 Xian Zhang1,2
Xian Zhang1,2 Xueduan Liu1,2
Xueduan Liu1,2 Yili Liang1,2
Yili Liang1,2 Yunhua Xiao1Liyuan Ma1Xue Guo1Bo Miao1,2
Yunhua Xiao1Liyuan Ma1Xue Guo1Bo Miao1,2 Hongwei Liu1,2Deliang Peng3Wenkun Huang3
Hongwei Liu1,2Deliang Peng3Wenkun Huang3 Huaqun Yin1,2*
Huaqun Yin1,2*- 1School of Minerals Processing and Bioengineering, Central South University, Changsha, China
- 2Key Laboratory of Biometallurgy of Ministry of Education, Central South University, Changsha, China
- 3State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
The spatial-temporal distribution of populations in various econiches is thought to be potentially related to individual differences in the utilization of nutrients or other resources, but their functional roles in the microbial communities remain elusive. We compared differentiation in gene repertoire and metabolic profiles, with a focus on the potential functional traits of three commonly recognized members (Acidithiobacillus caldus, Leptospirillum ferriphilum, and Sulfobacillus thermosulfidooxidans) in bioleaching heaps. Comparative genomics revealed that intra-species divergence might be driven by horizontal gene transfer. These co-occurring bacteria shared a few homologous genes, which significantly suggested the genomic differences between these organisms. Notably, relatively more genes assigned to the Clusters of Orthologous Groups category [G] (carbohydrate transport and metabolism) were identified in Sulfobacillus thermosulfidooxidans compared to the two other species, which probably indicated their mixotrophic capabilities that assimilate both organic and inorganic forms of carbon. Further inspection revealed distinctive metabolic capabilities involving carbon assimilation, nitrogen uptake, and iron-sulfur cycling, providing robust evidence for functional differences with respect to nutrient utilization. Therefore, we proposed that the mutual compensation of functionalities among these co-occurring organisms might provide a selective advantage for efficiently utilizing the limited resources in their habitats. Furthermore, it might be favorable to chemoautotrophs' lifestyles to form mutualistic interactions with these heterotrophic and/or mixotrophic acidophiles, whereby the latter could degrade organic compounds to effectively detoxify the environments. Collectively, the findings shed light on the genetic traits and potential metabolic activities of these organisms, and enable us to make some inferences about genomic and functional differences that might allow them to co-exist.
Introduction
Unraveling the ecological and functional roles of microorganisms in biological communities is an important but still elusive issue (Prosser et al., 2007), even though these microbes are thought to be crucial to the function ecosystems (Harris, 2009; Jiao et al., 2010; Hua et al., 2015). As stated by Sogin et al. (2006), there is a surprisingly wide biodiversity of microbial communities in pristine environments. In their study, the dominate populations are numerically significant, but the members of the rare biosphere account for the majority of the phylogenetic diversity. Similar results were generally observed in other natural and anthropogenic environments based on metagenomic and metatranscriptomic analyses (Chen et al., 2015; Goltsman et al., 2015; Xiao et al., 2016; Zhang et al., 2016d). Genomes of microbial members in various communities have been reconstructed with the benefit of cultivation-independent sequencing (Tyson et al., 2004; Mason et al., 2012; Wu et al., 2016), providing a first glimpse of their functional roles in situ. Additionally, several bioinformatics-based strategies have been attempted to obtain genomic assemblies from metagenomic datasets (Dick et al., 2009; Hua et al., 2015). Considerable efforts have been made to expand the scope of microbial genetics and ecophysiology on a global scale; however, relatively little is known about how these populations co-exist within the same microbial community.
Acidophilic microorganisms are widely distributed in both pristine environments (e.g., acid rock drainage and volcanic, geothermal areas) and acidic environments of anthropogenic origin (e.g., acid mine drainage and bioleaching heaps, Baker and Banfield, 2003; Denef et al., 2010; Bonnefoy and Holmes, 2012; Zhang et al., 2016c,d). Many acidophiles that are metabolically active in anthropogenic environments are obligate chemolithoautotrophs capable of assimilating atmospheric CO2 and deriving energy from the aerobic oxidation of ferrous iron and/or a variety of sulfur species (Hallberg and Johnson, 2001; Rawlings, 2005; Johnson and Hallberg, 2008). In addition, heterotrophs that assimilate organic carbon and mixotrophs that utilize both organic and inorganic forms of carbon are found in these acidic settings (Johnson and Hallberg, 2003).
Until recently, numerous pieces of evidence suggested that Acidithiobacillus and Leptospirillum are considered to be the common inhabitants of acidophilic, metal-tolerant microbial consortia in many sulfide-rich mining environments (Kock and Schippers, 2006, 2008; Breuker et al., 2009; Chen et al., 2015; Xiao et al., 2016; Zhang et al., 2016d). Species of Acidithiobacillus isolates are demonstrated iron- and/or sulfur-oxidizing acidophiles that are phylogenetically affiliated with the class Acidithiobacillia (Williams and Kelly, 2013), which mainly includes A. ferrooxidans (Valdés et al., 2008), A. thiooxidans (Yin et al., 2014), A. caldus (Valdes et al., 2009), and A. ferrivorans (Liljeqvist et al., 2011). Four recognized members of iron-oxidizing Leptospirillum bacteria (Zhang et al., 2016c), including Group I (L. ferrooxidans, Fujimura et al., 2012), Group II (L. rubarum and L. ferriphilum, Jiang et al., 2015), Group III (L. ferrodiazotrophum), and Group IV (Goltsman et al., 2013), have been documented. In contrast to these obligate chemolithoautotrophs, heterotrophic and/or mixotrophic acidophiles, such as Sulfobacillus spp., have been also characterized from these environments (Guo et al., 2014; Justice et al., 2014). Despite relatively poor understanding of the moderately thermophilic Sulfobacillus, it was believed to play an important role in the biogeochemical cycle of sulfur (Justice et al., 2014). Several isolated Sulfobacillus species, including S. benefaciens (Johnson et al., 2008), S. thermosulfidooxidans (Justice et al., 2014), S. sibiricus (Melamud et al., 2003), and S. thermotolerans (Bogdanova et al., 2006), have been used to demonstrate their key metabolic features.
Interactions occur ubiquitously among co-existing microbes in laboratory cultures and natural environments (Li and Gu, 2007; Summers et al., 2010; Wintermute and Silver, 2010; Gupta and Schuster, 2013; Yin et al., 2015). Recently, a striking example of an inter-species interaction was found between Arthrobacter sp. and Sphingopyxis sp. in a co-culture (Liang et al., 2017) where Mn(II)-oxidizing activity was presumed to be triggered by contact-dependent interactions of two investigated bacteria, although neither of them has the ability to oxidize Mn(II). As in other environments, microbial interactions in mixed communities of acidic environments were also described (Johnson, 1998; Baker and Banfield, 2003). The spatial-temporal distribution of different populations might be correlated with the differences in environmental conditions of individual ecological niches, and in nutrients or in other resources that they could utilize (Yelton et al., 2013). Thus, it is important to investigate how these physiologically different acidophilic microorganisms interact, thereby allowing them to co-exist.
Here, we present the detailed analyses of gene repertoire, metabolic features, and potential functional roles of three acidophilic species, including Acidithiobacillus caldus, Sulfobacillus thermosulfidooxidans, and Leptospirillum ferriphilum, which were isolated from disparate bioleaching heaps located in Dexing Copper Mine (Jiangxi Province, China) and Zijinshan Copper Mine (Fujian Province, China), respectively. A comparative survey based on the bacterial genomes was performed to delineate the genomic and functional differences among these co-occurring acidophiles that potentially contribute to a mutualistic relationship rather than competitive exclusion.
Materials and Methods
Sampling, DNA Extraction, Genome Sequencing, and Assembly
Samples were collected from mine tailings heaps in Dexing Copper Mine (Jiangxi, China) and Zijinshan Copper Mine (Fujian, China). Leaching solution that harbored plentiful microorganisms was pumped from leaching pools and sprayed on the leaching heaps periodically in the process of industrial bioleaching operations. Environmental attributes of these two sampling sites have been elaborated on in earlier studies (Yin et al., 2008; Xiao et al., 2016). All bioleaching samples were repeatedly washed with distilled water (pH 2.0), and then they were filtered through the filter membrane with a 0.22-μm pore-size as described earlier (Zhang et al., 2016d). Gradient dilution was employed to isolate the pure bacteria from these environmental samples according to individual growth conditions of targeted isolates. In general, all of the strains were grown in liquid 9K medium on a shaking table at 170 rpm. Additional details are as follows: autoclave-sterilized elemental sulfur (10 g/L), 45°C, and pH 2.0 for Acidithiobacillus caldus; and 50 mM of ferrous [Fe(II)], 40°C, and pH 1.5 for Leptospirillum ferriphilum. The culture conditions of S. thermosulfidooxidans were previously documented (Zhang et al., 2017).
Strains were cultivated aerobically at specific conditions as above. Bacterial cells were harvested at the stationary phase by centrifugation (12,000 g) for 10 min at 4°C. Genomic DNA was extracted using a TIANamp Bacteria DNA Kit (Tiangen, China) following the manufacturer's instructions. The Illumina paired-end libraries with an average of 300 bp inserts were prepared from bacterial genomes and sequenced using the Illumina MiSeq sequencer (Illumina, San Diego, USA). The NGS QC Toolkit v2.3.1 (Patel and Jain, 2012) was used to screen the high-quality (HQ) read pairs with the following parameters: the cut-off read length for HQ was 70% and the cut-off quality score was 20. HQ filtered reads were then de novo assembled using Velvet (Zerbino and Birney, 2008) with various k-mers, and the best resulting assembly was chosen based on contiguity statistics. Finally, the completeness of the newly assembled genomes presented here was evaluated by the CheckM package (Parks et al., 2015).
Taxonomic and Functional Analysis
The 16S ribosomal RNA (rRNA) gene sequences dispersed in new genome assemblies were obtained using RNAmmer (Lagesen et al., 2007) in order to determine the bacterial phylogeny. Similar sequences were identified using the online BLAST search tool. Multiple sequence alignment for the complete 16S rRNA sequences of the novel strains and similar sequences retrieved from GenBank was performed using ClustalX v1.81. The nucleotide pairwise genetic distances were calculated by implementing the Tamura-Nei model of nucleotide substitution. The maximum likelihood tree was constructed from the ClustalX results using MEGA v5.05 (Tamura et al., 2011). The node support was evaluated using 1,000 bootstrap replications. To further infer the phylogenetic relationship, in silico DNA-DNA hybridization was performed using JSpecies v1.2.1 software (Richter and Rosselló-Móra, 2009) by calculating the average nucleotide identity (ANI) based on the BLAST algorithm (ANIb, Goris et al., 2007) and tetranucleotide frequency correlation coefficient (TETRA, Teeling et al., 2004). The ANI calculation was conducted using the default and evaluated parameters: sequence identity (%) ≥ 30%, alignment (%) ≥ 70%, and query length = 1,020 bp. In addition, the default values of –X(150), −q (−1), −F (F), −e (1e−15), and –a (2) were applied for the ANIb calculation.
The prediction of putative protein-coding sequences (CDS) and automatic annotation were achieved using the NCBI Prokaryotic Annotation Pipeline. In-house Perl scripts were used to extract protein sequences from the GenBank files; these sequences were then aligned against the specialized databases, such as the extended Clusters of Orthologous Groups (COG, Franceschini et al., 2013), with a BLASTP algorithm and an E-value cut-off of 1e−5. Visualization was performed using HemI (Deng et al., 2014) for the percentage of CDS assigned to the COG categories. The metabolic potentials of all of the strains were reconstructed using the KEGG Automatic Annotation Server (KAAS) with the BLAST algorithm against the manually curated KEGG GENES database (Moriya et al., 2007). ISFinder (Siguier et al., 2006) was used to identify the putative transposable elements, including transposons and insertion sequences. Additionally, tRNA genes were identified by tRNAscan-SE (Lowe and Eddy, 1997).
Identification of Orthologous Proteins and Comparison of Genome Architectures
A BLASTP all-vs-all comparison of protein sequences extracted from the GenBank files was conducted. BLAST results with a tabular format were used to identify the orthologous groups implementing the program Pan-genome Ortholog Clustering Tool (PanOCT) v3.18 (Fouts et al., 2012). The following parameters were applied: E ≤ 0.001, percent identity ≥ 30, and length of match ≥ 65 bp, reference to a previous study (Zhang and Sievert, 2014). In consideration of the lineage-specific expansions (Carretero-Paulet et al., 2015), the transposable elements were filtered out in this study. Finally, the comparative results were manually checked.
Protein sequences shared by all strains and unique to individual species were extracted from the PanOCT results and they were then aligned against the extended COG as described above. Furthermore, the intra-species divergence was investigated by BLASTN-based whole genome comparisons with the following parameters: E ≤ 1e−5 and sequence identity ≥ 50%. Circular visualization of genomic data was performed using Circos software (Krzywinski et al., 2009).
Availability of Supporting Data
The Whole Genome Shotgun projects have been deposited at DDBJ/ENA/GenBank under the accession MPOJ00000000 (Leptospirillum ferriphilum DX) and MPOK00000000 (L. ferriphilum ZJ). The versions described in this paper are version MPOJ01000000 and MPOK01000000. Additionally, the data sets regarding Acidithiobacillus caldus DX (LZYE00000000), A. caldus ZJ (LZYG00000000), S. thermosulfidooxidans DX (MDZD00000000), and S. thermosulfidooxidans ZJ (MDZF00000000) are available in the NCBI repository. The corresponding versions described in this article are version LZYE02000000, LZYG02000000, MDZD02000000, and MDZF02000000, respectively.
Results
Phylogeny of Newly Sequenced Strains and Overall Genome Statistics
Two novel strains in this study were isolated from Dexing Copper Mine and Zijinshan Copper Mine. There were 16S rRNA gene sequences that were extracted from these two newly sequenced genomes using RNAmmer, and they were then used for the identification of phylogeny. Phylogenetic analysis showed that the novel strains were clustered on a distinct branch within Leptospirillum sp. Group II, but they were obviously distinguished from other Leptospirillum groups, suggesting a close relationship between new isolates and Leptospirillum sp. Group II (Figure 1). Accordingly, eight Leptospirillum genomes that are available in the GenBank database were chosen for phylogenomic analysis (Table 1). The phylogenetic relationship among all putative or recognized Leptospirillum strains was inferred by the comparison of the average nucleotide identity (ANI) based on BLAST (ANIb) and tetranucleotide frequency correlation coefficient (TETRA). The values of ANIb (≥ 97.96%) and TETRA (≥ 0.999) that were evaluated by JSpecies strongly indicated that strains DX and ZJ analyzed in our study were very closely related to L. ferriphilum strains but separate from other Leptospirillum spp. (Table 1), further supporting the prior notion of 16S rRNA gene-based phylogenetic analysis. In addition, these two newly sequenced strains (L. ferriphilum DX and ZJ) shared high values of ANIb (98.56) and TETRA (0.999) with each other (Table 1). Collectively, we inferred that the novel strains DX and ZJ inspected in this study were phylogenetically affiliated with L. ferriphilum.
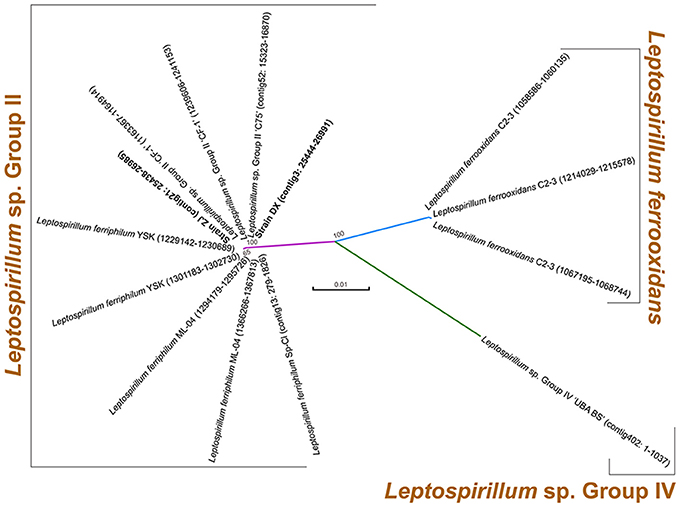
Figure 1. Phylogenetic tree based on 16S rRNA genes of two newly sequenced strains and other known Leptospirillum spp. For each strain, 16S rRNA gene sequence was extracted from complete or draft genome using RNAmmer, and its genomic locus was shown.
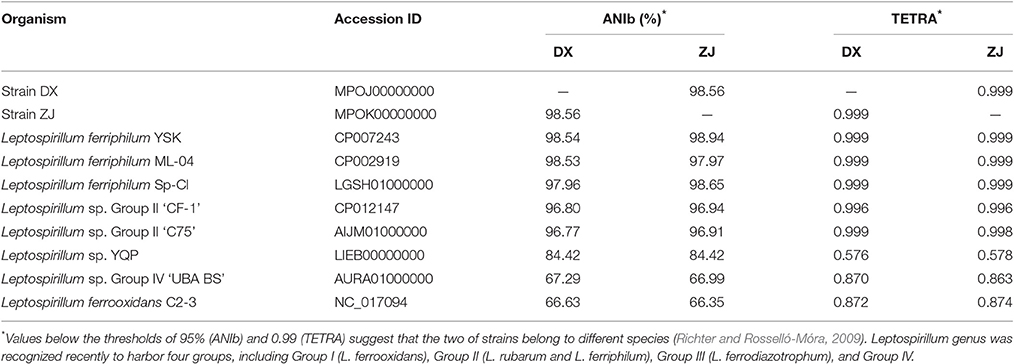
Table 1. Genome-based phylogenetic indicators of two novel strains compared to other recognized Leptospirillum spp.
Genomic features of L. ferriphilum, A. caldus (Zhang et al., 2016b), and S. thermosulfidooxidans (Zhang et al., 2017), the latter two of which were also used as part of this study, are summarized in Table 2. Each genome had between 20 to 396 contigs with coverage ranging from 52 to 165 ×. Genome completeness assessed by CheckM suggested the near-complete genomes of all six strains, although the genome coverage of L. ferriphilum ZJ was relatively low compared to the other strains. A. caldus and S. thermosulfidooxidans had larger genomes (between 3.12 and 3.18 Mbp) than L. ferriphilum (between 2.34 and 2.36 Mbp). A. caldus strains with higher GC contents (DX with 61.01% and ZJ with 61.00%) were observed, while the GC contents in L. ferriphilum and S. thermosulfidooxidans ranged from 48.47 to 54.70%.
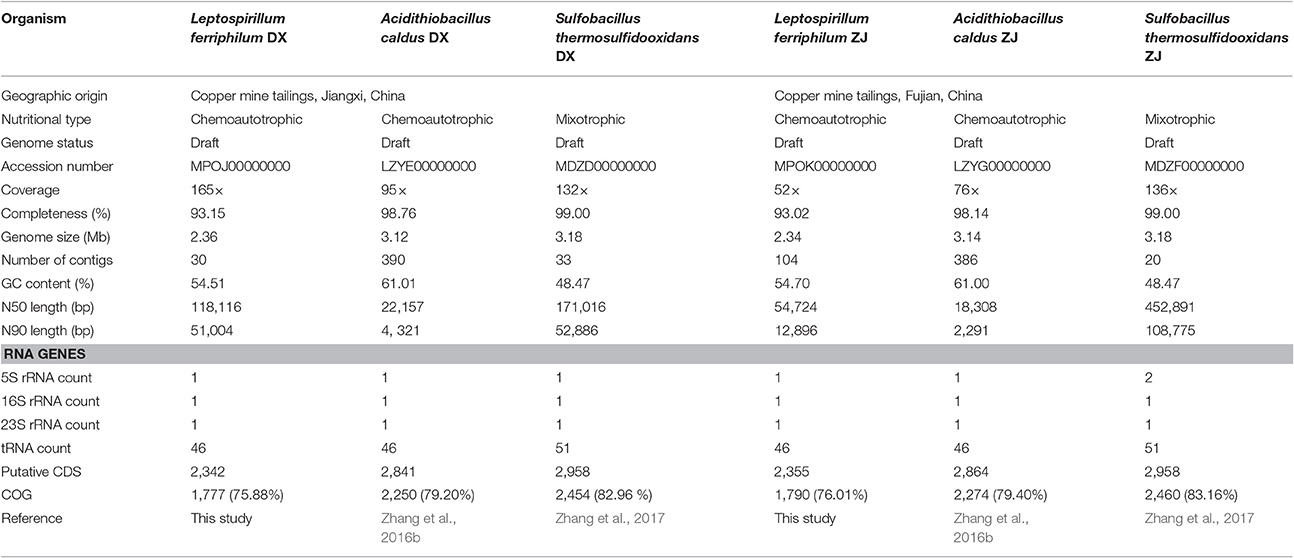
Table 2. Comparison of genomic and phenotypic features of co-occurring bacteria isolated from two distinctive copper mines.
All of the genome assemblies had the full suite of tRNA that covered all of the 20 amino acids. Bacterial genomes were predicted to contain between 2,342 and 2,958 protein-coding sequences (CDS). The Clusters of Orthologous Groups (COG) annotation suggested that between 75.88 and 83.16% of the CDS were matched to putative proteins with known functions (Table 2). Of the 25 COG categories, CDS were assigned to 21 (L. ferriphilum and S. thermosulfidooxidans) or 22 (A. caldus) of them. In addition to COG categories [S] (Function unknown) and [R] (General function prediction only), “Energy production and conversion (C),” “Amino acid transport and metabolism (E),” and “Cell wall/membrane/envelope biogenesis (M)” were commonly abundant in all of the genomes (Figure S1). It is important to note that the proportion of CDS assigned to “carbohydrate transport and metabolism (G)” in S. thermosulfidooxidans (5.17% in DX and 5.21% in ZJ) was relatively large compared to the other two species (between 2.83 and 3.12%; Figure S1).
Identification of Core Genes and Flexible Genes
There were 8,114 CDS that were clustered using the PanOCT with a 30% sequence identity cut-off in order to infer the shared and flexible genes among the tested strains. Comparative analysis showed that 419 putative orthologous genes were shared by all of the strains (Figure 2), representing the low proportions compared to the entire CDS of the individual genome: 17.89% in L. ferriphilum DX, 17.79% in L. ferriphilum ZJ, 14.75% in A. caldus DX, 14.63% in A. caldus ZJ, 14.16% in S. thermosulfidooxidans DX, and 14.16% in S. thermosulfidooxidans ZJ. These shared genes were mainly associated with “translation, ribosomal structure, and biogenesis (J),” “amino acid transport and metabolism (E),” “energy production and conversion (C),” and “nucleotide transport and metabolism (F).” Aside from the core genes that are common in all of the strains, the flexible genes, including genes present in some but not all of the genomes and strain-specific genes unique to the individual genomes are also depicted (Figure 2). Notably, the CDS only shared by each species (1,302 in L. ferriphilum, 2,023 in A. caldus, and 2,262 in S. thermosulfidooxidans) were much more than the core genes, clearly suggesting the genomic difference among these organisms. Of these sequences, the COG classification revealed that many were annotated as hypothetical proteins or proteins with unknown functions. In addition, the CDS involved in the COG categories [M] (cell wall/membrane/envelope biogenesis) and [C] were numerically significant, highlighting the distinguishing features among these species. Similar to the results shown in Figure S1, the percentage of the CDS related to the COG category [G] in the S. thermosulfidooxidans species (5.13%) were larger than that in the others (2.53% in L. ferriphilum and 1.93% in A. caldus). Of genes unique in S. thermosulfidooxidans, many were involved in the biosynthesis of amino acid, such as cysteine, arginine, glycine, as well as alanine (unpublished).
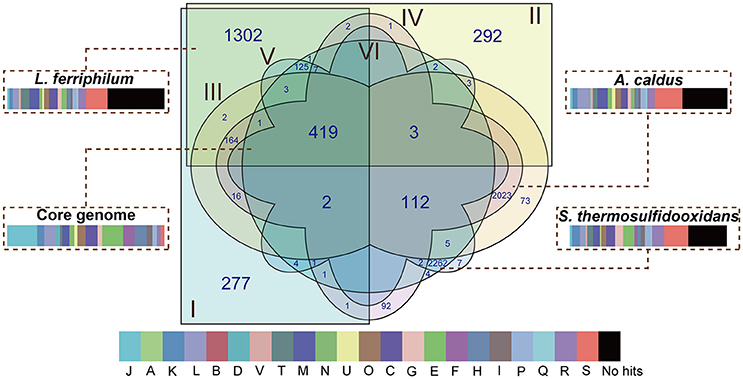
Figure 2. Venn diagram depicting orthologous and non-orthologous genes. L. ferriphilum DX (I), L. ferriphilum ZJ (II), A. caldus DX (III), A. caldus ZJ (IV), S. thermosulfidooxidans DX (V), and S. thermosulfidooxidans ZJ (VI) are shown in different colors. The numbers shown in the Venn diagram indicate the number of orthologous or strain-specific genes. If no number is given in certain patterns, it suggests there is no orthologous protein among these strains. The core genome and unique genes in each species were used for functional analysis based on COG classification. The descriptions of the COG categories are provided in Figure S1. The width of rectangle indicates the percentage of CDS compared to the entire genome of individual strains.
BLASTN-based whole genome comparisons of these targeted strains isolated from distinct areas were performed and visualized using Circos (Figure 3) to acquire some insights into intra-species differences. An examination of the architecture and global structural genomes revealed that a number of non-shared genomic regions were found between the strains belonging to the same species, although they shared a high-sequence identity. Further inspection revealed that many genes distributed in the genomic regions (sections 1–10 in Figure 3) of each L. ferriphilum strain were predicted to encode putative proteins with unclear functions (Table S1). In addition, several mobile genetic elements, such as transposase, integrase, and phage-associated protein, were identified in the genomic neighborhoods. Similar results were also reported in A. caldus (Zhang et al., 2016b) and S. thermosulfidooxidans (Zhang et al., 2017).
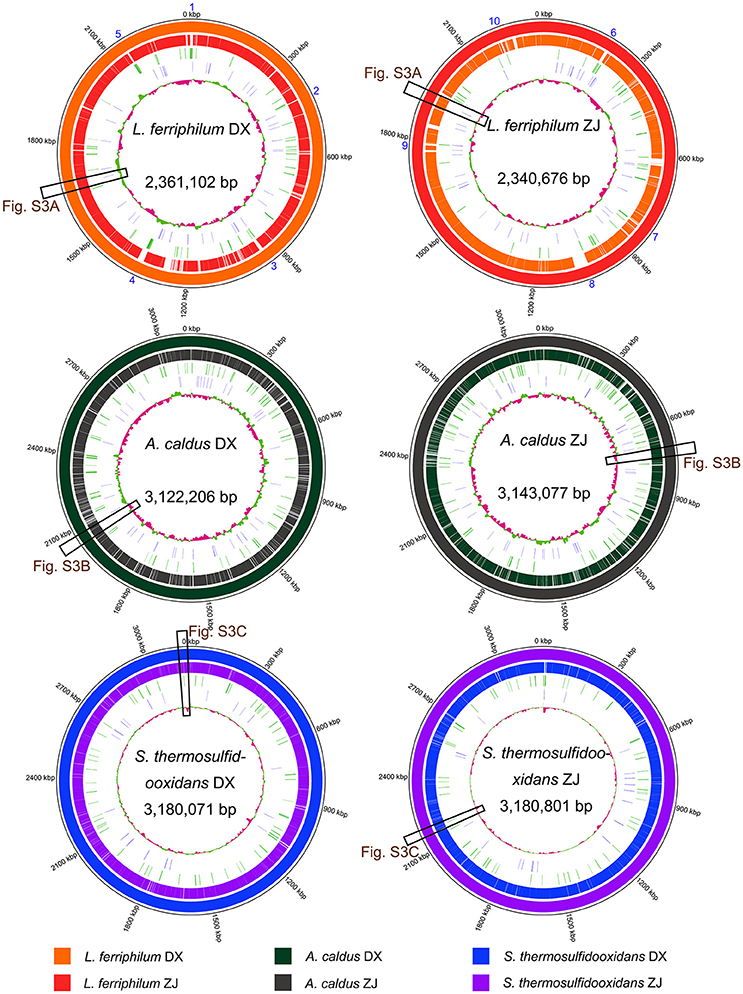
Figure 3. Reference-based whole genome comparisons and visualization of each strain. GC contents of individual genomes are indicated in the center of the figure. In addition, tRNA and transposases are shown on the second and the third rings from the inside. Matches to the reference genomes (≥ 50% sequence identity) are shown with different colors. Cross references showing further analysis of certain genomic regions are linked to other figures. Detailed description for genomic regions 1–10 in L. ferriphilum isolates are shown in Table S1.
Comparison of Inferred Metabolic Profiles
The KAAS online server was employed to investigate the metabolic potential of the microorganisms in this study (Table S2). In general, four types of metabolism having the most abundant genes in all of the strains were: “carbohydrate metabolism,” “amino acid metabolism,” “energy metabolism,” and “metabolism of cofactors and vitamins.” Metabolism-related CDS in S. thermosulfidooxidans with a larger genome (1,124 in DX and 1,125 in ZJ) were more than that in the other two species (between 818 and 885). Furthermore, the key metabolisms of each genome are listed and comparisons are presented in Table 3. In the following section, we discuss the metabolic differences among the species.
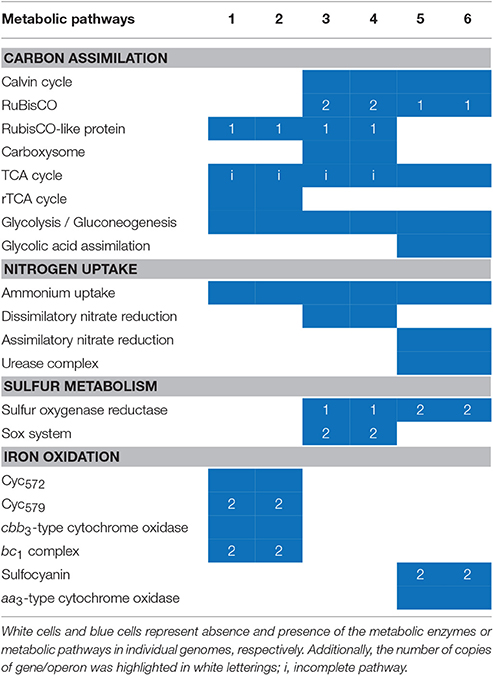
Table 3. Overall comparisons of the sequenced genomes of L. ferriphilum DX (1), L. ferriphilum ZJ (2), A. caldus DX (3), A. caldus ZJ (4), S. thermosulfidooxidans DX (5), and S. thermosulfidooxidans ZJ (6) with reference to key metabolic potentials.
Central Carbon Metabolism
As previously noted by Berg et al. (2010), six autotrophic carbon fixation mechanisms that assimilate CO2 into cellular material have been well-documented, including the Calvin-Benson-Bassham (CBB) cycle, the reductive tricarboxylic acid (rTCA) cycle, the reductive acetyl-coenzyme A pathway, the 3-Hydroxypropionate bicycle, the hydroxypropionate-hydroxybutyrate cycle, and the dicarboxylate-hydroxybutyrate cycle. Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO), one of the key enzymes associated with the first rate-limiting step for the CBB cycle, has been classified into four forms: I-IV (Zhang et al., 2016c). In previous studies, the CBB cycle was considered to be the way to fix carbon in Leptospirillum genus (Coram and Rawlings, 2002). Our present results showed that no gene encoding putative RuBisCO was identified in L. ferriphilum isolates, but non-canonical RuBisCO-like protein was predicted (Table S3). However, L. ferriphilum genomes were predicted to harbor a full suite of genes encoding canonical enzymes, which were involved in the rTCA cycle for carbon fixation. Similar results could be also found in several other reviews and papers (Levicán et al., 2008; Mi et al., 2011; Zhang et al., 2016c). In particular, genes encoding pyruvate ferredoxin oxidoreductase and 2-oxoglutarate ferredoxin oxidoreductase were predicted to exist in a common genome region (Figure S2A). In contrast, A. caldus strains were reported to harbor distinct forms of RuBisCO, and S. thermosulfidooxidans only had type I RuBisCO. For the A. caldus genomes, genes encoding type I RuBisCO were located in a gene cluster involved in carboxysome formation (Figure S2B). Notably, A. caldus isolates were predicted to harbor a carboxysome-associated carbonic anhydrase; however, no genome region related to carboxysome was identified in S. thermosulfidooxidans, despite the existence of a gene encoding the putative carbonic anhydrase.
All of the isolates were also anticipated to share a variety of genes predicted to be involved in the synthesis of lipopolysaccharides, peptidoglycan, and, potentially, exopolysaccharides (Table S3). In all of the isolates, the accumulated 3-phosphoglycerate from carbon fixation was predicted to be converted via glycolysis/gluconeogenesis to produce the major precursors (glucose-6-phosphate and glucose-1-phosphate) for the biosynthesis of cell envelope polysaccharides. In light of an earlier report using “Ferrovum” genomes (Ullrich et al., 2016a), a core set of genes that are potentially associated with amino sugar and nucleotide sugar metabolism (ko00520) were predicted in all of the investigated strains (Table S3). A variety of intermediate metabolites, such as UDP-glucose, UDP-glucuronate, UDP-galactose, and UDP-N-acetyl-glucosamine, within these metabolic processes might be potential precursors for the biosynthesis of cell envelope polysaccharides in all of the strains. Several genes that are potentially involved in the formation of precursors, such as UDP-mannose and UDP-N-acetyl-mannosamine, were only identified in the S. thermosulfidooxidans genomes, while L. ferriphilum and A. caldus were found to share the potential to produce alternative precursors, such as ADP-glucose and UDP-galacturonate. Furthermore, only the A. caldus strains appeared to catalyze GDP-mannose to GDP-4-dehydro-6-deoxy-D-mannose by GDP-mannose 4,6-dehydratase (EC 4.2.1.47).
Leptospirillum ferriphilum and Acidithiobacillus caldus were found to harbor a slightly larger enzyme repertoire related to the aforementioned biosynthesis, suggesting the production of additional macromolecules with species-specific monosaccharide compositions in comparison with S. thermosulfidooxidans. For mechanisms potentially involved in the export of cell envelope polysaccharides, the ABC-2-type polysaccharide transport system was identified in all strains, while the Lpt-type lipopolysaccharide export system was found solely in L. ferriphilum and A. caldus. Compared to the other microorganisms (Haft et al., 2006; Craig et al., 2011; Ullrich et al., 2016a), only L. ferriphilum strains shared the genes predicted to encode the PEP-CTERM exosortase system that was presumed to be involved in the export of exopolysaccharides during biofilm formation. In several earlier studies, biofilm was proposed to provide a reaction space between the sulfide surfaces and bacterial cells, thereby accelerating sulfide mineral dissolution (Watling, 2006; Rohwerder and Sand, 2007; González et al., 2013).
Acidithiobacillus caldus and Leptospirillum ferriphilum were predicted to exhibit an incomplete TCA cycle (Table S3). However, S. thermosulfidooxidans strains harbored a full suite of genes encoding the tricarboxylic acid cycle (TCA), which was thought to be essential for heterotrophic growth (Wood et al., 2004). In addition, S. thermosulfidooxidans strains exhibited the potential ability to degrade glycolic acid, as genes encoding putative glycolate oxidase and malate synthase were found in both genomes (Table S3). In particular, S. thermosulfidooxidans strains were predicted to harbor genomic regions potentially associated with glycolate oxidase (Figure S2C). A pairwise comparison of these gene clusters revealed the identical order and orientation of all of the genes, as well as the nucleotide sequence identities of up to 100%, suggesting that the gene clusters involved in glycolate oxidase might evolve from a common ancestor.
As for comparison and analysis, genome sequences of these six strains were used for the identification of carbohydrate-active enzymes (CAZymes), which represented an enormous number of enzymes that were responsible for the assembly, modification, and breakdown of oligo- and polysaccharides (Lombard et al., 2013). As shown in Table S4, the gene repertoire of potentially encoding CAZymes, including glycoside hydrolases (GHs), glycosyltransferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), auxiliary activities (AAs), and carbohydrate-binding modules (CBMs), were identified in all of the genomes. Apparently, more abundant CAZymes were identified in the S. thermosulfidooxidans genomes (156 in DX and 157 in ZJ) compared to the others (between 80 and 92), especially the GHs that are responsible for the hydrolysis of glycosidic bonds and the CEs that hydrolyze the carbohydrate esters. The most abundant GH classes in the S. thermosulfidooxidans isolates were strongly biased toward the catabolism of oligo- or polysaccharides, such as chitinase (GH18), cellobiohydrolase (GH74), glucoamylase (GH15), and α-amylase (GH13).
Similar to an earlier study (Yelton et al., 2013), another potential organic carbon source for the S. thermosulfidooxidans strains might be the lipids from the lysed cells. They were predicted to harbor a full set of putative proteins involved in fatty acid degradation (Table S3), including long-chain acyl-CoA synthetase, acyl-CoA dehydrogenase, enoyl-CoA hydratase, and acetyl-CoA acyltransferase. Additionally, lactate permease and lactate utilization of protein B/C was identified in the S. thermosulfidooxidans isolates (Table S3), suggesting their heterotrophic growth on lactate.
Nitrogen Uptake
Acidithiobacillus caldus strains harbor the complete genes required for dissimilatory nitrate reduction, which were observed as NarGHJI and nirBD operons (Table S3). Thus, it was hypothesized that the A. caldus strains in this study might utilize nitrate and nitrite as electron acceptors under anaerobic environmental conditions. As for assimilatory nitrate reduction, A. caldus was predicted to harbor narA potentially as an encoding assimilatory nitrate reductase catalytic subunit; however, there was no NasB subunit, which was reported to transfer electrons from NADH to nitrate (Lin and Stewart, 1997), thereby making the electron donor unclear. Additionally, no nirA gene was identified in the A. caldus genomes. In contrast, S. thermosulfidooxidans share the set of genes that encode assimilatory nitrate reduction. We identified the genes encoding the assimilatory nitrate reductase catalytic subunit (NasA) and ferredoxin-nitrite reductase (NirA). Although putative genes potentially encoding dissimilatory nitrite reductase small subunit (NirD) and ferredoxin-nitrite reductase (NirA) were found in the L. ferriphilum genomes, their functional roles need to be studied further. Additionally, all of the strains share the potential to utilize ammonium via the Amt family transporter (Table S3), and to assimilate it into the central metabolic pathways via glutamine synthetase or glutamate synthase. Given that dissimilatory nitrate reduction and assimilatory nitrate reduction were absent in L. ferriphilum, they might utilize ammonium as the sole nitrogen source. In addition, S. thermosulfidooxidans was the only species that contains gene cluster encoding urease subunits (UreABC) and accessory proteins (UreDEFG; Table S3). Accordingly, it seems that S. thermosulfidooxidans could utilize extracellular urea, although there is still no direct evidence showing the existence of these compounds in the bioleaching heaps.
Iron-Sulfur Cycling
Both L. ferriphilum and S. thermosulfidooxidans are demonstrated iron oxidizers (Guo et al., 2014; Justice et al., 2014; Zhang et al., 2016c). These two species were predicted to harbor membrane-associated c-type cytochromes that are potentially involved in iron oxidation. In L. ferriphilum, the electrons might be transferred from an outer-membrane cytochrome c (Cyc572) to a periplasmic cytochrome c (Cyt579), and then across periplasmic cytochromes c either reducing oxygen via cbb3-type terminal oxidase (downhill), or reducing NAD+ passing through the bc1 complex, quinone pool, and NADH dehydrogenase (uphill, Bonnefoy and Holmes, 2012). Similar to Cyt579 in L. ferriphilum, the periplasmic sulfocyanins identified in S. thermosulfidooxidans (Table S3) were postulated to be the branch point at which electrons derived from membrane c-type cytochromes might be channeled either downhill or uphill. As for the uphill pathway, the bc1 complex in Leptospirillum spp. was proposed to utilize the proton motive force (PMF) to push electrons uphill against the thermodynamically unfavorable gradient (Bonnefoy and Holmes, 2012). Herein, the outflux of protons catalyzed by the cbb3-type terminal oxidase contributes to the generation of the PMF during electron transfer to oxygen via a downhill pathway. However, genes encoding the putative bc1 complex were absent in S. thermosulfidooxidans. It seems that this species may not require reverse electron transfer to generate reduction equivalents (e.g., NADH and NADPH), but depend on the oxidation of organic compounds, such as TCA cycle, oxidative pentose phosphate pathway, to produce these compounds, since it was reported to be mixotroph capable of the assimilation of both inorganic and organic carbon compounds. In addition, A. caldus and S. thermosulfidooxidans analyzed here were replete with metabolic enzymes related to sulfur oxidation (Table S3), and they have the ability to oxidize minerals, which produce sulfuric acid (Figure 4). Their activities facilitate the dissolution of base metals in certain acid-soluble sulfide minerals, such as chalcocite, via an oxidative route (Johnson and Hallberg, 2003). Oddly, gene encoding putative thiosulfate:quinone oxidoreductase (TQO) was absent in both A. caldus strains, while the corresponding gene was identified in the homologous strains such as SM-1 and ATCC 51756 (Zhang et al., 2016b). In addition, gene encoding sulfate adenylyltransferase (SAT) was not found in S. thermosulfidooxidans genomes analyzed in this study, however, the homologous strain ST was reported to harbor the sat gene (Guo et al., 2014; Zhang et al., 2017). In A. caldus (Chen et al., 2012), electrons from TQO, sulfide quinone reductase, sulfur oxidizing protein, and heterodisulfide reductase were mediated by the quinol pool, and then (i) either were transferred to oxygen via bd-type or bo3-type terminal oxidases, (ii) or were transferred to NADH complex I (Table S3). Similarly, genes encoding putative bd-type terminal oxidase and NADH complex I were identified in S. thermosulfidooxidans genomes.
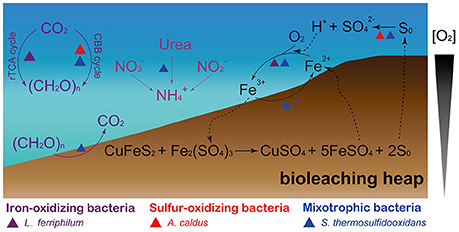
Figure 4. Schematic representation showing the microbe-mediated biogeochemical cycle of main elements (carbon, nitrogen, iron, and sulfur) in bioleaching heaps. The figure was adapted from Johnson (1998), Baker and Banfield (2003), and Chen et al. (2016).
Discussion
General Comparisons of the Gene Repertoire among Bacterial Genomes
The overview of bacterial genomes is shown in Table 2. Compared to the others, the GC contents of the A. caldus genomes were much higher; a plausible explanation for this finding is that the optimal growth temperature (Topt) is regarded as one of the environmental factors that positively influences genomic GC content in prokaryotes (Musto et al., 2004, 2006) given that A. caldus is the primary sulfur oxidizer in bioleaching operations at temperatures above 40°C (Acuña et al., 2013). Nevertheless, this inference was widely divergent from our recollection since the moderate thermophile S. thermosulfidooxidans harbors relatively low GC contents. In fact, the GC contents of the bacterial genomes varied dramatically, and they were influenced by multiple factors (Hildebrand et al., 2010), such as genome size (Bentley and Parkhill, 2004), environment (Foerstner et al., 2005), nitrogen utilization (Mcewan et al., 1998), and aerobiosis (Naya et al., 2002). However, more evidence should be provided to further determine whether the relatedness between the genomic GC contents and multifactorial mechanisms is a stochastic process or a result of natural selection.
An analysis of the gene contents showed that all of the genomes were predicted to harbor a large number of CDS that were potentially assigned to the COG categories [C], [E], and [M]. This finding was similar to our previous study (Zhang et al., 2016a). The COG categories [C] and [E] are involved in the metabolic pathways of energy and amino acids, and COG category [M] is related to the synthesis of cell membranes, membrane channel proteins, and extracellular polymeric substances. Thus, we infer that the bacterial lifestyle requires efficient utilization of energy and nutrients from external environments and specialized cellular structures to adapt to changing environments. In addition, the abundant CDS in these bacterial genomes were assigned to COG category [L] (replication, recombination, and repair), which contains many enzymes associated with DNA repair. The habitats that these acidophilic microorganisms inhabit are characterized by extreme acidity and an abundance of heavy metals. When cell damage occurs, the corresponding proteins may play an important role in DNA repair (Mi et al., 2011; Chen et al., 2013). Of note, many CDS assigned to COG category [E] were only identified in S. thermosulfidooxidans genomes. We therefore hypothesize that S. thermosulfidooxidans strains exhibited additional abilities to synthesize amino acids, suggesting that they depend more heavily on organic compound assimilation than the others.
Inter- and Intra-Species Genome Comparison
A comparative survey of three species (S. thermosulfidooxidans, L. ferriphilum, and A. caldus) that are commonly recognized in many acidic and sulfur-containing environments was performed. Each species was distinctively different from the others, which strongly suggests the genomic differences among these isolates. It is important to note that the number of CDS associated with carbohydrate transport and metabolism in S. thermosulfidooxidans was relatively large in comparison with its counterparts. Hence, we interpreted this as an indication that the S. thermosulfidooxidans strains were mixotrophic acidophiles that are capable of assimilating of inorganic and organic carbon. Collectively, the findings presented herein imply that genomic and functional differentiation of these acidophilic species in the same community might allow them to co-exist.
Acidophilic bacteria analyzed in this study were isolated from different tailings samples located at Dexing Copper Mine and Zijinshan Copper Mine. Since mobile genetic elements, such as transposases and integrases, were recognized as the signatures of potential horizontal gene transfer (HGT) events (Waack et al., 2006; Juhas et al., 2009; Acuña et al., 2013), the identification of mobile genetic elements in genomic neighborhoods of L. ferriphilum strains implied that the bacterial genomes were likely to undergo the HGT event, resulting in the coincidental acquisition of novel functionalities that might be advantageous under certain conditions (Gogarten et al., 2002). The finding was similar to our earlier studies concerning the intra-species divergence of A. caldus (Zhang et al., 2016b) and S. thermosulfidooxidans (Zhang et al., 2017) at the genome level. Thus, in this study we underscore the important role of HGT in shaping the genetic content of bacterial genomes and contributing to intra-species differentiation.
Differences in Metabolic Profiles of Acidophilic Microorganisms
Our genomic analysis focused on the differences of key metabolic pathways, including carbon, nitrogen, iron, and sulfur metabolism (Figure 4). The species with different metabolic capacities may efficiently utilize nutrients or other resources in the same community, likely allowing them to co-exist in bioleaching heaps and thus, avoid competitive exclusion.
Central Carbon Metabolism
An earlier study revealed that Acidithiobacillus spp. and/or Leptospirillum spp. were observed to be the major carbon fixers in acid mine drainage (AMD) communities (Chen et al., 2015). Previous studies showed detailed evidence supporting the fixation of carbon dioxide via the classical CBB cycle in A. caldus (You et al., 2011; Zhang et al., 2016b) and S. thermosulfidooxidans (Guo et al., 2014; Justice et al., 2014; Zhang et al., 2017). Apart from autotrophic growth, S. thermosulfidooxidans isolates have been reported to grow heterotrophically via the catabolism of various organic carbon compounds (Guo et al., 2014; Justice et al., 2014). Unlike S. thermosulfidooxidans, A. caldus was predicted to elevate the concentration of carbon dioxide near RuBisCO by a carboxysome-associated carbonic anhydrase, which was responsible for the conversion of accumulated cytosolic bicarbonate into CO2 (Zhang et al., 2016e). Thus, carboxysome within A. caldus strains might be a critical component that contributes to their ability to grow autotrophically through the efficient utilization of scarce carbon availability from acidic environments.
Certain organic compounds, such as cell exudates or lysates that originated from the autotrophic primary producers might be utilized by heterotrophic prokaryotes (Johnson and Hallberg, 2003). Glycolic acid (C00160) was reported to be an exudate originating from autotrophic acidophiles, such as Leptospirillum (Justice et al., 2014). In contrast to Sulfobacillus acidophilus (Justice et al., 2014), no genes encoding putative isocitrate lyase (EC 4.1.3.1) were identified in S. thermosulfidooxidans. Accordingly, we surmise that S. thermosulfidooxidans might not have the ability to assimilate isocitrate to malate via glyoxylate bypass, a pathway closely related to the TCA cycle (Yelton et al., 2013). In addition, a gene cluster involved in glycolate oxidase was predicted in S. thermosulfidooxidans. A putative transposase identified in its downstream region suggested that this cluster might be introduced by HGT, thereby recruiting a novel ability to degrade the organic carbon (Figure S2C). Taken together, we presented a potential route of glycolic acid assimilation that enables S. thermosulfidooxidans to utilize organic acid as an alternative carbon source (Figure S3). In this pathway, glycolate oxidase coupled with malate synthase may degrade glycolic acid to produce malate, and the latter then enters the conventional TCA cycle. Chemolithotrophic acidophiles were sensitive to organic acids in their habitats (Johnson, 1998); therefore, it might be favorable for chemoautotrophs to form a mutualistic relationship with other heterotrophic and/or mixotrophic acidophiles because the latter could effectively detoxify the environments by degrading organic compounds (Johnson and Hallberg, 2003).
In addition, CDS assigned to chitinases (GH18) were found in S. thermosulfidooxidans. The GH18 family contained chitinolytic enzymes involved in chitin degradation, and chitin is known to be a substantial component of fungal cell walls. An intriguing explanation for the abundant chitinolytic enzymes in Trichoderma is that chitinases are regarded as an integral part of the mycoparasitic lifestyle via lysis of the prey's cell walls (Kubicek et al., 2011). In our study, we could not exclude the possibility that putative chitinases only present in S. thermosulfidooxidans might endow them with an additional ability to degrade the lysates of fungi in bioleaching systems. This scenario might be reasonable as fungi were found in several acidic environments, such as AMD ecosystems (Mosier et al., 2016), although their functional roles in bioleaching heaps remain elusive (Cárdenas et al., 2016).
Nitrogen Uptake
Like other microorganisms in AMD that are characterized by scarce nitrogen availability (Parro et al., 2007; Chen et al., 2015), the habitants in the bioleaching systems may share nitrogen assimilation strategies to cope with the nitrogen-limited conditions. In general, microorganisms commonly utilize atmospheric nitrogen (N2), ammonium, nitrite, and nitrate as their inorganic nitrogen sources (Arrigo, 2005). As a member of the Acidithiobacillus genus, A. ferrooxidans has been predicted to harbor nitrogenase, which is responsible for N2 fixation (Levicán et al., 2008; Zhang et al., 2016e). Likewise, nitrogen fixation was also reported in the Leptospirillum groups, except for Group II (Goltsman et al., 2013). Under the low nitrogen level, the diazotrophic lifestyle of these bacteria in the microbial communities might be a striking feature to maintain the concentration of nitrogen availability in the habitat environments.
Although the three species analyzed in this study do not have the ability to fix atmospheric nitrogen, they were predicted to utilize environmental ammonium, nitrite, and even nitrate as an alternative nitrogen source. L. ferriphilum was predicted to merely harbor the ability of ammonium uptake, but the other two species could utilize other forms of nitrogen, such as nitrate or nitrite as the alternative nitrogen source, thereby, to some extent, avoiding competitive exclusion. Notably, microbial ureases have been reported to hydrolyze environmental urea to produce ammonia and bicarbonate (Mobley and Hausinger, 1989; Ullrich et al., 2016a,b), and ammonia at circumneutral cytoplasmic pH is present as ammonium (Ullrich et al., 2016b). Thus, the S. thermosulfidooxidans strains appear to be able to incorporate cytoplasmic ammonium derived from the hydrolysis of urea as a nitrogen source (Figure 4), and utilize the released bicarbonate as an alternative carbon source via carbonic anhydrase.
Iron-Sulfur Cycling
The models for potential iron-sulfur cycling were well-studied in acidic and high-sulfur environments (Johnson, 1998; Baker and Banfield, 2003; Méndez-García et al., 2015; Chen et al., 2016; Zhang et al., 2016e). In these settings, ferrous iron oxidation implemented by iron-oxidizing bacteria, such as L. ferriphilum and S. thermosulfidooxidans, is the primary biochemical transformation of iron. In addition, the oxidation of sulfide minerals, elemental sulfur, and tetrathionate has been well-documented in A. caldus and S. thermosulfidooxidans (Mangold et al., 2011; Chen et al., 2012; Guo et al., 2014; Zhang et al., 2016b). Fe(III) under anoxic conditions could accept electrons originating from inorganic (e.g., sulfur and/or hydrogen) and/or organic donors (e.g., glucose and/or glycerol), which occurs in the case of chemolithotrophic and heterotrophic acidophiles, respectively (Méndez-García et al., 2015). When exposed to moisture and air, sulfur-containing mineral ores oxidize spontaneously in situations where molecular oxygen or Fe(III) act as an oxidant (Johnson and Hallberg, 2003). The formation of acidic environments in turn provides advantages to stabilizing Fe(II), even in the presence of atmospheric oxygen (Bonnefoy and Holmes, 2012); this is an opportunity for iron-oxidizing microorganisms inhabiting these settings to utilize Fe(II) as a source of energy.
Acidithiobacillus caldus and Sulfobacillus thermosulfidooxidans strains used in this study were predicted to lack TQO and SAT encoding genes, respectively. One plausible explanation for this issue was that missing genes might be artifacts of assembly or gene calling procedure. On the other hand, we could not exclude the possibility that gene tqo in homologous strains of A. caldus (SM-1 and ATCC 51756) and sat in homologous strains of S. thermosulfidooxidans (ST) might be introduced by HGT. With respect to ferric iron [Fe(III)] reduction, previous studies have shown that Sulfobacillus spp. harbored the ability to reduce ferric iron (Melamud et al., 2003; Bogdanova et al., 2006; Johnson et al., 2008). Although Fe(III) reduction activities under oxygen-limiting conditions were observed in acidophilic microorganisms, the determination of a precise enzymatic system is still an important goal for future studies.
Comparative genomics of three typical co-occurring acidophiles in bioleaching heaps yields valuable insights into the differences in gene content, metabolic profiles, and potential functional roles. In short, it could present a scenario whereby certain essential functions are partitioned in co-occurring members of microbial communities, and individual members harboring different metabolic functions generate not merely commensalistic but mutualistic interactions. Thus, it might provide a selective advantage for the efficient utilization of limited resources in their habitats in which they benefit from each other to maintain their respective lifestyle.
Author Contributions
XZ, XL, YL, and HY conceived and designed the experiments. XZ, YX, LM, XG, BM, and HL performed the experiments. XZ analyzed the data. XZ wrote the manuscript. XL, LY, DP, WH, and HY revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31570113 and 41573072) and the Fundamental Research Funds for the Central Universities of Central South University (2016zzts102). In addition, we thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00790/full#supplementary-material
Figure S1. Heat map depicting the percentages of genes assigned to the COG classification in A. caldus DX (I), A. caldus ZJ (II), L. ferriphilum DX (III), L. ferriphilum ZJ (IV), S. thermosulfidooxidans DX (V), and S. thermosulfidooxidans ZJ (VI). The five abundant functional categories within all of the bacterial genomes are highlighted in blue.
Figure S2. Homologous genome regions in individual strains associated with pyruvate ferredoxin oxidoreductase and 2-oxoglutarate ferredoxin oxidoreductase (A), carboxysome (B), and glycolate oxidase (C). porG, pyruvate ferredoxin oxidoreductase gamma subunit; porB, pyruvate ferredoxin oxidoreductase beta subunit; porA, pyruvate ferredoxin oxidoreductase alpha subunit; hp, hypothetical protein; forG, 2-oxoglutarate ferredoxin oxidoreductase gamma subunit; forB, 2-oxoglutarate ferredoxin oxidoreductase beta subunit; forA, 2-oxoglutarate ferredoxin oxidoreductase alpha subunit; cbbL, Rubisco large chain; cbbS, Rubisco small chain; CsoS2, carboxysome shell protein CsoS2; CsoSCA, carboxysome-associated carbonic anhydrase; CsoS4, carboxysome shell protein CsoS4; Tnp, transposase; pucR, PucR family transcriptional regulator; IclR, IclR family transcriptional regulator; glcF, glycolate oxidase iron-sulfur subunit; gcl, glyoxylate carboligase; hyi, hydroxypyruvate isomerase; glxR, 2-hydroxy-3-oxopropionate reductase; glcE, glycolate oxidase FAD binding subunit; glcD, glycolate oxidase subunit GlcD.
Figure S3. Putative route for glycolic acid assimilation within S. thermosulfidooxidans strains.
Table S1. Details for genomic regions of interest in Leptospirillum ferriphilum strains.
Table S2. Comparisons of the inferred metabolic profiles of bacterial genomes.
Table S3. Detailed information for putative genes associated with key metabolic pathways of co-occurring bacteria in bioleaching heaps.
Table S4. Comparisons of predicted CAZymes in the genomes of L. ferriphilum, A. caldus, and S. thermosulfidooxidans.
References
Acuña, L. G., Cárdenas, J. P., Covarrubias, P. C., Haristoy, J. J., Flores, R., Nuñez, H., et al. (2013). Architecture and gene repertoire of the flexible genome of the extreme acidophile Acidithiobacillus caldus. PLoS ONE 8:e78237. doi: 10.1371/journal.pone.0078237
Arrigo, K. R. (2005). Marine microorganisms and global nutrient cycles. Nature 437, 349–355. doi: 10.1038/nature04159
Baker, B. J., and Banfield, J. F. (2003). Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 44, 139–152. doi: 10.1016/S0168-6496(03)00028-X
Bentley, S. D., and Parkhill, J. (2004). Comparative genomic structure of prokaryotes. Annu. Rev. Genet. 38, 771–791. doi: 10.1146/annurev.genet.38.072902.094318
Berg, I. A., Kockelkorn, D., Ramos-Vera, W. H., Say, R. F., Zarzycki, J., Hügler, M., et al. (2010). Autotrophic carbon fixation in archaea. Nat. Rev. Microbiol. 8, 447–460. doi: 10.1038/nrmicro2365
Bogdanova, T. I., Tsaplina, I. A., Kondrat'eva, T. F., Duda, V. I., Suzina, N. E., Melamud, V. S., et al. (2006). Sulfobacillus thermotolerans sp. nov., a thermotolerant, chemolithotrophic bacterium. Int. J. Syst. Evol. Microbiol. 56, 1039–1042. doi: 10.1099/ijs.0.64106-0
Bonnefoy, V., and Holmes, D. S. (2012). Genomic insights into microbial iron oxidation and iron uptake strategies in extremely acidic environments. Environ. Microbiol. 14, 1597–1611. doi: 10.1111/j.1462-2920.2011.02626.x
Breuker, A., Blazejak, A., Bosecker, K., and Schippers, A. (2009). Diversity of iron oxidizing bacteria from various sulfidic mine waste dumps. Adv. Mater. Res. 71–73, 47–50. doi: 10.4028/www.scientific.net/AMR.71-73.47
Cárdenas, J. P., Quatrini, R., and Holmes, D. S. (2016). Genomic and metagenomic challenges and opportunities for bioleaching: a mini-review. Res. Microbiol. 167, 529–538. doi: 10.1016/j.resmic.2016.06.007
Carretero-Paulet, L., Librado, P., Chang, T., Ibarra-Laclette, E., Herrera-Estrella, L., Rozas, J., et al. (2015). High gene family turnover rates and gene space adaptation in the compact genome of the carnivorous plant Utricularia gibba. Mol. Biol. Evol. 32, 1284–1295. doi: 10.1093/molbev/msv020
Chen, L. X., Huang, L. N., Méndez-García, C., Kuang, J., Hua, Z., Liu, J., et al. (2016). Microbial communities, processes and functions in acid mine drainage ecosystems. Curr. Opin. Biotechnol. 38, 150–158. doi: 10.1016/j.copbio.2016.01.013
Chen, L., Ren, Y., Lin, J., Liu, X., Pang, X., and Lin, J. (2012). Acidithiobacillus caldus sulfur oxidation model based on transcriptome analysis between the wild type and sulfur oxygenase reductase defective mutant. PLoS ONE 7:e39470. doi: 10.1371/journal.pone.0039470
Chen, L. X., Hu, M., Huang, L. N., Hua, Z. S., Kuang, J. L., Li, S. J., et al. (2015). Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J. 9, 1579–1592. doi: 10.1038/ismej.2014.245
Chen, L. X., Li, J. T., Chen, Y. T., Huang, L. N., Hua, Z. S., Hu, M., et al. (2013). Shifts in microbial community composition and function in the acidification of a lead/zinc mine tailings. Environ. Microbiol. 15, 2431–2444. doi: 10.1111/1462-2920.12114
Coram, N. J., and Rawlings, D. E. (2002). Molecular relationship between two groups of the genus Leptospirillum and the finding that Leptospirillum ferriphilum sp. nov. dominates South African commercial biooxidation tanks that operate at 40°C. Appl. Environ. Microbiol. 68, 838–845. doi: 10.1128/AEM.68.2.838-845.2002
Craig, J. W., Cherry, M. A., and Brady, S. F. (2011). Long-chain N-acyl amino acid synthases are linked to the putative PEP-CTERM/exosortase protein-sorting system in gram-negative bacteria. J. Bacteriol. 193, 5707–5715. doi: 10.1128/JB.05426-11
Denef, V. J., Mueller, R. S., and Banfield, J. F. (2010). AMD biofilms: using model communities to study microbial evolution and ecological complexity in nature. ISME J. 4, 599–610. doi: 10.1038/ismej.2009.158
Deng, W., Wang, Y., Liu, Z., Cheng, H., and Xue, Y. (2014). HemI: a toolkit for illustrating heatmaps. PLoS ONE 9:e111988. doi: 10.1371/journal.pone.0111988
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton, A. P., et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10:R85. doi: 10.1186/gb-2009-10-8-r85
Foerstner, K. U., von Mering, C., Hooper, S. D., and Bork, P. (2005). Environments shape the nucleotide composition of genomes. EMBO Rep. 6, 1208–1213. doi: 10.1038/sj.embor.7400538
Fouts, D. E., Brinkac, L., Beck, E., Inman, J., and Sutton, G. (2012). PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res. 40:e172. doi: 10.1093/nar/gks757
Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., Roth, A., et al. (2013). STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. doi: 10.1093/nar/gks1094
Fujimura, R., Sato, Y., Nishizawa, T., Oshima, K., Kim, S., Hattori, M., et al. (2012). Complete genome sequence of Leptospirillum ferrooxidans strain C2-3, isolated from a fresh volcanic ash deposit on the island of Miyake, Japan. J. Bacteriol. 194, 4122–4123. doi: 10.1128/JB.00696-12
Gogarten, J. P., Doolittle, W. F., and Lawrence, J. G. (2002). Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 19, 2226–2238. doi: 10.1093/oxfordjournals.molbev.a004046
Goltsman, D. S. A., Comolli, L. R., Thomas, B. C., and Banfield, J. F. (2015). Community transcriptomics reveals unexpected high microbial diversity in acidophilic biofilm communities. ISME J. 9, 1014–1023. doi: 10.1038/ismej.2014.200
Goltsman, D. S., Dasari, M., Thomas, B. C., Shah, M. B., VerBerkmoes, N. C., Hettich, R. L., et al. (2013). New group in the Leptospirillum clade: cultivation-independent community genomics, proteomics, and transcriptomics of the new species “Leptospirillum group IV UBA BS”. Appl. Environ. Microbiol. 79, 5384–5393. doi: 10.1128/AEM.00202-13
González, A., Bellenberg, S., Mamani, S., Ruiz, L., Echeverría, A., Soulère, L., et al. (2013). AHL signaling molecules with a large acyl chain enhance biofilm formation on sulfur and metal sulfides by the bioleaching bacterium Acidithiobacillus ferrooxidans. Appl. Microbiol. Biotechnol. 97, 3729–3737. doi: 10.1007/s00253-012-4229-3
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Guo, X., Yin, H., Liang, Y., Hu, Q., Zhou, X., Xiao, Y., et al. (2014). Comparative genome analysis reveals metabolic versatility and environmental adaptations of Sulfobacillus thermosulfidooxidans strain ST. PLoS ONE 9:e99417. doi: 10.1371/journal.pone.0099417
Gupta, R., and Schuster, M. (2013). Negative regulation of bacterial quorum sensing tunes public goods cooperation. ISME J. 7, 2159–2168. doi: 10.1038/ismej.2013.109
Haft, D. H., Paulsen, I. T., Ward, N., and Selengut, J. D. (2006). Exopolysaccharide-associated protein sorting in environmental organisms: the PEP-CTERM/EpsH system. Application of a novel phylogenetic profiling heuristic. BMC Biol. 4:29. doi: 10.1186/1741-7007-4-29
Hallberg, K. B., and Johnson, D. B. (2001). Biodiversity of acidophilic prokaryotes. Adv. Appl. Microbiol. 49, 37–84. doi: 10.1016/S0065-2164(01)49009-5
Harris, J. (2009). Soil microbial communities and restoration ecology: facilitators or followers? Science 325, 573–574. doi: 10.1126/science.1172975
Hildebrand, F., Meyer, A., and Eyre-Walker, A. (2010). Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 6:e1001107. doi: 10.1371/journal.pgen.1001107
Hua, Z. S., Han, Y. J., Chen, L. X., Liu, J., Hu, M., Li, S. J., et al. (2015). Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 9, 1280–1294. doi: 10.1038/ismej.2014.212
Jiang, H., Liang, Y., Yin, H., Xiao, Y., Guo, X., Xu, Y., et al. (2015). Effects of arsenite resistance on the growth and functional gene expression of Leptospirillum ferriphilum and Acidithiobacillus thiooxidans in pure culture and coculture. Biomed Res. Int. 2015, 1–13. doi: 10.1155/2015/792904
Jiao, N., Herndl, G. J., Hansell, D. A., Benner, R., Kattner, G., Wilhelm, S. W., et al. (2010). Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 8, 593–599. doi: 10.1038/nrmicro2386
Johnson, D. B. (1998). Biodiversity and ecology of acidophilic microorganisms. FEMS Microbiol. Ecol. 27, 307–317. doi: 10.1111/j.1574-6941.1998.tb00547.x
Johnson, D. B., and Hallberg, K. B. (2003). The microbiology of acidic mine waters. Res. Microbiol. 154, 466–473. doi: 10.1016/S0923-2508(03)00114-1
Johnson, D. B., and Hallberg, K. B. (2008). Carbon, iron and sulfur metabolism in acidophilic micro-organisms. Adv. Microb. Physiol. 54, 201–255. doi: 10.1016/S0065-2911(08)00003-9
Johnson, D. B., Joulian, C. D., Hugues, P., and Hallberg, K. B. (2008). Sulfobacillus benefaciens sp. nov., an acidophilic facultative anaerobic Firmicute isolated from mineral bioleaching operations. Extremophiles 12, 789–798. doi: 10.1007/s00792-008-0184-4
Juhas, M., van der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Justice, N. B., Norman, A., Brown, C. T., Singh, A., Thomas, B. C., and Banfield, J. F. (2014). Comparison of environmental and isolate Sulfobacillus genomes reveals diverse carbon, sulfur, nitrogen, and hydrogen metabolisms. BMC Genomics 15:1107. doi: 10.1186/1471-2164-15-1107
Kock, D., and Schippers, A. (2006). Geomicrobiological investigation of two different mine waste tailings generating acid mine drainage. Hydrometallurgy 83, 167–175. doi: 10.1016/j.hydromet.2006.03.022
Kock, D., and Schippers, A. (2008). Quantitative microbial community analysis of three different sulfidic mine tailing dumps generating acid mine drainage. Appl. Environ. Microbiol. 74, 5211–5219. doi: 10.1128/AEM.00649-08
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kubicek, C. P., Herrera-Estrella, A., Seidl-Seiboth, V., Martinez, D. A., Druzhinina, I. S., Thon, M., et al. (2011). Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 12:R40. doi: 10.1186/gb-2011-12-4-r40
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Levicán, G., Ugalde, J. A., Ehrenfeld, N., Maass, A., and Parada, P. (2008). Comparative genomic analysis of carbon and nitrogen assimilation mechanisms in three indigenous bioleaching bacteria: predictions and validations. BMC Genomics 9:581. doi: 10.1186/1471-2164-9-581
Li, J., and Gu, J. (2007). Complete degradation of dimethyl isophthalate requires the biochemical cooperation between Klebsiella oxytoca Sc and Methylobacterium mesophilicum Sr Isolated from Wetland sediment. Sci. Total Environ. 380, 181–187. doi: 10.1016/j.scitotenv.2006.12.033
Liang, J., Bai, Y., Men, Y., and Qu, J. (2017). Microbe–microbe interactions trigger Mn (II)-oxidizing gene expression. ISME J. 11, 67–77. doi: 10.1038/ismej.2016.106
Liljeqvist, M., Valdes, J., Holmes, D. S., and Dopson, M. (2011). Draft genome of the psychrotolerant acidophile Acidithiobacillus ferrivorans SS3. J. Bacteriol. 193, 4304–4305. doi: 10.1128/JB.05373-11
Lin, J. T., and Stewart, V. (1997). Nitrate assimilation by bacteria. Adv. Microb. Physiol. 39, 1–30. doi: 10.1016/S0065-2911(08)60014-4
Lombard, V., Ramulu, H. G., Drula, E., Coutinho, P. M., and Henrissat, B. (2013). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
Mangold, S., Valdés, J., Holmes, D. S., and Dopson, M. (2011). Sulfur metabolism in the extreme acidophile Acidithiobacillus caldus. Front. Microbiol. 2:17. doi: 10.3389/fmicb.2011.00017
Mason, O. U., Hazen, T. C., Borglin, S., Chain, P. S. G., Dubinsky, E. A., Fortney, J. L., et al. (2012). Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 6, 1715–1727. doi: 10.1038/ismej.2012.59
Mcewan, C. E., Gatherer, D., and Mcewan, N. R. (1998). Nitrogen-fixing aerobic bacteria have higher genomic GC content than non-fixing species within the same genus. Hereditas 128, 173–178. doi: 10.1111/j.1601-5223.1998.00173.x
Melamud, V. S., Pivovarova, T. A., Tourova, T. P., Kolganova, T. V., Osipov, G. A., Lysenko, A. M., et al. (2003). Sulfobacillus sibiricus sp. nov., a new moderately thermophilic bacterium. Microbiology 72, 605–612. doi: 10.1023/A:1026007620113
Méndez-García, C., Peláez, A. I., Mesa, V., Sánchez, J., Golyshina, O. V., and Ferrer, M. (2015). Microbial diversity and metabolic networks in acid mine drainage habitats. Front. Microbiol. 6:475. doi: 10.3389/fmicb.2015.00475
Mi, S., Song, J., Lin, J., Che, Y., Zheng, H., and Lin, J. (2011). Complete genome of Leptospirillum ferriphilum ML-04 provides insight into its physiology and environmental adaptation. J. Microbiol. 49, 890–901. doi: 10.1007/s12275-011-1099-9
Mobley, H. L., and Hausinger, R. P. (1989). Microbial ureases: significance, regulation, and molecular characterization. Microbiol. Mol. Biol. Rev. 53, 85–108.
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Mosier, A. C., Miller, C. S., Frischkorn, K. R., Ohm, R. A., Li, Z., LaButti, K., et al. (2016). Fungi contribute critical but spatially varying roles in nitrogen and carbon cycling in acid mine drainage. Front. Microbiol. 7:238. doi: 10.3389/fmicb.2016.00238
Musto, H., Naya, H., Zavala, A., Romero, H., Alvarez-Valín, F., and Bernardi, G. (2006). Genomic GC level, optimal growth temperature, and genome size in prokaryotes. Biochem. Biophys. Res. Commun. 347, 1–3. doi: 10.1016/j.bbrc.2006.06.054
Musto, H., Naya, H., Zavala, A., Romero, H., Alvarez-Valín, F., and Bernardi, G. (2004). Correlations between genomic GC levels and optimal growth temperatures in prokaryotes. FEBS Lett. 573, 73–77. doi: 10.1016/j.febslet.2004.07.056
Naya, H., Romero, H., Zavala, A., Alvarez, B., and Musto, H. (2002). Aerobiosis increases the genomic guanine plus cytosine content (GC%) in prokaryotes. J. Mol. Evol. 55, 260–264. doi: 10.1007/s00239-002-2323-3
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parro, V., Moreno-Paz, M., and González-Toril, E. (2007). Analysis of environmental transcriptomes by DNA microarrays. Environ. Microbiol. 9, 453–464. doi: 10.1111/j.1462-2920.2006.01162.x
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. doi: 10.1371/journal.pone.0030619
Prosser, J. I., Bohannan, B. J. M., Curtis, T. P., Ellis, R. J., Firestone, M. K., Freckleton, R. P., et al. (2007). The role of ecological theory in microbial ecology. Nat. Rev. Microbiol. 5, 384–392. doi: 10.1038/nrmicro1643
Rawlings, D. E. (2005). Characteristics and adaptability of iron-and sulfur-oxidizing microorganisms used for the recovery of metals from minerals and their concentrates. Microb. Cell Fact. 4:13. doi: 10.1186/1475-2859-4-13
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rohwerder, T., and Sand, W. (2007). Oxidation of inorganic sulfur compounds in acidophilic prokaryotes. Eng. Life Sci. 7, 301–309. doi: 10.1002/elsc.200720204
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Summers, Z. M., Fogarty, H. E., Leang, C., Franks, A. E., Malvankar, N. S., and Lovley, D. R. (2010). Direct exchange of electrons within aggregates of an evolved syntrophic coculture of anaerobic bacteria. Science 330, 1413–1415. doi: 10.1126/science.1196526
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Teeling, H., Meyerdierks, A., Bauer, M., Amann, R., and Glöckner, F. O. (2004). Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 6, 938–947. doi: 10.1111/j.1462-2920.2004.00624.x
Tyson, G. W., Chapman, J., Hugenholtz, P., Allen, E. E., Ram, R. J., Richardson, P. M., et al. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428, 37–43. doi: 10.1038/nature02340
Ullrich, S. R., González, C., Poehlein, A., Tischler, J. S., Daniel, R., Schlömann, M., et al. (2016a). Gene loss and horizontal gene transfer contributed to the genome evolution of the extreme acidophile “Ferrovum”. Front. Microbiol. 7:797. doi: 10.3389/fmicb.2016.00797
Ullrich, S. R., Poehlein, A., Tischler, J. S., González, C., Ossandon, F. J., Daniel, R., et al. (2016b). Genome analysis of the biotechnologically relevant acidophilic iron oxidising strain JA12 indicates phylogenetic and metabolic diversity within the novel genus “Ferrovum”. PLoS ONE 11:e0146832. doi: 10.1371/journal.pone.0146832
Valdés, J., Pedroso, I., Quatrini, R., Dodson, R. J., Tettelin, H., Blake, R., et al. (2008). Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications. BMC Genomics 9:597. doi: 10.1186/1471-2164-9-597
Valdes, J., Quatrini, R., Hallberg, K., Dopson, M., Valenzuela, P. D. T., and Holmes, D. S. (2009). Draft genome sequence of the extremely acidophilic bacterium Acidithiobacillus caldus ATCC 51756 reveals metabolic versatility in the genus Acidithiobacillus. J. Bacteriol. 191, 5877–5878. doi: 10.1128/JB.00843-09
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142
Watling, H. R. (2006). The bioleaching of sulphide minerals with emphasis on copper sulphides - a review. Hydrometallurgy 84, 81–108. doi: 10.1016/j.hydromet.2006.05.001
Williams, K. P., and Kelly, D. P. (2013). Proposal for a new class within the phylum Proteobacteria, Acidithiobacillia classis nov., with the type order Acidithiobacillales, and emended description of the class Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 63, 2901–2906. doi: 10.1099/ijs.0.049270-0
Wintermute, E. H., and Silver, P. A. (2010). Emergent cooperation in microbial metabolism. Mol. Syst. Biol. 6:407. doi: 10.1038/msb.2010.66
Wood, A. P., Aurikko, J. P., and Kelly, D. P. (2004). A challenge for 21st century molecular biology and biochemistry: what are the causes of obligate autotrophy and methanotrophy? FEMS Microbiol. Rev. 28, 335–352. doi: 10.1016/j.femsre.2003.12.001
Wu, X., Holmfeldt, K., Hubalek, V., Lundin, D., Åström, M., Bertilsson, S., et al. (2016). Microbial metagenomes from three aquifers in the Fennoscandian shield terrestrial deep biosphere reveal metabolic partitioning among populations. ISME J. 10, 1192–1203. doi: 10.1038/ismej.2015.185
Xiao, Y., Liu, X., Ma, L., Liang, Y., Niu, J., Gu, Y., et al. (2016). Microbial communities from different subsystems in biological heap leaching system play different roles in iron and sulfur metabolisms. Appl. Microbiol. Biotechnol. 100, 6871–6880. doi: 10.1007/s00253-016-7537-1
Yelton, A. P., Comolli, L. R., Justice, N. B., Castelle, C., Denef, V. J., Thomas, B. C., et al. (2013). Comparative genomics in acid mine drainage biofilm communities reveals metabolic and structural differentiation of co-occurring archaea. BMC Genomics 14:485. doi: 10.1186/1471-2164-14-485
Yin, H., Niu, J., Ren, Y., Cong, J., Zhang, X., Fan, F., et al. (2015). An integrated insight into the response of sedimentary microbial communities to heavy metal contamination. Sci. Rep. 5:14266. doi: 10.1038/srep14266
Yin, H., Qiu, G., Wu, L., Xie, M., Zhou, J., Dai, Z., et al. (2008). Microbial community diversity and changes associated with a mine drainage gradient at the Dexing copper mine, China. Aquat. Microb. Ecol. 51, 67–76. doi: 10.3354/ame01172
Yin, H., Zhang, X., Liang, Y., Xiao, Y., Niu, J., and Liu, X. (2014). Draft genome sequence of the extremophile Acidithiobacillus thiooxidans A01, isolated from the wastewater of a coal dump. Genome Announc. 2:e00222–14. doi: 10.1128/genomeA.00222-14
You, X. Y., Guo, X., Zheng, H. J., Zhang, M. J., Liu, L. J., Zhu, Y. Q., et al. (2011). Unraveling the Acidithiobacillus caldus complete genome and its central metabolisms for carbon assimilation. J. Genet. Genomics 38, 243–252. doi: 10.1016/j.jgg.2011.04.006
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, X., Feng, X., Tao, J., Ma, L., Xiao, Y., Liang, Y., et al. (2016a). Comparative genomics of the extreme acidophile Acidithiobacillus thiooxidans reveals intraspecific divergence and niche adaptation. Int. J. Mol. Sci. 17:1355. doi: 10.3390/ijms17081355
Zhang, X., Liu, X., He, Q., Dong, W., Zhang, X., Fan, F., et al. (2016b). Gene turnover contributes to the evolutionary adaptation of Acidithiobacillus caldus: insights from comparative genomics. Front. Microbiol. 7:1960. doi: 10.3389/fmicb.2016.01960
Zhang, X., Liu, X., Liang, Y., Fan, F., Zhang, X., and Yin, H. (2016c). Metabolic diversity and adaptive mechanisms of iron- and/or sulfur-oxidizing autotrophic acidophiles in extremely acidic environments. Environ. Microbiol. Rep. 8, 738–751. doi: 10.1111/1758-2229.12435
Zhang, X., Liu, X., Liang, Y., Guo, X., Xiao, Y., Ma, L., et al. (2017). Adaptive evolution of extreme acidophile Sulfobacillus thermosulfidooxidans potentially driven by horizontal gene transfer and gene loss. Appl. Environ. Microbiol. 83, e03098–16. doi: 10.1128/aem.03098-16
Zhang, X., Niu, J., Liang, Y., Liu, X., and Yin, H. (2016d). Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet. 17:21. doi: 10.1186/s12863-016-0330-4
Zhang, X., She, S., Dong, W., Niu, J., Xiao, Y., Liang, Y., et al. (2016e). Comparative genomics unravels metabolic differences at species and/or strain level and extremely acidic environmental adaptation of ten bacteria belonging to the genus Acidithiobacillus. Syst. Appl. Microbiol. 39, 493–502. doi: 10.1016/j.syapm.2016.08.007
Keywords: bioleaching heaps, co-occurring bacteria, comparative genomics, functional roles, mutualistic interaction
Citation: Zhang X, Liu X, Liang Y, Xiao Y, Ma L, Guo X, Miao B, Liu H, Peng D, Huang W and Yin H (2017) Comparative Genomics Unravels the Functional Roles of Co-occurring Acidophilic Bacteria in Bioleaching Heaps. Front. Microbiol. 8:790. doi: 10.3389/fmicb.2017.00790
Received: 11 February 2017; Accepted: 18 April 2017;
Published: 05 May 2017.
Edited by:
Axel Schippers, Federal Institute for Geosciences and Natural Resources, GermanyReviewed by:
Li-Nan Huang, Sun Yat-sen University, ChinaSophie R. Ullrich, Technische Universität Bergakademie Freiberg, Germany
Copyright © 2017 Zhang, Liu, Liang, Xiao, Ma, Guo, Miao, Liu, Peng, Huang and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huaqun Yin, eWluaHVhcXVuQGdtYWlsLmNvbQ==