 Susanne Kramer1†‡
Susanne Kramer1†‡ Dörte Dibbern2‡
Dörte Dibbern2‡ Julia Moll3,4‡
Julia Moll3,4‡ Maike Huenninghaus5‡
Maike Huenninghaus5‡ Robert Koller5†
Robert Koller5† Dirk Krueger3
Dirk Krueger3 Sven Marhan1
Sven Marhan1 Tim Urich6
Tim Urich6 Tesfaye Wubet3,7
Tesfaye Wubet3,7 Michael Bonkowski5François Buscot3,4,7
Michael Bonkowski5François Buscot3,4,7 Tillmann Lueders2*Ellen Kandeler1
Tillmann Lueders2*Ellen Kandeler1- 1Institute of Soil Science and Land Evaluation, University of Hohenheim, Stuttgart, Germany
- 2Institute of Groundwater Ecology, Helmholtz Zentrum München - German Research Center for Environmental Health, Neuherberg, Germany
- 3Department of Soil Ecology, Helmholtz Centre for Environmental Research Leipzig-Halle, Halle, Germany
- 4Institute of Biology, University of Leipzig, Leipzig, Germany
- 5Department of Terrestrial Ecology, Institute of Zoology, University of Cologne, Köln, Germany
- 6Department of Bacterial Physiology, Institute for Microbiology, Ernst-Moritz-Arndt University of Greifswald, Greifswald, Germany
- 7German Centre for Integrative Biodiversity Research (iDiv), Leipzig, Germany
The flow of plant-derived carbon in soil is a key component of global carbon cycling. Conceptual models of trophic carbon fluxes in soil have assumed separate bacterial and fungal energy channels in the detritusphere, controlled by both substrate complexity and recalcitrance. However, detailed understanding of the key populations involved and niche-partitioning between them is limited. Here, a microcosm experiment was performed to trace the flow of detritusphere C from substrate analogs (glucose, cellulose) and plant biomass amendments (maize leaves, roots) in an agricultural soil. Carbon flow was traced by rRNA stable isotope probing and amplicon sequencing across three microbial kingdoms. Distinct lineages within the Actinobacteria, Bacteroidetes, Gammaproteobacteria, Basidiomycota, Ascomycota as well as Peronosporomycetes were identified as important primary substrate consumers. A dynamic succession of primary consumers was observed especially in the cellulose treatments, but also in plant amendments over time. While intra-kingdom niche partitioning was clearly observed, distinct bacterial and fungal energy channels were not apparent. Furthermore, while the diversity of primary substrate consumers did not notably increase with substrate complexity, consumer succession and secondary trophic links to bacterivorous and fungivorous microbes resulted in increased food web complexity in the more recalcitrant substrates. This suggests that rather than substrate-defined energy channels, consumer succession as well as intra- and inter-kingdom cross-feeding should be considered as mechanisms supporting food web complexity in the detritusphere.
Introduction
Microbes perform many crucial functions in soil, such as the primary decomposition of plant litter and the turnover of soil organic matter (Berg and McClaugherty, 2008; Schmidt et al., 2011). Despite the importance of these processes for global carbon budgets, factors controlling the roles and activities of distinct soil microbiota in the detritusphere are still poorly understood. Bacteria are classically thought to be more active in the degradation of labile organic matter as well as in early phases of litter decomposition (“bacterial energy channel”). In contrast, fungi are assumed to be more important in the degradation of complex and recalcitrant substrates, as well as in later stages of litter decomposition (“fungal energy channel”; de Boer et al., 2005; Paterson et al., 2008). However, recent work suggests that the role of bacteria in the mineralization of recalcitrant substrates as well as that of fungi in early stages of litter decomposition may also be significant (Bastian et al., 2009; Poll et al., 2010; España et al., 2011). This has fueled an ongoing debate on the specific roles and quantitative contributions of distinct soil microbiota in overall litter degradation (Strickland and Rousk, 2010; Rousk and Frey, 2015).
Resource partitioning between microbes consuming complex substrates is also considered an important driver of soil microbial diversity (Zhou et al., 2002). This has been demonstrated for primary substrate degraders in soil (Goldfarb et al., 2011), but can also be assumed for higher trophic levels, i.e., that of protists. These are known to exert important top-down controls on the bacterial energy channel (Ekelund and Ronn, 1994), but can also harbor primary detritus decomposers (Termorshuizen and Jeger, 2008). This complicates the quantitative understanding of their role in detritusphere energy channels. Protistan grazing on fungal hyphae and yeasts (Adl and Gupta, 2006) may represent an under-investigated cross-link between both energy channels. Hence, niche partitioning and the contributions of distinct key populations to detritusphere carbon flow across microbial kingdoms remains a matter of investigation.
The amendment of isotopically labeled substrates is an important experimental approach to tackle such questions. Especially in combination with nucleic acid-based stable isotope probing (SIP), this strategy makes it possible to trace substrate-derived carbon flow and to identify key populations involved. A number of studies have used 13C-labeled detritusphere substrate analogs such as cellulose or derived di- and monosaccharides to follow their uptake by soil bacteria, fungi, and/or protists (Haichar et al., 2007; Schellenberger et al., 2010; Eichorst and Kuske, 2012; Štursová et al., 2012; Chatzinotas et al., 2013; Verastegui et al., 2014; Wang et al., 2015; Pepe-Ranney et al., 2016). However, few studies have addressed the degradation of actual plant biomass by soil bacteria, e.g., with labeled residues of wheat (Bernard et al., 2007, 2009), maize (Fan et al., 2014), or rice root callus (Li et al., 2011). Carbon flow from the latter has been probed up to the levels of fungi and protists (Murase et al., 2012). Results of this relatively small number of studies suggest that the partitioning of natural detritusphere substrates between microbial kingdoms in soil warrants further elaboration.
Here, a SIP microcosm experiment with soil from an experimental maize field (Kramer et al., 2012; Scharroba et al., 2012) is reported. The site, dominated by Cambisols in conjunction with Luvisols, represents one of the most common and productive agricultural soil types in Central Europe (Akça et al., 2005). Treatments included the amendment of U-13C-labeled glucose and cellulose as defined mono- and polymeric substrates, and U-13C-maize leaves and roots as complex detritusphere substrates. Key label-assimilating bacteria, fungi, and protists were traced by rRNA-SIP at early and later stages of decomposition. We hypothesized that (i) distinct bacterial and fungal substrate utilization channels could be identified, (ii) the number of primary substrate consumers should increase with substrate complexity, and (iii) niche partitioning and succession should influence the relative contributions of distinct key populations to detritusphere carbon flow across microbial kingdoms. These objectives were addressed using a combination of PLFA- and rRNA-SIP as well as amplicon sequencing, providing an exceptional level of detail on the identities, succession and niche partitioning of key microbial populations in the detritusphere of an agricultural soil.
Materials and Methods
Soil
The soil originated from an agricultural field experiment located near Göttingen (Germany), which was designed to trace the flow of plant-derived carbon into the soil food web via C3 to C4 plant exchange (Kramer et al., 2012; Scharroba et al., 2012). A representative composite topsoil sample (0-10 cm) was taken from an area of ∼2 m2 within one control plot under wheat in October 2010. The C and N contents of the soil were 1.37 and 0.14%, respectively; soil pHCaCl2 was 6.0. Topsoil texture comprised 7% clay, 87% silt, and 6% sand. Further soil parameters can be found in Kramer et al. (2012). A homogeneous horizontal distribution of bacterial and fungal communities across replicate treatment plots of maize and wheat was previously shown for the site (Scharroba et al., 2012; Moll et al., 2015, 2016). Moreover, rapid and pronounced incorporation of plant-derived C into both bacterial and fungal biomass has been demonstrated for the soil (Kramer et al., 2012; Müller et al., 2016; Pausch et al., 2016).
Microcosm Setup for SIP
Soil corresponding to 50 g dry weight was filled into steel cylinders (diameter = 5.5 cm, height = 4 cm). The four different substrates (glucose, cellulose, senescent maize leaves, and roots) were mixed into the soil and water content was adjusted to 60% water holding capacity. 13C treatments were set up with 13C-labeled substrates (>97 atom %; as determined by the supplier) as well as soil microcosms with unlabeled substrates (∼ natural abundance of 13C in maize plants; 12C treatments) and microcosms without substrate amendment (control). All substrates were purchased from IsoLife (Wageningen, Netherlands) and were derived from equivalent maize plants, with the exception of unlabeled glucose (Sigma-Aldrich, St. Louis, MO, USA). Amendments were added to the soil to a final amount of 12 mg C microcosm-1 (240 μg C g-1 soil), which represented only ∼2% of the intrinsic organic carbon (Kramer et al., 2012). To ensure a homogenous distribution of plant biomass amendments in the microcosms, materials were milled to <1 mm by the supplier. Cylinders were placed into air-tight glasses containing a small vessel attached to the lid to hold 1 M NaOH for absorbing evolved CO2. The microcosms were incubated in a climate chamber at 12°C, representing the long term autumn mean temperature at the site. Microcosms were sampled destructively after 2, 8, 16, and 32 days. For each time point, 12C treatments and controls were set up in triplicate, while 13C treatments were only set up with one microcosm per time point. See the SI for further details on the setup and substrate composition and also a scheme of the experimental setup and downstream workflow (Supplementary Figure S1).
Inference of Substrate-Derived C-Pools
Carbon dioxide production, microbial biomass carbon (Cmic), phospholipid fatty acids (PLFA) as well as the δ13C in CO2, Cmic, and PLFAs were determined from 12C-microcosms as described in the SI. The following mixing model was used for calculation of the relative amounts of substrate-derived C in CO2, Cmic, and PLFAs:
δsample was the δ13C value of the respective sample and δreference was the mean δ13C value of controls (soil without substrate amendment). δsubstrate was the δ13C value of the amended material, and δsoil the δ13C value of the Corg at the beginning of the experiment.
RNA Extraction and rRNA Stable Isotope Probing (rRNA-SIP)
RNA was extracted from soil as described by Lueders et al. (2004) with minor modifications (see SI). RNA extracts of two time points were selected for SIP based on substrate mineralization and assimilation data. These were days 8 (high substrate use) and 32 (later stage of decomposition) for all treatments. Soil from triplicate 12C-microcosms was pooled and extracted as a composite sample for SIP. 13C-labeled rRNA was extracted from single microcosms per time point and treatment. Isopycnic centrifugation and gradient fractionation was done as previously described (Glaubitz et al., 2009; Kleindienst et al., 2014) with 750 ng of total RNA loaded into each gradient and collection of 12 RNA fractions after centrifugation.
Fingerprinting and Sequencing of Density Resolved rRNA
Bacterial, fungal and protist rRNA in density-resolved SIP fractions (fractions 2 to 10 of all gradients) were first analyzed by T-RFLP fingerprinting following published protocols (Edel-Hermann et al., 2008; Euringer and Lueders, 2008; Pilloni et al., 2012). See the SI for full methodological detail. Based on rRNA fingerprinting results of gradient fractions, fractions 3 and 8 of all gradients were selected as representative for “heavy” and “light” rRNA and subjected to 454 amplicon sequencing (Pilloni et al., 2012; Dibbern et al., 2014; Kleindienst et al., 2014). All sequenced fractions were from a buoyant density of either ∼1.79 or ∼1.82 g ml-1 CsTFA, which are typical for light and heavy rRNA, respectively (Lueders et al., 2004). Processing and quality filtering of pyrosequencing reads was done as described in the SI. T-RFs for dominant taxa were predicted in silico via assembled contigs of amplicon reads as previously described (Pilloni et al., 2012). The sequencing of protist rRNA was restricted to gradients from one labile (glucose) and one complex (leaves) detritusphere substrate. All sequencing raw data have been deposited with the NCBI sequence read archive under SRA accession numbers SRP031455 (bacterial 16S rRNA reads), SRP031774 (protist 18S rRNA), and SRP033337 (fungal 18S).
Calculation of Taxon-Specific Enrichment Factors in Labeled rRNA
To identify taxa involved in the assimilation of 13C from amended substrates within the different groups (bacteria, fungi, protists), sequencing read ‘enrichment factors’ (EF) in heavy rRNA fractions were inferred. EFs were calculated for all taxa with >2% read abundance in heavy rRNA fractions of at least one treatment and time point. This cutoff was chosen to cautiously constrain data interpretation to taxa with more notable rRNA enrichment and abundance in our experiment, and not to over-interpret EFs for taxa with low total read numbers. For protists only, all taxa were included in the calculation irrespective of their relative abundance in heavy fractions. The enrichment factors were calculated as follows:
where 13Cheavy and 13Clight is the relative abundance of taxon-specific reads in heavy and light rRNA fractions of 13C treatments, and 12Cheavy and 12Clight is the same for respective 12C-controls. This approach is a variation of the ‘subtraction values’ of T-RFs in heavy vs. light rRNA used by Zumsteg et al. (2013), or of the ‘differential abundance’ approach for sequencing OTUs recently introduced for DNA-SIP (Pepe-Ranney et al., 2015). All taxa that showed an EF >0.5 in at least one treatment or time point were considered as 13C-labeled. For graphical display, the EFs were combined with relative read abundance of labeled taxa in heavy 13C-rRNA. Our EF-approach was supported by ‘classical’ labeling patterns as evident via abundance shifts of taxon-associated T-RFs across gradient fractions.
Statistical Analysis
Cumulative CO2 production in 12C vs. 13C treatments were compared by one-way ANOVA. Substrate-derived C in CO2 over time was analyzed via ANOVA with date as repeated factor. A factorial ANOVA was done for comparison of relative incorporation of substrate-derived C into total microbial biomass as well as fungal and bacterial PLFAs (for each date separately). A post hoc test (Tukey HSD) was conducted for significance testing. Data were transformed if necessary to increase homogeneity of variance (tested by Levene‘s test). Similarity of biologically replicated PLFA community patterns for the different treatments and time points was assessed by multidimensional scaling (MDS) and discriminant function analysis (DFA; Marhan et al., 2007). All statistics were performed using STATISTICA 6.0 (Statsoft, Tulsa, OK, USA), details are described in the SI.
Results
Substrate Mineralization and 13C-Assimilation during Microcosm Incubation
Total turnover of 13C-labeled substrates as well as carbon flow into Cmic and CO2 depended on the complexity and recalcitrance of the added substrates. Approximately two-thirds of added glucose-C and cellulose-C were mineralized after 32 days, while only ∼45 and ∼12% of leaf- and root-C, respectively, were mineralized during the same time period (Supplementary Figure S2). Mineralization of 12C and 13C substrate amendments was not significantly different between microcosms (F1,14 = 0.004; p = 0.95).
Substrate mineralization and assimilation was quantified for replicated 12C treatments. This was possible via the ‘natural’ 13C-pulse of C4 plant material amended to a soil cultivated solely with C3 plants over the last decades (Kramer et al., 2012). On day 2, almost 70% of the CO2 produced originated from glucose in the respective treatments but was reduced to ∼7% by the end of the experiment (Figure 1A). In contrast, mineralization of cellulose and maize leaves peaked at day 8, with 64 and 48 %, respectively, of substrate-derived CO2. In the root treatment, the proportion of substrate-derived CO2 remained low (12-17 %) over time (Figure 1A; substrate effect: F3,7= 14.77, p = 0.002, date effect: F3,21= 71.49, p < 0.001). Resource-derived C in Cmic was highest for glucose (∼40%) after only 2 days of incubation (Figure 1B). Assimilation efficiency appeared much lower for the other substrates, and substrate-derived C was at a maximum of ∼15% for cellulose on day 8, and of ∼11 and ∼5% for leaf and root, respectively, at the end of the experiment (Figure 1B; substrate effect: F3,32= 236.54, p < 0.001, date effect: F3,32= 8.42, p < 0.001).
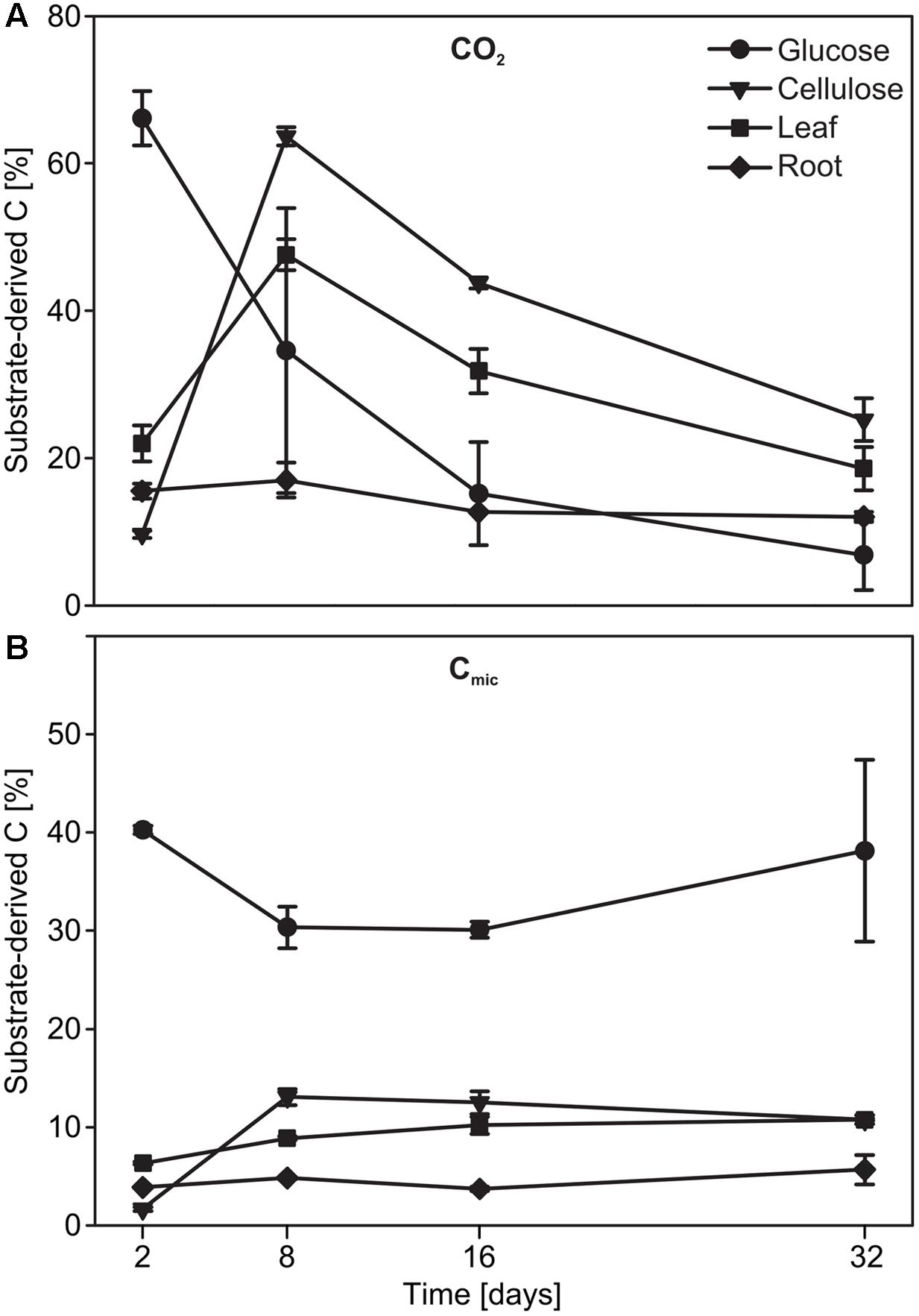
FIGURE 1. Time course of the evolution of substrate-derived C in (A) CO2 and (B) Cmic of microcosms during incubation.
These distinctions in substrate-dependent carbon flow were also apparent in the allocation of substrate-derived C to bacteria and fungi as inferred via δ13C analyses of PLFAs in the 12C treatments. The fungal pool technically also included potential protist-derived fatty acids, as well as plant-derived fatty acids in the plant biomass treatments (Ruess and Chamberlain, 2010; Kaiser et al., 2015). Relative incorporation of substrate-derived C decreased with substrate complexity and recalcitrance (Figure 2, 8 days: F3,15= 4.79, p = 0.02; 32 days: F3,14= 24.41, p < 0.001). In general, a much higher incorporation into Cfung of substrate-derived C was observed (8 days: F1,15= 34.86, p < 0.001; 32 days: F1,14= 71.00, p < 0.001). Between the individual treatments, a slightly but significantly lower relative incorporation into Cbact vs. Cfung was observed for the root treatment after 32 days. However, pronounced quantitative distinctions in comparative kingdom-level substrate utilization were not observed for the labile vs. recalcitrant amendments (Figure 2).
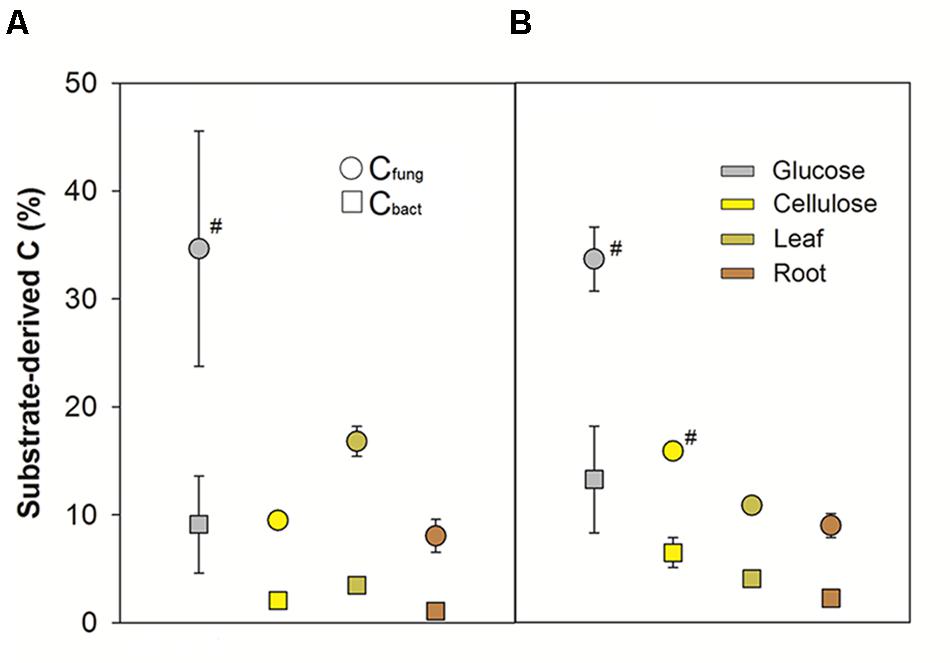
FIGURE 2. Relative substrate-derived C in bacterial and fungal PLFAs after (A) 8 and (B) 32 days of incubation. SE of measurements for triplicate microcosms per treatment and time point are indicated, except #: n = 2.
Total PLFA community patterns were used to substantiate overall microbial community similarity between triplicate 12C- and singular 13C-microcosms per time point. Multivariate analyses of PLFA patterns showed a highly consistent overall community composition within replicate microcosms of all treatments and time points (Supplementary Figure S3). A significant separation (p < 0.001) was only observed between PLFA patterns of the day 8 glucose microcosms and all other samples (MDS axis 1). The secondary MDS axis discriminated earlier and later time points of incubation, but not treatments. More importantly, the grouping of all PLFA patterns from 13C-microcosms amongst the replicated 12C-microcosms (Supplementary Figure S3) indicated a comparable overall community composition between 12C- and 13C-amended microcosms.
rRNA Stable Isotope Probing
The incorporation of 13C from all substrates into bacterial and microeukaryote populations was confirmed by our initial screening of rRNA-SIP gradient fractions by RT-qPCR (SI) and by T-RFLP fingerprinting (Supplementary Figures S4–S6). For all substrates and time points, distinct T-RFs of bacterial, fungal, and even protist rRNA enriched in heavy vs. light fractions of 13C-gradients were observed. Such shifts were not observed in 12C-control gradients, a key criterion for substantiating 13C-labeling (Lueders et al., 2016). Subsequently, amplicon pyrosequencing of rRNA from representative heavy vs. light fractions was used to infer taxon-specific read EF for rRNA of labeled detritusphere microbes. We want to stress that the EFs as inferred here from non-replicated SIP gradients do not allow for a strict quantitative interpretation of taxon-specific buoyant density shifts as recently introduced for DNA-SIP (Hungate et al., 2015; Pepe-Ranney et al., 2016). Still, we are confident that they allow at least for a relative comparison of labeling efficiencies for distinct taxa between treatments and time points, and to identify the most important substrate consumers. Consistent with PLFA patterns (Supplementary Figure S3), fingerprinting (Supplementary Figures S4–S6) and sequencing analysis across all light rRNA fractions also supported a high similarity of total soil microbiota between treatments. EFs were also largely consistent for many labeled taxa between leaf and root treatments (Figures 3 and 4), which was not unexpected given the similar nature of these biomass amendments. To further support the inference of labeling via sequencing-based EFs, almost all taxa with a marked read enrichment could subsequently also be linked to respective T-RFs enriched in heavy 13C-rRNA (Table 1).
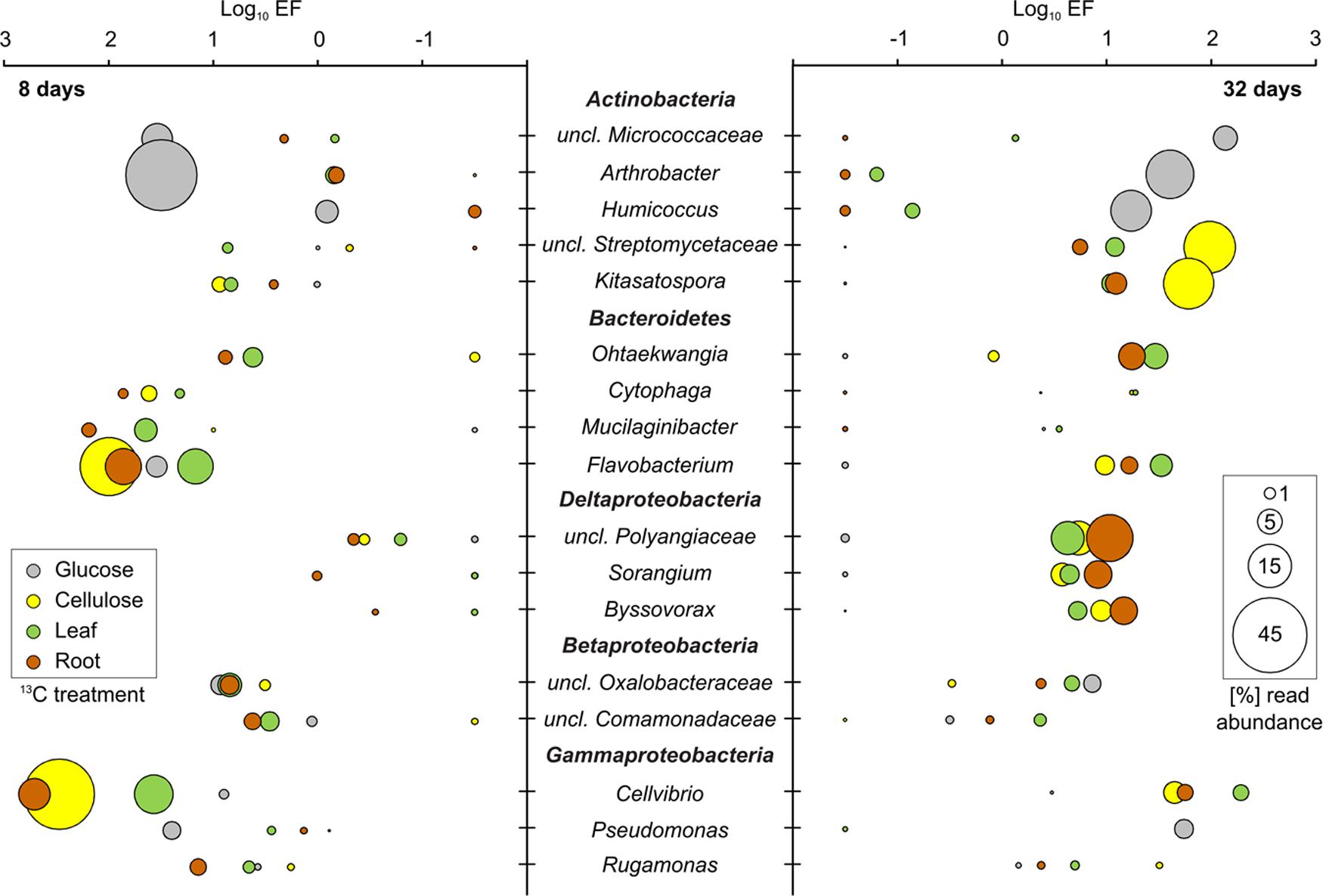
FIGURE 3. 13C-labeled bacterial taxa identified in SIP after 8 and 32 days of incubation. Labeling was inferred via comparative sequencing read enrichment factors (EF) in heavy vs. light rRNA gradient fractions of 13C- and 12C-treatments. All bacterial taxa that showed an EF >0.5 in at least one treatment or time point were considered as 13C-labeled. Other taxa identified in sequencing libraries are not shown. EFs were combined with relative read abundance of labeled taxa in heavy 13C-rRNA. The EF of taxa with a negative read enrichment for given time points or treatments, but labeled in others, was manually set to a log10 of -1.5 for graphical display.
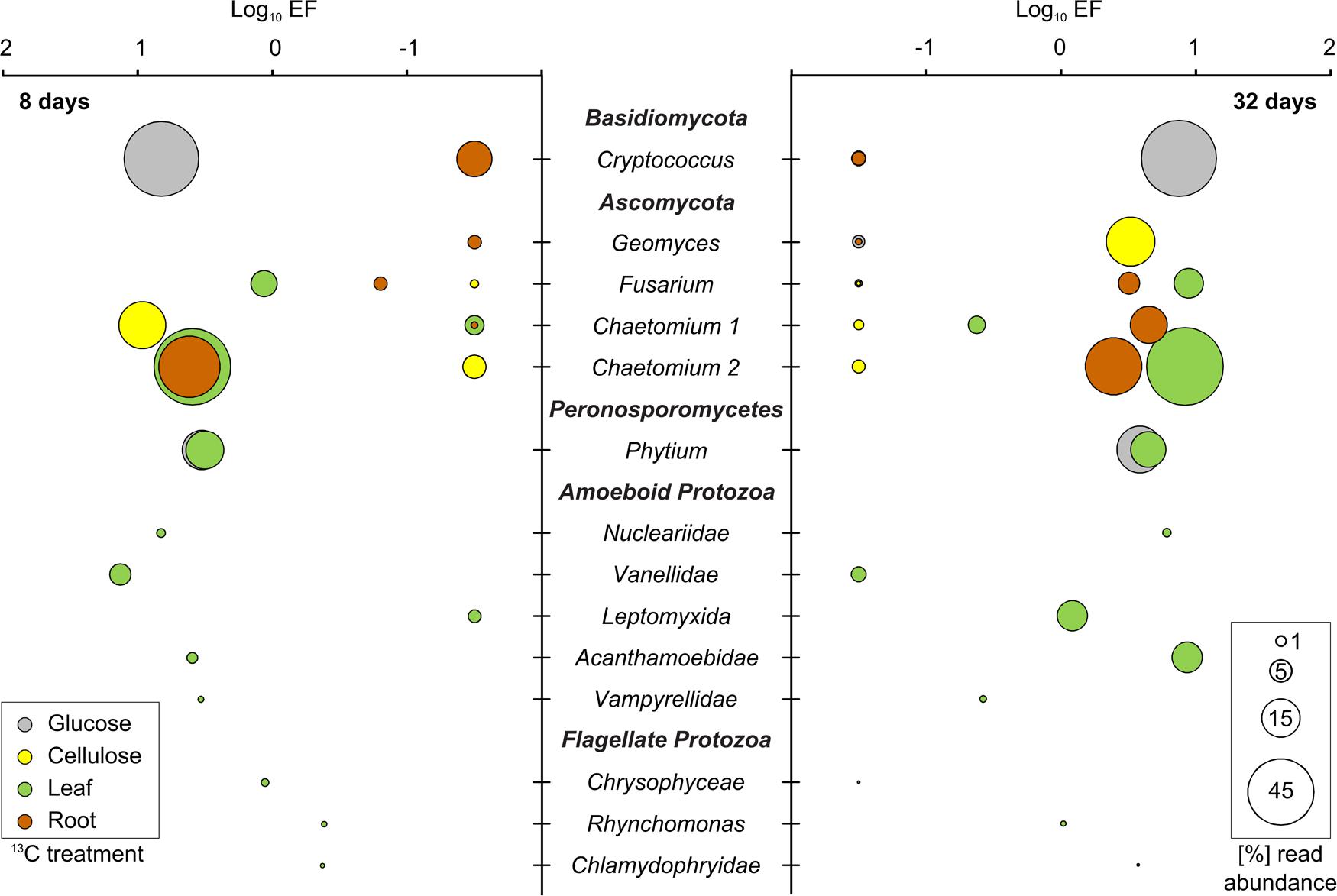
FIGURE 4. 13C-labeled fungal and protist taxa identified in SIP after 8 and 32 days of incubation. All further details: see legend of Figure 3.
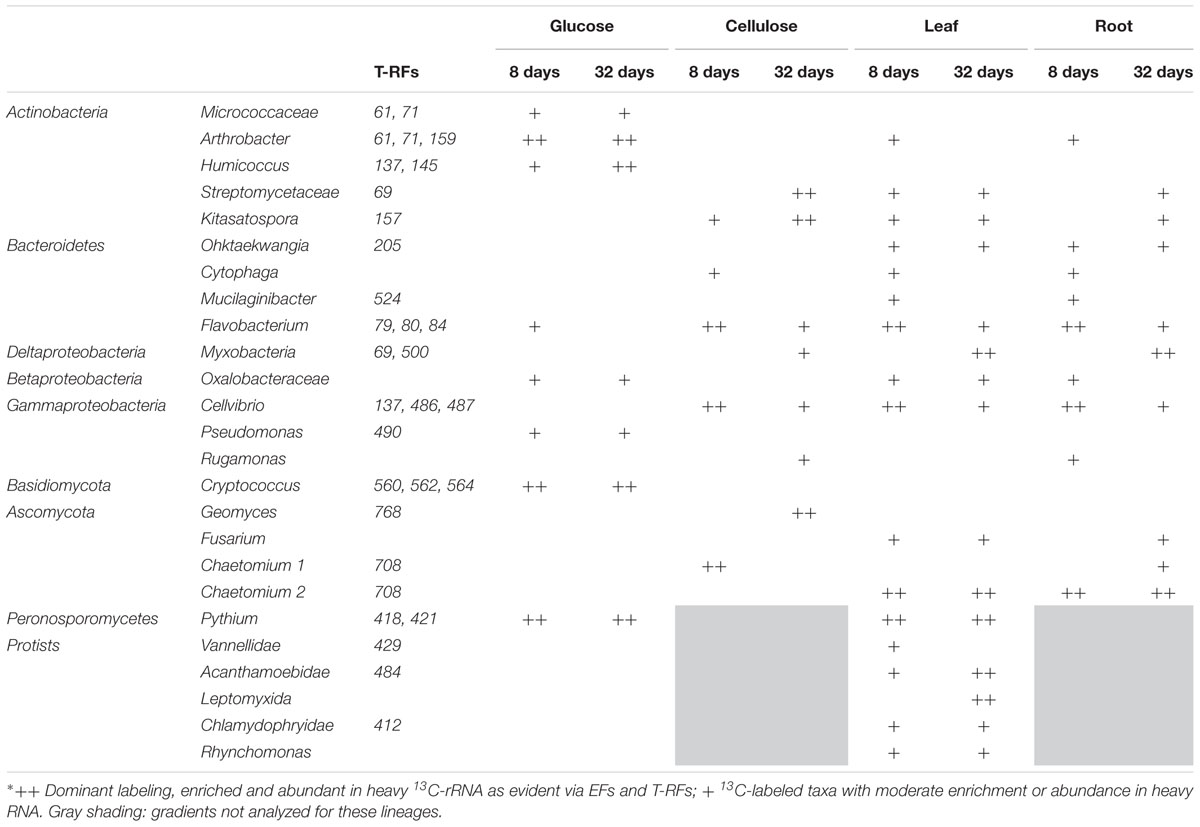
TABLE 1. Overview of 13C-labeled taxa identified in the detritusphere SIP experiment∗.
Labeled Bacterial rRNA
The different bacterial taxa incorporating 13C-label belonged mainly to three bacterial phyla: Actinobacteria, Bacteroidetes and Proteobacteria (Figure 3). Amongst the latter, mostly Gammaproteobacteria but also Beta- and Deltaproteobacteria were labeled. At day 8 of incubation, reads affiliated with Arthrobacter spp. were strongly enriched and abundant in heavy rRNA of the glucose treatment. However, unclassified Micrococcaceae, Flavobacterium spp., unclassified Oxalobacteraceae and Pseudomonas spp. were also enriched, albeit at lower read abundances. Interestingly, although glucose mineralization activity was mostly complete after 8 days (Figure 1), a dynamic labeling pattern was observed. After 32 days, reads related to Flavobacterium spp. were no longer enriched; while the EF of unclassified Micrococcaceae, Humicoccus and Pseudomonas spp. increased. However, Arthrobacter spp. remained the dominant taxon in heavy rRNA. Cellvibrio- and Flavobacterium-related reads were most highly enriched and abundant in heavy rRNA of the cellulose treatment after 8 days (Figure 3). While both were reduced after 32 days, sequences of unclassified Streptomycetaceae and Kitasatospora spp. became more important in labeled rRNA at this later time point. Cytophaga and Rugamonas spp. were identified as less enriched and/or abundant taxa after 8 and 32 days, respectively.
In leaf and root treatments, the highest enrichment was also observed for rRNA of Flavobacterium and Cellvibrio spp., but also for the less frequent Mucilaginibacter and Cytophaga spp. after 8 days. Strong enrichment was detected after 32 days for Cellvibrio, Flavobacterium, and Ohtaekwangia spp. in both leaf and root treatments. Interestingly, labeled rRNA of unclassified Polyangiaceae as well as of other Myxobacteria became noticeably more abundant and enriched within the leaf, root, and cellulose amendments after 32 days. Only one taxon (Ohtaekwangia spp., Bacteroidetes) appeared specifically enriched in the leaf and root treatments, especially after 32 days. Hardly any labeling was observed for this taxon under glucose or cellulose amendment. A summary of the most markedly labeled taxa identified in SIP is given in Table 1. Associated T-RFs, supportive of labeling for these lineages (Supplementary Figures S4–S6), are also listed.
Labeled Fungal rRNA
Compared to bacteria, labeled fungi were less diverse and showed less pronounced enrichment in heavy rRNA. However, they showed more pronounced preferences for specific substrates. All fungal degraders of the added detritusphere substrates belonged to Basidiomycota and Ascomycota (Figure 4; Table 1). Glucose carbon was strongly assimilated by Cryptococcus spp. Surprisingly, other fungi were not labeled in this treatment. In turn, Cryptococcus yeasts were not labeled in any other treatment. Under cellulose amendment, enrichment was observed only for the Chaetomium-related phylotype 1 after 8 days, while Geomyces spp. were the only labeled fungi after 32 days. In the leaf and root treatments, a second Chaetomium-related phylotype was enriched and highly abundant after 8 days. Labeling was also observed for Fusarium spp. in both plant litter treatments after 32 days, as well as for the first Chaetomium phylotype in the root amendment. The labeling of associated T-RFs (Table 1) was also apparent in fingerprinting of density-resolved fungal rRNA (Supplementary Figure S6).
Labeled Protist rRNA
Sequences of fungus-like protists within the Peronosporomycetes (Pythium spp.) were highly enriched in heavy rRNA in both the glucose and leaf treatments (Figure 4). Only these treatments were investigated for protists. Read abundance of protists other than Pythium was low in heavy rRNA of the glucose treatment and no further labeling was observed. In contrast, five amoeboid and three flagellate protozoan taxa appeared enriched in the leaf treatment (Figure 4). After 8 days, enrichment was found especially for the amoeboid Vannellidae (Amoebozoa) but also for the less abundant Nucleariidae (Choanozoa), Acanthamoebidae (Amoebozoa) and Vampyrellidae (Cercozoa). Labeling of Vannellidae was reduced after 32 days, while it was now more clearly detectable for Acanthamoebidae and Leptomyxida (Amoebozoa). Among the flagellate protozoa, members of the Chrysophyceae (Bacillariophyceae) and Rhynchomonas spp. (Kinetoplastida) also showed some rRNA enrichment, which was again also apparent for some associated T-RFs (Table 1; Supplementary Figure S6).
Discussion
Mineralization and Assimilation of Detritusphere Substrates
The diversity and succession of specific bacteria and microeukaryotes actively involved in the degradation of detritusphere substrates was investigated in a typical agricultural soil. Compared to several previous SIP studies that have focused on the degradation of cellulose and other substrate analogues in soil (Haichar et al., 2007; Schellenberger et al., 2010; Eichorst and Kuske, 2012; Štursová et al., 2012; Verastegui et al., 2014; Wang et al., 2015; Pepe-Ranney et al., 2016), the present work addressed distinct substrate complexities and their mineralization by detritusphere microbes up to the level of actual plant biomass. Additionally, detritusphere carbon flow and consumer succession was traced across microbial kingdoms. We show that overall mineralization was not significantly influenced by the isotopic composition of the amendments (Supplementary Figure S2), demonstrating that C utilization of the 12C- and 13C-substrate pairs was comparable over the experimental period. Compared to glucose (Figure 1A), the much less efficient mineralization and assimilation of C from plant biomass amendments (Figures 1 and 2; Supplementary Figure S2) appeared, as expected, directly related to the higher complexity of these materials (de Boer et al., 2005; Bertrand et al., 2006).
13C-PLFA analyses suggested that fungi incorporated more substrate-derived C than bacteria, consistent with the rapid and pronounced incorporation of plant-derived C into fungi already reported for the investigated soil (Kramer et al., 2012; Müller et al., 2016; Pausch et al., 2016), and also for other rhizosphere (Denef et al., 2007; Tavi et al., 2013) and detritusphere systems (Paterson et al., 2008; Alfredsson et al., 2016). The fungal 18:2ω6,9 PLFA biomarker (Ruess and Chamberlain, 2010) is also present in plants (Kaiser et al., 2015), which is why we must caution our quantitative interpretation especially for the leaf and root treatments after 8 days (Figure 2). However, we have evidence that plant-derived PLFA contributed only a minor fraction to the fungal pool from related studies with the same soil. Müller et al. (2016) followed the incorporation of 13C into fungal PLFA as well as ergosterol, a biomarker exclusive to fungal cell membranes. They reported a strict correlation of relative 13C incorporation into these independent fungal signature molecules. Moreover, our present study shows a higher relative incorporation of substrate-derived C into fungal PLFA also for the glucose and cellulose treatments, where plant-derived PLFA were not amended. Therefore, our data on 13C incorporation into bacterial and fungal PLFAs suggest that distinct bacterial and fungal energy channels were not apparent for labile vs. recalcitrant substrates, rejecting our first initial hypothesis for the investigated soil.
Bacterial Key-Players in the Detritusphere
Actinobacteria have been previously shown to dominate sugar utilization for a number of soils (Schellenberger et al., 2010; Verastegui et al., 2014), thus our identification of Arthrobacter spp. and members of the Micrococcaceae as key glucose consumers was not surprising. Pseudomonas spp. are also well known to utilize glucose but more recalcitrant compounds as well, including lignin in soil (Goldfarb et al., 2011). A marked succession of bacterial glucose utilizers over time was not observed. This was in strong contrast to cellulose degradation, where an early cellulose degrading community dominated by Cellvibrio and Flavobacterium spp. profoundly shifted towards Actinobacteria after 32 days. While members of the Bacteroidetes and Cellvibrio spp. have been identified previously as cellulose consumers for a whole range of soils (Haichar et al., 2007; Schellenberger et al., 2010; Verastegui et al., 2014), such dynamic successions have not been observed (Pepe-Ranney et al., 2016). The secondary involvement of Streptomycetes in cellulose degradation seems plausible (Kämpfer, 2006), possible mechanisms of this succession will be discussed further down.
Most of the important cellulose decomposers were consistently apparent under plant biomass amendment, as well as their succession. This was not unexpected, given that cellulose was a major component of these amendments. After 32 days, unclassified Polyangiaceae, Sorangium and Byssovorax spp. (all Deltaproteobacteria) also emerged as labeled in the cellulose, leaf and root treatments. These Myxobacteria are known as saprotrophs but also as bacterial micropredators in soil (Johnke et al., 2014). Their strict secondary labeling could be indicative of feeding on labeled bacterial biomass (Lueders et al., 2006) of primary detritusphere components, which will also be discussed below.
Detritusphere Fungi
In contrast to bacteria, detritusphere fungi showed more marked distinctions between glucose, cellulose and plant residue-utilizing taxa. Cryptococcus spp. dominated glucose utilization in our experiment, highlighting the role of these fast-growing yeasts as important competitors for labile carbon in the detritusphere. However, they remained unlabeled, surprisingly, in all other treatments. This is in contrast to Štursová et al. (2012), who reported Cryptococcus spp. as the main cellulolytic fungi in a forest soil. Either soil type or the use of bacterial vs. plant-derived cellulose amendments could potentially explain this distinction.
Early cellulose utilization was dominated by a Chaetomium phylotype and a marked succession to Geomyces spp. was observed after 32 days. Both genera have been previously identified by SIP as cellulose utilizers for a range of soils (Eichorst and Kuske, 2012; Štursová et al., 2012), but their succession has not been reported. A second Chaetomium-phylotype as well as Fusarium spp. were found as dominant fungi in plant residue degradation. The distinct substrate utilization patterns observed suggest that these fungi may harbor distinct exoenzymatic capabilities (Romani et al., 2006). Cryptococcus and Chaetomium spp. were also abundant in amplicon sequencing libraries generated directly from the investigated agricultural field (Moll et al., 2016), thus strengthening the link between labeling results obtained in laboratory microcosms and the detritusphere food web in situ.
Labeled Protists
The flow of detritusphere carbon into protists was investigated only for one labile (glucose) and one complex (leaves) substrate (see SI). We are aware that this limits our interpretation of the effects of substrate complexity and recalcitrance on food web succession. However, due to the large overlaps of labeled bacteria detected in the non-glucose treatments (Table 1), we argue that this selection still makes it possible to infer the most fundamental distinctions in protist labeling. The only protists labeled in the glucose treatment were Pythium spp. These oomycetes are fungus-like protists within the Stramenopiles and known as important plant pathogens (Hendrix and Campbell, 1973), but can also act as pioneer saprotrophs on fresh plant residues in soil (Deacon, 1997). rRNA of Pythium spp. was labeled at both time points for maize leaf degradation also, indicating an important role of these protists in the detritusphere. Similarly, Pythium spp. has been found to be involved in carbon flow from rice root callus (Murase et al., 2012).
In contrast to the glucose treatment, labeling of diverse protozoa was observed in the leaf treatment. This labeling suggests a substantial flow of carbon to these higher trophic levels, despite the low C assimilation efficiency of protozoa and concomitant high respiration losses of 13CO2 (Crotty et al., 2012). 13C enrichment was mainly found in rRNA of amoeboid protozoa, again with marked succession over time. The amoeboid life style was likely more competitive in our soil microcosms, a distinction which seems warranted given the more marked labeling of flagellates reported for cellulose degradation in soil slurries (Chatzinotas et al., 2013). At least some of the labeled protozoa are known as fungivores, such as members of the Leptomyxida (Chakraborty and Old, 1982), the Vampyrellidae (Hess et al., 2012) and the Chlamydophryidae (Dumack et al., 2015). Although such fungivores are increasingly recognized as widespread in soils (Geisen et al., 2015), they are not yet embedded in food web concepts. Our study is the first to demonstrate a direct involvement of such fungivorous protozoa in detritusphere carbon flow.
Substrate Complexity, Consumer Diversity, and Succession
The total number of dominantly labeled primary substrate consumers was low and not noticeably larger for any of the administered treatments (Table 1). Despite the fact that the investigated field site has been shown to host a horizontally homogeneous, but diverse soil microbiota (Scharroba et al., 2012; Dibbern et al., 2014; Moll et al., 2015; Moll et al., 2016), only a minority of taxa seemed to be involved in detritusphere decomposition. Thus in contrast to our second initial hypothesis, substrate complexity and recalcitrance were not directly correlated with consumer diversity. Although effects of substrate recalcitrance on overall microbial diversity have been reported (Nicolardot et al., 2007; Goldfarb et al., 2011), our results show that this does not necessarily apply to primary detritusphere consumers.
In contrary, we demonstrate that consumer succession and secondary trophic labeling increased with substrate complexity. Successional labeling was pronounced for most of the secondary consumers, which we believe discriminates them from primary consumers with low incorporation rates of labeled substrate. Marked successions of both bacterial and fungal key players were observed during cellulose decomposition and also for leaf and root treatments, albeit at decreasing kinetics (reflecting increasing substrate recalcitrance). Degradative succession has been intensively discussed for detritusphere fungi (Frankland, 1998) and recently also for bacteria (Pepe-Ranney et al., 2016) and is assumed to be primarily substrate-driven. In the present work, however, top-down controls may also have been involved in the observed successions. It is known that Gram-positive Actinobacteria are a non-preferred prey of protists due to their rigid cell walls and hyphal growth (Jousset, 2012). Potentially, the initial bursts of Gram-negative Cellvibrio and Flavobacterium populations in the cellulose and plant treatments may have been subject to intensive grazing by the diverse protozoa identified in the leaf treatment. Thus, niche space for more grazing-resistant actinobacterial cellulose consumers could have been generated. Consistently, the surprising lack of labeled protozoan rRNA in the glucose treatment may also reflect the dominant role of Actinobacteria in this amendment.
We also identified a prominent secondary succession of labeling within the Myxobacteria. These are known as saprotrophs but also as intra-bacterial micropredators in soil (Lueders et al., 2006; Johnke et al., 2014). Their consistent secondary labeling in the cellulose and plant biomass treatments does not allow to clearly differentiate both roles. Nevertheless, while intra-bacterial predation could indeed have occurred in the detritusphere, this question certainly merits further investigation.
Bacterial and Fungal Energy Channels
Bacteria, fungi, and even protists were identified as primary consumers of all amendments, irrespective of substrate complexity or recalcitrance. This was suggested not only via rRNA-, but also PLFA-labeling, as also yeasts such as Cryptococcus (Marumo and Aoki, 1990) and protists such as Pythium (Erwin, 1973) harbor respective marker fatty acids. A preferential allocation of C from the labile amendments to bacteria was clearly not observed (Figure 2), thus rejecting the hypothesis of distinct bacterial and fungal energy channels for the investigated soil. Substrate partitioning may vary for different soils, as both a faster and more pronounced incorporation of C from labile and soluble amendments into bacterial PLFAs (Paterson et al., 2008), and also of labile root exudates into fungal PLFAs (Butler et al., 2003; Denef et al., 2007) have been reported. For the agricultural soil investigated here, our results promote the perspective of simultaneous and overlapping substrate utilization by bacteria, fungi, and protists in the detritusphere, irrespective of resource quality.
Conclusion
Substrate complexity and recalcitrance were identified as primary drivers of consumer succession and secondary trophic labeling over time. If the detritusphere is envisioned as a continuum of substrate inputs and degradation in discrete niches, such successional effects will essentially increase consumer diversity for more recalcitrant substrates. However, the mechanisms are distinct from a direct substrate-driven selection of primary consumers and may be partly under top-down control. Our results suggest a role of protists not only as bacterivores, but also as fungivores and even primary saprotrophs in the investigated detritusphere food web. Likewise, Myxobacteria may also have been active as bacterivores. Such intra- and trans-kingdom feedbacks and successions await better incorporation into conceptual models of soil food webs. Here, the direct linking of key microbial populations to relevant decomposition processes is a major challenge (Trivedi et al., 2013) which can be addressed using SIP, as shown here. Trophic interactions and succession rather than substrate-defined energy channels may be a vital asset to existing ecosystem models (Moore et al., 2005; McGuire and Treseder, 2010), possibly facilitating more accurate predictions of soil carbon cycling in the future.
Author Contributions
EK, TL, and SM designed the experiments. SK, DD, JM, and MH conducted the experiments. SK, DD, JM, MH, RK, DK, SM, TU, and TW analyzed data. SK, EK, MB, FB, and TL wrote the manuscript with contributions from all authors.
Funding
This study was funded by grants of the DFG (Deutsche Forschungsgemeinschaft) to EK, TL, FB, and MB within the Research Unit “Carbon flow in belowground food webs assessed with isotope tracers” (FOR 918). Further support was provided by the Helmholtz Society.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Kathleen Regan (University of Hohenheim) for English language editing. We thank M. Engel, B. Schloter-Hai, and M. S. Granitsiotis for assistance in pyrosequencing at the Helmholtz Zentrum München. We also thank L. Ruess (University of Berlin) and S. Scheu (University of Göttingen) for coordinating the Research Unit and for valuable discussions. An early version of this manuscript has been published as part of the Ph.D. thesis of SK accepted at the University of Hohenheim (http://opus.uni-hohenheim.de/volltexte/2014/990/).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01524
References
Adl, M. S., and Gupta, V. S. (2006). Protists in soil ecology and forest nutrient cycling. Can. J. Forest Res. 36, 1805–1817. doi: 10.1139/x06-056
Akça, E., Alba, S. D., Álvarez, A. G., Bialousz, S., Berger, B., Bielek, P., et al. (2005). Soil Atlas of Europe. Luxembourg: European Soil Bureau Network.
Alfredsson, H., Clymans, W., Stadmark, J., Conley, D., and Rousk, J. (2016). Bacterial and fungal colonization and decomposition of submerged plant litter: consequences for biogenic silica dissolution. FEMS Microbiol. Ecol. 92:fiw011. doi: 10.1093/femsec/fiw011
Bastian, F., Bouziri, L., Nicolardot, B., and Ranjard, L. (2009). Impact of wheat straw decomposition on successional patterns of soil microbial community structure. Soil Biol. Biochem. 41, 262–275. doi: 10.1016/j.soilbio.2008.10.024
Bernard, L., Maron, P. A., Mougel, C., Nowak, V., Leveque, J., Marol, C., et al. (2009). Contamination of soil by copper affects the dynamics, diversity, and activity of soil bacterial communities involved in wheat decomposition and carbon storage. Appl. Environ. Microbiol. 75, 7565–7569. doi: 10.1128/aem.00616-09
Bernard, L., Mougel, C., Maron, P.-A., Nowak, V., Leveque, J., Henault, C., et al. (2007). Dynamics and identification of soil microbial populations actively assimilating carbon from 13C-labelled wheat residue as estimated by DNA- and RNA-SIP techniques. Environ. Microbiol. 9, 752–764. doi: 10.1111/j.1462-2920.2006.01197.x
Bertrand, I., Chabbert, B., Kurek, B., and Recous, S. (2006). Can the biochemical features and histology of wheat residues explain their decomposition in soil? Plant Soil 281, 291–307. doi: 10.1007/s11104-005-4628-7
Butler, J., Williams, M., Bottomley, P., and Myrold, D. (2003). Microbial community dynamics associated with rhizosphere carbon flow. Appl. Environ. Microbiol. 69, 6793–6800. doi: 10.1128/AEM.69.11.6793-6800.2003
Chakraborty, S., and Old, K. M. (1982). Mycophagous soil amoeba: interactions with three plant pathogenic fungi. Soil Biol. Biochem. 14, 247–255. doi: 10.1016/0038-0717(82)90034-7
Chatzinotas, A., Schellenberger, S., Glaser, K., and Kolb, S. (2013). Assimilation of cellulose-derived carbon by microeukaryotes in oxic and anoxic slurries of an aerated soil. Appl. Environ. Microbiol. 79, 5777–5781. doi: 10.1128/aem.01598-13
Crotty, F. V., Adl, S. M., Blackshaw, R. P., and Murray, P. J. (2012). Using stable isotopes to differentiate trophic feeding channels within soil food webs. J Eukar. Microbiol. 59, 520–526. doi: 10.1111/j.1550-7408.2011.00608.x
de Boer, W., Folman, L. B., Summerbell, R. C., and Boddy, L. (2005). Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev. 29, 795–811. doi: 10.1016/j.femsre.2004.11.005
Denef, K., Bubenheim, H., Lenhart, K., Vermeulen, J., Cleemput, O. V., Boeckx, P., et al. (2007). Community shifts and carbon translocation within metabolically-active rhizosphere microorganisms in grasslands under elevated CO2. Biogeosciences 4, 769–779. doi: 10.5194/bg-4-769-2007
Dibbern, D., Schmalwasser, A., Lueders, T., and Totsche, K. U. (2014). Selective transport of plant root-associated bacterial populations in agricultural soils upon snowmelt. Soil Biol. Biochem. 69, 187–196. doi: 10.1016/j.soilbio.2013.10.040
Dumack, K., Mueller, M., and Bonkowski, M. (2015). Description of a soil dwelling fungi and algae feeding protist, Lecythium terrestris sp. nov. (Rhizaria, Cercozoa, Tectofilosida, Chlamydophryidae), based on morphology and SSU sequence data. Protist 167, 93–105. doi: 10.1016/j.protis.2016.01.001
Edel-Hermann, V., Gautheron, N., Alabouvette, C., and Steinberg, C. (2008). Fingerprinting methods to approach multitrophic interactions among microflora and microfauna communities in soil. Biol. Fertil. Soils 44, 975–984. doi: 10.1007/s00374-008-0287-1
Eichorst, S. A., and Kuske, C. R. (2012). Identification of cellulose-responsive bacterial and fungal communities in geographically and edaphically different soils by using stable isotope probing. Appl. Environ. Microbiol. 78, 2316–2327. doi: 10.1128/AEM.07313-11
Ekelund, F., and Ronn, R. (1994). Notes on protozoa in agricultural soil with emphasis on heterotrophic flagellates and naked amoebae and their ecology. FEMS Microbiol. Rev. 15, 321–353. doi: 10.1016/0168-6445(94)90068-X
Erwin, J. (1973). “Comparative biochemistry of fatty acids in eukaryotic microorganisms,” in Lipids and Biomembranes of Eukaryotic Microorganisms, ed. J. A. Erwin (Cambridge, MA: Academic Press), 41–143.
España, M., Rasche, F., Kandeler, E., Brune, T., Rodriguez, B., Bending, G. D., et al. (2011). Assessing the effect of organic residue quality on active decomposing fungi in a tropical vertisol using 15N-DNA stable isotope probing. Fungal Ecol. 4, 115–119. doi: 10.1016/j.funeco.2010.09.005
Euringer, K., and Lueders, T. (2008). An optimised PCR/T-RFLP fingerprinting approach for the investigation of protistan communities in groundwater environments. J. Microbiol. Methods 75, 262–268. doi: 10.1016/j.mimet.2008.06.012
Fan, F., Yin, C., Tang, Y., Li, Z., Song, A., Wakelin, S. A., et al. (2014). Probing potential microbial coupling of carbon and nitrogen cycling during decomposition of maize residue by 13C-DNA-SIP. Soil Biol. Biochem. 70, 12–21. doi: 10.1016/j.soilbio.2013.12.002
Frankland, J. C. (1998). Fungal succession–unravelling the unpredictable. Mycol. Res. 102, 1–15. doi: 10.1017/S0953756297005364
Geisen, S., Tveit, A. T., Clark, I. M., Richter, A., Svenning, M. M., Bonkowski, M., et al. (2015). Metatranscriptomic census of active protists in soils. ISME J. 9, 2178–2190. doi: 10.1038/ismej.2015.30
Glaubitz, S., Lueders, T., Abraham, W.-R., Jost, G., Jürgens, K., and Labrenz, M. (2009). 13C-isotope analyses reveal that chemolithoautotrophic gamma- and epsilonproteobacteria feed a microbial food web in a pelagic redoxcline of the central Baltic Sea. Environ. Microbiol. 11, 326–337. doi: 10.1111/j.1462-2920.2008.01770.x
Goldfarb, K. C., Karaoz, U., Hanson, C. A., Santee, C. A., Bradford, M. A., Treseder, K. K., et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol 2:94. doi: 10.3389/fmicb.2011.00094
Haichar, F. E. Z., Achouak, W., Christen, R., Heulin, T., Marol, C., Marais, M.-F., et al. (2007). Identification of cellulolytic bacteria in soil by stable isotope probing. Environ. Microbiol. 9, 625–634. doi: 10.1111/j.1462-2920.2006.01182.x
Hendrix, F. F. J., and Campbell, W. A. (1973). “Pythiums as plant pathogens,” in Annual Review Of Phytopathology, Vol. 11, ed. F. Baker Kenneth (Palo Alto, CA: Annual Reviews Inc), 77–98.
Hess, S., Sausen, N., and Melkonian, M. (2012). Shedding light on vampires: the phylogeny of vampyrellid amoebae revisited. PLoS ONE 7:e31165. doi: 10.1371/journal.pone.0031165
Hungate, B. A., Mau, R. L., Schwartz, E., Caporaso, J. G., Dijkstra, P., van Gestel, N., et al. (2015). Quantitative microbial ecology through stable isotope probing. Appl. Environ. Microbiol. 81, 7570–7581. doi: 10.1128/aem.02280-15
Johnke, J., Cohen, Y., de Leeuw, M., Kushmaro, A., Jurkevitch, E., and Chatzinotas, A. (2014). Multiple micro-predators controlling bacterial communities in the environment. Curr. Opin. Biotechnol. 27, 185–190. doi: 10.1016/j.copbio.2014.02.003
Jousset, A. (2012). Ecological and evolutive implications of bacterial defences against predators. Environ. Microbiol. 14, 1830–1843. doi: 10.1111/j.1462-2920.2011.02627.x
Kaiser, C., Kilburn, M. R., Clode, P. L., Fuchslueger, L., Koranda, M., Cliff, J. B., et al. (2015). Exploring the transfer of recent plant photosynthates to soil microbes: mycorrhizal pathway vs direct root exudation. New Phytol. 205, 1537–1551. doi: 10.1111/nph.13138
Kämpfer, P. (2006). “The family Streptomycetaceae, Part I: taxonomy,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 538–604.
Kleindienst, S., Herbst, F.-A., Stagars, M., von Netzer, F., von Bergen, M., Seifert, J., et al. (2014). Diverse sulfate-reducing bacteria of the Desulfosarcina/Desulfococcus clade are the key alkane degraders at marine seeps. ISME J. 8, 2029–2044. doi: 10.1038/ismej.2014.51
Kramer, S., Marhan, S., Ruess, L., Armbruster, W., Butenschoen, O., Haslwimmer, H., et al. (2012). Carbon flow into microbial and fungal biomass as a basis for the belowground food web of agroecosystems. Pedobiologia 55, 111–119. doi: 10.1016/j.pedobi.2011.12.001
Li, Y., Lee, C. G., Watanabe, T., Murase, J., Asakawa, S., and Kimura, M. (2011). Identification of microbial communities that assimilate substrate from root cap cells in an aerobic soil using a DNA-SIP approach. Soil Biol. Biochem. 43, 1928–1935. doi: 10.1016/j.soilbio.2011.05.016
Lueders, T., Dumont, M. G., Bradford, L., and Manefield, M. (2016). RNA-stable isotope probing: from carbon flow within key microbiota to targeted transcriptomes. Curr. Opin. Biotechnol. 41, 83–89. doi: 10.1016/j.copbio.2016.02.023
Lueders, T., Kindler, R., Miltner, A., Friedrich, M. W., and Kaestner, M. (2006). Identification of bacterial micropredators distinctively active in a soil microbial food web. Appl. Environ. Microbiol. 72, 5342–5348. doi: 10.1128/AEM.00400-06
Lueders, T., Manefield, M., and Friedrich, M. W. (2004). Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ. Microbiol. 6, 73–78. doi: 10.1046/j.1462-2920.2003.00536.x
Marhan, S., Kandeler, E., and Scheu, S. (2007). Phospholipid fatty acid profiles and xylanase activity in particle size fractions of forest soil and casts of Lumbricus terrestris L. (Oligochaeta, Lumbricidae). Appl. Soil Ecol. 35, 412–422. doi: 10.1016/j.apsoil.2006.06.003
Marumo, K., and Aoki, Y. (1990). Discriminant analysis of cellular fatty acids of candida species, Torulopsis glabrata, and Cryptococcus neoformans determined by gas-liquid chromatography. J. Clin. Microbiol. 28, 1509–1513.
McGuire, K. L., and Treseder, K. K. (2010). Microbial communities and their relevance for ecosystem models: decomposition as a case study. Soil Biol. Biochem. 42, 529–535. doi: 10.1016/j.soilbio.2009.11.016
Moll, J., Goldmann, K., Kramer, S., Hempel, S., Kandeler, E., Marhan, S., et al. (2015). Resource type and availability regulate fungal communities along arable soil profiles. Microb. Ecol. 70, 390–399. doi: 10.1007/s00248-015-0569-8
Moll, J., Hoppe, B., König, S., Wubet, T., Buscot, F., and Krüger, D. (2016). Spatial distribution of fungal communities in an arable soil. PLoS ONE 11:e0148130. doi: 10.1371/journal.pone.0148130
Moore, J. C., McCann, K., and de Ruiter, P. C. (2005). Modeling trophic pathways, nutrient cycling, and dynamic stability in soils. Pedobiologia 49, 499–510. doi: 10.1016/j.pedobi.2005.05.008
Müller, K., Kramer, S., Haslwimmer, H., Marhan, S., Scheunemann, N., Butenschön, O., et al. (2016). Carbon transfer from maize roots and litter into bacteria and fungi depends on soil depth and time. Soil Biol. Biochem. 93, 79–89. doi: 10.1016/j.soilbio.2015.10.015
Murase, J., Shibata, M., Lee, C. G., Watanabe, T., Asakawa, S., and Kimura, M. (2012). Incorporation of plant residue–derived carbon into the microeukaryotic community in a rice field soil revealed by DNA stable-isotope probing. FEMS Microbiol. Ecol. 79, 371–379. doi: 10.1111/j.1574-6941.2011.01224.x
Nicolardot, B., Bouziri, L., Bastian, F., and Ranjard, L. (2007). A microcosm experiment to evaluate the influence of location and quality of plant residues on residue decomposition and genetic structure of soil microbial communities. Soil Biol. Biochem. 39:1631. doi: 10.1016/j.soilbio.2007.01.012
Paterson, E., Osler, G., Dawson, L. A., Gebbing, T., Sim, A., and Ord, B. (2008). Labile and recalcitrant plant fractions are utilised by distinct microbial communities in soil: independent of the presence of roots and mycorrhizal fungi. Soil Biol. Biochem. 40, 1103. doi: 10.1016/j.soilbio.2007.12.003
Pausch, J., Kramer, S., Scharroba, A., Scheunemann, N., Butenschoen, O., Kandeler, E., et al. (2016). Small but active–pool size does not matter for carbon incorporation in below-ground food webs. Funct. Ecol. 30, 479–489. doi: 10.1111/1365-2435.12512
Pepe-Ranney, C., Campbell, A. N., Koechli, C. N., Berthrong, S., and Buckley, D. H. (2016). Unearthing the ecology of soil microorganisms using a high resolution DNA-SIP approach to explore cellulose and xylose metabolism in soil. Front. Microbiol. 7:703. doi: 10.3389/fmicb.2016.00703
Pepe-Ranney, C., Koechli, C., Potrafka, R., Andam, C., Eggleston, E., Garcia-Pichel, F., et al. (2015). Non-cyanobacterial diazotrophs mediate dinitrogen fixation in biological soil crusts during early crust formation. ISME J. 10, 287–298. doi: 10.1038/ismej.2015.1106
Pilloni, G., Granitsiotis, M. S., Engel, M., and Lueders, T. (2012). Testing the limits of 454 pyrotag sequencing: reproducibility, quantitative assessment and comparison to T-RFLP fingerprinting of aquifer microbes. PLoS ONE 7:e40467. doi: 10.1371/journal.pone.0040467
Poll, C., Brune, T., Begerow, D., and Kandeler, E. (2010). Small-scale diversity and succession of fungi in the detritusphere of rye residues. Microb. Ecol. 59, 130–140. doi: 10.1007/s00248-009-9541-9
Romani, A. M., Fischer, H., Mille-Lindblom, C., and Tranvik, L. J. (2006). Interactions of bacteria and fungi on decomposing litter: differential extracellular enzyme activities. Ecology 87, 2559–2569. doi: 10.1890/0012-9658(2006)87[2559:IOBAFO]2.0.CO;2
Rousk, J., and Frey, S. D. (2015). Revisiting the hypothesis that fungal-to-bacterial dominance characterizes turnover of soil organic matter and nutrients. Ecol. Monogr. 85, 457–472. doi: 10.1890/14-1796.1
Ruess, L., and Chamberlain, P. M. (2010). The fat that matters: soil food web analysis using fatty acids and their carbon stable isotope signature. Soil Biol. Biochem. 42, 1898–1910. doi: 10.1016/j.soilbio.2010.07.020
Scharroba, A., Dibbern, D., Hünninghaus, M., Kramer, S., Moll, J., Butenschoen, O., et al. (2012). Effects of resource availability and quality on the structure of the micro-food web of an arable soil across depth. Soil Biol. Biochem. 50, 1–11. doi: 10.1016/j.soilbio.2012.03.002
Schellenberger, S., Kolb, S., and Drake, H. L. (2010). Metabolic responses of novel cellulolytic and saccharolytic agricultural soil bacteria to oxygen. Environ. Microbiol. 12, 845–861. doi: 10.1111/j.1462-2920.2009.02128.x
Schmidt, M. W. I., Torn, M. S., Abiven, S., Dittmar, T., Guggenberger, G., Janssens, I. A., et al. (2011). Persistence of soil organic matter as an ecosystem property. Nature 478, 49–56. doi: 10.1038/nature10386
Strickland, M. S., and Rousk, J. (2010). Considering fungal:bacterial dominance in soils methods, controls, and ecosystem implications. Soil Biol. Biochem. 42, 1385–1395. doi: 10.1016/j.soilbio.2010.05.007
Štursová, M., Žifčáková, L., Leigh, M. B., Burgess, R., and Baldrian, P. (2012). Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol. Ecol. 80, 735–746. doi: 10.1111/j.1574-6941.2012.01343.x
Tavi, N. M., Martikainen, P. J., Lokko, K., Kontro, M., Wild, B., Richter, A., et al. (2013). Linking microbial community structure and allocation of plant-derived carbon in an organic agricultural soil using 13CO2 pulse-chase labelling combined with 13C-PLFA profiling. Soil Biol. Biochem. 58, 207–215. doi: 10.1016/j.soilbio.2012.11.013
Termorshuizen, A. J., and Jeger, M. J. (2008). Strategies of soilborne plant pathogenic fungi in relation to disease suppression. Fungal Ecol. 1, 108–114. doi: 10.1016/j.funeco.2008.10.006
Trivedi, P., Anderson, I. C., and Singh, B. K. (2013). Microbial modulators of soil carbon storage: integrating genomic and metabolic knowledge for global prediction. Trends Microbiol. 21, 641–651. doi: 10.1016/j.tim.2013.09.005
Verastegui, Y., Cheng, J., Engel, K., Kolczynski, D., Mortimer, S., Lavigne, J., et al. (2014). Multisubstrate isotope labeling and metagenomic analysis of active soil bacterial communities. mBio 5, e1157–e1114. doi: 10.1128/mBio.01157-14
Wang, X., Sharp, C. E., Jones, G. M., Grasby, S. E., Brady, A. L., and Dunfield, P. F. (2015). Stable-isotope probing identifies uncultured planctomycetes as primary degraders of a complex heteropolysaccharide in soil. Appl. Environ. Microbiol. 81, 4607–4615. doi: 10.1128/AEM.00055-15
Zhou, J., Xia, B., Treves, D. S., Wu, L. Y., Marsh, T. L., O’Neill, R. V., et al. (2002). Spatial and resource factors influencing high microbial diversity in soil. Appl. Environ. Microbiol 68, 326–334. doi: 10.1128/AEM.68.1.326-334.2002
Keywords: plant litter decomposition, rRNA-SIP, stable isotope probing, amplicon sequencing, energy channels
Citation: Kramer S, Dibbern D, Moll J, Huenninghaus M, Koller R, Krueger D, Marhan S, Urich T, Wubet T, Bonkowski M, Buscot F, Lueders T and Kandeler E (2016) Resource Partitioning between Bacteria, Fungi, and Protists in the Detritusphere of an Agricultural Soil. Front. Microbiol. 7:1524. doi: 10.3389/fmicb.2016.01524
Received: 29 June 2016; Accepted: 12 September 2016;
Published: 26 September 2016.
Edited by:
Paul Bodelier, Nederlands Instituut voor Ecologie, NetherlandsReviewed by:
Claudia Knief, University of Bonn, GermanyOsnat Gillor, Ben-Gurion University of the Negev, Israel
Copyright © 2016 Kramer, Dibbern, Moll, Huenninghaus, Koller, Krueger, Marhan, Urich, Wubet, Bonkowski, Buscot, Lueders and Kandeler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tillmann Lueders, dGlsbG1hbm4ubHVlZGVyc0BoZWxtaG9sdHotbXVlbmNoZW4uZGU=
†Present address: Susanne Kramer, Leibniz Institute of Vegetable and Ornamental Crops, Großbeeren, Germany Robert Koller, Institute of Bio- and Geosciences: Plant Sciences, Forschungszentrum Jülich, Jülich, Germany
‡These authors have contributed equally to this work.