 Julie L. Meyer
Julie L. Meyer Valerie J. Paul
Valerie J. Paul Laurie J. Raymundo3
Laurie J. Raymundo3 Max Teplitski
Max Teplitski- 1Soil and Water Science Department, University of Florida-Institute of Food and Agricultural Sciences, Gainesville, FL, USA
- 2Smithsonian Marine Station, Fort Pierce, FL, USA
- 3University of Guam Marine Laboratory, Mangilao, Guam
Black Band Disease (BBD), the destructive microbial consortium dominated by the cyanobacterium Roseofilum reptotaenium, affects corals worldwide. While the taxonomic composition of BBD consortia has been well-characterized, substantially less is known about its functional repertoire. We sequenced the metagenomes of Caribbean and Pacific black band mats and cultured Roseofilum and obtained five metagenome-assembled genomes (MAGs) of Roseofilum, nine of Proteobacteria, and 12 of Bacteroidetes. Genomic content analysis suggests that Roseofilum is a source of organic carbon and nitrogen, as well as natural products that may influence interactions between microbes. Proteobacteria and Bacteroidetes members of the disease consortium are suited to the degradation of amino acids, proteins, and carbohydrates. The accumulation of sulfide underneath the black band mat, in part due to a lack of sulfur oxidizers, contributes to the lethality of the disease. The presence of sulfide:quinone oxidoreductase genes in all five Roseofilum MAGs and in the MAGs of several heterotrophs demonstrates that resistance to sulfide is an important characteristic for members of the BBD consortium.
Introduction
Black Band Disease (BBD) is a globally distributed coral disease that devastates dozens of species of corals, including large, reef-building scleractinians (Sato et al., 2016). It is easily recognized by the appearance of a dense dark purple or black band, which is the visible accumulation of phycoerythrin-rich filamentous cyanobacteria. In the Caribbean, known as a “hotbed” of coral disease (Weil et al., 2006), BBD is more prevalent in warmer, shallower waters (Kuta and Richardson, 2002), and manipulative experiments have demonstrated that BBD progresses faster at higher light levels and temperature (Kuehl et al., 2011; Sato et al., 2011). The cyanobacterium recently renamed Roseofilum reptotaenium has been implicated as the causative agent of BBD within the polymicrobial disease consortium (Casamatta et al., 2012). Strains of Roseofilum have been cultivated in the laboratory, but like other filamentous cyanobacteria, Roseofilum cannot be fully isolated, only grown in non-axenic, unicyanobacterial cultures (Richardson et al., 2014). This conserved interdependence of Roseofilum and heterotrophic bacteria may be linked to metabolic requirements and may explain why this cyanobacterium is found within apparently healthy coral microbiomes (Meyer et al., 2016). In many ways, BBD consortia resemble microbial mats found in tropical lagoons (Echenique-Subiabre et al., 2015), mangroves (Guidi-Rotani et al., 2014), and modern marine stromatolites (Ruvindy et al., 2016) that are characterized by steep physicochemical gradients and the presence of Cyanobacteria, Bacteroidetes, and Proteobacteria (especially Alpha-, Delta-, and Gammaproteobacteria; Bolhuis et al., 2014). With the large size of filamentous cyanobacteria relative to other bacterial cells, their production of exopolysaccharides that consolidate the mat, and their critical roles in converting inorganic carbon, nitrogen, and sulfur to organic forms, Cyanobacteria are the pioneering microorganisms of microbial mats (Bolhuis et al., 2014). Likewise, Roseofilum is the engineer of the black band consortium, creating a narrow polymicrobial band on the surface of reef-building corals (Miller and Richardson, 2011; Casamatta et al., 2012; Richardson et al., 2014).
While the composition of BBD has been studied for decades (Miller and Richardson, 2011), the recent application of high-throughput sequencing to characterize the microbial community structure in both the black band layer and adjacent healthy coral epimicrobiome has uncovered two important factors in understanding this disease. First, BBD is highly localized, and in as little as 10 cm from the leading edge of the black band mat, the microbial community in the surface mucus layer is indistinguishable from that of healthy corals (Meyer et al., 2016). Second, Roseofilum is a rare but ubiquitous member of healthy Caribbean coral microbiota, implying that growth of Roseofilum is constrained in healthy tissue until undefined restrictions are removed (Meyer et al., 2016). Together, this suggests that while Roseofilum is capable of engineering the highly altered black band layer, its influence is spatially limited and its pathogenesis is contextual. To uncover the mechanisms through which Roseofilum proliferates and engineers a new environment on the surface of corals during BBD, we examined the metagenomic potential of members of black band consortia in situ and in vitro.
Methods
Samples of coral surface microbiota and the BBD mat were collected by aspiration with sterile, needle-less syringes from the Florida Keys, Belize, Honduras, and Guam (Table S1), as in Meyer et al. (2016).
Enrichment Culturing of the Black Band Disease Consortium
The black band layer of an infected Montastraea cavernosa coral was collected at Looe Key Reef, Florida on April 23, 2014 and used to isolate Cyanobacteria from the disease consortium. Roseofilum was grown in unicyanobacterial, but not axenic cultures, in a medium containing four parts artificial seawater made from 36 g/L Red Sea Coral Pro Salts and 1 part Cyanobacterial BG-11 media (ATCC Medium 616), at pH 7, room temperature, with 12 h of light and dark per day. Cyanobacterial cultures were confirmed as unicyanobacterial by amplifying extracted community DNA with cyanobacterial specific 16S rRNA primers (Nübel et al., 1997) and directly sequencing the cleaned PCR product by Sanger sequencing at the DNA Lab of Arizona State University (GenBank Accession No. KP689103).
16S Illumina Tag Sequencing
Genomic DNA for 16S Illumina tag sequencing was extracted with a PowerSoil DNA isolation kit (MoBio, Carlsbad, CA). The V6 region of 16S rRNA genes was amplified in triplicate for each sample with previously reported Illumina-compatible primers designed for bacteria (Eren et al., 2013), using previously described conditions for amplicon library preparation and analysis (Meyer et al., 2016). Sequencing of amplicon libraries was performed at the Genomics Core Facility at Pennsylvania State University on an Illumina MiSeq with a 150-bp paired-end protocol, using single indexing. Sequencing reads are publicly available through NCBI's Sequence Read Archive (SRA) under the BioProject ID PRJNA269585. Analysis of the 16S amplicon libraries was performed using the previously described parameters (Meyer et al., 2016). Briefly, sequences were clustered at 97% using the subsampled open reference OTU picking method (Rideout et al., 2014) with no removal of singletons in Qiime v.1.8 (Caporaso et al., 2010), and assigned taxonomy with the Greengenes database v.13.8 (DeSantis et al., 2006). Community structure was analyzed in R with Phyloseq (McMurdie and Holmes, 2013) and plotted with ggplot2 (Wickham, 2009).
Metagenomic Library Construction and Sequencing
For metagenomic library construction, DNA and RNA were simultaneously extracted from the BBD samples from infected corals using a Qiagen AllPrep DNA/RNA Micro Kit (Germantown, MD) from the Belize and Florida samples. Only DNA was extracted from the Guam sample due to limited biomass using a PowerSoil DNA isolation kit (MoBio, Carlsbad, CA). Metagenomic libraries were constructed with a TruSeq DNA Sample Preparation Kit (Illumina, San Diego, CA) and sequenced at the University of Maryland Institute for Bioscience and Biotechnology Research on an Illumina HiSeq with a 100-bp paired-end protocol.
Metagenomic Data Analysis
Sequencing reads were quality-filtered by trimming adaptors with cutadapt (Martin, 2011) and filtering reads for a minimum quality score of 30, minimum length of 100 bp, and discarding all sequences with ambiguous base calls using Sickle (Joshi and Fass, 2011), and interlaced with the shuffleSequences_fastq.pl script from velvet (Zerbino and Birney, 2008) prior to assembly. The unassembled, quality-filtered reads are publicly available through NCBI's Sequence Read Archive (SRA) under the BioProject ID PRJNA269585. Metagenomes were assembled individually with IDBA-UD (Peng et al., 2012) with k-mer sizes of 30–80 and submitted to IMG-MER (Markowitz et al., 2006) for annotation. Annotated metagenomic assemblies are publicly available in IMG under the IMG Genome IDs 3300003272, 3300003311, 3300003317, 3300003641, 3300003309 (Table S2). Unassembled reads were mapped to the assembled contigs with bowtie2 (Langmead and Salzberg, 2012) and alignment statistics were recovered with samtools (Li et al., 2009). The metagenomic sequencing coverage was assessed with non-pareil (Rodriguez and Konstantinidis, 2014).
Metagenome-assembled genomes (MAGs) were retrieved from the assembled metagenomic contigs by binning based on tetranucleotide frequencies using Emergent Self-Organizing Maps (ESOM) as in Dick et al. (2009), using the protocol described at https://github.com/tetramerFreqs/Binning. The training algorithm was K-batch, the starting radius was 50, and the map sizes were as follows: BLZ4 181 rows × 362 columns, BLZD 144 × 288, Cyano 244 × 488, LKpool 199 × 398, and Guam 180 × 360. Within each MAG, any scaffolds with unusually high or low coverage of mapped reads were removed from the bin. Summary statistics for each MAG were determined with QUAST v. 2.3 (Gurevich et al., 2013). Genome completeness and the presence of contamination within MAGs was determined with Anvi'o v. 2.1.0 (Eren et al., 2015), using four different sets of single-copy genes. Antibiotic and secondary metabolite biosynthetic genes in MAGs were identified using antiSMASH v. 3.0 (Weber et al., 2015). The two-way average nucleotide identity (ANI) between closely related genomes was calculated as in Goris et al. (2007) through the online tools of the Environmental Microbial Genomics Laboratory (enve-omics lab) at the Georgia Institute of Technology. MAGs were submitted to both IMG-ER (Markowitz et al., 2006) and RAST (Aziz et al., 2008) for annotation and the IMG-annotated MAGs are publically available under the accession numbers listed in Table 1. Statistical analyses of SEED subsystems annotation from RAST profiles of MAGs were performed with STAMP (Parks et al., 2014), using Storey FDR multiple test correction, q < 0.05 and effect size >0.6. Summary figures were created in R with ggplot2. Pan-genome analysis of Roseofilum MAGs was performed with Roary v.3.4.1 (Page et al., 2015). A second pan-genome analysis of the five Roseofilum MAGs described here, along with the recently published MAGs of Roseofilum from the Great Barrier Reef (Sato et al., 2016), a Roseofilum culture from the Great Barrier Reef (Buerger et al., 2016), and a Geitlerinema culture from Caribbean black band mat (Den Uyl et al., 2016) was performed with Anvi'o v.2.1.0, in which proteins were clustered with the MCL algorithm (van Dongen and Abreu-Goodger, 2012) using 2 for the MCL inflation parameter and identified by NCBI's blastp (Altschul et al., 1990).
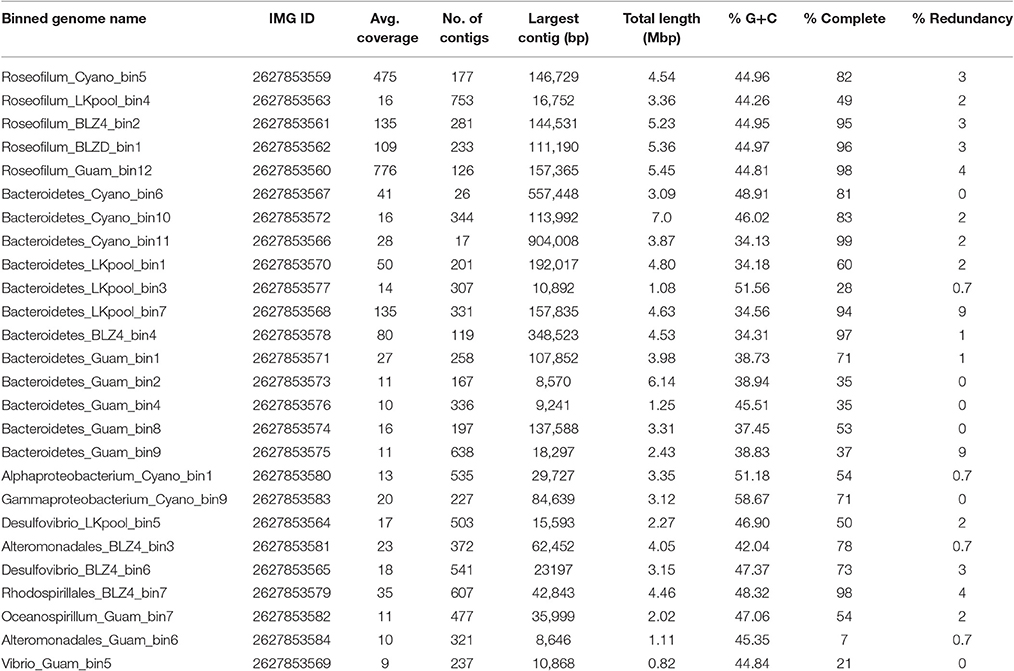
Table 1. Characteristics of metagenome-assembled genomes from Black Band Disease mats and cultured Roseofilum reptotaenium.
Results and Discussion
Microbial Community Structure from 16S rRNA Gene Sequencing
Bacterial community structure was determined for 24 BBD consortia, 30 healthy coral surface microbiomes, and one Roseofilum culture by amplification and Illumina sequencing of the V6 hypervariable region of 16S rRNA genes (Table S1). Consistent with previous results from Caribbean corals only (Meyer et al., 2016), the reanalysis of the bacterial community structure with both Caribbean and Pacific corals demonstrates the prevalence of Roseofilum, Rhodobacteraceae, Bacteroidales, and Desulfovibrio in black band consortia in contrast to the high proportions of Gammaproteobacteria (such as Halomonas and Moritella) and Actinobacteria (such as Renibacterium) in healthy epimicrobiomes (Figure 1). Comparison of the composition of 24 in situ BBD samples and a unicyanobacterial, but not axenic Roseofilum culture revealed that three proteobacterial genera were depleted in the culture. The relative abundance of Vibrio in the culture was two orders of magnitude lower (0.001% of reads in the Roseofilum culture vs. ~0.1% in BBD), Arcobacter was three orders of magnitude lower (0.0004% vs. ~0.1%), and Desulfovibrio was three orders of magnitude lower (0.0002% vs. ~0.01%).
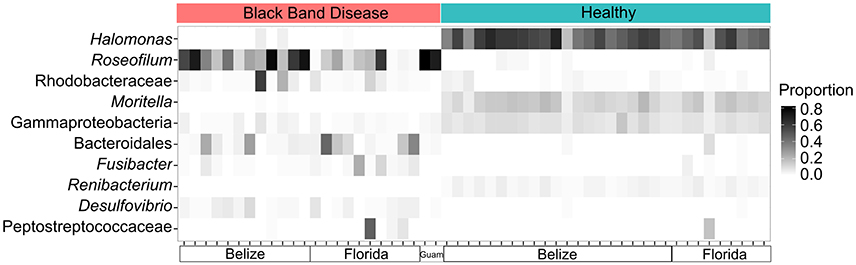
Figure 1. Community shifts in Black Band Disease (BBD) consortia and healthy coral epimicrobiomes. Relative abundance of the 10 most common bacterial genera in 16S rRNA gene libraries from 54 coral surfaces. Each column in the heatmap represents an individual coral surface microbiome sample.
The remarkable degree of conservation of taxa in the Roseofilum culture compared to in situ BBD metagenomes suggests a strong interdependence of members of the disease consortium that is unrelated to the coral host. Specifically, no major BBD taxonomic groups were entirely missing in the Roseofilum culture and the proportions of Bacteroidetes, Deltaproteobacteria, and Gammaproteobacteria were relatively unchanged between the Roseofilum culture and in situ BBD. However, three proteobacterial genera (Vibrio, Arcobacter, and Desulfovibrio) were detected in lower relative abundance in the culture compared to in situ BBD, based on 16S rDNA amplicon libraries. Although these genera may not be the most abundant organisms within the BBD consortium, each has the potential to impact the progression of the disease, as both Vibrio and Arcobacter are genera containing pathogens and both Arcobacter and Desulfovibrio are involved in sulfur cycling. Recent metatranscriptomic analysis of BBD consortia in the Red Sea demonstrated that the most highly expressed functional gene among BBD vibrios was a thiamin transporter (Arotsker et al., 2016), suggesting that Roseofilum, as a producer of thiamin, has the capacity to encourage the growth of vibrios that lyse coral tissue (Arotsker et al., 2009) during the colonization of adjacent healthy tissue. However, interactions between Roseofilum and vibrios are likely complex, as we previously demonstrated mutual exclusion between Roseofilum and Vibrio spp., that may be mediated through production of lyngbic acid by Roseofilum that is capable of disrupting the CAI-1 vibrio quorum-sensing pathway (Meyer et al., 2016). While previous work revealed a signficant co-occurrence of Roseofilum and Desulfovibrio in BBD consortia (Meyer et al., 2016), Desulfovibrio was not co-enriched with Roseofilum in the culture. This indicates that the co-occurrence of these two genera is related to opportunistic interactions on the coral surface, rather than a specific interactive relationship. Furthermore, it suggests the two groups contribute individually to disease progression.
Assembled Metagenomes and Metagenome-Assembled Genomes from Black Band Disease Consortia
Four metagenomes of the disease consortia were generated from the black band community on Pseudodiploria strigosa and Orbicella annularis corals from Belize, Goniopora fruticosa from Guam, and a pooled sample from M. cavernosa and O. faveolata from Florida. A fifth metagenome was generated from a unicyanobacterial, but not axenic, culture of R. reptotaenium isolated from a black band layer on M. cavernosa collected in Florida. Each metagenomic dataset contained 16,853,081–44,199,247 read pairs after quality filtering (Table S2). Annotated metagenomic assemblies contained 69,548–612,757 protein-coding genes (Table S2). The coverage of metagenomic sequencing was assessed with non-pareil using the unassembled quality-filtered sequencing reads and ranged from 87 to 99%, indicating that this sequencing depth was adequate to capture most of the unique sequences in the extracted community DNA, even if not all of this diversity was captured in the metagenomic assemblies. To assess the quality of the metagenomic assemblies, the unassembled sequencing reads were mapped to the metagenomic assemblies, with an overall alignment rate that ranged from 41% (BLZD) to 92% (Cyano), where a higher alignment rate indicates that the metagenomic assembly encompassed more of the diversity present in the sequencing reads (Table S2). Between 2 and 24 unique genomes were detected within each metagenomic assembly (Table S2) by Anvi'o (Eren et al., 2015), using four different databases of single copy genes as in Delmont and Eren (2016).
The number of protein-coding genes in each metagenome ranged from 69,548 to 612,757 (Table S2). Of the annotated genes in the assembled metagenomes, 12 to 47% could be assigned to bacterial phyla (16,674–79,576 bacterial genes per metagenome). Less than 1% of annotated genes in any metagenomic assembly could be assigned to archaeal phyla. The remaining genes were either unassigned or identified as eukaryotic. A total of 53 bacterial phyla and 6 archaeal phyla were identified in the metagenomic assemblies (Table S3). The four most abundant bacterial phyla (Cyanobacteria, Bacteroidetes, Proteobacteria, and Firmicutes) constituted 80–95% of the annotated bacterial genes in the four environmental metagenomic assemblies (Figure S1). In the Roseofilum culture, these four phyla constituted 68% of annotated genes with taxonomic assignment, and Planctomycetes were more abundant than Firmicutes (Figure S1). The proportions of functional genes assigned to Cyanobacteria and Bacteroidetes in the metagenomic assemblies matched the proportion of these phyla detected by 16S rRNA gene surveys, while the proportion of Proteobacteria was higher in 16S gene surveys than in metagenomic assemblies (Figure S1).
Binning of the assembled metagenomic datasets produced a total of 26 MAGs, with estimated genome coverage ranging from 9 × to 776 × (Table 1). The annotated MAGs are publicly available in IMG and under the accession numbers listed in Table 1. MAGs had varying levels of estimated genome completeness and redundancy (Table 1). With the exception of the LKpool Roseofilum, which was assembled from pooled coral hosts, the MAGs of Roseofilum were relatively complete and contained low redundancy (4% or less of the single-copy genes in the Roseofilum MAGs were present in more than one copy). Only one cyanobacterial MAG and one full-length cyanobacterial 16S rRNA gene were retrieved from each assembled metagenome. To corroborate the low diversity of cyanobacterial genomes, contigs identified as containing cyanobacterial genes based on IMG annotation from each of the five metagenomic assemblies were pooled in silico and analyzed for the number of genomes present. Assembled contigs annotated as Cyanobacteria from the Belize Orbicella metagenome (BLZ4) contained up to three copies of single copy genes, while cyanobacterial contigs from the other metagenomes contained one to two copies of single copy genes, indicating a low diversity of Cyanobacteria in the black band layer. The cyanobacterial 16S rRNA gene sequence was identical in metagenomic assemblies from the three Caribbean samples and the unicyanobacterial culture, while minor variation (98.9% similarity) was detected in the cyanobacterial 16S rRNA gene sequence from Guam. The 16S rRNA gene sequences recovered from the Caribbean cyanobacterial MAGs are identical to the reported sequence in R. reptotaenium (EU743965) with the exception of one nucleotide at the 5′ end of the published sequence for the type species. The two-way average nucleotide identity of the four Caribbean Roseofilum MAGs was >99% similarity, while the Guam Roseofilum MAG showed around 94% similarity to the Caribbean Roseofilum MAGs. In addition to the five Roseofilum MAGs, MAGs of 12 Bacteroidetes, and 9 Proteobacteria were recovered by binning, including an unclassified gammaproteobacterium, Bacteroidales, and Desulfovibrio, which have been detected as common in BBD consortia through 16S amplicon surveys (Figure 1; Miller and Richardson, 2011; Meyer et al., 2016).
Functional Characteristics of Black Band Disease Metagenomes and Metagenome-Assembled Genomes
The five Roseofilum MAGs had similar functional profiles based on KEGG Ontology (Figure S2), as well as similar gene synteny (Figure S3) in contrast to the more taxonomically and functionally diverse Bacteroidetes and Proteobacteria MAGs (Figure S2). Pan-genomic clustering of protein-coding genes in Roseofilum and Geitlernema MAGs (Figure 2) reveals a conserved core of Roseofilum functional genes that is distinct from the Geitlerinema MAG based on sequence variation, as expected for taxa assigned to different genera. However, the overall functional profile of all eight MAGs, based on SEED subsystems, is well-conserved (with the exception of the less-complete LKpool Roseofilum; Figure 2). Clustering of Roseofilum MAGs based on the relative abundance of proteins showed a clear geographic signal, with Caribbean MAGs clustering together and Pacific MAGs (from three different research groups) clustering together (Figure 2). A total of 36,649 genes in 8,690 protein clusters were identified in the eight MAGs. The Roseofilum core indicated in Figure 2 contained 27,153 genes in 3,470 protein clusters, while the Geitlerinema specific bin contained 2,702 genes. Clusters in the Roseofilum core were defined as being present in at least five of the seven Roseofilum MAGs. The remaining, less-conserved regions contained a total of 6,794 genes in 2,634 protein clusters. Of these less-conserved protein clusters, 40% (1055 clusters) contained only one gene and 18% (485 clusters) contained only two genes.
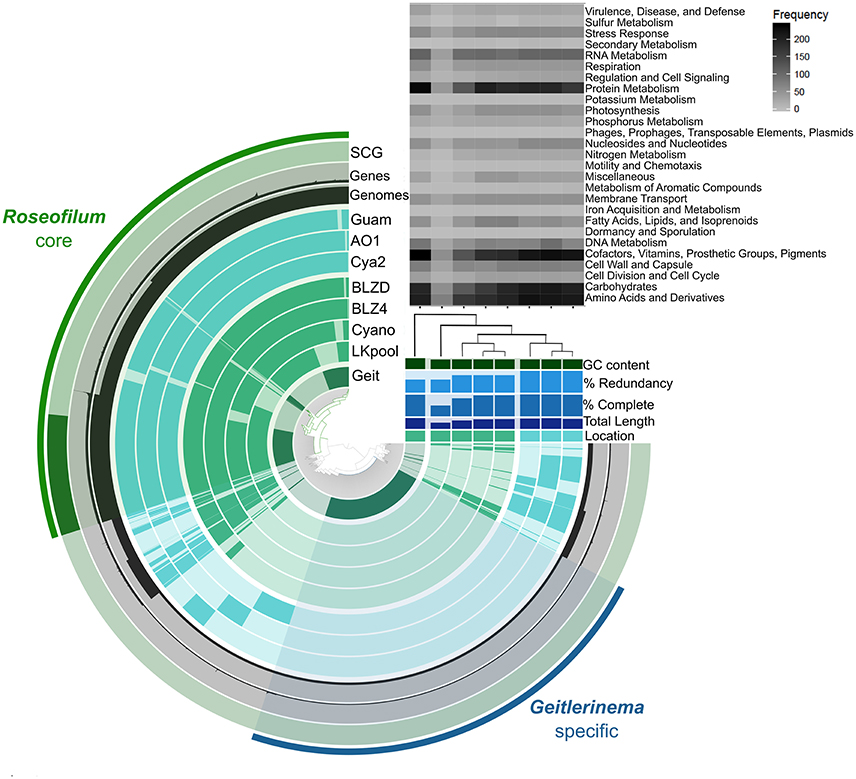
Figure 2. Roseofilum pan-genome. Protein clusters from seven Roseofilum and one Geitlerinema metagenome-assembled genomes (MAGs) are displayed in the central dendrogram and genes present in each MAG are indicated on concentric rings. The outer-most ring displays Single Copy Genes present in the Roseofilum core genome. The next two rings display the number of genes and number of MAGs, respectively, for each protein cluster. Upper right: Relative abundance of genes in each MAG assigned to Level 1 SEED subsystems annotation. The % completeness and % redundancy of each MAG is based on the presence/absence of single-copy genes. Clustering of MAGs (right) was based on the relative abundance of proteins and reflected the location: Green for Caribbean samples and Teal for Pacific samples. “Geit” is the cultured Geitlerinema sp. BBD 1991 MAG isolated from Orbicella annularis in Florida (Den Uyl et al., 2016), “LKpool” is the Roseofilum MAG from a pooled sample from Montastraea cavernosa and O. faveolata from Florida, “Cyano” is the cultured Roseofilum MAG isolated from M. cavernosa collected in Florida, “BLZ4” is the Roseofilum MAG from Orbicella annularis in Belize, “BLZD” is the Roseofilum MAG from Pseudodiploria strigosa in Belize, “Cya2” is the Roseofilum MAG from Montipora hispida on the central Great Barrier Reef (Sato et al., 2016), “AO1” is the cultured Roseofilum MAG isolated from Pavona duerdeni on the central Great Barrier Reef (Buerger et al., 2016), and “Guam” is the Roseofilum MAG from Goniopora fruticosa in Guam.
To determine the potential roles and interactions between different members of the polymicrobial disease consortium, we tested for differences in the relative abundances of protein-coding genes assigned to SEED subsystems (Overbeek et al., 2014) level 3 functions. Examination of differentially represented functions among the three predominant phyla (Cyanobacteria, Bacteroidetes, Proteobacteria) revealed 87 subsystems with statistically different abundances (Table S3). Of these differentially represented functions, 13 had an effect size >0.6, meaning that the difference in the mean relative abundance was >60% and encompassed mostly functions that were present solely in the Roseofilum MAGs and not in the MAGs of heterotrophs (Figure 3). Genes associated with carbon fixation (Subsystem CO2 uptake, carboxysome), micronutrient acquisition (Subsystem ECF class transporters), and phototrophy (Subsystems Cytochrome B6-F complex, Photosystem II, and Phycobilisome) were more abundant in the Roseofilum MAGs than the MAGs of heterotrophs (Figure 3). In addition, functions related to carbon respiration (Subsystems Cyanobacterial bypass in the TCA, Maltose and maltodextrin utilization), and the redox-regulated molecular chaperone Hsp33 (Subsystem Steptococcus pyogenes recombinatorial Zone) were primarily unique to the Roseofilum MAGs (Figure 3).
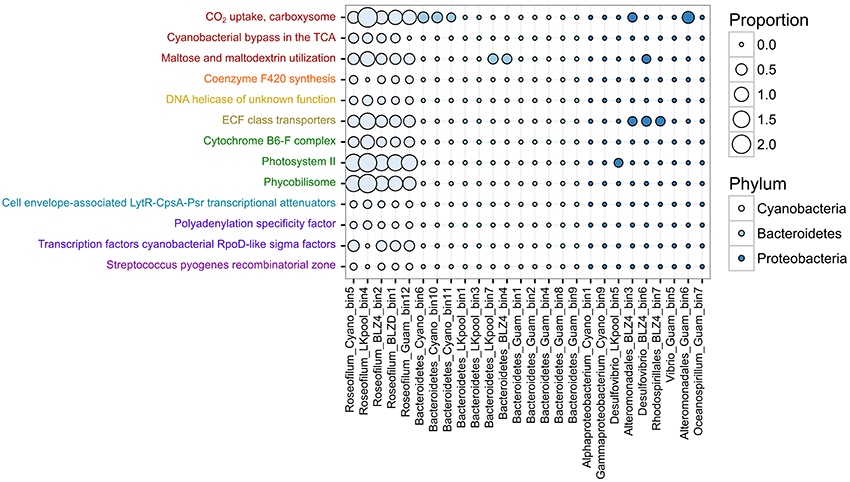
Figure 3. Functional differences in metagenome-assembled genomes from Black Band Disease consortia. Differences in the relative abundance of Level 3 SEED subsystems annotations among metagenome-assembled genomes from three phyla. Functional annotations with the same lettering color belong to the same Level 1 SEED subsystems category. Only features passing the filtering criteria are shown (q < 0.05, effect size > 0.6, multiple test correction with Storey FDR).
Differentially abundant functions between the three phyla also suggest a strong internal cycling of organic nitrogen within the black band layer in which Bacteroidetes and Proteobacteria degrade amino acids to produce urea and urea is degraded by Roseofilum. Both Bacteroidetes and Proteobacteria MAGs contained genes for the degradation of branched chain amino acids (leucine, isoleucine, valine) that were not present in the five Roseofilum MAGs, and the Roseofilum MAGs had more genes associated with the degradation of urea than Bacteroidetes and Proteobacteria MAGs (Table S4). In addition, genes for the production and degradation of cyanophycin, a polypeptide-like nitrogen storage compound, were present in the five Roseofilum MAGs and in the Cyano_bin9_Gammaproteobacteria MAG (Table S5). Genes for the assimilation of ammonia and nitrate were common in the MAGs of both Roseofilum and heterotrophs, demonstrating that each member of the consortium utilizes multiple pathways to assimilate nitrogen in the nitrogen-limited coral reef ecosystem. Genes for nitrogen fixation (nifHDK) were detected in the five Roseofilum MAGs as well as in two Proteobacteria MAGs (BLZ4_bin3_Alteromonadales, BLZ4_bin7_Rhodospirillales; Table S5). The diversity of methods utilized by Roseofilum for the acquisition of nitrogen mirrors those found in the Geitlerinema strain isolated from BBD (Den Uyl et al., 2016).
The multiple, diverse Bacteroidetes MAGs recovered here from both Caribbean and Pacific BBD underscore the previously underappreciated predominance of Bacteroidetes in BBD. While Bacteroidetes have been detected in 16S rRNA gene surveys of BBD and have been hypothesized to play a role in the pathogenesis of BBD (Frias-Lopez et al., 2004), the abundance of Bacteroidetes MAGs and high relative abundance of genes assigned to Bacteroidetes in the metagenomic assemblies (Table S3) suggests this group has a substantial role in the BBD consortia. Marine Bacteroidetes are known for their ability to degrade complex polymers (Fernández-Gómez et al., 2013) and may thus play a critical role in the progression of BBD. Whether these Bacteroidetes are simply opportunistic in the degradation of coral tissue under a BBD mat or whether they contribute to the invasion of adjacent healthy tissue is yet to be determined. Recent work has demonstrated that benthic cyanobacterial mats in Caribbean coral reef ecosystems are stimulated by the localized degradation of organic matter in sediments (Brocke et al., 2015), further suggesting that microbial mat-forming filamentous cyanobacteria like Roseofilum require the release of nutrients by heterotrophs to proliferate. Here, we found that both Proteobacteria and Bacteroidetes in the five metagenomes carry glycoside hydrolase families for the degradation of starch/glycogen, oligosaccharides, and chitin (data not shown). Future metatranscriptomic studies may reveal which groups are most actively degrading complex carbon in the black band layer.
Akin to other cyanobacterial mats, a gradient of decreasing oxygen and increasing sulfide with depth in the black band layer has been detected (Glas et al., 2012), and the accumulation of sulfide may have an important role in BBD as sulfate reduction appears to be necessary for the initiation of the disease but not for the progression of already established BBD (Richardson et al., 2009; Brownell and Richardson, 2014). Previous investigations of key sulfur-cycling genes in BBD have identified dissimilatory sulfate reductase genes in Desulfovibrio (Bourne et al., 2011) and sulfur oxidation genes in Rhodobacteraceae (Bourne et al., 2013). However, the low levels and lack of diversity in sulfur oxidation genes associated with BBD imply that sulfide is not broken down, but rather accumulates beneath the mat (Bourne et al., 2013). These results are consistent with the current metagenomic study. Dissimilatory sulfate reduction genes (dsrA) were detected only in the two Desulfovibrio MAGs and not in the other major groups found in the black band consortium (Table S5). No sulfur oxidation genes of the sox pathway were detected in any of the five BBD metagenomes nor in the 26 MAGs, indicating that this pathway is likely rare in the black band layer, allowing the accumulation of sulfide during anaerobic phases of the diel cycle (Glas et al., 2012).
While few members of the disease consortium may be capable of oxidizing sulfide, it is possible that sulfide is detoxified through the action of sulfide:quinone oxidoreductase (sqr) genes. Each of the Roseofilum MAGs contains two unique copies of sqr, with the exception of the less complete Florida MAG (LKpool) that has only one assembled copy (Figure 4). The Roseofilum MAGs contain both Type VI and Type II sqr genes, which are adapted to high sulfide concentrations and low sulfide affinity, respectively (Marcia et al., 2010). The detoxification of sulfide appears to be a common feature in genomes of filamentous cyanobacteria that form mats, as this lifestyle promotes the growth of sulfate reducers and the sulfide they produce inhibits photosynthesis through the irreversible blockage of Photosystem I (Voorhies et al., 2012). The MAG of the Geitlerinema strain cultured from BBD (Den Uyl et al., 2016) also contains one copy of a Type VI sqr, that is about 58% similar to the Type VI sqr gene in the Caribbean Roseofilum MAGs (Figure 4). Neither Roseofilum nor Geitlerinema from black band contained the third type of sqr found in Cyanobacteria, Type I that has a high sulfide affinity and is used in anoxygenic photosynthesis in the filamentous cyanobacterium Geitlerinema sp. PCC 9228 (Grim and Dick, 2016). Single copies of sqr were also found in the unclassified gammaproteobacterium from the Roseofilum culture (IMG gene id 2628135161), in Bacteroidetes MAGs from Florida (2628091167), Belize (2628117824), and Guam (2628109795), and in the Rhodospirillales MAG from Belize (2628122119), indicating that the detoxification of sulfide is a common feature in members of the BBD polymicrobial consortium.
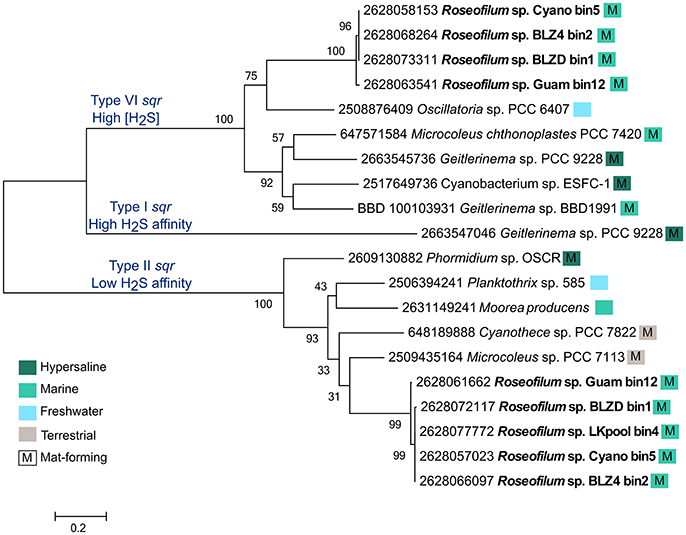
Figure 4. Maximum likelihood tree of sulfide:quinone oxidoreductase (sqr) genes from filamentous cyanobacteria. The five metagenome-assembled genomes of Roseofilum contained both Type VI and Type II sqr genes and are compared to sqr genes from other Oscillatoriales genomes. Branch labels are preceded by the IMG gene id and colored boxes indicate the habitat from which the cyanobacteria were isolated and whether or not it forms mats, based on publicly available metadata.
Metabolism of DMSP (dimethylsulfoniopropionate) has been implicated as an important process in the marine sulfur cycle, particularly in coral reefs, where DMSP functions as an osmolyte and is rapidly degraded by bacteria in the coral surface microbiome (Raina et al., 2009). No genes in the five metagenomes were annotated as DMSP lyases, which may reflect either the use of alternative pathways for DMSP degradation or the replacement of native coral commensals that would normally degrade DMSP with bacteria that are opportunistic generalists, lacking specialized adaptations to the coral epimicrobiome niche. In the light of extensive coral tissue death beneath the cyanobacterial mat and the lack of DMSP production genes in Roseofilum, it is unlikely that significant quantities of DMSP would be available in the black band layer, though this has not been directly measured.
Production of Secondary Metabolites by Black Band Disease Consortia
Cyanobacteria are known for their ability to produce diverse natural products using non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) pathways, including toxins and siderophores (Leão et al., 2012; Shih et al., 2015). At least four unique NRPS/PKS biosynthetic clusters were detected per Roseofilum MAG, along with biosynthetic clusters for terpenes, bacteriocins, and cyanobactins (Figure 5). The abundance of biosynthetic clusters in the Roseofilum MAGs is consistent with the distribution of biosynthetic clusters in cyanobacterial genomes, as uncovered in a recent large-scale comparative genomics study (Shih et al., 2015). Natural products from marine cyanobacteria have been extensively explored for bioactive properties in the development of anti-inflammatory and anti-cancer drugs (Nunnery et al., 2010), though the intended purpose of these compounds from the perspective of cyanobacteria is potentially as grazing deterrents (Nagle and Paul, 1999), for UV protection (Sorrels et al., 2009) and allelopathy (Leão et al., 2012), or for interference in bacterial communication (quorum sensing inhibition; Dobretsov et al., 2010; Meyer et al., 2016). The Roseofilum MAGs contained the highest number of biosynthetic clusters, followed by Bacteroidetes MAGs, and the fewest clusters were detected in Proteobacteria MAGs. Two of the Proteobacteria MAGs (Desulfovibrio from the Belize Orbicella and Vibrio from the Guam Goniopora) had no detectable biosynthetic clusters for secondary metabolites. The role of secondary metabolites in the establishment and persistence of BBD consortia is an area of ongoing active research.
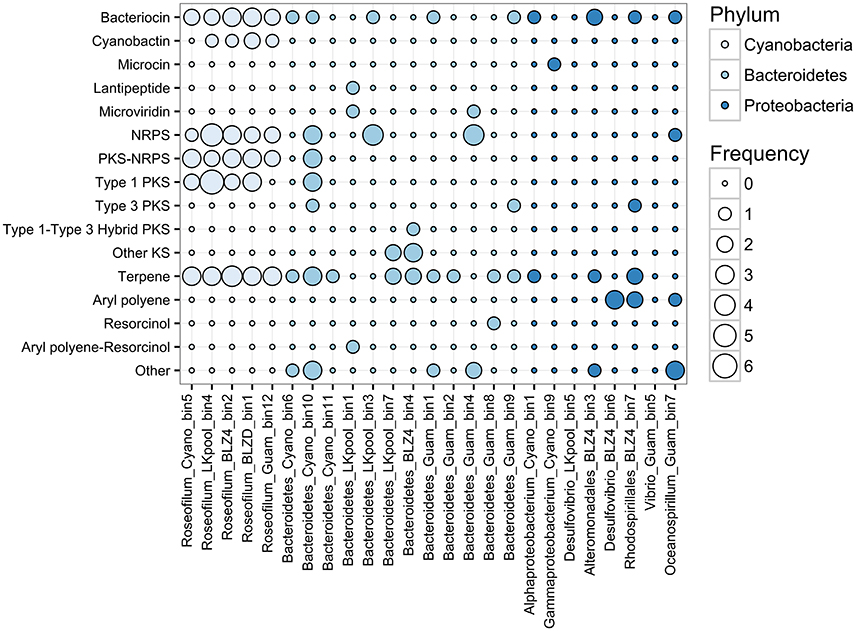
Figure 5. Frequency of antibiotic and secondary metabolite biosynthetic clusters in binned genomes from Black Band Disease consortia. Biosynthetic clusters were identified with AntiSMASH. Each column represents one metagenome-assembled genome and bubbles represent the relative abundance of biosynthetic clusters. NRPS, Non-ribosomal peptide synthase; PKS, polyketide synthase; Other KS, PKS other than type 1, 2, 3, or trans-AT.
Conclusions
The functional repertoire of BBD microbial communities highlights the role of the cyanobacterium R. reptotaenium as the engineer of the disease consortium through the production of organic carbon and nitrogen, as well as novel secondary metabolites. The Roseofilum MAGs recovered here from five different coral species and two different oceans contain a suite of both ribosome-dependent and non-ribosomal peptide and polyketide biosynthetic clusters, at copy numbers similar to or greater than other filamentous cyanobacteria (Shih et al., 2015). These secondary metabolites may be the key to understanding microbe-microbe interactions in the BBD microbial community, such as previously demonstrated for the role of lyngbic acid in inhibiting QS in Vibrio species (Meyer et al., 2016).
The genomic content of Bacteroidetes and Proteobacteria in BBD consortia suggests that heterotrophs fill the role of degrading and recycling organic matter within the mat, fueling the continued growth of Roseofilum. This dependence of Roseofilum on heterotrophic partners is consistent with the fact that filamentous cyanobacteria cannot be easily cultured axenically. However, the relationship between Roseofilum and its heterotrophic partners is not exclusively beneficial. For example, despite its potential for sulfide detoxification, Roseofilum migrates away from the build-up of sulfide within in situ BBD mats (Glas et al., 2012). In aerated cultures where Desulfovibrio is absent (and therefore not creating sulfide), Roseofilum can easily be overwhelmed by heterotrophic growth if the culture medium is too rich and must be transferred regularly to maintain active growth (personal observation). A similar accumulation of heterotrophic cells over time was documented in two different unicyanobacterial cultures of mats from Hot Lake, Washington (Cole et al., 2014).
The conservation of taxonomic groups in the minimal saltwater culture strongly suggests that once the disease consortium is established, the BBD microbial mat does not require the coral host tissue to thrive. This suggests that the pathogenesis of Roseofilum and other disease consortium members is contextual and has important implications for the mitigation of BBD in coral reef systems, primarily that if the conditions initiating the establishment of the microbial mat can be prevented, the disease can be thwarted.
Author Contributions
MT and VP conceived the project. MT, VP, and LR collected samples. JM performed the culturing and molecular work. All authors analyzed data and wrote the manuscript.
Funding
JM is supported by the L'Oréal USA for Women in Science Fellowship and MT by the George E. Burch Fellowship in Theoretic Medicine and Affiliated Sciences from the Smithsonian Institution. This research was supported by Mote Marine Laboratory Protect Our Reefs grants POR 2012-1, POR 2013-2, and POR 2014-10 (MT, and VP).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank E. Bartels, C. Walters, and C. Thacker for assistance with sample collection and L. J. Houk for assistance in cultivation of Roseofilum. We thank the Florida Keys National Marine Sanctuary (FKNMS-2013-023), the Belize Fisheries Department, and the Guam Department of Agriculture's Division of Aquatic and Wildlife Resources for permits to collect BBD. This is contribution no. 996 of the Smithsonian Caribbean Coral Reef Ecosystems Program and contribution no. 1060 of the Smithsonian Marine Station.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00618/full#supplementary-material
Figure S1. Comparison of (A) the coverage (number of sequencing reads mapped to assembled metagenomes) of functional genes assigned to phyla in the non-axenic Roseofilum culture and four Black Band Disease mats and (B) the relative abundance of 16S amplicon sequences assigned to major bacterial phyla in the same samples.
Figure S2. NMDS plot of the Bray-Curtis similarity of functional profiles based on the presence or absence of genes assigned KEGG Orthology numbers in 26 binned genomes. The phylum of each metagenome-assembled genome (MAG) is indicated by color and the metagenome origin of each MAG is indicated by the symbol.
Figure S3. Pairwise dot-plot comparisons of the contigs in Roseofilum metagenome-assembled genomes demonstrates high gene synteny among the four Caribbean-based samples and greater genome rearrangements in the Guam sample.
Table S1. Summary of sample characteristics for 54 coral surface microbiomes and 1 Roseofilum culture used to assess bacterial community structure based on the V6 hypervariable region of 16S rRNA genes.
Table S2. Characteristics of metagenome assemblies from Black Band Disease mats and cultured Roseofilum reptotaenium.
Table S3. Phylogenetic distribution of genes in metagenome assemblies from Black Band Disease mats and cultured Roseofilum reptotaenium.
Table S4. Differential represented SEED subsystems (Level 3) functions between three bacterial phyla in Black Band Disease metagenome-assembled genomes.
Table S5. Genes associated with nitrogen and sulfur cycling in Black Band Disease metagenome-assembled genomes.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Arotsker, L., Kramarsky-Winter, E., Ben-Dov, E., and Kushmaro, A. (2016). Microbial transcriptome profiling of black band disease in a Faviid coral during a seasonal disease peak. Dis. Aquat. Org. 118, 77–89. doi: 10.3354/dao02952
Arotsker, L., Siboni, N., Ben-Dov, E., Kramarsky-Winter, E., Loya, Y., and Kushmaro, A. (2009). Vibrio sp. as a potentially important member of the Black Band Disease (BBD) consortium in Favia sp. corals. FEMS Microbiol. Ecol. 70, 515–524. doi: 10.1111/j.1574-6941.2009.00770.x
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bolhuis, H., Cretoiu, M. S., and Stal, L. J. (2014). Molecular ecology of microbial mats. FEMS Microbiol. Ecol. 90, 335–350. doi: 10.1111/1574-6941.12408
Bourne, D. G., Muirhead, A., and Sato, Y. (2011). Changes in sulfate-reducing bacterial populations during the onset of black band disease. ISME J. 5, 559–564. doi: 10.1038/ismej.2010.143
Bourne, D. G., van der Zee, M. J. J., Botté, E. S., and Sato, Y. (2013). Sulfur-oxidizing bacterial populations within cyanobacterial dominated coral disease lesions. Environ. Microbiol Rep. 5, 518–524. doi: 10.1111/1758-2229.12055
Brocke, H. J., Polerecky, L., de Beer, D., Weber, M., Claudet, J., and Nugues, M. M. (2015). Organic matter degradation drives benthic cyanobacterial mat abundance on Caribbean coral reefs. PLoS ONE 10:e0125445. doi: 10.1371/journal.pone.0125445
Brownell, A. C., and Richardson, L. L. (2014). Sulfate reducing bacteria as secondary and necessary pathogens in black band disease of corals. Rev. Biol. Trop. 62, 249–257.
Buerger, P., Wood-Charlson, E. M., Weynberg, K. D., Willis, B. L., and van Oppen, M. J. H. (2016). CRISPR-Cas defense system and potential prophages in Cyanobacteria associated with the coral Black Band Disease. Front. Microbiol. 7:2077. doi: 10.3389/fmicb.2016.02077
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Casamatta, D., Stanic, D., Gantar, M., and Richardson, L. L. (2012). Characterization of Roseofilum reptotaenium (Oscillatoriales, Cyanobacteria) gen. et sp nov isolated from Caribbean black band disease. Phycologia 51, 489–499. doi: 10.2216/11-10.1
Cole, J. K., Hutchison, J. R., Renslow, R. S., Kim, Y. M., Chrisler, W. B., Engelmann, H. E., et al. (2014). Phototrophic biofilm assembly in microbial-mat-derived unicyanobacterial consortia: model systems for the study of autotroph-heterotroph interactions. Front. Microbiol. 5:109. doi: 10.3389/fmicb.2014.00109
Delmont, T. O., and Eren, A. M. (2016). Identifying contamination with advanced visualization and analysis practices: metagenomics approaches for eukaryotic genome assemblies. PeerJ 4:e1839. doi: 10.7717/peerj.1839
Den Uyl, P. A., Richardson, L. L., Jain, S., and Dick, G. J. (2016). Unraveling the physiological roles of the cyanobacterium Geitlerinema sp. BBD and other Black Band Disease community members through genomic analysis of a mixed culture. PLoS ONE 11:e0157953. doi: 10.1371/journal.pone.0157953.
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton, A. P., et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10:R85. doi: 10.1186/gb-2009-10-8-r85
Dobretsov, S., Teplitski, M., Alagely, A., Gunasekera, S. P., and Paul, V. J. (2010). Malyngolide from the cyanobacterium Lyngbya majuscula interferes with quorum sensing circuitry. Environ. Microbiol. Rep. 2, 739–744. doi: 10.1111/j.1758-2229.2010.00169.x
Echenique-Subiabre, I., Villeneuve, A., Golubic, S., Turquet, J., Humbert, J.-F., and Gugger, M. (2015). Influence of local and global environmental parameters on the composition of cyanobacterial mats in a tropical lagoon. Microb. Ecol. 69, 234–244. doi: 10.1007/s00248-014-0496-0
Eren, A. M., Esen, Ö. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Eren, A. M., Vineis, J. H., Morrison, H. G., and Sogin, M. L. (2013). A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS ONE 8:e66643. doi: 10.1371/journal.pone.0066643
Fernández-Gómez, B., Richter, M., Schüler, M., Pinhassi, J., Acinas, S. G., González, J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Frias-Lopez, J., Klaus, J. S., Bonheyo, G. T., and Fouke, B. W. (2004). Bacterial community associated with black band disease in corals. Appl. Environ. Microbiol. 70, 5955–5962. doi: 10.1128/AEM.70.10.5955-5962.2004
Glas, M. S., Sato, Y., Ulstrup, K. E., and Bourne, D. G. (2012). Biogeochemical conditions determine virulence of black band disease in corals. ISME J. 6, 1526–1534. doi: 10.1038/ismej.2012.2
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grim, S. L., and Dick, G. J. (2016). Photosynthetic versatility in the genome of Geitlerinema sp. PCC 9228 (Formerly Oscillatoria limentica ‘Solar Lake’), a model anoxygneic photosynthetic cyanobacterium. Front. Microbiol. 7:1546. doi: 10.3389/fmicb.2016.01546
Guidi-Rotani, C., Jean, M. R. N., Gonzalez-Rizzo, S., Bolte-Kluge, S., and Gros, O. (2014). Description of new filamentous toxic Cyanobacteria (Oscillatoriales) colonizing the sulfidic periphyton mat in marin mangroves. FEMS Microbiol. Lett. 359, 173–181. doi: 10.1111/1574-6968.12551
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Joshi, N., and Fass, J. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33). [software]. Available online at: https://github.com/najoshi/sickle
Kuehl, K., Jones, R., Gibbs, D., and Richardson, L. (2011). The roles of temperature and light in black band disease (BBD) progression on corals of the genus Diploria in Bermuda. J. Invertebr. Pathol. 106, 366–370. doi: 10.1016/j.jip.2010.12.012
Kuta, K. G., and Richardson, L. L. (2002). Ecological aspects of black band disease of corals: relationships between disease incidence and environmental factors. Coral Reefs 21, 393–398. doi: 10.1038/nmeth.1923
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leão, P. N., Engene, N., Antunes, A., Gerwick, W. H., and Vasconcelos, V. (2012). The chemical ecology of cyanobacteria. Nat. Prod. Rep. 29, 372–391. doi: 10.1038/nmeth.1923
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Marcia, M., Ermler, U., Peng, G., and Hartmut, M. (2010). A new structure-based classification of sulfide:quinone oxidoreductases. Proteins 78, 1073–1083. doi: 10.1002/prot.22665
Markowitz, V. M., Korzeniewski, F., Palaniappan, K., Szeto, E., Werner, G., Padki, A., et al. (2006). The integrated microbial genomes (IMG) system. Nucleic Acids Res. 34, D344–D348. doi: 10.1002/prot.22665
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17, 10–12. doi: 10.14806/ej.17.1.200
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Meyer, J. L., Gunasekera, S. P., Scott, R. M., Paul, V. J., and Teplitski, M. (2016). Microbiome shifts and the inhibition of quorum sensing by Black Band Disease cyanobacteria. ISME J. 10, 1204–1216. doi: 10.1038/ismej.2015.184
Miller, A. W., and Richardson, L. L. (2011). A meta-analysis of 16S rRNA gene clone libraries from the polymicrobial black band disease of corals. FEMS Microbiol. Ecol. 75, 231–241. doi: 10.1111/j.1574-6941.2010.00991.x
Nagle, D. G., and Paul, V. J. (1999). Production of secondary metabolites by filamentous tropical marine cyanobacteria: ecological functions of the compounds. J. Phycol. 35, 1412–1421. doi: 10.1046/j.1529-8817.1999.3561412.x
Nübel, U., Garcia-Pichel, F., and Muyzer, G. (1997). PCR primers to amplify 16S rRNA genes from Cyanobacteria. Appl. Environ. Microb. 63, 3327–3332.
Nunnery, J. K., Mevers, E., and Gerwick, W. H. (2010). Biologically active secondary metabolites from marine cyanobacteria. Curr. Opin. Biotech. 21, 787–793. doi: 10.1016/j.copbio.2010.09.019
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Peng, Y., Leung, H. C., Yiu, S. M., and Chin, F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Raina, J. B., Tapiolas, D., Willis, B. L., and Bourne, D. G. (2009). Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 75, 3492–3501. doi: 10.1128/AEM.02567-08
Richardson, L. L., Miller, A. W., Broderick, E., Kaczmarsky, L., Gantar, M., Stanic, D., et al. (2009). Sulfide, microcystin, and the etiology of black band disease. Dis. Aquat. Org. 87, 79–90. doi: 10.3354/dao02083.
Richardson, L. L., Stanic, D., May, A., Brownell, A., Gantar, M., and Campagna, S. R. (2014). Ecology and physiology of the pathogenic cyanobacterium Roseofilum reptotaenium. Life 4, 968–987. doi: 10.3390/life4040968
Rideout, J. R., He, Y., Navas-Molina, J. A., Walters, W. A., Ursell, L. K., Gibbons, S. M., et al. (2014). Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2:e545. doi: 10.7717/peerj.545
Rodriguez, R. L., and Konstantinidis, K. T. (2014). Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30, 629–635. doi: 10.1093/bioinformatics/btt584
Ruvindy, R., White, R. A., Neilan, B. A., and Burns, B. P. (2016). Unravelling core microbial metabolisms in the hypersaline microbial mats of Shark Bay using high-throughput metagenomics. ISME J. 10, 183–196. doi: 10.1038/ismej.2015.87
Sato, Y., Bourne, D. G., and Willis, B. L. (2011). Effects of temperature and light on the progression of black band disease on the reef coral, Montipora hispida. Coral Reefs 30, 753–761. doi: 10.1007/s00338-011-0751-5
Sato, Y., Civiello, M., Bell, S. C., Willis, B. L., and Bourne, D. G. (2016). Integrated approach to understanding the onset and pathogenesis of black band disease in corals. Environ. Microbiol. 18, 752–765. doi: 10.1111/1462-2920.13122
Shih, P. M., Wu, D., Latifi, A., Axen, S. D., Fewer, D. P., Talla, E., et al. (2015). Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, 1053–1058. doi: 10.1073/pnas.1217107110
Sorrels, C. M., Proteau, P. J., and Gerwick, W. H. (2009). Organization, evolution, and expression analysis of the biosynthetic gene cluster for scytonemin, a cyanobacterial UV-absorbing pigment. Appl. Environ. Microb. 75, 4861–4869. doi: 10.1128/AEM.02508-08
van Dongen, S., and Abreu-Goodger, C. (2012). Using MCL to extract clusters from networks. Methods Mol. Biol. 804, 281–295. doi: 10.1007/978-1-61779-361-5_15
Voorhies, A. A., Biddanda, B. A., Kendall, S. T., Jain, S., Marcus, D. N., Nold, S. C., et al. (2012). Cyanobacterial life at low O2: community genomics and function reveal metabolic versatility and extremely low diversity in a Great Lakes sinkhole mat. Geobiology 10, 250–267. doi: 10.1111/j.1472-4669.2012.00322.x
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Weil, E., Smith, G., and Gil-Agudelo, D. L. (2006). Status and progress in coral reef disease research. Dis. Aquat. Org. 69, 1–7. doi: 10.3354/dao069001
Keywords: coral microbiology, coral disease, cyanobacteria, microbiome, Roseofilum reptotaenium
Citation: Meyer JL, Paul VJ, Raymundo LJ and Teplitski M (2017) Comparative Metagenomics of the Polymicrobial Black Band Disease of Corals. Front. Microbiol. 8:618. doi: 10.3389/fmicb.2017.00618
Received: 27 December 2016; Accepted: 27 March 2017;
Published: 18 April 2017.
Edited by:
Télesphore Sime-Ngando, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Fabiano Thompson, Federal University of Rio de Janeiro, BrazilTom O. Delmont, University of Chicago, USA
Copyright © 2017 Meyer, Paul, Raymundo and Teplitski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie L. Meyer, anVsaWVtZXllckB1ZmwuZWR1