent foci of LacI-mKate2 is 25% enhanced with the same excitation/emission filters as compared to regular far-red proteins. The photostability of the two proteins is similar. In both cases after 300 s of exposure, 95% of all spots were still detectable through our automated software, showing that LacI-mCherry or LacI-mKate2 are both excellent candidates for operator-repressor system in time-lapse imaging. Furthermore, we engineered a LacI-mKO2 fusion based on strain RL189, and its foci are five times brighter in the orange spectral range than in the red spectral range (Figure 2F, Supplementary Figure S2). The photobleaching half-time of LacI-mKO2 is, however, 2.5 times shorter at the orange spectral range, likely due to the strong light absorbance.
Our comparison between all the orange-red and far-red fluorescent fusion tested here shows that TagRFP-T is indeed superior in photostability, agreeing with the quantitative data from the purified proteins and from fluorescent fusions in yeast (Shaner et al., 2008; Lee et al., 2013). Its far-red derivative mKate2 is indeed brighter than mChrrey in bacteria, but less significantly as previously reported in purified form and as fusion tag in yeast, which can be resulted from a slower maturation of mKate2 compared to mCherry.
Expanding the spectral range of fluorescence proteins for cytosolic label is also advantageous for studies using microstructures. Microstructures and microfluidics are emerging tools for studying bacterial physiology (Wang et al., 2010; Männik et al., 2012; Hol and Dekker, 2014; Wu et al., 2015). While these tools provide many advantages in manipulating the chemical and physical environment, they can impose challenges for imaging. In particular, the materials for the confining microstructures can make a great difference for visualizing cell body without fluorescence. For example, the structures made from agarose gel are transparent and allow phase contrast microscopy to visualize cell boundaries, owing to the fact that an agarose gel has lower refractive index (1.33, same as water) than bacteria (∼1.4; Figure 2G). By contrast, polydimethylsiloxane (PDMS), the most commonly used polymer for microfluidic applications, has a reflective index of around 1.4, which is almost identical to that of a bacterial cell. When imaging bacteria in between PDMS structures and agarose, the cell boundaries display much less contrast (Figure 2H). This type of interference between the microfluidic channel boundary and the bacterial boundary is even worsened for PDMS–PDMS interfaces. Furthermore, silicon structures are non-transparent, thus only allowing bright-field or DIC microscopy, in reflection rather than transmission, and bacteria in ∼micron-sized channels are difficult to distinguish (Figure 2I). In all these scenarios, the availability of cytosolic labels at the near-infrared (eqFP670) spectral range becomes particularly useful as a third or fourth color (Figures 2G–I, bottom panels).
A Bright and Photostable FtsZswTagRFP-T Fusion for Multi-Color Imaging of Cell Division
As shown above, TagRFP-T can be spectrally well-separable from the near-infrared protein eqFP670 as well as from GFPs/YFPs. This can potentially lead to three-colors imaging at the long-wavelength regime, where phototoxicity can be reduced by avoiding near-UV excitations (such as necessary for CFPs or BFPs). Such an approach is in fact essential when frequent time-lapse imaging is required for tracking dynamic protein localizations over long time. Here, we describe the construction of new FtsZ and MinD fusions for time-lapse imaging of division site selection in E. coli with high temporal resolution.
An FtsZ molecule has a globular structure composed of independently folded N-terminal and C-terminal domains both essential for polymerization, followed by an unstructured flexible linker, and a conserved C-terminal tail responsible for interacting with its membrane-bound partners FtsA and ZipA as well as lateral interactions between FtsZ polymers (Figure 3A; Oliva et al., 2004; Osawa and Erickson, 2005; Shen and Lutkenhaus, 2009; Erickson et al., 2010; Buske and Levin, 2013). Biochemical and genetic studies also indicated that the C-terminal tail is the primary target of two negative regulators of FtsZ polymerization, MinC and SlmA (Shen and Lutkenhaus, 2009; Du and Lutkenhaus, 2014). So far, none of the realized fluorescent fusions of FtsZ have been shown to be fully functional, indicating that both the C-terminal tail and the globular domain are sensitive to spatial perturbations. Two close-to-functional examples are an EYFP-FtsZ fusion at temperatures below 28°C though with a very small fraction of cleaved FtsZ (Alexeeva et al., 2010), and an FtsZswVenus fusion (where Venus is inserted into the unstructured linker region of FtsZ) that rescued FtsZ deletion at 42°C after several rounds of passage likely involving mutations in the genome (Osawa and Erickson, 2005).
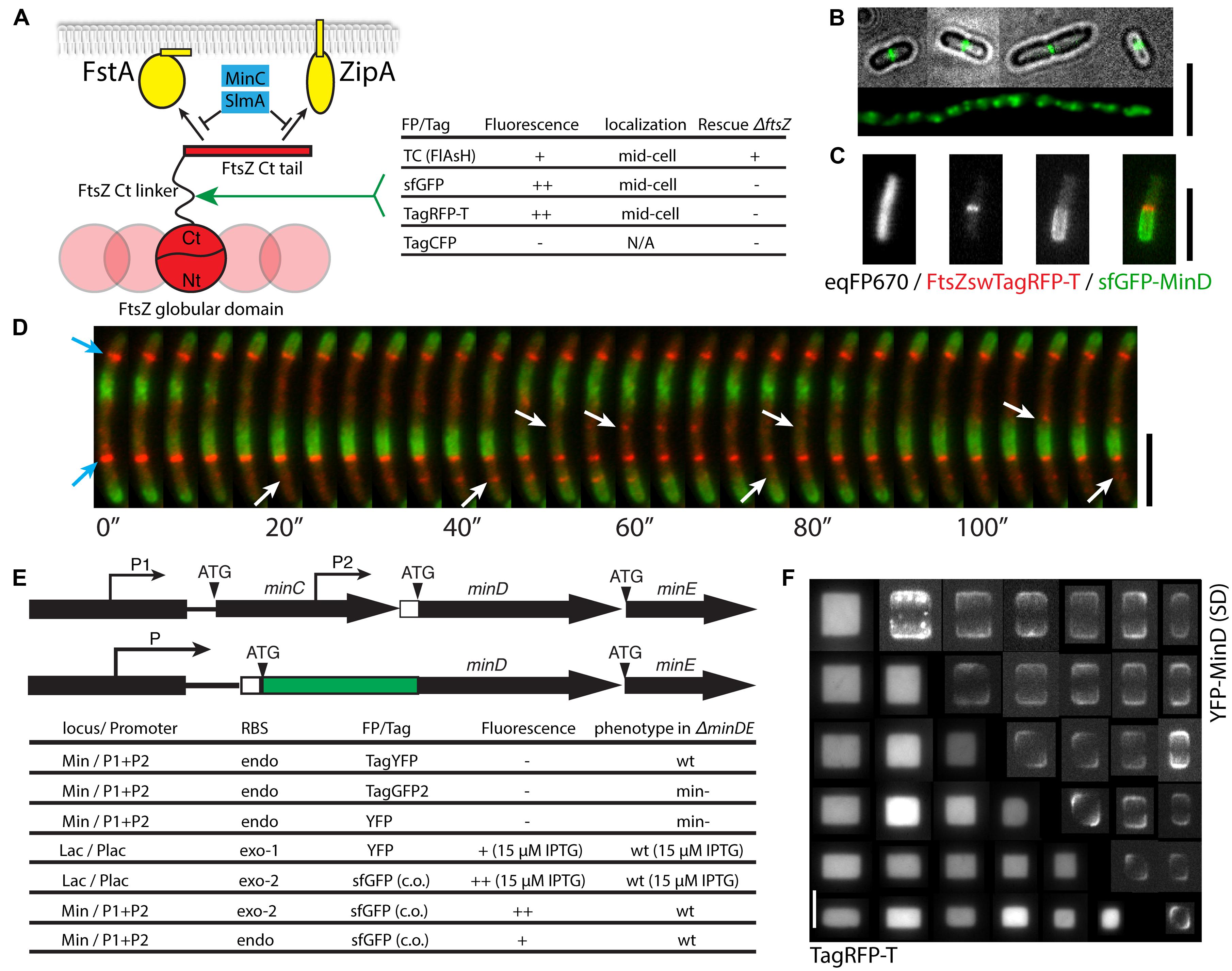
FIGURE 3. Multi-color imaging of cell division at the green, yellow, orange, and near-infrared spectral range. (A) A sandwich fusion strategy for labeling FtsZ and the effectiveness of using various fluorescent tags or proteins. These fluorescent probes were inserted within the flexible linker between the extreme C-terminal tail of FtsZ that is responsible for the direct interactions with FtsA, ZipA, MinC and SlmA, and the globular structure responsible for polymerization. (B) Fluorescence microscopy images of a strain expressing FtsZswTC labeled by the FlAsH dye (in green). The top four cells show regular rod-shaped cells (cell boundary shown in gray scale) with central Z-ring (green), while the bottom one is part of a filamentous cell in the same culture showing overexpressed FtsZ fusion proteins (green). (C) A combination of near infrared, orange-red and green FPs for three-colors imaging (strain FW1559 with pFWZ4, induced with 0.0002% arabinose). The FtsZswTagRFP-T was expressed from an ectopic copy under the arabinose promoter PBAD. (D) Time-lapse images showing the dynamics of FtsZswTagRFP-T and sfGFP-MinD in an elongated cell (strain FW1554 with pFWZ4, induced with 0.0002% arabinose) treated with cephalexin. Blue arrows show stable Z-rings; white arrows indicate the locations where FtsZ proteins polymerize and depolymerize. All scale bars indicate 5 μm. (E) Schematic and table showing the effectiveness of the fluorescent labeling of MinD at different genomic loci with different promoters and RBSs. Note that for simplicity, the RBSs of MinC and MinE are not shown. P, promoter; ATG, start codon; the white box denotes the RBS; exo, exogenous; endo, endogenous. (F) Fluorescent images of strain FW1503 co-expressing cytosolic TagRFP-T and YFP-MinD used in a cell shaping experiment, where single cells grow into the shapes of nanofabricated chambers, as described in Wu et al. (2015). This shows that orange-red and yellow FPs can be combined for multi-color imaging. The bottom left show cytosolic eqFP670 fluorescence and the top right images are YFP-MinD standard-deviation image calculated from the 25 images taken in 2 min.
Since directed replacement and random insertion assays both indicated that the unstructured linker region of FtsZ can be perturbed to some extent (Osawa and Erickson, 2005; Buske and Levin, 2013), we set out to probe the possibilities of inserting other fluorescent tags at the amino acid 333 of FtsZ by adopting the strains and selection processes for FtsZswVenus (see Osawa and Erickson, 2005 and Materials and Methods; Figure 3A). We hoped that a FP derived from another origin could perform differently during the folding of the sandwich fusion, thus have potential to success in sandwich fusion. In order to confirm the suitability of the custom-designed flexible linkers and insertion site, we first inserted a tetracysteine (TC) sequence (FLNCCPGCCVEP) flanked by the short flexible linkers (GSGSGS-TC-GGSGSS) into the ftsZ gene, yielding pFWZ7. This TC tag can later be labeled by Fluorescein Arsenical Hairpin (FlAsH; Griffin et al., 1998). When induced with 0.2% arabinose, the construct pFWZ7 was able to both co-exist with the native FtsZ, and rescue the lethal effect of FtsZ deletion (Figure 3B). Under this growth condition, however, the ΔftsZ strain with pFWZ7 always showed a significant fraction of filamentous cells (30% in biomass). To label the FtsZswTC with the FlAsH dye, we optimized the protocol for live cell staining in E. coli. By incubating an exponentially growing bacterial culture in LB with 2 μM FlAsH dye for 3 h followed by a gentle wash step with EDT2 buffer, we were able to observe the fluorescence of Z-rings localizing at the cell middle in the regular rod-shaped cells (Figure 3B). By contrast, the filamentous cells show patches of fluorescence over the whole cell body, indicating that the filamentation is mainly caused by the overproduction of FtsZswTC, which disrupted the ratio between FtsA and FtsZ. It was shown previously that an overexpression of only FtsZ or only FtsA can lead to a filamentous phenotype (Dai and Lutkenhaus, 1992). In order to confirm that filamentation is caused by an incorrect protein expression level, we engineered pFWZ0 (Para::ftsZ) and transformed into the ΔftsZ strain, which rescued the cell growth upon induction but also showed a similar morphological heterogeneity. We were, however, unable to reduce the proportion of filamentous cells by lowering arabinose concentration, likely due to the inherent all-or-none expression pattern of PBAD promoter. Because the FlAsH tag showed very weak fluorescence and poor photostability, we did not proceed further to optimize the construct for homogeneous induction or insertion into the genome.
We further replaced the TC peptide in the pFWZ7 construct with sfGFP, TagRFP-T, and TagCFP, which are derived from three different protein origins (Figure 3A). The FtsZswsfGFP and FtsZswTagRFP-T were able to form fluorescent Z-rings with or without the existence of native, untagged FtsZ, but they were not able to rescue the lethal effect of FtsZ deletion. The FtsZswTagCFP, on the other hand, did not fluoresce and was not investigated further. Nevertheless, the former two constructs can serve as alternative ectopic FtsZ probes in addition to the existing ones.
The FtsZswTagRFP-T is bright and photostable, and can be combined with eqFP670 and sfGFP-MinD for three-colors imaging (Figure 3C). The superior brightness and photostability of both FtsZswTagRFP-T and sfGFP-MinD allowed us to probe the dynamic relations between the localization of FtsZ and MinD with a frame rate of 1–4 s for 2 min. Figure 3D shows that FtsZ filaments can be assembled and disassembled within a time frame of 20–30 s, in a pattern which anti-correlates with sfGFP-MinD localization patterns. Besides the above-mentioned advantages, the use of FtsZswTagRFP-T also results in less phototoxicity due to its long excitation/emission wavelength, in contrast to the previous YFP-MinD/FtsZ-CFP combination (Shih et al., 2005).
The Endogenous minD Fusion Reveals the Importance of Codon-Dependent Translational Read-Through for N-Terminal Fusions
Min proteins in E. coli oscillate between the two cell poles, forming a time-averaged concentration gradient of MinC, an FtsZ antagonist, with a minimum at the cell middle to allow Z-ring formation (Figure 1F). Without the Min system, E. coli often divide asymmetrically, producing anucleated minicells. The Min operon is composed of minC, minD, and minE genes which are positioned sequentially in the genome, with one promoter in front of MinC that drives the transcription of all three genes, and another promoter embedded in the minC gene to only drive MinD and MinE expression (de Boer et al., 1989; Figure 3E). Co-transcription of minD and minE ensures the ratio of their protein products to be rather constant, which is essential for the localization patterns of all Min proteins. Previously, gfp-minDE and yfp-minDE constructs were shown to be able to complement the ΔminDE phenotype when placed under the lac promoter in medium copy plasmids and induced with low concentrations of IPTG (Raskin and de Boer, 1999; Shih et al., 2002). These constructs were widely used for studying the Min protein dynamics. However, it is difficult to match inducible expression to native protein levels at all physiological conditions, and the lac promoter is known to show inhomogeneity and leaky expression.
We set out to engineer a strain where the expression of a MinD fusion will be driven by the endogenous promoter and RBS such that the expression level of the fusion will most closely reflect the native status (Figure 3E; Wu et al., 2015). We first constructed a few N-terminal minD fusion constructs that are followed by the minE gene and a kanamycin resistance gene. The tags are TagYFP and TagGFP2, purchased from Evrogen for their claimed photostability and fast-folding property, and YFP, from the original construct mentioned above. Unfortunately, when inserted into the min locus replacing a ΔminDE::sacB-cat cassette, none of them resulted in a strain that showed fluorescence, and only TagYFP-MinD appeared to be functional in complementing the minicell phenotype. The non-functional endogenous yfp-minD fusion must be caused by an insufficient expression level rather than the functionality of the fusion proteins, since YFP-MinD fusion was proven functional and fluorescent when expressed from a plasmid. On the other hand, TagYFP-MinD appeared to be expressed and functional but do not fluoresce. We next inserted the Plac::yfp-minDE construct and a new Plac::sfGFP-minDE construct into the genomic lac operon, keeping the RBS from the original plasmids, and these cells showed YFP-MinD/sfGFP-MinD oscillations, when induced using 15 μM IPTG in both wild-type and ΔminDE background. The sfGFP used here is a codon-optimized version. This further confirms that the difficulty in the endogenous fusion of minD is caused by the translational read-through when the endogenous minD RBS is combined with the other FP genes.
To test whether the endogenous minD RBS can be functional when combined with codon-optimized version of the sfGFP gene, we first replaced the RBS of the Plac::sfGFP-MinD construct with the minD RBS, which successfully result in sfGFP-MinD oscillations under IPTG induction. Finally, we replaced the ΔminDE::sacB-cat region with the sfGFP-minDE construct either with only the endogenous minD RBS or with an exogenous RBS from the plasmid behind the endogenous minD RBS. Both strains appeared to completely rescue the minicell phenotype (Figure 3E). The sfGFP-MinD fusion under the endogenous RBS was expressed at a level almost identical to the wild-type (Wu et al., 2015).
The endogenous fusion of the sfGFP-minD, in combination with cytosolic eqFP670 label, allowed us to study the Min pattern formation in diverse cell shapes defined by microstructures (Wu et al., 2015). By inoculating the E. coli cells into microchambers, we were able to mold them into defined shapes across a large range of sizes. Studying sfGFP-MinD oscillation patterns in these cells revealed that Min proteins are able to orient their oscillations according to the symmetry and scale of the cell boundary, and scale their concentration gradients with the cell dimension within a length range of 3–6 microns (Wu et al., 2015). Here, we show that also the ectopically expressed YFP-MinD fusion (in a wild-type minDE+ background, strain FW1503) can be combined with cytosolic TagRFP-T (strain FW1503) for the same applications, yielding the same localization patterns as sfGFP-MinD (Figure 3F). This confirms that the effect of symmetry and scale on the Min pattern formation is intrinsic and not dependent on the FPs, and verifies that the previous studies with modest YFP-MinD levels indeed represent well the native Min protein behavior. However, strain FW1503 appeared to be more sensitive to osmotic shock due to unknown reasons and thus less suitable for quantitative studies or for producing cells larger than 4.5 μm × 4.5 μm × 1 μm using the same shaping method.
TagBFP Directly Interacts with TagRFP-T
From the characteristics of all FPs above, we expected that TagBFP and TagRFP-T would be excellent, mutually exclusive partners for multi-color imaging, in combination with the broadly available bright GFPs/YFPs variants. Thus, we transformed pFWZ4 (FtsZswTagRFP-T) into strain FW1359 for co-imaging of FtsZ and chromosome. To our surprise, we observed patchy blobs in the orange channel, which colocalize with the fluorescence signals from the TagBFP channel (Figure 4). It appears that the HU-TagBFP recruites FtsZswTagRFP-T to the nucleoid. To confirm such a strong interaction between TagBFP with TagRFP-T, we transduced a tagRFP-T cassette into strain FW1359, and found that HU-TagBFP is also able to recruit the freely diffusing cytosolic TagRFP-T molecules to the nucleoid (Figure 4). Moreover, this recruitment induced elongated cell morphology and chromosome-segregation defects (Figure 4).
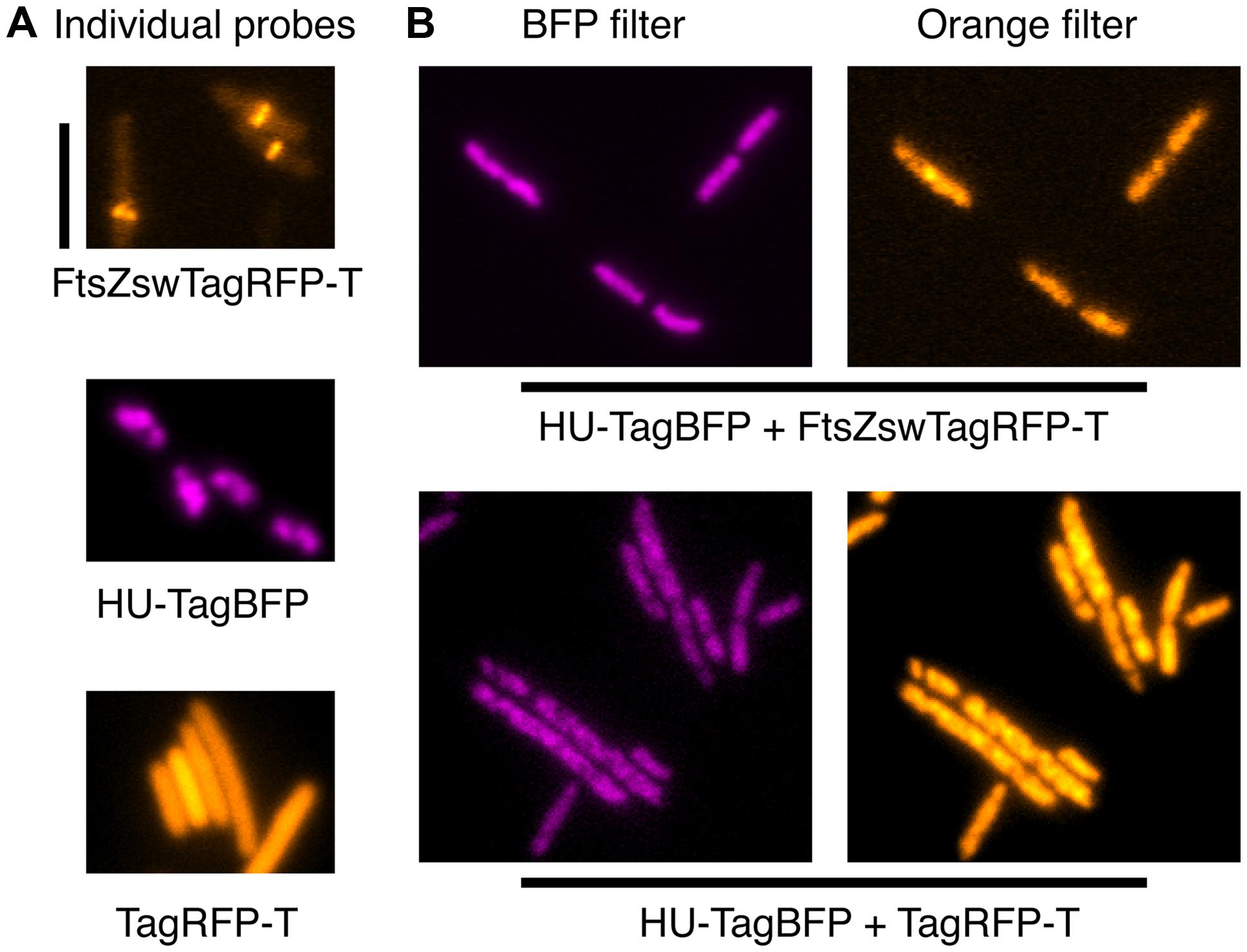
FIGURE 4. HU-TagBFP interferes with the localizations of TagRFP-T and FtsZswTagRFP-T. (A) False-color images of the individual fluorescent fusions. (B) The recruitment of both the FtsZswTagRFP-T and cytosolic TagRFP-T by the HU-TagBFP to the nucleoid, when these probes were combined. Scale bar at the top left indicates 5 μm.
Discussion
We have introduced recently developed blue (TagBFP), orange (TagRFP-T and mKO2), red (mKate2), and near-infrared FPs (eqFP670) into localization studies in bacteria. These proteins generally showed the superior brightness and photostability in bacteria, consistent with previous reports of their in vitro behavior. This indicates that their folding properties are not the limiting factor for their general application in bacteria, contrasting earlier versions of proteins in the blue and orange spectral range, such as EBFP2 and mOrange. The near-infrared protein eqFP670 exhibited a strong brightness under our imaging conditions, presenting itself as an excellent candidate for cell body labeling for long-term and/or frequent imaging. Its extremely far-red excitation at 610 nm can minimize phototoxicity during imaging compared to all other FPs presented here. The addition of these FPs now leads to the ability to re-shuffle optimal choices of fluorescent probes in vivo for various combinations of colors. At the far-red spectral range, mKate2 showed a 25% improvement over mCherry. Since the latter is often chosen for its super-fast folding property, it is yet to be determined whether mKate2 and TagRFP-T can outperform mCherry in all aspects, such as for tagging periplasmic proteins.
For single-color imaging with high temporal resolution, TagRFP-T appears to be an excellent probe for bacterial studies. Besides its extremely strong photostability, it is excited with a much longer wavelength compared to the common GFPs. Using TagRFP-T reduces the phototoxicity compared to GFPs, allowing imaging at higher frequency and intensity. Adding to a previous study which successfully used its ancestor TagRPF as N-terminal tag (Alexeeva et al., 2010), we showed here that the brightness and photostability of TagRF-T is not perturbed by fusions to proteins either in the middle or at their C terminus, thus proving that TagRFP-T can be generally applicable in protein fusions.
For two-colors imaging, we have shown that a combination of TagRFP-T and sfGFP can allow imaging at high frequency without significant photobleaching. For three-colors protein tagging, TagBFP/GFP/mKO2 can be a good alternative to the commonly used set of CFP/YFP/mCherry. In particular, cyan FPs are mostly dimmer and less photostable than GFPs, whereas TagBFP is a fast-folding, bright and photostable variant. However, we note that the excitation spectrum of TagBFP is closer to the UV range compare to that of CFPs, which imposes a trade off between brightness and phototoxicity in time-lapse imaging. When a fourth color is required, one can add eqFP670 to the former combination (Table 4), whereas SBFP2 can supplement the latter. However, eqFP670 is a dimer, and thus often unsuitable for protein tagging, while SBFP2 shows a very low brightness, thus requiring either high expression levels, or strong excitation at around 380 nm.
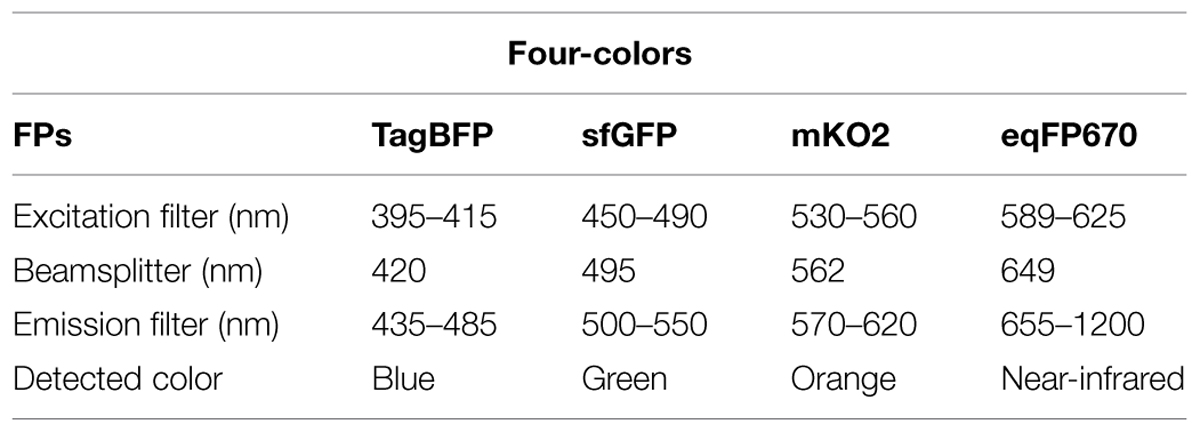
TABLE 4. Suggestions of four-colors combinations and excitation/emission filter sets.
For five-colors imaging, it is possible to combine SBFP2/mCerulean/mYPet/TagRFP-T/eqFP670 (see Table 5). Note that the above-mentioned limitations regarding the brightness of SBFP2 and the dimer property of eqFP670 are evident. The brightness in the blue/cyan spectrum can be improved by either engineering a bright FP that is more blue-shifted than TagBFP, or by engineering a cyan FP with a narrower excitation/emission spectrum that reduces spectral cross-talk with TagBFP. Regarding monomeric near-infrared or infrared FPs, there have been reports of newly evolved versions with an improved brightness (Yu et al., 2014). They were, however, mainly developed for deep-tissue imaging, and are yet to be tested for fluorescent tagging in bacteria for their brightness and folding properties.
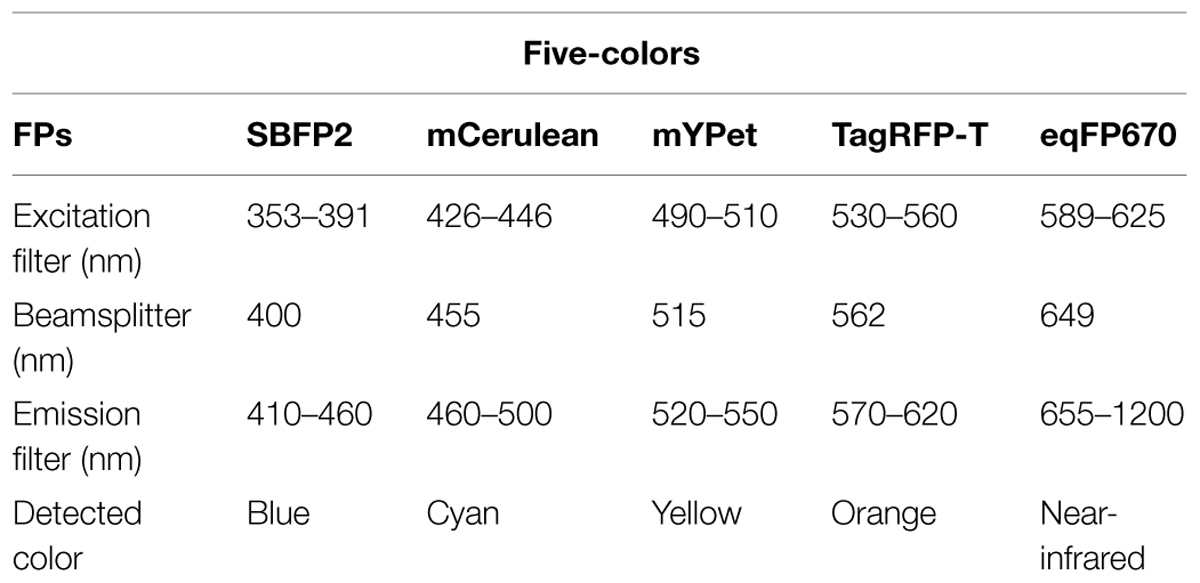
TABLE 5. Suggestions of five-colors combinations and excitation/emission filter sets.
The observed direct interaction between TagBFP and TagRFP-T prohibits the combination of these two proteins in the same cell. While the dimerization or aggregation of monomeric FPs at high concentration has been recognized (Snaith et al., 2010), the possibility of two monomeric proteins interacting with each other is not commonly tested. For example, by constructing a library of individual fluorescent fusions to the same protein in yeast, Lee et al. (2013) proposed the combination of the above two proteins for multi-color imaging. Our findings emphasize that the evolution of future fluorescent probes should take the interspecies interactions into account.
Our efforts to produce HU-2, FtsZ, and MinD fusion proteins for fully replacing the endogenous ones exemplify the multi-faceted challenge regarding fluorescent tagging, which naturally depend on the structural properties of the native proteins and the transcriptional/translational read-through at their genomic loci. The fact that a short TC peptide insertion into the unstructured region of FtsZ successfully rescued the lethal effect of ΔftsZ but not in the case of sfGFP and TagRFP-T agrees with the finding that the unstructured region of the FtsZ has a limited tolerance to the size of a insertion (Buske and Levin, 2013). For visualizing MinD, we showed that it is possible to carry out N-terminal fusions at an endogenous genetic locus, albeit typically requiring independent sampling effort for individual genes due to the sensitivity of the transcription and translation read-through at the coding sequence close to the RBS (Kudla et al., 2009). Regarding a valid representation of a native localization pattern, a previous example showed that, while the super-fast folders sfGFP and mCherry were shown to be particularly advantageous in N-terminal or sandwich fusions and studies in periplasm (Bendezú et al., 2009; Dinh and Bernhardt, 2011; Wu et al., 2015), they were shown to form non-native foci when fused to ClpXP (Landgraf et al., 2012). Here we assessed the advantage of the recently developed FPs for studies in bacteria based on quantitative comparisons on a number of targets. We suggest that their performance as fluorescent tags be further tested in comparison to the native proteins in other future studies.
The expansion and detailed study of the FP reported in this paper will help imaging cell division in bacteria. The understanding of the processes involved in chromosome organization and cell division in bacteria has seen great development owing to the use of FPs. Current challenges in microscopy imaging of these processes lie in increasing the spatiotemporal resolution, uncovering the native behavior, and simultaneously inspecting the multiple interacting sub-components. We expect that our quantitative evaluation of novel FPs and the fusion strategies will facilitate tackling these challenges.
Author Contributions
FW, JK, and CD conceived the experiments, FW, EVR, and BVS did the experiments, FW analyzed the data, FW and CD wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Louis Kuijpers, Margot Guurink, and Kevin Felter for their assistance in part of the strain construction. We thank Roger Tsien and Hugo Snippert for the pTagRFP-T plasmid, Dorus Gadella for the pSBFP2-C2 plasmid and the spectral information of SBFP2, Conrad Woldringh for HupA plasmids which we used for initial inspection of HU labels, Masaki Osawa for the JKD7-1 strain and pKD3ftsQAZ, Rodrigo Reyes-Lamothe, and David Sherratt for the strain RRL189 and a mYpet construct, Sriram Krishnan for a mYpet construct, Yu-Ling Shih for pFX40, and Piet de Boer for pFB174. This work was partly supported by the Netherlands Organisation for Scientific Research (NWO/OCW) as part of the Frontiers of Nanoscience program, NanoNextNL program 3B (FW), and European Research Council NanoforBio No. 247072 (CD).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00607/abstract
References
Ai, H.-W., Shaner, N. C., Cheng, Z., Tsien, R. Y., and Campbell, R. E. (2007). Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry 46, 5904–5910. doi: 10.1021/bi700199g
Alexeeva, S., Gadella, T. W. J., Verheul, J., Verhoeven, G. S., and Den Blaauwen, T. (2010). Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol. Microbiol. 77, 384–398. doi: 10.1111/j.1365-2958.2010.07211.x
Ali Azam, T., Iwata, A., Nishimura, A., Ueda, S., and Ishihama, A. (1999). Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 181, 6361–6370.
Ausmees, N., Kuhn, J. R., and Jacobs-Wagner, C. (2003). The bacterial cytoskeleton: an intermediate filament-like function in cell shape. Cell 115, 705–713. doi: 10.1016/S0092-8674(03)00935-8
Bailey, M. W., Bisicchia, P., Warren, B. T., Sherratt, D. J., and Männik, J. (2014). Evidence for divisome localization mechanisms independent of the min system and SlmA in Escherichia coli. PLoS Genet. 10:e1004504. doi: 10.1371/journal.pgen.1004504
Bendezú, F. O., and de Boer, P. A. J. (2008). Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J. Bacteriol. 190, 1792–1811. doi: 10.1128/jb.01322-07
Bendezú, F. O., Hale, C. A., Bernhardt, T. G., and De Boer, P. A. J. (2009). RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 28, 193–204. doi: 10.1038/emboj.2008.264
Buske, P. J., and Levin, P. A. (2013). A flexible C-terminal linker is required for proper FtsZ assembly in vitro and cytokinetic ring formation in vivo. Mol. Microbiol. 89, 249–263. doi: 10.1111/mmi.12272
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W., and Prasher, D. (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. doi: 10.1126/science.8303295
Charles, M., Pérez, M., Kobil, J. H., and Goldberg, M. B. (2001). Polar targeting of Shigella virulence factor IcsA in Enterobacteriacae and Vibrio. Proc. Natl. Acad. Sci. U.S.A. 98, 9871–9876. doi: 10.1073/pnas.171310498
Dai, K., and Lutkenhaus, J. (1991). ftsZ is an essential cell division gene in Escherichia coli. J. Bacteriol. 173, 3500–3506.
Dai, K., and Lutkenhaus, J. (1992). The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. J. Bacteriol. 174, 6145–6151.
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
de Boer, P. A. J., Crossley, R. E., and Rothfield, L. I. (1989). A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56, 641–649. doi: 10.1016/0092-8674(89)90586-2
Dinh, T., and Bernhardt, T. G. (2011). Using superfolder green fluorescent protein for periplasmic protein localization studies. J. Bacteriol. 193, 4984–4987. doi: 10.1128/jb.00315-311
Domínguez-Escobar, J., Chastanet, A., Crevenna, A. H., Fromion, V., Wedlich-Söldner, R., and Carballido-López, R. (2011). Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science 333, 225–228. doi: 10.1126/science.1203466
Du, S., and Lutkenhaus, J. (2014). SlmA antagonism of FtsZ assembly employs a Two-pronged mechanism like MinCD. PLoS Genet. 10:e1004460. doi: 10.1371/journal.pgen.1004460
Erickson, H. P., Anderson, D. E., and Osawa, M. (2010). FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol. Mol. Biol. Rev. 74, 504–528. doi: 10.1128/MMBR.00021-10
Garner, E. C., Bernard, R., Wang, W., Zhuang, X., Rudner, D. Z., and Mitchison, T. (2011). Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science 333, 222–225. doi: 10.1126/science.1203285
Griffin, B. A., Adams, S. R., and Tsien, R. Y. (1998). Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269–272. doi: 10.1126/science.281.5374.269
Hayashi, K., Morooka, N., Yamamoto, Y., Fujita, K., Isono, K., Choi, S., et al. (2006). Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2, 2006.0007. doi: 10.1038/msb4100049
Hol, F. J. H., and Dekker, C. (2014). Zooming in to see the bigger picture: microfluidic and nanofabrication tools to study bacteria. Science 346:1251821. doi: 10.1126/science.1251821
Hu, Z., and Lutkenhaus, J. (1999). Topological regulation of cell division in Escherichia coli involves rapid pole to pole oscillation of the division inhibitor MinC under the control of MinD and MinE. Mol. Microbiol. 34, 82–90. doi: 10.1046/j.1365-2958.1999.01575.x
Jones, L. J. F., Carballido-López, R., and Errington, J. (2001). Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell 104, 913–922. doi: 10.1016/S0092-8674(01)00287-2
Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H., et al. (2006). Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299. doi: 10.1093/dnares/dsi012
Kremers, G.-J., Goedhart, J., Van Den Heuvel, D. J., Gerritsen, H. C., and Gadella, T. W. J. (2007). Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry 46, 3775–3783. doi: 10.1021/bi0622874
Kremers, G.-J., Goedhart, J., Van Munster, E. B., and Gadella, T. W. J. (2006). Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET förster radius. Biochemistry 45, 6570–6580. doi: 10.1021/bi0516273
Kudla, G., Murray, A. W., Tollervey, D., and Plotkin, J. B. (2009). Coding-sequence determinants of gene expression in Escherichia coli. Science 324, 255–258. doi: 10.1126/science.1170160
Kuwada, N. J., Traxler, B., and Wiggins, P. A. (2015). Genome-scale quantitative characterization of bacterial protein localization dynamics throughout the cell cycle. Mol. Microbiol. 95, 64–79. doi: 10.1111/mmi.12841
Landgraf, D., Okumus, B., Chien, P., Baker, T. A., and Paulsson, J. (2012). Segregation of molecules at cell division reveals native protein localization. Nat Meth. 9, 480–482. doi: 10.1038/nmeth.1955
Lau, I. F., Filipe, S. R., Søballe, B., Økstad, O.-A., Barre, F.-X., and Sherratt, D. J. (2003). Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol. Microbiol. 49, 731–743. doi: 10.1046/j.1365-2958.2003.03640.x
Lee, S., Lim, W. A., and Thorn, K. S. (2013). Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS ONE 8:e67902. doi: 10.1371/journal.pone.0067902
Li, G., and Young, K. D. (2012). Isolation and identification of new inner membrane-associated proteins that localize to cell poles in Escherichia coli. Mol. Microbiol. 84, 276–295. doi: 10.1111/j.1365-2958.2012.08021.x
Lindner, A. B., Madden, R., Demarez, A., Stewart, E. J., and Taddei, F. (2008). Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc. Natl. Acad. Sci. U.S.A. 105, 3076–3081. doi: 10.1073/pnas.0708931105
Ma, X., Ehrhardt, D. W., and Margolin, W. (1996). Colocalization of cell division proteins FtsZ and FtsA to cytoskeletal structures in living Escherichia coli cells by using green fluorescent protein. Proc. Natl. Acad. Sci. U.S.A. 93, 12998–13003. doi: 10.1073/pnas.93.23.12998
Männik, J., Wu, F., Hol, F. J. H., Bisicchia, P., Sherratt, D. J., Keymer, J. E., et al. (2012). Robustness and accuracy of cell division in Escherichia coli in diverse cell shapes. Proc. Natl. Acad. Sci. U.S.A. 109, 6957–6962. doi: 10.1073/pnas.1120854109
Morozova, K. S., Piatkevich, K. D., Gould, T. J., Zhang, J., Bewersdorf, J., and Verkhusha, V. V. (2010). Far-Red fluorescent protein excitable with red lasers for flow cytometry and superresolution STED nanoscopy. Biophys. J. 99, L13–L15. doi: 10.1016/j.bpj.2010.04.025
Nguyen, A. W., and Daugherty, P. S. (2005). Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 23, 355–360. doi: 10.1038/nbt1066
Oliva, M. A., Cordell, S. C., and Lowe, J. (2004). Structural insights into FtsZ protofilament formation. Nat. Struct. Mol. Biol. 11, 1243–1250. doi: 10.1038/nsmb855
Osawa, M., and Erickson, H. P. (2005). Probing the domain structure of FtsZ by random truncation and insertion of GFP. Microbiology 151, 4033–4043. doi: 10.1099/mic.0.28219-0
Pedelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C., and Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88. doi: 10.1038/nbt1172
Prasher, D. C., Eckenrode, V. K., Ward, W. W., Prendergast, F. G., and Cormier, M. J. (1992). Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111, 229–233. doi: 10.1016/0378-1119(92)90691-H
Raskin, D. M., and de Boer, P. A. J. (1997). The MinE ring: an FtsZ-Independent cell structure required for selection of the correct division site in E. coli. Cell 91, 685–694. doi: 10.1016/S0092-8674(00)80455-9
Raskin, D. M., and de Boer, P. A. J. (1999). Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 96, 4971–4976. doi: 10.1073/pnas.96.9.4971
Reyes-Lamothe, R., Possoz, C., Danilova, O., and Sherratt, D. J. (2008). Independent positioning and action of Escherichia coli replisomes in live cells. Cell 133, 90–102. doi: 10.1016/j.cell.2008.01.044
Rizzo, M. A., Springer, G. H., Granada, B., and Piston, D. W. (2004). An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 22, 445–449. doi: 10.1038/nbt945
Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., and Tsien, R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572. doi: 10.1038/nbt1037
Shaner, N. C., Lin, M. Z., Mckeown, M. R., Steinbach, P. A., Hazelwood, K. L., Davidson, M. W., et al. (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5, 545–551. doi: 10.1038/nmeth.1209
Shcherbo, D., Murphy, C. S., Ermakova, G. V., Solovieva, E. A., Chepurnykh, T. V., Shcheglov, A. S., et al. (2009). Far-red fluorescent tags for protein imaging in living tissues. Biochem. J. 418, 567–574. doi: 10.1042/BJ20081949
Shcherbo, D., Shemiakina, I. I., Ryabova, A. V., Luker, K. E., Schmidt, B. T., Souslova, E. A., et al. (2010). Near-infrared fluorescent proteins. Nat. Methods 7, 827–829. doi: 10.1038/nmeth.1501
Shen, B., and Lutkenhaus, J. (2009). The conserved C-terminal tail of FtsZ is required for the septal localization and division inhibitory activity of MinC(C)/MinD. Mol. Microbiol. 72, 410–424. doi: 10.1111/j.1365-2958.2009.06651.x
Shih, Y.-L., Fu, X., King, G. F., Le, T., and Rothfield, L. (2002). Division site placement in E.coli: mutations that prevent formation of the MinE ring lead to loss of the normal midcell arrest of growth of polar MinD membrane domains. EMBO J. 21, 3347–3357. doi: 10.1093/emboj/cdf323
Shih, Y.-L., Kawagishi, I., and Rothfield, L. (2005). The MreB and Min cytoskeletal-like systems play independent roles in prokaryotic polar differentiation. Mol. Microbiol. 58, 917–928. doi: 10.1111/j.1365-2958.2005.04841.x
Snaith, H. A., Anders, A., Samejima, I., and Sawin, K. E. (2010). “New and old reagents for fluorescent protein tagging of microtubules in fission yeast: experimental and critical evaluation,” in Methods in Cell Biology, Chap. 9, eds C. Lynne and T. Phong (Waltham, MA: Academic Press), 147–172. doi: 10.1016/s0091-679x(10)97009-x
Sourjik, V., and Berg, H. C. (2000). Localization of components of the chemotaxis machinery of Escherichia coli using fluorescent protein fusions. Mol. Microbiol. 37, 740–751. doi: 10.1046/j.1365-2958.2000.02044.x
Subach, O. M., Gundorov, I. S., Yoshimura, M., Subach, F. V., Zhang, J., Grüenwald, D., et al. (2008). Conversion of red fluorescent protein into a bright blue probe. Chem.Bbiol. 15, 1116–1124. doi: 10.1016/j.chembiol.2008.08.006
Sun, Y., Booker, C. F., Kumari, S., Day, R. N., Davidson, M., and Periasamy, A. (2009). Characterization of an orange acceptor fluorescent protein for sensitized spectral fluorescence resonance energy transfer microscopy using a white-light laser. J. Biomed. Opt. 14:054009. doi: 10.1117/1.3227036
Takeuchi, S., Diluzio, W. R., Weibel, D. B., and Whitesides, G. M. (2005). Controlling the shape of filamentous cells of Escherichia coli. Nano Lett. 5, 1819–1823. doi: 10.1021/nl0507360
Taniguchi, Y., Choi, P. J., Li, G.-W., Chen, H., Babu, M., Hearn, J., et al. (2010). Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538. doi: 10.1126/science.1188308
van Teeffelen, S., Wang, S., Furchtgott, L., Huang, K. C., Wingreen, N. S., Shaevitz, J. W., et al. (2011). The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc. Natl. Acad. Sci. U.S.A. 108, 15822–15827. doi: 10.1073/pnas.1108999108
Wang, P., Robert, L., Pelletier, J., Dang, W. L., Taddei, F., Wright, A., et al. (2010). Robust growth of Escherichia coli. Curr. Biol. 20, 1099–1103. doi: 10.1016/j.cub.2010.04.045
Werner, J. N., Chen, E. Y., Guberman, J. M., Zippilli, A. R., Irgon, J. J., and Gitai, Z. (2009). Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proc. Natl. Acad. Sci. U.S.A. 106, 7858–7863. doi: 10.1073/pnas.0901781106
Wery, M., Woldringh, C. L., and Rouviere-Yaniv, J. (2001). HU-GFP and DAPI co-localize on the Escherichia coli nucleoid. Biochimie 83, 193–200. doi: 10.1016/S0300-9084(01)01254-8
Wu, F., Van Schie, B., Keymer, J. E., and Dekker, C. (2015). Symmetry and scale orient Min protein patterns in bacterial shape sculptures. Nat. Nanotechnol. doi: 10.1038/nnano.2015.126
Keywords: fluorescent proteins, bacterial cell division, bacterial chromosome, MinD, HU, FtsZ, TagRFP-T, TagBFP
Citation: Wu F, Van Rijn E, Van Schie BGC, Keymer JE and Dekker C (2015) Multi-color imaging of the bacterial nucleoid and division proteins with blue, orange, and near-infrared fluorescent proteins. Front. Microbiol. 6:607. doi: 10.3389/fmicb.2015.00607
Received: 30 March 2015; Accepted: 02 June 2015;
Published: 17 June 2015
Edited by:
Conrad L. Woldringh, University of Amsterdam, NetherlandsReviewed by:
Joe Lutkenhaus, University of Kansas Medical Center, USAYu-Ling Shih, Institute of Biological Chemistry – Academia Sinica, Taiwan
Copyright © 2015 Wu, Van Rijn, Van Schie, Keymer and Dekker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cees Dekker, Department of Bionanoscience, Kavli Institute of Nanoscience, Delft University of Technology, Lorentzweg 1, 2628CJ Delft, Netherlands,Yy5kZWtrZXJAdHVkZWxmdC5ubA==
†Present address: Juan E. Keymer, Institute of Ecology and Biodiversity and Department of Ecology, School of Biological Sciences, Pontifical Catholic University, Santiago, Chile