 Bruno M. Di Genova
Bruno M. Di Genova Renata R. Tonelli
Renata R. Tonelli- 1Departamento de Microbiologia e Imunologia, Universidade Federal de São Paulo, São Paulo, Brazil
- 2Instituto de Ciências Ambientais, Químicas e Farmacêuticas, Departamento de Ciências Biológicas, Universidade Federal de São Paulo, Diadema, Brazil
Giardia lamblia, Cryptosporidium sp., and Entamoeba histolytica are important pathogenic intestinal parasites and are amongst the leading causes worldwide of diarrheal illness in humans. Diseases caused by these organisms, giardiasis, cryptosporidiosis, and amoebiasis, respectively, are characterized by self-limited diarrhea but can evolve to long-term complications. The cellular and molecular mechanisms underlying the pathogenesis of diarrhea associated with these three pathogens are being unraveled, with knowledge of both the strategies explored by the parasites to establish infection and the methods evolved by hosts to avoid it. Special attention is being given to molecules participating in parasite–host interaction and in the mechanisms implicated in the diseases’ pathophysiologic processes. This review focuses on cell mechanisms that are modulated during infection, including gene transcription, cytoskeleton rearrangements, signal transduction pathways, and cell death.
Introduction
Intestinal infection is the most common cause of diarrhea in humans worldwide and although presenting low mortality rates, complications are not uncommon, with some cases requiring hospital care. Diarrhea may be caused by viruses, bacteria, helminths and protozoa, most of which are disseminated with feces-contaminated water and food. Amongst protozoan parasites, Giardia lamblia, Cryptosporidium parvum, and Entamoeba histolytica are the three most common etiological agents of diarrhea and other related diseases (giardiasis, cryptosporidiosis, and amoebiasis, respectively) characterized as acute and self-limited dysentery. Nevertheless, in some patients disease may become chronic with long-term effects such as malnutrition, growth delays, and cognitive impairment.
Diarrhea is an increase in the volume or liquidity of stool and it may or may not be accompanied by frequent evacuations (Viswanathan et al., 2009). Disorders of both the small and large intestines can result in diarrhea which, based on the duration, may be classified as acute (≤14 days), persistent (from 15 to 29 days) or chronic (≥30 days). This classification is clinically important to determine the etiological agent for diagnostic and treatment purposes (Guerrant et al., 2001). Diarrhea may also be classified into five categories based on the pathophysiological mechanisms as osmotic, secretory, exudative, inflammatory and resulting from motility disturbances (Field, 2003). Osmotic diarrhea is triggered when healthy individuals (with normal gut functions) ingest large amounts of poorly absorbed substrates, usually carbohydrates (polyethylene glycol, mannitol, lactulose) and divalent ions (MgSO4, MgOH2; Hammer et al., 1989; Izzo et al., 1994). An increase in intraluminal unabsorbed nutrients associated with epithelial damage and reduction of the intestinal absorptive surface also characterizes osmotic diarrhea. Secretory diarrhea results from overstimulation of intestinal tract secretory capacity. Exposure to enterotoxins from several types of bacteria (e.g., Escherichia coli heat-labile toxin, Cholera toxin), excessive bile acid synthesis, low levels of short-chain fatty acids and intestinal inflammation (seen in autoimmune diseases like inflammatory bowel disease and celiac disease) can trigger this type of diarrhea (Sullivan et al., 2009; Walters et al., 2009; Binder, 2010). When the intestinal barrier is compromised due to loss of epithelial cells or disruption of tight junctions (TJs), diarrhea is referred as exudative. Finally, increased or decreased propulsion of stools relates to diarrhea caused by motility problems (Field, 2003).
In many gastrointestinal infectious diseases, more than one of the five pathophysiological mechanisms is involved in the development of diarrhea. This is the case for giardiasis, cryptosporidiosis, and amoebiasis that, in spite of sharing similar pathophysiological mechanisms of diarrhea, have different initiating events. The early events triggered by the interaction of these three protozoans with their respective hosts are the focus of this review, with special attention to gene transcription, signal transduction pathways, cytoskeleton rearrangements, and cell death in host cells (Figure 1).
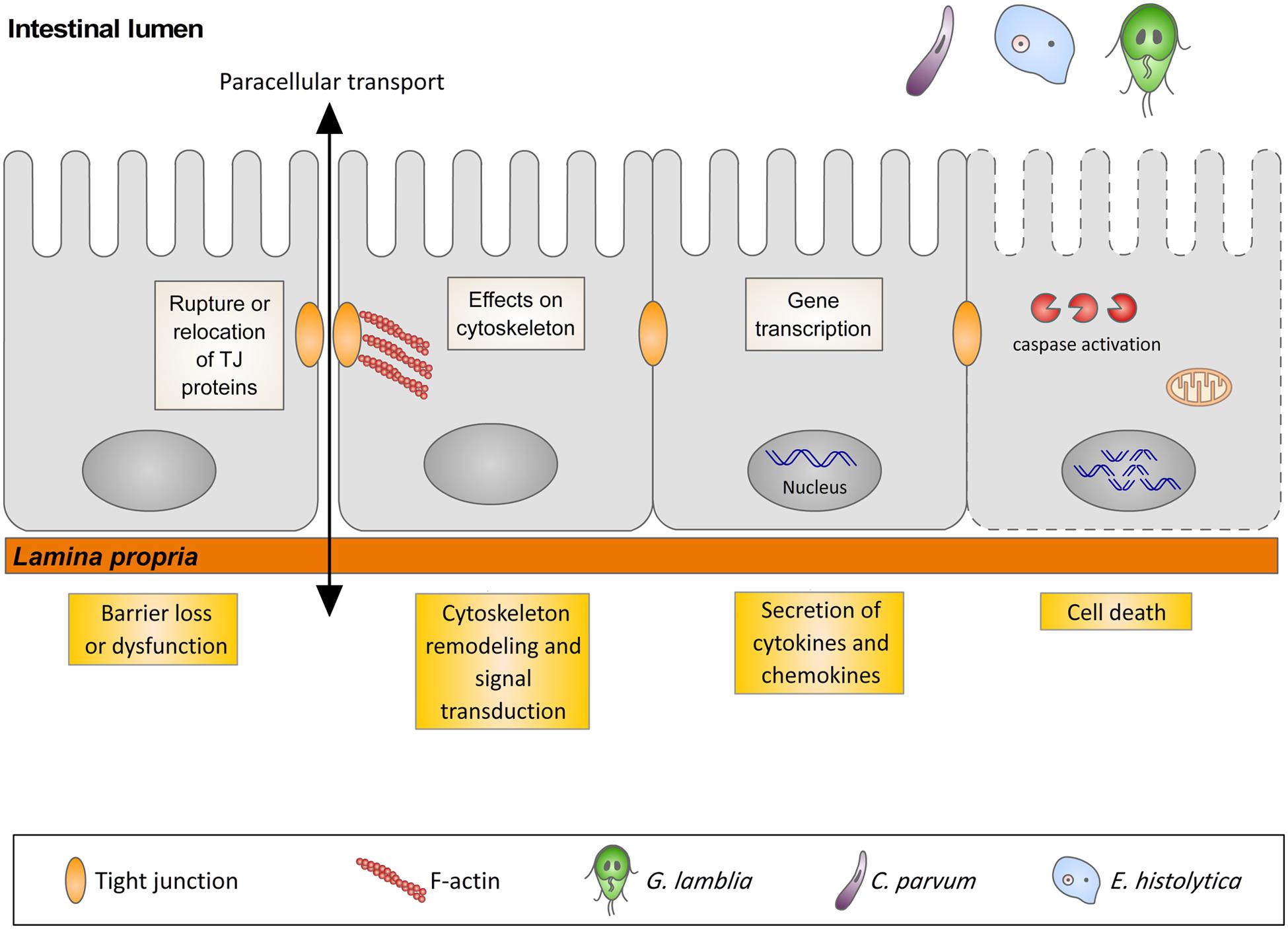
FIGURE 1. Illustration representing the interaction of G. lamblia, C. parvum and E. histolytica with the intestinal epithelium and the host cells responses.
Giardia lamblia and Giardiasis
The genus Giardia comprises many species that inhabit the intestinal tract of a series of vertebrate hosts including domestic animals, rodents, dogs, cats, livestock, and wildlife. However, one species, G. lamblia (synonyms G. duodenalis and G. intestinalis), is known to infect and cause giardiasis in humans and mammals, suggesting a zoonotic transmission (Ryan and Cacciò, 2013).
Giardia lamblia has a simple lifecycle comprising two morphogenetic stages, the infectious and environmentally resistant cyst stage and the vegetative trophozoite stage, which colonizes the small intestine epithelium and causes the disease. Infection initiates when a host ingests viable cysts directly or with contaminated water and food (the infective dose for a symptomatic infection is about 10–100 cysts). After passing through the stomach, cysts begin excysting (excystation process), releasing two trophozoites in the upper part of the small intestine where they adhere to the cells lining the intestinal lumen (enterocytes) through an adhesive ventral disk and multiply by binary fission. Under suitable environmental conditions (i.e., increased bile salt concentration and cholesterol deprivation), trophozoites transform into cysts (encystation process) that are excreted and passed with the feces, thus completing their lifecycle (Gillin et al., 1988; Luján et al., 1996).
Host Cell Transcription during Giardia lamblia Infection
As mentioned, G. lamblia trophozoites are confined to the lumen of the intestinal tract of humans and animals. To cause disease they must “swim” through the intraluminal fluid flow, overcome peristaltic motions, evade host immunological defense mechanisms and replicate attached to the small intestine mucosal surface. Colonization of the host intestinal epithelium by trophozoites is mediated by an adhesive, microtubule-based organelle denominated the ventral disk (Holberton, 1973; Adam, 2001; Schwartz et al., 2012). Specific molecular mechanisms may be involved, with a range of Giardia cell-surface constituents, including lectins and saccharides, being described as ligands for host cell attachment (Farthing et al., 1986; Inge et al., 1988; Ward et al., 1988; Céu Sousa et al., 2001). Membrane rafts present at the surface of trophozoites are also implicated in trophozoite adhesion to human enterocyte-like cells, as parasite treatment with methyl-β-cyclodextrin (a lipid raft disorganizing agent) resulted in diminished attachment of G. lamblia to Caco-2/TC7 cells (Humen et al., 2011). Independent of whether adhesion results from a mere mechanical adhesion through the ventral disk or involves ligand–receptor interactions, the host cells are not passive recipients for Giardia attachment but are active participants, having evolved specialized strategies to resist infection. These include, for example, the amplification and regulation of the expression of genes, many of which are involved in the immunological defense of host cells to Giardia (Ferella et al., 2014). The importance of the early transcription of genes coding for components of intestinal mucosal immunity can be appreciated by studies using animal models. For example, it has been shown that infection of C57BL/6 mice with G. muris induces upregulation of interleukin 17A starting 1 week post-infection (Dreesen et al., 2014). The immune-modulating cytokine interleukin-6 (IL-6) is also involved in the control of G. lamblia infection, as IL-6-deficient mice were not able to handle the acute phase of the disease and developed chronic giardiasis (Zhou et al., 2003). Importantly, reverse transcription-PCR-based quantitation of cytokine mRNA levels in peripheral lymph node cells exhibited a short-term upregulation of IL-4 expression in IL-6-deficient mice that seemed to be associated with failure to control the parasite population (Zhou et al., 2003).
Recently, host gene expression of mice whole small intestinal tissue following G. lamblia infection has been analyzed using oligonucleotide arrays (Tako et al., 2013). The results from this analysis indicated that genes associated with antibodies, mast cell proteases and matrix metalloprotease 7 (Mmp7) were upregulated (Tako et al., 2013). The role of Mmp7 was then confirmed in vivo, as Mmp7-deficient mice presented increased numbers of trophozoites in the small intestine when compared to control animals (Tako et al., 2013). In mice, the Mmp7 gene encodes the processing proteinase of murine Paneth cell defensins, a class of antimicrobial peptides important in the innate immune response of the small intestines (Ostaff et al., 2013). G. lamblia trophozoites are lyzed by α-defensin peptides in vitro (Aley et al., 1994), making it plausible to consider that Mmp7-deficient mice were unable to clear the infection with Giardia probably due to their inability to release or to process antimicrobial defensins from intestinal epithelia.
In vitro as well as in vivo models for genome-wide analysis of gene expression have also contributed to understanding the epithelial cell response to G. lamblia infection. Using differentiated Caco-2 human cell line as a model of the intestinal epithelium, Roxström-Lindquist et al. (2005) demonstrated that co-incubation of cells with G. lamblia resulted in the upregulation of genes coding for chemokines (CCL2, CCL20, CXCL1, CXCL2, CXCL3) and stress-induced genes like c-Fos, c-Jun and immediate-early response 3 (IER3), to cite a few examples. In addition, genes involved in cellular proliferation (G0S2, PCNA, ORC5L, MCM2, MCM3) had their expression reduced after 6 and 18 h of co-incubation of host cells with trophozoites. Therefore, it appears that infection with Giardia interferes with the transcription of host genes involved in innate immunity and prevents cell turnover, probably to maintain a stable niche for colonization as suggested by Stadelmann et al. (2012).
Giardia lamblia Infection and Host Cell Death
At a certain point, the physical attachment of trophozoites to epithelial cells may target specific signaling networks, provoking downstream events that impair normal organ function and lead to associated signs and symptoms of giardiasis. In this scenario, the most striking outcome of Giardia–host interaction may be considered the activation of cell death mechanisms such as apoptosis. Apoptosis is described as a regulated and controlled process of autonomous cell death that avoids eliciting inflammation (Elmore, 2007). It can be activated extrinsically by receptor-mediated signaling (through death receptor ligation and DISK assembly) or intrinsically by disruption of the mitochondria. In the first case, the major effectors of apoptosis are a family of aspartic acid-specific proteases known as caspases (Thornberry and Lazebnik, 1998). These are normally synthesized as inactive precursors, but become activated at the onset of apoptosis by activation signals. At the initiation of apoptosis, different sets of caspases are activated depending on whether cell death was triggered extrinsically (caspases 8 and 10) or intrinsically (caspases 9 and 2). However, propagation of the apoptosis signal relies on the direct cleavage of a downstream effector caspase such as caspase-3 that, in turn, cleaves key substrates, producing many of the cellular and biochemical events of apoptosis (Thornberry and Lazebnik, 1998; Slee et al., 2001; Elmore, 2007).
In the gastrointestinal tract, epithelial cells are organized as a single cell layer that covers the entire tissue. They are originated at the crypt and migrate up to the villous tip where they are constantly renewed by extrusion to the lumen. At this point, the turnover of cells is extremely fast (5–7 days) and crucial for the maintenance of normal organ morphology and function. A number of studies have demonstrated that this process is highly regulated and can only be maintained by balancing the levels of cell death and proliferation (Günther et al., 2013). In the gut, cell death occurs through the activation of apoptosis and, although important for gut homeostasis, excessive enterocyte death has been associated with different disorders of the gastrointestinal tract including ulcerative colitis, celiac disease and Crohn’s disease (Ciccocioppo et al., 2001; Di Sabatino et al., 2003; Turner, 2009). Infection of human ileocecal adenocarcinoma cell line HCT-8 with G. lamblia can also induce host cell apoptosis. In this case, signs of chromatin condensation were observed within the nuclei of intestinal cell monolayers exposed to different G. lamblia assemblages (A, B, and E) or with a combination of these assemblages (Koh et al., 2013). In addition, caspase-3 activation was found to occur in G. lamblia-induced apoptosis, as nuclear fragmentation and cell death was effectively suppressed by a caspase-3 inhibitor (Koh et al., 2013). Interestingly, the apoptotic response elicited by Giardia was not dependent on co-incubation of host cells with live trophozoites, as sonicated parasites induced caspase-3-dependent apoptosis of non-transformed human duodenal epithelial cell line (SCBN; Chin et al., 2002). Confirming these data, morphological changes consistent with apoptosis and activation of caspase-3 were also observed by others (Panaro et al., 2007). The results from this work indicated that initiation of apoptosis occurred through the activation of caspase-8 and caspase-9, demonstrating that both the intrinsic and extrinsic pathways were triggered during Giardia infection (Panaro et al., 2007). At the same time, a significant downregulation of Bcl-2 and increased expression of the pro-apoptotic protein Bax were observed, being the first demonstration of the participation of members from the Bcl-2 family in the induction of enterocyte apoptosis during Giardia–host cell interaction (Panaro et al., 2007). Bcl-2 family proteins, which have either pro-apoptotic (Bax and Bak) or anti-apoptotic activities (Bcl-2, Bcl-XL, and induced myeloid leukemia cell differentiation protein 1 or Mcl-1), are critical regulators of caspase activation and apoptosis (Adams and Cory, 1998, 2001). In mammalian cells, overexpression of Bax was associated with loss of mitochondrial membrane potential, cytosolic accumulation of cytochrome c, caspase activation, cleavage of poly(ADP-ribose)-polymerase (PARP), DNA fragmentation, and cell death (Oliver et al., 1998). Interestingly, during host cell infections with G. lamblia, cleavage of PARP occurred upon activation of caspase-3 (Panaro et al., 2007). Taken together, these data indicate that downregulation of Bcl-2 and upregulation of Bax is associated with the activation of both the intrinsic and extrinsic apoptotic pathways during G. lamblia infection. In this scenario, the activated initiators caspase-8 and caspase-9 trigger executioner caspase-3, leading to the proteolytic cleavage of PARP and induction of apoptosis (Panaro et al., 2007).
While the critical role of apoptosis in the pathophysiology of giardiasis is well-documented, host responses to prevent cell death during Giardia infection remain poorly understood. In an attempt to shed light on this issue, some authors postulated that inhibition of apoptosis in Giardia-infected cells would involve the upregulation of sodium-dependent glucose cotransporter (SGLT)-1 as demonstrated by bacteria. In this work, Yu et al. (2005) described that activation and enhanced glucose uptake into enterocytes rescued cells from lipopolysaccharide (LPS)-induced apoptosis via SGLT-1. In G. lamblia infections, it was shown that, when SGLT-1-transfected Caco-2 cells were exposed to trophozoite products in high (25 mM) glucose media, host cell apoptosis was abolished (Yu et al., 2008). In addition, a soluble proteolytic fraction of G. lamblia was found to upregulate SGLT-1-mediated glucose uptake in association with increased apical SGLT-1 expression in epithelial cells (Yu et al., 2008). These findings indicated that SGLT-1-dependent glucose uptake might represent a novel epithelial cell rescue mechanism against G. lamblia-induced apoptosis.
Disassembly of Tight Junctions and Cytoskeleton Reorganization during Giardia lamblia Infection
The intestinal epithelium is formed by a single layer of epithelial cells that function as a physical barrier between the lumen and the subepithelial tissue. It prevents the entrance of microorganisms, luminal antigens, and toxins to the mucosal tissue; controls the paracellular movement (transport in the space between epithelial cells) of water, solutes, and macromolecules; and regulates cell proliferation, polarization, and differentiation (Turner, 2009). Within the barrier, epithelial cells are held together by complex structures of tetraspan (claudins, occludins, and tricellulin) and single-span transmembrane proteins known as TJs or zonulae occludentes (ZOs; Balda and Matter, 2008; Turner, 2009). TJs form a continuous belt-like structure that completely encircles the cell at the apical region of the plasma membrane. They also work as signaling platforms, as junctional proteins like ZO-1 interact with cytoskeleton actin through a plaque of cytoplasmic proteins localized under the junction (Matter and Balda, 2003; Turner, 2009). Due to their importance in providing adhesive contacts between neighboring cells and in controlling the permeability of the intestinal barrier, it is not surprising that disruption or reduced expression of TJ proteins and loss of epithelial barrier function have been associated with many intestinal disorders including giardiasis (Teoh et al., 2000; Buret et al., 2002; Chin et al., 2002; Müller and von Allmen, 2005; Buret, 2007; Troeger et al., 2007; Koh et al., 2013). Indeed, some in vitro studies demonstrated that co-incubation of different cell lines (SCBN and HCT-8) with trophozoites results in the disruption of the TJ protein ZO-1 which, in some cases, leads to increased cell permeability (Buret et al., 2002; Chin et al., 2002; Koh et al., 2013). Disruption of ZO-1 by Giardia was associated with caspase-3-dependent apoptosis, as the loss of this protein was abolished in cells treated with caspase-3 inhibitors prior to infection (Chin et al., 2002; Koh et al., 2013). In line with these observations, apoptosis has been observed in many diseases involving TJ disruption (Zeissig et al., 2007; Su et al., 2013). However, whereas in some of these diseases apoptosis is a downstream response to loss of junctional proteins, in giardiasis this fact has yet to be determined.
In recent years, a body of evidence has indicated that loss of TJs may result not only from reduced expression of junctional components but also as a consequence of their relocation/reorganization within cells. For example, in Alzheimer’s disease, it was shown that endothelial cells exposed to β-amyloid peptide (Aβ) display a disrupted plasma membrane pattern of claudin-5 and ZO-2, which are relocated to the cytoplasm (Marco and Skaper, 2006). During enteropathogenic E. coli infections, loss of occludin association with claudin-1 and ZO-1 was observed to occur due to the translocation of apically localized TJ proteins to the lateral membrane (Muza-Moons et al., 2004). In this case, the E. coli-induced reorganization of junctional complexes resulted in decreased transepithelial electrical resistance (TEER) and disruption of the intestinal barrier (Muza-Moons et al., 2004). In G. lamblia infections, relocation of TJ proteins ZO-1 and claudin-1 from the cell–cell contact region to the cytoplasm were shown to occur during co-incubation of Caco-2 cells with trophozoites. Moreover, F-actin was retracted and concentrated near cellular contacts, resulting in microvillous atrophy as observed by scanning electron microscopy (Maia-Brigagão et al., 2012). This is in accordance with previous observations describing that infections of colonic cells with Giardia induced localized condensation of F-actin, loss of perijunctional α-actinin and increased cell permeability (Teoh et al., 2000). The mechanism by which epithelial TJs and cytoskeleton were disassembled involved the post-translational modification of the myosin light chain (MLC) of myosin II by MLC kinase (MLCK), as exposition of cells with Giardia triggered MLC phosphorylation (Scott et al., 2002). The importance of MLCK in TJ and cytoskeleton disassembly was further reinforced by the observation that co-incubation of cells with a specific MLCK inhibitor blocked the effects of Giardia on epithelial permeability, F-actin, and ZO-1 (Scott et al., 2002).
The hypothesis that TJ disruption, either by reduced expression or relocalization of junctional components, has an important role in barrier function in giardiasis is supported by in vivo data. In this case, analysis of duodenal biopsy specimens from patients with chronic giardiasis has shown reduced claudin-1 expression and serious villous shortening (Troeger et al., 2007). These were accompanied by a decrease in the absorptive capacity of the duodenum and active anion secretion as evidenced by reduced Na2+-coupled D-glucose absorption and electrophysiological measurements, respectively (Troeger et al., 2007).
Therefore, it can be concluded that the activation of the MLCK signaling pathway during giardiasis relates to loss of TJ proteins, cytoskeleton rearrangement and barrier dysfunction, which can contribute to the pathophysiological mechanisms underlying diarrhea such as electrolyte secretion and malabsorption. A summary of the studies about the pathophysiology of Giardia infections is listed in Table 1.
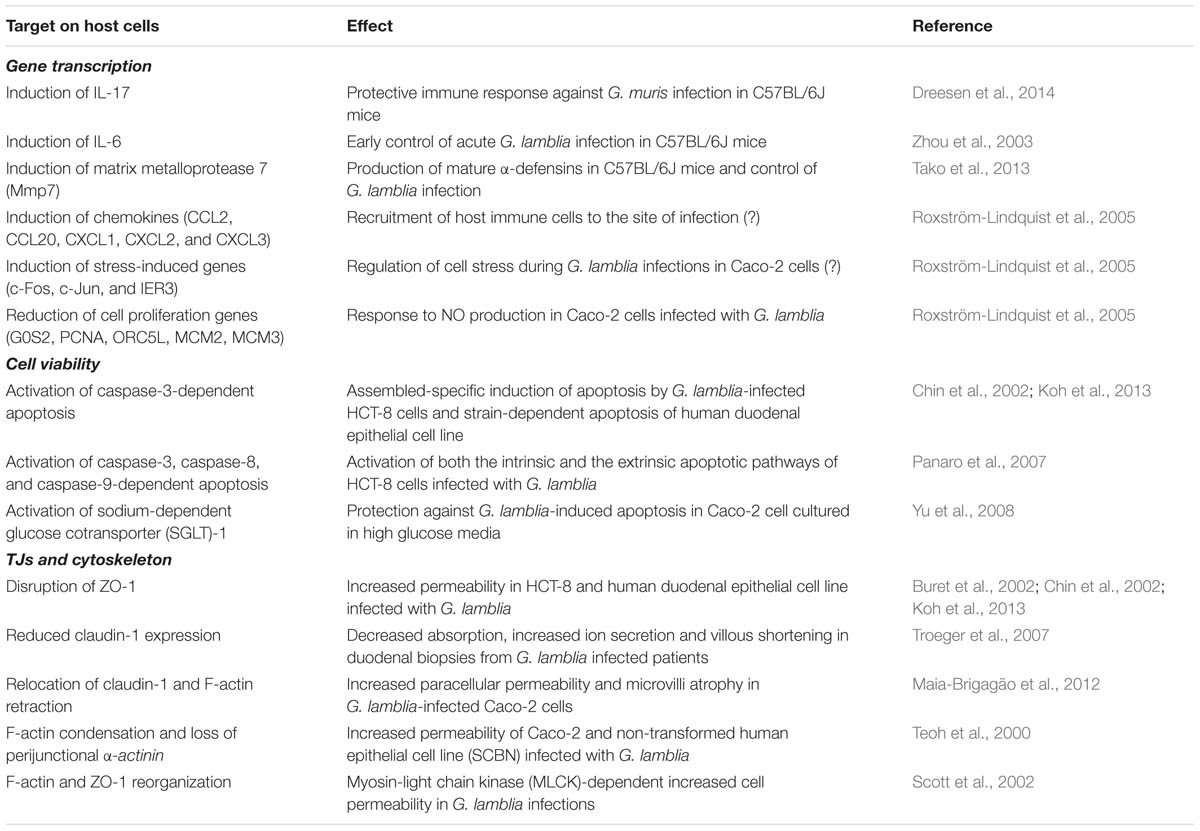
TABLE 1. Summary on studies describing the effect of Giardia lamblia infection on host cell responses.
Cryptosporidium parvum
Cryptosporidium parvum is an obligatory intracellular intestinal parasite of humans and animals and is responsible for many cases of cryptosporidiosis worldwide. Cryptosporidiosis is characterized as a watery diarrhea and is a potentially life-threatening disease in both immunocompetent and immunosuppressed hosts. The parasite exists in the environment as an oocyst that contains four sporozoites. When ingested by a host (by fecal-oral contact or by contaminated drinking or recreational water), the oocyst travels through the gut lumen to the small intestine where the sporozoites are released by excystation. Motile sporozoites then attach to the intestinal epithelium and are enveloped by the host cell apical membrane forming an extracytoplasmic parasitophorous vacuole inside which the parasite undergoes asexual multiplication. The sporozoites then enter a sexual reproductive stage and develop into female macrogamonts and male microgamonts. After fertilization, the zygote can develop into two types of oocysts: (a) a thick-walled oocyst that is excreted into the environment or (b) a thin-walled oocyst that can auto-infect the host (Petersen, 1993).
Cryptosporidiosis is characterized by watery diarrhea, malabsorption and wasting. The pathophysiology of cryptosporidiosis is multifactorial, and three pathophysiological mechanisms have been proposed to occur during infection: first, infiltration of the lamina propria by host immune cells (lymphocytes, macrophages, and neutrophils), responsible for inflammatory diarrhea; increased transepithelial permeability, villous atrophy, crypt hyperplasia and cell death, characteristic of exudative diarrhea; and malabsorption due to loss of the intestinal architecture relating to osmotic diarrhea. In the following section, the events preceding the development of diarrhea during C. parvum infection are described.
Host Cell Transcription during Cryptosporidium Infection
As a member of the phylum Apicomplexa, which also includes Plasmodium and Toxoplasma sp., C. parvum is equipped with a specialized apical apparatus named the apical complex. This complex is composed of secretory organelles such as rhoptries, micronemes and dense granules, which play distinct roles during Cryptosporidium–host cell interaction. As an obligatory intracellular parasite, invasion of cells by Cryptosporidium is mandatory and is preceded by the adhesion of parasites to intestinal epithelial cells. Adherence allows the parasite to anchor itself to the epithelial layer, and this process has been shown to be mediated by the presence of adhesins like thrombospondins (TSPs) and thrombospondin-related adhesive protein (TRAP)-C1, and glycoproteins such as mucins and mucin-like proteins (GP900), to cite a few examples (Wanyri and Ward, 2006). Following adherence, parasites are internalized, resulting in subtle changes of host cell gene expression such as the development of a mechanism for evading the host cell immune response. Indeed, several studies have described changes in certain selected host cellular genes due to Cryptosporidium infection (Castellanos-Gonzalez et al., 2008; Zhou et al., 2009). For example, microarray analysis of human ileal mucosa explants infected with C. parvum or C. hominis demonstrated increased expression of osteoprotegerin (OPG) mRNA compared to uninfected cells (Castellanos-Gonzalez et al., 2008). The relevance of this finding was further extended, as jejunal biopsy specimens obtained from a volunteer (before and after experimental infection with C. meleagridis) displayed a 1281-fold increase in OPG mRNA post-infection (Castellanos-Gonzalez et al., 2008). OPG is a soluble glycoprotein produced by osteoblasts, intestine cells, hematopoietic and immune cells (dendritic cells and lymphocytes; Simonet et al., 1997; Vidal et al., 2004). It is a member of the TNF superfamily which includes proteins such as TNF-α, Fas/FasL and TRAIL, which are involved in cell differentiation, proliferation, survival, apoptosis and in immune responses (Simonet et al., 1997). Various human intestinal epithelial cell lines were reported to constitutively express OPG, especially during inflammation, suggesting that it may play an important role as a mucosal immunoregulatory factor (Vidal et al., 2004). Whether OPG exerts its biological effect in response to cell inflammation during C. parvum infection remains to be elucidated. Nonetheless, data obtained by Castellanos-Gonzalez et al. (2008) demonstrated that OPG is produced during the early stages of C. parvum infection blocking TRAIL-mediated apoptosis of host cells, indicating OPG as a protective factor in cryptosporidiosis.
Cytokines and chemokines are proteins that regulate inflammation and modulate cellular activities such as growth, survival and differentiation (Zlotnik, 2000; Dinarello, 2007). They may act as pro- or anti-inflammatory factors (cytokines) or as chemotactic attractants (chemokines) to leukocytes and in trafficking of immune cells (Dinarello, 2007). They can be produced by a series of cells such as T helper cells (Th) and macrophages but also by intestinal epithelial cells (Stadnyk, 2002). Reports on the pathogenesis of cryptosporidiosis have shown increased mRNA levels for cytokines (IL-1β, IL-4, IL-8, IL-14, IL-15, IFN-γ, TGF-β) and chemokines (C-C and C-X-C subfamilies) in human and murine intestinal cells and xenografts infected with C. parvum (Laurent et al., 1997; Seydel et al., 1998b; Robinson et al., 2000, 2001; Lacroix-Lamandé et al., 2002; Deng et al., 2004; Tessema et al., 2009). These results are consistent with previous observations showing the recruitment of effector cells to the site of inflammation in the intestinal lamina propria from Cryptosporidium-infected patients (Robinson et al., 2001), as chemokines may control the localization of immune cells throughout the body (Griffith et al., 2014). Therefore, these data suggest that expression of multiple cytokines and chemokines by host cells may play an important role in the control of inflammation during Cryptosporidium infections. More recently, fractalkine or CX3CL1, a membrane-bound chemokine of the CX3C family, was shown to be upregulated in human biliary epithelial cells following C. parvum infection. Induction of CX3CL1 expression involved downregulation of microRNAs (miR-424 and miR-503) both known to target the CX3CL1 3′ UTR, suppressing its translation and inducing RNA degradation (Zhou et al., 2013). MicroRNAs are non-coding, single-stranded RNAs that negatively regulate gene expression through interactions with 3′ UTRs of the target mRNA (Bartel, 2009).
Defensins are a family of antimicrobial peptides expressed by different cells including Paneth cells in the epithelium of the small intestine. They are subdivided into two families, the α-defensins (also known as cryptdins in mice) and the β-defensins, both displaying microbicidal activity (Bevins and Salzman, 2011). Experiments with human colonic (HT29) and murine rectal adenocarcinoma (CMT-93) cell lines infected with C. parvum have shown differential β-defensin gene expression (Zaalouk et al., 2004). Indeed, using reverse transcription-PCR, a reduction in human-defensin-1 (hBD-1) and induction of hBD-2 were observed in Cryptosporidium-infected colonic cells. Furthermore, enterocytes infected with C. parvum and treated with recombinant hBD-1 and hBD-2 showed a reduction in the percentage of viable sporozoites, indicating that these peptides may have an important role in the host’s innate response against infection (Zaalouk et al., 2004). Upregulation of inducible nitric oxide synthase (iNOS) was also shown to occur in neonatal piglets during acute C. parvum infection. Curiously, expression of iNOS was not restricted to infected cells, possibly indicating a non-specific response against Cryptosporidium infection, although the importance of iNOS in the control of tissue parasitism was further confirmed, as inhibition of iNOS activity resulted in increased parasite burden in intestinal epithelial cells (Gookin et al., 2006). Finally, iNOS induction was shown to be NF-κB dependent, as iNOS activity was abolished when infected cells were incubated with lactacystin (a proteasome inhibitor that prevents degradation of IκBα; Gookin et al., 2006). These data, together with the demonstration that iNOS expression by macrophages and other cell types occurs in tissues from patients with a wide variety of infectious diseases (Bogdan, 2001), may suggest a protective role for nitric oxide in cryptosporidiosis. Further experiments are needed to confirm this hypothesis in the human disease.
Cryptosporidium parvum Infection and Host Cell Death
The first report on the occurrence of apoptosis in C. parvum-infected human biliary epithelial cells (H69 cells) was issued by Chen et al. (1998). Later, nuclear condensation and DNA fragmentation (as markers of apoptosis) during C. parvum infection were shown to be caspase-dependent and induced by Fas/FasL, as caspase inhibitors or neutralizing antibodies to either the Fas receptor (Fas) or Fas ligand (FasL) blocked these events (Chen et al., 1999; Ojcius et al., 1999). Cryptosporidium-induced apoptosis was documented to occur independent of cell line (CaCo-2, MDBK, and HCT-8 cells) and resulted in impaired C. parvum development in vitro, suggesting a host–cell mechanism to control the spread of infection (Widmer et al., 2000). Further studies, however, demonstrated that apoptotic changes of intestinal epithelial cells were modulated by the C. parvum development stages and displayed biphasic activation with early inhibition (at the trophozoite stage) and late moderate promotion (at the sporozoite and merozoite stages; Mele et al., 2004; Liu et al., 2009). On the basis of these data, it has been suggested that Cryptosporidium precisely regulates host cell apoptosis to favor its growth and development at initial stages of infection, and to promote its propagation later on.
In line with this hypothesis, namely that Cryptosporidium can exert control on the processes that regulate apoptosis in the host, Chen et al. (2001) have shown that C. parvum-infected biliary cells activated the NF-κB signaling cascade, leading to secretion of the pro-inflammatory cytokine IL-8 and inhibition of cell apoptosis. Moreover, these events were restricted to infected cells given that C. parvum-induced apoptosis was limited to bystander uninfected cells (Chen et al., 2001). Inhibition of host cell apoptosis during Cryptosporidium infection has also been reported to involve the expression of members of the IAP family (inhibitors of apoptosis proteins) such as c-IAP1, c-IAP2, XIAP, and survivin. In this case, it was demonstrated that knockdown of survivin (but not that of c-IAP1, c-IAP2, or XIAP) by siRNA enhanced caspase-3/7 activity and resulted in increased host cell apoptosis and decreased C. parvum infection (Liu et al., 2008). The role of IAPs in the control of cell death in cryptosporidiosis is reinforced by a study showing that XIAP mediated proteasome-dependent inhibition of activated caspase-3 in C. parvum infection (Foster et al., 2012).
Disassembly of Tight Junctions during Cryptosporidium Infection
Dysfunction of the epithelial barrier during in vivo intestinal infections with Cryptosporidium has been documented in humans and animals. Villous atrophy, hyperplasia of the crypt epithelium and increased transepithelial permeability are some of the abnormalities reported in cryptosporidiosis (Genta et al., 1993; Adams et al., 1994; Gookin et al., 2002). In vitro studies on Cryptosporidium andersoni-infected human (Caco-2) and bovine (MDBK and NBL-1) epithelial cells reported disruption of ZO-1 and nuclear fragmentation during infection (Buret et al., 2003). Interestingly, both events were reversed by pretreatment of host cells with recombinant human epidermal growth factor (rhEGF), and significantly reduced infection rates in bovine and human enterocytes (Buret et al., 2003). The relationship, if any, between C. andersoni-induced ZO-1 disruption and loss of barrier function is still unknown. Moreover, how EGF exerts its biological effect during Cryptosporidium infection deserves further investigation. Table 2 summarizes work on Cryptosporidium.
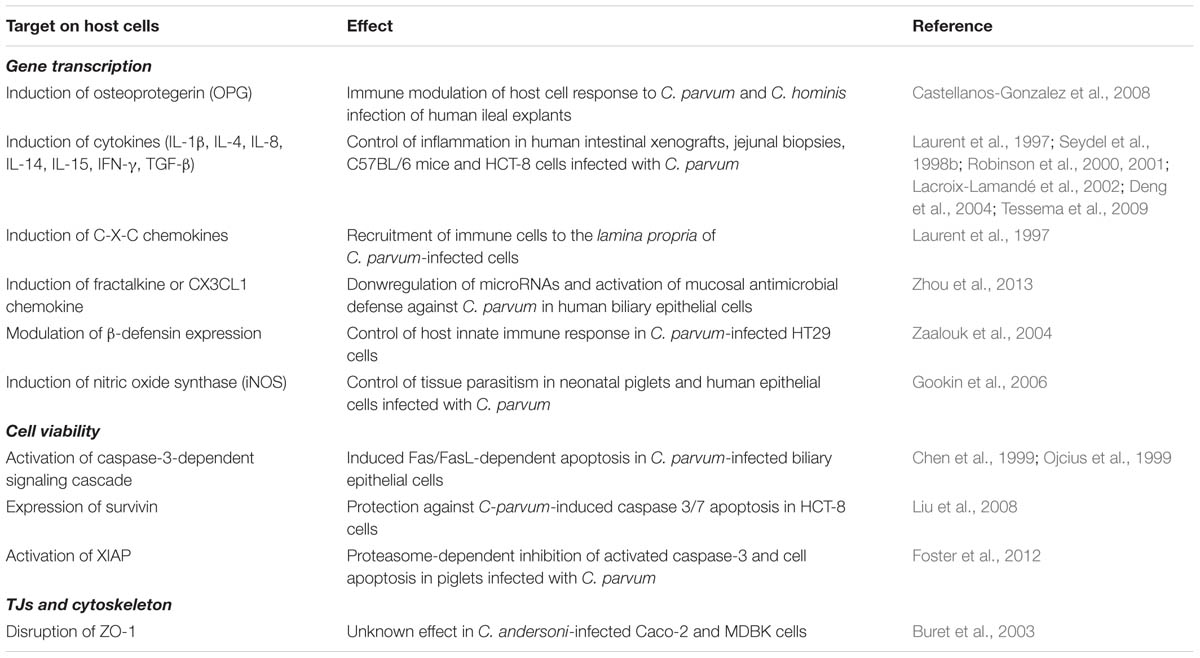
TABLE 2. Summary on studies describing the effect of Cryptosporidium infection on host cell responses.
Entamoeba histolytica
Entamoeba histolytica is a protozoan parasite that colonizes the large intestine of humans causing amoebiasis. Although most infections with E. histolytica are asymptomatic, some patients may experience clinical manifestations of invasive amoebiasis such as amoebic colitis and amoebic liver abscess (Marie and Petri, 2014).
Entamoeba histolytica has a simple lifecycle that involves two distinct morphogenetic stages, the amoeboid and proliferative trophozoite, and the infectious cyst form. Human infections begin with ingestion of the viable cysts in food or water that has been contaminated by feces. Excystation occurs in the small intestine, and released trophozoites migrate to the colon where they multiply by binary fission. In the end, trophozoites encyst, completing the lifecycle when they are excreted into the environment in stool (Marie and Petri, 2014). Trophozoite adhesion to colonic mucus and epithelial cells is a critical step in the colonization of the large intestine by E. histolytica and a Gal/GalNAc lectin (260 kDa) expressed at the parasite surface was shown to mediate its binding to host mucins and cell surface carbohydrates (Frederick and Petri, 2005).
Host Cell Transcription during Entamoeba histolytica Infection
It is well-known that infection with the protozoan parasite E. histolytica results in significant inflammatory responses that contribute to tissue damage and invasion. In vitro studies using co-culture of human epithelial and stromal cells and cell lines (HeLa, HT29, and T84) demonstrated an upregulation of IL-8 transcripts during E. histolytica infections that correlated with increased secretion of this pro-inflammatory cytokine and others such as GROα, GM-CSF, IL-6, and IL-1α (Eckmann et al., 1995). Increased mRNA for both IL-1β and IL-8 were also reported to occur in vivo when mouse–human intestinal xenografts (SCID-HU-INT) were infected with E. histolytica trophozoites (Seydel et al., 1997). The relevance of these findings was further reinforced when intraluminal administration of an antisense oligonucleotide (to block the production of IL-1β and IL-8) inhibited the gut inflammatory response to E. histolytica infection (Seydel et al., 1998a).
In human patients with acute or convalescent amoebiasis, gene expression profiles obtained by microarray analysis of intestinal biopsies clearly demonstrated upregulation of REG1A and REG1B genes (Peterson et al., 2011). REG1A and REG1B belong to the regenerating islet-derived (REG) gene family encoding for C-type lectin-like proteins (Parikh et al., 2012). They are involved in the proliferation and differentiation of diverse cell types and are well-known to be highly expressed in some pathologies as inflammatory diseases, cancer and diabetes (Parikh et al., 2012). One member of this family, the REG1α protein, was also shown to mediate the anti-apoptotic effect of STAT3 in cancer cells (Sekikawa et al., 2008). During E. histolytica infections, expression of REG1α and REG1β were shown to inhibit parasite-induced apoptosis in vitro as REG1-/- mice were found to be more susceptible to cell death.
Entamoeba histolytica Infection and Host Cell Death
As the name suggests, E. histolytica (histo: tissue and lytica: destroyer) is a tissue-destroying amoeba (Pinilla et al., 2008) and host cell apoptosis is one of the most common events associated with infections with this parasite (Huston et al., 2000; Christy and Petri, 2011; Marie and Petri, 2014). The mechanisms leading to host cell death in E. histolytica infections are not completely understood. However, apoptosis and trogocytosis have been reported to occur in amoebiasis without parasite penetration within host cells. Apoptosis was first suggested to occur during E. histolytica infections as DNA fragmentation was observed after trophozoite adhesion to a murine myeloid cell line (FDC-P1; Ragland et al., 1994). Cell killing by E. histolytica was further shown to occur via a Bcl-2-independent mechanism, as FDC-P1 cells transfected with a retrovirus construct to express the Bcl-2 protein were susceptible to amoeba contact-dependent killing (Ragland et al., 1994). Later, using Jurkat cells it was demonstrated that infection with E. histolytica rapidly activated caspase-3, independently of caspase-8 and -9 activation (Huston et al., 2000). Interestingly, E. histolytica activation of caspase-3 was followed by phagocytosis of host cells, suggesting that cell killing precedes ingestion by trophozoites (Huston et al., 2003). Over the past years, diverse studies have tried to elucidate the pathways explored by E. histolytica to trigger host cell apoptosis. For example, in hepatocytes, live E. histolytica was shown to induce an apoptosis-like death without the participation of both Fas and TNF-α pathways (Seydel and Stanley, 1998). Teixeira and Mann have observed that adhesion of E. histolytica trophozoites to Jurkat cells induced a contact-dependent protein dephosphorylation by host cell protein tyrosine phosphatases (PTPs) such as SHP-1 and SHP-2 (Kim et al., 2010), since pretreatment of cells with a PTP inhibitor inhibited amoeba-induced dephosphorylation and cell apoptosis (Teixeira and Mann, 2002). Activation of host cell PTP occurred through a calcium-dependent calpain protease responsible for PTP1B cleavage that led, at last, to cell death (Teixeira and Mann, 2002). Reinforcing these data, activation of host cell calpain by E. histolytica was also observed by others (Kim et al., 2007; Jang et al., 2011) and was shown to modulate the degradation of STAT proteins (STAT3 and STAT5) and NF-κB (p65) in Caco-2 cells (Kim et al., 2014). Furthermore, pretreatment of Caco-2 cells with calpeptin (a calpain inhibitor) or calpain silencing partially reduced Entamoeba-induced DNA fragmentation (Kim et al., 2014).
In recent years, a number of studies have shown that oxidative stress could cause cellular apoptosis via both the extrinsic and intrinsic pathways in health and pathological conditions (Lin and Beal, 2006). In amoebiasis, for example, incubation of human neutrophils with E. histolytica trophozoites triggered NADPH oxidase-dependent production of reactive oxygen species (ROS) and cell apoptosis (Sim et al., 2005). The mechanism involved in Entamoeba-induced ROS generation and apoptosis was associated with ERK1/2 activation, possibly through β2-integrin, as cells pretreated with a MEK1 inhibitor (PD98059) and with a monoclonal antibody to CD18 (anti-integrin β2 subunit) prevented cell apoptosis (Sim et al., 2005, 2007). Phosphatidylinositol-3-kinase (PI-3-kinase) was also involved in ROS production and apoptosis during Entamoeba infection, suggesting that signaling molecules may be key factors in E. histolytica-induced, ROS-dependent apoptosis (Sim et al., 2007). Similar results were reported in colonic Caco-2 and HT-29 cells, as increased levels of intracellular ROS were reported to occur through NOX1 oxidase after cell exposure to trophozoites (Kim et al., 2011, 2013). In this case, cell death was shown to be caspase-independent and the signaling cascade activated during this event is still unknown.
The historical concept that E. histolytica kills cells by apoptosis was recently challenged by Ralston et al. (2014). Using both Caco-2 and Jurkat cells, the authors demonstrated that, immediately after contact with human cells, E. histolytica ingests small fragments of the cell membrane, some containing cellular components like cell cytoplasm and mitochondria (Ralston et al., 2014). Surprisingly, host cells were alive when ingestion of fragments was initiated, and resulted in the elevation of the intracellular amount of calcium before the eventual death of cells as trophozoites detached from corpses. The internalization of cell fragments by E. histolytica was named as amoebic trogocytosis (from the Greek trogo, for nibble) and only occurred with live cells as pre-killed cells are ingested intact (Ralston et al., 2014).
On the whole, the results of these studies demonstrated that E. histolytica infections might result in cell death both by apoptosis and trogocytosis, and that these events might contribute to tissue invasion by the parasite.
Disassembly of Tight Junctions during Entamoeba Infection
It is widely accepted that tissue invasion by E. histolytica is preceded by the interaction of trophozoites with intestinal epithelial cells, and a series of studies have shown that this interaction impacts cell morphology, intercellular contacts and regulation of paracellular transport of molecules across the intestinal epithelium. Indeed, in vitro studies have shown a rapid decrease in transepithelial resistance (TER) and increased mannitol flux during trophozoite interaction with polarized human intestinal Caco-2 and T84 cells (Martinez-Palomo et al., 1985; Li et al., 1994; Leroy et al., 2000). Apical injury of host cells such as loss of brush border in regions of contact between epithelial cells and amoebae was also reported. Importantly, these changes were only observed when Caco-2 cells were co-incubated with live trophozoites but not with amoeba lysates or conditioned medium, indicating that they were not mediated by soluble amoebic cytotoxins (Li et al., 1994; Leroy et al., 2000). In human enteric T84 cells co-cultured with amoebae, decreased TER was associated with changes in TJ proteins like release of ZO-1 from ZO-2, degradation of ZO-1 and dephosphorylation of ZO-2 (Leroy et al., 2000).
Besides E. histolytica-host cell contact, amoebic products also have been shown to be crucial for cellular barrier dysfunctions during parasite infections. For example, prostaglandin E2 (PGE2) secreted by E. histolytica was shown to alter the spatial localization of claudin-4 that resulted in increased sodium ion permeability through TJs (Lejeune et al., 2011). EhCP112, an E. histolytica-secreted cysteine protease, has been shown to digest gelatin, collagen type I, fibronectin, hemoglobin and, most importantly, to destroy MDCK cell monolayers (Ocádiz et al., 2005). When complexed with EhADH112 adhesin, the formed EhCPADH112 complex was shown to co-localize with claudin-1 and occludin at the TJs after the incubation of epithelial MDCK cells with trophozoite extracts. Furthermore, EhCPADH112 induced progressive disruption of the paracellular barrier as measured by TER. Importantly, these effects were reversed when co-cultures were incubated with a protease inhibitor cocktail or a monoclonal antibody against the EhCPADH112 complex (Betanzos et al., 2013). Cysteine proteases are important virulence factors in E. histolytica, and 20 genes encoding for these proteases have been identified on the genome (Bruchhaus et al., 2003). Their role in parasite loss of host cell integrity is highlighted in a study showing that calpain (a calcium-dependent cysteine protease) activation resulted in degradation of paxillin, Cas, vimentin, vinculin, talin, and α- or β-spectrin in Jurkat T cells infected with E. histolytica (Lee et al., 2011). In addition to proteases, Goplen et al. (2013) have shown that E. histolytica expressed a cognate “occludin-like” protein of the host, as revealed by confocal microscopy using antibodies for human occludin. Apical administration of “occluding-like” protein to T84 human colonic epithelial cells resulted in epithelial disruption and decreased TER, suggesting the involvement of this protein in the pathophysiology of amoebiasis. The exact mechanism by which “occluding-like” protein exerts its effects is not completely understood but the authors suggested that it might compete for epithelial occludin–occludin interactions, a hypothesis that needs further investigation. Studies on E. histolytica are summarized in Table 3.
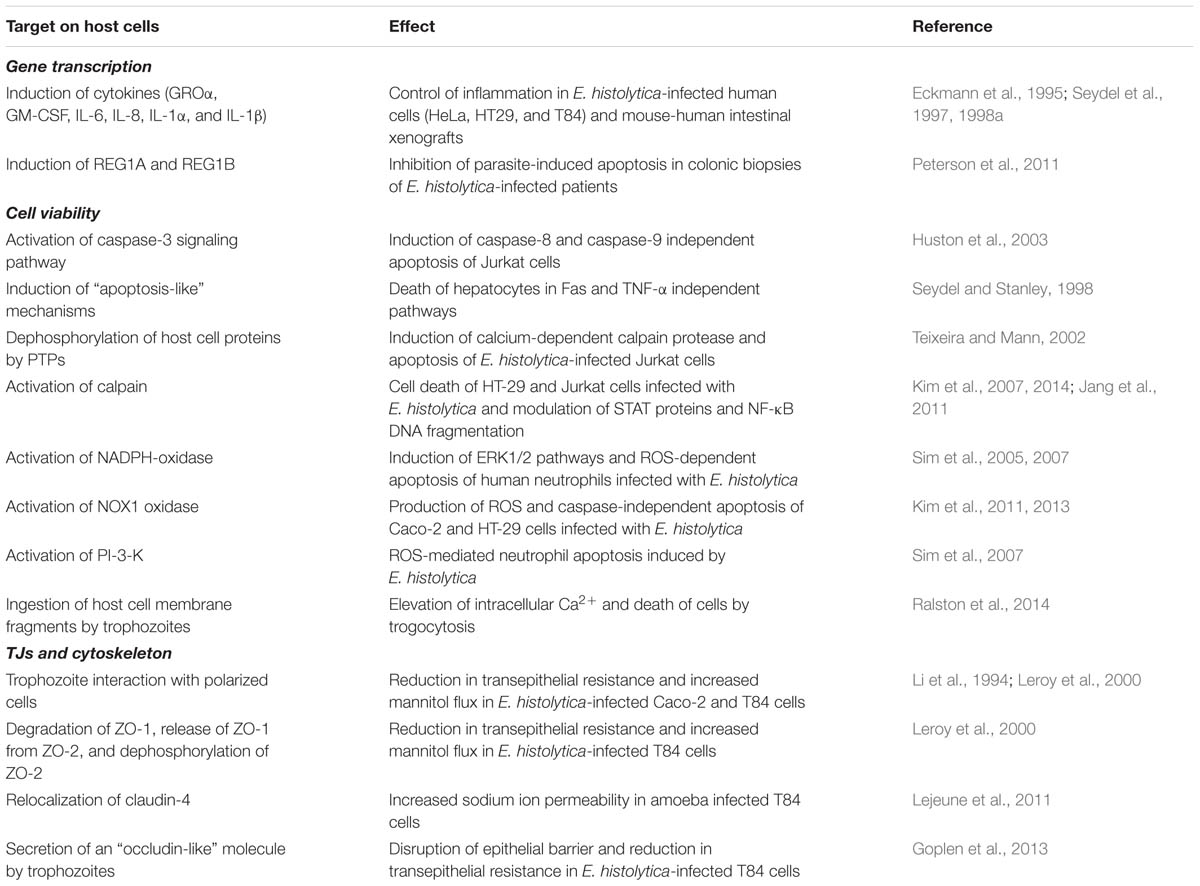
TABLE 3. Summary on studies describing the effect of Entamoeba histolytica infection on host cell responses.
Giardiasis, Cryptosporidiosis, and Amoebiasis: Mechanistic Similarities and Differences
The pathophysiology of diarrhea caused by G. lamblia, Cryptosporidium sp., and E. histolytica is multifactorial and, despite depending on the microbiological agent causing it, these diseases share some mechanistic features (Table 4).
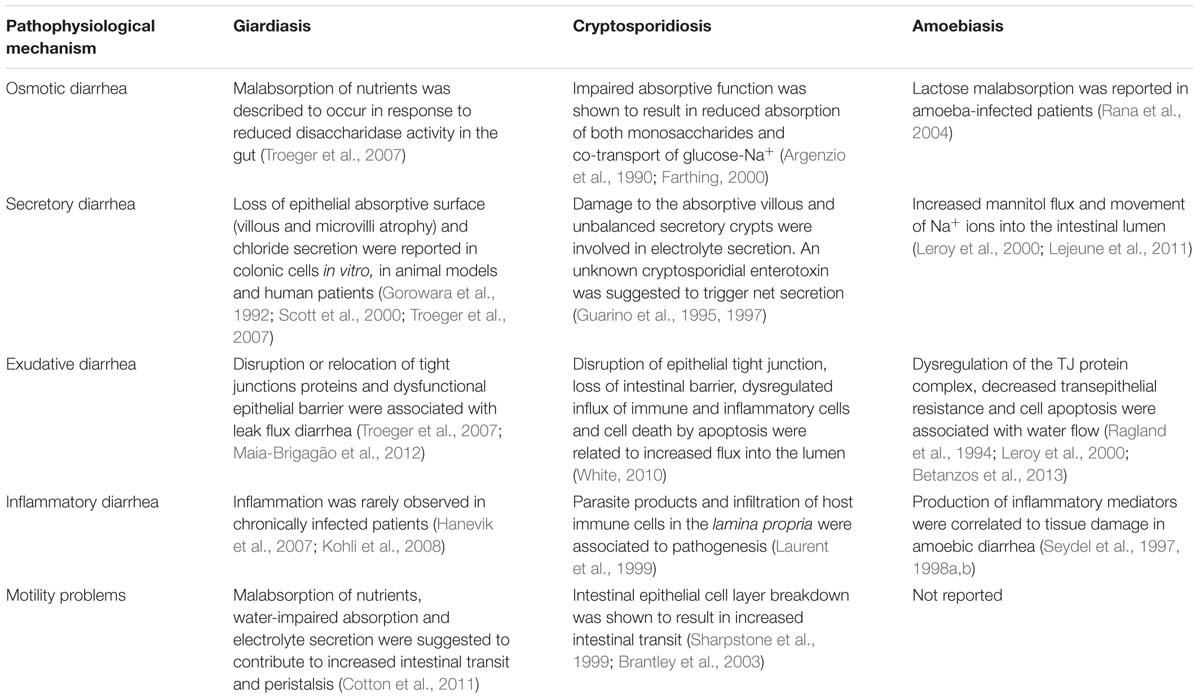
TABLE 4. Pathophysiological mechanisms implicated in diarrhea caused by G. lamblia, Cryptosporidium sp., and E. histolytica.
The exact mechanism leading to giardiasis is unknown, although research points to a combination between osmosis, active secretion, exudation, inflammation and altered motility as drivers of Giardia-induced diarrhea. In molecular terms, disruption, reduced expression and/or relocation of TJ and cytoskeleton proteins (such as ZO-1, claudin-1, F-actin, and α-actinin) were shown to result in increased intestinal permeability and a drop in TER, indicating that infection can cause paracellular leakage (exudative diarrhea). The events leading to this class of diarrhea are similar to cryptosporidiosis and amoebiasis. Accordingly, disruption of ZO-1 was reported in colonic cells infected with Cryptosporidium (Buret et al., 2003) while in cells infected with E. histolytica, contact-dependent degradation of TJ proteins ZO-1 and ZO-2, dephosphorylation of ZO-2, relocation of claudin-4 and reduction in TER were shown to underlie exudative diarrhea (Leroy et al., 2000; Lejeune et al., 2011). However, while Giardia causes TJ disruption without penetrating the epithelium, E. histolytica kills (through apoptosis and trogocytosis), invades and destroys host tissues. In cryptosporidiosis, further studies are needed to assess whether cell invasion or parasitic products initiate these alterations.
Tight junction alterations were also observed to indirectly increase the luminal Cl- concentration (secretory diarrhea) as a consequence of the loss of absorptive function (villous shortening, microvilli atrophy and increased cell death) and/or increased secretion (destruction of the epithelial barrier) in Giardia-infected cells (Troeger et al., 2007; Maia-Brigagão et al., 2012). Similarly, damage to the absorptive villi and enhanced fluid secretion from the crypts have been documented in cryptosporidiosis, supporting diarrhea by active secretion (Guarino et al., 1995, 1997). In amoebiasis, increased mannitol flux and movement of sodium ions into the intestinal lumen were reported (Leroy et al., 2000; Lejeune et al., 2011).
As digestion of nutrients in the small intestine depends on hydrolytic enzymes (disaccharidases such as sucrose, maltase, lactase, and peptidase) produced by the brush border membrane of microvilli, dysfunctional microvilli may interfere significantly with the absorption of nutrients. In giardiasis, loss of microvilli brush border, combined with villous atrophy, is responsible for disaccharidase insufficiencies and malabsorption of nutrients, ultimately causing osmotic diarrhea (Buret, 2007, 2008; Troeger et al., 2007). Likewise, in enteric cryptosporidiosis, villous atrophy and crypt hyperplasia were shown to account for impaired monosaccharide and glucose-Na+ absorption while lactose malabsorption was described in individuals infected with E. histolytica (Rana et al., 2004).
In some diarrheal infections, the association between impaired absorption and increased secretion may contribute to accelerated intestinal transit. Indeed, in giardiasis and cryptosporidiosis, increased motility was reported, which in turn may contribute to the exacerbation of weight loss observed in Giardia-infected patients. On the contrary, whether motility dysfunction occurs and its importance on the development of amoebiasis it are unclear. However, it cannot be ruled out, as increased secretion and malabsorption are triggered by E. histolytica infection.
Despite the similarities in the events leading to osmotic, secretory and exudative diarrhea, there are some differences between giardiasis, cryptosporidiosis, and amoebiasis when considering the immunological and inflammatory response of the host (inflammatory diarrhea). For example, while several lines of evidence support the hypothesis that infections with Giardia are rarely accompanied by inflammation (Hanevik et al., 2007; Morken et al., 2008), a parasite extract was shown to be a poor cytokine inducer (inducing only small amounts of IL-6 and TNF-α; Zhou et al., 2003, 2007). On the contrary, a hallmark of amoebiasis and cryptosporidiosis is acute intestinal inflammation dominated by NF-κB-mediated secretion of inflammatory cytokines produced by host cells (Eckmann et al., 1995; Seydel et al., 1997; McCole et al., 2000; Chen et al., 2001; Hou et al., 2010). For example, IL-1β, IL-6, IL-8, TNF-α, and IFN-γ are key factors in the inflammatory response elicited by host cells after contact with amoebae (Eckmann et al., 1995; Seydel et al., 1997; Hou et al., 2010). However, whether production of pro-inflammatory cytokines influences the permeability of epithelial cell TJs and gut absorption is not known. Similar to amoebiasis, upon Cryptosporidium infection, epithelial cells release pro-inflammatory cytokines (IL-1β, IL-8, TNF-α, IFN-γ) and chemokines (C-X-C and fractalkine) to the site of infection, which in turn may contribute to increased epithelial permeability, impaired intestinal absorption and enhanced secretion (Seydel et al., 1998a; Farthing, 2000; Lacroix-Lamandé et al., 2002).
Collectively, these observations suggest that malabsorption, secretion of electrolytes and impairment of TJs may underlie luminal fluid accumulation during G. lamblia infection. Marked mucosal inflammation, decreased absorptive surface and malabsorption are thought to contribute to the pathogenesis of Cryptosporidium-induced diarrhea, while in E. histolytica-infected cells, epithelial destruction and inflammation infection appears to be the basis of the disease.
Concluding Remarks
Intestinal parasitism is extremely common, with G. lamblia, C. parvum, and E. histolytica being the most important intestinal protozoan parasites of humans worldwide. Infections begin when a person ingests the infective stage of the parasite with contaminated food or water. Once inside the host, parasites lodge in the intestinal tract causing acute and self-limited diarrhea. However, in some patients, the disease can progress to chronic diarrhea and related complications such as malnutrition, growth delays and cognitive impairment.
Significant progress has been made in understanding the processes by which G. lamblia, C. parvum, and E. histolytica trigger diarrhea and how the host cell responds to infection. Disruption of TJ barrier function, alterations of host cell architecture, and transcription of genes involved in host immunity and cell death are some of the events elicited in the host cell when interacting with these parasites.
Future elucidation of the processes that integrate these events and eliminate the disease may lead to novel therapeutic approaches for diarrhea caused by enteropathogenic parasites.
Author Contributions
Conceived and wrote the paper RT. Wrote the paper BG.
Funding
The work herein mentioned was supported by grants of the Fundação de Amparo à Pesquisa do Estado de São Paulo (RT 2010/15042-2; BDG 2013/04272-5).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adam, R. D. (2001). Biology of Giardia lamblia. Clin. Microbiol. Rev. 14, 447–475. doi: 10.1128/CMR.14.3.447-475.2001
Adams, J. M., and Cory, S. (1998). The Bcl-2 protein family: arbiters of cell survival. Science 281, 1322–1326. doi: 10.1126/science.281.5381.1322
Adams, J. M., and Cory, S. (2001). Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 26, 61–66. doi: 10.1016/S0968-0004(00)01740-0
Adams, R. B., Guerrant, R. L., Zu, S., Fang, G., and Roche, J. K. (1994). Cryptosporidium parvum infection of intestinal epithelium: morphologic and functional studies in an in vitro model. J. Infect. Dis. 169, 170–177. doi: 10.1093/infdis/169.1.170
Aley, S. B., Zimmerman, M., Hetsko, M., Selsted, M. E., and Gillin, F. D. (1994). Killing of Giardia lamblia by cryptdins and cationic neutrophil peptides. Infect. Immun. 62, 5397–5403.
Argenzio, R. A., Liacos, J. A., Levy, M. L., Meuten, D. J., Lecce, J. G., and Powell, D. W. (1990). Villous atrophy, crypt hyperplasia, cellular infiltration, and impaired glucose-Na absorption in enteric cryptosporidiosis of pigs. Gastroenterology 98, 1129–1140.
Balda, M. S., and Matter, K. (2008). Tight junctions at a glance. J. Cell Sci. 121, 3677–3682. doi: 10.1242/jcs.023887
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Betanzos, A., Javier-Reyna, R., García-Rivera, G., Bañuelos, C., González-Mariscal, L., Schnoor, M., et al. (2013). The EhCPADH112 complex of Entamoeba histolytica interacts with tight junction proteins occludin and claudin-1 to produce epithelial damage. PLoS ONE 8:e65100. doi: 10.1371/journal.pone.0065100
Bevins, C. L., and Salzman, N. H. (2011). Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9, 356–368. doi: 10.1038/nrmicro2546
Binder, H. J. (2010). Role of colonic short-chain fatty acid transport in diarrhea. Annu. Rev. Physiol. 72, 297–313. doi: 10.1146/annurev-physiol-021909-135817
Bogdan, C. (2001). Nitric oxide and the immune response. Nat. Immunol. 2, 907–916. doi: 10.1038/ni1001-907
Brantley, R. K., Williams, K. R., Silva, T. M., Sistrom, M., Thielman, N. M., Ward, H., et al. (2003). AIDS-associated diarrhea and wasting in Northeast Brazil is associated with subtherapeutic plasma levels of antiretroviral medications and with both bovine and human subtypes of Cryptosporidium parvum. Braz. J. Infect. Dis. 7, 16–22. doi: 10.1590/S1413-86702003000100003
Bruchhaus, I., Loftus, B. J., Hall, N., and Tannich, E. (2003). The intestinal protozoan parasite Entamoeba histolytica contains 20 cysteine protease genes, of which only a small subset is expressed during in vitro cultivation. Eukaryot. Cell 2, 501–509. doi: 10.1128/EC.2.3.501-509.2003
Buret, A. G. (2007). Mechanisms of epithelial dysfunction in giardiasis. Gut 56, 316–317. doi: 10.1136/gut.2006.107771
Buret, A. G. (2008). Pathophysiology of enteric infections with Giardia duodenalis. Parasite 15, 262–265. doi: 10.1051/parasite/2008153261
Buret, A. G., Chin, A. C., and Scott, K. G. E. (2003). Infection of human and bovine epithelial cells with Cryptosporidium andersoni induces apoptosis and disrupts tight junctional ZO-1: effects of epidermal growth factor. Int. J. Parasitol. 33, 1363–1371. doi: 10.1016/S0020-7519(03)00138-3
Buret, A. G., Mitchell, K., Muench, D. G., and Scott, K. G. (2002). Giardia lamblia disrupts tight junctional ZO-1 and increases permeability in non-transformed human small intestinal epithelial monolayers: effects of epidermal growth factor. Parasitology 125, 11–19. doi: 10.1017/S0031182002001853
Castellanos-Gonzalez, A., Yancey, L. S., Wang, H. C., Pantenburg, B., Liscum, K. R., and Lewis, D. (2008). Cryptosporidium infection of human intestinal epithelial cells increases expression of osteoprotegerin: a novel mechanism for evasion of host defenses. J. Infect. Dis. 197, 916–923. doi: 10.1086/528374
Céu Sousa, M., Gonçalves, C. A., Bairos, V. A., and Poiares-Da-Silva, J. (2001). Adherence of Giardia lamblia trophozoites to Int-407 human intestinal cells. Clin. Diagn. Lab. Immunol. 8, 258–265.
Chen, X. M., Gores, G. J., Paya, C. V., and Larusso, N. F. (1999). Cryptosporidium parvum induces apoptosis in biliary epithelia by a Fas/Fas ligand-dependent mechanism. Am. J. Physiol. 277, G599–G608.
Chen, X. M., Levine, S. A., Splinter, P. L., Tietz, P. S., Ganong, A. L., Jobin, C., et al. (2001). Cryptosporidium parvum activaes nuclear factor κB in biliary epithelia preventing epithelial cell apoptosis. Gastroenterology 120, 1774–1783. doi: 10.1053/gast.2001.24850
Chen, X. M., Levine, S. A., Tietz, P., Krueger, E., McNiven, M. A., Jefferson, D. M., et al. (1998). Cryptosporidium parvum is cytopathic for cultured human biliary epithelia via an apoptotic mechanism. Hepatology 28, 906–913. doi: 10.1002/hep.510280402
Chin, A. C., Teoh, D. A., Scott, K. G.-E., Meddings, J. B., MacNaughton, W. K., and Buret, A. G. (2002). Strain-dependent induction of enterocyte apoptosis by Giardia lamblia disrupts epithelial barrier function in a caspase-3-dependent manner. Infect. Immun. 70, 3673–3680. doi: 10.1128/IAI.70.7.3673-3680.2002
Christy, N. C., and Petri, W. A. Jr. (2011). Mechanisms of adherence, cytotoxicity and phagocytosis modulate the pathogenesis of Entamoeba histolytica. Future Microbiol. 6, 1501–1519. doi: 10.2217/fmb.11.120
Ciccocioppo, R., Di Sabatino, A., Parroni, R., Muzi, P., D’Alò, S., Ventura, T., et al. (2001). Increased enterocyte apoptosis and Fas-Fas ligand system in celiac disease. Am. J. Clin. Pathol. 115, 494–503. doi: 10.1309/UV54-BHP3-A66B-0QUD
Cotton, J. A., Beatty, J. K., and Buret, A. G. (2011). Host parasite interactions and pathophysiology in Giardia infections. Int. J. Parasitol. 41, 925–933. doi: 10.1016/j.ijpara.2011.05.002
Deng, M., Lancto, C. A., and Abrahamsen, M. S. (2004). Cryptosporidium parvum regulation of human epithelial cell gene expression. Int. J. Parasitol. 34, 73–82. doi: 10.1016/j.ijpara.2003.10.001
Di Sabatino, A., Ciccocioppo, R., Luinetti, O., Ricevuti, L., Morera, R., Cifone, M. G., et al. (2003). Increased enterocyte apoptosis in inflamed areas of Crohn’s disease. Dis. Colon Rectum 46, 1498–1507. doi: 10.1007/s10350-004-6802-z
Dinarello, C. A. (2007). Historical review of cytokines. Eur. J. Immunol. 37, S34–S45. doi: 10.1002/eji.200737772
Dreesen, L., Bosscher, K. D., Grit, G., Staels, B., Lubberts, E., Bauge, E., et al. (2014). Giardia muris infection in mice is associated with a protective interleukin 17A response and induction of peroxisome proliferator-activated receptor alpha. Infect. Immun. 82, 3333–3340. doi: 10.1128/IAI.01536-14
Eckmann, L., Reed, S. L., Smith, J. R., and Kagnoff, M. F. (1995). Entamoeba histolytica trophozoites induce an inflammatory cytokine response by cultured human cells through the paracrine action of cytolytically released interleukin-1 alpha. J. Clin. Invest. 96, 1269–1279. doi: 10.1172/JCI118161
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Farthing, M. J., Pereira, M. E., and Keusch, G. T. (1986). Description and characterization of a surface lectin from Giardia lamblia. Infect. Immun. 51, 661–667.
Farthing, M. J. G. (2000). “Clinical aspects of human cryptosporidiosis,” in Cryptosporidiosis and Microsporidiosis, ed. F. Petry (Basel: Karger), 1–268.
Ferella, M., Davids, B. J., Cipriano, M. J., Birkeland, S. R., Palm, D., Gillin, F. D., et al. (2014). Gene expression changes during Giardia–host cell interactions in serum-free medium. Mol. Biochem. Parasitol. 197, 21–23. doi: 10.1016/j.molbiopara.2014.09.007
Field, M. (2003). Intestinal ion transport and the pathophysiology of diarrhea. J. Clin. Invest. 111, 931–943. doi: 10.1172/JCI200318326
Foster, D. M., Stauffer, S. H., Stone, M. R., and Gookin, J. L. (2012). Proteasome inhibition of pathologic shedding of enterocytes to defend barrier function requires X-linked inhibitor of apoptosis protein and nuclear factor κB. Gastroenterology 143, 133–144. doi: 10.1053/j.gastro.2012.03.030
Frederick, J. R., and Petri, W. A. Jr. (2005). Roles for the galactose-/N-acetylgalactosamine-binding lectin of Entamoeba in parasite virulence and differentiation. Glycobiology 15, 53R–59R. doi: 10.1093/glycob/cwj007
Genta, R. M., Chappell, C. L., White, A. C. Jr., Kimball, K. T., and Goodgame, R. W. (1993). Duodenal morphology and intensity of infection in AIDS-related intestinal cryptosporidiosis. Gastroenterology 105, 1769–1775.
Gillin, F. D., Reiner, D. S., and Boucher, S. E. (1988). Small-intestinal factors promote encystation of Giardia lamblia in vitro. Infect. Immun. 56, 705–707.
Gookin, J. L., Chiang, S., Allen, J., Armstrong, M. U., Satuffer, S. H., Finnegan, C., et al. (2006). NF-κB-mediated expression of iNOS promotes epithelial defense against infection by Cryptosporidium parvum in neonatal piglets. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G164–G174. doi: 10.1152/ajpgi.00460.2004
Gookin, J. L., Nordone, S. K., and Argenzio, R. A. (2002). Host response to Cryptosporidium infection. J. Vet. Intern. Med. 16, 12–21. doi: 10.1111/j.1939-1676.2002.tb01602.x
Goplen, M., Lejeune, M., Cornick, S., Moreau, F., and Chadee, K. (2013). Entamoeba histolytica contains an occludin-like protein that can alter colonic epithelial barrier function. PLoS ONE 8:e73339. doi: 10.1371/journal.pone.0073339
Gorowara, S., Ganguly, N. K., Mahajan, R. C., and Walia, B. N. (1992). Study on the mechanism of Giardia lamblia induced diarrhoea in mice. Biochim. Biophys. Acta 1138, 122–126. doi: 10.1016/0925-4439(92)90051-N
Griffith, J. W., Sokol, C. L., and Luster, A. D. (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 32, 659–702. doi: 10.1146/annurev-immunol-032713-120145
Guarino, A., Canani, R. B., Casola, A., Pozio, E., Russo, R., Bruzzese, E., et al. (1995). Human intestinal cryptosporidiosis: secretory diarrhea and enterotoxic activity in Caco-2 cells. J. Infect. Dis. 171, 976–983. doi: 10.1093/infdis/171.4.976
Guarino, A., Castaldo, A., Russo, S., Spagnuolo, M. I., Canani, R. B., Tarallo, L., et al. (1997). Enteric cryptosporidiosis in pediatric HIV infection. J. Pediatr. Gastroenterol. Nutr. 25, 187–192. doi: 10.1097/00005176-199708000-00009
Guerrant, R. L., Van Gilder, T., Steiner, T. S., Thielman, N. M., Slutsker, L., Tauxe, R. V., et al. (2001). Practice guidelines for the management of infectious diarrhea. Clin. Infect. Dis. 32, 331–351. doi: 10.1086/318514
Günther, C., Neumann, H., Neurath, M. F., and Becker, C. (2013). Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut 62, 1062–1071. doi: 10.1136/gutjnl-2011-301364
Hammer, H. F., Santa Ana, C. A., Schiller, L. R., and Fordtran, J. S. (1989). Studies of osmotic diarrhea induced in normal subjects by ingestion of polyethylene glycol and lactulose. J. Clin. Invest. 84, 1056–1062. doi: 10.1172/JCI114267
Hanevik, K., Hausken, T., Morken, M., Strand, E., Morch, K., Coll, P., et al. (2007). Persisting symptoms and duodenal inflammation related to Giardia duodenalis infection. J. Infect. 55, 524–530. doi: 10.1016/j.jinf.2007.09.004
Holberton, D. V. (1973). Fine structure of the ventral disk apparatus and the mechanism of attachment in the flagellate Giardia muris. J. Cell Sci. 13, 11–41.
Hou, Y., Mortimer, L., and Chadee, K. (2010). Entamoeba histolytica cysteine proteinase 5 binds integrin on colonic cells and stimulates NFkappaB-mediated pro-inflammatory responses. J. Biol. Chem. 285, 35497–35504. doi: 10.1074/jbc.M109.066035
Humen, M. A., Pérez, P. F., and Moal, V. L. L. (2011). Lipid raft-dependent adhesion of Giardia intestinalis trophozoites to a cultured human enterocyte-like Caco-2/TC7 cell monolayer leads to cytoskeleton-dependent functional injuries. Cell. Microbiol. 13, 1683–1702. doi: 10.1111/j.1462-5822.2011.01647.x
Huston, C. D., Boettner, D. R., Miller-Sims, V., and Petri, W. A. Jr. (2003). Apoptotic killing and phagocytosis of host cells by the parasite Entamoeba histolytica. Infect. Immun. 71, 964–972. doi: 10.1128/IAI.71.2.964-972.2003
Huston, C. D., Houpt, E. R., Mann, B. J., Hahn, C. S., and Petri, W. A. Jr. (2000). Caspase 3-dependent killing of host cells by the parasite Entamoeba histolytica. Cell. Microbiol. 2, 617–625. doi: 10.1046/j.1462-5822.2000.00085.x
Inge, P. M., Edson, C. M., and Farthing, M. J. (1988). Attachment of Giardia lamblia to rat intestinal epithelial cells. Gut 29, 795–801. doi: 10.1136/gut.29.6.795
Izzo, A. A., Gaginella, T. S., Mascolo, N., and Capasso, F. (1994). Nitric oxide as a mediator of the laxative action of magnesium sulphate. Br. J. Pharmacol. 113, 228–232. doi: 10.1111/j.1476-5381.1994.tb16198.x
Jang, Y. S., Song, K. J., Kim, J. Y., Lee, Y. A., Kim, K. A., Lee, S. K., et al. (2011). Calpains are involved in Entamoeba histolytica-induced death of HT-29 colonic epithelial cells. Korean J. Parasitol. 49, 177–180. doi: 10.3347/kjp.2011.49.2.177
Kim, K. A., Kim, J. Y., Lee, Y. A., Min, A., Bahk, Y. Y., and Shin, M. H. (2013). Entamoeba histolytica induces cell death of HT29 colonic epithelial cells via NOX1-derived ROS. Korean J. Parasitol. 51, 61–68. doi: 10.3347/kjp.2013.51.1.61
Kim, K. A., Kim, J. Y., Lee, Y. A., Song, K. J., Min, D., and Shin, M. H. (2011). NOX1 participates in ROS-dependent cell death of colon epithelial Caco2 cells induced by Entamoeba histolytica. Microbes Infect. 13, 1052–1061. doi: 10.1016/j.micinf.2011.06.001
Kim, K. A., Lee, Y. A., and Shin, M. H. (2007). Calpain-dependent calpastatin cleavage regulates caspase-3 activation during apoptosis of Jurkat T cells induced by Entamoeba histolytica. Int. J. Parasitol. 37, 1209–1219. doi: 10.1016/j.ijpara.2007.03.011
Kim, K. A., Lee, Y. A., and Shin, M. H. (2010). Calpain-dependent cleavage of SHP-1 and SHP-2 is involved in the dephosphorylation of Jurkat T cells induced by Entamoeba histolytica. Parasite Immunol. 32, 176–183. doi: 10.1111/j.1365-3024.2009.01175.x
Kim, K. A., Min, A., Lee, Y. A., and Shin, M. H. (2014). Degradation of the transcription factor NF-κB, STAT3, and STAT5 is involved in Entamoeba histolytica-induced cell death in Caco-2 colonic epithelial cells. Korean J. Parasitol. 52, 459–469. doi: 10.3347/kjp.2014.52.5.459
Koh, W. H., Geurden, T., Paget, T., O’Handley, R., Steuart, R. F., Thompson, R. C. A., et al. (2013). Giardia duodenalis assemblage-specific induction of apoptosis and tight junction disruption in human intestinal epithelial cells: effects of mixed infections. J. Parasitol. 99, 353–358. doi: 10.1645/GE-3021.1
Kohli, A., Bushen, O. Y., Pinkerton, R. C., Houpt, E., Newman, R. D., Sears, C. L., et al. (2008). Giardia duodenalis assemblage, clinical presentation and markers of intestinal inflammation in Brazilian children. Trans. R. Soc. Trop. Med. Hyg. 102, 718–725. doi: 10.1016/j.trstmh.2008.03.002
Lacroix-Lamandé, S., Mancassola, R., Naciri, M., and Laurent, F. (2002). Role of gamma interferon in chemokine expression in the ileum of mice and in a murine intestinal epithelial cell line after Cryptosporidium parvum infection. Infect. Immun. 70, 2090–2099. doi: 10.1128/IAI.70.4.2090-2099.2002
Laurent, F., Eckmann, L., Savidge, T. C., Morgan, G., Theodos, C., Naciri, M., et al. (1997). Cryptosporidium parvum infection of human intestinal epithelial cells induces the polarized secretion of C-X-C chemokines. Infect. Immun. 65, 5067–5073.
Laurent, F., McCole, D., Eckmann, L., and Kagnoff, M. F. (1999). Pathogenesis of Cryptosporidium parvum infection. Microbes Infect. 2, 141–148. doi: 10.1016/S1286-4579(99)80005-7
Lee, Y. A., Kim, K. A., and Shin, M. H. (2011). Calpain mediates degradation of cytoskeletal proteins during Jurkat T-cell death induced by Entamoeba histolytica. Parasite Immunol. 33, 349–356. doi: 10.1111/j.1365-3024.2011.01290.x
Lejeune, M., Moreau, F., and Chadee, K. (2011). Prostaglandin E2 produced by Entamoeba histolytica signals via EP4 receptor and alters claudin-4 to increase ion permeability of tight junctions. Am. J. Pathol. 179, 807–818. doi: 10.1016/j.ajpath.2011.05.001
Leroy, A., Lauwaet, T., De Bruyne, G., Cornelissen, M., and Mareel, M. (2000). Entamoeba histolytica disturbs the tight junction complex in human enteric T84 cell layers. FASEB J. 14, 1139–1146.
Li, W., Stenson, W. F., Kunz-Jenkins, C., Swanson, P. E., Duncan, R., and Stanley, S. L. Jr. (1994). Entamoeba histolytica interactions with polarized human intestinal Caco-2 epithelial cells. Infect. Immun. 62, 5112–5119.
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Liu, J., Deng, M., Lancto, C. A., Abrahamsen, M. S., Rutherford, M. S., and Enomoto, S. (2009). Biphasic modulation of apoptotic pathways in Cryptosporidium parvum-infected human intestinal epithelial cells. Infect. Immun. 77, 837–849. doi: 10.1128/IAI.00955-08
Liu, J., Enomoto, S., Lancto, C. A., Abrahamsen, M. S., and Rutherford, M. S. (2008). Inhibition of apoptosis in Cryptosporidium parvum-infected intestinal epithelial cells is dependent on survivin. Infect. Immun. 76, 3784–3792. doi: 10.1128/IAI.00308-08
Luján, H. D., Mowatt, M. R., Byrd, L. G., and Nash, T. E. (1996). Cholesterol starvation induces differentiation of the intestinal parasite Giardia lamblia. Proc. Natl. Acad. Sci. U.S.A. 93, 7628–7633. doi: 10.1073/pnas.93.15.7628
Maia-Brigagão, C., Morgado-Díaz, J. A., and De Souza, W. (2012). Giardia disrupts the arrangement of tight, adherens and desmosomal junction proteins of intestinal cells. Parasitol. Int. 61, 280–287. doi: 10.1016/j.parint.2011.11.002
Marco, S., and Skaper, S. D. (2006). Amyloid β-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci. Lett. 401, 219–224. doi: 10.1016/j.neulet.2006.03.047
Marie, C., and Petri, W. A. Jr. (2014). Regulation of virulence of Entamoeba histolytica. Annu. Rev. Microbiol. 68, 493–520. doi: 10.1146/annurev-micro-091313-103550
Martinez-Palomo, A., Gonzalez-Robles, A., Chavez, B., Orozco, E., Fernadez-Castelo, S., and Cervantes, A. (1985). Structural bases of the cytolytic mechanisms of Entamoeba histolytica. J. Protozool. 32, 166–175. doi: 10.1111/j.1550-7408.1985.tb03033.x
Matter, K., and Balda, M. (2003). Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 4, 225–236. doi: 10.1038/nrm1055
McCole, D. F., Eckmann, L., Laurent, F., and Kagnoff, M. F. (2000). Intestinal epithelial cell apoptosis following Cryptosporidium parvum infection. Infect. Immun. 68, 1710–1713. doi: 10.1128/IAI.68.3.1710-1713.2000
Mele, R., Gomez Morales, M. A., Tosini, F., and Pozio, E. (2004). Cryptosporidium parvum at different developmental stages modulates host cell apoptosis in vitro. Infect. Immun. 72, 6061–6067. doi: 10.1128/IAI.72.10.6061-6067.2004
Morken, M. H., Nysaeter, G., Strand, E. A., Hausken, T., and Berstad, A. (2008). Lactulose breath test results in patients with persistent abdominal symptoms following Giardia lamblia infection. Scand. J. Gastroenterol. 43, 141–145.
Müller, N., and von Allmen, N. (2005). Recent insights into the mucosal reactions associated with Giardia lamblia infections. Int. J. Parasitol. 35, 1339–1347. doi: 10.1016/j.ijpara.2005.07.008
Muza-Moons, M. M., Schneeberger, E. E., and Hecht, G. A. (2004). Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cell. Microbiol. 6, 783–793. doi: 10.1111/j.1462-5822.2004.00404.x
Ocádiz, R., Orozco, E., Carrillo, E., Quintas, L. I., Ortega-López, J., García-Pérez, R. M., et al. (2005). EhCP112 is an Entamoeba histolytica secreted cysteine protease that may be involved in the parasite-virulence. Cell. Microbiol. 7, 221–232. doi: 10.1111/j.1462-5822.2004.00453.x
Ojcius, D. M., Perfettini, J. L., Bonnin, A., and Laurent, F. (1999). Caspase-dependent apoptosis during infection with Cryptosporidium parvum. Microbes Infect. 1, 1163–1168. doi: 10.1016/S1286-4579(99)00246-4
Oliver, F. J., De La Rubia, G., Rolli, V., Ruiz-Ruiz, M. C., Murcia, G., and Murcia, J. M. (1998). Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J. Biol. Chem. 273, 33533–33539.
Ostaff, M. J., Stange, E. F., and Wehkamp, J. (2013). Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 5, 1465–1483. doi: 10.1002/emmm.201201773
Panaro, M. A., Cianciulli, A., Mitolo, V., Mitolo, C. I., Acquafredda, A., Brandonisio, O., et al. (2007). Caspase-dependent apoptosis of the HCT-8 epithelial cell line induced by the parasite Giardia intestinalis. FEMS Immunol. Med. Microbiol. 51, 302–309. doi: 10.1111/j.1574-695X.2007.00304.x
Parikh, A., Stephan, A. F., and Tzanakakis, E. S. (2012). Regenerating proteins and their expression, regulation and signaling. Biomol. Concepts 3, 57–70. doi: 10.1515/bmc.2011.055
Petersen, C. (1993). Cellular biology of Cryptosporidium parvum. Parasitol. Today 9, 87–91. doi: 10.1016/0169-4758(93)90211-W
Peterson, K. M., Guo, X., Elkahloun, A. G., Mondal, D., Bardhan, P. K., Sugawara, A., et al. (2011). The expression of REG 1A and REG 1B is increased during acute amebic colitis. Parasitol. Int. 60, 296–300. doi: 10.1016/j.parint.2011.04.005
Pinilla, A. E., Lopez, M. C., and Viasus, D. F. (2008). [History of the Entamoeba histolytica protozoan]. Rev. Med. Chile 136, 118–124. doi: 10.4067/S0034-98872008000100015
Ragland, B. D., Ashley, L. S., Vaux, D. L., and Petri, W. A. (1994). Entamoeba histolytica: target cells killed by trophozoites undergo DNA fragmentation which is not blocked by Bcl-2. Exp. Parasitol. 79, 460–467. doi: 10.1006/expr.1994.1107
Ralston, K. S., Solga, M. D., Mackey-Lawrence, N. M., Bhattacharya, A., and Petri, W. A. (2014). Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasio. Nature 508, 526–530. doi: 10.1038/nature13242
Rana, S. V., Bhasin, D. K., and Vinayak, V. K. (2004). Prospective evaluation of lactose malabsorption by lactose hydrogen breath test in individuals infected with Entamoeba histolytica and passing cysts. Br. J. Nutr. 92, 207–208. doi: 10.1079/BJN20041194
Robinson, P., Okhuysen, P. C., Chappell, C. L., Lewis, D. E., Shahab, I., and Lahoti, S. (2000). Transforming growth factor β1 is expressed in the jejunum after experimental Cryptosporidium parvum infection in humans. Infect. Immun. 68, 5405–5407. doi: 10.1128/IAI.68.9.5405-5407.2000
Robinson, P., Okhuysen, P. C., Chappell, C. L., Lewis, D. E., Shahab, I., and Lahoti, S. (2001). Expression of IL-15 and IL-4 in IFN-gamma-independent control of experimental human Cryptosporidium parvum infection. Cytokine 15, 39–46. doi: 10.1006/cyto.2001.0888
Roxström-Lindquist, K., Ringqvist, E., Palm, D., and Svard, S. (2005). Giardia lamblia-induced changes in gene expression in differentiated Caco-2 human intestinal epithelial cells. Infect. Immun. 73, 8204–8208. doi: 10.1128/IAI.73.12.8204-8208.2005
Ryan, U., and Cacciò, S. M. (2013). Zoonotic potential of Giardia. Int. J. Parasitol. 43, 943–956. doi: 10.1016/j.ijpara.2013.06.001
Schwartz, C. L., Heumann, J. M., Dawson, S. C., and Hoenger, A. (2012). A detailed, hierarchical study of Giardia lamblia’s ventral disc reveals novel microtubule-associated protein complexes. PLoS ONE 7:e43783. doi: 10.1371/journal.pone.0043783
Scott, K., Meddings, J., Kirk, D., Lees-Miller, S., and Buret, A. (2002). Intestinal infection with Giardia spp. reduces epithelial barrier function in a myosin light chain kinase-dependent fashion. Gastroenterology 123, 1179–1190. doi: 10.1053/gast.2002.36002
Scott, K. G.-E., Logan, M. R., Klammer, G. M., Teoh, D. A., and Buret, A. G. (2000). Jejunal brush border microvillous alterations in Giardia muris-infected mice: role of T lymphocytes and interleukin-6. Infect. Immun. 68, 3412–3418. doi: 10.1128/IAI.68.6.3412-3418.2000
Sekikawa, A., Fukui, H., Fujii, S., Ichikawa, K., Tomita, S., Imura, J., et al. (2008). REG Ialpha protein mediates an anti-apoptotic effect of STAT3 signaling in gastric cancer cells. Carcinogenesis 29, 76–83. doi: 10.1093/carcin/bgm250
Seydel, K. B., Li, E., Swanson, P. E., and Stanley, S. L. Jr. (1997). Human intestinal epithelial cells produce proinflammatory cytokines in response to infection in a SCID mouse-human intestinal xenograft model of amebiasis. Infect. Immun. 65, 1631–1639.
Seydel, K. B., Li, E., Zhang, Z., and Stanley, S. L. Jr. (1998a). Epithelial cell-initiated inflammation plays a crucial role in early tissue damage in amebic infection of human intestine. Gastroenterology 115, 1446–1453. doi: 10.1016/S0016-5085(98)70023-X
Seydel, K. B., and Stanley, S. L. Jr. (1998). Entamoeba histolytica induces host cell death in amebic liver abscess by a non-Fas-dependent, non-tumor necrosis factor alpha-dependent pathway of apoptosis. Infect. Immun. 66, 2980–2983.
Seydel, K. B., Zhang, T., Champion, G. A., Fichtenbaum, C., Swanson, P. E., Tzipori, S., et al. (1998b). Cryptosporidium parvum infection of human intestinal xenografts in SCID mice induces production of human tumor necrosis factor alpha and interleukin-8. Infect. Immun. 66, 2379–2382.
Sharpstone, D., Neild, P., Crane, R., Taylor, C., Hodgson, C., Sherwood, R., et al. (1999). Small intestinal transit, absorption, and permeability in patients with AIDS with and without diarrhoea. Gut 45, 70–76. doi: 10.1136/gut.45.1.70
Sim, S., Park, S. J., Yong, T. S., Im, K. I., and Shin, M. H. (2007). Involvement of beta 2-integrin in ROS-mediated neutrophil apoptosis induced by Entamoeba histolytica. Microbes Infect. 9, 1368–1375. doi: 10.1016/j.micinf.2007.06.013
Sim, S., Yong, T. S., Park, S. J., Im, K. I., Kong, Y., Ryu, J. S., et al. (2005). NADPH oxidase-derived reactive oxygen species-mediated activation of ERK1/2 is required for apoptosis of human neutrophils induced by Entamoeba histolytica. J. Immunol. 174, 4279–4288. doi: 10.4049/jimmunol.174.7.4279
Simonet, W. S., Lacey, D. L., Dunstan, C. R., Kelley, M., Chang, M. S., Lüthy, R., et al. (1997). Osteoprotogerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319. doi: 10.1016/S0092-8674(00)80209-3
Slee, E. A., Adrain, C., and Martin, S. J. (2001). Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276, 7320–7326. doi: 10.1074/jbc.M008363200
Stadelmann, B., Merino, M. C., Persson, L., and Svärd, S. G. (2012). Arginine consumption by the intestinal parasite Giardia intestinalis reduces proliferation of intestinal epithelial cells. PLoS ONE 7:e45325. doi: 10.1371/journal.pone.0045325
Stadnyk, A. W. (2002). Intestinal epithelial cells as a source of inflammatory cytokines and chemokines. Can. J. Gastroenterol. 16, 241–246.
Su, L., Nalle, S. C., Shen, L., Turner, E. S., Singh, G., Breskin, L. A., et al. (2013). TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology 145, 407–415. doi: 10.1053/j.gastro.2013.04.011
Sullivan, S., Alex, P., Dassopoulos, T., Zachos, N. C., Iacobuzio-Donahue, C., Donowitz, M., et al. (2009). Down-regulation of sodium transporters and NHERF proteins in IBD patients and mouse colitis models: potential contributors to IBD-associated diarrhea. Inflamm. Bowel Dis. 15, 261–274. doi: 10.1002/ibd.20743
Tako, E. A., Hassimi, M. F., and Singer, S. M. (2013). Transcriptomic analysis of the host response to Giardia duodenalis infection reveals redundant mechanisms for parasite control. mBio 4:e660-13. doi: 10.1128/mBio.00660-13
Teixeira, J. E., and Mann, B. J. (2002). Entamoeba histolytica-induced dephosphorylation in host cells. Infect. Immun. 70, 1816–1823. doi: 10.1128/IAI.70.4.1816-1823.2002
Teoh, D., Kamieniecki, D., Pang, G., and Buret, A. (2000). Giardia lamblia rearranges F-actin and alpha-actinin in human colonic and duodenal monolayers and reduces transepithelial electrical resistance. J. Parasitol. 86, 800–806. doi: 10.1645/0022-3395(2000)086[0800:GLRFAA]2.0.CO;2
Tessema, T. S., Schwamb, B., Lochner, M., Förster, I., Jakobi, V., and Petry, F. (2009). Dynamics of gut mucosal and systemic Th1/Th2 cytokine responses in interferon-gamma and interleukin-12p40 knock out mice during primary and challenge Cryptosporidium parvum infection. Immunobiology 214, 454–466. doi: 10.1016/j.imbio.2008.11.015
Thornberry, N. A., and Lazebnik, Y. (1998). Caspases: enemies within. Science 281, 1312–1316. doi: 10.1126/science.281.5381.1312
Troeger, H., Epple, H.-J., Schneider, T., Wahnschaffe, U., Ullrich, R., Burchard, G.-D., et al. (2007). Effect of chronic Giardia lamblia infection on epithelial transport and barrier function in human duodenum. Gut 56, 328–335. doi: 10.1136/gut.2006.100198
Turner, J. (2009). Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809. doi: 10.1038/nri2653
Vidal, K., Serrant, P., Schlosser, B., Van Den Broek, P., Lorget, F., and Donnet-Hughes, A. (2004). Osteoprotegerin production by human intestinal epithelial cells: a potential regulator of mucosal immune responses. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G836–G844. doi: 10.1152/ajpgi.00428.2003
Viswanathan, V. K., Hodges, K., and Hecht, G. (2009). Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat. Rev. Microbiol. 7, 110–119. doi: 10.1038/nrmicro2053
Walters, J. R. F., Tasleem, A. M., Omer, O. S., Brydon, W. G., Dew, T., and Le Roux, C. W. (2009). A new mechanism for bile acid diarrhea: defective feedback inhibition of bile acid biosynthesis. Clin. Gastroenterol. Hepatol. 7, 1189–1194. doi: 10.1016/j.cgh.2009.04.024
Wanyri, J., and Ward, H. (2006). Molecular basis of Cryptosporidium-host cell interactions: recent advances and future prospects. Future Microbiol. 1, 201–208. doi: 10.2217/17460913.1.2.201
Ward, H. D., Alroy, J., Lev, B. I., Keusch, G. T., and Pereira, M. E. (1988). Biology of Giardia lamblia. Detection of N-acetyl-D-glucosamine as the only surface saccharide moiety and identification of two distinct subsets of trophozoites by lectin binding. J. Exp. Med. 167, 73–88. doi: 10.1084/jem.167.1.73
White, C. (2010). “Cryptosporidium species,” in Principles and Practice of Infectious Diseases, 7th Edn, eds G. L. Mandell, J. E. Bennett, and R. Dolin (Philadelphia: Churchill Livingstone, Elsevier).
Widmer, G., Corey, E. A., Stein, B., Griffiths, J. K., and Tzipori, S. (2000). Host cell apoptosis impairs Cryptosporidium parvum development in vitro. J. Parasitol. 86, 922–928. doi: 10.1645/0022-3395(2000)086[0922:HCAICP]2.0.CO;2
Yu, L. C., Flynn, A. N., Turner, J. R., and Buret, A. G. (2005). SGLT-1-mediated glucose uptake protects intestinal epithelial cells against LPS-induced apoptosis and barrier defects: a novel cellular rescue mechanism? FASEB J. 19, 1822–1835. doi: 10.1096/fj.05-4226com
Yu, L. C. H., Huang, C. Y., Kuo, W., Sayer, H., Turner, J. R., and Buret, A. G. (2008). SGLT-1-mediated glucose uptake protects human intestinal epithelial cells against Giardia duodenalis-induced apoptosis. Int. J. Parasitol. 38, 923–934. doi: 10.1016/j.ijpara.2007.12.004
Zaalouk, T. K., Bajaj-Elliott, M., George, J. T., and McDonald, V. (2004). Differential regulation of β-defensin gene expression during Cryptosporidium parvum infection. Infect. Immun. 72, 2772–2779. doi: 10.1128/IAI.72.5.2772-2779.2004
Zeissig, S., Bürge, N., Günzel, D., Richter, J., Mankertz, J., Wahnschaffe, U., et al. (2007). Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56, 61–72. doi: 10.1136/gut.2006.094375
Zhou, P., Li, E., Robertson, J., Nash, T. E., and Singer, S. M. (2003). Role of interleukin-6 in the control of acute and chronic Giardia lamblia infections in mice. Infect. Immun. 71, 1566–1568. doi: 10.1128/IAI.71.3.1566-1568.2003
Zhou, P., Li, E., Shea-Donohue, T., and Singer, S. M. (2007). Tumour necrosis factor alpha contributes to protection against Giardia lamblia infection in mice. Parasite Immunol. 29, 367–374. doi: 10.1111/j.1365-3024.2007.00953.x
Zhou, R., Gong, A., Chen, D., Miller, R. E., Eischeid, A. N., and Chen, X. M. (2013). Histone deacetylases and NF-kB signaling coordinate expression of CX3CL1 in epithelial cells in response to microbial challenge by suppressing miR-424 and miR-503. PLoS ONE 8:e65153. doi: 10.1371/journal.pone.0065153
Zhou, R., Hu, G. Z., Liiu, J., Gong, A., Drescher, K. M., and Chen, X. M. (2009). NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog. 5:e1000681. doi: 10.1371/journal.ppat.1000681
Keywords: protozoan parasites, intestinal infection, diarrhea, gastrointestinal tract, intestinal epithelial barrier, parasite–host interaction
Citation: Di Genova BM and Tonelli RR (2016) Infection Strategies of Intestinal Parasite Pathogens and Host Cell Responses. Front. Microbiol. 7:256. doi: 10.3389/fmicb.2016.00256
Received: 25 November 2015; Accepted: 16 February 2016;
Published: 03 March 2016.
Edited by:
Olivier Dussurget, University Paris Diderot, Institut Pasteur, INSERM, INRA, FranceReviewed by:
Thomas Dandekar, University of Wuerzburg, GermanyAlexandre Morrot, Federal University of Rio de Janeiro, Brazil
Copyright © 2016 Di Genova and Tonelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renata R. Tonelli, ci50b25lbGxpQHVuaWZlc3AuYnI=