 Nan Zhang1†
Nan Zhang1† Dongqing Yang1†Joshua R. A. Kendall1,2Rainer Borriss3
Dongqing Yang1†Joshua R. A. Kendall1,2Rainer Borriss3 Irina S. Druzhinina4Christian P. Kubicek4Qirong Shen1Ruifu Zhang1,5*
Irina S. Druzhinina4Christian P. Kubicek4Qirong Shen1Ruifu Zhang1,5*- 1Jiangsu Key Lab and Engineering Center for Solid Organic Waste Utilization, National Engineering Research Center for Organic-based Fertilizers, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, Nanjing Agricultural University, Nanjing, China
- 2Department of Science and Technology, Evangel University, Springfield, IL, USA
- 3Fachgebiet Phytomedizin, Institut für Agrar- und Gartenbauwissenschaften, Humboldt- Universität zu Berlin, Germany
- 4Research Area Biotechnology and Microbiology, Institute of Chemical Engineering, Vienna University of Technology, Vienna, Austria
- 5Key Laboratory of Microbial Resources Collection and Preservation, Ministry of Agriculture, Institute of Agricultural Resources and Regional Planning, Chinese Academy of Agricultural Sciences, Beijing, China
Bacillus subtilis and its sister species B. amyloliquefaciens comprise an evolutionary compact but physiologically versatile group of bacteria that includes strains isolated from diverse habitats. Many of these strains are used as plant growth-promoting rhizobacteria (PGPR) in agriculture and a plant-specialized subspecies of B. amyloliquefaciens—B. amyloliquefaciens subsp. plantarum, has recently been recognized, here we used 31 whole genomes [including two newly sequenced PGPR strains: B. amyloliquefaciens NJN-6 isolated from Musa sp. (banana) and B. subtilis HJ5 from Gossypium sp. (cotton)] to perform comparative analysis and investigate the genomic characteristics and evolution traits of both species in different niches. Phylogenomic analysis indicated that strains isolated from plant-associated (PA) habitats could be distinguished from those from non-plant-associated (nPA) niches in both species. The core genomes of PA strains are more abundant in genes relevant to intermediary metabolism and secondary metabolites biosynthesis as compared with those of nPA strains, and they also possess additional specific genes involved in utilization of plant-derived substrates and synthesis of antibiotics. A further gene gain/loss analysis indicated that only a few of these specific genes (18/192 for B. amyloliquefaciens and 53/688 for B. subtilis) were acquired by PA strains at the initial divergence event, but most were obtained successively by different subgroups of PA stains during the evolutional process. This study demonstrated the genomic differences between PA and nPA B. amyloliquefaciens and B. subtilis from different niches and the involved evolutional traits, and has implications for screening of PGPR strains in agricultural production.
Introduction
As a group of Gram-positive, aerobic, and endospore-forming Firmicutes bacteria, commonly known as the “Bacillus subtilis group” or B. subtilis sensu lato is of importance in both basic and applied microbiology (Fritze, 2004). Members of this group, B. subtilis sensu stricto and a recently distinguished sister species B. amyloliquefaciens, have been both widely used as producers of commercial chemicals in industry (Harwood, 1992; Geng et al., 2011), and beneficial agents for plant growth promotion and suppression of soil-borne diseases in agriculture (Chen et al., 2007, 2009; Fan et al., 2012).
B. subtilis and B. amyloliquefaciens are frequently isolated from various niches, including soil, animal feces, human food, aquatic environments, and so on (Earl et al., 2008). It is known that bacterial adaptation to different environments during the evolution process would lead to differentiation as indicated by different genomic and physiological characteristics (Earl et al., 2008; de Wit et al., 2012). For instance, B. amyloliquefaciens FZB42T isolated from the rhizosphere of Beta vulgaris (sugar beet) is widely used as a commercial plant growth-promoting rhizobacteria (PGPR), based on its abilities for plant root-colonizing and antibiotics production (Borriss et al., 2011; Chowdhury et al., 2015); while B. amyloliquefaciens LL3, isolated from fermented vegetables (Korean bibimbap), has been applied as an industrial strain for poly-γ-glutamic acid production (Geng et al., 2011). Recently, whole-genome sequencing of numerous Bacillus strains has uncovered the molecular basis for their versatile performance under different environments (Kunst et al., 1997; Deng et al., 2011; Blom et al., 2012; He et al., 2012). Based on a comparative genomic analysis within Bacillus spp., Alcaraz et al. indicated that the pathogen B. cereus possesses enriched genes involved in defense mechanisms, while the soil dweller B. subtilis harbors over-represented genes relevant to carbohydrates degradation (Alcaraz et al., 2010). Recently, it was proposed that B. amyloliquefaciens comprises two subspecies: The plant-associated B. amyloliquefaciens subsp. plantarum, and the non-plant-associated B. amyloliquefaciens subsp. amyloliquefaciens, based on both phylogenic analysis and physiological characteristics such as abilities for root colonization and production of plant growth hormones/antibiotics (Borriss et al., 2011); however, the evolution events leading to such divergence has not yet been investigated in more detail. In addition, for B. subtilis it is unclear whether they can be distinguished by different characteristics typical for plant-associated and non-plant-associated life style (Yi et al., 2014).
Based on this background, for better understanding of the genomic differences between B. subtilis and B. amyloliquefaciens strains isolated from plant-associated niches (rhizosphere) and non-plant-associated niches, we try to answer three questions: (i) Are there any significant genomic differences between plant-associated (PA) B. amyloliquefaciens/B. subtilis strains and non-plant-associated (nPA) strains? (ii) If yes, what are the specific genes caused the differences and potentially involved in rhizosphere adaptation? (iii) And, what's the evolution process of these specific genes, i.e., did the specific genes were absent in the ancestor but were acquired by the PA strains during the history (simultaneously or continuously), or they were shared by all strains but were discarded by the nPA strains? In this study, 29 previously published B. amyloliquefaciens and B. subtilis genomes, and two newly sequenced rhizospheric strains: B. amyloliquefaciens NJN-6 isolated from Musa sp. (banana) and B. subtilis HJ5 from Gossypium sp. (cotton) were collected to perform the comparative genomic and dynamic gene gain/loss analysis. Our results revealed genomic differences between PA and nPA B. amyloliquefaciens/B. subtilis, and indicated that the PA strains possess additional genes involved in utilization of plant-derived substrates and synthesis of antibiotics, which have arisen via horizontal gene transfer (HGT) events during the evolutionary process.
Results
Genome Structure of Two Newly Isolated Rhizosphere Strains of Bacillus spp.
The whole genome sequencing of two newly isolated rhizospheric PGPR strains, B. amyloliquefaciens NJN-6 and B. subtilis HJ5, was performed as described in Materials and Methods. The genome sizes of NJN-6 and HJ5 were found to be similar (4.05 and 4.01 Mb, respectively; Table 1). The number of predicted protein-coding genes of NJN-6 and HJ5 were 3894 and 3917, respectively (Table 1). The annotated sequences of NJN-6 and HJ5 were submitted to the NCBI GenBank database with the accession number of CP007165 and CP007173, respectively.
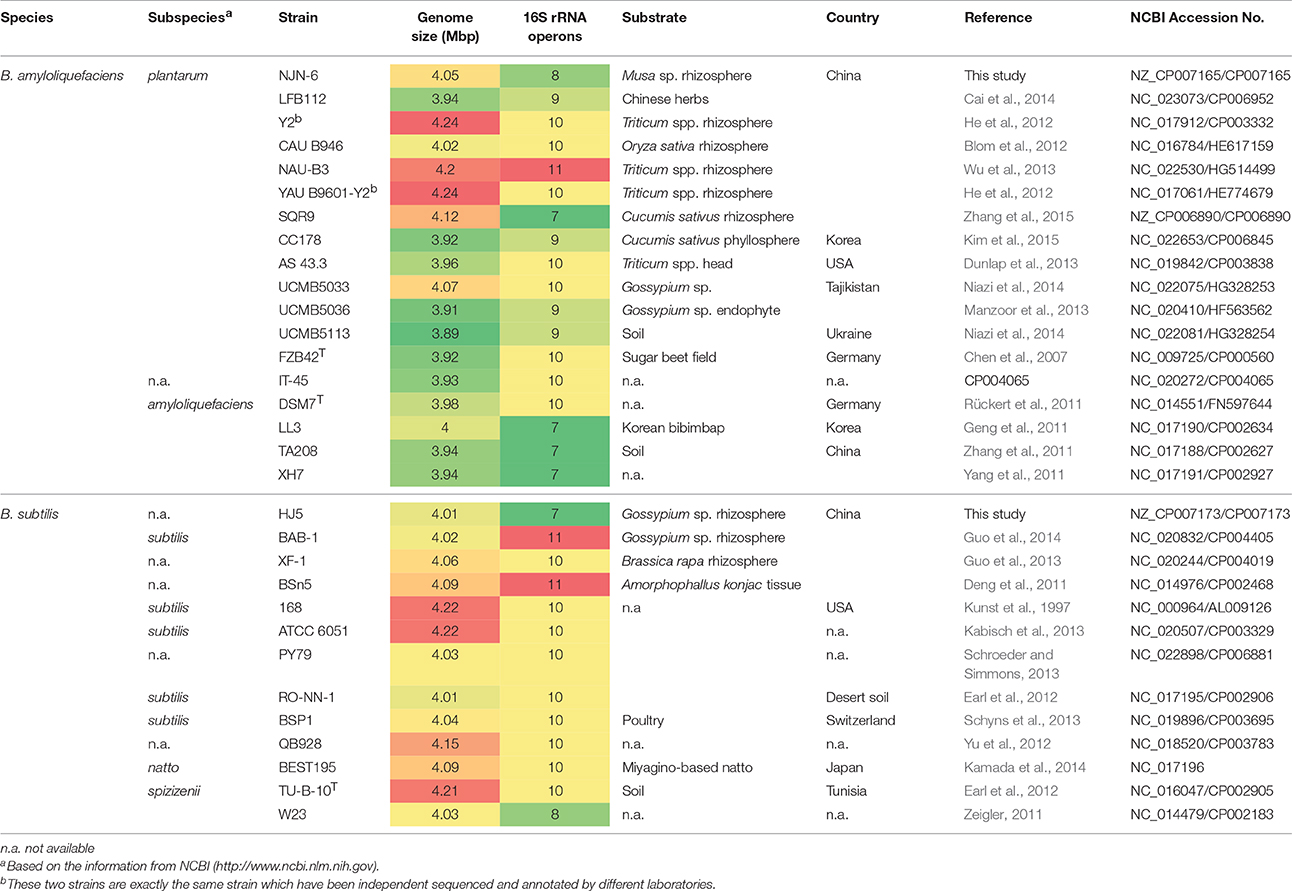
Table 1. Summary statistics and information on the genome and the isolation of the 31 Bacillus genomes in this study.
The Core- and Pan-Genomes of B. amyloliquefaciens and B. subtilis
The global gene repertoire of B. amyloliquefaciens and B. subtilis was determined by comparing the 31 genomes (Table 1). This resulted in a total of 123,555 genes, of which 121,056 (~97%) were clustered into 6596 orthologous groups (OGs) by OrthoMCL (Data Sheet S1). Of these OGs, 60 contained three or more gene copies, 279 contained two gene copies (doublets), and 6257 contained only a single gene copy (singletons).
The numbers corresponding to the core genome genes of B. amyloliquefaciens and B. subtilis respectively, decreased continually with the addition of new stains, while the pan-genome showed the opposite trend (Figure 1; Table S1). Both curves tended to be saturated when all 31 genomes were included. The B. amyloliquefaciens and B. subtilis core genomes consisted of 2409 OGs, representing around 60% of the average genome size (N = 31). The pan-genome for all lineages consisted of 9037 genes, corresponding to more than two-fold the average size of these 31 genomes (Figure 1).
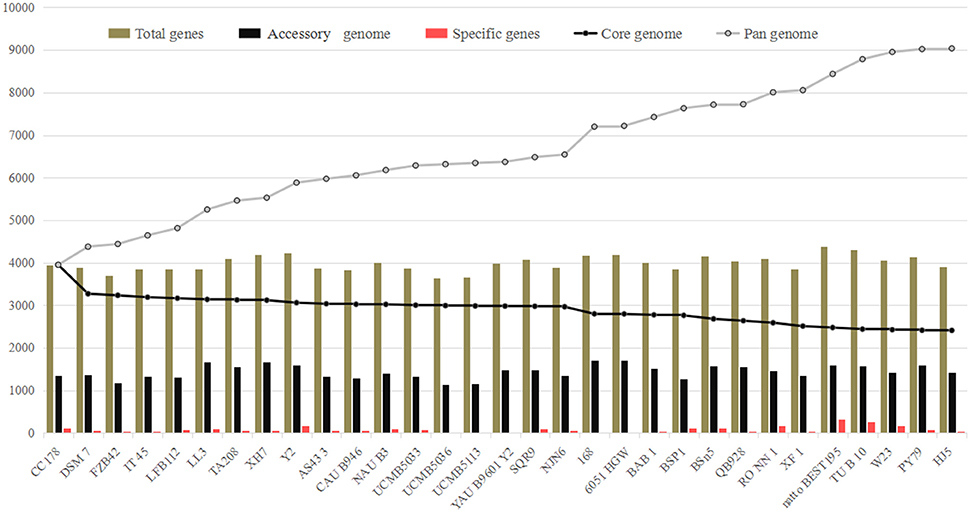
Figure 1. Pan- and core-genome analysis of 18 Bacillus amyloliquefaciens and 13 B. subtilis strains. The lines in gray and black represent the pan- and core-genomes, respectively. The pan-genome increased with addition of new strains to the study (9037 OGs), while the core genome decreased at a slow rate with added strains (2409).
Phylogenomic and Hierarchal Clustering Analysis Demonstrated the Differentiation between Plant-Associated (PA) and Non-Plant-Associated (nPA) B. amyloliquefaciens/B. subtilis
Phylogenetic relationship based on the core-genomes of B. amyloliquefaciens and B. subtilis was estimated from 1836 concatenated proteins, which resulted in a matrix of 500,457 amino acids residues. Figure 2A shows the consensus maximum likelihood phylogram rooted against B. pumilus SAFR-032 as an appropriate outgroup (Gioia et al., 2007). The resulting phylogenetic tree grouped the 31 genomes into two species clades with high bootstrap support values (Figure 2A) that supported the diversification of B. subtilis sensu lato into two monophyletic phylogenetic species, B. subtilis sensu stricto and B. amyloliquefaciens, respectively. Thirteen B. amyloliquefaciens strains that were isolated from plant-associated habitats (mainly rhizosphere) and IT-45, the one strain whose origin is unknown, but of which definition in NCBI organism overview has been reported as a PGPR strain (12th June 2013, http://www.ncbi.nlm.nih.gov/genome/848?genome_assembly_id=169345), formed a supported subclade corresponding to B. amyloliquefaciens subsp. plantarum (Figure 2A, plant-associated B. amyloliquefaciens, BA-PA group). Four strains that were isolated from none-plant-associated habitats formed another subclade corresponding to B. amyloliquefaciens subsp. amyloliquefaciens (non-plant-associated B. amyloliquefaciens, BA-nPA group). The phylogenetic structure of B. subtilis sensu stricto appeared to be more complex. Two strains, TU-B-10 and W23 formed a statistically supported basal subclade corresponding to the taxonomic definition of B. subtilis subsp. spizizenii (Zeigler, 2011), while the other eleven strains shared the same common ancestor and thus belonged to another subclade as B. subtilis subspecies subtilis (referred to B. subtilis in the rest of the article). Similar to another species, three Chinese rhizospheric strains being HJ5, BAB-1, and XF-1, formed an individual subclade (plant-associated B. subtilis, BS-PA). Interestingly this subclade did not include BSn5 strain isolated from calli tissue (Amorphophallus konjac) (Figure 2A). The remaining eight B. subtilis strains formed another clade defined as non-plant-associated B. subtilis group (BS-nPA). This phylogenetic structure indicates that within B. subtilis, rhizospheric strains share the same evolutionary pathway that was also observed for B. amyloliquefaciens.
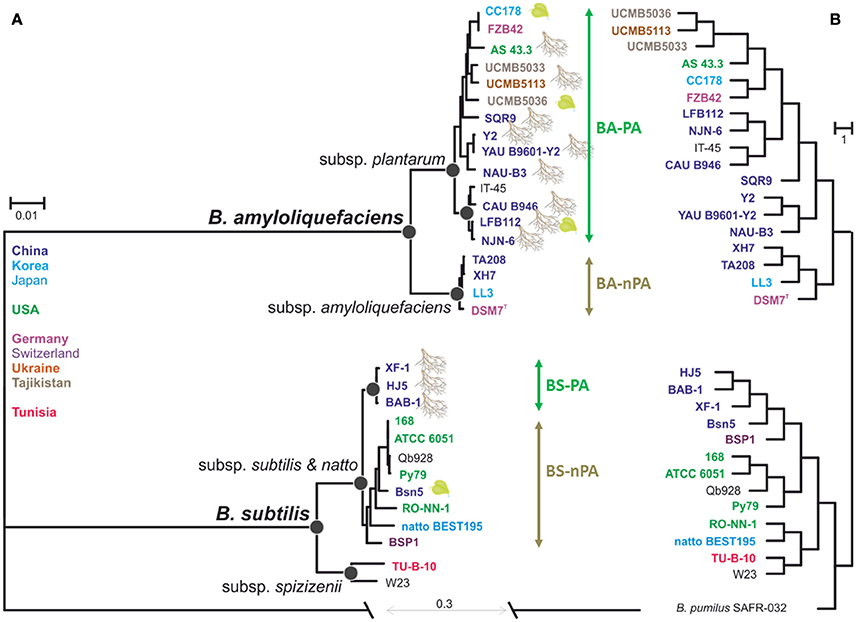
Figure 2. Evolutionary relationship between the 31 Bacillus strains and hierarchal clustering. (A) The maximum-likehood phylogeny derived from the alignment of 1835 concatenation core genes. Leaf clipart indicates association with plants. Colors indicate biogeographic origin of the strains. All nodes shown on the phylogram are supported by >85% bootstrap values and >0.94 posterior probabilities obtained after the 0.2 M mcmc generations in Bayesian analysis. Black circles indicate support for the most important nodes. (B) Hierarchal clustering among genomes using presence/absence of orthologous groups Almost all of nodes on the phylogram are supported by 97% >approximately unbiased P-values.
In order to find out whether adaptation to the rhizosphere habitat went through convergent evolution in both species, we also looked at the genomic features of respective strains and compared them with saprotrophic members of each species (divergence of PA and nPA strains). For this reason we performed the hierarchal clustering analysis based on gene presence/absence among each strain and found it recovered most of the major groups found in the species tree (Figure 2B), which also clearly distinguished the PA and nPA groups within B. amyloliquefaciens and B. subtilis, respectively, only with some discrepancies.
Core Genome of Plant-Associated B. amyloliquefaciens and B. subtilis Strains are More Abundant in Genes Involved in Metabolism of Plant-Derived Substrates and Synthesis of Antibiotics
The phylogenomic analysis (vide supra) revealed that in both Bacillus species, the plant-associated isolates formed their own clades. Subsequently, OrthoMCL was used to delimit the core genomes of PA and nPA strains respectively based on phylogenetic groups (Table S2). The core genome of the PA strains contained 2756 orthologous groups, while nPA strains contained 2714. Of these, 342 orthologous groups in the core genome of PA strains were identified not belong to core genome of the whole 31 strains, while there are 234 orthologous groups different for nPA (Tables S3, S4), which were defined as PA and nPA-specific core genome, respectively. It was found that 228 genes from PA-specific core genome and 183 from the nPA could be classified into multiple COGs. Among these, categories G (carbohydrate transport and metabolism) and E (amino acid transport and metabolism) were found to be more abundant in the PA-specific core genome as compared to that of nPA strains (24 vs. 15 for category G and 29 vs. 15 for E, respectively, Figure 3). In detail, the bglC/eglS (GP103033, encoding endo-1, 4-β-glucanase) and bglS (GP102126, endo-β-1,3-1,4 glucanase) involved in cellulose degradation, xylA (GP102236, xylose isomerase) and xynB (GP102238, β-xylosidase) involved in xylan degradation, amyE (GP103025, α-amylase) for saccharifying starch, and pelB (GP102863, pectate lyase) for pectate hydrolysis, were all identified in category G of the PA-specific core genome (Tables S3, S4). Category E mainly included genes encoding ABC-type amino acid transporters, such as glnQHMP (GP102791-94) for glutamine transport, and opuCB (GP102371) for glycine transport. Another two over-represented categories in the PA-specific core genome were Q (secondary metabolites biosynthesis, transport, and catabolism, 7 vs. 2 genes for PA and nPA, respectively) and I (lipid transport and metabolism, 14 vs. 7 genes, respectively) (Figure 3), which include genes involved in synthesis of polyketide antibiotics bacillaene (bae cluster, GP100000, 100027, and 102850-57) and difficidin (dfnL, GP100038) (Chen et al., 2009; Xu et al., 2014).
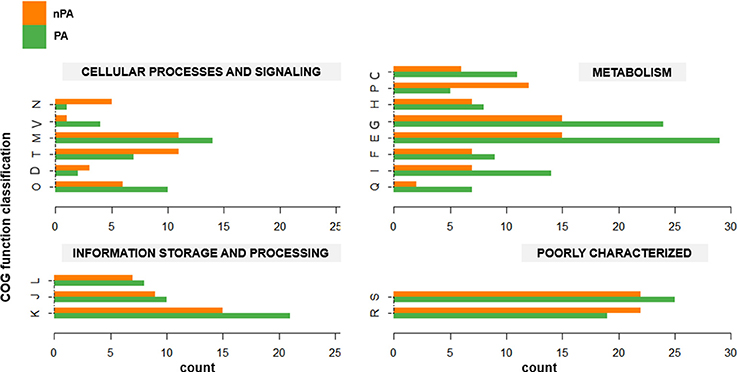
Figure 3. Gene counts for each COG category in the core genome for non-plant-associated (nPA) vs. plant-associated (PA) groups. Each pair of columns represents the COG category of the specific genes. Cellular process and signaling: D, cell cycle control, cell division, chromosome partitioning; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, chaperones; T, signal transduction mechanisms; V, defense mechanisms. Metabolism: C, energy production and conversion; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport and catabolism. Information storage and processing: J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination and repair. Poorly characterized: R, general function prediction only; S, function unknown.
Interestingly, it was found that some sporulation genes were more frequently found among PA strains [e.g., gerBA (GP102918), spoIIE (GP102731), and cotA (GP102745), etc.; Table S3] while others more frequently among nPA strains [e.g., gerPA (GP102165), spoIVB (GP102275), and cotJA (GP102930), etc., Table S4]. This observation might be attributed to the complex and redundancy of sporulation genes (Tan and Ramamurthi, 2014), but the detailed comparison of sporulation abilities between PA and nPA strains, as well as the ecological significance in the rhizosphere, still need further exploration.
Since the limitation of core genome comparison (e.g., genes identified in PA-specific core-genome is probably because they are missing in one or a few nPA strains, but actually they are still encoded by many nPA strains), we further identified the genes that are present in all fourteen BA-PA strains but absent in all four BA-nPA strains (BA-PA specific genes), and those that occur in all three BS-PA strains but are absent in all ten BS-nPA strains (BS-PA specific genes). The 56 genes that only existed in the fourteen BA-PA strains included genes belonging to the dfn (dfnBCJKMXY, GP103464, and GP103547-52) and mln clusters (mlnH and mlnI, GP103539 and GP103540, respectively), which participate in nonribosomal synthesis of difficidin and macrolactin, respectively (Chen et al., 2009). Importantly, amyE (GP103025, α-amylase) and bglC/eglS (GP103033, endo-β-1, 4-glucanase) were also occurred in the PA-specific core genome summarized above (Table S5). This finding is in agreement with a previous study (Borriss et al., 2011) and implicated the importance of utilization of plant-derived polysaccharides and antibiotics production in rhizosphere competition. Most of the BS-PA specific genes were annotated as hypothetical proteins and their relationship to rhizosphere adaptation can therefore not be predicted; only GP105349 shows similarity to putative methyl-accepting transducer for sensing specific ligands possibly governing chemotaxis behavior of the bacterial cell in the rhizosphere (Table S6).
PA-Preferential Genes Were Acquired Successively by Different Subgroups of PA Strains through Horizontal Gene Transfer (HGT)
Based on the comparative genomic analysis (vide supra), in order to further explore the dynamic events contributing to the differentiation between PA and nPA strains, AnGST was used to reconcile observed differences between gene and species phylogenetic trees (the phylogeny tree is same with that in Figure 2A) to identify the genomic events concomitant with diversification of both PA Bacillus clades (subsp.) during the evolutionary process. We detected 453 and 1058 genes that were gained on the branch leading to the last common ancestors of PA/nPA B. amyloliquefaciens and B. subtilis, respectively (Figure 4). Of these genes, 192 in the BA-PA and 688 in the BS-PA cluster were predicted to have been gained through HGT from other strains included in this analysis. The remaining genes were attributed to gene duplication and speciation (David and Alm, 2011) (Table S7).
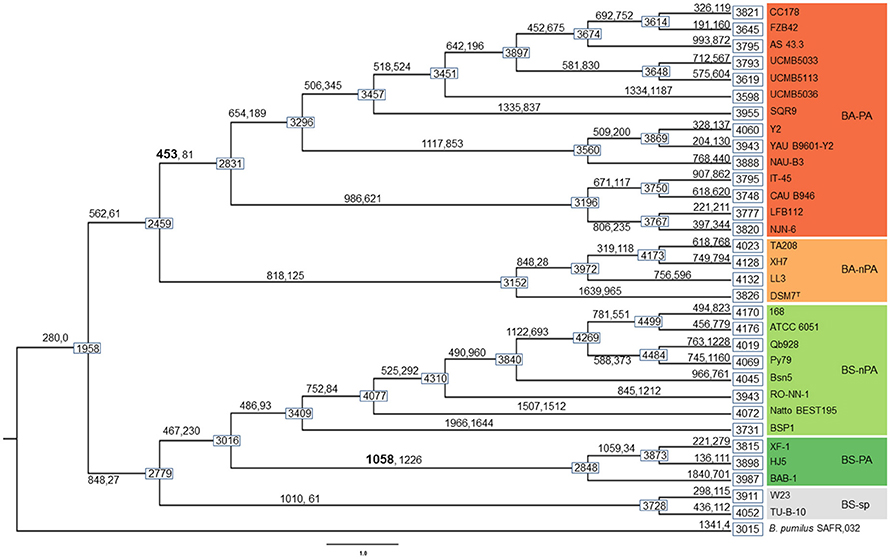
Figure 4. Results of gene gain/loss analysis. Boxes on nodes and tips of the phylogeny show genome size (genome size calculations exclude one of the orthologous groups that was excluded from the analysis, see Results). Numbers on branches indicate the number of genes gained (left, through HGT, gene duplication, or gene birth) and lost (right) at the divergence event.
The genes were taken from the divergence gene pool of PA/nPA in both species and then assigned into COGs (Figure 4; Tables S8–S11). BS-PA acquired most genes (total 688, and 537 with COG assignment), followed by BA-nPA (233/355), BS-nPA (212/286), and BA-PA (129/192). Clustering of the PA and nPA species suggested that adaptation of both B. subtilis and B. amyloliquefaciens revealed the same tendency (Figure 5). A Venn diagram based on the four HGT pools was also generated but no obvious trends for gene distribution could be observed (Figure S1). Genes acquired at the divergence event by the four groups (BA-PA vs. BA-nPA, and BS-PA vs. BS-nPA) mainly belonged to categories E (amino acid transport and metabolism, 9.0–11.8%), G (carbohydrate transport and metabolism, 4.7–9.9%), and K (transcription, 8.4–15.6%), implying that new features in intermediary metabolism and transcriptional regulation were important events during evolution under different environmental conditions. Further, the BA-PA acquired relatively more genes in categories C (energy production and conversion, 4.7 vs. 3.9%), G (9.3 vs. 8.6%), and K (15.5 vs. 9.4%), than did in BA-nPA. Genes in categories E (11.7 vs. 9.0%), G (9.9 vs. 4.7%), and N (cell motility, genes involved in flagellum synthesis and chemotaxis, 2.0 vs. 1.4%) were over-represented in BS-PA compared with BS-nPA (Tables S8–S11). Additionally, the antibiotic resistance genes (ARGs) in the four HGT pools acquired by BA-PA, BA-nPA, BS-PA, and BA-nPA were identified using the Resfams HMM profiles (E-value: 1e-5), and 87, 131, 312, and 100 ARGs were detected in the four pools, respectively (Tables S12–S15). Numerous ARGs belonging to various types could be detected, including those encoding major facilitator superfamily (MFS)-type transporter, acetyltransferase, glyoxalase, β-lactamase, ABC transporter/efflux, and so on (Tables S12–S15). Interestingly, the proportion of ARGs to total HGT genes in BA-PA (Table S12; 87/192, 45.3%) and BS-PA (Table S14, 312/688, 45.3%) was higher than that in BA-nPA (Table S13; 131/355, 36.9%) and BS-nPA (Table S15; 100/286, 35.0%), suggesting that the plant-associated strains preferred to acquiring ARGs as compared with non-plant-associated strains at the divergence event.
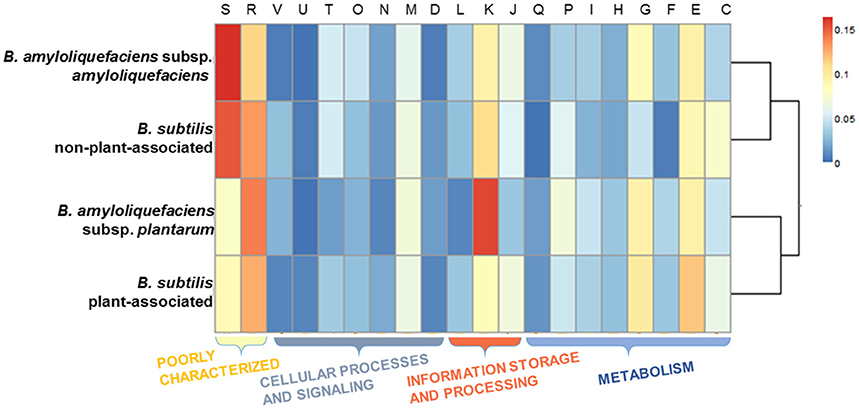
Figure 5. Heat map representing the COG categories across the four HGT gene pools acquired by BA-nPA (non-plant-associated B. amyloliquefaciens), BS-nPA (non-plant-associated B. subtilis), BA-PA (plant-associated B. amyloliquefaciens), and BS-PA (plant-associated B. subtilis). The dendrogram was constructed using the pairwise Pearson's r correlation calculated for the protein cluster membership distributions. The cell color in the heatmap represents the percentage of gene numbers of each COG category, as revealed by the legend.
Only a few genes were shared by the PA-specific core genome and the group of genes acquired by HGT at the divergence event. Of the 192 genes acquired by the BA-PA, only 18 genes (9.4%) were also present in the PA-specific core genome, including mtlA (GP102111, mannitol transporter) and glnQ (GP102791, glutamine transporter) that might be relevant to rhizosphere adaptation (Table S8). For BS-PA, the overlap of genes between HGT and specific core genome was found to be 53 of 688 (7.7%), which included bglC/eglS (GP103033, cellulase C) and glnM (GP102793, glutamine ABC transporter) (Table S10).
We then pay attention to the last question raised in the introduction: What's the evolutionary process of the 96 PA-specific genes (56 BA-PA specific genes and 40 BS-PA specific genes, Tables S5, S6), did they were absent in the ancestor but were acquired by the PA strains during the history or were shared by all strains but were subsequently discarded by the nPA strains? Analysis based on the AnGST model indicated that all the PA-specific genes were absent in both ancestors of B. amyloliquefaciens or B. subtilis, and only nine genes (including bglC/eglS) were predicted to be obtained by all PA strains at the initial divergence event via HGT, being BA-PA or BS-PA branches (Table S16). Most of the remaining PA-specific genes were primarily acquired by subgroups of the PA strains (probably from outgroup organisms) at the divergence event (BA-PA or BS-PA), and were then introduced to other strains through HGT during the following evolutionary process (Data Sheet S2). In detail, taking gene mlnH (polyketide macrolactin synthase, GP103539) for example, it firstly occurred in the ancestor of group “Y2-YAB9601-NAUB3-CC178-FZB42-AS43-UCMB5033-UCMB5113-UCMB5036-SQR9,” thereafter it was transferred from the ancestor of group “CC178-FZB42-AS43-UCMB5033-UCMB5113-UCMB5036” to the ancestor of group “IT45-CAB946,” followed by another HGT from IT45 to the ancestor of group “LFB112-NJN6” (Data Sheet S2). Evolutionary history of other PA-specific genes revealed similar patterns with that of mlnH, suggesting that they were not obtained by a single divergence event between PA and nPA strains, but were obtained successively by different subgroups of the PA stains during the evolution process (Data Sheet S2).
Finally, a variety of acquired genes relevant to rhizosphere adaptation at the divergence event, including those involved in polysaccharide utilization (Beauregard et al., 2013), antibiotics production (Ongena and Jacques, 2008), and stress resistance (Raaijmakers et al., 2008), were also identified. The HGT gene pools of BA-PA branch included NJN-6|AW02_009760 (GP100108) encoding polysaccharide deacetylase, IT-45|451347442 (GP102036) encoding arabinogalactan endo-1, 4-beta-galactosidase, and two genes involved in stress tolerance [AS 43.3|429503728 (GP102479) encoding an antibiotic transport system ATP-binding protein and yusP (UCMB5036|452856902, GP100085) encoding a putative multidrug-efflux transporter] (Table S8). In the BS-PA branch, genes involved in plant polysaccharides degradation [BAB-1|472330442 (GP102774) and BAB-1|472331232 (GP102331) both encoding endo-β-1, 4-xylanase, and bglC/eglS (XF-1|449094502, GP103033)], antibiotic biosynthesis [srfAD (XF-1|449093047, GP100275) and BAB-1|472330387 (GP100001) participate in the synthesis of lipopeptide surfactin, and plipastatin, respectively], and antibiotic resistance [BAB-1|472328995 (GP103444) encoding glyoxalase/bleomycin resistance protein/dioxygenase, and HJ5|AW03_002620 (GP100347) encoding a drug-export protein], were all predicted to have been acquired by HGT (Table S10).
Discussion
Factors Driving the Divergent Evolution of PA and nPA Strains
Genomic and evolutionary characteristics of B. amyloliquefaciens and B. subtilis, especially the root-colonizing strains, are gaining great interest right at present because of their potential in agriculture. In this study, we performed a comparative genomics analysis based on 29 previously published B. amyloliquefaciens and B. subtilis genomes, and two newly sequenced rhizospheric strain B. amyloliquefaciens NJN-6 and B. subtilis HJ5. The phylogeny based on core genomes indicated that strains isolated from the rhizosphere (PA strains) are distinguished from the nPA strains, the hierarchal clustering of OGs based on the presence/absence matrix in both B. amyloliquefaciens and B. subtilis revealed the similar pattern (Figure 2). These findings generally suggests that important changes in the genome of these Bacillus strains have occurred during adaptation to different habitats; more precisely a co-evolution with plants could be a driving factor (Hartmann et al., 2008). This implies that a specific niche, such as plant-associated habitats, influences HGT [plant-associated habitats, e.g., rhizosphere, are often conducive to HGT processes (van Elsas et al., 2003)] and highlights the consistency between the molecular clock mutation (nucleotide sequence, species phylogenetic tree) and gene gain/loss (hierarchal clustering) (Hao and Golding, 2006), which is in accordance with data demonstrating HGT among numerous Streptococcus species within a shared bovine environment (Richards et al., 2014).
In both trees the differentiation within B. amyloliquefaciens is quite clear and consistent with their habitats (Figure 2A), while in B. subtilis the clustering in the two trees is different and not fully coincided with their origins (Figure 2B). Since B. subtilis 168, PY79, ATCC 6051, and QB928 were all derived from root-colonizing strain NCIB3610 (Zeigler et al., 2008; Yu et al., 2012), it seemed that they should be clustered with the PA strains in the phylogenetic tree, as well as BSn5 isolated from calli tissue. The reason for this contradiction is still unclear: For the four domesticated strains, the subsequent mutation in laboratory might lead to differentiations, but actually the extent of genetic changes caused by domestication appears to be limited; for BSn5, maybe bacterium isolated from a given environment does not guarantee it has adapted to that particular habitat. For the clustering relationship of B. subtilis observed in the hierarchal clustering based on the matrix agrees, the three PA strain and four domesticated strains as well as BSn5 were clustered in one branch, but BSP1 that isolated from poultry was also included (Figure 2B). In general, the two trees for Bacillus still suggested that the three most representative plant-relevant strains (HJ5, BAB-1, and XF-1) could be distinguished from other B. subtilis strains, but further investigation including newly sequenced wild strains and eliminating the lab-domesticated strains is importantly needed for exploring the genomic characteristics between PA and nPA isolates in a more comprehensive perspective.
PA Strains Possess More Abundant Genes Involved in Intermediary Metabolism and Antibiotics Production as Compared to PA Strains
As the existence of root exudates, the rhizosphere represents a highly dynamic front for interactions and competitions between various organisms; microbes within this niche have to contend with each other for resources, through nutrient/space competition and direct suppression by “weapons” such as antibiotics and extracellular hydrolase (Bais et al., 2006). Comparison of the core genome between PA and nPA strains based on COGs analysis revealed that the genes involved in intermediary metabolism (categories E and G) and biosynthesis of secondary metabolites (Q) were more abundant in PA strains (Figure 3). Alcaraz et al. reported that B. subtilis inhabiting soil harbors a higher proportion of genes related to carbohydrate transport and metabolism (G) when compared to other Bacillus spp. (Alcaraz et al., 2010). Barret et al. also reviewed the bacterial genes expressed during PGPR-plant interactions in the rhizosphere that reported by previous studies, and indicated that the genes relevant to rhizosphere competence were mainly involved in central metabolism, detoxification and stress, and secretion (Barret et al., 2011). Therefore, the presence of the genes mentioned above could have the advantageous results of increasing the capacity to utilize various plant-derived substrates (e.g., polysaccharides as main components of plant cell wall and amino acids in root exudates) and synthesizing antibiotics, which would contribute to competitive edge and rhizosphere adaptation (Borriss et al., 2011).
Noticing that some genes identified in PA-specific core-genome are only absent in one or a few nPA strains but still exist in most of the nPA strains, a further PA-specific gene analysis was performed for exploring the categorical genomic differences between PA and nPA strains. Several genes involved in plant-derived polysaccharides degradation (bglC/eglS and amyE) and antibiotics production (dfn and mln clusters) were identified specifically from BA-PA strains (Table S5), suggesting that these two groups of genes are probably the most important characteristic that contribute to rhizosphere adaption of PA strains and their differences from nPA stains (Borriss et al., 2011; Wu et al., 2015). However, the BS-PA specific genes revealed limited relationship with rhizosphere adaptation, maybe further analysis based on more wild type Bacillus genomes could provide significant information.
Successively HGT of Rhizospheric-Specific Genes Acquired by Different Subgroups of PA Strains
HGT is recognized as an important factor that drives evolution and differentiation of different organisms. Itakura et al., demonstrated that the Bradyrhizobium japonicum strains acquired additional DNA fragments (e.g., genomic islands) through HGT performed superior symbiotic nitrogen-fixation capability than other relative strains (Itakura et al., 2009). Recently, Richards et al. investigated the dynamics of genome evolution of Streptococcus spp. by using AnGST, and found that eight GO groups were acquired during the early evolution/divergence events, thus enabling a better adaptation to new habitats (Richards et al., 2014). In this study, we performed an AnGST analysis to explore the gene gain/lost events during the evolutionary process of the two subspecies of Bacillus spp. (Figure 4). It was found that the major genes acquired by both BA-PA and BS-PA are involved in metabolism (categories E and G), transcription/signal transduction (K or KT), and synthesis of secondary metabolites (Q) (Tables S8, S10), which are all potentially important for rhizosphere survival and competition (Alcaraz et al., 2010). Especially, the higher proportion of ARGs in the HGT pools of PA strains than nPA strains at the divergence event (Tables S12–S15) suggested their roles in adaptation to various antibiotic stress in the rhizosphere.
Interestingly, only a few of the PA-specific genes could be recovered from the HGT pools of BA-PA and BS-PA branches (Tables S8, S10, S12), while most of them were acquired by subgroups of PA strains at the initial divergence event, and then transferred to other groups of PA strains successively (Data Sheet S2). This finding suggested that the rhizosphere-specific genes were not obtained by a single event, but rather were acquired by members of subpopulation that might have first migrated to the rhizosphere. Transfer of these genes to subsequently arising populations, and maintenance of acquired genes resulted in the contemporary genomic differences currently observed between PA and nPA. This phenomenon provides new information for better understanding the evolutional process of Bacillus spp. isolated from different niches.
Transcriptional Profiling in Response to Maize Root Exudates
Recently, the transcriptome analysis of B. amyloliquefaciens SQR9 was reported (a member of the BA-PA that also isolated by our laboratory) incubated with maize root exudates [which partially mimicked the presence of a rhizosphere (Zhang et al., 2015)]. Thirteen genes that belong to PA-specific core genome and were gained through HGT at the divergence event between PA and nPA (BA-PA branch), were observed to be induced by root exudates (Tables S3, S8, S10), including mtlA (GP102111, mannitol transporter) and glnQ/M (GP102791/GP102793, glutamine transporters) involved in transport of important compounds in root exudates (Badri and Vivanco, 2009). Another significant gene was ycbA that encodes a transcriptional regulator involved in biofilm formation (GP102097) (Stanley et al., 2003) (Tables S3, S8, S10).
Additionally, partial genes in PA-specific core genome or in the BA-PA/BS-PA HGT group also revealed positive response to root exudates, including those involved in plant-derived carbohydrates transport and metabolism, cell motility and chemotaxis, biofilm formation, and transcriptional regulation (Tables S3, S8, S10). Unfortunately, most of the significant genes belong to the BA-PA specific genes were identified as hypothetical proteins (Tables S3, S8, S10). In general, the data from the transcriptomic analysis implicated the genes in the PA-specific core genome and HGT-derived pool could be involved in rhizospheric interactions, which suggests that genes were specifically acquired by the plant-colonizing Bacillus spp. strains.
Conclusion
Comparative genomic analysis of B. amyloliquefaciens and B. subtilis indicated that the plant-associated (PA) and non-plant-associated (nPA) strains were clearly distinguished. Genes relevant to plant-derived polysaccharides utilization and antibiotics synthesis are more abundant in the core genome of PA strains. Only a few of these rhizospheric-specific genes were acquired by PA strains at the initial divergence event, but most were obtained successively by different subgroups of PA stains. Our study offers new information regarding genomic differences between PA and nPA B. subtilis and B. amyloliquefaciens from different habitats and the relevant evolutionary traits, and has implications for screening of PGPR strains for application in agriculture production.
Materials and Methods
Strains Used in this Study
Two newly isolated rhizospheric strains, B. amyloliquefaciensNJN-6 (from the banana rhizosphere) and B. subtilis HJ5 (from the cotton rhizosphere), were used in this study. They are both PGPR strains with outstanding plant growth promotion and biocontrol performance (such as suppression of Fusarium wilt of banana and Verticillium wilt of cotton, respectively) (Li et al., 2013; Yuan et al., 2013). NJN-6 produces several antagonistic compounds, including homolog of the cyclic lipopeptide iturin A (bacillomycin D, macrolactin A/E) and numerous volatile compounds with antifungal activity (Yuan et al., 2011, 2012a,b, 2014). HJ5 colonizes cotton roots and forms a robust biofilm (Li et al., 2013). These two strains were then selected for whole genome sequencing to obtain the relevant molecular genetic information.
Genome Sequencing and Annotation
The genome sequences were obtained using shotgun sequencing with a combination of Sanger sequencing and 454 FLX system with 28 × coverage and 30.5 × coverages for B. amyloliquefaciens NJN-6 and B. subtilis HJ5, respectively. Assemblies were performed using Newbler, version 2.3 (Margulies et al., 2005) resulting in 53 and 50 contigs for NJN-6 and HJ5, respectively. Gaps in the assembly and regions where the sequence was uncertain were completed by Sanger sequencing. Predictions of protein-coding genes were implemented in Prodigal (Hyatt et al., 2010). Functional annotation was carried out using BLASTP, with B. amyloliquefaciens subsp. plantarum FZB42T and B. subtilis 168 as references for NJN-6 and HJ5, respectively, and against GenBank's non-redundant protein databases (nr) with an E value of <1e-5. Protein clusters and domains were assigned by an RPS-BLAST search against the COG (Tatusov et al., 2003) and PFAM databases (Punta et al., 2012), with an E value of <1e-5. Transfer RNA genes (tRNA) and rRNA were identified separately using tRNAscan-SE (Lowe and Eddy, 1997) and RNAmmer 1.2 server (Lagesen et al., 2007).
Reference Genomes and Identification of Gene Orthologous Groups
Twenty nine complete genome sequences, and associated protein functions, for B. amyloliquefaciens and B. subtilis were downloaded from NCBI (12th June 2013, ftp://ftp.ncbi.nih.gov/genomes/Bacteria/).
Orthologous groups were delimited using OrthoMCL version 2.9 (Li et al., 2003), in which all the proteins sequences were compared using a BLASTP all-against-all search with an E value cutoff of <1e-05 and percent match cutoff of > 70%. Then, the Markov Cluster (MCL) algorithm was used to delineate proteins into orthologous groups (OGs) using an inflation value of 1.8 (Enright et al., 2002), which is robust to fluctuations of inflation (Brohee and van Helden, 2006). The putative pan-genome, core genome and lineage-specific gene sets were extracted from the OrthoMCL output.
Phylogenomic Analyses
A phylogenetic tree for the set of 31 genomes from the two species was inferred using a core genome alignment concatenation approach. 1836 OGs, which were present in all genomes as single-copy genes, were considered as phylogenetic markers. The multiple sequence alignments were constructed in the AUQA software (Muller et al., 2010), which relies on MUSCLE (Edgar, 2004) and MAFFT (Katoh and Standley, 2013) as aligners. The resulting 1836 amino acid alignments were combined, which resulted in a 551,710 amino acid concatenated dataset. The Gblocks server with defaults settings (Castresana, 2000) was used to remove non-alignable regions resulting in 500,457 amino acids final alignment. A maximum likelihood tree was built with IQ-TREE (version 1.1.0) (Minh et al., 2013) using the LG+I+G4+F substitution model (Le and Gascuel, 2008), and a consensus tree was constructed from 10,000 bootstrap trees. A Bayesian phylogram was obtained in MrBayes 3.2.1. (http://mrbayes.sourceforge.net/) run on CIPRES server (https://www.phylo.org/) with 200,000 mcmc generations (maximum achievable for such a large dataset); every 100 trees were sampled and a consensus tree was obtained after the first 100 generations were removed with burnin command. Gene phylogenetic trees were constructed using OGs containing three or more genes in IQ-TREE (Minh et al., 2013) with the GTR + I + G substitution model and 1000 bootstrap replicates.
Hierarchal Clustering
To evaluate the uncertainty of speciation from the OGs that were not shared among all the genomes analyzed in this study, we first constructed a 0/1 matrix with 1 indicating that the OG was present in a genome and 0 indicating the absence of that OG. OGs that were only shared by partial strains were probably obtained through HGT (Zhu et al., 2014). The hierarchical clustering was used to analyze the 0/1 matrix as implemented in the R package pvclust (Suzuki and Shimodaira, 2006) with 500 bootstrap replicates.
Reconciliation between Species Tree and Gene Tree
A parsimony-based reconciliation approach, AnGST, was used to understand the history of the PA genomic evolution events (David and Alm, 2011). The reconciliation was obtained by inferring a minimum set of evolutionary events between the gene- and species-trees, including horizontal gene transfer (HGT), gene duplication (DUP), gene loss (LOS), speciation (SPC), and one gene birth or genesis event (GEN). All the OGs composed of more than two proteins were analyzed by AnGST using the species tree and default parameters. Inference errors due to phylogenetic uncertainty were minimized by incorporating 1000 bootstrap replicates per gene. For those OGs containing only two genes, one of the genes was observed once in two separate taxa or twice in one taxa. For these genes, we followed the methods of Richards (Richards et al., 2014) to overlay the position of the two genes onto the species tree, using the same set of evolutionary events and event penalties as described above.
The antibiotic resistance genes in the four HGT pools acquired by BS-PA, BS-nPA, BA-nPA, BA-PA were identified by searching against ResFams (v1.2) (Gibson et al., 2015) using HMMER (v3.1b2; E-value: 1e-5) (Eddy, 2011). Venn diagram based on the four gene pools was generated with R package VennDiagram (Chen and Boutros, 2011).
Author Contributions
Conceived and designed experiments: ID, CK, QS, and RZ. Performed the experiments: NZ. Performed bioinformatic analysis: DY and JK. Wrote the paper: NZ, DY, RB, ID, and RZ. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was financially supported by National Key Basic Research Program of China (973 program, 2015CB150505), National Natural Science Foundation of China (31330069, 41271271, and 31301845), the Chinese Ministry of Science and Technology (2013AA102802), and the Fundamental Research Funds for the Central Universities (KYZ201307). RZ and QS were also supported by the 111 Project (B12009) and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions. ID. was supported by the Austrian Science Fund (FWF): project number P 25745. We also acknowledge the help of Miss Komal Chenthamara Kariyankode, Vienna University of Technology, for her support with phylogeny.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02039/full#supplementary-material
Table S1. Statistics for the core-genome and pan-genome of the 31 Bacillus strains.
Table S2. Groups divided into plant-associated (PA) and non-plant-associated (nPA) based on the species phylogenetic tree.
Table S3. Gene products and COG assignments of the PA-specific core genome. The genes that showed homology with SQR9's were appended with the different gene expression patterns regulated by root exudate at 24 and 48 h post-inoculation.
Table S4. Gene products and COG assignments of the nPA-specific core genome. The genes that showed homology with SQR9's were appended with the different gene expression patterns regulated by root exudate at 24 and 48 h post-inoculation.
Table S5. Genes that existed in all Bacillus amyloliquefaciens subsp. plantarum (BA-PA) but not in Bacillus amyloliquefaciens subsp. amyloliquefaciens (BA-nPA).
Table S6. Genes that existed in Bacillus subtilis plant-associated (BS-PA) but not in Bacillus subtilis non-plant-associated (BS-nPA).
Table S7. Statistics of evolutionary events derived from AnGST along each branch. hgt (total), total gene count of genes involved in horizontal gene transfer, speciation and gene duplication. spc, speciation; dup, gene duplication; los, gene loss; brn, gene birth; cur, gene counts in current strain.
Table S8. Gene products and COG assignments of predicted HGT in Bacillus amyloliquefaciens subsp. plantarum (BA-PA). The genes that showed homology with SQR9's were appended with the different gene expression patterns regulated by root exudate at 24 and 48 h post-inoculation.
Table S9. Gene product and COG assignments of predicted HGT in B. amyloliquefaciens subsp. amyloliquefaciens (BA-nPA). The genes that showed homology with SQR9's were appended with the different gene expression patterns regulated by root exudate at 24 and 48 h post-inoculation.
Table S10. Gene products and COG assignments of predicted HGT in B. subtilis plant-associated (BS-PA). The genes that showed homology with SQR9's were appended with the different gene expression patterns regulated by root exudate at 24 and 48 h post-inoculation.
Table S11. Gene products and COG assignments of predicted HGT in B. subtilis non-plant-associated (BS-nPA). The genes that showed homology with SQR9's were appended with the different gene expression patterns regulated by root exudate at 24 and 48 h post-inoculation.
Table S12. Antibiotic resistance genes (ARGs) of HGT on the plant-associated Bacillus amyloliquefaciens (BA-PA) predicated by ResFams.
Table S13. Antibiotic resistance genes (ARGs) of HGT on the non-plant-associated Bacillus amyloliquefaciens (BA-nPA) predicated by ResFams.
Table S14. Antibiotic resistance genes (ARGs) of HGT on the plant-associated Bacillus subtilis (BS-PA) predicated by ResFams.
Table S15. Antibiotic resistance genes (ARGs) of HGT on the non-plant-associated Bacillus subtilis (BS-nPA) predicated by ResFams.
Table S16. List of genes that were BA/BS-PA specific and were involved in HGT at the divergence event of PA and nPA.
Figure S1. Venn diagram based on the four HGT gene pools acquired by BA-PA (plant-associated B. amyloliquefaciens), BS-PA (plant-associated B. subtilis), BS-nPA (non-plant-associated B. subtilis), and BA-nPA (non-plant-associated B. amyloliquefaciens).
Data Sheet 1. Orthologous groups derived from OrthoMCL.
Data Sheet 2. The evolutionary history of each gene copy for genes related to rhizosphere adaptation.
References
Alcaraz, L. D., Moreno-Hagelsieb, G., Eguiarte, L. E., Souza, V., Herrera-Estrella, L., and Olmedo, G. (2010). Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genomics 11:332. doi: 10.1186/1471-2164-11-332
Badri, D. V., and Vivanco, J. M. (2009). Regulation and function of root exudates. Plant Cell Environ. 32, 666–681. doi: 10.1111/j.1365-3040.2009.01926.x
Bais, H. P., Weir, T. L., Perry, L. G., Gilroy, S., and Vivanco, J. M. (2006). The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 57, 233–266. doi: 10.1146/annurev.arplant.57.032905.105159
Barret, M., Morrissey, J. P., and O'Gara, F. (2011). Functional genomics analysis of plant growth-promoting rhizobacterial traits involved in rhizosphere competence. Biol. Fertil. Soils 47, 729–743. doi: 10.1007/s00374-011-0605-x
Beauregard, P. B., Chai, Y., Vlamakis, H., Losick, R., and Kolter, R. (2013). Bacillus subtilis biofilm induction by plant polysaccharides. Proc. Natl. Acad. Sci. U.S.A. 110, E1621–E1630. doi: 10.1073/pnas.1218984110
Blom, J., Rueckert, C., Niu, B., Wang, Q., and Borriss, R. (2012). The complete genome of Bacillus amyloliquefaciens subsp. plantarum CAU B946 contains a gene cluster for nonribosomal synthesis of iturin A. J. Bacteriol. 194, 1845–1846. doi: 10.1128/JB.06762-11
Borriss, R., Chen, X., Rueckert, C., Blom, J., Becker, A., Baumgarth, B., et al. (2011). Relationship of Bacillus amyloliquefaciens clades associated with strains DSM 7T and FZB42T: a proposal for Bacillus amyloliquefaciens subsp. amyloliquefaciens subsp. nov. and Bacillus amyloliquefaciens subsp. plantarum subsp. nov. based on complete genome sequence comparisons. Int. J. syst. Evol. Microbiol. 61, 1786–1801. doi: 10.1099/ijs.0.023267-0
Brohee, S., and van Helden, J. (2006). Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 7:488. doi: 10.1186/1471-2105-7-488
Cai, J., Liu, F., Liao, X., and Zhang, R. (2014). Complete genome sequence of Bacillus amyloliquefaciens LFB112 isolated from Chinese herbs, a strain of a broad inhibitory spectrum against domestic animal pathogens. J. Biotechnol. 175, 63–64. doi: 10.1016/j.jbiotec.2014.01.013
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552
Chen, H., and Boutros, P. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35
Chen, X., Koumoutsi, A., Scholz, R., Eisenreich, A., Schneider, K., Heinemeyer, I., et al. (2007). Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 25, 1007–1014. doi: 10.1038/nbt1325
Chen, X., Koumoutsi, A., Scholz, R., Schneider, K., Vater, J., Sussmuth, R., et al. (2009). Genome analysis of Bacillus amyloliquefaciens FZB42 reveals its potential for biocontrol of plant pathogens. J. Biotechnol. 140, 27–37. doi: 10.1016/j.jbiotec.2008.10.011
Chowdhury, S. P., Hartmann, A., Gao, X., and Borriss, R. (2015). Biocontrol mechanism by root-associated Bacillus amyloliquefaciens FZB42 - a review. Front. Microbiol. 6:780. doi: 10.3389/fmicb.2015.00780
David, L. A., and Alm, E. J. (2011). Rapid evolutionary innovation during an Archaean genetic expansion. Nature 469, 93–96. doi: 10.1038/nature09649
Deng, Y., Zhu, Y., Wang, P., Zhu, L., Zheng, J., Li, R., et al. (2011). Complete genome sequence of Bacillus subtilis BSn5, an endophytic bacterium of Amorphophallus konjac with antimicrobial activity for the plant pathogen Erwinia carotovora subsp. carotovora. J. Bacteriol. 193, 2070–2071. doi: 10.1128/JB.00129-11
de Wit, P. J., van der Burgt, A., Okmen, B., Stergiopoulos, I., Abd-Elsalam, K. A., Aerts, A. L., et al. (2012). The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 8:e1003088. doi: 10.1371/journal.pgen.1003088
Dunlap, C. A., Bowman, M. J., and Schisler, D. A. (2013). Genomic analysis and secondary metabolite production in Bacillus amyloliquefaciens AS 43.3: a biocontrol antagonist of Fusarium head blight. Biol. Control 64, 166–175. doi: 10.1016/j.biocontrol.2012.11.002
Earl, A. M., Eppinger, M., Florian Fricke, W., Rosovitz, M. J., Rasko, D. A., Daugherty, S., et al. (2012). Whole-genome sequences of Bacillus subtilis and close relatives. J. Bacteriol. 194, 2378–2379. doi: 10.1128/JB.05675-11
Earl, A. M., Losick, R., and Kolter, R. (2008). Ecology and genomics of Bacillus subtilis. Trends Microbiol. 16, 269–275. doi: 10.1016/j.tim.2008.03.004
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Enright, A. J., van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Fan, B., Carvalhais, L. C., Becker, A., Fedoseyenko, D., von Wiren, N., and Borriss, R. (2012). Transcriptomic profiling of Bacillus amyloliquefaciens FZB42 in response to maize root exudates. BMC Genomics 12:116. doi: 10.1186/1471-2180-12-116
Fritze, D. (2004). Taxonomy of the genus Bacillus and related genera: the aerobic endospore-forming bacteria. Phytopathology 94, 1245–1248. doi: 10.1094/PHYTO.2004.94.11.1245
Geng, W., Cao, M., Song, C., Xie, H., Liu, L., Yang, C., et al. (2011). Complete genome sequence of Bacillus amyloliquefaciens LL3, which exhibits glutamic acid-independent production of poly-γ-glutamic acid. J. Bacteriol. 193, 3393–3394. doi: 10.1128/JB.05058-11
Gibson, M. K., Forsberg, K. J., and Dantas, G. (2015). Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 9, 207–216. doi: 10.1038/ismej.2014.106
Gioia, J., Yerrapragada, S., Qin, X., Jiang, H., Igboeli, O. C., Muzny, D., et al. (2007). Paradoxical DNA repair and peroxide resistance gene conservation in Bacillus pumilus SAFR-032. PLoS ONE 2:e928. doi: 10.1371/journal.pone.0000928
Guo, S., Mao, Z., Wu, Y., Hao, K., He, P., and He, Y. (2013). Genome sequencing of Bacillus subtilis strain XF-1 with high efficiency in the suppression of Plasmodiophora brassicae. Genome Announc. 1:e00066–13. doi: 10.1128/genomeA.00066-13
Guo, Q., Li, S., Lu, X., Zhang, X., Wang, P., and Ma, P. (2014). Complete genome sequence of Bacillus subtilis BAB-1, a biocontrol agent for suppression of tomato gray mold. Genome Announc. 2:e00744–14. doi: 10.1128/genomeA.00744-14
Hao, W., and Golding, G. B. (2006). The fate of laterally transferred genes: life in the fast lane to adaptation or death. Genome Res. 16, 636–643. doi: 10.1101/gr.4746406
Hartmann, A., Schmid, M., van Tuinen, D., and Berg, G. (2008). Plant-driven selection of microbes. Plant Soil 321, 235–257. doi: 10.1007/s11104-008-9814-y
Harwood, C. R. (1992). Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends Biotechnol. 10, 247–256. doi: 10.1016/0167-7799(92)90233-L
He, P., Hao, K., Blom, J., Ruckert, C., Vater, J., Mao, Z., et al. (2012). Genome sequence of the plant growth promoting strain Bacillus amyloliquefaciens subsp. plantarum B9601-Y2 and expression of mersacidin and other secondary metabolites. J. Biotechnol. 164, 281–291. doi: 10.1016/j.jbiotec.2012.12.014
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Itakura, M., Saeki, K., Omori, H., Yokoyama, T., Kaneko, T., Tabata, S., et al. (2009). Genomic comparison of Bradyrhizobium japonicum strains with different symbiotic nitrogen-fixing capabilities and other Bradyrhizobiaceae members. ISME J. 3, 326–339. doi: 10.1038/ismej.2008.88
Kabisch, J., Thurmer, A., Hubel, T., Popper, L., Daniel, R., and Schweder, T. (2013). Characterization and optimization of Bacillus subtilis ATCC 6051 as an expression host. J. Biotechnol. 163, 97–104. doi: 10.1016/j.jbiotec.2012.06.034
Kamada, M., Hase, S., Sato, K., Toyoda, A., Fujiyama, A., and Sakakibara, Y. (2014). Whole genome complete resequencing of Bacillus subtilis Natto by combining long reads with high-quality short reads. PLoS ONE 9:e109999. doi: 10.1371/journal.pone.0109999
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, B.-Y., Lee, S.-Y., Ahn, J.-H., Song, J., Kim, W.-G., and Weon, H.-Y. (2015). Complete genome sequence of Bacillus amyloliquefaciens subsp. plantarum CC178, a phyllosphere bacterium antagonistic to plant pathogenic fungi. Genome Announc. 3:e01368-14. doi: 10.1128/genomeA.01368-14
Kunst, F., Ogasawara, N., Moszer, I., Albertini, A. M., Alloni, G., Azevedo, V., et al. (1997). The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256. doi: 10.1038/36786
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Li, L. Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, S., Zhang, N., Zhang, Z., Luo, J., Shen, B., Zhang, R., et al. (2013). Antagonist Bacillus subtilis HJ5 controls Verticillium wilt of cotton by root colonization and biofilm formation. Biol. Fertil. Soils 49, 295–303. doi: 10.1007/s00374-012-0718-x
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964.
Manzoor, S., Niazi, A., Bejai, S., Meijer, J., and Bongcam-Rudloffa, E. (2013). Genome sequence of a plant-associated bacterium, Bacillus amyloliquefaciens strain UCMB5036. Genome Announc. 1:e00111–13. doi: 10.1128/genomeA.00111-13
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Minh, B. Q., Nguyen, M. A., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Muller, J., Creevey, C. J., Thompson, J. D., Arendt, D., and Bork, P. (2010). AQUA: automated quality improvement for multiple sequence alignments. Bioinformatics 26, 263–265. doi: 10.1093/bioinformatics/btp651
Niazi, A., Manzoor, S., Bejai, S., Meijer, J., and Bongcam-Rudloff, E. (2014). Complete genome sequence of a plant associated bacterium Bacillus amyloliquefaciens subsp. plantarum UCMB5033. Stand. Genome. Sci. 9, 718–725. doi: 10.4056/sigs.4758653
Ongena, M., and Jacques, P. (2008). Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol. 16, 115–125. doi: 10.1016/j.tim.2007.12.009
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkr1065
Raaijmakers, J. M., Paulitz, T. C., Steinberg, C., Alabouvette, C., and Moënne-Loccoz, Y. (2008). The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 321, 341–361. doi: 10.1007/s11104-008-9568-6
Richards, V. P., Palmer, S. R., Pavinski Bitar, P. D., Qin, X., Weinstock, G. M., Highlander, S. K., et al. (2014). Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol. Evol. 6, 741–753. doi: 10.1093/gbe/evu048
Rückert, C., Blom, J., Chen, X., Reva, O., and Borriss, R. (2011). Genome sequence of B. amyloliquefaciens type strain DSM7T reveals differences to plant-associated B. amyloliquefaciens FZB42. J. Biotechnol. 155, 78–85. doi: 10.1016/j.jbiotec.2011.01.006
Schroeder, J. W., and Simmons, L. A. (2013). Complete genome sequence of Bacillus subtilis strain PY79. Genome Announc. 1:e01085–13. doi: 10.1128/genomeA.01085-13
Schyns, G., Serra, C. R., Lapointe, T., Pereira-Leal, J. B., Potot, S., Fickers, P., et al. (2013). Genome of a gut strain of Bacillus subtilis. Genome Announc. 1:e00184–12. doi: 10.1128/genomeA.00184-12
Stanley, N. R., Britton, R. A., Grossman, A. D., and Lazazzera, B. A. (2003). Identification of catabolite repression as a physiological regulator of biofilm formation by Bacillus subtilis by use of DNA microarrays. J. Bacteriol. 185, 1951–1957. doi: 10.1128/jb.185.6.1951-1957.2003
Suzuki, R., and Shimodaira, H. (2006). Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542. doi: 10.1093/bioinformatics/btl117
Tan, I. S., and Ramamurthi, K. S. (2014). Spore formation in Bacillus subtilis. Environ. Microbiol. Rep. 6, 212–225. doi: 10.1111/1758-2229.12130
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
van Elsas, J. D., Turner, S., and Bailey, M. J. (2003). Horizontal gene transfer in the phytosphere. New Phytol. 157, 525–537. doi: 10.1007/s10142-013-0345-0
Wu, H., Qiao, J., Blom, J., Rueckert, C., Reva, O., Gao, X., et al. (2013). The rhizobacterium Bacillus amyloliquefaciens subsp. plantarum NAU-B3 contains a large inversion within the central portion of the genome. Genome Announc. 1:e00941–13. doi: 10.1128/genomeA.00941-13
Wu, L., Wu, H., Chen, L., Yu, X., Borriss, R., and Gao, X. (2015). Difficidin and bacilysin from Bacillus amyloliquefaciens FZB42 have antibacterial activity against Xanthomonas oryzae rice pathogens. Sci. Rep. 5:12975. doi: 10.1038/srep12975
Xu, Z., Zhang, R., Wang, D., Qiu, M., Feng, H., Zhang, N., et al. (2014). Enhanced control of cucumber wilt disease by Bacillus amyloliquefaciens SQR9 by altering the regulation of its DegU phosphorylation. Appl. Environ. Microbiol. 80, 2941–2950. doi: 10.1128/AEM.03943-13
Yang, H., Liao, Y., Wang, B., Lin, Y., and Pan, L. (2011). Complete genome sequence of Bacillus amyloliquefaciens XH7, which exhibits production of purine nucleosides. J. Bacteriol. 193, 5593–5594. doi: 10.1128/JB.05880-11
Yi, H., Chun, J., and Cha, C. J. (2014). Genomic insights into the taxonomic status of the three subspecies of Bacillus subtilis. Syst. Appl. Microbiol. 37, 95–99. doi: 10.1016/j.syapm.2013.09.006
Yu, C., Yim, K., Tsui, S. K., and Chan, T. (2012). Complete genome sequence of Bacillus subtilis strain QB928. J. Bacteriol. 194, 6308–6309. doi: 10.1128/JB.01533-12
Yuan, J., Li, B., Zhang, N., Waseem, R., Shen, Q., and Huang, Q. (2012a). Production of bacillomycin- and macrolactin-type antibiotics by Bacillus amyloliquefaciens NJN-6 for suppressing soilborne plant pathogens. J. Agric. Food Chem. 60, 2976–2981. doi: 10.1021/jf204868z
Yuan, J., Raza, W., Huang, Q., and Shen, Q. (2011). Quantification of the antifungal lipopeptide iturin A by high performance liquid chromatography coupled with aqueous two-phase extraction. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 879, 2746–2750. doi: 10.1016/j.jchromb.2011.07.041
Yuan, J., Raza, W., Shen, Q., and Huang, Q. (2012b). Antifungal activity of Bacillus amyloliquefaciens NJN-6 volatile compounds against Fusarium oxysporum f. sp. cubense. Appl. Environ. Microbiol. 78, 5942–5944. doi: 10.1128/AEM.01357-12
Yuan, J., Ruan, Y., Wang, B., Zhang, J., Waseem, R., Huang, Q., et al. (2013). Plant growth-promoting rhizobacteria strain Bacillus amyloliquefaciens NJN-6-enriched bio-organic fertilizer suppressed Fusarium wilt and promoted the growth of banana plants. J. Agric. Food Chem. 61, 3774–3780. doi: 10.1021/jf400038z
Yuan, J., Zhang, F., Wu, Y., Zhang, J., Raza, W., Shen, Q., et al. (2014). Recovery of several cell pellet-associated antibiotics produced by Bacillus amyloliquefaciens NJN-6. Lett. Appl. Microbiol. 59, 169–176. doi: 10.1111/lam.12260
Zeigler, D. R. (2011). The genome sequence of Bacillus subtilis subsp. spizizenii W23: insights into speciation within the B. subtilis complex and into the history of B. subtilis genetics. Microbiology 157, 2033–2041. doi: 10.1099/mic.0.048520-0
Zeigler, D. R., Pragai, Z., Rodriguez, S., Chevreux, B., Muffler, A., Albert, T., et al. (2008). The origins of 168, W23, and other Bacillus subtilis legacy strains. J. Bacteriol. 190, 6983–6995. doi: 10.1128/JB.00722-08
Zhang, N., Yang, D., Wang, D., Miao, Y., Shao, J., Zhou, X., et al. (2015). Whole transcriptomic analysis of the plant-beneficial rhizobacterium Bacillus amyloliquefaciens SQR9 during enhanced biofilm formation regulated by maize root exudates. BMC Genomics 16:685. doi: 10.1186/s12864-015-1825-5
Zhang, G., Deng, A., Xu, Q., Liang, Y., Chen, N., and Wen, T. (2011). Complete genome sequence of Bacillus amyloliquefaciens TA208, a strain for industrial production of guanosine and ribavirin. J. Bacteriol. 193, 3142–3143. doi: 10.1128/JB.00440-11
Keywords: Bacillus subtilis and Bacillus amyloliquefaciens, genome evolution, horizontal gene transfer, plant-associated, phylogenomics, rhizosphere adaptation
Citation: Zhang N, Yang D, Kendall JRA, Borriss R, Druzhinina IS, Kubicek CP, Shen Q and Zhang R (2016) Comparative Genomic Analysis of Bacillus amyloliquefaciens and Bacillus subtilis Reveals Evolutional Traits for Adaptation to Plant-Associated Habitats. Front. Microbiol. 7:2039. doi: 10.3389/fmicb.2016.02039
Received: 27 July 2016; Accepted: 05 December 2016;
Published: 20 December 2016.
Edited by:
Rakesh Sharma, Institute of Genomics and Integrative Biology (CSIR), IndiaReviewed by:
Dong-Woo Lee, Kyungpook National University, Republic of KoreaZhuofei Xu, Huazhong Agricultural University, China
Copyright © 2016 Zhang, Yang, Kendall, Borriss, Druzhinina, Kubicek, Shen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruifu Zhang, cmZ6aGFuZ0BuamF1LmVkdS5jbg==
†These authors have contributed equally to this work.