 Zulema Gómez-Lunar1
Zulema Gómez-Lunar1 Ismael Hernández-González1María-Dolores Rodríguez-Torres1
Ismael Hernández-González1María-Dolores Rodríguez-Torres1 Valeria Souza2
Valeria Souza2 Gabriela Olmedo-Álvarez1*
Gabriela Olmedo-Álvarez1*- 1Laboratorio de Biología Molecular y Ecología Microbiana, Departamento de Ingeniería Genética, Unidad Irapuato, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Irapuato, Mexico
- 2Laboratorio de Evolución Molecular y Experimental, Departamento de Ecología Evolutiva, Instituto de Ecología, Universidad Nacional Autónoma de México, México City, Mexico
Bacterial genomes undergo numerous events of gene losses and gains that generate genome variability among strains of the same species (microevolution). Our aim was to compare the genomes and relevant phenotypes of three Bacillus coahuilensis strains from two oligotrophic hydrological systems in the Cuatro Ciénegas Basin (México), to unveil the environmental challenges that this species cope with, and the microevolutionary differences in these genotypes. Since the strains were isolated from a low P environment, we placed emphasis on the search of different phosphorus acquisition strategies. The three B. coahuilensis strains exhibited similar numbers of coding DNA sequences, of which 82% (2,893) constituted the core genome, and 18% corresponded to accessory genes. Most of the genes in this last group were associated with mobile genetic elements (MGEs) or were annotated as hypothetical proteins. Ten percent of the pangenome consisted of strain-specific genes. Alignment of the three B. coahuilensis genomes indicated a high level of synteny and revealed the presence of several genomic islands. Unexpectedly, one of these islands contained genes that encode the 2-keto-3-deoxymannooctulosonic acid (Kdo) biosynthesis enzymes, a feature associated to cell walls of Gram-negative bacteria. Some microevolutionary changes were clearly associated with MGEs. Our analysis revealed inconsistencies between phenotype and genotype, which we suggest result from the impossibility to map regulatory features to genome analysis. Experimental results revealed variability in the types and numbers of auxotrophies between the strains that could not consistently be explained by in silico metabolic models. Several intraspecific differences in preferences for carbohydrate and phosphorus utilization were observed. Regarding phosphorus recycling, scavenging, and storage, variations were found between the three genomes. The three strains exhibited differences regarding alkaline phosphatase that revealed that in addition to gene gain and loss, regulation adjustment of gene expression also has contributed to the intraspecific diversity of B. coahuilensis.
Introduction
Bacterial genomes are highly dynamic and continually undergo numerous events of gains and losses of genes, which contribute to variation in genome size, gene content, and order (Koonin and Wolf, 2008). These genome rearrangements provide genome plasticity and generate new genome variants within the same species (Radnedge et al., 2002; Dobrindt et al., 2004). Modifications of gene repertoire arise via different mechanisms. For example, bacterial genomes acquire material from other organisms by horizontal gene transfer (HGT). The HGT can result in the addition of a completely new gene, the replacement of an existing gene, or genetic redundancy if a homologous gene is already present in the recipient genome (xenolog genes; Mira et al., 2002; Abby and Daubin, 2007). Gene duplication is an additional mechanism, which results in the formation of paralog genes and increase in genome size (Mira et al., 2001). The mechanisms that result in gene loss are gene excision and formation of pseudogenes, which arise when mutations (i.e., point mutations, insertions, deletions) accumulate and result in function loss (Ochman and Davalos, 2006). After this, evolutionary forces allow genes to remain or to be lost, and variants occur over time and space that promote individual adaptation to the environment (Ochman, 2001; Ochman and Moran, 2001; Maughan et al., 2006). On the other hand, genome arrangements and nucleotide polymorphisms among strains also confer high genomic diversity upon prokaryotic species (Ochman and Davalos, 2006; Abby and Daubin, 2007).
Evolutionary changes that occur below the level of species are referred to as microevolution (Abby and Daubin, 2007). Comparative genomic studies have shown that even bacterial strains belonging to the same species exhibit considerable variation in gene content as a result of events of DNA gains and losses (Tettelin et al., 2008; Rouli et al., 2015). The pangenome concept arose from the comparison of genomes from a given study group to define the gene repertoire (Tettelin et al., 2008). The core genome concept defines the subgroup of genes shared between all compared genomes (Mira et al., 2010; Vernikos et al., 2015). Genes present in only one or two strains are considered part of the accessory genome, it may some times be located in genomic islands (GI), and the genetic material is usually derived from HGT. The accessory genome may represent a survival strategy when it provides sets of genes from other organisms that may allow the recipient to adapt to new environments (Cohan and Koeppel, 2008). In this sense, recent studies of a bacterial species genomes suggest that the strain-specific genes contribute to individual adaptation to specific environmental conditions (Math et al., 2012; Penn and Jensen, 2012; Tian et al., 2012).
An analysis of the genome of Bacillus coahuilensis m4-4 (Alcaraz et al., 2008), a strain isolated from a desiccation lagoon in the Churince system in the Cuatro Ciénegas Basin (CCB; Cerritos et al., 2008) revealed events of gene gain and loss. Particularly, two adaptations to an oligotrophic environment were found. The first one is the presence of genes that encode sulfolipid biosynthesis (sqd1 and sqdX), which likely increase fitness by reducing phosphorus utilization. The second adaptation is the occurrence of a high number of ATP-binding cassette (ABC) transporter systems for amino acids and oligopeptides, that suggests a high dependency on nutrients synthetized by other organisms in the microbial community (Alcaraz et al., 2008). Another finding was that B. coahuilensis has one of the smallest genomes of the Bacillus genus (3.38 Mpb; Alcaraz et al., 2008). Only three strains of this species have been recovered in the recent years of extensive isolation of thousands of Bacillus sp. from the CCB hydrological systems, and thus only relatively small populations of B. coahuilensis are likely present in the CCB.
The CCB is located in the Chihuahua Desert (Coahuila, North Central México). The basin is surrounded by high mountains (>3,000 m altitude). Stratigraphic evidence supports the theory that this valley had a marine origin (Mckee et al., 1990). Exceptional biodiversity and large numbers of endemic plant and animal species are characteristic of this area (Minckley, 1969; Johnson, 2005; Carson and Dowling, 2006). Within the basin, a large number of highly diverse springs (>300), spring-fed streams and evaporation ponds form an inverse archipelago, in which aquatic systems are separated by sparse desert vegetation, and salty soils (Souza et al., 2008). A high aquatic microbial diversity has also been described, and almost 50% of the bacteria isolated from CCB ponds were related to marine bacteria (Souza et al., 2006; Cerritos et al., 2011). The abundance of taxa related to marine bacteria leads to the hypothesis that some portion of the biota and water of CCB may have been derived from microbes and water entrapped in the Mesozoic strata; which have been released recently during ongoing subsurface karstification (Souza et al., 2006). An outstanding characteristic of the CCB is the extremely low nutrient levels in the hydrological systems (Souza et al., 2008). The phosphorus levels, in particular, appear to be low in the CCB (i.e., ∼0.60 μMol of total P; Elser et al., 2005). Bacteria may have several strategies to survive in low phosphorus environments, such as the replacement of membrane phospholipids for sulfolipids, small genome size, and enzymes to scavenge for alternative sources of phosphorus, like phosphonates (Dufresne et al., 2005; Dyhrman et al., 2006; Van Mooy et al., 2009; Villarreal-Chiu et al., 2012). The particular geological characteristics and low nutrient availability of CCB make it a suitable location to describe intraspecific genomic diversity in species that have survived in an ancient oligotrophic environment.
The aim of this study was to analyze the genomic and phenotypic variation between three B. coahuilensis strains to describe their genome plasticity. How many differences would there be among strains that are free living, but have a small genome and are exposed to similar environmental conditions? We sequenced, assembled, and annotated the genomes of B. coahuilensis m2-6, and p1.1.43; and compared them with the already published B. coahuilensis m4-4 genome. Since the strains were isolated from the same low P environment, we placed emphasis on the search of different strategies for phosphorus metabolism. Experimental assays revealed genotype-phenotype inconsistencies, probably the result of regulatory differences. We describe microevolutionary changes of the B. coahuilensis strains globally and, in particular, differences in phosphorus acquisition strategies among the strains.
Materials and Methods
Sample Locations and Culture Media
Three B. coahuilensis strains were isolated from water samples from different hydrological systems in the CCB. B. coahuilensis m4-4, was isolated from a desiccation lagoon (Churince system) with a C:N:P ratio of 50:7.5:1, and is located at the west side of the valley (26° 50.830′N, 102° 09.335′W; Cerritos et al., 2008). In this side of the valley the dominant soils are gypsisols (IUSS Working Group WRB, 2014). The strain B. coahuilensis m2-6 was also isolated from the Churince system (26° 51.199′N, 102° 09.009′W; Cerritos et al., 2011). Finally, the strain B. coahuilensis p1.1.43 was isolated from within the east side of the basin in the spring pool Poza Azul I (26°49.642′N, 102°01.458′E) with a C:N:P ratio of 51:1.8:1 (Cerritos et al., 2011). In this side of the valley the dominant soils are calcisols (IUSS Working Group WRB, 2014). Marine Medium (MM) was used for general culture of all afore mentioned strains [5% casein hydrolysate, 1% yeast extract, 85.55 mM NaCl, 7.04 mM Na2SO4, 3.60 mM CaCl, 2.68 mM KCl, 0.94 mM Na2CO3, 0.29 mM C6H5FeO7, 40 mM MgSO4, 0.67 mM KBr, 0.35 mM H3BO3, 0.57 mM NaF, 0.19 mM NO3NH4, and 0.56 mM Na2HPO4].
Comparative Genomics
Genome Sequencing, Assembly, and Annotation
Cultures of the B. coahuilensis strains were grown in liquid MM overnight at 37°C. Genomic DNA was obtained using the modified rapid method for the isolation of genomic DNA (Hoffman and Winston, 1987). Genomic DNA was quantified using a NanoDrop spectrophotometer (ND-1000, Thermo Fisher Scientific, Wilmington, DE, USA). Two sequencing technologies were used to sequence the strain genomes: shotgun libraries sequenced with the GS-FLX Titanium® pyrosequencer (454 Life Sciences, Roche, Branford, CT, USA), located at the Cinvestav–Langebio (Irapuato, México), and a mate pair libraries sequencing analysis (5-kb inserts) with an Illumina® genome analyzer (Illumina, San Diego, CA, USA), located at the Instituto de Biotecnología-UNAM (Cuernavaca, México). Pyrosequence reads were assembled using a Celera assembler v8.2 (Celera Genomics, Rockville, MD, USA; Myers et al., 2000).
Contigs were ordered and oriented by mapping the Illumina mate-pair reads to the Celera contigs using the alignment program BOWTIE v1.1.2 (Langmead et al., 2009). To optimize genome scaffolding, we only used the illumina reads that mapped once in each contig. The B. coahuilensis m4-4 genome was re-sequenced, using the mate-pair library sequencing with 5-kb inserts and the Illumina analyzer. The Illumina mate-pair reads were also mapped to the previously reported contigs (Alcaraz et al., 2008). The quality assessment for genome assemblies was done using QUAST and CheckM (Gurevich et al., 2013; Parks et al., 2015). The complete genomic sequences of B. coahuilensis strains m2-6 and p1.1.43 have been deposited in GenBank under the accession numbers LDYF00000000.1 and LDYG0000000.1, respectively. The average nucleotide identity (ANI) was calculated according to Goris et al. (2007). The open reading frames (ORFs) were predicted and annotated using RAST v2.0 (Rapid Annotation using Subsystem Technology; Aziz et al., 2008). The genome-scale metabolic models were obtained using Model SEED v1.0 (Overbeek et al., 2005). The results for the metabolic models of amino acid biosynthesis, carbohydrate utilization, ability to swarm and swim, and phosphate metabolism, were compared with the results obtained from experimental data.
Determination of Accessory Genome and Genomic Islands
Bacillus coahuilensis core genome and accessory genes were identified searching for orthologous genes, according to a previously described methodology (Moreno-Hagelsieb and Latimer, 2008). The genome alignment was performed and visualized using the Mauve software tool v2.4 (Darling et al., 2004). GI were identified using the Genomic Island Suite of Tools GIST v1.0 (Hasan et al., 2012) and determination of GC% in these genomic regions. Identification of mobile genetic elements (MGE) was performed using PHAST web server for the phage sequences and ISsaga v1.0 for the insertion sequences (ISs; Varani et al., 2011; Zhou et al., 2011).
To search for genes encode phosphonate metabolism, we built profile hidden Markov models using the software HMMER v3.1 (Finn et al., 2011). We built profiles based on protein alignments of sequences obtained from the Genbank1 for each protein, including phosphonopyruvate hydrolase (PalA), phosphonoacetate hydrolase (PhnA), transcriptional regulator (PhnW), repressor protein (PhnF), phosphonate lyase (PhnGHM), phosphonate transporters (PhnCDE1E2), and phosphonoaldehyde dehydrogenase (PhnXY).
Inferring Horizontal Gene Transfer (HGT) of Phosphonate Transporter and Proteins of 2-Keto-3-Deoxymannooctulosonic Acid (Kdo) Biosynthesis
To analyze whether the genes encoding the phosphonate transporter and the Kdo biosynthesis proteins had been transferred by HGT into B. coahuilensis m2-6 genome, we first searched for genomic signatures of: (a) significant difference in GC content for the genome segments that can be an indication of foreign DNA, and (b) analysis of the genomic context, searching for flanking repeat sequences and presence of nearby integrases or transposases genes that could suggest a non-native region (Ravenhall et al., 2015). We also inferred HGT by analyzing the phylogeny where we compared gene tree with their associated species tree to search for unusual distribution of the genes that probably are from HGT (Ravenhall et al., 2015). This was the case for the phosphonate transporter gene, for which we did a phylogenetic reconstruction. Multiple alignment of the Phn amino acid sequences of several species of the class Bacilli (PhnC, PhnD, PhnE1, and PhnE2) were assembled using Muscle v3.8.31 (Edgar, 2004). The alignments were further concatenated and used in the phylogenetic analyses, which in turn were performed using maximum likelihood estimation software PhyML v3.0 (Guindon et al., 2010). The LG substitution model was supported by the test results (–lnL = 7221.82; Abascal et al., 2005). Clostridium ultunense Phn transporter sequences were used as outgroup. Bootstrap values were calculated after 100 pseudo-replicates to estimate nodal support. We used the same phylogenetic inference analysis strategy to determine the KdsB protein (3-deoxy-manno-octulosanate cytidylyltransferase) associated with Kdo biosynthesis. The JTT substitution model was supported by the test results (–lnL = 9752.74). The Arabidopsis thaliana KdsB sequence was used as outgroup. The species tree was based on the phylogenetic reconstruction of the 16S rRNA gene. The GTR substitution model was supported by the test results (–lnL = 11838.64). The Clostridium ultunense 16S rRNA sequence was used as outgroup.
Experimental Assays for Phenotypic Diversity
Amino Acid Requirements
The three B. coahuilensis strains were plated on modified phosphate defined medium (PDM) and incubated at 37°C for 48 h to determine amino acid requirements (Muller et al., 1997). The PDM medium contained 50 mM Tris, pH 7.5, 85.55 mM NaCl, 3.6 mM CaCl2, 3.98 mM MgSO4, 0.19 mM NH4NO3, 7.71 mM Na3C6H5O7, 1.58 mM MnCl2, 0.001 mM ZnCl2, 1.66 mM FeCl3, 1.39 mM KH2PO4, and 49.95 mM glucose. We added for each amino acid: 50 μg/mL of D-alanine, L-arginine, L-asparagine, L-glutamine, L-glycine, L-histidine, L-isoleucine, L-leucine, L-lysine, L-methionine, L-phenylalanine, L-proline, L-serine, L-threonine, and L-valine; 500 μg/mL of L-aspartic acid, and L-glutamic acid; 40 μg/mL of L-cysteine; 20 μg/mL of L-tryptophan, and L-tyrosine (Harwood and Cutting, 1990). Separate plates were inoculated to perform three biological replicates.
Differences in Carbon Source Utilization
The B. coahuilensis strains were plated on Biolog Universal Agar (BUGTM Agar; Biolog, Inc., Hayward, CA, USA). MM salts were added to improve strain growth and thioglycolate was added to decrease bacterial capsule biosynthesis. The plates were incubated at 30°C for 24 h. Subsequent steps were performed according to the manufacturer’s instructions. Three replicate of each strain were prepared. The absorbance values were normalized by calculating dual wavelength data (DWD) according to the manufacturer’s formula.
Swarming and Swimming
The ability to move to search for nutrients or respond to favorable or unfavorable conditions is another important phenotypic characteristic of microorganisms living in oligotrophic environments. For swarming assays, plates (9 cm) containing 25 mL MM (0.6% agar) were prepared 1 h before inoculation, and dried with the lids open for 15 min in a laminar-flow hood. The plates were centrally inoculated with 2 × 107 cells in 10 μL, the lid was left open for another 10 min, and the plates were then incubated at 37°C for 24 h. Swimming/motility assays were performed on MM with 0.3% agar by inoculating cells in a 10 μL (at the center of each plate), and incubating at 37°C for 24 h (Kearns and Losick, 2003).
Differences in Phosphorus Source Utilization
To determine growth when bacteria were exposed to different phosphorus (P) sources, the three strains were plated on PDM solid medium at 37°C for 48 h. They were then re-streaked onto PDM media containing different phosphorus sources 1.4 mM KH2PO4, 0.1 mM Ca3(PO4)2, 0.1 mM 5-bromo-4-chloro-3-indolyl phosphate (X-P), 0.1 mM Na2HPO3, 0.1 mM 2-aminoethylphosphonic acid, 0.1 mM phosphonoacetaldehyde, 0.1% DNA, and 0.1% RNA. After 48 h of incubation, the strains were then streaked again on the identical medium and the same phosphorus source to confirm the growth phenotypes. Separate plates were inoculated with three biological replicates.
Alkaline Phosphatase Activity
Alkaline phosphatase activity was assayed at high and low concentrations of phosphorus. The high PDM (HPDM) contained 1.4 mM KH2PO4. No KH2PO4 was added to the low PDM (LPDM), so the bacteria were exposed only to the trace phosphorus present in the reagents used for the preparation of the medium. A molybdate colorimetric method after ascorbic acid reduction was used to determine the final concentration of phosphorus in the media (147.1 mg P mL-1 HPDM and 3.3 mg P mL-1 LPDM). Strains were inoculated in liquid MM at 37°C for 24 h. Strains were inoculated on LPDM or HPDM solid medium containing 40 μg/mL 5-Bromo-4-chloro-3-indolyl phosphate (X-Pi) as an indicator of alkaline phosphatase activity (Metcalf and Wanner, 1991). Separate plates were inoculated with three biological replicates.
Results
Characteristics of the B. coahuilensis m2-6 and B. coahuilensis p1.1.43 Genomes
The B. coahuilensis m2-6 and B. coahuilensis p1.1.43 genomes were sequenced using 454 technology at 62x and 64x coverage, respectively. The assembly using 454 and Illumina data resulted in 71 contigs for B. coahuilensis m2-6, and 59 contigs for B. coahuilensis p1.1.43. The results obtained and comparisons with the results of B. coahuilensis m4-4 genome (accession number NZ_ABFU00000000.1; Alcaraz et al., 2008) are presented in Table 1. The results of the quality assessment for genome assemblies indicated that the three genomes could be considered near-complete with low contamination (Table 1). The ANI value between B. coahuilensis m4-4 and B. coahuilensis m2-6 was the highest (99.37%). The ANI value between B. coahuilensis m2-6 and B. coahuilensis p1.1.43 was 98.93%. And for B. coahuilensis m4-4 and B. coahuilensis p1.1.43, the ANI was the lowest with 98.88%. All the ANI values are in agreement with those reported for strains of the same species. The results indicated that the B. coahuilensis strains had small differences in Coding DNA Sequences (CDS) number (Table 1). Furthermore, there were minor differences in the percent values for guanine–cytosine (GC) content. Strain B. coahuilensis m2-6 had the highest percent GC value (37.97%), followed by strain p1.1.43 (37.96%) and strain m4-4 (37.92 %). The results indicated that there were differences in genome size. B. coahuilensis p1.1.43 had the largest (3.40 Mbp), and B. coahuilensis m2-6 had the smallest (3.21 Mbp) genome.
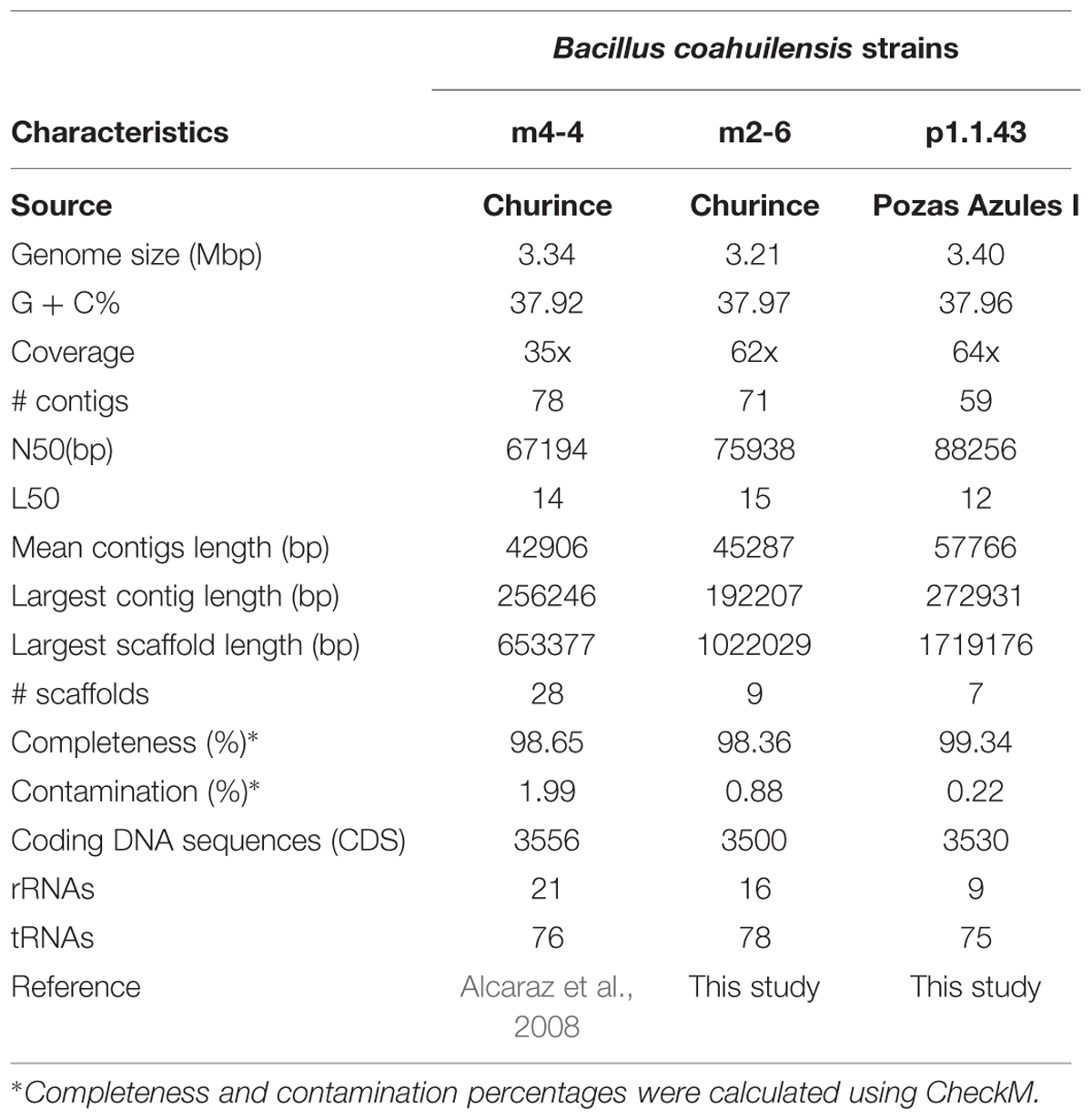
TABLE 1. General characteristics of the Bacillus coahuilensis genomes.
Pangenome and Mobile Genetic Element Diversity
The core genome shared between the B. coahuilensis strains was determined using previously published methods (Moreno-Hagelsieb and Latimer, 2008). Comparison of the orthologous genes revealed that the pangenome consisted of 4,416 genes, and the core genome consisted of 2,893 orthologous genes (82%). The accessory genome was 18% of the pangenome. Strain p1.1.43 had the largest number of strain-specific genes (4.1%), followed by strain m4-4 (3.6%) and m2-6 (3.0%; Figure 1). B. coahuilensis m4-4 and m2-6 (both isolated from the Churince pond) shared more genes between them than with strain p1.1.43 (isolated from Pozas Azules; Figure 1). The search for MGE revealed that IS and phage-related genes were present in all three genomes. The m4-4 and m2-6 genomes had clustered regularly interspaced short palindromic repeat (CRISPR) genes, in contrast; we did not find CRISPR in the p1.1.43 genome. The B. coahuilensis m2-6 genome had a higher number of ORFs related to ISs (52) compared with the other two genomes. The B. coahuilensis m4-4 is the only one genome that possesses genes coding for Tn7 (8). The p1.1.43 genome had more phage-related genes (25) than the m2-6 (5) and m4-4 genomes (11) (Figure 2). We also investigated what was encoded in the accessory genes and, in particular, if there were MGEs that could explain the differences in the B. coahuilensis genomic repertoire. Some MGEs were shared by the m4-4 and the m2-6 strains (e.g., CRISPR, ISs). Strains m4-4 and p1.143 shared phage-related genes. As previously described in Figure 1, strain p1.1.43 had more strain-specific genes compared with the other two genomes of which 5.3% were MGEs. For example, some of the MGEs were phage-related genes, and some were ISs (IS660, IS1182, ISShvi3, ISBsp1, ISArsp6, TnShfr1, ISXc4, and ISSwi1). Approximately 5% of the strain-specific genes present in strain m2-6 were MGEs; the remainder were mostly ISs (ISBspe1, ISBsp3, ISCth6, ISSep2, ISEnfa110, ISPa38, and ISAcsp1). We also found that 8.79% of the strain-specific genes present in strain m4-4 were phage-related genes, ISs (IS1326, ISSm4, ISBce19, ISBse1, ISBaps1, and ISCth6), and that transposons were also present.
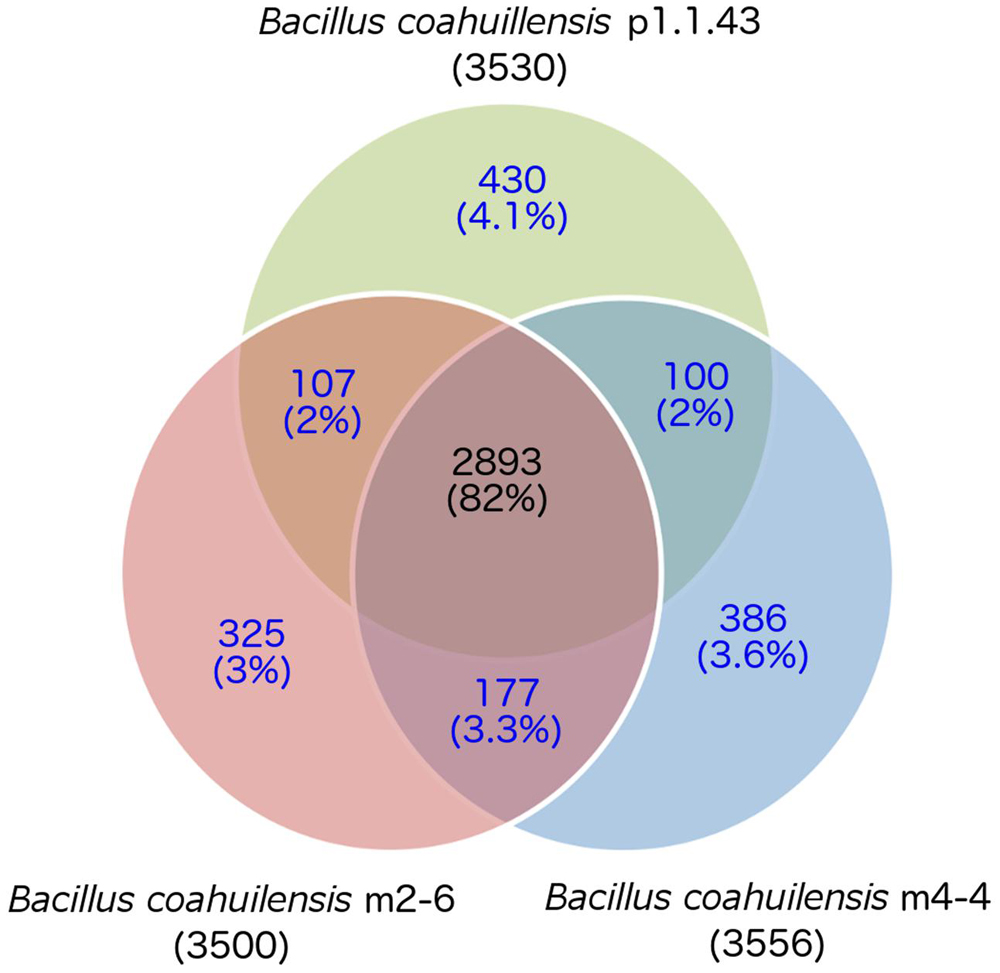
FIGURE 1. Core and accessory genomes of three Bacillus coahuilensis strains (m4-4, m2-6, and p1.1.43). Ortholog identification was performed using the bidirectional best hit (BBH) approach with mutual coverage or a shared length of 60% and an e-value ≥10-6. The core genome is indicated by the numbers in black font. Accessory gene numbers are in blue font. The number of predicted genes is shown in parenthesis for each genome.
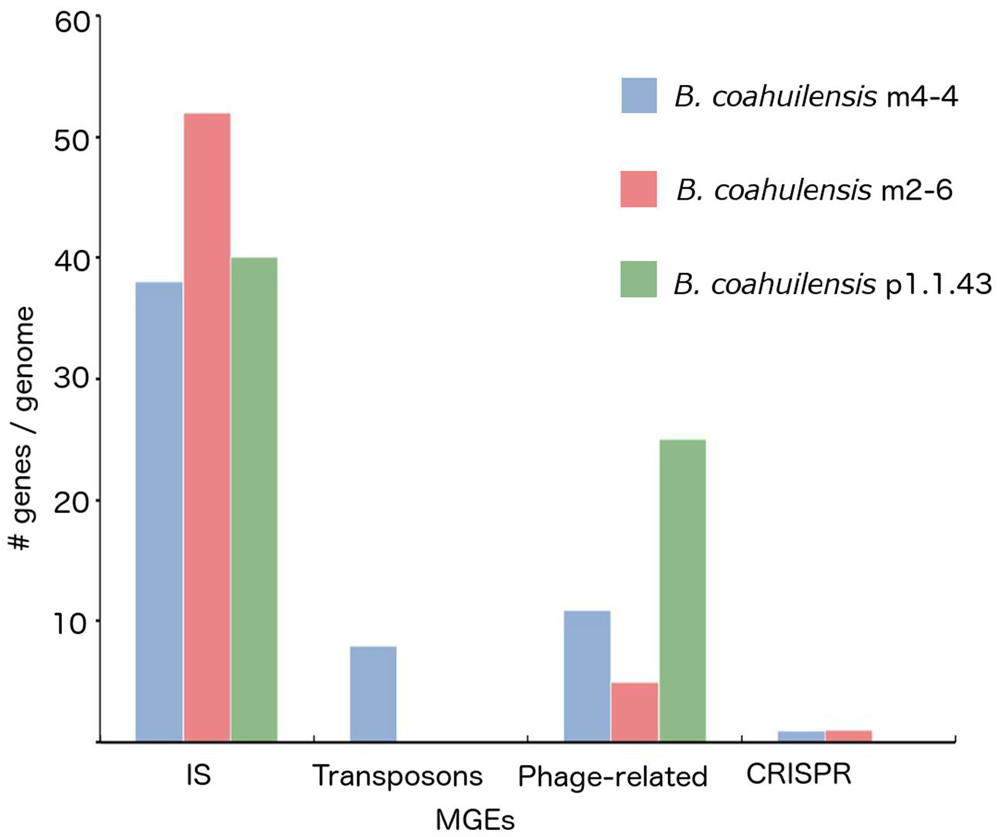
FIGURE 2. Mobile genetic elements (MGEs) identified in the B. coahuilensis genomes (strains m4-4, m2-6, and p1.1.43). Axis Y indicates the number of genes related to MGEs per genome, axis X indicates different MGEs coding in the genomes (IS, transposons, phage-related genes, and CRISPR).
Synteny and Identification of Genomic Islands
The genomes shared a high amount of genetic material and exhibited a high level of synteny. However, differences in gene content indicated that the chromosomes had suffered changes from rearrangement and HGT events. We confirmed the identity of the GI using the GIST to determine whether the unique segments present in the genomes were associated with MGEs (Hasan et al., 2012). Several unique areas (i.e., GIs) were observed in the three genomes. Two GIs associated with transposons (GIm4a and GIm4c) and other containing phage-related genes (GIm4b) were present in the m4-4 genome. Two GIs (GIp1a and GIp1b) associated with an IS, and phage-related genes, respectively, were present in strain p1.1.43. Two GIs associated with ISs (GIm2a and GIm2c) and two phage-related genes (GIm2b and GIm2e) were present in strain m2-6 (Figure 3). Unexpectedly, a GI was present in the m2-6 genome (GIm2d) that had a significantly lower percentage value for GC content (31.68%) than that observed for this genome (37.97%; Figure 4A). Analysis of this region indicated that it contained genes associated with the biosynthesis of lipopolysaccharide (LPS). We found genes for the biosynthetic pathway for saccharide 2-keto-3-deoxymannooctulosonic acid (Kdo), which is an important component of the cell wall of Gram-negative bacteria. We also found that sequences encoding several hypothetical genes, and ISs were present in this GI (Figure 4A). We performed a phylogenetic analysis using a sequence alignment for the KdsB protein to explore the evolutionary origin of the genes associated with Kdo biosynthesis that were identified in GIm2d of strain m2-6. The alignment of deduced amino acid sequences provided a total of 260 aligned residues, 42 of which were unambiguous. The KdsB proteins from the Fusobacteria class were a group with a high support value (100). The kdsB gene products from the Negativicutes class group that included some of the Clostridia class also had high support values (86). Another well-supported group (70) included the KdsB proteins from the Deltaproteobacteria. The KdsB from strain m2-6 formed a well-supported group (bootstrap of 100) with Bacillus cibi (isolated from traditional Korean fermented seafood), and these two protein sequences were most closely related to KdsB proteins from the Gammaproteobacteria class (Figure 4B).
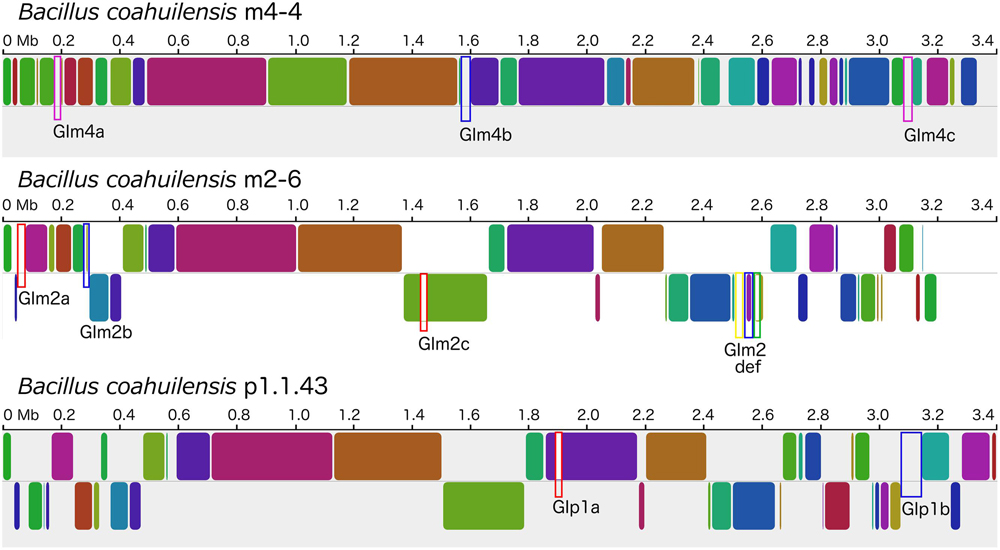
FIGURE 3. Mauve alignment of the B. coahuilensis genomes (strains m4-4, m2-6, and p1.1.43). Colored rounded boxes represent syntenic regions and white/gray areas represent unique regions in the three genomes. Blue rectangular boxes indicate genomic islands that include phage-related genes. Rectangular pink boxes indicate unique areas that include genes related to transposons. Rectangular red boxes indicate unique areas that include insertion sequences. The rectangular yellow box indicates a genomic island with genes associated with the biosynthesis of cell wall lipopolysaccharide, common only in Gram-negative bacteria. The rectangular green box indicates a unique region encoding genes related to phosphonate transport in the B. coahuilensis m2-6 genome.
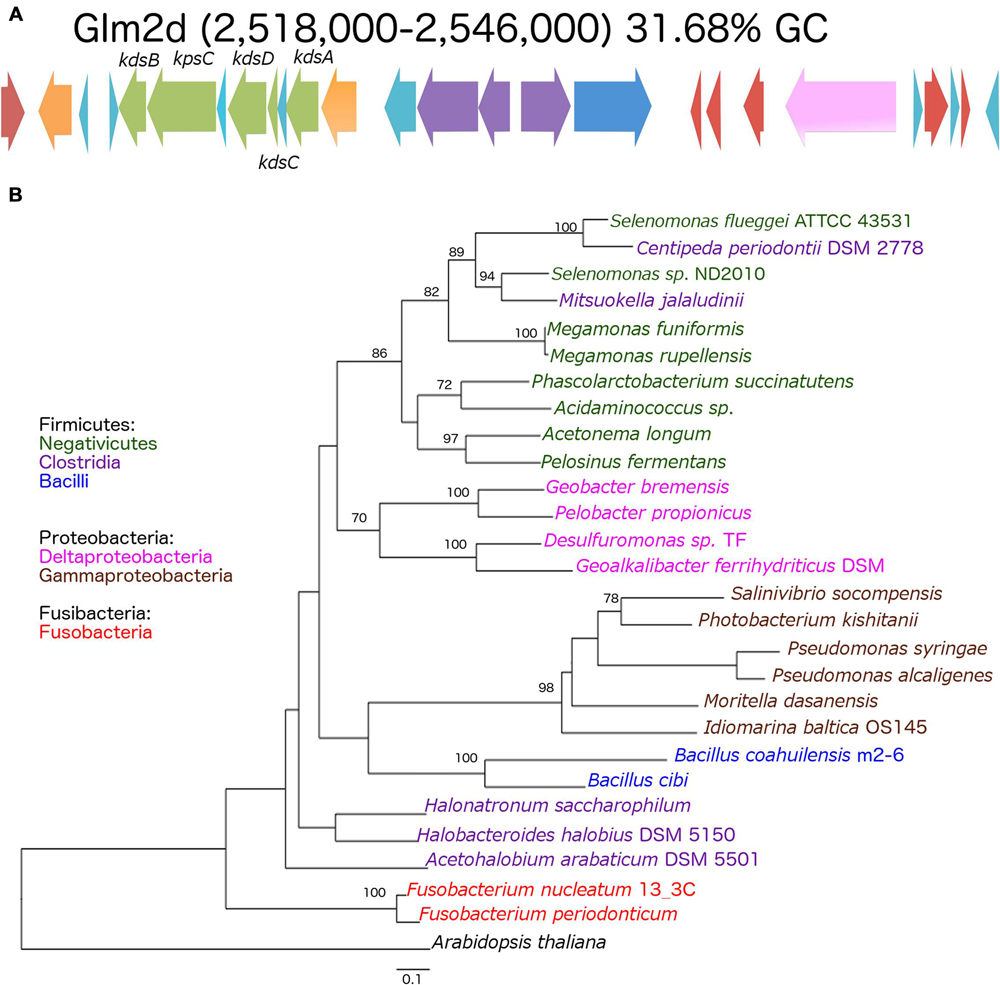
FIGURE 4. Genomic island (GIm2d) of Bacillus coahuilensis m2-6 and phylogenetic reconstruction of KdsB. (A) Genes arrangement in GIm2d. Red arrows indicate insertion sequences (IS643); orange arrows indicate genes coding for glycosyltransferase; light blue arrows indicate genes encoding hypothetical proteins; green arrows indicate genes associated with Kdo biosynthesis; purple arrows indicate genes coding for membrane proteins; the dark blue arrow indicates a gene coding for N-acetylmuramoyl-L-alanine amidase; the pink arrow indicates a gene coding for gamma-D-glutamyl-L-diamino acid endopeptidase I. Name of the genes associated with Kdo biosynthesis: kdsB, CMP-Kdo synthetase; kpsC, capsular polysaccharide export system protein; kdsD, D-arabinose-5-phosphate isomerase; kdsC, Kdo-8-P phosphatase; kdsA, Kdo-8-P synthase. (B) The phylogenetic reconstruction of the KdsB associated with Kdo biosynthesis was based on a maximum likelihood method. Numbers next to the branches represent bootstrap values expressed as percentages of 100 replications; only the values >70% are indicated. Bar represents 0.1 substitutions per nucleotide position. The Arabidopsis thaliana KdsB was used as outgroup. Colors represent bacterial lineages.
Reduction of Amino Acid Biosynthetic Capabilities
Experiment to determine amino acid biosynthesis capabilities revealed that there were numerous auxotrophies to amino acids. Strain m2-6 had more auxotrophies (11) than strains p1.1.43 (8) and m4-4 (7). In all three strains, arginine auxotrophy was congruent with a predicted incomplete urea cycle. Valine, leucine, and isoleucine were required by the three strains. These results were consistent with the metabolic model, which revealed that the pyruvate pathway was extensively eroded. Auxotrophies that varied between strains were proline, cysteine, aspartic, threonine, and lysine. Glutamic and phenylalanine were required by all three strains. Despite this, with the metabolic models, we did not find a defective pathway to explain the auxotrophies for these seven amino acids. This result suggested that there were strain-specific regulatory differences in the expression of genes involved in the biosynthesis of amino acid. In silico metabolic models also revealed that strain m4-4 had an alternative pathway for the serine biosynthesis via transformation of pyruvate into serine by L-serine ammonia-lyase. An optional methionine biosynthesis pathway using a salvage pathway in which methylthioadenosine was recycled as methionine was also present (Table 2).
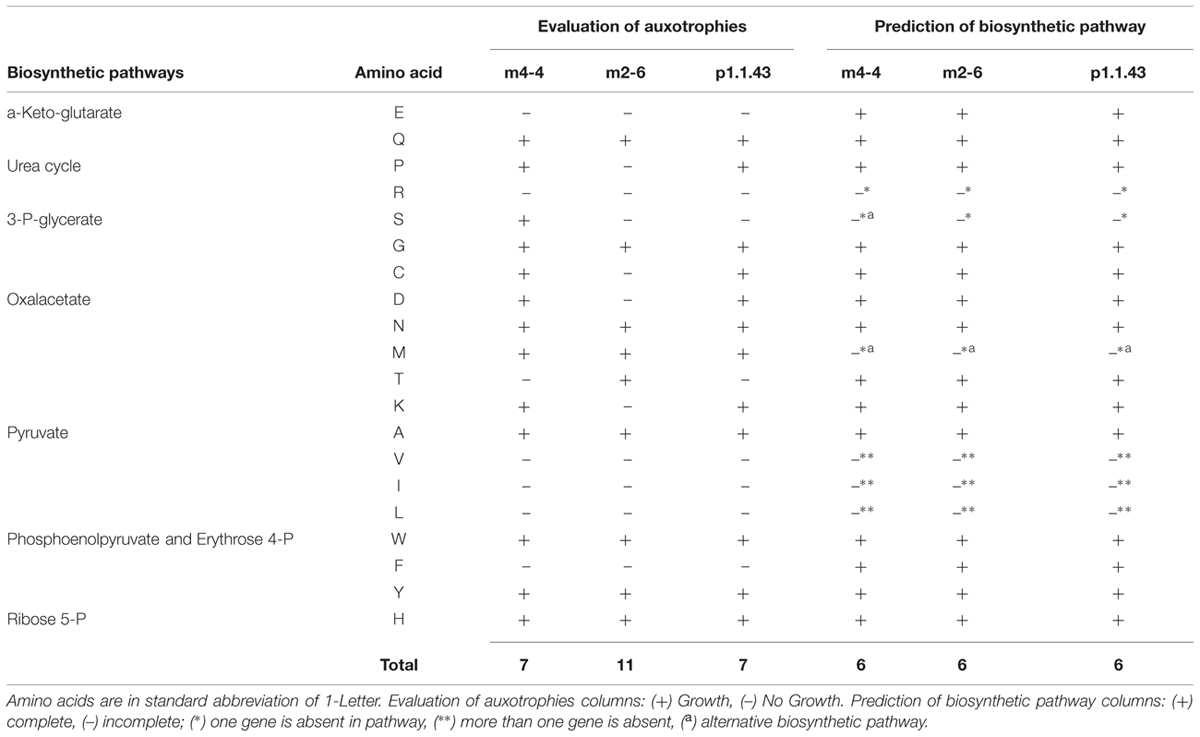
TABLE 2. Utilization of different amino acid sources by each Bacillus coahuilensis strain.
Diversification in Carbohydrate Scavenging Capabilities
Our results showed that strain p1.1.43 was more versatile in the use of carbon sources; however, all the strains used poly- and oligosaccharides of D-glucose, D-fructose, D-mannose, and D-turanose, which are mostly linked by alpha bonds. Moreover, our metabolic models suggested that genes encoding alpha- and beta-glucosidases for the hydrolysis of several di- and oligosaccharides of D-glucose and D-fructose were present, but we did not detect genes encoding galactosidase for the hydrolysis of disaccharides of D-galactose. In addition, specific transport systems for raffinose, melibiose, and stachyose were identified. However, experimental analysis using Biolog to determine the strain’s capabilities to utilize different carbon sources did not reveal evidence that any of the strains could use these saccharides (Figure 5A). The Biolog results and metabolic models indicated that all three strains could use the monosaccharides D-ribose, D-fructose, and alpha-D-glucose. Strain m4-4 could use a higher number of monosaccharides, compared with strains m2-6 and p1.1.43. Only strains m4-4 and m2-6 efficiently used the precursors of hemicellulose (L-arabinose and D-xylose). However, when we performed the in silico metabolic models, we found apparently incomplete pathways for L-arabinose, D-xylose, and for other reactions required for the synthesis of some other monosaccharides (Figure 5B). The Biolog results also revealed that strain m4-4 had more flexibility in the assimilation of carboxylic acids (pyruvic acid, acetic acid, L-malic acid, propionic acid, succinic acid, and valeric acid) compared with the other two strains. Strain p1.1.43 was the only strain capable of using the aromatic compounds inosine, adenosine, thymidine, uridine, and 2′-deoxy adenosine. However, results for in silico metabolic models suggested that all genomes had genes for membrane transport and assimilation of the carboxylic acids and the aromatic compounds tested (Figure 5C).
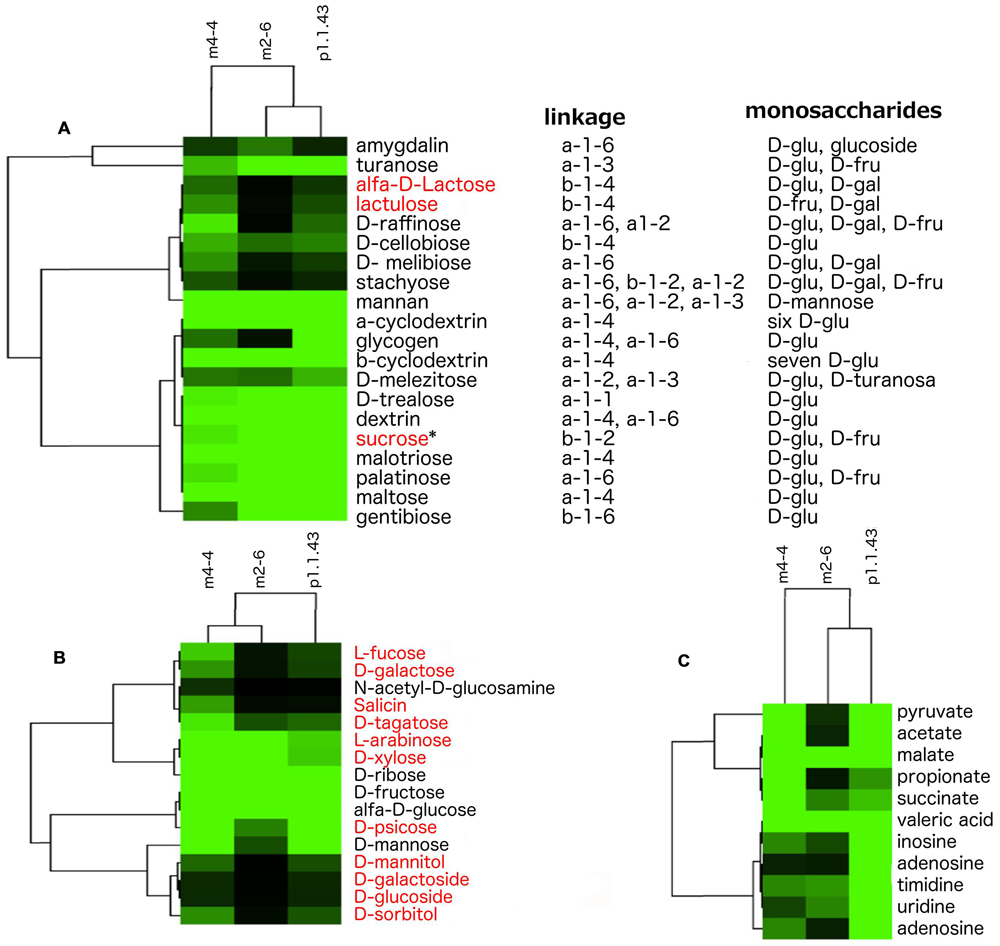
FIGURE 5. Observed variability in the utilization of carbon sources in three B. coahuilensis strains. (A) Biolog results for the use of poly- and oligosaccharides. Compounds were clustered according to how the strains (m4-4, m2-6, and p1.1.43) used poly- and oligosaccharides. (B) Biolog results for the use of monosaccharides. Data were clustered according to bacterial utilization, as in (A). (C) Biolog results for the use of carboxylic acids and aromatic compounds. Data were clustered according to bacterial utilization, as in (A). Carbohydrates names in red font indicate that in silico metabolic models have missing genes in the three genomes. ∗Indicates that genes are missing only in B. coahuilensis m4-4. In all cases, lighter green areas indicate greater evidence of source utilization, and black green indicates no utilization for poly- and oligosaccharides (A), monosaccharides (B), and carboxylic acids and aromatic compounds (C), respectively.
Between-Strain Variability in Motility
Our assays revealed that strains m2-6 and p1.1.43 had the ability to swim while m4-4 lacked this ability (Figure 6). When we compared these results with the in silico metabolic model, we found that the genome of all three had the same number of genes associated with flagellar biosynthesis and bacterial chemotaxis. The results of the swarming tests were negative for all three strains (Figure 6). Swarming depends on the flagellar synthesis and on surfactin production. All strains lacked the srfABCD operon that encodes surfactin, a lipopeptide required for swarming (Supplementary Table S1).

FIGURE 6. Differences in swim capabilities and lack of swarm ability of three B. coahuilensis strains. The control is marine medium (MM) containing 2% agar. Swarm was evaluated on MM containing 0.6% agar. Swim was evaluated on MM containing 0.3% agar.
Different Strategies for Metabolism of Inorganic and Organic P Sources
The ability to synthesize membrane sulfolipids is used by several organisms to maintain membrane phospholipids as a P reserve (Alcaraz et al., 2008). In this sense, we found that both B. coahuilensis m2-6 and B. coahuilensis p1.1.43 genomes had sulfolipid biosynthesis genes, encoding sulfoquinovose synthase (Sqd1) and glycosyltransferase (SqdX). The experiments revealed that the membranes in all three strains had both sulfolipids and phospholipids (data not shown). We also analyzed other characteristics that could result advantageous in a low P environment, such as growth in media with different inorganic and organic P sources, and alkaline phosphatase activity. Our results indicated that the strains exhibited some differences in the assimilation of P sources. All three strains preferred to use the inorganic P source, KH2PO4. Only strain p1.1.43 used Ca3(PO4)2. None used phosphite (Na2HPO3; Table 3). On the other hand, consistent with the experimental analysis, the metabolic model analysis identified genes encoding the phosphate transport system (pstABCS), and the metabolism of phosphates, but not the genes ptxABC for phosphite transport and oxidation (Supplementary Table S2).
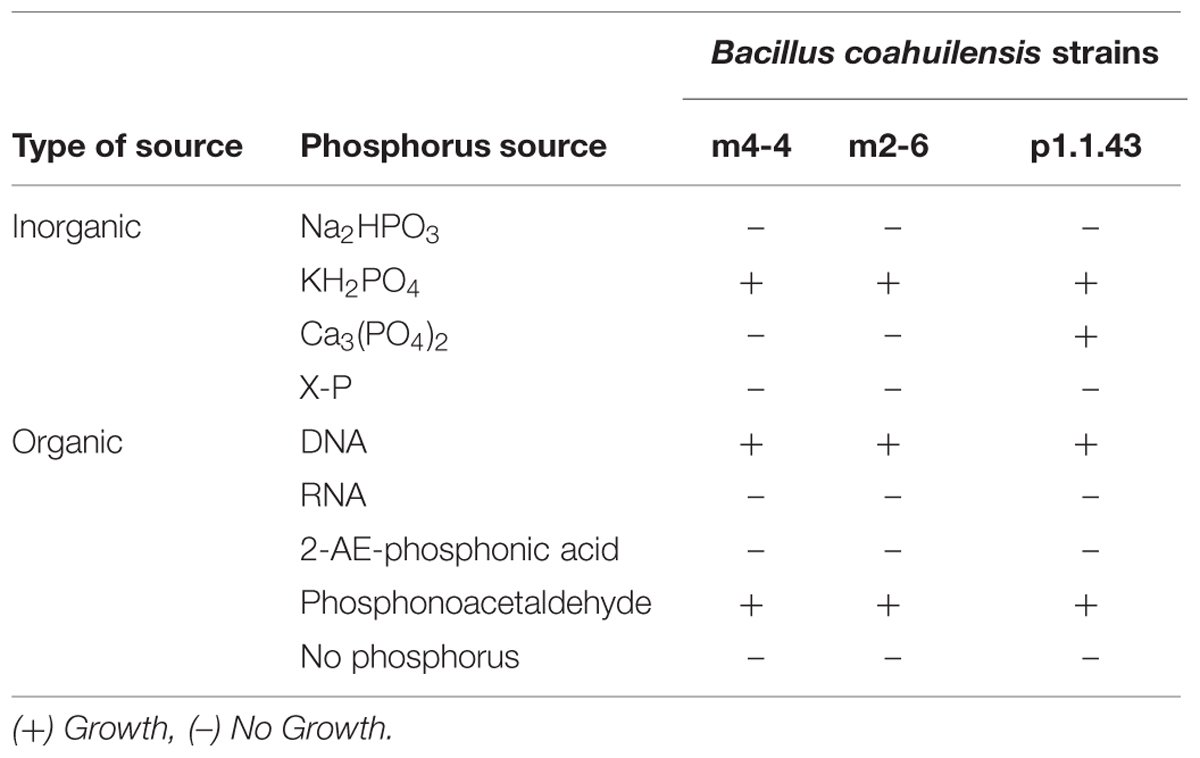
TABLE 3. Utilization of different phosphate sources by each Bacillus coahuilensis strain.
Additionally, we found that all three strains are able to recycle organic phosphorus, and scavenging phosphorus from phosphonates. The results for the use of organic P sources, particularly the nucleic acids (DNA and RNA), revealed that the strains were DNA, but not RNA, as the sole source of P (Table 3). Our results of the metabolic model analysis indicated that the possible genes involved in the metabolism of organic P included genes for an alkaline phosphodiesterase, and a 2′, 3′- cyclic-nucleotide 2′-phosphodiesterase (Table 4). The results from a BLAST analysis using the SubtiList database2 indicated that the 2′, 3′- cyclic-nucleotide 2′-phosphodiesterase gene had an identity of 29% with gene yfkN, which encodes a secreted phosphodiesterase in B. subtilis. A third gene encoded an alkaline phosphomonoesterase (PhoA).
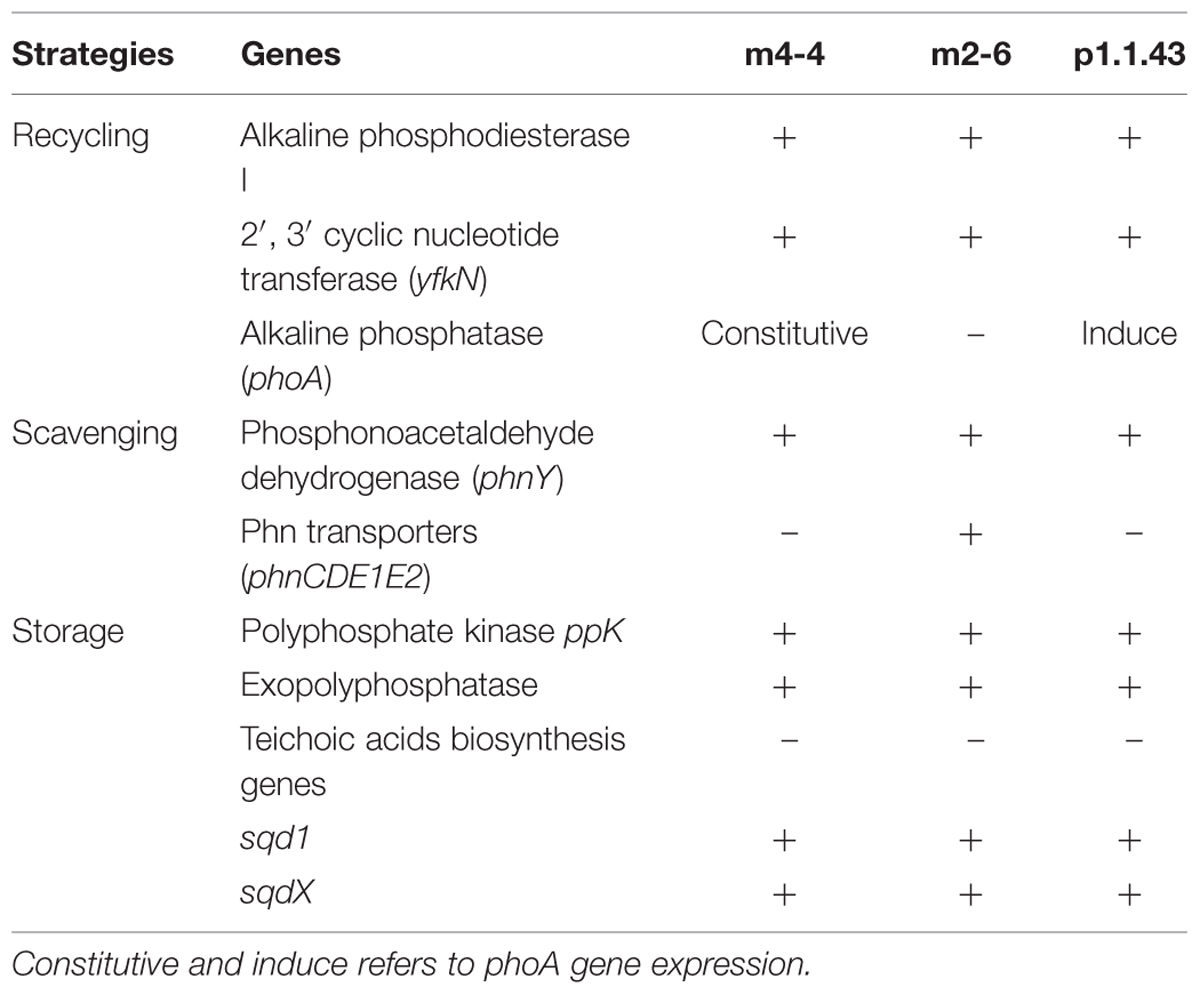
TABLE 4. Strategies for the recycling, scavenging, and storage of phosphorus.
Some bacteria can use C-P containing organic P sources (e.g., phosphonates). All three strains were able to grow in the presence of phosphonoacetaldehyde, but not in the presence of 2-aminoethylphosphonic acid (Table 3). A comparison of these experimental results with the in silico metabolic model indicated that there were no genes associated with phosphonate utilization. We developed hidden Markov models for different enzymes associated with phosphonate metabolism. We identified in all three genomes a gene encoding an aldehyde dehydrogenase that matched to a phosphonoacetaldehyde dehydrogenase gene (phnY), with a significant E-value (1e-111 to 3.8 e-111; Supplementary Table S2). This enzyme (PhnY) catalyzes the transformation of phosphonoacetaldehyde to phosphonoacetate (Villarreal-Chiu et al., 2012). In addition, a gene cluster associated with phosphonate transport that was present in strain m2-6 was absent in the other two genomes. We also searched for genes related to phosphorus storage (biosynthesis of polyphosphate and teichoic acids). We observed in all three genomes the genes coding for the synthesis and hydrolysis of polyphosphates, but not genes related to teichoic acids biosynthesis (Table 4).
The three strains exhibited differences in the presence of the alkaline phosphate gene and its regulation. Consistent with this result was the alkaline phosphatase activity assay that showed constitutive alkaline phosphatase activity for strain m4-4 (expressed both in high and low KH2PO4 concentration). Alkaline phosphatase activity also occurred in strain p1.1.43, but this activity was induced only in low phosphate concentration conditions. No activity was observed for B. coahuilensis strain m2-6 (Figure 7). These results suggested that microevolutionary changes had affected phosphorus utilization strategies, both through gene loss (e.g., the alkaline phosphatase gene in m2-6) and through changes in gene regulation (Table 4).

FIGURE 7. Intraspecific differences in alkaline phosphatase activity in three B. coahuilensis strains (m4-4, m2-6, and p1.1.43). LPDM, low phosphate defined medium; HPDM, high phosphate defined medium with KH2PO4. X-Pi, 5-Bromo-4-chloro-3-indolyl phosphate (40 μM/mL) was used as an indicator of alkaline phosphatase activity.
HGT Origin of Phosphonate Transport in B. coahuilensis m2-6
To explore the possible evolutionary origin of the phosphonate (phn) transport cluster identified in B. coahuilensis m2-6, we performed a phylogenetic analysis using the multiple protein sequence alignments for the phn transport genes (phnC, phnD, phnE1, and phnE2). The concatenated alignments of the deduced amino acid sequences provided a total of 1159 aligned residues, of which 425 were unambiguous. The Phn transporters from members of the Bacillus cereus group form another well-supported clade. The Phn gene products from the Alicyclobacillus pohliae were associated with some species of the genus Paenibacillus with high support (>90). Another well-supported group (100) included lineages of phn genes from Enterococcus and Lactobacillus species. The Phn transporter from strain m2-6 formed a well-supported group (100) with other Bacillus species isolated from aquatic environments, and with Salsuginibacillus kocurii, Caldibacillus debilis, and Gracilibacillus halophilus (Figure 8A). The presence of some well-supported monophyletic groups formed by different bacterial lineages suggested that several HGT events occurred in the class Bacilli (Figure 8A). The comparison of the phylogeny of Phn transporter with the species phylogeny (16S rRNA; Supplementary Figure S1) indicated discordances between Phn transporter and species tree that can be suggestive of an HGT event. Finally, to understand the genomic context and the biological role of the phn transport cluster in strain m2-6, we analyzed the genomic neighborhood of the phn transport cluster. We found that it was located within the trehalose operon (treP, treA, and treR). The results of a comparison of the genomic neighborhood of the phn transport cluster of strain m2-6 with Bacillus sp. m3-13 (isolated from the Churince system in the CCB) indicated that both gene clusters maintained a similar organization, and each had a gene coding for a 2′ 3′-cyclic-nucleotide 2′-phosphodiesterase (Figure 8B). The results of a BLAST analysis using the SubtiList database indicated that this gene was highly similar to B. subtilis yfkN, which encodes a secreted phosphodiesterase (27% identity). The biological role of this enzyme in the metabolism of phosphonates by B. coahuilensis is unknown.
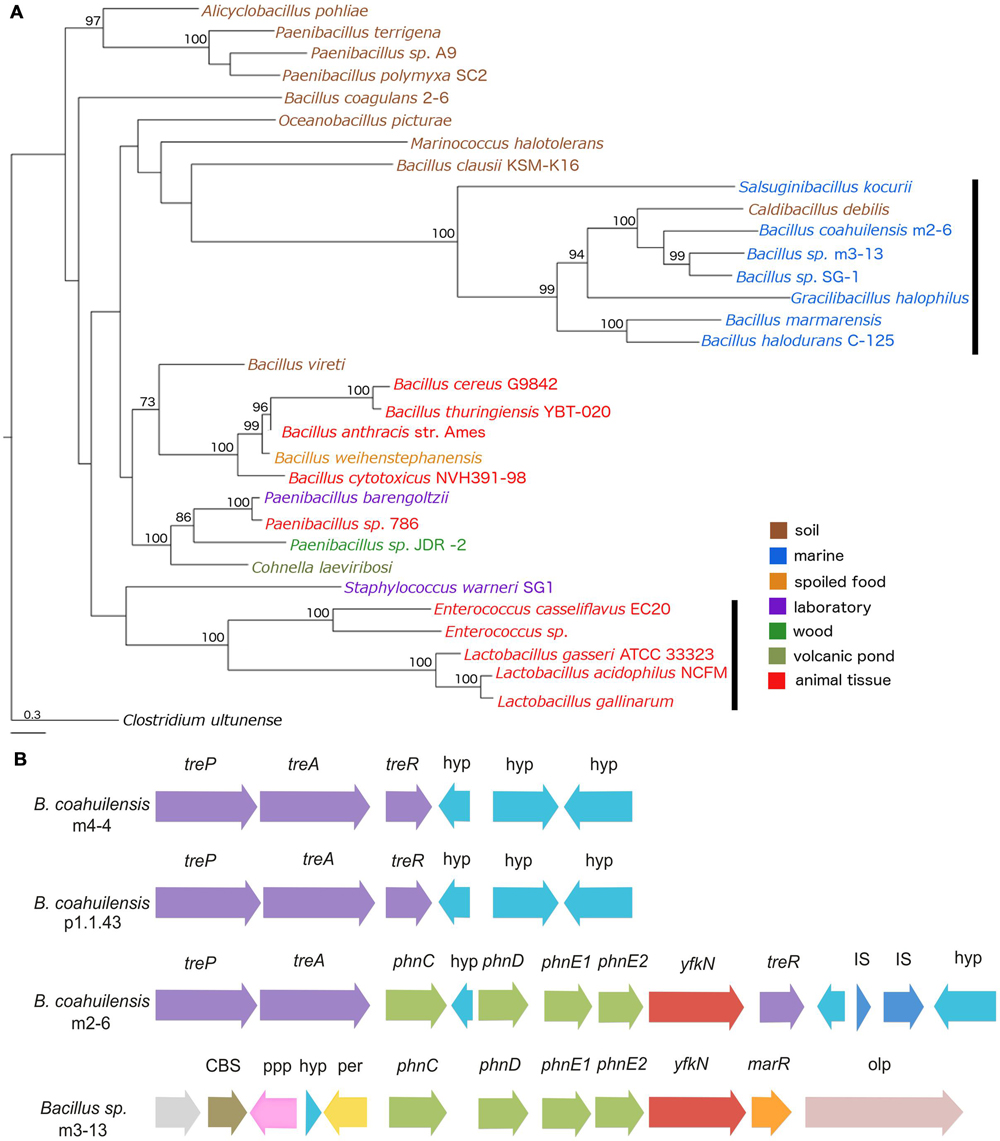
FIGURE 8. Phylogeny of Phn transport cluster and genomic context identified in B. coahuilensis strain m2-6. (A) Phylogenetic tree of concatenated Phn sequences (PhnC, PhnD, PhnE1, and PhnE2) was performed with a Maximum Likelihood method. Numbers next to the branches represent bootstrap values expressed as percentages of 100 replications; only values >70% are indicated. Bar represents 0.3 substitutions per nucleotide position. Clostridium ultunense was used as outgroup. Colors represent bacterial isolation environments. The black vertical bars indicate events of HGT, as suggested for Lactobacillus and Enterococcus (Huang et al., 2005). (B) Genomic neighborhood of phosphonate transport cluster (phnC, phnD, phnE1, and phnE2) in B. coahuilensis m2-6. treP, PTS system trehalose-specific IIB component; treA, trehalose-6-phosphate hydrolase; treR, trehalose operon transcriptional repressor; hyp, hypothetical protein; yfkN, 2′ 3′-cyclic-nucleotide 2′-phosphodiesterase; IS, insertion sequences; CBS, cystathionine beta-synthase domain protein; ppp, putative periplasmic protein; per, zinc/iron permease; marR, transcriptional regulator of MarR family; olp, oligopeptidase F.
Discussion
Genomic Diversity and Variation Associated to MGEs
Comparative genomics is a valuable approach to reveal in great detail the genetic variation among bacteria at species, strain, and individual levels, as well as to answer evolutionary questions on gene loss, gain, and transfer. In the present study, we performed a phenotypic-genotypic association analysis of the B. coahuilensis strains to understand the genomic and metabolic diversity. We wanted to explore the differences between strains with a relatively small genome, and exposed to similar oligotrophic conditions. We explored their ability to use different carbon sources, in silico and using Biolog and, since the strains were isolated from the same low P environment we emphasized the search of different strategies for phosphorus transport, metabolism, storage and regulation.
We observed high ANI values between B. coahuilensis genomes (98.8 and higher), which reflects the genetic distance. These highly ANI values have been observed in other organisms that have an ecological constrains (Konstantinidis et al., 2006), and is consistent with the oligotrophic environment from which the strains were isolated. The core genome and pangenome was informative of the potential gene pool available to B. coahuilensis. Overall, the three B. coahuilensis genomes shared 82% of their genes. This result was consistent with other intraspecific comparative genomics studies (Deng et al., 2010; Peña et al., 2010; Mann et al., 2013; Patiño-Navarrete et al., 2013). Most of the strain-specific genes encoded hypothetical genes and 5 to 8% of their genes encoded MGEs, mainly related to phages, CRISPS, transposons, and ISs. Our results agree with studies of other bacterial lineages, which found that the genomes are shaped by the presence of MGEs (Peña et al., 2010; Kalhoefer et al., 2011; Meyer and Huber, 2014). On the other hand, the alignment of the three B. coahuilensis genomes exhibited high synteny, probably as a consequence of very few genomic rearrangements. This result was consistent with the results of comparative genomic analysis of co-occurring environmental Lebetimonas acidiphila strains (Meyer and Huber, 2014). Also in genome alignment, we observed different GIs, in which the strain-specific genes were concentrated. The GIm2d found just in strain m2-6 encoded genes involved in the biosynthesis of Kdo, an important component of the LPS of the cell wall of Gram-negative bacteria. The phylogenetic reconstruction of KdsB from strain m2-6 indicated that the genes for Kdo biosynthesis were acquired by HGT. The acquisition of novel functional genes via HGT increases the potential for specific adaptations and microbial speciation in the CCB. This is an example of a microevolutionary change in this species, and it is also an unexpected finding for the Firmicutes that is worth describing. It will be interesting to elucidate the function of this cell-surface component.
Metabolic Analysis Uncovers that B. coahuilensis is Highly Dependent on Nutrients and a Diversification in Carbohydrate Scavenging Capabilities
The phenotypic and genotypic analysis revealed that the B. coahuilensis strains had lost numerous capabilities related to amino acid biosynthesis. This species probably is highly dependent on nutrients available in the sediment microbial community. The loss of some biosynthetic pathways in all three strain genomes may be due to a genome-size reduction, and/or an adaptation to a unique particular niche (Alcaraz et al., 2008), which can be referred as streamlining, a term used to describe gene loss occurring in bacteria (Giovannoni et al., 2005, 2014). It has been suggested that gene loss occurs particularly in microbial communities, as the result of changes of availability of public goods (Morris et al., 2012).
However, we found that some auxotrophies detected experimentally could not be explained by gene loss as the in silico metabolic models revealed complete amino acid biosynthetic pathways. Other phenotypic-genotypic association inconsistencies observed were related to swimming ability in strain m4-4, supporting the idea that regulation in gene expression also has contributed to the intraspecific diversity of B. coahuilensis. Therefore, we suggest that gene loss is probably not the only strategy to achieve energy efficiency because it implies a high risk, given the lack of environmental stability for free-living bacteria. Changes in gene expression allow efficient energy utilization, and also to conserve genetic information. Similar inconsistencies between in silico metabolic models and auxotrophies have been observed in other species of Bacillus genus isolated from CCB hydrological systems (data not shown). Nevertheless, these phenotypic-genotypic inconsistencies support the idea that the magnitude of the differences between genomes cannot be inferred merely from genomic comparison. Phenotypic data brings the true dimension of a cell’s function. Metabolic models performed so far require feedback from experimental data, to connect genome predictions to phenotypes. The main difficulty of metabolic predictions is precisely the near impossibility to include predicted regulatory features. Regulatory information gathered from the literature is crucial to improve metabolic models. We believe that in all organisms under study, the evolution through changes in gene regulation is yet under-appreciated given the need to uncover it experimentally. Understand the phenotype to genotype correlation is one of the most important issues of the post genomic era.
On the other hand, the genotypic-phenotypic association was more consistent for carbohydrate utilization, where most of the metabolic models agreed with the results of carbohydrate utilization obtained through Biolog analysis. However, we found that some pathways (L-arabinose and D-xylose) could not be fully elucidated, suggesting that unlike other model bacteria, B. coahuilensis may use alternative enzymes. Similar results have also been obtained in studies of other non-model bacteria such as Roseobacter litoralis (Kalhoefer et al., 2011). Based on the results of the utilization of alternative C sources (e.g., carboxylic acids and aromatic compounds), we suggest that there has been a diversification in carbohydrate scavenging capabilities among the B. coahuilensis strains.
Differences in Phosphorus Acquisition Strategies and Their Regulation
Strategies to survival in a low P environment are key in the CCB, as revealed by previous analysis of the B. coahuilensis m4-4 genome (Alcaraz et al., 2008). Our results showed similarities and differences in the abilities of the three B. coahuilensis strains for phosphorus recycling, scavenging, and storage. The first similarity among all three strains was the ability to use DNA as a phosphorus source, consistent with the occurrence of genes coding for an alkaline phosphodiesterase I, and the 2′ 3′-cyclic-nucleotide 2′-phosphodiesterase, YfkN. Both enzymes are associated with the recycling of phosphorus from extracellular RNA and DNA, and are induced in response to phosphorus starvation (Vershinina and Znamenskaya, 2002; Singh et al., 2015). Another similarity was that all three genomes encoded a glycerolphosphodiesterase (GlpQ), which is an important enzyme that hydrolyzes deacylated phospholipids to glycerol-3-phosphate. In B. subtilis, this phosphodiesterase is strongly induced by phosphorus starvation and is regulated by the PhoP–PhoR system (Antelmann et al., 2000). The third similarity is the ability to scavenge phosphorus from phosphonate. The three genomes have the gene encoded a putative phosphonoacetaldehyde dehydrogenase (PhnY). This enzyme has been described in Sinorhizobium meliloti 1021 as part of a novel pathway for degradation of 2-aminoethylphosphonate, catalyzed transformation of phosphonoacetaldehyde to phosphonoacetate (Borisova et al., 2011). A fourth similarity is the presences of genes that encode polyphosphates biosynthesis. A recent study in Sargasso Sea plankton indicated that the enrichment in polyphosphate coincided with enhanced alkaline phosphatase activity and substitution of sulfolipids for phospholipids, which are both indicators of phosphorus stress (Martin et al., 2014). A fifth coincidence is the absence of the genes related to teichoic acids biosynthesis. These polymers of the cell walls of Gram-positive bacteria are considered as phosphorus storage molecules, given that they have repeating polyol or glycosylpolyol residues linked by phosphodiester bonds (Kulakovskaya, 2014). It is likely that B. coahuilensis lost the capacity to produce teichoic acid because of the P-limited environment of CCB (Alcaraz et al., 2008).
As for the differences, B. coahuilensis p1.1.43 was the only strain with the ability to use Ca3(PO4)2. The results of many studies have indicated that the ability to use this mineralized phosphorus source is accompanied by the ability to solubilize it by secreting organic acids (e.g., succinate, fumarate, gluconic, and malate; Deubel and Merbach, 2005; Khan et al., 2009; Sharma et al., 2013). Other between-strain differences in the P recycling, and was related to gene content, and expression of the gene phoA encoding alkaline phosphomonoesterase. Finally, the last difference was related to the presence of a phosphonate (phn) transport cluster in the strain m2-6 genome. Phosphonates have an increasingly acknowledged role in phosphorus cycling in nature. Biosynthetic and metabolic pathways that include phosphonates have been discovered (Quinn et al., 2007). The ability of microbes to cleave the C–P bond confers organisms the possibility to use a wide range of phosphonates as nutrients. The phosphonates scavenging as P reservoir in the oceans has been well established as well as the strategies that marine microorganisms employ for their utilization (Villarreal-Chiu et al., 2012). Furthermore, alternative phosphonate utilization pathways were recently described in representatives of Proteobacteria, Planctomycetes, and Cyanobacteria, commonly found in marine plankton (Martinez et al., 2010).
The genomic neighborhood analysis and comparison with the phn transport cluster of the Bacillus sp. m3-13 genome revealed variation in structure and gene composition. Unexpectedly, we also found a gene, yfkN, which was consistently associated with the phn gene cluster in the B. coahuilensis m2-6 and the Bacillus sp. m3-13 genomes. In B. subtilis, this gene is induced in response to P starvation. The gene product exhibits 2′, 3′ cyclic nucleotide phosphodiesterase, 2′ (or 3′) nucleotidase, and 5′ nucleotidase activities to provide pathways for the recycling of P from extracellular DNA and RNA (Allenby et al., 2005). A recent study revealed that in Rhizobium leguminosarum, an enzyme that originally was reported to have a phosphodiesterase function could also hydrolyze phosphonate monoesters (Jonas et al., 2008). The observation of this dual function suggested that this gene could have phosphonatase activity in strain m2-6.
Recycling phosphorus from DNA, scavenging from phosphonates, and phosphate storage are possibly acquisition strategies that, along with the ability to synthesize membrane sulfolipids, help B. coahuilensis to survive in a harsh environment. The evolutionary force that could explain the diversification in the phosphorus acquisition strategies could be genetic drift. Sometimes restricted dispersal creates geographically structured subpopulations (each with a reduced Ne), promoting their genetic diversification (Luo and Moran, 2015). In this sense, only three strains of B. coahuilensis have been recovered in the last 8 years of extensive isolation efforts of Bacillus sp., suggesting that this species might be structured in small subpopulation with a restricted dispersal that could promote microevolutionary change. Finally, most comparative genomic studies of bacteria focus on pathogens and symbionts of humans, animals, and plants. The determination of the genomic and metabolic diversity of free-living bacteria such as B. coahuilensis represents an effort to understand what drives bacterial microevolution and adaptation in natural oligotrophic environments. Further transcriptomic and proteomic studies could help to reconcile the differences observed between the genotypic and phenotypic results. Another avenue open for research is the cross metabolic regulation between strains co-occurring in microbial communities.
Conclusion
Our results revealed that an important source of the B. coahuilensis strains’ genome variation is associated with their numerous MGEs. We also identified the loss of some amino acid biosynthetic pathways, variability in motility ability, and a diversification in carbohydrate and phosphorus scavenging capabilities, and suggest that this microevolutionary changes result not just from gene loss and/or gain, but also from gene regulation affecting specific pathways. Particularly, we found some phosphorus acquisition strategies that enable the B. coahuilensis strains to recycle phosphorus, scavenging, and phosphate storage, all important strategies for survival in an environment with low concentration of this essential element.
Author Contributions
ZG-L contributed to the conception and design of the study, to data acquisition and analysis, interpretation of the results, and preparation of the manuscript; IH-G and M-DR-T contributed to data acquisition and analysis, and to interpretation of the results; GO-A participated in the conception and design of the study, and to manuscript preparation; VS participated in a critical revision of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a CONACyT fellowship (to ZG-L). The project was funded by a CINVESTAV multidisciplinary project award (to GO-A) and CONACYT CB-2013-01 No.220536 (to GO-A). We thank Africa Islas, Julio Cruz, José Luis Hernández, Susana de la Torre, and Rosalinda Serrato for specialized technical assistance in the laboratory. Special thanks for thoughtful comments go to Varinia López and Luis David Alcaraz.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00058
Footnotes
References
Abascal, F., Zardoya, R., and Posada, D. (2005). ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105. doi: 10.1093/bioinformatics/bti263
Abby, S., and Daubin, V. (2007). Comparative genomics and the evolution of prokaryotes. Trends Microbiol. 15, 135–141. doi: 10.1016/j.tim.2007.01.007
Alcaraz, L. D., Olmedo, G., Bonilla, G., Cerritos, R., Hernandez, G., Cruz, A., et al. (2008). The genome of Bacillus coahuilensis reveals adaptations essential for survival in the relic of an ancient marine environment. Proc. Natl. Acad. Sci. U.S.A. 105, 5803–5808. doi: 10.1073/pnas.0800981105
Allenby, N. E. E., O’Connor, N., Pragai, Z., Ward, A. C., Wipat, A., and Harwood, C. R. (2005). Genome-wide transcriptional analysis of the phosphate starvation stimulon of Bacillus subtilis. J. Bacteriol. 187, 8063–8080. doi: 10.1128/JB.187.23.8063-8080.2005
Antelmann, H., Scharf, C., and Hecker, M. (2000). Phosphate starvation-inducible proteins of Bacillus subtilis: proteomics and transcriptional analysis. J. Bacteriol. 182, 4478–4490. doi: 10.1128/JB.182.16.4478-4490.2000
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Borisova, S. A., Christman, H. D., Metcalf, M. E. M., Zulkepli, N. A., Zhang, J. K., van der Donk, W. A., et al. (2011). Genetic and biochemical characterization of a pathway for the degradation of 2-aminoethylphosphonate in Sinorhizobium meliloti 1021. J. Biol. Chem. 286, 22283–22290. doi: 10.1074/jbc.M111.237735
Carson, E. W., and Dowling, T. E. (2006). Influence of hydrogeographic history and hybridization on the distribution of genetic variation in the pupfishes Cyprinodon atrorus and C. bifasciatus. Mol. Ecol. 15, 667–679. doi: 10.1111/j.1365-294X.2005.02763.x
Cerritos, R., Eguiarte, L. E., Avitia, M., Siefert, J., Travisano, M., Rodriguez-Verdugo, A., et al. (2011). Diversity of culturable thermo-resistant aquatic bacteria along an environmental gradient in Cuatro Cienegas, Coahuila, Mexico. Antonie Van Leeuwenhoek 99, 303–318. doi: 10.1007/s10482-010-9490-9
Cerritos, R., Vinuesa, P., Eguiarte, L. E., Herrera-Estrella, L., Alcaraz-Peraza, L. D., Arvizu-Gomez, J. L., et al. (2008). Bacillus coahuilensis sp. nov., a moderately halophilic species from a desiccation lagoon in the Cuatro Cienegas Valley in Coahuila, Mexico. Int. J. Syst. Evol. Microbiol. 58, 919–923. doi: 10.1099/ijs.0.64959-0
Cohan, F. M., and Koeppel, A. F. (2008). The origins of ecological diversity in prokaryotes. Curr. Biol. 18, R1024–R1034. doi: 10.1016/j.cub.2008.09.014
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Deng, X., Phillippy, A. M., Li, Z., Salzberg, S. L., and Zhang, W. (2010). Probing the pan-genome of Listeria monocytogenes: new insights into intraspecific niche expansion and genomic diversification. BMC Genomics 11:500. doi: 10.1186/1471-2164-11-500
Deubel, A., and Merbach, W. (2005). “Influence of microorganisms on phosphorus bioavailability in soils,” in Microorganisms in Soils: Roles in Genesis and Functions, eds A. Varma and F. Buscot (Berlin: Springer), 177–191. doi: 10.1007/b137872
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Dufresne, A., Garczarek, L., and Partensky, F. (2005). Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 6, R14–R14. doi: 10.1186/gb-2005-6-2-r14
Dyhrman, S. T., Chappell, P. D., Haley, S. T., Moffett, J. W., Orchard, E. D., Waterbury, J. B., et al. (2006). Phosphonate utilization by the globally important marine diazotroph Trichodesmium. Nature 439, 68–71. doi: 10.1038/nature04203
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Elser, J. J., Schampel, J. H., Garcia-Pichel, F., Wade, B. D., Souza, V., Eguiarte, L., et al. (2005). Effects of phosphorus enrichment and grazing snails on modern stromatolitic microbial communities. Freshw. Biol. 50, 1808–1825. doi: 10.1111/j.1365-2427.2005.01451.x
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Giovannoni, S. J., Cameron Thrash, J., and Temperton, B. (2014). Implications of streamlining theory for microbial ecology. ISME J. 8, 1553–1565. doi: 10.1038/ismej.2014.60
Giovannoni, S. J., Tripp, H. J., Givan, S., Podar, M., Vergin, K. L., Baptista, D., et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245. doi: 10.1126/science.1114057
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Harwood, C. R., and Cutting, S. M. (1990). Molecular Biological Methods for Bacillus, 1st Edn. Chichester: John Wyley and Sons.
Hasan, M. S., Liu, Q., Wang, H., Fazekas, J., Chen, B., and Che, D. (2012). GIST: genomic island suite of tools for predicting genomic islands in genomic sequences. Bioinformation 8, 203–205. doi: 10.6026/97320630008203
Hoffman, C. S., and Winston, F. (1987). A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57, 267–272. doi: 10.1016/0378-1119(87)90131-4
Huang, J., Su, Z., and Xu, Y. (2005). The evolution of microbial phosphonate degradative pathways. J. Mol. Evol. 61, 682–690. doi: 10.1007/s00239-004-0349-4
IUSS Working Group WRB (2014). World Reference Base for Soil Resources 2014. International Soil Classification System for Naming Soils and Creating Legends for Soil Maps. Rome: Food and Agriculture Organization of the United Nations.
Johnson, S. G. (2005). Age, phylogeography and population structure of the microendemic banded spring snail, Mexipyrgus churinceanus. Mol. Ecol. 14, 2299–2311. doi: 10.1111/j.1365-294x.2005.02580.x
Jonas, S., van Loo, B., Hyvonen, M., and Hollfelder, F. (2008). A new member of the alkaline phosphatase superfamily with a formylglycine nucleophile: structural and kinetic characterisation of a phosphonate monoester hydrolase/phosphodiesterase from Rhizobium leguminosarum. J. Mol. Biol. 384, 120–136. doi: 10.1016/j.jmb.2008.08.072
Kalhoefer, D., Thole, S., Voget, S., Lehmann, R., Liesegang, H., Wollher, A., et al. (2011). Comparative genome analysis and genome-guided physiological analysis of Roseobacter litoralis. BMC Genomics 12:324. doi: 10.1186/1471-2164-12-324
Kearns, D. B., and Losick, R. (2003). Swarming motility in undomesticated Bacillus subtilis. Mol. Microbiol. 49, 581–590. doi: 10.1046/j.1365-2958.2003.03584.x
Khan, A. A., Jilani, G., Akhtar, M. S., Naqvi, S. M. S., and Rasheed, M. (2009). Phosphorus solubilizing bacteria: occurrence, mechanisms and their role in crop production. J. Agric. Biol. Sci. 1, 48–58.
Konstantinidis, K. T., Ramette, A., and Tiedje, J. M. (2006). The bacterial species definition in the genomic era. Philos. Trans. R. Soc. B Biol. Sci. 361, 1929–1940. doi: 10.1098/rstb.2006.1920
Koonin, E. V., and Wolf, Y. I. (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719. doi: 10.1093/nar/gkn668
Kulakovskaya, T. (2014). Phosphorus storage in Microorganisms: diversity and evolutionary insight. Biochem. Physiol. 4, 1–4. doi: 10.4172/2168-9652.1000e130
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25
Luo, H., and Moran, M. A. (2015). How do divergent ecological strategies emerge among marine bacterioplankton lineages? Trends Microbiol. 23, 577–584. doi: 10.1016/j.tim.2015.05.004
Mann, R. A., Smits, T. H., Buhlmann, A., Blom, J., Goesmann, A., Frey, J. E., et al. (2013). Comparative genomics of 12 strains of Erwinia amylovora identifies a pan-genome with a large conserved core. PLoS ONE 8:e55644. doi: 10.1371/journal.pone.0055644
Martin, P., Dyhrman, S. T., Lomas, M. W., Poulton, N. J., and Van Mooy, B. A. S. (2014). Accumulation and enhanced cycling of polyphosphate by Sargasso Sea plankton in response to low phosphorus. Proc. Natl. Acad. Sci. U.S.A. 111, 8089–8094. doi: 10.1073/pnas.1321719111
Martinez, A., Tyson, G. W., and DeLong, E. F. (2010). Widespread known and novel phosphonate utilization pathways in marine bacteria revealed by functional screening and metagenomic analyses. Environ. Microbiol. 12, 222–238. doi: 10.1111/j.1462-2920.2009.02062.x
Math, R. K., Jin, H. M., Kim, J. M., Hahn, Y., Park, W., Madsen, E. L., et al. (2012). Comparative genomics reveals adaptation by Alteromonas sp. SN2 to marine tidal-flat conditions: cold tolerance and aromatic hydrocarbon metabolism. PLoS ONE 7:e35784. doi: 10.1371/journal.pone.0035784
Maughan, H., Callicotte, V., Hancock, A., Birky, C. W., Nicholson, W. L., and Masel, J. (2006). The population genetics of phenotypic deterioration in experimental populations of Bacillus subtilis. Evolution 60, 686–695. doi: 10.1111/j.0014-3820.2006.tb01148.x
Mckee, J. W., Jones, N. W., and Long, L. E. (1990). Stratigraphy and provenance of strata along the San Marcos fault, central Coahuila, Mexico. Geol. Soc. Am. Bull. 102, 593–614. doi: 10.1130/0016-7606(1990)102<0593:SAPOSA>2.3.CO;2
Metcalf, W. W., and Wanner, B. L. (1991). Involvement of the Escherichia coli phn (psiD) gene cluster in assimilation of phosphorus in the form of phosphonates, phosphite, Pi esters, and Pi. J. Bacteriol. 173, 587–600.
Meyer, J. L., and Huber, J. A. (2014). Strain-level genomic variation in natural populations of Lebetimonas from an erupting deep-sea volcano. ISME J. 8, 867–880. doi: 10.1038/ismej.2013.206
Minckley, W. L. (1969). Environments of the Bolson of Cuatro Ciénegas, Coahuila, México, with Special Reference to the Aquatic Biota. El Paso, TX: Texas Western Press.
Mira, A., Klasson, L., and Andersson, S. G. E. (2002). Microbial genome evolution: sources of variability. Curr. Opin. Microbiol. 5, 506–512. doi: 10.1016/S1369-5274(02)00358-2
Mira, A., Martin-Cuadrado, A. B., D’Auria, G., and Rodriguez-Valera, F. (2010). The bacterial pan-genome: a new paradigm in microbiology. Int. Microbiol. 13, 45–57.
Mira, A., Ochman, H., and Moran, N. A. (2001). Deletional bias and the evolution of bacterial genomes. Trends Genet. 17, 589–596. doi: 10.1016/S0168-9525(01)02447-7
Moreno-Hagelsieb, G., and Latimer, K. (2008). Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24, 319–324. doi: 10.1093/bioinformatics/btm585
Morris, J. J., Lenski, R. E., and Zinser, E. R. (2012). The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio 3, e00036–12. doi: 10.1128/mBio.00036-12
Muller, J. P., An, Z., Merad, T., Hancock, I. C., and Harwood, C. R. (1997). Influence of Bacillus subtilis phoR on cell wall anionic polymers. Microbiology 143, 947–956. doi: 10.1099/00221287-143-3-947
Myers, E. W., Sutton, G. G., Delcher, A. L., Dew, I. M., Fasulo, D. P., Flanigan, M. J., et al. (2000). A whole-genome assembly of Drosophila. Science 287, 2196–2204. doi: 10.1126/science.287.5461.2196
Ochman, H. (2001). Lateral and oblique gene transfer. Curr. Opin. Genet. Dev. 11, 616–619. doi: 10.1016/S0959-437X(00)00243-4
Ochman, H., and Davalos, L. M. (2006). The nature and dynamics of bacterial genomes. Science 311, 1730–1733. doi: 10.1126/science.1119966
Ochman, H., and Moran, N. A. (2001). Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. Science 292, 1096–1099. doi: 10.1126/science.1058543
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patiño-Navarrete, R., Moya, A., Latorre, A., and Peretó, J. (2013). Comparative genomics of Blattabacterium cuenoti: the frozen legacy of an ancient endosymbiont genome. Genome Biol. Evol. 5, 351–361. doi: 10.1093/gbe/evt011
Peña, A., Teeling, H., Huerta-Cepas, J., Santos, F., Yarza, P., Brito-Echeverria, J., et al. (2010). Fine-scale evolution: genomic, phenotypic and ecological differentiation in two coexisting Salinibacter ruber strains. ISME J. 4, 882–895. doi: 10.1038/ismej.2010.6
Penn, K., and Jensen, P. R. (2012). Comparative genomics reveals evidence of marine adaptation in Salinispora species. BMC Genomics 13:86. doi: 10.1186/1471-2164-13-86
Quinn, J. P., Kulakova, A. N., Cooley, N. A., and McGrath, J. W. (2007). New ways to break an old bond: the bacterial carbon–phosphorus hydrolases and their role in biogeochemical phosphorus cycling. Environ. Microbiol. 9, 2392–2400. doi: 10.1111/j.1462-2920.2007.01397.x
Radnedge, L., Agron, P. G., Worsham, P. L., and Andersen, G. L. (2002). Genome plasticity in Yersinia pestis. Microbiology 148, 1687–1698. doi: 10.1099/00221287-148-6-1687
Ravenhall, M., Škunca, N., Lassalle, F., and Dessimoz, C. (2015). Inferring horizontal gene transfer. PLoS Comput. Biol. 11:e1004095. doi: 10.1371/journal.pcbi.1004095
Rouli, L., Merhej, V., Fournier, P.-E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Sharma, S. B., Sayyed, R. Z., Trivedi, M. H., and Gobi, T. A. (2013). Phosphate solubilizing microbes: sustainable approach for managing phosphorus deficiency in agricultural soils. Springerplus 2, 587. doi: 10.1186/2193-1801-2-587
Singh, D. N., Gupta, A., Singh, V. S., Mishra, R., Kateriya, S., and Tripathi, A. K. (2015). Identification and characterization of a novel phosphodiesterase from the metagenome of an Indian coalbed. PLoS ONE 10:e0118075. doi: 10.1371/journal.pone.0118075
Souza, V., Eguiarte, L. E., Siefert, J., and Elser, J. J. (2008). Microbial endemism: does phosphorus limitation enhance speciation? Nat. Rev. Microbiol. 6, 559–564. doi: 10.1038/nrmicro1917
Souza, V., Espinosa-Asuar, L., Escalante, A. E., Eguiarte, L. E., Farmer, J., Forney, L., et al. (2006). An endangered oasis of aquatic microbial biodiversity in the Chihuahuan desert. Proc. Natl. Acad. Sci. U.S.A. 103, 6565–6570. doi: 10.1073/pnas.0601434103
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Tian, C. F., Zhou, Y. J., Zhang, Y. M., Li, Q. Q., Zhang, Y. Z., Li, D. F., et al. (2012). Comparative genomics of rhizobia nodulating soybean suggests extensive recruitment of lineage-specific genes in adaptations. Proc. Natl. Acad. Sci. U.S.A. 109, 8629–8634. doi: 10.1073/pnas.1120436109
Van Mooy, B. A. S., Fredricks, H. F., Pedler, B. E., Dyhrman, S. T., Karl, D. M., Koblizek, M., et al. (2009). Phytoplankton in the ocean use non-phosphorus lipids in response to phosphorus scarcity. Nature 458, 69–72. doi: 10.1038/nature07659
Varani, A. M., Siguier, P., Gourbeyre, E., Charneau, V., and Chandler, M. (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12, R30. doi: 10.1186/gb-2011-12-3-r30
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Vershinina, O. A., and Znamenskaya, L. V. (2002). The pho regulons of bacteria. Microbiology 71, 497–511. doi: 10.1023/a:1020547616096
Villarreal-Chiu, J. F., Quinn, J. P., and McGrath, J. W. (2012). The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol. 3:19. doi: 10.3389/fmicb.2012.00019
Keywords: microevolution, comparative genomics, intraspecific diversity, phenotypic-genotypic association, mobile genetic elements, phosphorus limitation, phosphonate transport, Kdo biosynthesis
Citation: Gómez-Lunar Z, Hernández-González I, Rodríguez-Torres M-D, Souza V and Olmedo-Álvarez G (2016) Microevolution Analysis of Bacillus coahuilensis Unveils Differences in Phosphorus Acquisition Strategies and Their Regulation. Front. Microbiol. 7:58. doi: 10.3389/fmicb.2016.00058
Received: 14 October 2015; Accepted: 13 January 2016;
Published: 08 February 2016.
Edited by:
Frank T. Robb, University of Maryland School of Medicine, USAReviewed by:
David John Studholme, University of Exeter, UKJuan M. Gonzalez, Consejo Superior de Investigaciones Científicas, Spain
Copyright © 2016 Gómez-Lunar, Hernández-González, Rodríguez-Torres, Souza and Olmedo-Álvarez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Olmedo-Álvarez, Z29sbWVkb0BpcmEuY2ludmVzdGF2Lm14