 Karan G. Kaval
Karan G. Kaval Birgitt Hahn1
Birgitt Hahn1 Nayana Tusamda
Nayana Tusamda Sven Halbedel
Sven Halbedel- 1FG11 Division of Enteropathogenic Bacteria and Legionella, Robert Koch Institute, Wernigerode, Germany
- 2Institute of Microbiology, University of Greifswald, Greifswald, Germany
Invasion of the bacterial pathogen Listeria monocytogenes into human host cells requires specialized surface molecules for attachment and induction of phagocytosis. However, efficient invasion is also dependent on factors with house-keeping functions, such as SecA2-dependent secretion of autolysins for post-divisional segregation of daughter cells. Mutations in this pathway prevent degradation of peptidoglycan cross-walls, so that long cell chains are formed that cannot be phagocytosed. The extreme chaining of such mutants manifests as rough colony phenotype. One rough clone was isolated from a transposon library with a transposon insertion in the uncharacterized lmo0720 gene (lftS) together with a spontaneous point mutation in the secA2 gene. We separated both mutations and demonstrated that this point mutation in the intramolecular regulator 2 domain of SecA2 was sufficient to inactivate the protein. In contrast, lftS deletion did not cause a ΔsecA2-like phenotype. lftS is located in an operon with lftR (lmo0719), encoding a PadR-like transcriptional regulator, and lftR deletion affected growth, invasion and day-light dependent coordination of swarming. Inactivation of lftS partially suppressed these phenotypes, suggesting a functional relationship between LftR and LftS. However, the invasion defect of the ΔlftR mutant was only marginally suppressed by lftS removal. LftR regulates expression of the lmo0979–0980 (lieAB) operon, encoding a putative multidrug resistance transporter and lieAB transcription was strongly upregulated in the absence of LftR. Deletion of lieAB in the ΔlftR background restores wild type-like invasion levels. Hence, we conclude that tight transcriptional repression of the lieAB operon is essential for efficient listerial host cell invasion.
Introduction
Listeria monocytogenes is an opportunistic pathogen, which can cause life-threatening gastrointestinal infections in humans upon ingestion of contaminated food. Cells of L. monocytogenes can actively invade non-phagocytic host cells and persist and multiply inside their cytoplasm (Freitag et al., 2009). Motility inside the host cell is facilitated by comet tail-like polymerization of host actin at one bacterial cell pole that generates the driving force to push the bacteria through the viscous cytoplasm (Tilney and Portnoy, 1989; Dabiri et al., 1990; Lambrechts et al., 2008). The same mechanism allows generation of membranous protrusions at the surface of the infected host cell, which are internalized by neighboring cells and finally mediates listerial spread from cell to cell (Tilney and Portnoy, 1989; Ireton et al., 2014). Using this strategy, L. monocytogenes manages to spread within host tissues and breaches all barriers of the human body, i.e., the gastrointestinal, the placental and the blood brain barrier, thereby causing gastrointestinal symptoms and infections of the brain or the fetus (Cossart and Toledo-Arana, 2008). Thus, invasion is the very first step in a sequence of events that eventually lead to manifestation of listeriosis, which, remarkably, can cause case-fatality rates of up to 30% (Swaminathan and Gerner-Smidt, 2007; Hsieh et al., 2009).
Listerial invasion depends on internalins, which are specialized bacterial surface proteins that contact receptors at the host cell surface. L. monocytogenes encodes 25 internalins (Gaillard et al., 1991; Dramsi et al., 1995; Bierne et al., 2007). However, the most prominent members of this protein class are InlA and InlB, which bind to E-cadherin and Met (Mengaud et al., 1996; Shen et al., 2000), respectively, at the host cell surface and these protein–protein interactions induce cytoskeletal re-arrangements in the host cells that lead to uptake of the bacterium in a phagocytosis-like process (Cossart and Toledo-Arana, 2008). Apart from these invasion-specific molecules, mutations in other genes with more general house-keeping functions have been shown to severely reduce the invasive potential of L. monocytogenes. Among these, factors contributing to synthesis, modification and degradation of the bacterial envelope are predominant (Seveau et al., 2007; Camejo et al., 2011; Pizarro-Cerda et al., 2012).
Here we characterize the functions of four so far unknown genes of this latter type that encode for obvious house-keeping functions but affect invasion. This includes the lmo0719–0720 genes, which are organized in a bi-cistronic operon. While lmo0720 codes for a gene of unknown function, lmo0719 shares homology with PadR-type transcriptional regulators. PadR-like transcriptional regulators can be found in many bacteria and are often associated with control of detoxification genes (Barthelmebs et al., 2000; Gury et al., 2004; Agustiandari et al., 2008). In contrast, genes homologous to lmo0720 are less wide-spread and are specific to the genus Listeria and a few other Gram-positive bacteria. We demonstrate that Lmo0719 controls transcription of an uncharacterized putative ABC multidrug resistance transporter, encoded by the lmo0979–0980 operon, which shares similarity with the LmrCD transporter of Lactococcus lactis (Lubelski et al., 2006). Strikingly, invasion of Δlmo0719 mutants into HeLa cells was strongly reduced and hence, lmo0719 was renamed lftR (listerial protein facilitating invasion/transcriptional regulator, and accordingly, lmo0720 was renamed lftS). This defect could be restored by deletion of the lmo0979–0980 operon, suggesting that tight repression of lmo0979–0980 transcription through LftR is necessary to ensure efficient invasion of eukaryotic host cells. Further experiments identified ethidium bromide as one artificial substrate, which is taken up by this transporter, and thus the lmo0979–0980 genes were renamed lieAB (listerial importer of ethidium bromide as artificial substrate).
Materials and Methods
Bacterial Strains and Growth Conditions
All strains used in this study are listed in Table 1. Cells of L. monocytogenes were generally cultivated in BHI broth or on BHI agar plates at 37°C and 200 rpm if not stated otherwise. Where required, antibiotics and supplements were added at the following concentrations: erythromycin (5 μg/ml), kanamycin (50 μg/ml) and X-Gal (100 μg/ml). Escherichia coli TOP10 was used as standard cloning host (Sambrook et al., 1989).
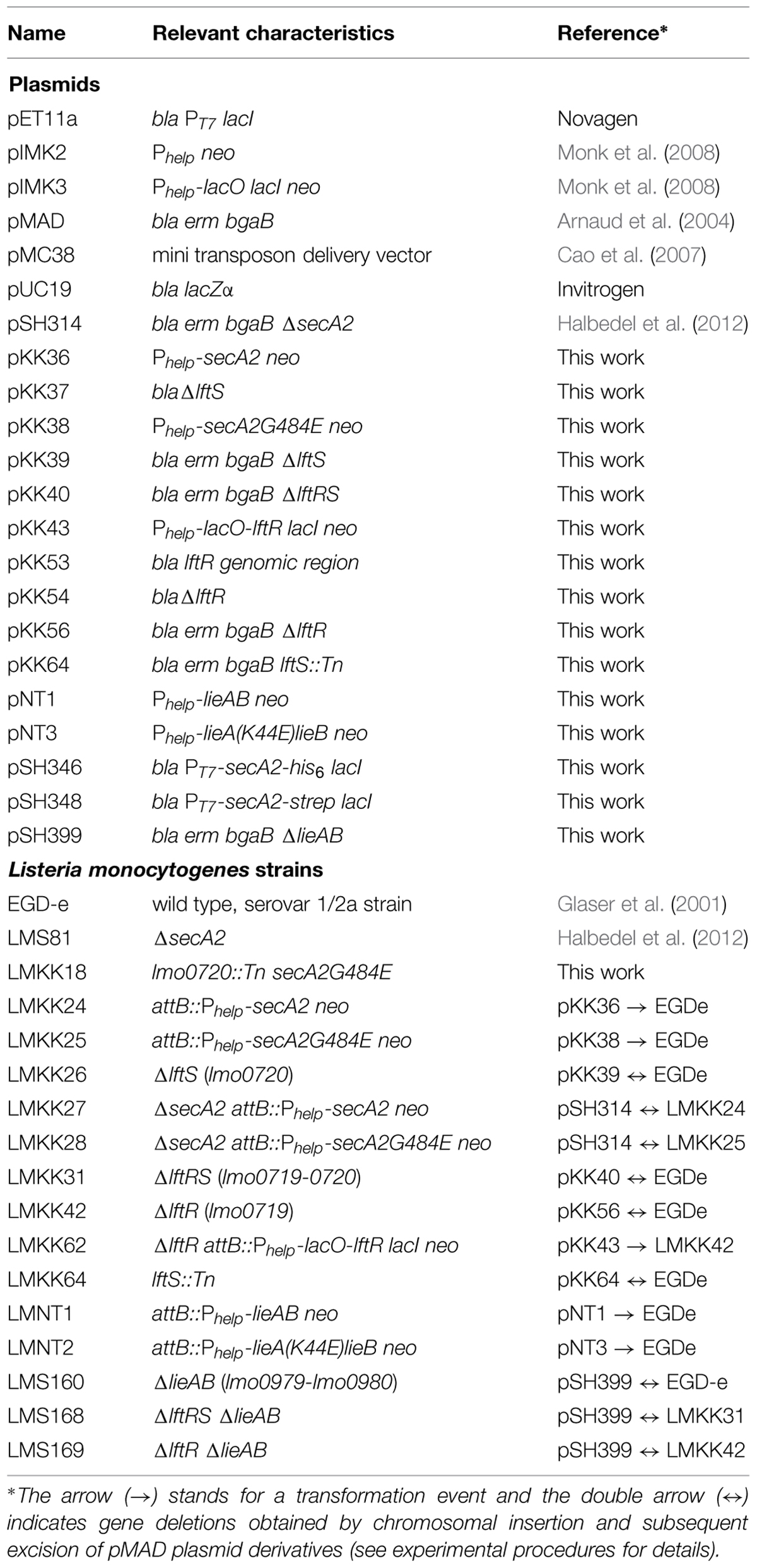
TABLE 1. Strains and plasmids used in this study.
General Methods, Oligonucleotides, and Manipulation of DNA
Transformation of E. coli, isolation of plasmid and chromosomal DNA was performed using standard methods (Sambrook et al., 1989). Preparation of electro-competent L. monocytogenes cells and their transformation were carried out as described elsewhere (Monk et al., 2008). Restriction and ligation of DNA was done as per the manufacturer’s instructions. For restriction free modification of plasmids an altered version of the original Quickchange mutagenesis protocol was employed (Zheng et al., 2004). All primer sequences are listed in Table 2.
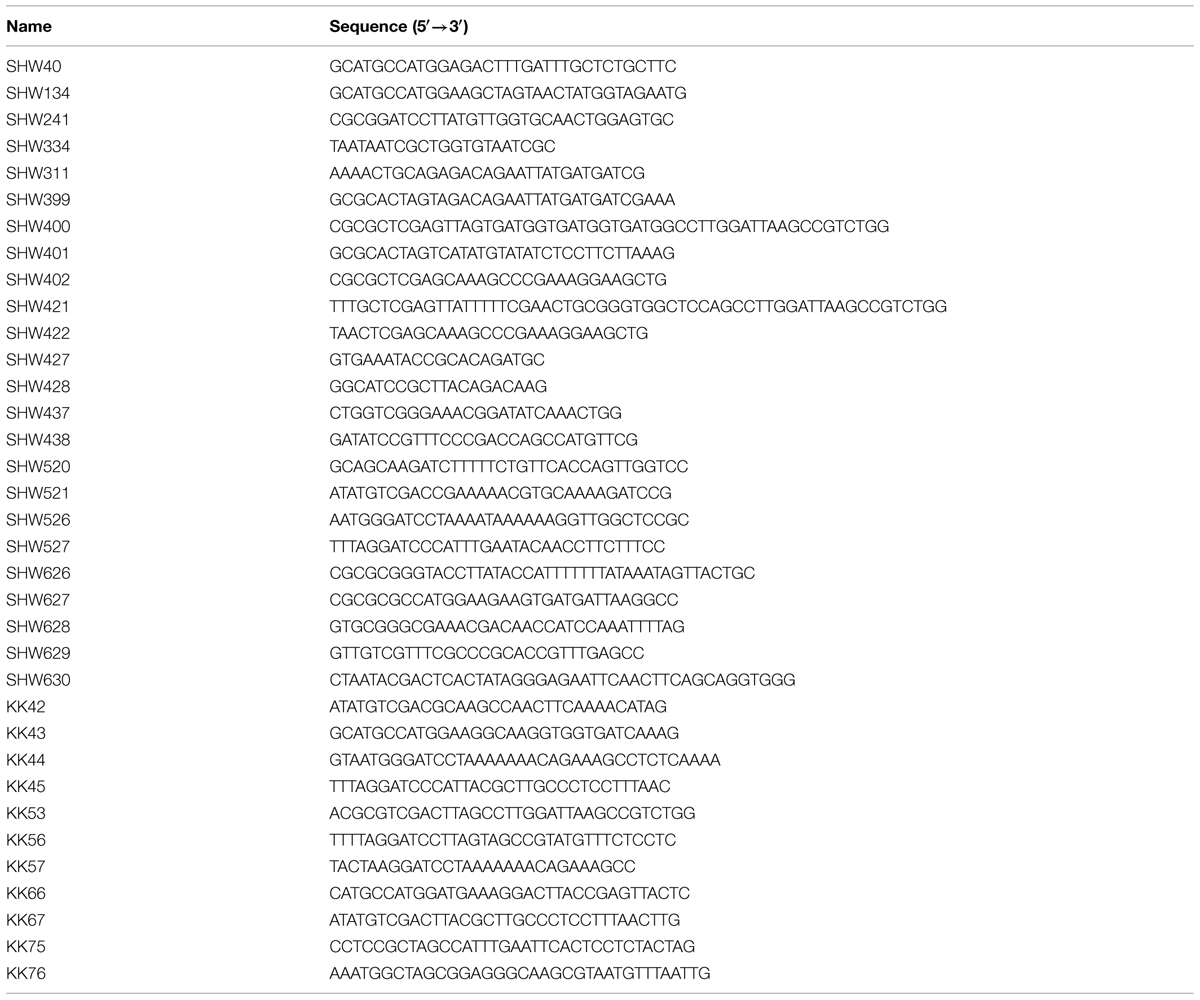
TABLE 2. Oligonucleotides used in this study.
Transposon Mutagenesis and Inverse PCR
The HimarI transposon delivery vector pMC38 (Cao et al., 2007) was introduced into L. monocytogenes by electroporation and erythromycin-resistant clones were selected at 30°C. Five colonies were randomly chosen and grown in BHI medium supplemented with kanamycin and erythromycin at 30°C overnight. These cultures were diluted 1:200 in fresh BHI broth containing erythromycin and incubated at 30°C for 1 h. Afterward, the temperature was increased to 42°C for 6 h until an OD600 of 0.5 was reached. These cultures were serially diluted and plated on both BHI erythromycin as well as BHI kanamycin plates to determine CFU/ml and rate of plasmid retention. Aliquots were mixed with 50% glycerol and frozen at -80°C.
Rough colonies were isolated and plated on BHI agar containing erythromycin and BHI agar plates containing kanamycin. Chromosomal DNA was isolated from ermR kanS clones, which were then screened for transposon insertions in secA2 or divIVA as determined by PCR using the primer pairs SHW241/SHW334 and SHW134/SHW40, respectively. The site of transposon insertion was determined by inverse PCR. For this purpose, chromosomal DNA of relevant clones was subjected to TaqI digestion for 1 h, followed by a 20 min heat inactivation step. Ligation of the resultant digested DNA was carried out for 1 h at room temperature using T4 DNA ligase. A PCR was set up using the primer pair SHW427/SHW428 and the ligation mixture as the template. This PCR product was purified and the transposon insertion site was determined by DNA sequencing.
Construction of Plasmids and Strains
In order to facilitate overexpression of SecA2-Strep, plasmid pSH348 was constructed. It was obtained in two steps. In the first step, the secA2 gene was amplified using SHW399/SHW400 (SHW400 introduced a C-terminal His6-tag) and the resulting fragment was digested with SpeI/XhoI and cloned into pET11a, which had been linearized using the primer pair SHW401/SHW402. The His6-tag present at the C-terminus in the resulting plasmid (pSH346) was then replaced by a C-terminal Strep-tag in a PCR using SHW421/SHW422 as the primers.
Plasmid pKK36 was constructed to allow for constitutive expression of the secA2 gene in L. monocytogenes. It was obtained by amplification of the secA2 gene using the primer pair SHW311/KK53. The resulting fragment was PstI/SalI cut and ligated with the similarly cut backbone of pIMK2. The G484E mutation was brought into this vector by quickchange mutagenesis using the oligonucleotides SHW437/SHW438, yielding plasmid pKK38.
Plasmid pKK39, allowing marker-less removal of the lftS gene was obtained in two steps: first, up- and downstream regions of the lftS gene were amplified in PCRs with the primer pairs KK42/KK45 and KK44/KK43, respectively. Both fragments were joined together by ligation after their ends had been made compatible by BamHI digestion. The desired ΔlftS fragment was then amplified from the ligation mixture in a second PCR using the primer pair KK42/KK43 and blunt end cloned into SmaI cut pUC19. The ΔlftS fragment of the resulting plasmid (pKK37) was finally sub-cloned into pMAD using NcoI/SalI restriction digestion. Plasmid pKK40 for deletion of the entire lftRS operon was obtained by deletion of the lftR gene from plasmid pKK39 using the primer pair KK56/KK57.
For deletion of the lftR gene, plasmid pKK56 was constructed in three steps. First, the chromosomal region encompassing the lftR gene was amplified with the oligonucleotides KK42/KK43 and blunt end cloned into SmaI cut pUC19. The lftR gene was then removed from the resulting plasmid (pKK53) by PCR using the primer pair KK75/KK76, which yielded plasmid pKK54. Finally, the NcoI/SalI digested ΔlftR fragment of this plasmid was sub-cloned into similarly cut pMAD.
Plasmid pKK64 was generated to replace the lftS gene of strain EGD-e by the lftS::Tn allele of the rough transposon insertion mutant LMKK18. For this purpose, the lftS::Tn fragment was amplified by PCR from LMKK18 chromosomal DNA using the primer pair KK42/KK43, NcoI/SalI cut and ligated with the backbone of pMAD digested with the same enzymes.
In order to remove the lieAB operon from the listerial chromosome, plasmid pSH399 was designed. Up- and downstream regions of the lieAB operon were amplified with the primer pair SHW520/SHW527 and SHW526/SHW521, respectively, BamHI cut and fused together by ligation. The desired ΔlieAB fragment was amplified from the ligation mixture in a PCR using SHW520/SHW521 as the primers and cloned into pMAD using BglII/SalI.
For IPTG-inducible expression of lftR, plasmid pKK43 was constructed. The lftR gene was amplified in a PCR using KK66/KK67 as the primers and the resulting fragment was cloned into pIMK3 via NcoI/SalI.
Plasmid pNT1 was constructed for overexpression of lieAB. It was obtained by amplification of the lieAB operon using the primer pair SHW627/SHW626 and cloned with NcoI/KpnI into pIMK2. The K44E mutation was brought into the Walker motif of the lieA gene on plasmid pNT1 in a quickchange reaction with the primer pair SHW628/SHW629. The resulting plasmid was named pNT3.
pIMK2 and pIMK3 plasmids were introduced into L. monocytogenes strains by electroporation and kanamycin resistant clones were selected. Plasmid insertion at the attB site of the tRNAArg locus was verified by PCR. For the marker-less removal of genes, pMAD derivatives were transformed into the respective L. monocytogenes recipient strains and the genes were removed according to a previously described protocol (Arnaud et al., 2004). All gene deletions were verified by PCR.
Purification of SecA2 and Generation of an Anti-SecA2 Antiserum
SecA2-Strep was overexpressed in E. coli BL21. The bacteria were cultivated in LB broth containing ampicillin (100 μg/ml) at 37°C. Expression of the SecA2 protein was induced at an optical density of OD600 = 0.5 by addition of 1 mM IPTG (final concentration) after the culture had been cooled down to 18°C. Cultivation was continued over night at 18°C and cells were harvested, washed once with ZAP buffer (10 mM Tris/HCl pH 7.5 and 200 mM NaCl), and finally disrupted in the same buffer using the emulsiflex homogenisator system (Avestin, Germany). Cell debris were removed from the lysate by centrifugation (6000 × g, 5 min, 4°C) and the supernatant was ultracentrifuged (100.000 × g, 30 min 4°C). Strep-tagged proteins were then purified from the cleared lysates using affinity chromatography and Strep-Tactin® sepharose (IBA Lifesciences, Germany). Elution fractions containing SecA2-Strep were stored at -20°C. Purified SecA2-Strep was used for immunization of one rabbit to generate a polyclonal antiserum recognizing SecA2 (Seqlab, Germany).
Isolation of Cellular Proteins and Protein Detection Techniques
Cells were harvested by centrifugation (6000 × g, 5 min, 4°C) and washed once with ZAP buffer. The cell pellet was resuspended in 1 ml ZAP buffer also containing 1 mM PMSF and disrupted by sonication. Cell debris were removed by centrifugation and the resulting supernatant was considered as total cellular protein extract. Aliquots of these samples were separated by standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE). Gels were either stained using the colloidal coomassie agent Roti®-Blue (Roth, Germany) or transferred onto positively charged polyvinylidene fluoride (PVDF) membranes using a semi-dry transfer unit. Proteins of interest were immune-stained using polyclonal rabbit antisera recognizing DivIVA (Marston et al., 1998) or SecA2 (this work) as the primary antibody and anti-rabbit immunoglobuline G conjugated to horseradish peroxidase as the secondary one. The ECL chemiluminescence detection system (Thermo Scientific) was used for detection of the peroxidase conjugates on the PVDF membrane in a chemiluminescence imager (Vilber Lourmat). Protein identification by mass spectroscopy was performed as described recently (Halbedel et al., 2014).
RNA Isolation and Northern Blotting
Bacteria were cultivated in BHI broth at 37°C and total RNA was extracted from cells obtained from 5 ml cultures grown to an optical density of 0.8 (λ = 600 nm) using the RNeasy Mini Kit (Qiagen). Northern blot analysis was performed as described by Wetzstein et al. (1992). The lieA-specific digoxigenin-labeled RNA probe was generated by in vitro transcription with T7 RNA polymerase (Roche Diagnostics) and a lieA-internal PCR fragment generated with the primer pair SHW627/SHW630 (the reverse primer introduced the T7 RNA polymerase recognition site). In vitro transcription was carried out using the DIG RNA labeling Kit (Roche). Hybridisation and signal detection were performed using the DIG wash and block buffer set, an anti-digoxigenin antibody conjugated to alkaline phosphatase and the CDP-Star reagent (all chemicals obtained from Roche) according to the manufacturer’s instructions.
Swarming Assays
LB soft agar plates containing 0.3% agar were stab inoculated with the respective strains of L. monocytogenes. Plates were incubated at 30°C for 24 h to observe swarming halos. In order to observe concentric ring formation in swarming halos associated with the circadian swarming rhythm, the plates were kept for an additional 6 days at room temperature and thus exposed to a natural course of daylight.
Drug Susceptibility Assays
Determination of minimal inhibitory concentrations was performed as described earlier (Rismondo et al., 2015). MIC test strips with the following concentration gradients were purchased from Bestbiondx (Germany): Tetracycline (0.016–256 μg/ml), gentamicin (0.016–256 μg/ml), chloramphenicol (0.016–256 μg/ml) and vancomycin (0.016–256 μg/ml). Filter disk assays were used for susceptibility tests against ethidium bromide and Hoechst 33342. Sterile whatman paper disks (Ø 6 mm) were soaked with 27 μl of a 1 mg/ml ethidium bromide solution or a solution containing 10 mg/ml Hoechst 33342 and laid on top of BHI agar plates, which had been swab-inoculated with a re-suspension of the L. monocytogenes strains to be tested. Zones of growth inhibition became visible after overnight incubation at 37°C.
Ethidium Bromide Uptake Assay
Ethidium bromide was used as model substrate for the LieAB transporter. Measurement of ethidium bromide uptake was performed as described elsewhere with minor modifications (Neyfakh et al., 1991). Briefly, L. monocytogenes strains were grown in BHI broth at 37°C until an OD600 of about 0.5 was reached. Cell were washed once in phosphate buffered saline (PBS), resuspendend in PBS to a final OD600 of 0.5 and 100 μl aliquots were pipetted into the wells of a black 96-well plate. Ethidium bromide was added to a final concentration of 5 μg/ml and fluorescence was measured over time in a Tecan infinite M1000 microplate reader (λexcitation = 500 nm, λemission = 580 nm).
Microscopy
Samples (0.4 μl) from exponentially growing cultures were spotted onto microscope slides, covered with a thin agarose film (1.5% in distilled water). Samples were air-dried, covered with a cover lid and subjected to phase contrast or fluorescence microscopy. Staining of membranes was performed by addition of nile red (1 μg/ml final concentration) to 100 μl of culture and shaking for 20 min at 37°C. Images were taken with a Nikon Eclipse Ti microscope coupled to a Nikon DS-MBWc CCD camera. Bacterial colonies were imaged with an inverse Nikon Diaphot 300 microscope connected to a Digital Sight DS-Fi1 camera. Images were processed using the NIS elements AR software package (Nikon).
Cell Culture Techniques
Experimental infections of HeLa cells and J774 mouse macrophages were performed as described earlier (Halbedel et al., 2012, 2014).
Results
Identification of the lftRS Genes
From a transposon mutagenesis screen, which initially aimed at the identification of mutants with a rough colony phenotype, we identified one rough isolate without transposon insertions in either secA2 or divIVA, the deletion of which typically results in the formation of rough colonies (Lenz and Portnoy, 2002; Halbedel et al., 2012). This clone was designated LMKK18 and inverse PCR revealed that the transposon had integrated into the 29th amino acid codon of the lftS gene (lmo0720) encoding a protein of unknown function with the uncharacterized DUF1048 domain (Figure 1A). The lftS ORF is part of a bicistronic operon (Toledo-Arana et al., 2009) also containing the lftR gene (lmo0719) that encodes a putative transcriptional regulator. DNA sequencing revealed that the divIVA gene of strain LMKK18 had the wild type sequence, whereas a point mutation in its secA2 gene changed the glycine at position 484 into glutamate (Supplementary Figure S1A). Strain LMKK18 showed a slight but significant growth defect in BHI broth at 37°C (Figure 1B) and grew as long cell chains as it is characteristically observed with ΔsecA2 mutant strains (Figure 1C; Lenz and Portnoy, 2002; Kaval et al., 2014).
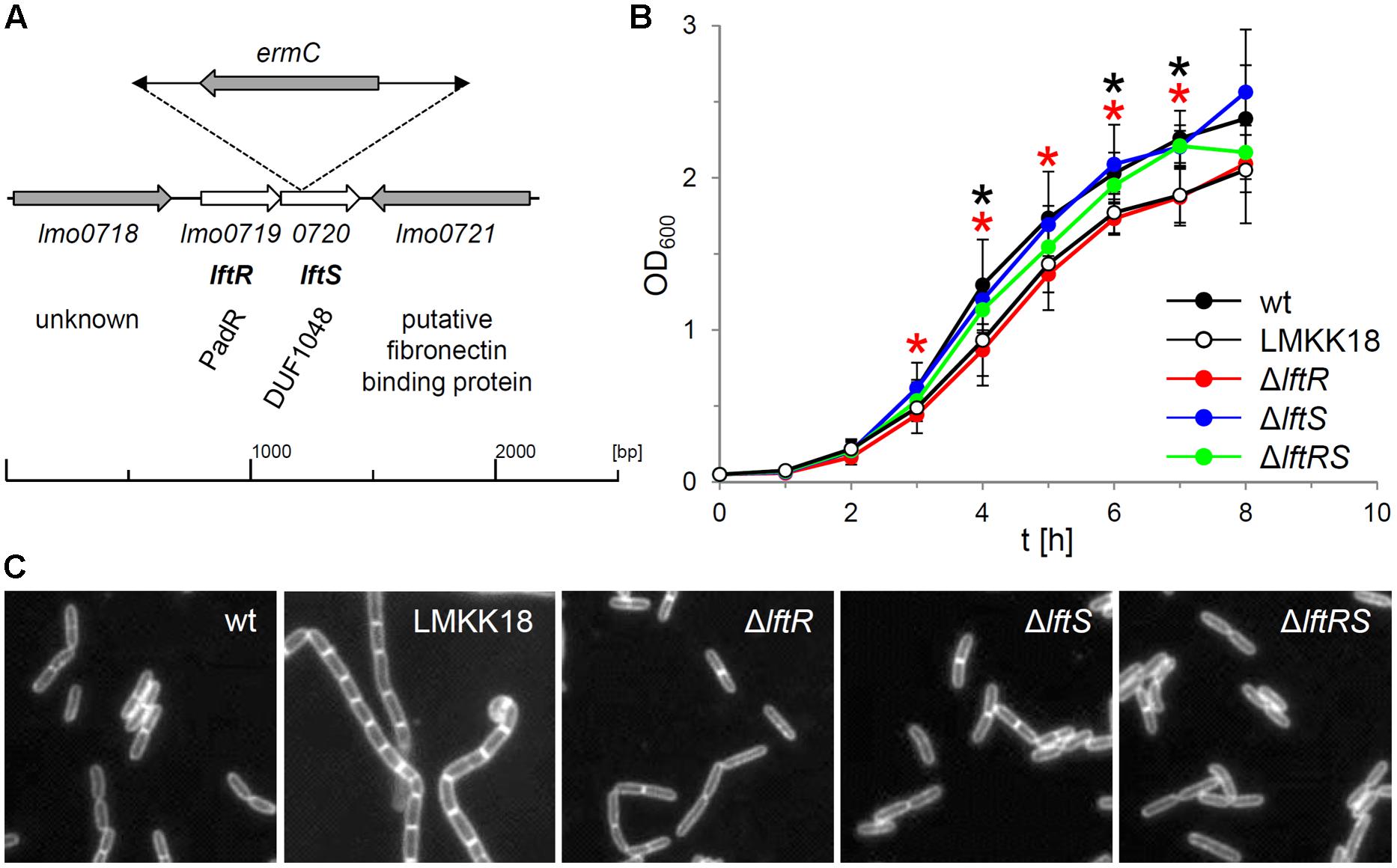
FIGURE 1. Growth phenotypes of lftRS mutant strains. (A) Schematic representation of the site of transposon insertion in the genome of strain LMKK18. Please note that the ermC gene located in the mini-transposon is oriented in the direction opposite to the lmo0719–0720 operon. (B) Growth of strains EGD-e (wt), LMKK18 (rough isolate, lftS::Tn secA2G484E), LMKK42 (ΔlftR), LMKK26 (ΔlftS) and LMKK31 (ΔlftRS) in BHI broth at 37°C. Values are averages from 5 repetitions. SD are indicated. Significance levels (P < 0.05) are indicated for strains LMKK18 (lftS::Tn secA2G484E, black asterisks) and LMKK42 (ΔlftR, red asterisks). (C) Micrographs showing nile red stained cells of the same set of strains cultivated in BHI broth at 37°C up to mid-logarithmic growth phase.
The G484 residue of L. monocytogenes SecA2 corresponds to G490 in Bacillus subtilis SecA and lies within the IRA2 domain that modulates ATP binding (Sianidis et al., 2001; Supplementary Figure S1B). This residue makes contacts with the α-phosphate of an ADP molecule (Hunt et al., 2002; Osborne et al., 2004; Supplementary Figure S1B) and should affect binding of ATP to SecA2, when mutated. In order to test this hypothesis, a complementation assay was established to analyze activity of secA2 mutant alleles. While the ΔsecA2 mutant grew as long chains of cells and formed rough colonies on BHI agar plates, the complemented strain (LMKK27) behaved like wild type (Supplementary Figures S1C,D). In contrast, strain LMKK28, expressing the mutated SecA2G484E protein, formed cell chains and rough colonies that were indifferent from the ΔsecA2 mutant (Supplementary Figures S1C,D) although SecA2G484E was expressed (Supplementary Figure S1E). Therefore, the presence of the secA2G484E mutation in strain LMKK18 explains the rough phenotype of this transposant.
Phenotype of lftRS Mutant Strains
We reasoned that the secA2G484E mutation might have arisen as a suppressor mutation in response to the transposon insertion in the lftS gene itself. In order to address this question, mutant strains lacking the lftR (LMKK42), the lftS (LMKK26) or the whole lftRS operon (LMKK31) were generated. These mutants were grown in BHI broth and analyzed by microscopy. This showed that all three mutant strains grew as wild type-like rods in contrast to strain LMKK18, which formed a chain of cells (Figure 1C). Cell length measurements of roughly 300 cells per strain furthermore demonstrated that neither LMKK18 nor any of the lftRS deletion mutants suffered from defective cell division (data not shown). During these experiments we repeatedly observed that strain LMKK42 (ΔlftR) showed the same slight but significant growth defect as the rough transposon mutant LMKK18 (Figure 1B), while the ΔlftS mutant (LMKK26) grew as fast as the wild type strain EGD-e and strain LMKK31, lacking the entire lftRS operon, grew with an intermediate kinetic (Figure 1B). Thus, growth of the rough transposon isolate LMKK18 is apparently identical to that of the ΔlftR deletion mutant. This indicates that the Tn insertion in lftS might exert a deleterious polar effect on expression of the lftR gene. In line with the absence of a rough phenotype in strains lacking lftR, lftS or both genes, no effects on expression of SecA2 or DivIVA were observed in these mutants (data not shown). In order to test the possibility whether the transposon insertion in the lftS gene of strain LMKK18 would generate a phenotype different from those observed with the clean deletion mutants, we replaced the lftS gene of strain EGD-e by the lftS::Tn allele of strain LMKK18. The resulting strain (LMKK64) formed smooth colonies (Supplementary Figure S2A). This shows that the lftRS genes are not involved in expression of the rough phenotype. Moreover, none of the constructed lftRS mutants was prone to spontaneous transition to the rough phenotype, also not during prolonged cultivation on BHI agar plates. Thus, we assumed that the secA2G484E mutation in LMKK18 occurred just by chance.
Daylight-Dependent Control of Swarming Motility Requires LftR
Mutants exhibiting the rough phenotype are unable to swarm on soft agar plates (Halbedel et al., 2012). Therefore, the strains lacking lftR, lftS, or lftRS were tested in a swarming assay. This revealed the absence of statistically significant differences in the formation of swarming halos obtained with these strains (Figure 2A). However, we observed that strain LMKK42 (ΔlftR), was almost unable to form the typical pattern of concentric rings, when cells were exposed to diurnal changes in daylight on swarming plates over several days at ambient room temperature (Figure 2B). These concentric rings correspond to alternating zones of opaque and translucent colony material, which are linked to the production of extracellular polymeric substance in opaque but not in translucent regions of the swarming halo (Tiensuu et al., 2013). This peculiar swarming phenotype was recently shown to be dependent on the coordinated action of the blue light receptor Lmo0799, on proteins controlling activation of the alternative sigma factor σB, as well as on several other proteins from different functional categories (Tiensuu et al., 2013). In good agreement with a previous report (Tiensuu et al., 2013), formation of these concentric swarming halos is only established under ambient light conditions and cannot be observed when the experiment is repeated in the dark or under constant light exposure (Supplementary Figures S3A,B). Deletion of lftS had no effect on this phenotype (Figure 2B), and likewise, the reconstituted lftS::Tn strain LMKK64 formed concentric swarming rings similar to the wild type (Supplementary Figures S2B,C). Interestingly, deletion of the lftS gene in the ΔlftR background restored concentric ring formation in the ΔlftRS double mutant strain LMKK31 (Figure 2B). That deletion of lftS suppresses a phenotype associated with deletion of the lftR gene again indicates that both genes must be functionally linked.
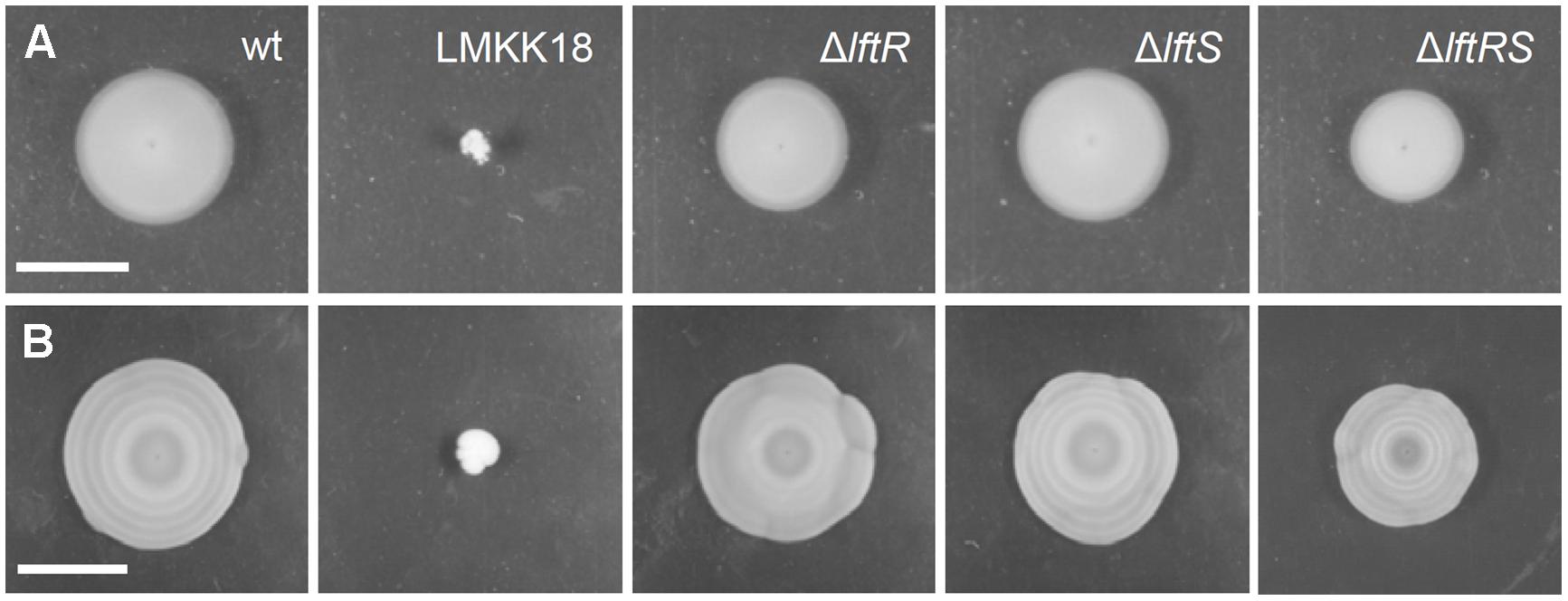
FIGURE 2. Swarming motility of mutants lacking the lftRS genes. (A,B) Swarming motility assay for EGD-e (wt), LMKK18 (lftS::Tn secA2G484E), LMKK42 (ΔlftR), LMKK26 (ΔlftS) and LMKK31 (ΔlftRS). (A) Soft LB agar plates were stab inoculated with the above strains from glycerol stocks and incubated at 30°C for 24 h and then documented. (B) The same plates photographed after 6 days of growth at room temperature under ambient light exposure. Scale bar in (A) indicates 1 cm, while that in (B) indicates 4 cm.
LftR Inhibits Expression of the LieAB Multidrug Resistance Transporter
The gene product of the lftR gene is annotated as a PadR-like transcriptional regulator and shares varying degrees of identity with recently crystallized members of this protein family, such as the Bce3449 protein of B. cereus (69% identity) or LmrR of Lactococcus lactis (28% identity) (Supplementary Figure S4A; Madoori et al., 2009; Fibriansah et al., 2012). We wondered as to whether LftR would act as a transcriptional regulator affecting gene expression and separated total cellular protein extracts of strains lacking lftR, lftS or both by conventional SDS PAGE, in order to identify de-repressed genes. This uncovered one clearly overexpressed band in the ΔlftR mutant (Figure 3A). Mass spectroscopy revealed that this band corresponds to the daunorubicin resistance ATP-binding protein Lmo0979 (which we have renamed here as LieA, Table 3). The membrane component of this putative multidrug transporter is encoded by the lmo0980 (lieB) gene, which is located in an operon together with lieA. LieA did not accumulate in the ΔlftS mutant, indicating that its up-regulation was an LftR-specific effect. In good agreement with this, LieA was still overexpressed in the ΔlftRS strain (Figure 3A). This suggests that expression of LieA is solely repressed by LftR – whether LftS is present or not. To confirm overexpression of LieA upon deletion of lftR, the lieAB operon was deleted from the ΔlftR mutant, resulting in strain LMS169 (ΔlftRΔlieAB). This strain, in fact, did not show overexpression of LieA anymore (Supplementary Figure S5). This demonstrated that identification of the overexpressed protein band as LieA was correct. Importantly, expression of LftR in the ΔlftR background (strain LMKK62, IlftR, I – designates inducible alleles) from an IPTG-dependent promoter cured the LieA overexpression effect in an inducer-dependent manner (Figure 3B).
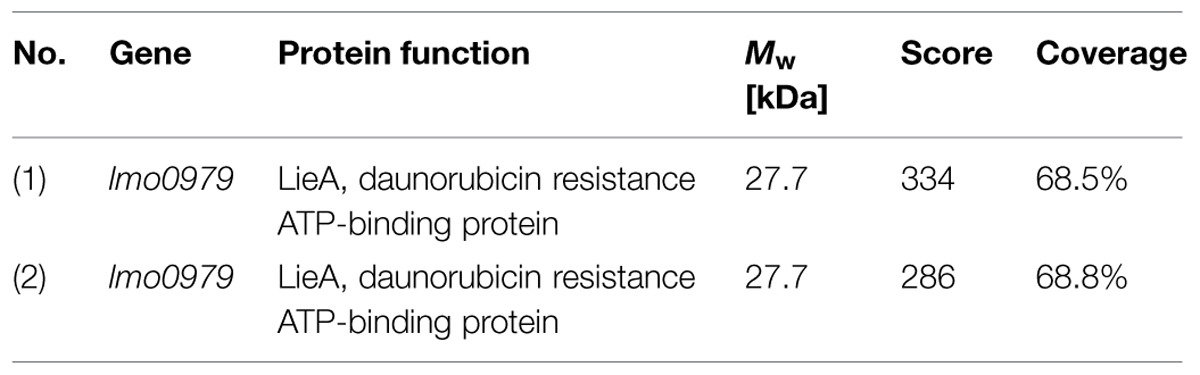
TABLE 3. Protein identification results.
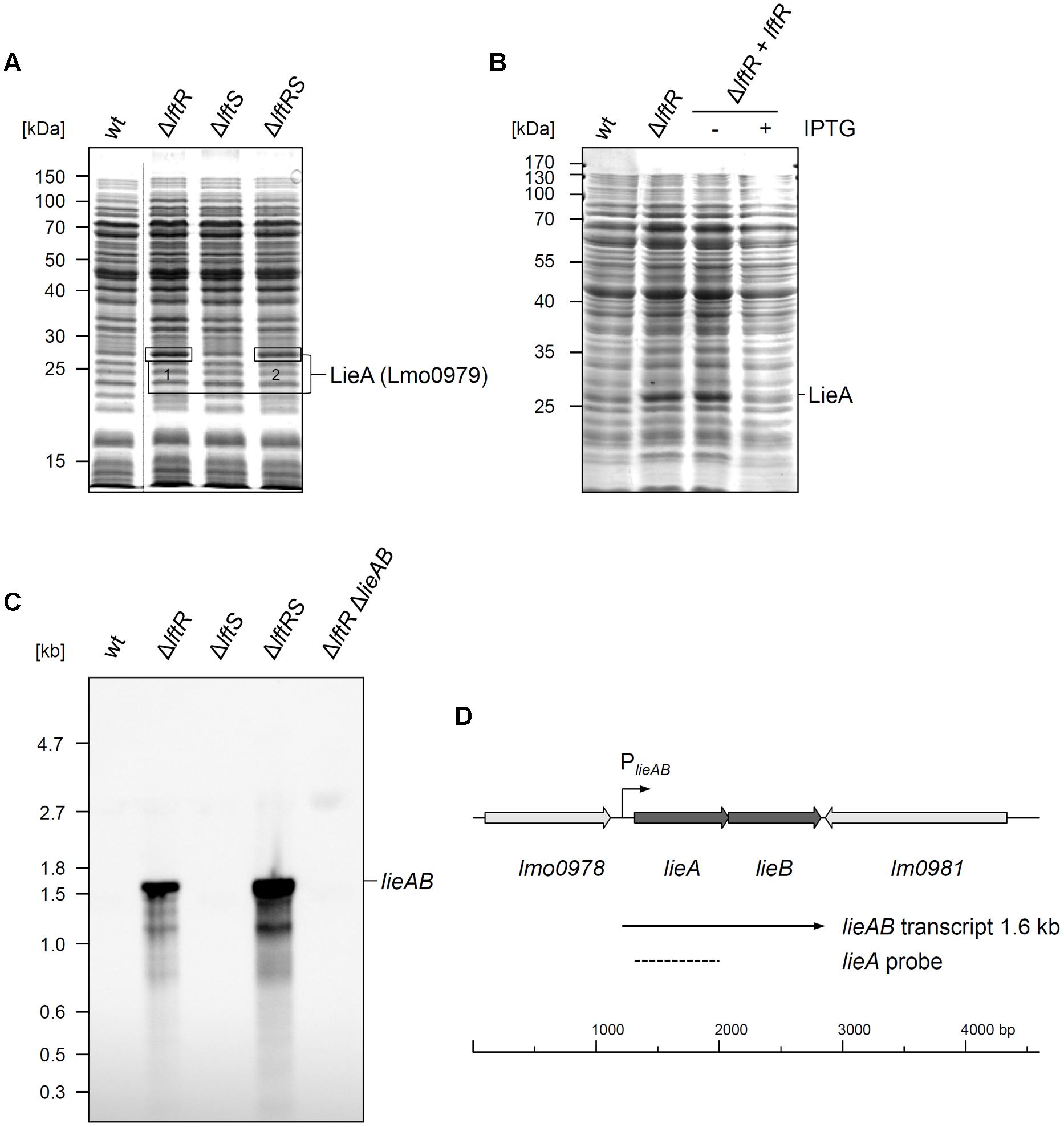
FIGURE 3. Derepression of lieAB in lftRS mutant strains. (A) Total cellular proteins were isolated from strains EGD-e (wt), LMKK42 (ΔlftR), LMKK26 (ΔlftS) and LMKK31 (ΔlftRS) that had been grown to OD600 = 1.0 in BHI broth at 37°C. Proteins were separated by SDS-PAGE and protein bands indicated by boxes were cut from the gel and identified by mass spectrometry. MS identification results are summarized in Table 3. The dashed line refers to non-relevant lanes, which were removed from the image. (B) Complementation of the LieA overexpression phenotype of the ΔlftR mutant by reintroduction of LftR. Total cellular proteins of strains EGD-e (wt), LMKK42 (ΔlftR) and LMKK62 (IlftR, I – designates IPTG-dependent alleles), grown in BHI (±1 mM IPTG), were separated by SDS-PAGE and the position of LieA is indicated. (C) Northern blot showing lieAB transcript levels in the same set of strains as in (A). Additionally, strain LMS169 (ΔlftR ΔlieAB) was included as control. Total RNA was isolated from logarithmically growing cells, separated in an agarose gel and probed with a DIG-labeled ribo-probe specific for lieA after transfer onto a nylon membrane. (D) Transcriptional organization of the L. monocytogenes lieAB locus. Positions of the PlieAB promoter, the detected lieAB transcript and the lieA probe are indicated.
LieA accumulation in the ΔlftR mutant is likely explained by repression of lieAB transcription through LftR. To test this hypothesis, total RNA preparations of L. monocytogenes EGD-e and the ΔlftR mutant were probed with a lieA-specific riboprobe in a Northern blot experiment. While no transcript signal at all was obtained with wild type RNA, a strong signal corresponding to a single mRNA with a size of roughly 1.6 kb was detected in RNA extracts of the ΔlftR mutant (Figure 3C). As expected, lieAB was not transcribed in the ΔlftS mutant, but strongly derepressed in the ΔlftRS strain (Figure 3C). This clearly demonstrates transcriptional derepression of lieAB in the absence of LftR. Furthermore, the observed transcript size of 1.6 kb would be in good agreement with a bicistronic lieAB mRNA (Figure 3D).
Repression of lieAB Expression through LftR is Crucial for Host Cell Invasion
The contribution of LftR to virulence of L. monocytogenes was tested in an in vitro infection assay using HeLa monolayers as host cells. This indicated that the absence of lftR, but not that of lftS affected invasion into non-phagocytic cells (Figure 4A, Supplementary Figure S2D). Interestingly, deletion of lftS in the lftR mutant suppressed this invasion defect, at least partially. Once inside the HeLa cells, all strains multiplied with an identical rate (Figure 4A). Apparently, lftR is critical for invasion into non-phagocytic human host cells but not for multiplication inside eukaryotic cells. This conclusion was further substantiated by the observation that all three strains grew similarly inside mouse macrophages (Figure 4B). We wondered as to whether the derepression of the putative multidrug resistance transporter encoded by lieAB operon might cause this effect, or whether other LftR-dependent factors affect host cell invasion of L. monocytogenes. This question was addressed using wild type, ΔlftR and ΔlftRS strains, in which the entire lieAB operon had been removed. Invasion efficiency of the resulting strains LMS160 (ΔlieAB), LMS169 (ΔlftR ΔlieAB) and LMS168 (ΔlftRSΔlieAB) was then tested in a separate HeLa cell infection experiment and compared to that of strains EGD-e (wt), LMKK42 (ΔlftR), LMKK26 (ΔlftS), and LMKK31 (ΔlftRS). In good agreement with the previous result, strains lacking lftR and lftRS showed reduced invasion rates, while invasion efficiency of the ΔlieAB mutant was unaffected (Figure 4C). Remarkably, the invasion defects associated with lack of lftR or lftRS genes were suppressed, when the lieAB operon was deleted in these strains (Figure 4C). This demonstrates that lftR contributes to efficient invasion of L. monocyctogenes into HeLa cells by preventing overexpression of the putative multidrug resistance transporter encoded by the lieAB genes. In contrast to this, introduction of the ΔlieAB deletion into the ΔlftR background did not restore wild type-like formation of concentric swarming rings in the resulting triple mutant (Supplementary Figure S3A).
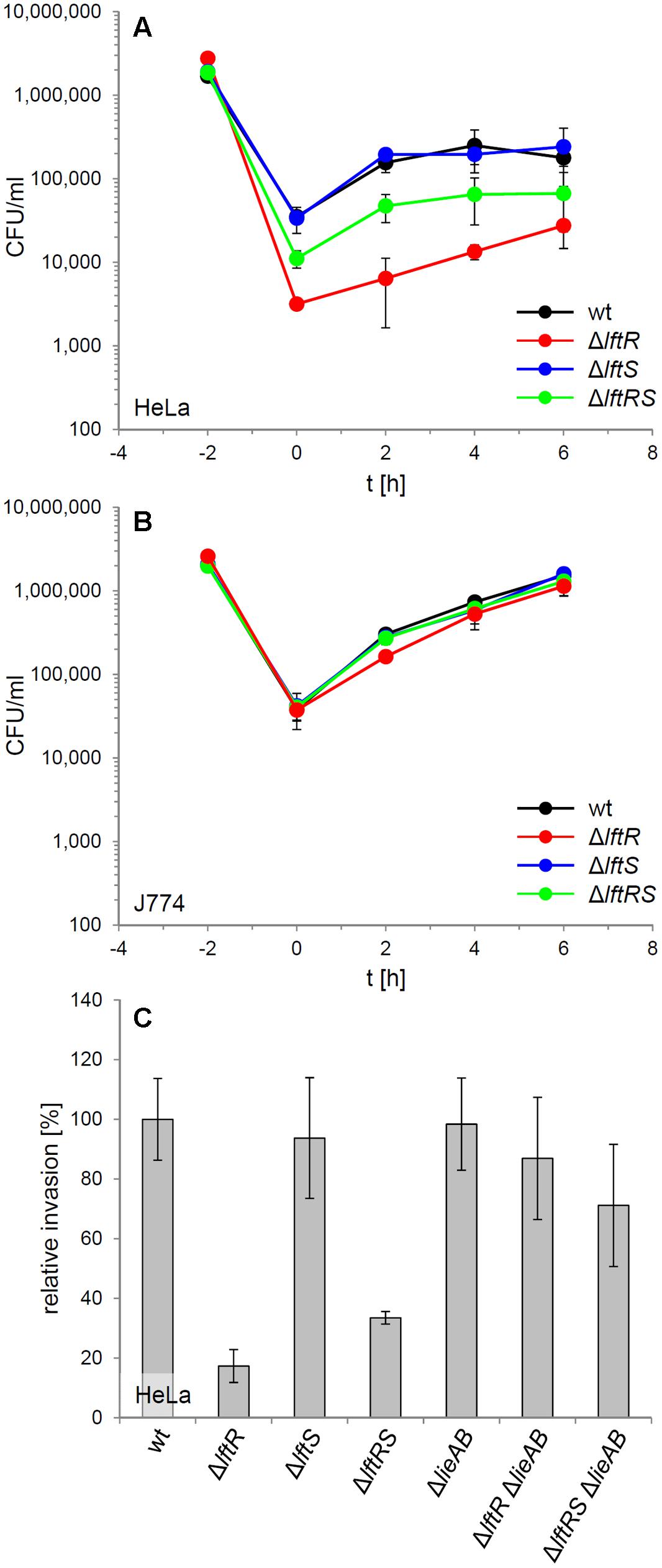
FIGURE 4. Attenuation of the lftRS mutants in in vitro infection experiments. (A) Intracellular multiplication of strains EGD-e (wt), LMKK42 (ΔlftR), LMKK26 (ΔlftS), and LMKK31 (ΔlftRS) was determined in an in vitro infection assay using HeLa cells. The bacterial inoculum was added to the HeLa cells at the time point -2 h, followed by infection for 1 h and killing of extracellular bacteria with gentamycin, in another 1 h incubation step. The number of intracellular multiplying bacteria (CFU/ml) was determined following sampling at 2 h intervals. (B) Intracellular growth of the same set of strains in J774 mouse macrophages. (C) Invasion of L. monocytogenes strains strains EGD-e (wt), LMKK42 (ΔlftR), LMKK26 (ΔlftS), LMKK31 (ΔlftRS), LMS160 (ΔlieAB), LMS169 (ΔlftR ΔlieAB), and LMS168 (ΔlftRSΔlieAB) into HeLa cells. Please note that the experiment shown in (A) was performed with bacteria grown to stationary phase, whereas the invasion experiment in (B) with bacteria from mid-logarithmic growth phase. SD were calculated from experiments performed in triplicate.
Contribution of the LieAB Transporter to Multidrug Resistance
The genes constituting the lieAB operon are annotated to encode an ATP-binding subunit and a transmembrane component, respectively, of a daunorubicin (=daunomycin) resistance ABC transporter. This designation was adapted from Lactococcus lactis LmrCD, which in fact contributes to resistance against this and a few other compounds, including Hoechst 33342, rhodamine and ethidium bromide (Lubelski et al., 2006). We hypothesized that LftR could contribute to multidrug resistance by controlling expression of the lieAB operon and tested this hypothesis by determination of the minimal inhibitory concentrations of several antibiotics with cytoplasmic targets (tetracycline, gentamicin, and chloramphenicol) and vancomycin as a control against ΔlftR, ΔlftS, ΔlftRS, and ΔlieAB mutant strains. This revealed the absence of any relevant changes in the susceptibilities of all tested strains against these antibiotics (Supplementary Table S1), indicating that the LieAB transporter does not mediate excretion of these compounds out of the cell. The resistance of the same set of strains against ethidium bromide was tested in a simple disk diffusion assay. A clear increase of the inhibition zones around the compound-soaked filter disks indicated an increased susceptibility of the ΔlftR and ΔlftRS strains against ethidium bromide (Figures 5A,B). In contrast, the ΔlftS mutant behaved like wild type, indicating that this was a mere LftR-dependent effect. However, deletion of the lieAB operon in the ΔlftR and ΔlftRS backgrounds corrected their increased susceptibilities against ethidium bromide back to normal wild type levels (Figures 5A,B). Moreover, the increased sensitivity of the ΔlftR mutant against ethidium bromide was also complemented back to normal wild type levels, when an ectopic allele of lftR was expressed in the ΔlftR background (strain LMKK62, Figure 5C). The susceptibility of the same set of strains against Hoechst 33342 was tested in a similar experimental set-up, but none of the strains was affected (Supplementary Figure S6), indicating specificity of the ethidium bromide effect.
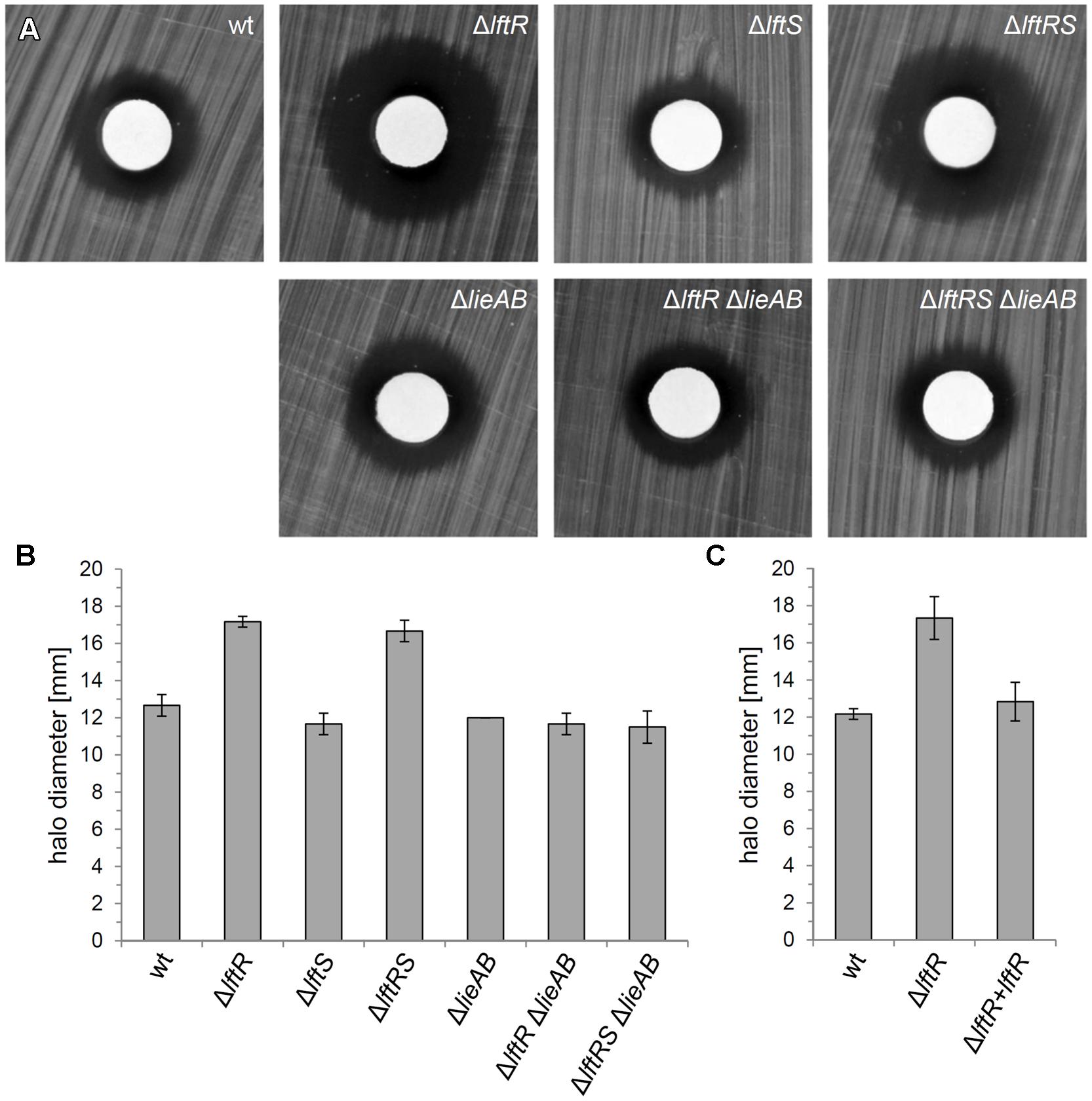
FIGURE 5. Reduced resistance of the L. monocytogenesΔlftR mutant against ethidium bromide. (A) Filter disks soaked with 1 mg/ml ethidium bromide solution were put on top of BHI agar plates, which had been inoculated with strains EGD-e (wt), LMKK42 (ΔlftR), LMKK26 (ΔlftS), LMKK31 (ΔlftRS), LMS160 (ΔlieAB), LMS169 (ΔlftR ΔlieAB), and LMS168 (ΔlftRSΔlieAB) and incubated overnight at 37°C. (B) Quantification of the experiment shown in (A). Diameters of inhibition zones were measured from three independent repetitions and average values and SD are shown. (C) Complementation of the ΔlftR phenotype in strain LMKK62. Ethidium bromide sensitivity of strains EGD-e (wt), LMKK42 (ΔlftR), and LMKK62 (ΔlftR + lftR) was determined and quantified as described for the experiment shown in (A,B).
The increased lieAB-dependent ethidium bromide susceptibility of the ΔlftR mutant suggested that the LieAB transporter possibly facilitates uptake of ethidium bromide into the cell, where it could exert its growth inhibiting effects. In order to test this possibility, uptake of ethidium bromide by the ΔlftR mutant cells was recorded by fluorescence measurements over time. This revealed a dramatic increase in ethidium bromide influx into ΔlftR cells as compared to the wild type strain (Figure 6A). Again, this effect was dependent on the presence of the lieAB operon, since its deletion corrected the increased ethidium bromide influx of the ΔlftR strain back to the normal wild type situation. This result demonstrates that LieAB acts as an importer, at least when ethidium bromide is considered as a substrate. In good agreement with this conclusion, we observed that artificial overexpression of the lieAB operon from an ectopic site was sufficient to increase the susceptibility of L. monocytogenes against ethidium bromide (Figure 6B). When a mutation was introduced into the ATP-binding site of the ectopically expressed lieA, changing the conserved lysine-44 residue into a glutamate, ethidium bromide susceptibility was corrected back, albeit not entirely, to the wild type level (Figure 6B). This shows that ethidium bromide influx through LieAB is an energy-dependent process, at least partially.
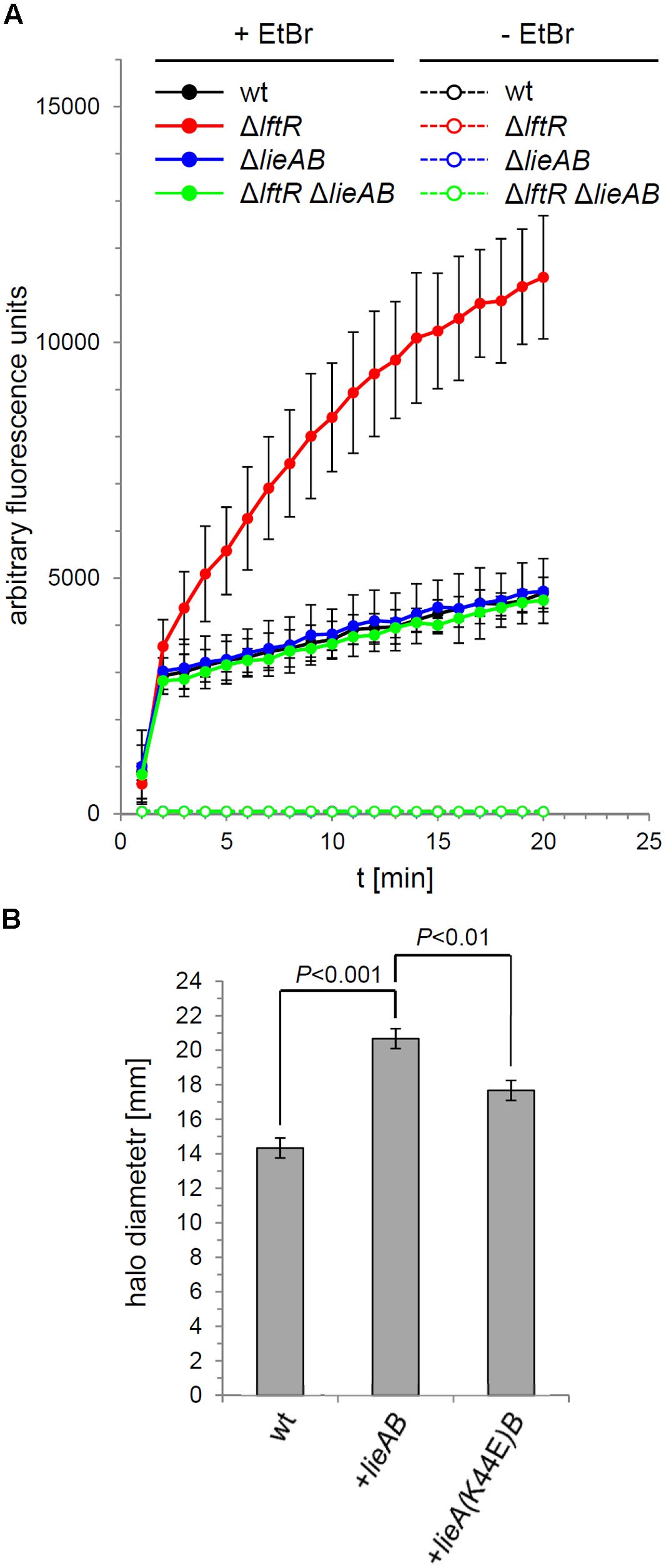
FIGURE 6. Ethidium bromide uptake through LieAB. (A) Intracellular accumulation of ethidium bromide in cells of strains EGD-e (wt), LMKK42 (ΔlftR), LMS160 (ΔlieAB) and LMS169 (ΔlftR ΔlieAB) was recorded by measurement of fluorescence over time (see Materials and Methods for details). As a control, the same experiment was performed in the absence of ethidium bromide. Average values and SD were calculated from an experiment performed in triplicate. (B) Energy dependence of ethidium bromide uptake by LieAB. Disk diffusion assays were used to test the susceptibility of strains EGD-e (wt), LMNT1 (+lieAB), and LMNT2 [+lieA(K44E)B] against ethidium bromide (n = 3). Significance levels are indicated.
Discussion
Invasion of L. monocytogenes into eukaryotic host cells is a multifactorial process that requires the concerted action of different attachment and internalization factors, together with the smooth functioning of background house-keeping cellular processes. With respect to LftR, we have identified a so far uncharacterized regulatory protein from the second class of invasion determinants that can disturb this process when deregulated. LftR belongs to the family of PadR-like transcriptional regulators and L. monocytogenes EGD-e encodes three more such proteins: LadR (Lmo1408, 26% identity), which represses transcription of the lmo1409 gene, encoding the multidrug efflux pump of the major facilitator-type MdrL, and the uncharacterized Lmo0599 protein (31% identity) (Mata et al., 2000; Huillet et al., 2006). LstR (encoded by the lmo0422 gene) is involved in heat shock response and was also annotated as a PadR-like transcriptional regulator of L. monocytogenes (Supplementary Figure S4B; Zhang et al., 2005).
PadR was first described in Lactobacillus plantarum and Pediococcus pentosaceus as a repressor of the phenolic acid decarboxylase gene (padA), which contributes to detoxification of phenolic acids (Barthelmebs et al., 2000; Gury et al., 2004). In a similar manner, B. subtilis PadR represses expression of the phenol acid decarboxylase padC gene (Tran et al., 2008). However, the best studied PadR homolog is LmrR from Lactococcus lactis, which binds to the promoter regions of its own gene and of the lmrCD operon, encoding a heterodimeric multidrug ABC transporter, to repress their transcription (Agustiandari et al., 2008). Crystallography showed that LmrR contains a helix-turn-helix motif for DNA binding and a C-terminal helix for dimerization (Supplementary Figure S4A; Madoori et al., 2009). LmrR binds to an inverted repeat in the promoter regions of the lmrR and lmrCD genes with the consensus sequence ATGT-N8-ACAT. Identical binding motifs were also described for PadR from P. pentosaceus and L. plantarum as well as for L. monocytogenes LadR (Gury et al., 2004; Huillet et al., 2006; Agustiandari et al., 2011). Binding of compounds like Hoechst 33342, daunomycin or ethidium is thought to induce conformational changes in LmrR, which prevent promoter recognition and thus lead to induction of the lmrR and lmrCD genes (Madoori et al., 2009; Agustiandari et al., 2011; Takeuchi et al., 2014). Most likely, LftR senses similar compounds.
LmrR does not seem to be the real LftR equivalent of Lactococcus lactis, since a higher degree of identity to LftR is observed with another, yet uncharacterized Lactococcus lactis transcriptional regulator (encoded by the llmg_2339 gene). Likewise, the listerial lieAB operon is only similar to but does not correspond to Lactococcus lactis lmrCD, which in turn shares the highest similarity with the lmo2751–2752 genes, coding for another putative multidrug ABC transporter of L. monocytogenes. Thus, the lftR lieAB genes are similar but not identical with the lmrR lmrCD module of Lactococcus lactis.
It is not clear, whether LftR represses transcription of lieAB genes directly, or whether this is an indirect effect. Sequence searches have not identified the typical ATGT-N8-ACAT PadR binding motif in the lmo0979–0980 promoter region or in front of the lftR gene. Also, a comparison of both promoters did not uncover another putative LftR binding site. Rather, an ideal ATGT-N8-ACAT motif is present in the PmdrL promoter, where it represents the binding site for LadR (Huillet et al., 2006). There is some degree of sequence variation in the DNA binding helices of the PadR proteins (Supplementary Figures S4A,B), so the DNA binding site for LftR might differ from the canonical PadR binding site. Alternatively, up-regulation of lieAB expression in the absence of LftR could be an indirect effect. We have looked for PadR binding sites in the whole L. monocytogenes genome and identified 68 matches in total, out of which 16 lie within a 400 bp range upstream of genes. However, these putative PadR sites overlap with potential promoter sequences only in a few cases (Supplementary Table S2). Among these are the promoters of the lmo0018 gene (encoding a β-galactosidase), the promoter of the lmo0748–0751 operon and the PmdrL promoter itself (Supplementary Table S2). The β-galactosidase encoded by lmo0018 is homologous to B. subtilis BglH (62% identity), which is necessary for catabolism of aryl-β-glycosides (Le Coq et al., 1995). In contrast, the lmo0748–0751 operon mainly encodes uncharacterized Listeria-specific genes with an interesting exception: the second gene of the operon (lmo0749) is annotated as a transcriptional regulator of the Cro-family of phage proteins, suggesting that LftR and/or other PadR-type proteins could be part of a hierarchical network of transcriptional regulators.
While PadR-type regulators such as LftR can be found in many bacteria, homologs of LftS are only present in the Listeria and some (but not all) Bacillus, Paenibacillus, Lactococcus, and Enterococcus species, in some Clostridia and a few actinobacterial species. Deletion of lftS alone has not resulted in any conspicuous phenotype in our hands. However, removal of lftS suppressed the swarming defect and (partially) the invasion defect of the ΔlftR mutant. If LftR – like LmrR of Lactococcus lactis – auto-represses transcription of the lftRS operon, then lftS would be de-repressed in the absence of lftR. Consequently, deletion of lftS in a ΔlftR mutant would remove all phenotypes that result from derepression of LftS. With this logic, we observe two classes of effects upon lftR deletion: LftS-dependent phenotypes that result from derepression of LftS. Such an effect is observed with the ΔlftR swarming phenotype, which is corrected back to wild type levels in a ΔlftRS double mutant. In contrast, the increased ethidium bromide sensitivity of the ΔlftR mutant constitutes a second class of effect, which is LftS-independent and merely caused by derepression of the LieAB transporter. The combination of both effects explains the invasion defect of the ΔlftR mutant, which is mainly caused by lieAB derepression, but which is also partially rescued by deletion of lftS. Presently, it is not clear, how LftS could contribute to invasion or concentric swarming ring formation, but the effect it exerts must be LftR-independent.
The pronounced invasion defect that results from the derepression of the lieAB operon in the ΔlftR mutant suggests that substrates of this transporter are detrimental for the infection process. There are 71 more lieA-like genes encoding ATP-binding proteins of ABC transporters present in the L. monocytogenes EGD-e genome, but the membrane component encoded by lieB is unique. The fact that multidrug resistance transporters are critical players in the infection process is not unprecedented in L. monocytogenes. Mdr transporters of the major facilitator type mediate extrusion of cyclic-di-AMP and this contributes to induction of the immune response in macrophages (Crimmins et al., 2008; Woodward et al., 2010; Kaplan Zeevi et al., 2013; Tadmor et al., 2014). Whether the LieAB transporter is a mere importer or whether it also excretes compounds is presently not clear. Its genuine substrate with relevance for invasion is also not known and ethidium bromide clearly has to be considered an artificial substrate. It is tempting to speculate, that uptake of natural LieAB substrates might be advantageous for L. monocytogenes during life as an environmental saprophyte, but disadvantageous during growth in rich media or during infection. Earlier studies have shown that lftR expression is upregulated during stationary phase, suggesting that even LftR levels are subject to control (Chatterjee et al., 2006; Toledo-Arana et al., 2009). Tightly controlled conditional expression of lieAB could adjust the LieAB transporter levels to the respective growth condition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by a DAAD/Siemens grant to KK and a DFG grant (HA 6830/1-1) as well as a grant of the Fonds der chemischen Industrie to SH. The authors would like to thank Steffi Walter (RKI Wernigerode) for help with some experiments and Katharina Riedel (University of Greifswald) for support.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00772
References
Agustiandari, H., Lubelski, J., Van Den Berg Van Saparoea, H. B., Kuipers, O. P., and Driessen, A. J. (2008). LmrR is a transcriptional repressor of expression of the multidrug ABC transporter LmrCD in Lactococcus lactis. J. Bacteriol. 190, 759–763. doi: 10.1128/JB.01151-07
Agustiandari, H., Peeters, E., De Wit, J. G., Charlier, D., and Driessen, A. J. (2011). LmrR-mediated gene regulation of multidrug resistance in Lactococcus lactis. Microbiology 157, 1519–1530. doi: 10.1099/mic.0.048025-0
Arnaud, M., Chastanet, A., and Debarbouille, M. (2004). New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004
Barthelmebs, L., Lecomte, B., Divies, C., and Cavin, J. F. (2000). Inducible metabolism of phenolic acids in Pediococcus pentosaceus is encoded by an autoregulated operon which involves a new class of negative transcriptional regulator. J. Bacteriol. 182, 6724–6731. doi: 10.1128/JB.182.23.6724-6731.2000
Bierne, H., Sabet, C., Personnic, N., and Cossart, P. (2007). Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect. 9, 1156–1166. doi: 10.1016/j.micinf.2007.05.003
Camejo, A., Carvalho, F., Reis, O., Leitao, E., Sousa, S., and Cabanes, D. (2011). The arsenal of virulence factors deployed by Listeria monocytogenes to promote its cell infection cycle. Virulence 2, 379–394. doi: 10.4161/viru.2.5.17703
Cao, M., Bitar, A. P., and Marquis, H. (2007). A mariner-based transposition system for Listeria monocytogenes. Appl. Environ. Microbiol. 73, 2758–2761. doi: 10.1128/AEM.02844-06
Chatterjee, S. S., Hossain, H., Otten, S., Kuenne, C., Kuchmina, K., Machata, S., et al. (2006). Intracellular gene expression profile of Listeria monocytogenes. Infect. Immun. 74, 1323–1338. doi: 10.1128/IAI.74.2.1323-1338.2006
Cossart, P., and Toledo-Arana, A. (2008). Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect. 10, 1041–1050. doi: 10.1016/j.micinf.2008.07.043
Crimmins, G. T., Herskovits, A. A., Rehder, K., Sivick, K. E., Lauer, P., Dubensky, T. W., et al. (2008). Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc. Natl. Acad. Sci. U.S.A. 105, 10191–10196. doi: 10.1073/pnas.0804170105
Dabiri, G. A., Sanger, J. M., Portnoy, D. A., and Southwick, F. S. (1990). Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc. Natl. Acad. Sci. U.S.A. 87, 6068–6072. doi: 10.1073/pnas.87.16.6068
Dramsi, S., Biswas, I., Maguin, E., Braun, L., Mastroeni, P., and Cossart, P. (1995). Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol. Microbiol. 16, 251–261. doi: 10.1111/j.1365-2958.1995.tb02297.x
Fibriansah, G., Kovacs, A. T., Pool, T. J., Boonstra, M., Kuipers, O. P., and Thunnissen, A. M. (2012). Crystal structures of two transcriptional regulators from Bacillus cereus define the conserved structural features of a PadR subfamily. PLoS ONE 7:e48015. doi: 10.1371/journal.pone.0048015
Freitag, N. E., Port, G. C., and Miner, M. D. (2009). Listeria monocytogenes - from saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 7, 623–628. doi: 10.1038/nrmicro2171
Gaillard, J. L., Berche, P., Frehel, C., Gouin, E., and Cossart, P. (1991). Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 65, 1127–1141. doi: 10.1016/0092-8674(91)90009-N
Glaser, P., Frangeul, L., Buchrieser, C., Rusniok, C., Amend, A., Baquero, F., et al. (2001). Comparative genomics of Listeria species. Science 294, 849–852.
Gury, J., Barthelmebs, L., Tran, N. P., Divies, C., and Cavin, J. F. (2004). Cloning, deletion, and characterization of PadR, the transcriptional repressor of the phenolic acid decarboxylase-encoding padA gene of Lactobacillus plantarum. Appl. Environ. Microbiol. 70, 2146–2153. doi: 10.1128/AEM.70.4.2146-2153.2004
Halbedel, S., Hahn, B., Daniel, R. A., and Flieger, A. (2012). DivIVA affects secretion of virulence-related autolysins in Listeria monocytogenes. Mol. Microbiol. 83, 821–839. doi: 10.1111/j.1365-2958.2012.07969.x
Halbedel, S., Reiss, S., Hahn, B., Albrecht, D., Mannala, G. K., Chakraborty, T., et al. (2014). A systematic proteomic analysis of Listeria monocytogenes house-keeping protein secretion systems. Mol. Cell. Proteomics 13, 3063–3081. doi: 10.1074/mcp.M114.041327
Hsieh, W. S., Tsai, L. Y., Jeng, S. F., Hsu, C. H., Lin, H. C., Hsueh, P. R., et al. (2009). Neonatal listeriosis in Taiwan, 1990-2007. Int. J. Infect. Dis. 13, 193–195. doi: 10.1016/j.ijid.2008.06.006
Huillet, E., Velge, P., Vallaeys, T., and Pardon, P. (2006). LadR, a new PadR-related transcriptional regulator from Listeria monocytogenes, negatively regulates the expression of the multidrug efflux pump MdrL. FEMS Microbiol. Lett. 254, 87–94. doi: 10.1111/j.1574-6968.2005.00014.x
Hunt, J. F., Weinkauf, S., Henry, L., Fak, J. J., Mcnicholas, P., Oliver, D. B., et al. (2002). Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science 297, 2018–2026. doi: 10.1126/science.1074424
Ireton, K., Rigano, L. A., Polle, L., and Schubert, W. D. (2014). Molecular mechanism of protrusion formation during cell-to-cell spread of Listeria. Front. Cell. Infect. Microbiol. 4:21. doi: 10.3389/fcimb.2014.00021
Kaplan Zeevi, M., Shafir, N. S., Shaham, S., Friedman, S., Sigal, N., Nir Paz, R., et al. (2013). Listeria monocytogenes multidrug resistance transporters and cyclic di-AMP, which contribute to type I interferon induction, play a role in cell wall stress. J. Bacteriol. 195, 5250–5261. doi: 10.1128/JB.00794-13
Kaval, K. G., Rismondo, J., and Halbedel, S. (2014). A function of DivIVA in Listeria monocytogenes division site selection. Mol. Microbiol. 94, 637–654. doi: 10.1111/mmi.12784
Lambrechts, A., Gevaert, K., Cossart, P., Vandekerckhove, J., and Van Troys, M. (2008). Listeria comet tails: the actin-based motility machinery at work. Trends Cell Biol. 18, 220–227. doi: 10.1016/j.tcb.2008.03.001
Le Coq, D., Lindner, C., Krüger, S., Steinmetz, M., and Stülke, J. (1995). New beta-glucoside (bgl) genes in Bacillus subtilis: the bglP gene product has both transport and regulatory functions similar to those of BglF, its Escherichia coli homolog. J. Bacteriol. 177, 1527–1535.
Lenz, L. L., and Portnoy, D. A. (2002). Identification of a second Listeria secA gene associated with protein secretion and the rough phenotype. Mol. Microbiol. 45, 1043–1056. doi: 10.1046/j.1365-2958.2002.03072.x
Lubelski, J., De Jong, A., Van Merkerk, R., Agustiandari, H., Kuipers, O. P., Kok, J., et al. (2006). LmrCD is a major multidrug resistance transporter in Lactococcus lactis. Mol. Microbiol. 61, 771–781. doi: 10.1111/j.1365-2958.2006.05267.x
Madoori, P. K., Agustiandari, H., Driessen, A. J., and Thunnissen, A. M. (2009). Structure of the transcriptional regulator LmrR and its mechanism of multidrug recognition. EMBO J. 28, 156–166. doi: 10.1038/emboj.2008.263
Marston, A. L., Thomaides, H. B., Edwards, D. H., Sharpe, M. E., and Errington, J. (1998). Polar localization of the MinD protein of Bacillus subtilis and its role in selection of the mid-cell division site. Genes Dev. 12, 3419–3430. doi: 10.1101/gad.12.21.3419
Mata, M. T., Baquero, F., and Perez-Diaz, J. C. (2000). A multidrug efflux transporter in Listeria monocytogenes. FEMS Microbiol. Lett. 187, 185–188. doi: 10.1111/j.1574-6968.2000.tb09158.x
Mengaud, J., Ohayon, H., Gounon, P., Mege, R. M., and Cossart, P. (1996). E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84, 923–932. doi: 10.1016/S0092-8674(00)81070-3
Monk, I. R., Gahan, C. G., and Hill, C. (2008). Tools for functional postgenomic analysis of Listeria monocytogenes. Appl. Environ. Microbiol. 74, 3921–3934. doi: 10.1128/AEM.00314-08
Neyfakh, A. A., Bidnenko, V. E., and Chen, L. B. (1991). Efflux-mediated multidrug resistance in Bacillus subtilis: similarities and dissimilarities with the mammalian system. Proc. Natl. Acad. Sci. U.S.A. 88, 4781–4785. doi: 10.1073/pnas.88.11.4781
Osborne, A. R., Clemons, W. M. Jr., and Rapoport, T. A. (2004). A large conformational change of the translocation ATPase SecA. Proc. Natl. Acad. Sci. U.S.A. 101, 10937–10942. doi: 10.1073/pnas.0401742101
Pizarro-Cerda, J., Kuhbacher, A., and Cossart, P. (2012). Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb. Perspect. Med. 2:a010009. doi: 10.1101/cshperspect.a010009
Rismondo, J., Möller, L., Aldridge, C., Gray, J., Vollmer, W., and Halbedel, S. (2015). Discrete and overlapping functions of peptidoglycan synthases in growth, cell division and virulence of Listeria monocytogenes. Mol. Microbiol. 95, 332–351. doi: 10.1111/mmi.12873
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning : A Laboratory Manual. Cold Spring Harbor, NY.: Cold Spring Harbor Laboratory Press.
Seveau, S., Pizarro-Cerda, J., and Cossart, P. (2007). Molecular mechanisms exploited by Listeria monocytogenes during host cell invasion. Microbes Infect. 9, 1167–1175. doi: 10.1016/j.micinf.2007.05.004
Shen, Y., Naujokas, M., Park, M., and Ireton, K. (2000). InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103, 501–510. doi: 10.1016/S0092-8674(00)00141-0
Sianidis, G., Karamanou, S., Vrontou, E., Boulias, K., Repanas, K., Kyrpides, N., et al. (2001). Cross-talk between catalytic and regulatory elements in a DEAD motor domain is essential for SecA function. EMBO J. 20, 961–970. doi: 10.1093/emboj/20.5.961
Swaminathan, B., and Gerner-Smidt, P. (2007). The epidemiology of human listeriosis. Microbes Infect. 9, 1236–1243. doi: 10.1016/j.micinf.2007.05.011
Tadmor, K., Pozniak, Y., Burg Golani, T., Lobel, L., Brenner, M., Sigal, N., et al. (2014). Listeria monocytogenes MDR transporters are involved in LTA synthesis and triggering of innate immunity during infection. Front. Cell. Infect. Microbiol. 4:16. doi: 10.3389/fcimb.2014.00016
Takeuchi, K., Tokunaga, Y., Imai, M., Takahashi, H., and Shimada, I. (2014). Dynamic multidrug recognition by multidrug transcriptional repressor LmrR. Sci. Rep. 4, 6922. doi: 10.1038/srep06922
Tiensuu, T., Andersson, C., Ryden, P., and Johansson, J. (2013). Cycles of light and dark co-ordinate reversible colony differentiation in Listeria monocytogenes. Mol. Microbiol. 87, 909–924. doi: 10.1111/mmi.12140
Tilney, L. G., and Portnoy, D. A. (1989). Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109, 1597–1608. doi: 10.1083/jcb.109.4.1597
Toledo-Arana, A., Dussurget, O., Nikitas, G., Sesto, N., Guet-Revillet, H., Balestrino, D., et al. (2009). The Listeria transcriptional landscape from saprophytism to virulence. Nature 459, 950–956. doi: 10.1038/nature08080
Tran, N. P., Gury, J., Dartois, V., Nguyen, T. K., Seraut, H., Barthelmebs, L., et al. (2008). Phenolic acid-mediated regulation of the padC gene, encoding the phenolic acid decarboxylase of Bacillus subtilis. J. Bacteriol. 190, 3213–3224. doi: 10.1128/JB.01936-07
Wetzstein, M., Volker, U., Dedio, J., Lobau, S., Zuber, U., Schiesswohl, M., et al. (1992). Cloning, sequencing, and molecular analysis of the dnaK locus from Bacillus subtilis. J. Bacteriol. 174, 3300–3310.
Woodward, J. J., Iavarone, A. T., and Portnoy, D. A. (2010). c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705. doi: 10.1126/science.1189801
Zhang, C., Nietfeldt, J., Zhang, M., and Benson, A. K. (2005). Functional consequences of genome evolution in Listeria monocytogenes: the lmo0423 and lmo0422 genes encode sigmaC and LstR, a lineage II-specific heat shock system. J. Bacteriol. 187, 7243–7253. doi: 10.1128/JB.187.21.7243-7253.2005
Keywords: multi drug resistance, circadian rhythm, swarming, ethidium bromide uptake, repressor proteins
Citation: Kaval KG, Hahn B, Tusamda N, Albrecht D and Halbedel S (2015) The PadR-like transcriptional regulator LftR ensures efficient invasion of Listeria monocytogenes into human host cells. Front. Microbiol. 6:772. doi: 10.3389/fmicb.2015.00772
Received: 16 June 2015; Accepted: 14 July 2015;
Published: 28 July 2015.
Edited by:
Weiwen Zhang, Tianjin University, ChinaReviewed by:
Fabian Moritz Commichau, University of Göttingen, GermanyJan Pané-Farré, Ernst-Moritz-Arndt-Universität Greifswald, Germany
Copyright © 2015 Kaval, Hahn, Tusamda, Albrecht and Halbedel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sven Halbedel, FG11 Division of Enteropathogenic Bacteria and Legionella, Robert Koch Institute, Burgstrasse 37, D-38855 Wernigerode, Germany,aGFsYmVkZWxzQHJraS5kZQ==